Предлагаемое изобретение имеет отношение к фармацевтической химии и особенно к N2-хинолил- или изохинолил-замещенным производным пурина, способам их получения, фармацевтическим соединениям, включающим указанные производные, и способам использования указанных производных в лечении рака.
Рак остается главной угрозой человеческому здоровью. Большинство раковых образований у людей вызваны факторами окружающей среды, и миллионы людей ежегодно умирают от рака во всем мире. Хотя многие способы лечения, такие как хирургия, лучевая терапия, химиотерапия и т.д., являются доступными, их лечебный эффект в общем достаточно низкий. Поэтому использование фармацевтических препаратов остается одним из самых эффективных способов профилактики и лечения раковых заболеваний.
Производные пурина или пиридина, как известно, обладают антивирусным или противоопухолевым действием. См., например, патентные документы ЕР 173624, ЕР 253412, ЕР 353955, WO 9201698, ЕР 481214 и др.
В природных или синтетических производных нуклеотидов пиридиновая или пуриновая или иная гетероциклическая группа находится в положении 1 сахаридного кольца (соответствующем положению 2 гидроксилзамещенных производных фурана). Эти соединения, как сообщалось, имеют противоопухолевое или антивирусное действие.
Другая группа производных, О6-алкилпроизводные пурина, известна их способностью ингибировать активность О6-алкилгуанин-ДНК алкилтрансферазы (АГТ), повышая таким образом эффективность алкилирующих агентов как препаратов противоопухолевой химиотерапии.
Хотя пока не ясен механизм разрушения раковой клетки О6-метилгуанином в клетках с дефицитом АТазы (аденилилтрансферазы), вообще понимается, что механизм действия О6-хлорэтилгуанина обусловлен присутствием циклоэтилиденгуанинового интермедиата, который образует поперечные сшивки нитей ДНК. Такое сшивание может быть устранено или предотвращено дихлорэтилированием при участии АТазы, например, с помощью АГТ. Способы блокирования О6-алкилгуанин-ДНК алкилтрансферазы (АГТ) в опухолевых клетках описаны, например, в патенте США №5091430 и в международной заявке WO 9113898.
N-Замещенные производные пурина известны. Например, N6-дизамещенные производные пурина, которые могут использоваться для лечения аллергии, описаны, например, в патенте США 4853386. Производные 6-циклопропиламино-9Н-пурина, обладающие антивирусными свойствами, описаны в патентах Японии JP 2003-55377 А и JP 2003-119197 А. Производные гликозилированного пурина, имеющие противовоспалительные свойства, описаны в журнале Org. Chem. (2004, 69:3212-3215). Производные N2-бутилфенил-2'-дезоксипурина, которые обладают свойствами ингибирования эукариотической ДНК-полимеразы α, описаны в журнале Med. Chem. (1984, 27:175-181). 2,6,9-Замещенные производные пурина описаны в журнале Tetrahedron Letters (1998, 39:1827-1830).
Однако ни об одном из вышеупомянутых соединений не было известно, чтобы они имели противоопухолевое действие или обладали способностью ингибировать ненормальный рост клетки. Соответственно, существует потребность в противоопухолевых лекарствах, которые могли бы быть использованы как противоопухолевые средства с низкой токсичностью, широким спектром антиканцерогенного действия и высокой стабильностью.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Предлагаемое изобретение имеет отношение к высокоустойчивым N2-замещенным производным пурина с низкой токсичностью и высокой противоопухолевой активностью.
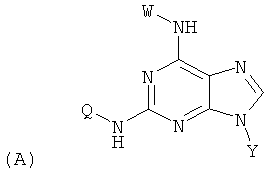
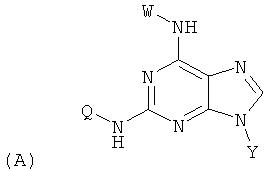
Изобретение относится к соединениям, имеющим следующую формулу (А):
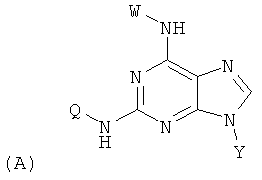
где W представляет собой водород, необязательно замещенный линейный или разветвленный C1-6 алкил, необязательно замещенный С3-6 циклоалкил или необязательно замещенный C1-6 галоалкил;
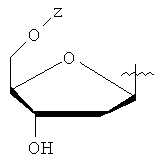
Y представляет собой водород или фармацевтически приемлемый сахарид любой из следующих формул:
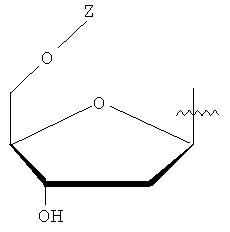 ,
,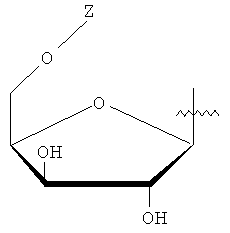 ,
,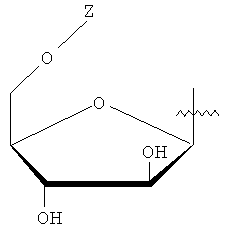 ,
,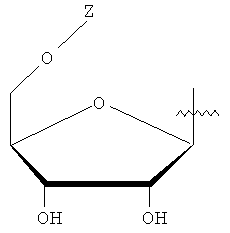 ;
;
Z представляет собой водород или заместитель, имеющий любую из следующих формул:
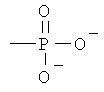 ,
,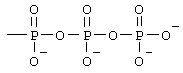 ,
,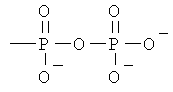 ;
;
Q представляет собой заместитель, имеющий любую из следующих формул:
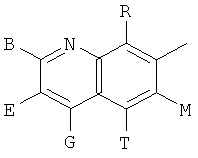 ,
,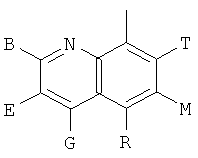 ,
,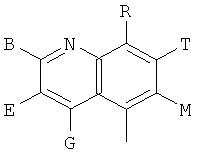 ,
,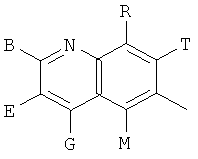 ,
,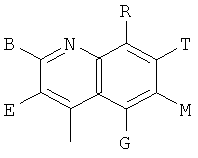 ,
,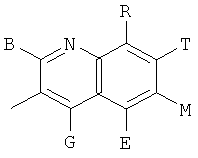 ,
,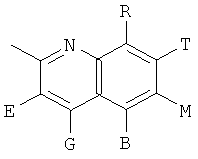 ,
,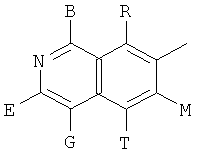 ,
,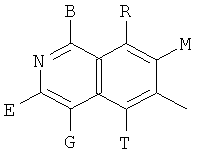 ,
,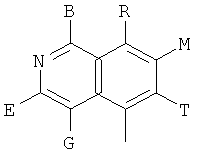 ,
,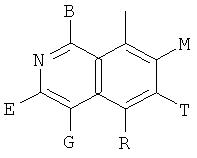 ,
,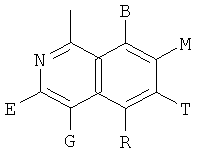 ,
,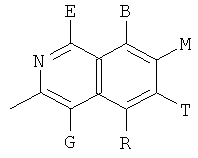 ,
,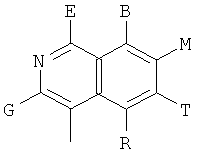
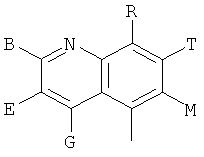
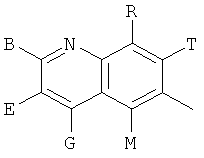
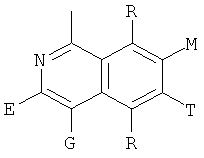
и В, Е, G, R, Т и М каждый независимо представляет собой водород, линейный или разветвленный C1-6алкил или галоалкил, С3-6циклоалкил, галоген, циано или амино.
Предпочтительно в соединении согласно предлагаемому изобретению, имеющем формулу (A), W представляет собой водород или заместитель, имеющий любую из следующих формул:
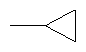 ,
, 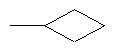 ,
, 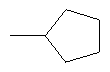 ,
, 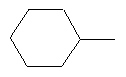 ,
,
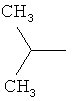 ,
, 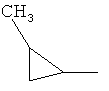 ,
, 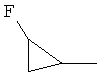 ,
, 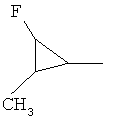 ,
,
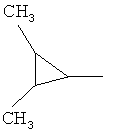 ,
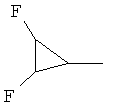
, 
Более предпочтительно W представляет собой одно из:
, , ,
Наиболее предпочтительно W представляет собой:
или
В одном из предпочтительных осуществлений соединения по предлагаемому изобретению, имеющего формулу (A), Q представляет собой:
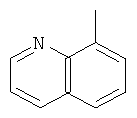 ,
,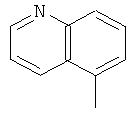 ,
,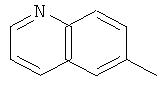 ,
,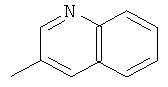 ,
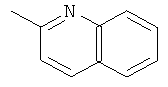
,
Более предпочтительно Q представляет собой следующую группу:
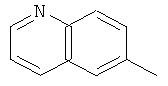 .
.
В одном из предпочтительных осуществлений соединения по предлагаемому изобретению, имеющего формулу (А), заместитель В, Е, G, R, Т или М каждый независимо представляет собой водород, фтор, метил, трифторметил, циано или амино, главным образом водород.
В одном из предпочтительных осуществлений соединения по предлагаемому изобретению, имеющего формулу (A), Y представляет собой водород.
