Область техники, к которой относится изобретение
Настоящее изобретение относится к новым тетралоновым или бензопираноновым производным и к способу получения этих производных. Новые тетралоновые или бензопираноновые производные по настоящему изобретению обладают ингибирующим действием в отношении 17β-гидроксистероиддегидрогеназы (в дальнейшем сокращенно 17β-ГСД) , поэтому такие производные могут быть использованы при приготовлении терапевтического средства для профилактики и/или лечения андроген- и эстрогензависимых заболеваний, в частности рака предстательной железы, доброкачественной гиперплазии предстательной железы, вирилизма, рака молочной железы, мастопатии, рака матки, эндометриоза, рака яичника и т.п.
Предпосылки создания изобретения
В последнее время в Японии вызывает тревогу рост заболеваемости андрогензависимыми болезнями, такими как рак предстательной железы и доброкачественная гиперплазия предстательной железы, и эстрогензависимыми болезнями, такими как рак молочной железы и эндометриоз. Так, например, по статистическим данным в 1984 г. смертность от рака предстательной железы составляла 3,9 человека на 100000 человек населения, что соответствовало приблизительно 1/10 для некавказцев в странах Запада. Однако увеличение продолжительности жизни благодаря улучшению медицинского обслуживания и характерному для западных стран пищевому рациону привело к постепенному росту этого показателя. В 1993 г. его доля составила 6,7 человек на 100000 человек населения и приближается к уровню европейских стран и США. Предполагается, что смертность от рака предстательной железы в 2015 г. будет в четыре раза выше, чем в 1990 г. Этот показатель роста является наихудшим из всех раковых заболеваний.
Если проанализировать многочисленные точки зрения, то становится ясно, что снижение содержания андрогена в крови при андрогензависимых заболеваниях приводит, вероятно, к улучшению субъективного состояния и объективного состояния. Таким образом, лечение этих заболеваний состоит в снижении содержания андрогена в крови путем кастрации, введения в организм вещества, обладающего сродством к LH-RH, для снижения уровня андрогена в крови до кастрационного уровня и введения антиандрогенных агентов, являющихся антагонистами андрогенного рецептора, для регулирования действия андрогена. Действительно, клинические результаты широко известны. Однако поскольку кастрация вызывает снижение QOL, к ней прибегают только в условиях очень ограниченного ряда заболеваний. Применение вещества, обладающего сродством к LH-RH, порождает проблемы, связанные с побочными эффектами, такими как боль в костях или дизурия, вызванная явлением, специфическим для вещества, обладающего сродством к рецептору (временное повышение уровня андрогена), и возобновление постоянного присутствия андрогена, продуцируемого надпочечниками. Более того, показано, что влияние антиандрогенных средств ослабляется расширением диапазона форм андрогенного рецептора в период введения в организм лекарственного средства. Следовательно, для более эффективной эндокринной терапии предписывается "метод полной блокады андрогена". Целью осуществления такого метода является полное подавление андрогена в крови за счет сочетания нескольких методов эндокринного лечения, от которого ожидают более эффективного лечения.
С участием 17β-ГСД из андростендиона в качестве субстрата возможен биосинтез тестостерона, проявляющего наиболее эффективное андрогенное действие среди C19 стероидов, обладающих андрогенным действием. Ингибирующее действие на эту 17β-ГСД приводит к такому непосредственному снижению концентрации тестостерона в крови, который, как предполагают, обеспечивает эффективное лечение вышеперечисленных андрогензависимых заболеваний. Кроме того, поскольку этот фермент представляет собой также фермент биосинтеза эстрадиола, проявляющего самое сильное эстрогенное действие среди C18 стероидов, обладающих эстрогенным действием, можно также рассчитывать на эффективное лечение эстрогензависимых заболеваний, таких как рак молочной железы и эндометриоз.
В качестве ингибиторов 17β-ГСД предложены стероидные соединения и нестероидные соединения. В качестве нестероидных соединений известны, например, флавоны и изофлавоны, которые описаны в Biochemical and Biophysical Research Communications, том 215, 1137-1144 (1995), а также жирные кислоты, которые описаны в Journal of Steroid Biochemistry, том 23, 357-363 (1985). Однако поскольку действие этих соединений оказывается неудовлетворительным, существует потребность в получении веществ, обладающих более сильным действием.
