Уровень техники
Изобретение относится к получению диизопинокамфеилхлорборана. Изобретение также относится к использованию сырого диизопинокамфеилхлорборанового продукта при восстановлении прохиральных кетонов.
Диизопинокамфеилхлорборан является хорошо известным хиральным восстанавливающим агентом и описано несколько способов его получения. В одной из ранних работ (Brown Н. С. and Jadhav P.К., J. Am. Chem. Soc., 1983, 105, 2092-2093), диизопинокамфеилхлорборан получают реакцией эфирата монохлорборана с α-пиненом в диэтиловом эфире. Эфират монохлорборана в свою очередь получают из боргидрида лития и трихлорида бора (Brown Н.С. and Ravindran N., J. Am. Chem. Soc., 1972, 94, 2112-3). В патенте США 5043479 сообщается, что вышеуказанный способ приводит к смеси продуктов, которая неудовлетворительна для достижения асимметричного восстановления кетонов с высоким энантиомерным избытком.
В патенте США 5043479 также описан новый способ получения диизопинокамфеилхлорборана. В этом способе вначале необходимо получить промежуточный диизопинокамфеилборан и выделить его кристаллизацией. Этот промежуточный продукт отличается высокой чувствительностью к кислороду и воде, что усложняет его выделение. Энантиомерная чистота диизопинокамфеилхлорборана была увеличена до > 99% после кристаллизации при исходной оптической чистоте α-пинена приблизительно 90%. Такое увеличение энантиомерной чистоты диизопинокамфеилхлорборана при кристаллизации считают предельным для получения максимальной энантиоселективности при восстановлении кетонов в спирты (См. Brown Н. С. et al, J. Org. Chem., 1987, 52, 5406 и ссылки, приведенные в этой работе; Brown Н.С. et al, J. Org. Chem., 1986, 51, 3394; Srebnick M. et al, J. Org. Chem., 1988, 53, 2916; и Brown H.C. et al, J. Am. Chem. Soc., 1988, 1539).
В патенте США 5292946 показано, что полученный in situ диизопинокамфеилхлорборан, без выделения или отдельной очистки как конечного продукта, так и промежуточного диизопинокамфеилборана, ведет себя аналогично выделенному реагенту. Оба описанных в патентах США 5043479 и 5292946 способа требуют использования коррозионного реагента хлористого водорода, и, следовательно, не идеальны с точки зрения промышленного применения.
В другом способе (King А.О. et al, J. Org. Chem., 1993, 58, 3731-5) диизопинокамфеилхлорборан получают из монохлорборандиметилсульфидного комплекса и α-пинена. Источник бора в этом способе содержит дурно пахнущее соединение диметилсульфид, что делает его неподходящим реагентом для применения в больших количествах.
Все известные ранее способы требуют использования или борана или монохлорборана, которые оба являются дорогостоящими реагентами и очень чувствительными к кислороду и влаге. Таким образом, существует необходимость в экономичном, удобном и эффективном способе получения диизопинокамфеилхлорборанового продукта, используемого при восстановлении прохиральных кетонов с получением гидроксисоединений с высокой оптической чистотой.
Краткое описание изобретения
Настоящее изобретение направлено на получение диизопинокамфеилхлорборана in situ и его использование при восстановлении прохиральных кетонов до спиртов с высокой оптической чистотой.
Подробное описание изобретения
Настоящее изобретение предлагает способ получения диизопинокамфеилхлорборана, который включает:
- контактирование боргидрида натрия и α-пинена с трихлоридом бора в инертном растворителе с получением композиции, содержащей диизопинокамфеилхлорборан.
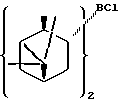
В предпочтительном варианте осуществления изобретения α-пинен представляет собой (1R)-(+)-α-пинен и диизопинокамфеилхлорборан имеет формулу I: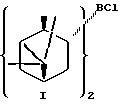
Используемое в данном описании выражение "композиция, содержащая диизопинокамфеилхлорборан" служит для того, чтобы показать, что названный продукт реакции диизопинокамфеилхлорборан никоим образом и ни в какой степени не выделяется из таких веществ, как растворитель, непрореагировавшие реагенты или возможных побочных реакционных продуктов, которые могут присутствовать в реакционном сосуде. Сокращение "ЭИ" ("ее") означает энантиомерный избыток. Термин "высокая оптическая чистота" означает соотношении энантиомерной пары, по меньшей мере, 95:5 в пользу требуемого энантиомера.