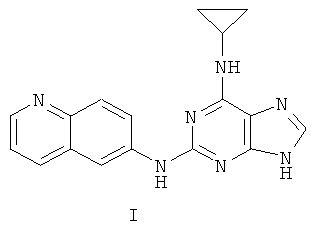
Наиболее предпочтительны следующие соединения:
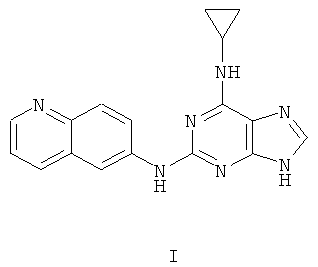
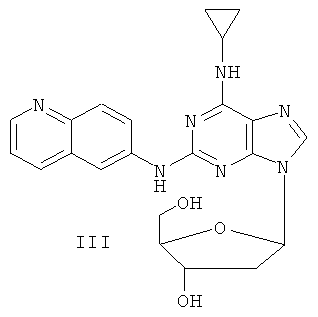
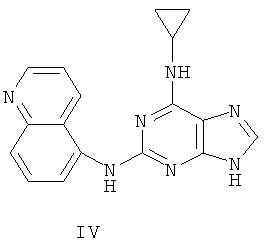
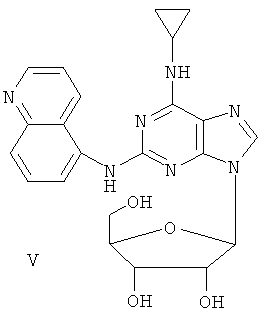
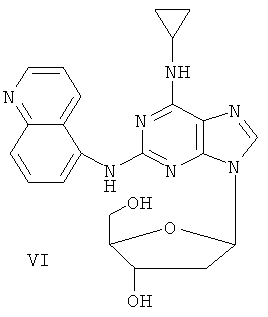
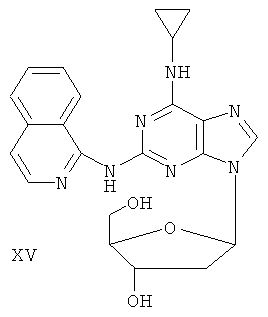
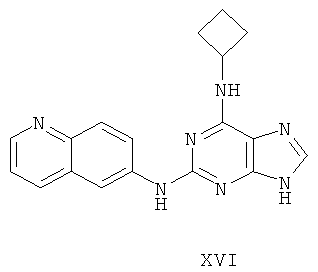
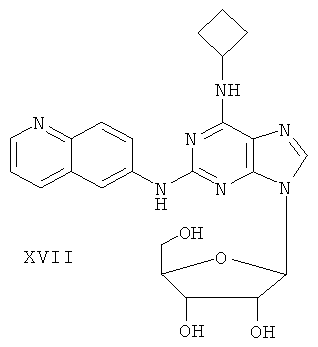
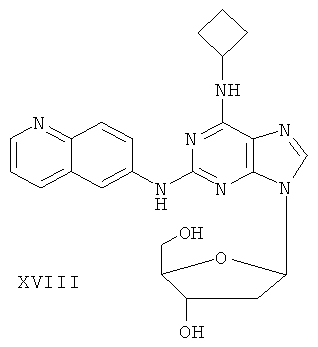
Соединение (I)
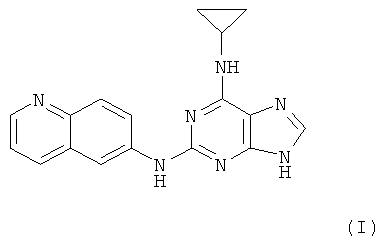
является особенно предпочтительным.
Другие осуществления предлагаемого изобретения представляют собой библиотеку соединений, которая включает любое описанное выше соединение, или его соль, или его гидрат.
Предлагаемое изобретение раскрывает также способ получения вышеописанных соединений. В одном из осуществлений соединения согласно предлагаемому изобретению могут быть получены в соответствии со следующими схемами.
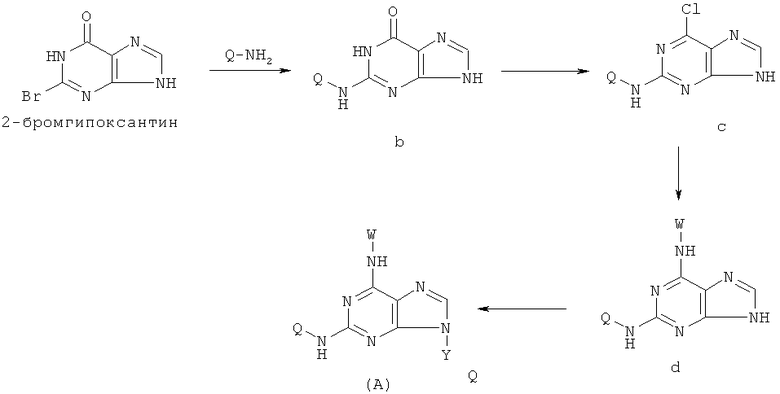
1) Реакция проводится с участием соединения формулы (a) и Q-NH2
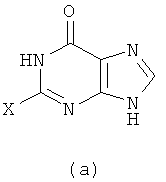
с получением в результате соединения формулы (b)
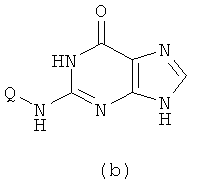
Вышеупомянутую реакцию проводят в органическом растворителе с участием соединения формулы (a) и Q-NH2 при их молярном соотношении 0,8-1,5; смесь нагревают при температуре 50-150°C, и реакция продолжается в течение 1-72 часов. Затем к реакционной смеси добавляют воду и реакционную смесь оставляют охлаждаться при комнатной температуры. Х представляет собой бром.
2) Приготовление соединения формулы (с)
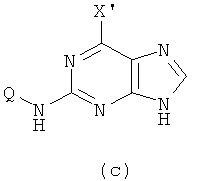
Реакцию проводят в органическом растворителе с участием соединения формулы (b) и галогенирующего агента; смесь нагревают при 50-150°C, и реакция продолжается в течение 1-72 часов, после чего реакционную смесь охлаждают. Затем добавляют воду и с помощью кислоты доводят рН смеси до значения 2-5, после чего смесь оставляют охлаждаться при комнатной температуре. X' представляет собой хлор.
(3) Приготовление соединения формулы (f)
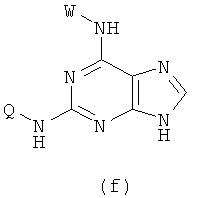
Реакцию проводят в органическом растворителе с участием соединения формулы (с) и W-NH2 при их молярном соотношении 0,8-1,5 в присутствии акцептора кислоты, смесь нагревают при 50-150°С, и реакция продолжается в течение 1-72 часов. Затем растворитель удаляют дистилляцией.
Согласно другому воплощению предлагаемое изобретение также обеспечивает способ получения соответствующей соли вышеупомянутого соединения.
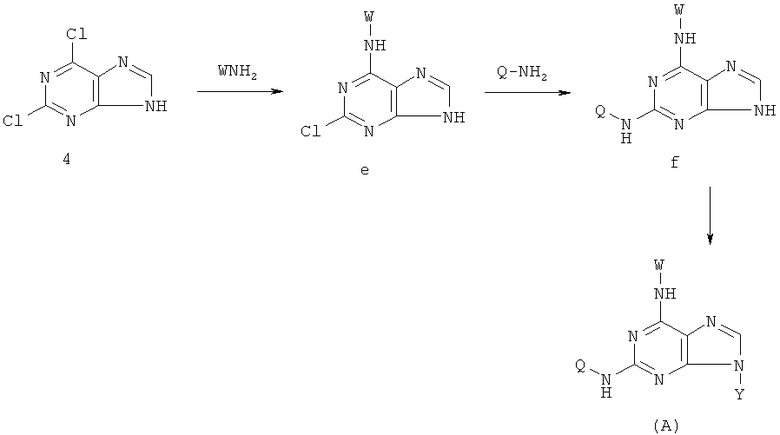
Соединения согласно изобретению могут быть приготовлены в соответствии со следующими схемами.
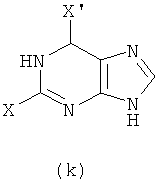
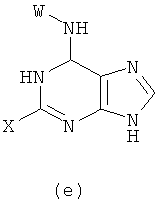
1) Реакцию проводят с участием соединения формулы (k) и W-NH2
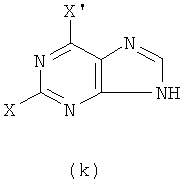
с получением в результате соединения формулы (е)
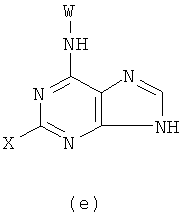
Реакцию проводят в органическом растворителе с участием соединения формулы (k) и W-NH2 при их молярном соотношении 0,8-1,5 в присутствии акцептора кислоты; смесь нагревают при температуре 30-120°С, и реакция продолжается в течение 1-72 часов. Затем растворитель удаляют дистилляцией. Х представляет бром, X' представляет хлор, a W - как указано выше.
2) Получение соединения формулы (f) реакцией соединения формулы (е) с Q-NH2
Реакция проводится в органическом растворителе с участием соединения формулы (е) и W-QH2 при их молярном соотношении 0,8-1,5 в присутствии акцептора кислоты. Смесь нагревают при температуре 70-170°С, и реакция продолжается в течение 1-72 часов. Затем растворитель удаляют дистилляцией. Солью соединения формулы (А) может быть любая фармацевтически приемлемая соль, известная специалистам в данной области. Аддитивные соли соединения могут быть синтезированы с участием органических или неорганических кислот, предпочтительно такие, как гидрохлорид, гидробромид, гидройодид, п-толуолсульфонат, фосфат, сульфат, перхлорид, ацетат, трифторацетат, пропионат, соль лимонной кислоты, малонат, сукцинат, лактат, оксалат, тартрат, бензоат. Соль также может быть образована в результате реакций с участием оснований, как неорганических так и органических, солей щелочноземельных металлов, таких как соли магния, соли кальция, органических аминов, таких как морфолин, пиперидин, диметиламин, диэтиламин и т.д.
В предпочтительном воплощении соединения по предлагаемому изобретению могут быть получены в соответствии со следующими путями А и В.
Путь А
Путь Б
Фармацевтическое соединение по предлагаемому изобретению можно вводить млекопитающим (включая людей) внутренне (например, перорально или ректально), местно или парентерально, или ингаляционным впрыскиванием, например, в ротовую полость, инъекцией, имплантацией или применять наружно. Подходящие формы для перорального введения включают таблетки (такие как обычные таблетки, буккальные таблетки, сублингвальные таблетки, оромукозальные таблетки (таблетки для использования в полости рта), жевательные таблетки, дисперсные таблетки, растворимые таблетки, шипучие таблетки, вагинальные таблетки, вагинальные шипучие таблетки, таблетки пролонгированного действия, таблетки с контролиремым высвобождением, таблетки с энтеросолюбильным покрытием, буккальные таблетки быстрого высвобождения), капсулы (твердые капсулы, мягкие капсулы, капсулы пролонгированного действия, капсулы с контролируемым высвобождением, капсулы с энтеросолюбильным покрытием и т.д.), пилюли (микропилюли, подслащенные пилюли, драже), жидкие лекарственные формы для перорального применения (сиропы, пероральные растворы, суспензии, эмульсии и т.д.), гранулы (гранулы для суспензий, шипучие гранулы, гранулы с энтеросолюбильным покрытием, гранулы пролонгированного действия, гранулы с контролируемым высвобождением и т.д.), порошки; инъекционные лекарственные формы, включая растворы для инъекций, стерильные порошки для инъекций или стерильные твердые частицы (включая препараты, полученные с использованием технологий кристаллизации растворителя, сушки распылением или сублимационной сушки (лиофилизации) и т.д.), концентрированные растворы для инъекций; импланты, а также лекарственные формы для наружного применения, такие как свечи, аэрозоли, аэрозольные порошки, спреи, пленки, гели, пластыри и т.д.