Описание изобретения
Принимая во внимание вышеуказанные проблемы, при создании настоящего изобретения были проведены тщательные исследования, в ходе которых было установлено, что на 17β-ГСД превосходное ингибирующее действие оказывают новые тетралоновые и бензопираноновые производные. Таким образом, задачей настоящего изобретения является создание новых тетралоновых и бензопираноновых производных и разработка способа получения этих производных.
Настоящее изобретение относится к новым тетралоновым и бензопираноновым производным и к способу получения этих производных. Предлагаемые по настоящему изобретению новые тетралоновые и бензопираноновые производные проявляют ингибирующее действие на 17β-гидроксистероиддегидрогеназу (17β-ГСД), поэтому такие производные могут быть использованы при приготовлении терапевтического средства для профилактики и/или лечения андроген- и эстрогензависимых заболеваний, в частности рака предстательной железы, доброкачественной гиперплазии предстательной железы, вирилизма, рака молочной железы, мастопатии, рака матки, эндометриоза, рака яичника и т.п.
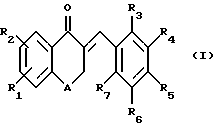
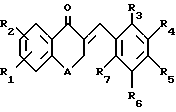
Предлагаемые по настоящему изобретению производные являются новыми тетралоновыми и бензопираноновыми производными следующей общей формулы (I):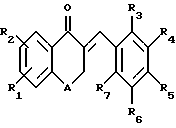 (I)
(I)
в которой
R1 и R2 каждый обозначает соответственно водород, гидроксильную группу, алкилоксигруппу или аралкилоксигруппу,
R3-R7 каждый обозначает водород, гидроксильную группу, прямую или разветвленную алкильную, алкилокси- или аралкилоксигруппу, содержащую 1-6 углеродных атомов, галоген, аминогруппу или алкилендиоксигруппу, присоединенную соответственно по месту положения R3 и R4, R4 и R5, R5 и R6 или R6 и R7, и
А обозначает метилен или кислород.
Кроме того, настоящее изобретение характеризуется тем, что для получения нового тетралонового или бензопиранонового производного в органическом растворителе растворяют тетралоновое или бензопираноновое соединение нижеследующей общей формулы (II), и бензальдегидное соединение нижеследующей общей формулы (III) и кипятят с обратным холодильником или проводят их взаимодействие в кислых условиях и очищают реакционный раствор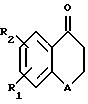 (II)
(II)
в которой R1 и R2 каждый обозначает соответственно водород, гидроксильную группу, алкилокси- или аралкилоксигруппу, а А обозначает метилен или кислород;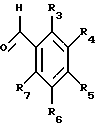 (III)
(III)
в которой R3-R7 каждый обозначает водород, гидроксильную группу, прямую или разветвленную алкильную, алкилокси- или аралкилоксигруппу, содержащую 1-6 углеродных атомов, галоген, аминогруппу или алкилендиоксигруппу, присоединенную соответственно по месту положения R3 и R4, R4 и R5, R5 и R6 или R6 и R7.