α-Пинен, используемый в настоящем способе, может представлять собой (1R)-(+)-α-пинен или (1S)-(-)-α-пинен, в котором энантиомерный избыток может быть низким, вплоть до приблизительно 70%. Таким образом, даже α-пинен с оптической чистотой приблизительно 70% обычно приводит к конечному спирту с оптической чистотой приблизительно 94%. α-Пинен с оптической чистотой менее приблизительно 70% также может быть использован с небольшой потерей оптической чистоты конечного спирта. В предпочтительном варианте осуществления изобретения используется (1R)-(+)-α-пинен с ЭИ 70% или выше.
Органический растворитель, используемый в настоящем способе, может быть любым, который практически не мешает проведению требуемой реакции. Предпочтительными растворителями являются полиоксигенированные эфиры. Примеры полиоксигенированных эфиров включают, но не ограничиваются только ими, 1,2-диметоксиэтан, диглим, триглим и им подобные соединения. Предпочтительным полиоксигенированным растворителем является 1,2-диметоксиэтан.
Реакцию можно проводить при температуре от приблизительно -40 до приблизительно 60oC. Предпочтительно во время добавления трихлорида бора к другим реагентам температуру реакционной смеси поддерживают при приблизительно 0oC или ниже; впоследствии температура может быть повышена до приблизительно 40oC. Реакции дают протекать по существу до завершения, что происходит за время от приблизительно 15 минут до приблизительно 2 часов; как правило, реакция по существу завершается за приблизительно 30 минут. Ход реакции можно контролировать способами, известными в данной области; например, потребления α-пинена может контролироваться с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) или путем определения отношения изопинокамфеола (получаемого окислением диизопинокамфеилхлорборана пероксидом водорода) к α-пинену с использованием газохроматографического анализа.
Реакцию предпочтительно проводят в инертной атмосфере, например в атмосфере азота.
Мольное отношение боргидрида натрия к трихлориду бора составляет предпочтительно 1:1.1. Мольное отношение α-пинена к боргидриду натрия и к трихлориду бора составляет по меньшей мере 4:1:1.1.
Полученная по описанному выше способу композиция, содержащая диизопинокамфеилхлорборан, может быть использована без дополнительной очистки для хирального восстановления прохиральных кетонов.
Таким образом, в соответствии с другим аспектом настоящего изобретения предлагается способ восстановления прохирального кетона с получением оптически активного спирта с высокой оптической чистотой, который включает:
(а) контактирование боргидрида натрия и трихлорида бора с α-пиненом, имеющим оптическую чистоту около 70% или выше, в инертном органическом растворителе с получением композиции, содержащей диизопинокамфеилхлорборан;
(б) взаимодействие прохирального кетона с композицией, полученной на стадии (а), с получением соответствующего оптически активного спирта с высокой оптической чистотой.
В предпочтительном варианте воплощения изобретения указанный α-пинен представляет собой (1R)-(+)-α-пинен, указанный диизопинокамфеилхлорборан имеет формулу I: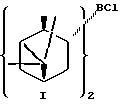
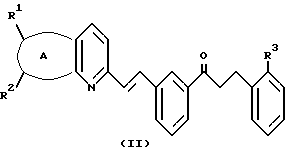
указанный кетон имеет формулу II: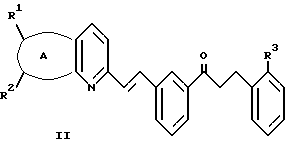
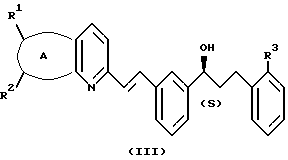
и указанный спирт имеет формулу III: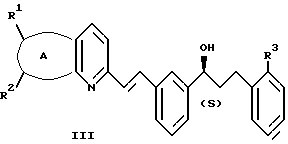
А представляет собой -CH=CH-S- или -CH=CH-CH=CH-;
заместители R1 и R2 независимо представляют собой водород или галоген;
заместитель R3 представляет собой CO2R6, COR6 или CO(R7)2-O-R8;
заместитель R6 представляет собой водород или низший алкил;
заместитель R7 представляет собой низший алкил; и
заместитель R8 представляет собой водород или гидроксизащитную группу; и указанный растворитель представляет собой полиоксигенированный эфир.