Фармацевтический препарат согласно предлагаемому изобретению может быть приготовлен любым известным в области фармацевтики способом подготовки лекарственных форм. Он может использоваться для лечения раковых опухолей и связанных с ними заболеваний как самостоятельно, так и в комбинации с другим или с другими противоопухолевыми лекарствами. Количество активного компонента, добавляемого к наполнителю, чтобы приготовить лекарственную форму, может быть различно в зависимости от объекта лечения и способа введения.
Соединения согласно предлагаемому изобретению могут использоваться для профилактики или ингибирования ненормальной пролиферации клеток, особенно в опухолях или раковых образованиях, включая рак легкого, рак печени, лейкемию, остеокарциному, рак поджелудочной железы, рак кожи, меланому, метрокарциному, оофорому, ректальную карциному, желудочную карциному, рак толстой кишки, рак молочной железы, сальпингокарциному, карциному оболочки матки, карциному шейки матки, карциному влагалища, карциному вульвы, карциномы пищевода, карциномы тонкой кишки, эндокринную карциному, саркому мягкой ткани, карциному уретры, рак простаты, лимфоцитому кожи, рак мочевого пузыря, рак почки или мочеточника, опухоли позвоночника, опухоли мозга и аденому гипофиза.
При исследованиях соединений согласно предлагаемому изобретению в экспериментах по ингибированию роста опухолевых клеток in vitro было обнаружено, что эти соединения демонстрируют замечательное ингибирующее действие на рост клеток рака легкого у людей, рака печени и лейкемии, инкубированных in vitro, а также установлена зависимость ингибирующего эффекта от дозы. Для лучшего соединение формулы I величина IC50=11,22 мг/мл для клеток рака печени человека, IC50=12,8 мг/мл для клеток лейкемии (рака крови) человека и IC50=10,24 мг/мл клеток рака легкого человека. Когда фармацевтическое соединение по изобретению вводилось мышам интраперитониальной инъекцией, значение LD50 для мышей составило 160 мг/кг.
В эксперименте по ингибированию опухоли Н22 рака печени мышей дозой 80 мг/кг ингибирующий эффект этих препаратов составил 69%.
Соединения могут найти применение при лечении опухолей в сочетании с химиотерапией, радиотерапией и биохимической терапией, чтобы повысить эффективность лечения и уменьшить фармацевтические побочные эффекты.
ФАРМАКОЛОГИЧЕСКИЕ ЭКСПЕРИМЕНТЫ
Противоопухолевая активность соединений определялась на примере соединения I.
Соединение I
1. Определение IC50 (рак легкого Льюиса).
(а) Клетки рака легкого Льюиса, выращенные в среде ДМЕМ, содержащей 15% телячей сыворотки, вносили в 96-луночный планшет с 1×104 клеток/лунка и планшет помещали при температуре 37°C в 5% CO2 - инкубатор. Соединение разводили до соответствующей концентрации средой DMEM, содержащей 15% телячьей сыворотки. Добавляли разведенный раствор в каждую лунку 96-луночного планшета, по 3 лунки для каждой концентрации и 6 лунок для чистого контроля. После 72 часов инкубации добавляли раствор МТТ и инкубировали 1 час, затем добавляли DMSO для усиления цвета и определяли значение оптической плотности на анализаторе с использованием ферментной метки. Вычисляли смертельную дозу каждой концентрации и оценивали IC50 графическим методом.
Рак легкого Льюиса: IC50=10,24 мг/мл.
(b) Определено IC50=11,22 мг/мл соединения I для клеток рака печени человека тем же самым вышеупомянутым методом тестирования.
(c) Определено IC50=12,8 мг/мл соединения I для клеток лейкемии (рака крови) человека тем же самым вышеупомянутым методом тестирования. Таким образом показано, что это соединение имеет способность ингибировать рост опухолевых клеток.
2. Замечательная противоопухолевая активность.
(а) Разделили произвольно 30 мышей (линии Kunming), мужских и женских особей поровну, с массой тела по 21-22 г, на 3 группы, каждая из которых состояла из 10 мышей. Сделали прививку клеток H22 рака печени по 0,2 мл каждой мыши, подкожно в правой стороне живота. Со следующего дня непрерывно в течение 7 дней вводили интраперитонеально один раз в день по 80 мг/кг и 40 мг/кг соединения I мышам соответственно каждой из двух групп в отдельности. Мышам контрольной группы вводили интраперитониально DMSO + физиологический раствор. На следующий день после прекращения введения мышей умертвили, определили вес тела и вес опухоли каждой мыши, и вычислили степень ингибирования роста опухоли. Соединение обладало очевидной противоопухолевой активностью по отношению к H22 со степенью ингибирования роста опухоли 69% и 40% при дозировке 80 мг/кг и 40 мг/кг соответственно.
(b) Ингибирование роста раковых клеток in vitro
Лечили клетки рака легкого человека (Льюис) и клетки лейкемии дозами соединения I по 8 мг/мл и 16 мг/мл, наблюдали динамику роста клеток в течение 4 дней. Различие между группами, подвергавшимися лечению, и контрольной группой в течение первых двух дней были незначительными, но начиная с третьего дня, количество клеток в группах, подвергавшихся лечению, резко понизилось, а раковые клетки в группе контроля непрерывно росли логарифмически. Такое различие между группами увеличивалось с течением времени.
Вышеописанные фармакологические эксперименты показывают, что упомянутое соединение согласно изобретению обладает очевидным эффектом ингибирования роста клеток рака печени, клеток лейкемии (твердая опухоль и мягкая опухоль) и соответствующим эффектом дозы.
Примеры осуществления изобретения
Предлагаемое изобретение иллюстрируется ниже примерами осуществления, которые используются только для того, чтобы раскрыть техническую сущность изобретения, но не ограничивают предлагаемое изобретение.
Примеры 1-3
Получение соединений I, II и III.
Путь А
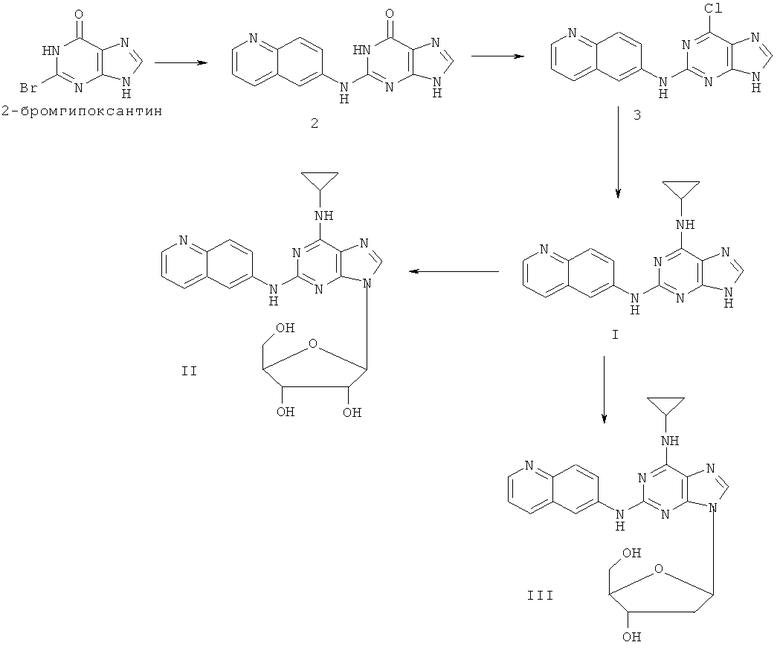
Пример 1. Получение соединения I
1) 200 мл воды, 20 г (93 ммоль) 2-бромзамещенного гипоксантина, 13 г (90 ммоль) 6-аминохинолина смешали с 60 мл моноэтилового эфира этиленгликоля и смесь кипятили с обратным холодильником в течение 48 часов, завершение реакции проверяли с помощью тонкослойной хроматографии (ТСХ). Затем реакционную смесь вылили в воду со льдом, отделили твердую фазу фильтрованием, промыли 200 мл концентрированной аммиачной воды и 3×50 мл метанола и высушили. Полученный сырой продукт очистили колоночной хроматографией на силикагеле с получением в результате 14,2 г соединения 2 (выход 57%).
2) 12 г (43 ммоль) соединения 2, 150 мл оксихлорида фосфора и 15 мл N,N-ксилидина смешали, и смесь кипятили с обратным холодильником в течение 30 минут. Затем реакционную смесь вылили в 2000 мл воды со льдом. Через 2 часа значение рН смеси отрегулировали уксусной кислотой до рН=3. Твердую фазу отделили фильтрованием. Полученный сырой продукт очистили колоночной хроматографией на силикагеле с получением в результате 11,5 г хлорида 3 (выход 90%).
3) Были смешаны 10 г (34 ммоль) хлорида 3, 10 мл (145 ммоль) циклопропиламина, 28 мл (200 ммоль) триэтиламина и 100 мл ДМФА (диметилформамида). Смесь перемешивали при 100°C для поддержания реакции в течение 3 часов. Завершение реакции проверили с помощью тонкослойной хроматографии (ТСХ). Затем растворитель удалили дистилляцией, а остаток растворили в диметиловом эфире этиленгликоля. Смесь отфильтровали, растворитель фильтрата удалили дистилляцией, а полученный остаток очистили колоночной хроматографией на силикагеле с получением в результате 7 г (22 ммоль) соединения I (т.пл.>250°C), (выход 65%).
Соединение I: 1Н ЯМР (DMSO-d6, δ): 0,640-0,642 (2Н, s), 0,845-0,860 (2H, m), 3,045 (1Н, s), 7,0411-7,432 (1Н, m), 7,632 (1H, s, пик исчез после добавления тяжелой воды), 7,865-7,888 (2Н, m), 7,997-8,018 (1Н, m), 8,116-8,136 (1Н, m), 8,623-8,682 (2Н, m), 9,242 (1Н, s, пик исчез после добавления тяжелой воды), 12,400 (1Н, s, пик исчез после добавления тяжелой воды). MS (ESI): 318 (М+Н+), 340 (М+Na+).
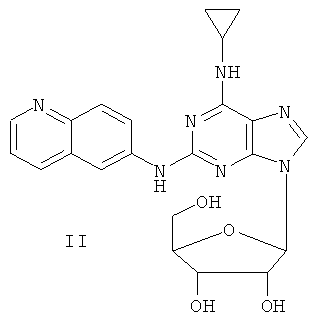
Пример 2. Получение соединения II
Смешали 10 мл безводного ацетонитрила, 4,76 г (15 ммоль) соединения I, полученного согласно примеру 1, и 6,3 мл (25 ммоль) N,O-бис(триметилсилил)ацетамида. Смесь перемешивали при комнатной температуре в течение 1 часа. Растворили 1,27 г (4 ммоль) тетраацетилрибофуранозы в 10 мл ацетонитрила, затем добавили к смеси 1,10 мл (16 ммоль) триметилсилилтрифлата (ТМСТФ) и кипятили с обратным холодильником в течение 5 часов. К смеси добавили 1,25 мл (5 ммоль) N,O-бис(триметилсилил)ацетамида и затем перемешивали в течение 24 часов. Растворитель удалили дистилляцией при пониженном давлении, а осадок растворили в 20 мл метанола. Реакционную смесь обрабатывали газообразным аммиаком в течение 2 часов. Растворитель удалили дистилляцией при пониженном давлении. Полученный остаток очистили колоночной хроматографией на силикагеле, с получением в результате 4,65 г соединения II (выход 72%).
Соединение II: MS (ESI): 450 (М+Н+), 472 (M+Na+).
Пример 3. Получение соединения III
Смешали 10 г (31,5 ммоль) 2-(6-аминохинолил)-6-циклопропилпурина, т.е. соединения I, полученного согласно примеру 1, 1,5 г (37,8 ммоль) 60% NaH и 150 мл безводного ацетонитрила. Смесь перемешивали в течение 30 минут в атмосфере азота. Затем в течение 20 минут к смеси порционно добавили 12 г (31,5 ммоль) 3,5-ди-пара-толуэнсульфонил-2-деокси-β-D-рибофуранозил-1-хлорида. Смесь выдерживали при комнатной температуре в течение 2 часов. Смесь отфильтровали, растворитель фильтрата удалили дистилляцией. Осадок очищали колоночной хроматографией на силикагеле, с получением в результате 9,8 г 2-(6-аминохинолил)-6-циклопропиламино-9-(3,5-ди-пара-толуэнсульфонил-2-деокси-β-D-рибофуранозил)пурина (выход 47%).
Вышеупомянутый продукт был добавлен к 25 ммоль метоксида натрия и 400 мл метанола. Смесь перемешивали при комнатной температуре в течение 5 часов. Затем значение рН смеси отрегулировали уксусной кислотой до рН=7. Растворитель удалили дистилляцией, а полученный остаток очищали колоночной хроматографией на силикагеле с получением в результате 5 г 2-(6-аминохилил)-6-циклопропиламино-9-(2-деокси-β-D-рибофуранозил)пурина, то есть соединения III (выход 80%). Соединение III: MS (ESI): 434 (M+H+), 456 (M+Na+).
Пример 4. Получение соединения I
Получение соединения I, описанного в примере 1, согласно следующему пути В:
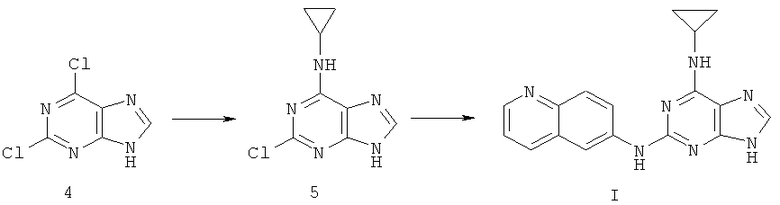
1) 3,78 г (20 ммоль) дихлорпурина 4 растворили в 50 мл ДМФ, добавили 1,4 мл (20 ммоль) циклопропиламина и 3,08 мл (22 ммоль) триэтиламина. Реакцию смеси осуществляли при температуре 80°C в течение 5 часов. Завершение реакции проверяли с помощью тонкослойной хроматографии (ТСХ). Затем растворитель удаляли дистилляцией при пониженном давлении, а полученный остаток очищали колоночной хроматографией на силикагеле с получением в результате 3,34 г соединения 5 (выход 80%).
2) 2,99 г (14,3 ммоль) соединения 5 смешали с 5,1 г (36,1 ммоль) 6-аминохинолина, 50 мл ДМФ и 2,4 мл (17,1 ммоль) триэтиламина. Смесь нагревали при 140°C с обратным холодильником в течение 72 часов. Анализ ТСХ подтвердил, что реакция была в основном закончена. Растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле, чтобы получить 3,54 г соединения I (выход 78%).
Примеры 5-7 иллюстрируют получение соединений IV, V, и VI.
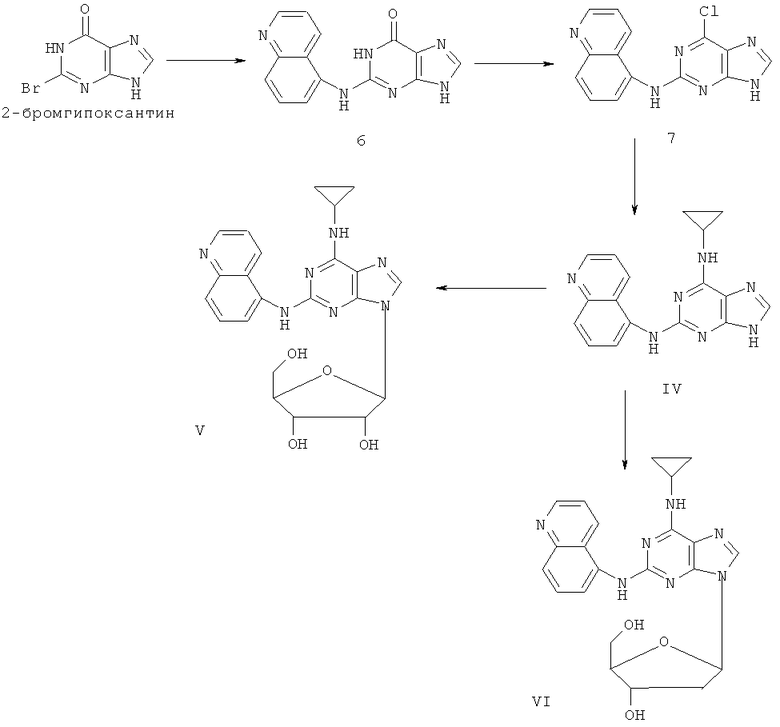
Пример 5. Получение соединения IV
1) Смешали 200 мл воды, 20 г (93 ммоль) 2-бромогипоксантина, 13 г (90 ммоль) 5-аминохинолина и 60 мл монометилового эфира этиленгликоля. Смесь кипятили с обратным холодильником в течение 48 часов. Завершение реакции проверили с помощью тонкослойной хроматографии (ТСХ). Реакционную смесь охладили до комнатной температуры и вылили в воду со льдом, осадок отделили фильтрацией, отмыли 200 мл концентрированной аммиачной воды и трижды 50 мл метанола, после чего высушили. Полученный остаток очищали колоночной хроматографией на силикагеле, чтобы получить 8 г продукта 6 (выход 32%).
2) Смешали 12 г (43 ммоль) продукта 6, 150 мл оксихлорида фосфора и 15 мл N,N-ксилидина. Смесь кипятили с обратным холодильником в течение 30 минут. Смесь охладили до комнатной температуры и затем через 2 часа вылили в 2000 мл воды со льдом, значение рН довели до 3 с помощью уксусной кислоты. Желтый осадок был выделен фильтрацией. Полученный остаток очищали колоночной хроматографией на силикагеле, чтобы получить 12,0 г хлорида 7 (выход 94%).
3) Смешали 10 г (34 ммоль) хлорида 7, 10 мл (145 ммоль) циклопропиламина, 28 мл (200 ммоль) триэтиламина и 100 мл ДМФ. Смесь нагревали при 100°C в течение 3 часов. Завершение реакции проверили с помощью тонкослойной хроматографии (ТСХ). Растворитель удалили дистилляцией, после чего остаток растворили в диэтиловом эфире этиленгликоля. Смесь отфильтровали, растворитель фильтрата дистиллировали, а полученный остаток очистили колоночной хроматографией на силикагеле, чтобы получить 9 г соединения IV (выход 84%).
Состав IV: MS (ESI): 318 (M+H+), 340 (M+Na+).
Пример 6. Получение соединения V
Смешали 10 мл безводного ацетонитрила, 4,76 г (15 ммоль) соединения IV, полученного согласно примеру 5, и 6,3 мл (25 ммоль) N,O-бис(триметилсилил)ацетамида. Смесь перемешивали при комнатной температуре в течение 1 часа. Растворили 1,27 г (4 ммоль) тетраацетилрибофуранозы в 10 мл ацетонитрила, затем добавили к смеси 1,10 мл (16 ммоль) ТМСТФ и смесь кипятили с обратным холодильником в течение 5 часов. После чего добавили к смеси 1,25 мл (5 ммоль) N,O-бис(триметилсилил)ацетамида и затем перемешивали в течение 24 часов. Завершение реакции проверили с помощью тонкослойной хроматографии (ТСХ). Растворитель удалили дистилляцией при пониженном давлении, а остаток растворили в 20 мл метанола. Реакционную смесь обрабатывали газообразным аммиаком в течение 2 часов при пониженном давлении. Растворитель удалили дистилляцией. Полученный остаток очищали колоночной хроматографией на силикагеле, чтобы получить 4,07 г соединения V (выход 63%).
Состав V: MS (ESI): (М+H+), 472 (M+Na+).
Пример 7. Получение соединения VI
Смешали 10 г (31,5 ммоль) соединения IV, то есть 2-(5-аминохинолил)-6-циклопропиламинопурина, полученного в примере 5, 1,5 г (37,8 ммоль) 60%-ого NaH и 150 мл безводного ацетонитрила. Смесь перемешивали в течение 30 минут в атмосфере азота. Затем в течение 20 минут порционно добавили к смеси 12 г (31,5 ммоль) 3,5-ди-пара-толуолсульфинил-2-деокси-β-D-рибофуранозил-1-хлорида. Смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь фильтровали и растворитель фильтрата удаляли дистилляцией. Остаток очищали колоночной хроматографией на силикагеле, получили 8,0 г 2-(5-аминохинолил)-6-циклопропиламино-9-(3,5-ди-пара-толуэнсульфинил-2-деокси-β-D-рибофуранозил)пурина (выход 38%).
Вышеупомянутый продукт добавили к 25 ммоль метоксида натрия и 400 мл метанола. Смесь перемешивали при комнатной температуре в течение 5 часов. Значение рН смеси довели до 7 с помощью уксусной кислоты. Растворитель удалили дистилляцией, а полученный остаток очищали колоночной хроматографией на силикагеле, чтобы получить 5,75 г 2-(5-аминохинолил)-6-циклопропиламино-9-(2-деокси-β-D-рибофуранозил)пурина, то есть соединения VI (выход 92%).