В более предпочтительном варианте осуществления изобретения заместители R1, R2 и бициклический гетероцикл, к которому они присоединены, вместе представляют собой 7-хлорхинолинильный фрагмент.
В способе настоящего изобретения стадию (а) проводят в соответствии с описанной ранее методикой. Стадию (б) способа предпочтительно проводят в эфирном растворителе. Более конкретно эфирные растворители включают, но не ограничиваются только ими, эфиры, такие как диэтиловый эфир, ди-н.-бутиловый и диизопентиловый эфиры, анизол, циклические эфиры, такие как тетрагидропиран, 4-метил-1,3-диоксан, дигидропиран, тетрагидрофуран, фуран, 2-метил-тетрагидрофуран и 2-этокситетрагидрофуран, наиболее предпочтительно использовать тетрагидрофуран.
Реакция может проводиться при температуре от -25 до 25oC, предпочтительно от приблизительно -20 до приблизительно 0oC. Реакции дают протекать по существу до завершения в течение от приблизительно 1 до 100 часов; при использовании прохирального кетона и диизопинокамфеилхлорборана предпочтительного варианта осуществления изобретения, конверсия до оптически активного спирта, как правило, составляет 90% в течение приблизительно 3 часов при температуре приблизительно -20oC; дальнейшее превращение может быть ускорено путем нагревания реакционной смеси до приблизительно 0oC. Реакцию предпочтительно проводят при давлении окружающей среды и в инертной атмосфере, например в азоте. По окончании требуемый оптически активный спирт может быть выделен обычными способами, например, с помощью кристаллизации, что приводит к желаемому оптическому изомеру с энантиомерным избытком до 99%.
Диизопинокамфеилхлорборан является полезным реагентом для восстановления прохиральных кетонов до соответствующих хиральных гидроксисоединений. Соединения формулы III иллюстрируют полезность хиральных гидроксисоединений. Соединения формулы III являются промежуточными при получении антагонистов лейкотриена формулы IV: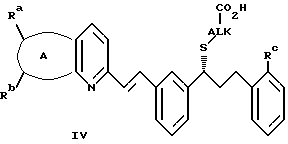
где А принимает значения, определенные ранее;
заместители Ra, Rb представляют собой, между прочим, водород или галоген; и
заместитель Rc может представлять собой CO2Rd, CORd или C(Re)2-OH;
заместитель Rd может представлять собой водород или низший алкил;
заместитель Re может представлять собой низший алкил;
ALK представляет собой, например, циклопропил-1,1-(бис)метилен, изопропил и т. д. Соединения формулы IV полезны как антиастматические, антиаллергические, противовоспалительные и цитозащитные терапевтические агенты. Получение этих антагонистов лейкотриена с использованием хиральных гидроксисоединений формулы III описано в патенте США 5270324 и в Европейской Патентной опубликованной заявке 604114, а также в находящейся на одновременном рассмотрении патентной заявке США с серийным номером 08/174931. Вышеупомянутые патент США, Европейская Патентная заявка и находящаяся на одновременном рассмотрении патентная заявка США также описывают получение предшественников прохиральных кетонов формулы II.
Следующие примеры представлены для иллюстрации настоящего изобретения и никоим образом не предназначены для ограничения объема изобретения, который определяется исключительно формулой изобретения.
Пример 1
Получение диизопинокамфеилхлорборана in situ
В круглодонную колбу объемом 250 мл помещают NaBH4 (1.89 г, 50.0 ммолей) и вытесняют азотом воздух. Добавляют диметоксиэтан (30 мл) и (+)-α-пинен (85% ЭИ, 31.8 мл, 200 ммолей) и полученную смесь охлаждают до -20oC. Добавляют раствор BCl3 (55 мл, 1.0 М в гептане, 55.0 ммолей) с такой скоростью, чтобы температура реакционной смеси не превышала 0oC (15 мин). Смесь выдерживают последовательно при 0oC в течение 15 минут, при комнатной температуре в течение 1 часа и при 40oC в течение 1 часа с получением хирального восстанавливающего агента диизопинокамфеилхлорборан.
Пример 2
Восстановление 2-(3-(3-(2-(7-хлор-2-хинолинил)этенил)- фенил)-3-оксопропил)бензоата до метил 2-(3-(3-(2-(7-хлор-2- хинолинил)этенил)фенил)-3(S)-гидроксипропил)бензоата.