Соединение VI: MS (ESI): 434 (М+H+), 456 (M+Na+).
Пример 8. Получение соединения IV, альтернатива примеру 5
Процесс получения соединения IV выглядел следующим образом:
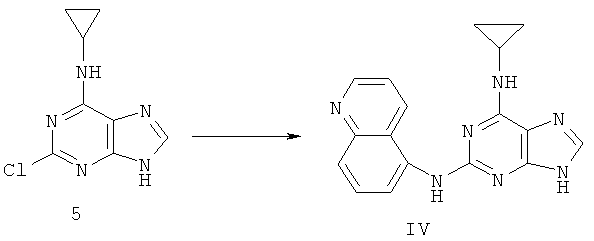
Смешали 2,99 г (14,3 ммоль) соединения 5, 5,1 г (36,1 ммоль) 5-аминохинолина, 50 мл ДМФ и 2,4 мл (17,1 ммоль) триэтиламина. Смесь нагревали при 140°C с обратным холодильником в течение 72 часов. Завершение реакции проверили с помощью тонкослойной хроматографии (ТСХ). Растворитель удалили дистилляцией при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле, чтобы получить 2,88 г соединения IV (выход 63%).
Примеры 9-11 иллюстрируют получение соединений VII, VIII и IX.
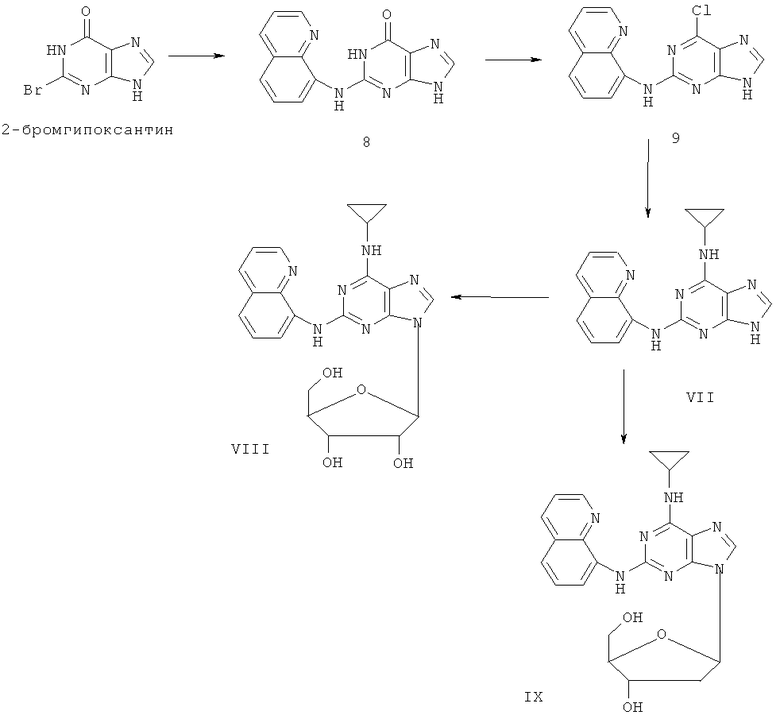
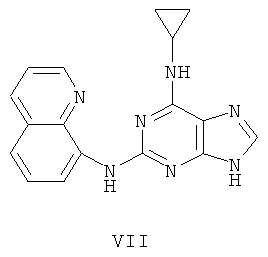
Пример 9. Получение соединения VII
1) В 200 мл воды смешали 20 г (93 ммоль) 2-бромгипоксантина, 13 г (90 ммоль) 8-аминохинолина и 60 мл монометилового эфира этиленгликоля, после чего смесь кипятили с обратным холодильником в течение 48 часов. Завершение реакции проверяли с помощью тонкослойной хроматографии (ТСХ). Затем реакционную смесь вылили в воду со льдом, отделили осадок фильтрацией, промыли его в 200 мл аммиачной воды и 3х50 мл метанола и высушили. Полученный остаток очищали колоночной хроматографией на силикагеле, чтобы получить 11,4 г продукта 8 (выход 46%).
2) Смешали 12 г (43 ммоль) соединения 8, 150 мл оксихлорида фосфора и 15 мл N,N-ксилидина и смесь кипятили с обратным холодильником в течение 30 минут. Затем охлаждали смесь до комнатной температуры в течение 2 часов, после чего вылили в 2000 мл воды со льдом. Значение рН смеси довели до 3 с помощью уксусной кислоты. Желтый осадок отделили фильтрацией. Полученный остаток очищали колоночной хроматографией на силикагеле, получили 10,3 г хлорида 9 (выход 81%).
3) Смешали 10 г (34 ммоль) хлорида 9 с 10 мл (145 ммоль) циклопропиламина, 28 мл (200 ммоль) триэтиламина и 100 мл ДМФ. Смесь перемешивали при 100°C в течение 3 часов. Растворитель удалили дистилляцией, а остаток растворили в диметиловом эфире этиленгликоля. Смесь профильтровали, растворитель фильтрата удалили дистилляцией, а полученный остаток очищали колоночной хроматографией на силикагеле, получили 6 г соединения VII (выход 56%).
Соединение VII: VII: MS (ESI):318 (M+H+), 340 (M+Na+).
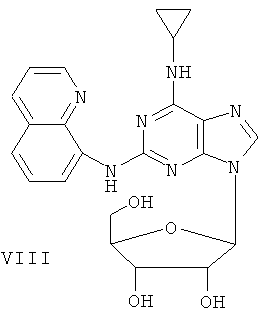
Пример 10. Получение соединения VIII
10 мл безводного ацетонитрила смешали с 4,76 г (15 ммоль) соединения VII и 6,3 мл (25 ммоль) N,O-бис(триметилсилил)ацетамида. Смесь перемешивали при комнатной температуре в течение 1 часа. Растворили 1,27 г (4 ммоль) тетраацетил-рибофуранозы в 10 мл ацетонитрила и затем добавили к смеси 1,10 мл (16 ммоль) ТМСТФ. Смесь кипятили с обратным холодильником в течение 5 часов. Добавили к смеси 1,25 мл (5 ммоль) N,O-бис(триметилсилил)ацетамида и затем перемешивали в течение 24 часов. Растворитель удалили дистилляцией при пониженном давлении, а остаток растворили в 20 мл метанола. Реакционную смесь поместили в атмосферу аммиачного газа на 2 часа. Растворитель удалили дистилляцией при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле, чтобы получить 4,2 г соединения VIII (выход 65%).
Соединение VIII: MS (ESI): 450 (M+H+), 472 (M+Na+).
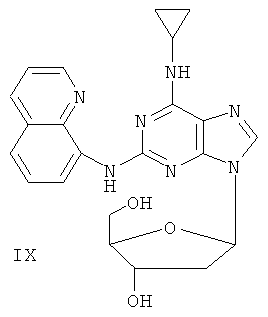
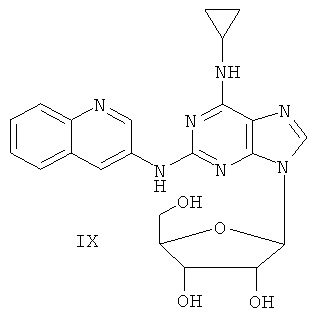
Пример 11. Получение соединения IX
10 г (31,5 ммоль) соединения VII, то есть 2-(8-аминохинолил)-6-циклопропиламинопурина, смешали с 1,5 г (37,8 ммоль) 60%-ного NaH и 150 мл безводного ацетонитрила. Смесь перемешивали в течение 30 минут в атмосфере азота. Затем в течение 20 минут к смеси порционно добавляли 12 г (31,5 ммоль) 3,5-ди-пара-толуолсульфонил-2-деокси-β-D-рибофуранозил-1-хлорида. Смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь фильтровали, и растворитель фильтрата удаляли дистилляцией. Маслянистый остаток очищали колоночной хроматографией на силикагеле, получили 7,4 г 2-(8-аминохинолил)-6-циклопропиламино-9-(3,5-ди-пара-толуолсульфонил-2-деокси-β-D-рибофуранозил)пурина (выход 35%).
Вышеупомянутый продукт добавили к 25 ммоль метоксида натрия и 400 мл метанола. Смесь перемешивали при комнатной температуре в течение 5 часов. Значение рН смеси довели до 7 с помощью уксусной кислоты. Растворитель удаляли дистилляцией и полученный остаток очищали колоночной хроматографией колонки на силикагеле, получили 3,2 г соединения IX (выход 51%).
Соединение IX: MS (ESI): 434 (М+Н+), 456 (M+Na+).
Пример 12. Получение соединения VII, альтернатива примеру 9
Процесс получения соединения осуществлялся следующим образом:
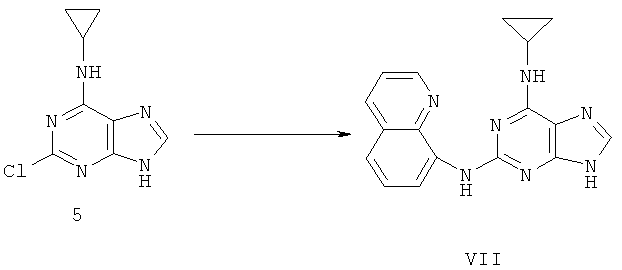
2,99 г (14,3 ммоль) соединения 5 смешивали с 5,1 г (36,1 ммоль) 8-аминохинолина, 50 мл ДМФ и 2,4 мл (17,1 ммоль) триэтиламина. Смесь нагревали при температуре 140°C с обратным холодильником в течение 72 часов. Завершение реакции проверяли методом ТСХ. Растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток очищали колоночной хроматографией колонки на силикагеле, получили 4,10 г соединения VII (выход 90%).
Примеры 13-15 иллюстрируют получение соединений X, XI и XII.
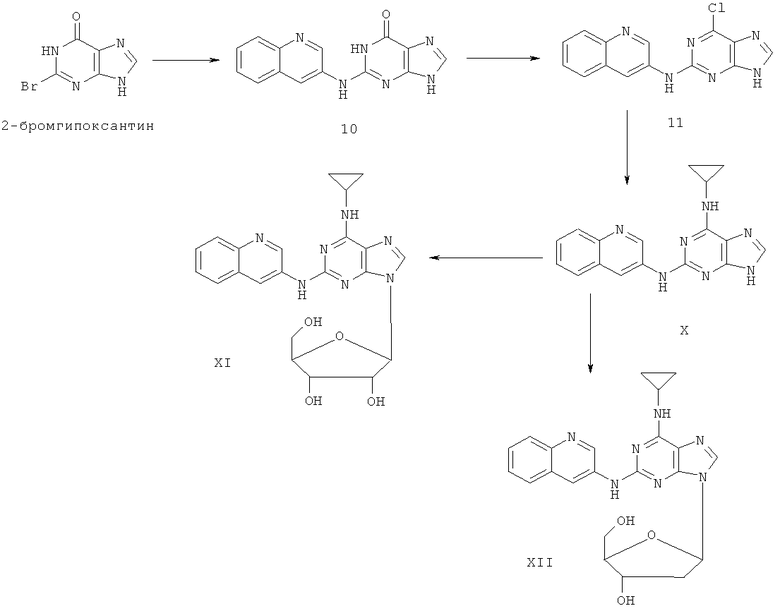
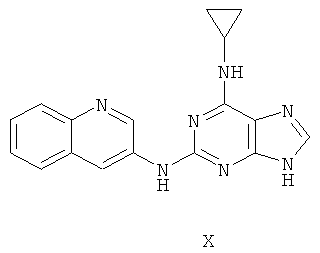
Пример 13. Получение соединения Х
1) В 200 мл воды смешивали 20 г (93 ммоль) 2-бромгипоксантина, 13 г (90 ммоль) 3-аминохинолина и 60 мл монометилового эфира этиленгликоля и смесь кипятили с обратным холодильником в течение 48 часов. Завершение реакции проверяли методом ТСХ. Затем реакционную смесь вылили в воду со льдом, твердый осадок отделяли фильтрацией, промывали аммиачной водой (200 мл) и три раза метанолом (3×50 мл) и сушили. Полученный остаток очищали колоночной хроматографией на силикагеле, получили 15,0 г соединения 10 (выход 60%).
2) Смешали 12 г (43 ммоль) соединения 10, 150 мл оксихлорида фосфора и 15 мл N,N-ксилидина и смесь кипятили с обратным холодильником в течение 30 минут. Затем смесь охлаждали при комнатной температуре в течение 2 часов. После чего реакционную смесь медленно вылили в 2000 мл воды со льдом, значение рН смеси довели до 3 с помощью уксусной кислоты. Желтый осадок был выделен фильтрацией. Полученный остаток очищали колоночной хроматографией на силикагеле, получили 10,9 г хлорида 11 (выход 85%).
3) Смешали 10 г (34 ммоль) хлорида 11 с 10 мл (145 ммоль) циклопропиламина, 28 мл (200 ммоль) триэтиламина и 100 мл ДМФ. Смесь перемешивали при 100°C в течение 3 часов, завершение реакции проверяли методом ТСХ. Растворитель удаляли дистилляцией, а осадок растворяли диметиловым эфиром этиленгликоля. Смесь фильтровали, растворитель фильтрата удаляли дистилляцией, а полученный остаток очищали колоночной хроматографией на силикагеле, получили 10,1 г соединения Х (выход 94%).
Соединение X: MS (ESI): 318 (M+H+), 340 (M+Na+).
Пример 14. Получение соединения XI
Смешали 10 мл безводного ацетонитрила, 4,76 г (15 ммоль) соединения Х и 6,3 мл (25 ммоль) N,O-бис(триметилсилил)ацетамида. Смесь перемешивали при комнатной температуре в течение 1 часа. Затем 1,27 г (4 ммоль) тетраацетил-рибофуранозы, растворенной в 10 мл ацетонитрила, и 1,10 мл (16 ммоль) ТМСТФ добавили к смеси и кипятили с обратным холодильником в течение 5 часов. После чего к смеси добавили 1,25 мл (5 ммоль) N,O-бис(триэтилсилил)ацетамида, затем перемешивали в течение 24 часов и завершение реакции проверяли методом ТСХ. Растворитель удаляли дистилляцией при пониженном давлением, а остаток растворили 20 мл метанола. Реакционную смесь выдерживали в атмосфере аммиачного газа в течение 2 часов. Растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле, получили 4,97 г соединения XI (выход 77%).
Соединение XI: MS (ESI): 450 (M+H+), 472 (M+Na+).
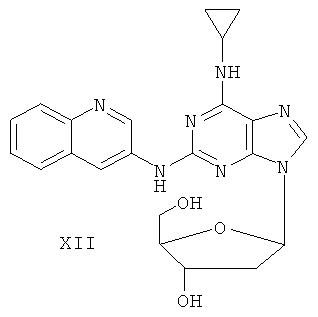
Пример 15. Получение соединения XII
Смешали 10 г (31,5 ммоль) соединения X, 1,5 г (37,8 ммоль) 60%-ного NaH и 150 мл безводного ацетонитрила. Смесь перемешивали в течение 30 минут в атмосфере азота. Затем в течение 20 минут к смеси добавляли порционно 12 г (31,5 ммоль) 3,5-ди-пара-толуолсульфонил-2-деокси-β-D-рибофуранозил-1-хлорида. Смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь фильтровали и растворитель фильтрата удаляли дистилляцией. Маслянистый остаток очищали колоночной хроматографией на силикагеле, получили 8,8 г 2-(3-аминоксинолил)-6-циклопропиламино-9-(3,5-ди-пара-толуолсульфонил-2-деокси-β-D-рибофуранозил)пурина (выход 42%).
Вышеупомянутый продукт добавили к 25 ммоль метоксида натрия и 400 мл метанола. Смесь перемешивали при комнатной температуре в течение 5 часов. Значение рН смеси довели до 7 с помощью уксусной кислоты. Растворитель удаляли дистилляцией и полученный остаток очищали колоночной хроматографией на силикагеле, получили 5,06 г соединения XII (выход 81%).
Соединение XII: MS (ESI): 434 (М+Н+), 456 (M+Na+).
Пример 16. Получение соединения X, альтернатива Примеру 9
Процесс получения соединения осуществляется следующим образом:
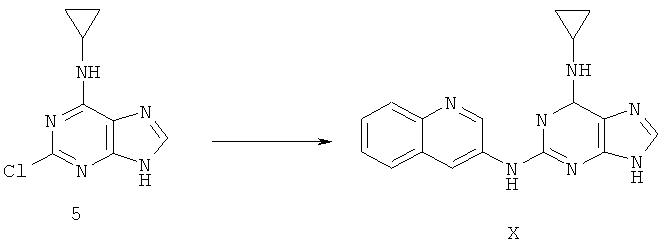
Смешали 2,99 г (14,3 ммоль) соединения 5 с 5,1 г (36,1 ммоль) 3-аминоксинолина, 50 мл ДМФ и 2,4 мл (17,1 ммоль) триэтиламина. Смесь нагревали с обратным холодильником при температуре 140°С в течение 72 часов. Завершение реакции проверяли методом ТСХ. Растворитель удаляли дистилляцией. Полученный остаток очищали колоночной хроматографией на силикагеле, получили 3,54 г соединения Х (выход 78%).
Примеры 17-19 иллюстрируют получение соединений XIII, XIV и XV.
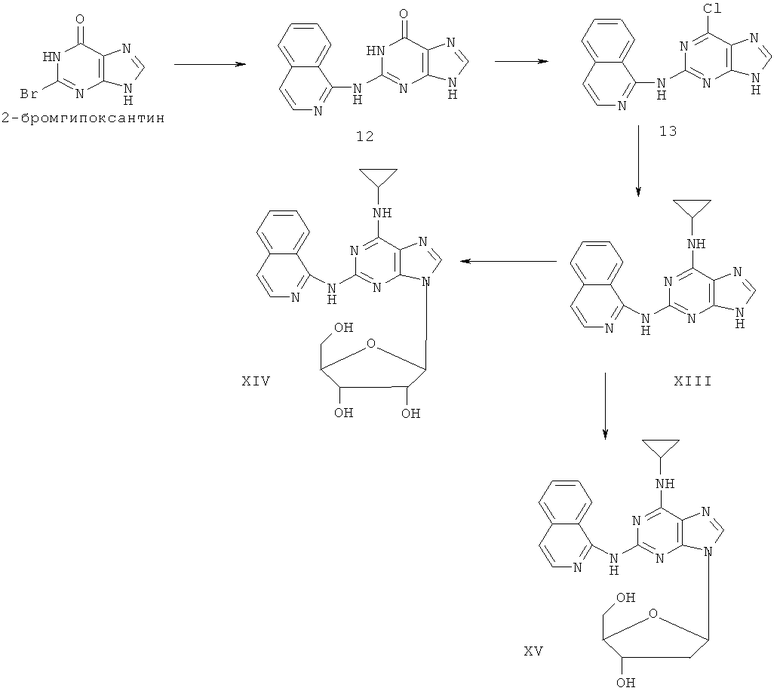
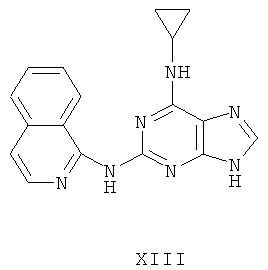
Пример 17. Получение соединения XIII
1) В 200 мл воды смешивали 20 г (93 ммоль) 2-бромгипоксантина, 13 г (90 ммоль) 1-аминохинолина и 60 мл монометилового эфира этиленгликоля и смесь кипятили с обратным холодильником в течение 48 часов. Завершение реакции проверяли методом ТСХ. Затем реакционную смесь вылили в воду со льдом, твердый осадок отделяли фильтрацией, промывали аммиачной водой (200 мл) и три раза метанолом (3×50 мл) и сушили. Полученный остаток очищали колоночной хроматографией на силикагеле, получили 14,2 г соединения 12 (выход 57%).
2) Смешали 12 г (43 ммоль) соединения 12, 150 мл оксихлорида фосфора и 15 мл N,N-ксилидина и смесь кипятили с обратным холодильником в течение 30 минут. Затем смесь охлаждали при комнатной температуре в течение 2 часов. После чего реакционную смесь вылили в 2000 мл воды со льдом, значение рН довели до 3 с помощью уксусной кислоты. Желтый осадок был выделен фильтрацией. Полученный остаток очищали колоночной хроматографией на силикагеле, получили 11,5 г хлорида 13 (выход 90%).
3) Смешали 10 г (34 ммоль) хлорида 13 с 10 мл (145 ммоль) циклопропиламина, 28 мл (200 ммоль) триэтиламина и 100 мл ДМФ. Смесь перемешивали при 100°C в течение 3 часов. Завершение реакции проверяли методом ТСХ. Растворитель удаляли дистилляцией, а осадок растворяли диметиловым эфиром этиленгликоля. Смесь фильтровали, растворитель фильтрата удаляли дистилляцией, а полученный остаток очищали колоночной хроматографией на силикагеле кварца, получили 4,2 г соединения XIII (выход 39%).