В отдельной колбе в атмосфере азота готовят суспензию метил 2-(3-(3-(2-(7-хлор-2-хинолинил)этенил)фенил)-3-оксопропил)-бензоата (в дальнейшем - кетоэфир, 25.77 г, 97.3% вес. , 55 ммолей) в тетрагидрофуране (200 мл) и затем охлаждают до -20oC. К кетоэфиру добавляют охлажденную суспензию хирального восстанавливающего реагента примера 1 (при -20oC) и полученную смесь выдерживают при -20oC в течение нескольких часов и затем при 0oC в течение 1 часа.
Реакцию гасят бензальдегидом (15 мл), смесь нагревают до 40oC и выдерживают в течение 1.5 часов. После охлаждения до 20oC смесь медленно вливают в интенсивно перемешиваемый водный раствор K2CO3 (30% вес., 100 мл). Перемешивание продолжают до тех пор, пока все твердое вещество не растворится. Органический слой отделяют, фильтруют и затем концентрируют до 1/3 его исходного объема (вакуум 20-23 дюйма (508-584.2 мм), температура бани 40-50oC). Добавляют гептан (120 мл), затем воду (3 мл), чтобы вызвать кристаллизацию. Добавляют еще гептан (120 мл) и смесь выдерживают при комнатной температуре в течение 4 часов для завершения кристаллизации. После фильтрования и промывки осадка смесью ТГФ/гептан (1/5, до тех пор пока фильтрат не станет почти бесцветным, 120 мл) и сушки в вакуумном сушильном шкафу при температуре 40oC, получают 25.3 г названного моногидрата (S)-гидроксиэфира в виде желтого твердого вещества (выход 94.7%, в пересчете на степень чистоты 98% вес. ) ЭИ% составляет ≥ 99.0%.
Настоящее изобретение относится к улучшенному способу получения диизопинокамфеилхлорборана формулы I в реакционной смеси, который включает взаимодействие боргидрида натрия и трихлорида бора с α-пиненом в инертном растворителе. Полученный таким образом дииазопинокамфеилхлорборан может быть использован без выделения для восстановления прохиральных кетонов до их соответствующих спиртов с высокой оптической чистотой. Предложен также способ восстановления прохирального кетона формулы (II) с получением оптически активного спирта формулы (III), где А является -СН=СН-S- или - СН = СH - СН = СН-; R1 и R2 являются водородом или галогеном; R3 являются группами CO2R6, СОR6, C(R7)2 - OR8; R6 является водородом или низшим алкилом; R7 является низшим алкилом; R8 является водородом или гидроксизащитной группой; с высокой оптической чистотой контактированием боргидрида натрия и трихлорида бора с α-пиненом, имеющим оптическую чистоту около 70% или выше, в инертном органическом растворителе с получением реакционной смеси, содержащей диизопинокамфеилхлорборан, которую затем подвергают взаимодействию с прохиральным кетоном. Вышеуказанный α-пинен представляет собой (1R)-(+)-α-пинен, растворитель представляет полиалкоксилированный эфир. 2 c. и 5 з.п.ф-лы. 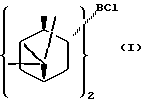
3. Способ по п.1, отличающийся тем, что растворитель представляет собой полиалкоксилированный эфир.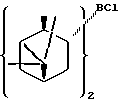
кетон имеет формулу II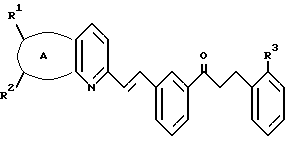
и спирт имеет формулу III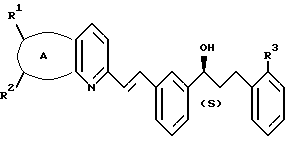
где А представляет собой -CH=CH-S- или -CH=CH-CH=CH-;
R1 и R2 независимо представляет собой водород или галоген;
R3 представляет собой CO2R6, COR6 или C(R7)2-O-R8;
R6 представляет собой водород или низший алкил;
R7 представляет собой низший алкил;
R8 представляет собой водород или гидроксизащитную группу,
и указанный растворитель представляет собой полиалкоксилированный эфир.
| US 5043479 A, 27.08.1991 | |||
| US 5292946 A, 08.03.1994 | |||
| US 5220072 A, 27.08.1991 | |||
| US 5162558 A, 27.10.1992 | |||
| RU 95115845 A1, 10.06.1997. |