Соединение XIII: MS (ESI): 318 (M+H+), 340 (M+Na+).
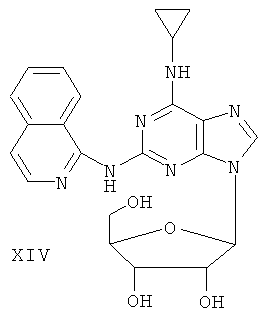
Пример 18. Получение соединения XIV
Смешали 10 мл безводного ацетонитрила, 4,76 г (15 ммоль) соединения XIII и 6,3 мл (25 ммоль) N,O-бис(триметилсилил)ацетамида. Смесь перемешивали при комнатной температуре в течение 1 часа. Затем 1,27 г (4 ммоль) тетраацетил-рибофуранозы, растворенной в 10 мл ацетонитрила, и 1,10 мл (16 ммоль) ТМСТФ добавили к смеси и кипятили с обратным холодильником в течение 5 часов. После чего к смеси добавили 1,25 мл (5 ммоль) N,O-бис(триэтилсилил)ацетамида, затем перемешивали в течение 24 часов, и завершение реакции проверяли методом ТСХ. Растворитель удаляли дистилляцией при пониженном давлением, а остаток растворили в 20 мл метанола. Реакционную смесь выдерживали в атмосфере аммиачного газа в течение 2 часов. Растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле, получили 4,2 г соединения XIV (выход 64%).
Соединение XIV: MS (ESI): 450 (M+H+), 472 (M+Na+).
Пример 19. Получение соединения XV
Смешали 10 г (31,5 ммоль) соединения XIII, 1,5 г (37,8 ммоль) 60%-ного NaH и 150 мл безводного ацетонитрила. Смесь перемешивали в течение 30 минут в атмосфере азота. Затем в течение 20 минут порционно добавили к смеси 12 г (31,5 ммоль) 3,5-ди-пара-толуолсульфонил-2-деокси-β-D-рибофуранозил-1-хлорида. Смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь фильтровали и растворитель фильтрата удалили дистилляцией. Маслянистый остаток очищали колоночной хроматографией на силикагеле, получили 7,09 г 2-(1-аминохинолил)-6-циклопропиламино-9-(3,5-ди-пара-толуэнсульфонил-2-деокси-β-D-рибофуранозил)пурина (выход 34%).
Вышеупомянутый продукт добавили к 25 ммоль метоксида натрия и 400 мл метанола. Смесь перемешивали при комнатной температуре в течение 5 часов. Значение рН смеси довели до 7 с помощью уксусной кислоты. Растворитель удаляли дистилляцией, и полученный остаток очищали колоночной хроматографией на силикагеле, получили 3,8 г соединения XV (выход 60,8%).
Соединение XV: MS (ESI): 434 (M+H+), 456 (M+Na+).
Пример 20. Получение соединения XIII, альтернатива примеру 17
Процесс получения соединения осуществляется следующим образом:
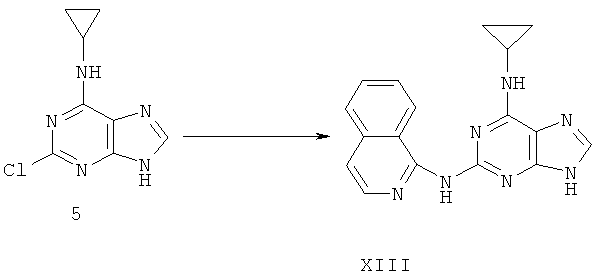
Смешали 2,99 г (14,3 ммоль) соединения 5 с 5,1 г (36,1 ммоль) 3-аминоксинолина, 50 мл ДМФ и 2,4 мл (17,1 ммоль) триэтиламина. Смесь нагревали с обратным холодильником при температуре 140°C в течение 72 часов. Завершение реакции проверяли методом ТСХ. Растворитель удаляли дистилляцией. Полученный остаток очищали колоночной хроматографией на силикагеле, получили 4,16 г соединения XIII (выход 92%).
Примеры 21-22 иллюстрируют получение соединений XVI, XVII и XVIII.
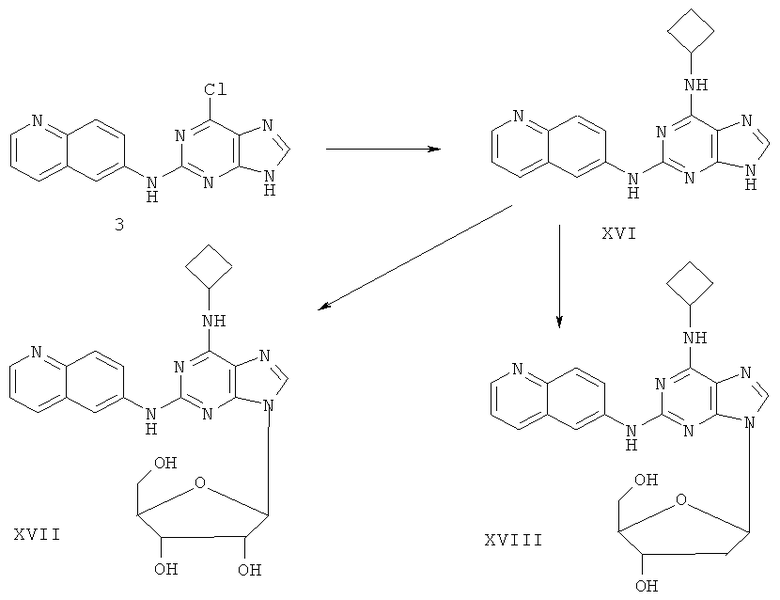
Пример 21. Получение соединения XVI
10 г (34 ммоль) соединения 3 смешали с 10,3 г (145 ммоль) циклобутиламина, 28 мл (200 ммоль) триэтиламина и 100 мл ДМФ. Смесь перемешивали при 100°C в течение 3 часов. Завершение реакции проверяли с помощью метода ТСХ. Растворитель удаляли дистилляцией, а остаток растворили с диметиловым эфиром этиленгликоля. Смесь фильтровали и растворитель фильтрата удаляли дистилляцией, а полученный остаток очищали колоночной хроматографией на силикагеле, получили 6,8 г соединения XVI (выход 60%).
Соединение XVI: MS (ESI): 332 (M+H+), 354 (M+Na+).
Пример 22. Получение соединения XVII
Смешали 10 мл безводного ацетонитрила, 4,97 г (15 ммоль) соединения XVI и 6,3 мл (25 ммоль) N,O-бис(триметилсилил)ацетамида. Смесь перемешивали при комнатной температуре в течение 1 часа. Затем 1,27 г (4 ммоль) тетраацетил-рибофуранозы, растворенной в 10 мл ацетонитрила, и 1,10 мл (16 ммоль) ТМСТФ добавили к смеси и кипятили с обратным холодильником в течение 5 часов. После чего к смеси добавили 1,25 мл (5 ммоль) N,O-бис(триэтилсилил)ацетамида, затем перемешивали в течение 24 часов. Завершение реакции проверяли методом ТСХ. Растворитель удаляли дистилляцией при пониженном давлением, а остаток растворили с 20 мл метанола. Реакционную смесь выдерживали в атмосфере аммиачного газа в течение 2 часов. Растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле, получили 4,71 г соединения XVII (выход 71%).
Соединение XVII: MS (ESI): 464 (M+H+), 486 (M+Na+).
Пример 23. Получение соединения XVIII
Смешали 10 г (31,5 ммоль) соединения XVI, то есть 2-(6-аминохинолил)-6-циклобутиламинопурина, 1,5 г (37,8 ммоль) 60%-ного NaH и 150 мл безводного ацетонитрила. Смесь перемешивали в течение 30 минут в атмосфере азота. Затем в течение 20 минут порционно добавили к смеси 12 г (31,5 ммоль) 3,5-ди-пара-толуолсульфонил-2-деокси-β-D-рибофуранозил-1-хлорида. Смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь фильтровали и растворитель фильтрата удаляли дистилляцией. Маслянистый остаток очищали колоночной хроматографией на силикагеле, получили 2-(6-аминохинолил)-6-циклобутиламино-9-(3,5-ди-пара-толуэнсульфонил-2-деокси-β-D-рибофуранозил)пурин.
Вышеупомянутый продукт добавили к 25 ммоль метоксида натрия и 400 мл метанола. Смесь перемешивали при комнатной температуре в течение 5 часов. Значение рН довели до 7 с помощью уксусной кислоты. Растворитель удаляли дистилляцией, а полученный остаток очищали колоночной хроматографией на силикагеле, получили 7 г соединения XVIII, т.е. 2-(6-аминохинолил)-6-циклобутиламино-9-(2-деокси-β-D-рибофуранозил)пурина (выход 51,8%).
Соединение XVIII: MS (ESI): 448 (M+H+), 470 (M+Na+).
Пример 24. Получение соединения XVI
Процесс получения соединения осуществляется следующим образом:
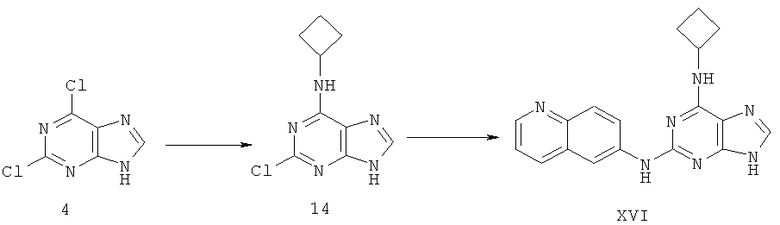
1) 3,78 г (20 ммоль) дихлорпурина 4, растворенного в 50 мл ДМФ, смешали с 1,4 мл (20 ммоль) циклобутиламина и 3,08 мл (22 ммоль) триэтиламина. Смесь перемешивали при температуре 80°C в течение 5 часов. Завершение реакции проверяли методом ТСХ. Растворитель удаляли дистилляцией при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле, получили 4,1 г соединения 14 (выход 92%).
2) Смешали 2,99 г (14,3 ммоль) соединения 14 с 5,1 г (36,1 ммоль) 3-аминоксинолина, 50 мл ДМФ и 2,4 мл (17,1 ммоль) триэтиламина. Смесь нагревали с обратным холодильником при температуре 140°C в течение 72 часов. Завершение реакции проверяли методом ТСХ. Растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле, получили 3,82 г соединения XVI (выход 81%).
Пример 25. Получение гидрохлорида и лактата соединения I
Получение гидрохлорида соединения I.
Смешали 10 г (31,54 ммоль) соединения I, 210 мл этанола и 25 мл воды. Смесь нагревали, чтобы растворить соединение I. Затем к смеси добавляли капельно 0,7 мл (37,85 ммоль) водного раствора HCl (2 моль/л). Смесь кипятили с обратным холодильником в течение 0,5 часа. Затем смесь оставляли медленно-медленно охлаждаться до комнатной температуры, а затем охлаждали смесь до температуры ниже 5°C в течение 5 часов. Бледно-желтый осадок отделяли фильтрацией. Бледно-желтый остаток после высушивания весил 10 г (выход 90%). Температура плавления бледно-желтого остатка была выше 270°C.
Получение лактата соединения I.
Смешали 5 г соединения I, 105 мл (15,77 ммоль) 95% этанола и 12,5 мл воды. Смесь нагревали, чтобы растворить соединение I. Затем к смеси добавляли капельно 10% раствор молочной кислоты в этаноле. Смесь кипятили с обратным холодильником в течение 1 часа. Затем смесь оставляли медленно-медленно охлаждаться до комнатной температуры, а затем охлаждали смесь до температуры ниже 5°C в течение 5 часов. Бледно-желтый осадок отделяли фильтрацией. Бледно-желтый остаток после высушивания весил 5,6 г (выход 87%). Температура плавления бледно-желтого остатка была 239-248°C.
Пример 26. Приготовление дозированных лекарственных форм
Приготовление таблеток, покрытых оболочкой
Состав ядра таблеток:
Технологический процесс. Смешивают точно по весу соединение I и лактозу, затем добавляют к смеси силикагель, чтобы улучшить текучесть. Добавляют и смешивают другие фармацевтические адъюванты, после чего выполняют прямое таблетирование.
Состав оболочки: покрытие Opadry 25 г в соответствующем количестве 80% этанола.
Приготовление инъекции
Состав инъекции:
Технологический процесс. Смешивают точно по весу соединение I и Tween-80 и растирают в порошок, затем добавляют к смеси 0,3% этилового спирта, чтобы растворить соединение I и Tween-80 с нагреванием. Жидкость фильтруют 0,22 µm мембранным фильтром в стерильных условиях, затем разливают в ампулы по 10 мл и стерилизуют в автоклаве при температуре 100°C.
Настоящее изобретение относится к соединениям общей формулы (А) или его фармацевтически приемлемым солям, где W - циклоалкил; Y - водород или фармацевтически приемлемый сахарид, имеющий формулу
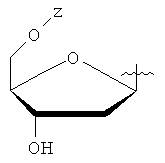 или
или 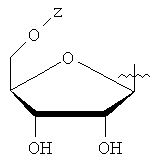 ;
;
Z - водород; Q - заместитель, выбранный из группы, состоящей из 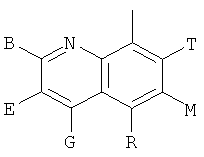 ;
;  ;
;  ;
;  ;
;  ;
;
и В, Е, O, К, Т и М каждый - водороды. Изобретение относится также к способам получения соединений (А) и их солей, к фармацевтической композиции на их основе для лечения раковых опухолей и связанных с ними заболеваний, а также к применению данных соединений и их солей при получении фармацевтических препаратов для профилактики и лечения опухолевых заболеваний. 5 н. и 10 з.п. ф-лы, 1 табл.
1. Соединение, имеющее следующую формулу (А):
или его фармацевтически приемлемые соли,
где W представляет собой С3-6 циклоалкил;
Y представляет собой водород или фармацевтически приемлемый сахарид, имеющий формулу, выбранную из группы, состоящей из:
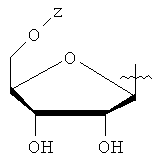
Z представляет собой водород;
Q представляет собой заместитель, выбранный из группы, состоящей из:
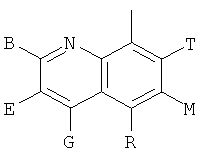
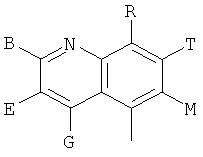
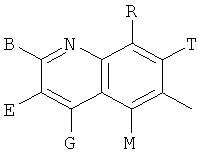
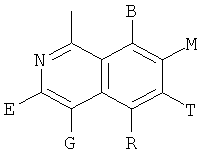
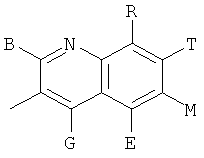
и В, E, G, R, Т и М каждый представляет собой водород.
2. Соединение по п.1 или его фармацевтически приемлемые соли, где W представляет собой заместитель, выбранный из группы, состоящей из:
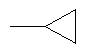
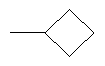
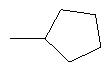
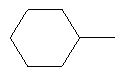
3. Соединение по п.1 или его фармацевтически приемлемые соли, где Y представляет собой водород.
4. Соединение по п.1 или его фармацевтически приемлемые соли, где W представляет собой заместитель, выбранный из группы, состоящей из:
а В, E, G, R, Т и М каждый представляет водород.
5. Соединение по п.1 или 3 или его фармацевтически приемлемые соли, где W выбран из группы, состоящей из:
или
a Q представляет собой заместитель, выбранный из группы, состоящей из:
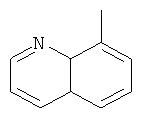
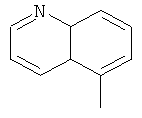
6. Соединение по п.1 или его фармацевтически приемлемые соли, где Q представляет собой
7. Соединение по п.1, отличающееся тем, что его соли представляют собой фармацевтически приемлемые соли, которые могут быть аддитивными солями, образуемыми с неорганическими или органическими кислотами, предпочтительно хлористоводородные соли, бромистоводородные соли, йодистоводородные соли, п-толуолсульфонатные соли, фосфатные соли, сульфатные соли, хлорнокислые соли, уксуснокислые соли, соли трифторуксусной кислоты, пропионаты, цитраты, малонатные соли, сукцинаты, соли молочной кислоты, оксалаты, соли тартрата и соли бензоата, а также соли, которые могут быть образованы с основаниями, включая неорганические или органические основания.
8. Соединение по п.5, отличающееся тем, что оно имеет формулу, выбранную из группы, состоящей из следующих формул (I-XVIII):
9. Соединение по п.5, отличающееся тем, что оно имеет формулу I
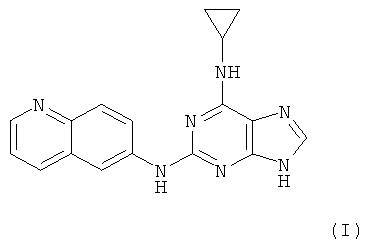
10. Фармацевтическая композиция для лечения раковых опухолей и связанных с ними заболеваний, содержащая соединение по любому из пп.1-9 или его соли в эффективном количестве, выбранном в зависимости от объекта лечения и способа введения, и фармацевтически приемлемый наполнитель.
11. Фармацевтическая композиция по п.10, отличающаяся тем, что она имеет форму таблетки, капсулы, пилюли, жидкого препарата для орального применения, гранулы, порошка, инъекции, импланта или препарата для наружного применения.
12. Способ получения соединения формулы (А) по любому из пп.1-9 и его солей, включающий следующие операции:
(1) проведение реакции соединения формулы (a) с Q-NH2 при их молярном отношении 0,8-1,5 в органическом растворителе при температуре приблизительно от 50 до 150°С в течение 1-72 ч с получением в результате первой реакционной смеси, добавление воды к первой реакционной смеси и охлаждение первой реакционной смеси при комнатной температуре, чтобы получить соединение формулы (b)
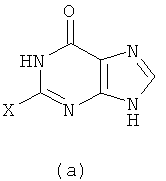
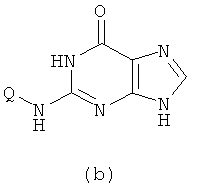
(2) проведение реакции соединения формулы (b) с галогенирующим агентом в органическом растворителе при температуре приблизительно от 50 до 150°С в течение 1-72 ч с получением в результате второй реакционной смеси, добавление воды ко второй реакционной смеси и регулирование значения рН второй реакционной смеси до приблизительно 2-5 с помощью кислоты, и охлаждение второй реакционной смеси при комнатной температуре, чтобы получить соединение формулы (с)
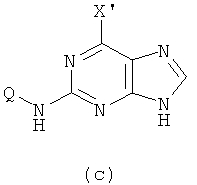
и
(3) проведение реакции соединения (с) с W-NH2 при их молярном отношении 0,8-1,5 в органическом растворителе в присутствии акцептора кислоты при температуре приблизительно от 50 до 150°С в течение 1-72 ч с последующим удалением растворителя дистилляцией, чтобы получить соединение формулы (f)
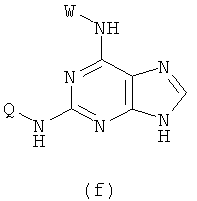
где Х представляет собой бром, X' представляет собой хлор, a W и Q являются такими, как определено в п.1.
13. Способ получения соединения формулы (А) по любому из пп.1-9 и его солей, включающий следующие операции:
(1) проведение реакции соединения формулы (k) с W-NH2 при их молярном отношении 0,8-1,5 в органическом растворителе в присутствии акцептора кислоты при температуре приблизительно от 30 до 120°С в течение 1-72 ч с последующим удалением растворителя дистилляцией, чтобы получить соединение формулы (е)
 и
и
(2) проведение реакции соединения формулы (е) с Q-NH2 при их молярном отношении 0,8-1,5 в органическом растворителе в присутствии акцептора кислоты при температуре приблизительно от 70 до 170°С в течение 1-72 ч с последующим удалением растворителя дистилляцией, чтобы получить соединение формулы (f)
где X представляет собой хлор, X' представляет собой хлор, a W и Q являются такими, как определено в п.1.
14. Применение соединений по любому из пп.1-9 и их солей при получении фармацевтических препаратов для профилактики и лечения опухолевых заболеваний.
15. Применение по п.14, отличающееся тем, что указанные опухолевые заболевания выбраны из группы, включающей рак легкого, рак печени, лейкемию, остеокарциному, рак поджелудочной железы, рак кожи, меланому, метрокарциному, оофорому, ректальную карциному, желудочную карциному, рак толстой кишки, рак молочной железы, сальпингокарциному, карциному оболочки матки, карциному шейки матки, карциному влагалища, карциному вульвы, карциномы пищевода, карциномы тонкой кишки, эндокринную карциному, саркому мягкой ткани, карциному уретры, рак простаты, лимфоцитому кожи, рак мочевого пузыря, рак почки или мочеточника, опухоли позвоночника, опухоли мозга и аденому гипофиза.
| ЕР 0704215 А2, 03.04.1996 | |||
| ПУРИНОВЫЕ ПРОИЗВОДНЫЕ КАК ИНГИБИТОРЫ ТИРОЗИНПРОТЕИНАЗЫ SYK | 2000 |
|
RU2248977C2 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |