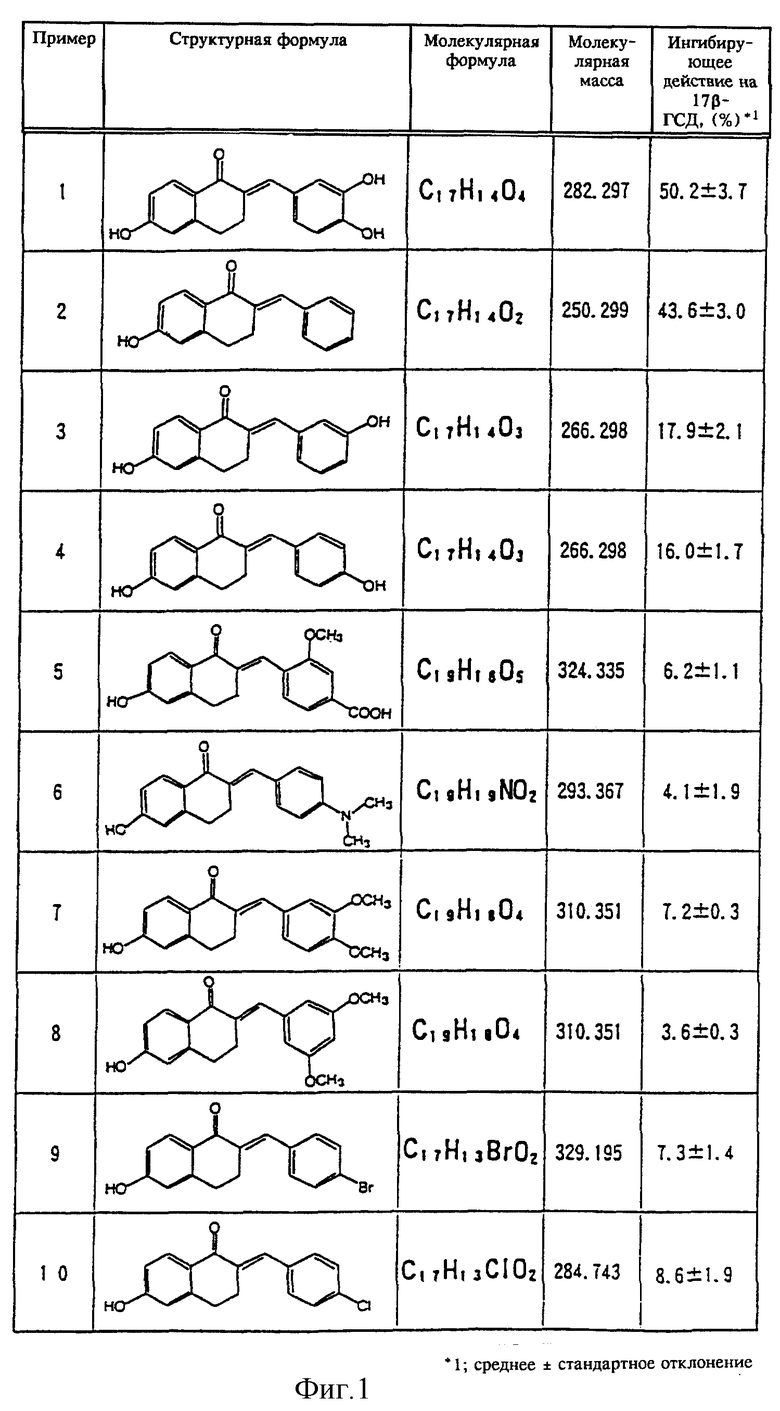
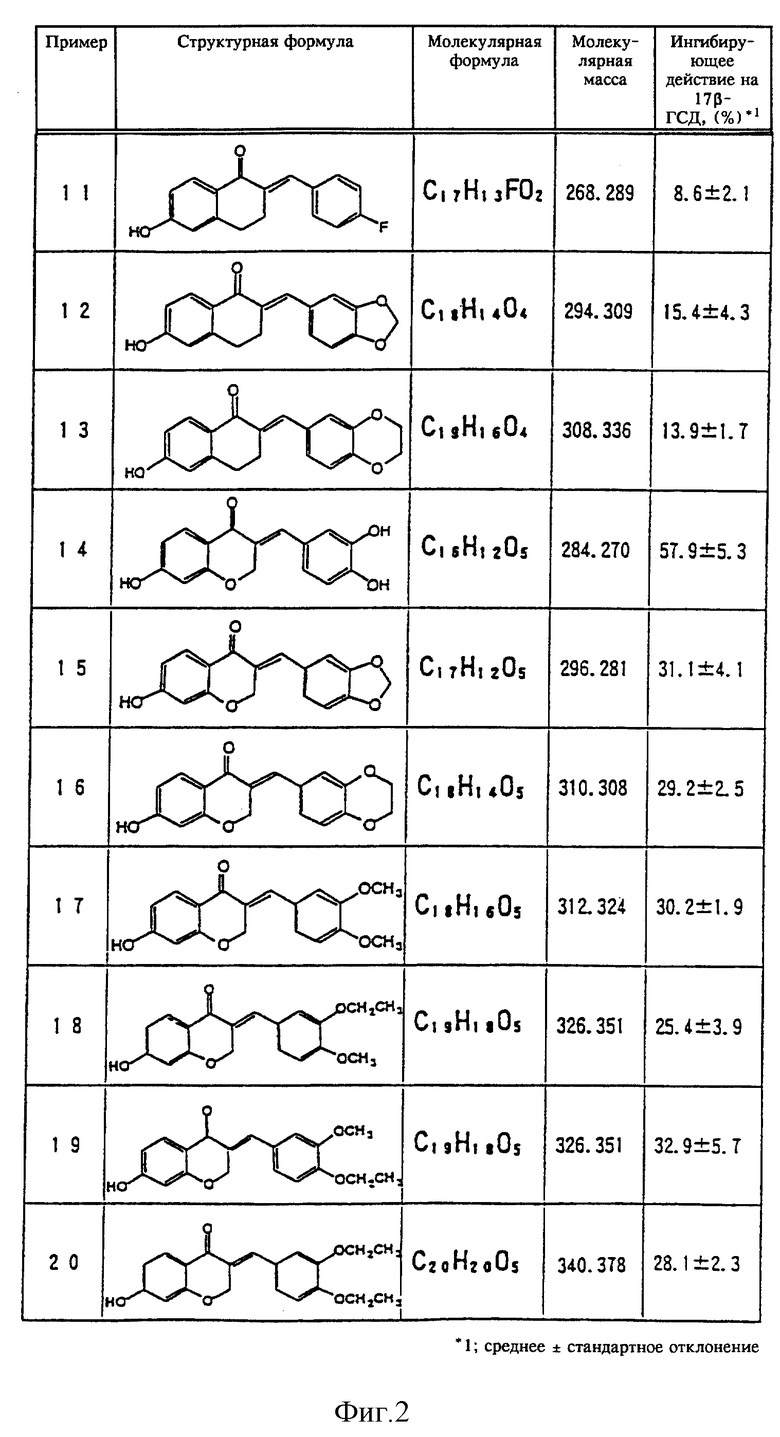
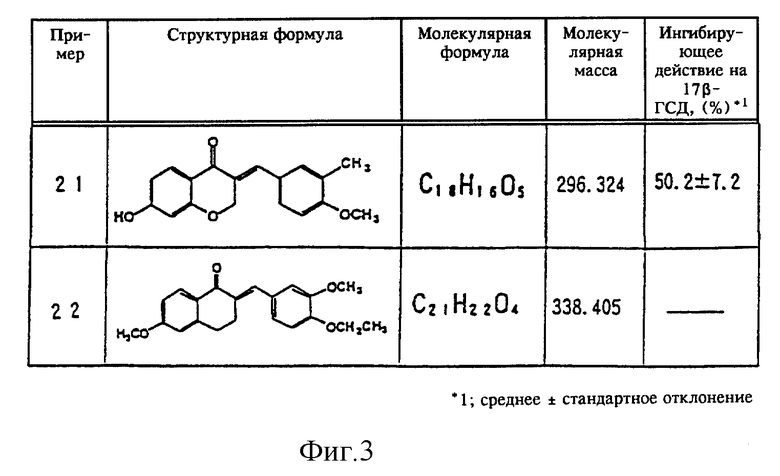
В качестве примеров новых тетралоновых и бензопираноновых производных общей формулы (I) можно привести следующие соединения (нумерация соединений и примеров, представленных на фиг. 1-3, совпадают):
(1) 2-[(3,4- дигидроксифенил)метилен]-6-гидрокси-1-тетралон,
(2) 2-(фенилметилен) -6-гидрокси-1-тетралон,
(3) 2-[(3-гидроксифенил)метилен]-6-гидрокси-1-тетралон,
(4) 2-[(4- гидроксифенил)метилен]-6-гидрокси-1-тетралон,
(5) 2-[(2- метоксифенил-4-карбоновая кислота)метилен] -6-гидрокси-1 - тетралон,
(6) 2-[(4-диметиламинофенил)метилен]-6-гидрокси-1-тетралон,
(7) 2-[(3,4-диметоксифенил)метилен]-6-гидрокси-1-тетралон,
(8) 2-[(3,5-диметоксифенил)метилен]-6-гидрокси-1-тетралон,
(9) 2-[(4-бромфенил)метилен]-6-гидрокси-1-тетралон,
(10) 2-[(4-хлорфенил)метилен]-6-гидрокси-1-тетралон,
(11) 2-[(4-фторфенил)метилен]-6-гидрокси-1-тетралон,
(12) 6-гидрокси-2-пиперонилиден-1-тетралон,
(13) 2-[(3,4-бензодиоксан)-6-метилен]-6-гидрокси-1-тетралон,
(14) 2-[(3,4-дигидроксифенил)метилен]-7- гидрокси-4(4H)бензопиранон,
(15) 7-гидрокси-3-пиперонилиден-4(4Н)бензопиранон,
(16) 3-[(1,4-бензодиоксан)-6-метилен] -7-гидрокси-4(4Н)бензопиранон,
(17) 3-[(3,4-диметоксифенил)метилен]-7-гидрокси-4(4Н)бензопиранон,
(18) 3-[(3-этокси-4-метоксифенил)метилен]-7-гидрокси-4(4Н)бензопиранон,
(19) 3-[(3-метокси-4-этоксифенил)метилен]-7-гидрокси-4(4Н)бензопиранон,
(20) 3-[(3,4-диэтоксифенил)метилен]-7-гидрокси-4(4Н)бензопиранон,
(21) 3-[(3-метил-4-метоксифенил)метилен]-7-гидрокси-4(4Н)бензопиранон,
(22) 2-метокси[(3-метокси-4-этоксифенил)метилен]-6-метокси-1-тетралон.
Помимо вышеперечисленных соединений, производные по настоящему изобретению включают стереоспецифические изомеры этих соединений и их соли, образуемые кислотами и основаниями. В качестве примеров солей оснований можно упомянуть, в частности, соли неорганических оснований, образуемых натрием, калием, магнием, кальцием и алюминием, соли органических оснований, образуемых низшими алкиламинами и низшими аминоспиртами, соли основных аминокислот, таких как лизин, альгинин и орнитин, а также аммониевые соли. Далее эти производные могут образовывать гидраты, сольваты низших спиртов и кристаллические полиморфы.
Производные по настоящему изобретению могут быть получены по следующим способам. Так, например, вышеупомянутые тетралоновые и бензопираноновые соединения (II) и вышеуказанные бензальдегидные соединения (III) растворяют в растворителе, таком как метанол, этанол или пропанол, добавляют концентрированную соляную кислоту, раствор кипятят с обратным холодильником в течение 1-24 ч и охлаждают, а выпавшие в осадок кристаллы отфильтровывают, получая целевые новые производные (I) по настоящему изобретению. Когда кристаллы не осаждаются, для осаждения кристаллов добавляют 100-400 мл воды, кристаллы отфильтровывают и сушат, получая целевые производные по настоящему изобретению. В другом варианте к соединениям формул (II) и (III) в растворителе, таком как метанол, этанол или пропанол, добавляют гидроксид натрия или гидроксид калия, раствор перемешивают в течение 1-24 ч, подкисляют соляной кислотой, а выпавшие в осадок кристаллы отфильтровывают, получая целевые производные по настоящему изобретению. Кроме того, целевые производные по настоящему изобретению могут быть получены растворением упомянутых соединений в насыщенном растворе газообразного хлористого водорода в органическом растворителе, таком как метанол, этанол, пропанол или диэтиловый эфир, после чего раствор охлаждают и оставляют стоять при комнатной температуре либо раствор нагревают, перемешивают в течение 1-24 ч, добавляют воды для осаждения целевых производных в виде кристаллов и выпавшие в осадок кристаллы отфильтровывают.
Эти производные по настоящему изобретению безвредны при пероральном и парентеральном введении в форме лекарственных средств людям и животным. В качестве примеров парентеральных путей введения этих лекарственных средств можно назвать внутривенную инъекцию, внутримышечную инъекцию, подкожную инъекцию, внутрибрюшинную инъекцию, введение через кожу, введение через легкие, интраназальное введение, введение через кишечник, введение через полость рта и введение через слизистую оболочку. В качестве примеров форм лекарственных средств можно упомянуть препараты для инъекций, суппозитории, аэрозольные средства, пластыри для абсорбции через кожу и т.п. В качестве примеров фармацевтически приемлемых препаративных лекарственных форм для перорального введения можно назвать таблетки (включая таблетки с сахарным покрытием, таблетки в оболочке и таблетки для трансбуккального введения), порошки, капсулы (включая мягкие капсулы), гранулы (включая гранулы с покрытием), пилюли, пастилки, жидкие препараты или препараты с непрерывным высвобождением таких лекарственных средств. Примерами жидких препаратов для перорального введения являются суспензии, эмульсии, сиропы (включая сухие сиропы) и эликсиры. Эти фармацевтические препараты готовят по обычным методам приготовления фармацевтической продукции; их вводят в организм в виде лекарственных композиций совместно с фармакологически приемлемыми носителями, наполнителями, разрыхлителями, замасливателями, красителями и т.п.
В качестве примеров носителей и наполнителей, используемых в составе этих фармацевтических препаратов, можно назвать лактозу, глюкозу, сахарозу, маннит, картофельный крахмал, кукурузный крахмал, карбонат кальция, фосфат кальция, сульфат кальция, кристаллическую целлюлозу, порошкообразный лакричник и порошкообразную горечавку. Примерами связующих веществ являются крахмал, трагант, аравийская камедь, желатин, сироп, поливиниловый спирт, поливиниловый эфир, поливинилпирролидон, гидроксипропилцеллюлоза, метилцеллюлоза, этилцеллюлоза и карбоксиметилцеллюлоза. Примерами разрыхлителей являются крахмал, агар, порошкообразный желатин, натрий- карбоксиметилцеллюлоза, кальций-карбоксиметилцеллюлоза, кристаллическая целлюлоза, карбонат кальция, бикарбонат кальция и натрийальгиновая кислота. В качестве замасливателей можно использовать стеарат магния, тальк, гидрогенизованное растительное масло и макрогол (macrogol). В качестве красителей можно использовать вещества, которые пригодны для введения в состав лекарственных средств.
Таблетки и гранулы можно покрывать сахарозой, желатином, гидроксипропилцеллюлозой, очищенным шеллаком, глицерином, сорбитом, этилцеллюлозой, гидроксипропилцеллюлозой, гидроксипропилметилцеллюлозой, поливинилпирролидоном, ацетилфталилцеллюлозой, фталатом гидроксипропилметилцеллюлозы, метилметакрилатом, полимером метакриловой кислоты и т.п., причем можно наносить одно или несколько покрытий. Можно использовать капсулы из этилцеллюлозы и желатина. Более того, при приготовлении препаратов для инъекций в основу по обычному методу можно вводить при необходимости агент для регулирования pH, буферный агент, стабилизатор, солюбилизирующую добавку и т.п.
При введении пациентам производных по настоящему изобретению доза конкретно не ограничивается вследствие различия условий, таких как состояние болезни, возраст пациента, относительное состояние здоровья и вес. Она составляет примерно 1-1000 мг/день для взрослого пациента, предпочтительно 50-200 мг, перорально или парентерально по одному или несколько раз в день.
Кроме того, производные по настоящему изобретению могут быть использованы при изготовлении полупроводниковых устройств и т.п. в составе композиции фоторезистов позитивного типа, которая обеспечивает высокочувствительное разрешение, обладает хорошими свойствами по проявлению, обладает теплостойкостью и позволяет получать фоторезистные изображения с исключительно высокой точностью.
Краткое описание чертежей
На фиг. 1 представлены результаты, полученные в примерах.
На фиг. 2 представлены результаты, полученные в примерах.
На фиг. 3 представлены результаты, полученные в примерах.
Предпочтительный вариант выполнения изобретения
Приведенные ниже примеры представлены только для иллюстрации сущности настоящего изобретения, и поэтому объем изобретения не ограничен этими примерами.
Пример 1
Синтез 2-[(3,4-дигидроксифенил)метилен]-6-гидрокси-1-тетралона
После добавления 1,0 г 6-гидрокси-1-тетралона и 0,85 г 3,4-дигидрокси-бензальдегида в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение 2,5 ч, охлаждали до комнатной температуры и добавляли 357 мл воды. Выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение шести часов под пониженным давлением с получением 1,25 г целевого соединения.
FAB-MC (М+1); 283.
1H-ЯМР (част./млн в ДМСО-d6): 2,83 (2H, m), 3,02 (2H, m), 6,66 (1H, d, J = 2,1 Гц), 6,75 (1H, dd, J = 8,5, 2,4 Гц), 6,79 (1H, d,J = 7,9 Гц), 6,83 (1H, dd, J = 8,3, 1,9 Гц), 6,94 (1H, d, J= 1,8 Гц), 7,49 (1H, s), 7,81 (1H, d, J = 8,5 Гц), 9,09 (1H, s), 9,35 (1H, s), 10,31 (1H, s).
Пример 2
Синтез 2-(фенилметилен)-6-гидрокси-1-тетралона
После добавления 1,0 г 6-гидрокси-1-тетралона и 0,42 мл бензальдегида в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение двух часов, охлаждали до комнатной температуры и добавляли 400 мл воды. Выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение шести часов под пониженным давлением с получением 0,756 г целевого соединения.
FAB-MC (М+1); 251.
1H-ЯМР (част./млн в ДМСО-d6): 2,83 (2H, m), 3,02 (2H, m), 6,66 (1H, s), 6,76 (1H, d, J = 8,6 Гц), 7,44 (4H, m), 7,62 (1H, s), 7,84 (1H, d, J = 8,8 Гц), 10,37 (1H, s).
Пример 3
Синтез 2-[(3-гидроксифенил)метилен]-6-гидрокси-1-тетралона
После добавления 1,0 г 6-гидрокси-1-тетралона и 0,903 г 3-гидроксибензальдегида в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение одного часа, охлаждали до комнатной температуры и добавляли 400 мл воды. Выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение шести часов под пониженным давлением с получением 0,92 г целевого соединения.
FAB-MC (М+1); 267.
1H-ЯМР (част./млн в ДМСО-d6): 2,83 (2H, m), 2,99 (2H, m), 6,66 (1H, d, J = 2,4 Гц), 6,76 (2H, m), 6,98 (2H, m), 7,23 (1H, t, J= 7,0 Гц), 7,53 (1H, s), 7,84 (1H, d, J= 8,4 Гц), 9,54 (1H, s), 10,39 (1H, s).
Пример 4
Синтез 2- [(4-гидроксифенил)метилен]-6-гидрокси-1-тетралона
После добавления 1,0 г 6-гидрокси-1-тетралона и 0,903 г 4-гидроксибензальдегида в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение одного часа, охлаждали до комнатной температуры, добавляли 200 мл воды и оставляли стоять в течение одного часа. Выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 0,941 г целевого соединения.
FAB-MC (М+1); 267.
1H-ЯМР(част./млн в ДМСО-d6: 2,81 (2H, m), 3,00 (2H, m), 6,65 (1H, d, J = 2,4 Гц), 6,75 (1H, dd, J = 8,5, 2,1 Гц), 6,84 (2H, d, J = 8,5 Гц), 7,35 (1H, d, J = 8,8 Гц), 7,55 (1H, s), 7,82 (1H, d, J = 8,5 Гц), 9,84 (1H, s), 10,33 (1H, s).
Пример 5
Синтез 2- [(2- метоксифенил-4-карбоновая кислота)метилен] -6-гидрокси-1- тетралона
После добавления 1,0 г 6-гидрокси-1-тетралона и 1,44 г 4-формил-2-метоксифенилацетата в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение одного часа, охлаждали до комнатной температуры, добавляли 400 мл воды и оставляли стоять в течение двух часов. Выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 0,671 г целевого соединения.
FAB-MC (М+1);325.
1H-ЯМР (част./млн в ДМСО-d6): 2,82 (2H, m), 3,05 (2H, m), 3,81 (3H, s), 6,66 (1H, d, J = 2,1 Гц), 6,75 (1H, dd, J = 8,2, 2,1 Гц), 6,66 (1H, d, J = 2,1 Гц), 6,98 (1H, dd, J = 8,2, 1,8 Гц), 7,07 (1H, d, J = 1,8 Гц), 7,58 (IH, s), 7,83 (1H, d, J = 8,5 Гц).
Пример 6
Синтез 2-[(4-диметиламиносфенил)метилен]-6-гидрокси-1-тетралона
После добавления 1,0 г 6-гидрокси-1- тетралона и 0,97 г 4-диметиламино-бензальдегида в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение 1,5 ч, охлаждали до комнатной температуры и добавляли 400 мл воды. Выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение шести часов под пониженным давлением с получением 0,885 г целевого соединения.
FAB-MC (М+1);294.
1H-ЯМР (част./млн в ДМСО-d6): 2,81 (2H, m), 2,98 (6H, s), 3,03 (2H, m), 6,66 (1H, d,J= 2,5 Гц), 6,75 (1H, dd, J = 8,5, 2,4 Гц), 6,83 (1H, d, J = 8,8 Гц), 7,40 (1H, d,J= 8,8 Гц), 7,58 (1H, s), 7,82 (1H, d, J= 8,5 Гц).
Пример 7
Синтез 2-[(3,4-диметоксифенил)метилен]-6-гидрокси-1-тетралона
После добавления 1,0 г 6-гидрокси-1-тетралона и 1,23 г 3,4-диметоксибензальдегида в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение 1,5 ч, охлаждали до комнатной температуры и выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение 5,5 ч под пониженным давлением с получением 1,60 г целевого соединения.
FAB-MC(М+1);311.
1H-ЯМР (част./млн в ДМСО-d6): 2,82 (2H, m), 3,06 (2H, m), 3,80 (6H, s), 6,66 (1H, d, J= 2,1 Гц), 6,76 (1H, dd, J = 8,5, 2,4 Гц), 7,01 (1H, d, J = 8,8 Гц), 7,07 (2H, m), 7,60 (1H, s), 7,84 (1H, d, J = 8,5 Гц), 10,19 (1H, s).
Пример 8
Синтез 2-[(3,5-диметоксифенил)метилен]-6-гидрокси-1-тетралона
После добавления 1,0 г 6-гидрокси-1-тетралона и 1,23 г 3,5-диметоксибензальдегида в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение 1,5 ч, охлаждали до комнатной температуры и выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 1,276 г целевого соединения.
FAB-MC (М+1);311.
1H-ЯМР (част./млн в ДМСО-d6): 2,82 (2H, m), 2,93 (2H, s), 3,83 (3H, s), 3,87 (3H, s), 6,57 (1H, dd, J = 8,2, 2,1 Гц), 6,63 (1H, dd, J = 8,6, 2,1 Гц), 6,77 (1H, dd, J = 8,5, 2,4 Гц), 7,29 (1H, d, J = 8,5 Гц), 7,83 (1H, s), 7,84 (1H, d, J = 8,5 Гц).
Пример 9
Синтез 2-[(4-бромфенил)метилен]-6-гидрокси-1-тетралона
После добавления 1,0 г 6-гидрокси-1-тетралона и 1,37 г 4-бромбензальдегида в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение одного часа, охлаждали до комнатной температуры и добавляли 250 мл воды. Выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 0,72г целевого соединения.
FAB-MC (М+1); 330.
1H-ЯМР (част./млн в ДМСО-d6):2,82 (2H, m), 2,97 (2H, m), 6,66 (1H, d, J = 1,5 Гц), 6,77 (1H, dd, J = 8,5, 2,4 Гц), 7,43 (2H, d, J = 8,5 Гц), 7,56 (1H, s), 7,61 (2H, d, J = 8,5 Гц), 7,85 (1H, d, J = 8,5 Гц), 10,42 (1H, s).
Пример 10
Синтез 2-[(4-хлорфенил)метилен]-6-гидрокси-1-тетралона
После добавления 1,0 г 6-гидрокси-1-тетралона и 1,03 г 4-хлорбензальдегида в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение 1,5 ч, охлаждали до комнатной температуры и добавляли 250 мл воды. Выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 0,77г целевого соединения.
FAB-MC (М+1); 285.
1H-ЯМР (част./млн в ДМСО-d6): 2,82 (2H, m), 2,98 (2H, m), 6,66 (1H, d, J = 2,1 Гц), 6,77 (1H, dd, J = 8,5, 2,2 Гц), 7,48 (5H, m), 7,59 (1H, s), 7,85 (1H, d, J = 8,5 Гц), 10,41 (1H, s).
Пример 11
Синтез 2-[(4-фторфенил)метилен]-6-гидрокси-1-тетралона
После добавления 1,0 г 6-гидрокси-1-тетралона и 0,79 г 4-фторбензальдегида в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение 1,5 ч, охлаждали до комнатной температуры и добавляли 300 мл воды. Выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 0,65 г целевого соединения.
FAB-MC (М+1); 269.
1H-ЯМР (част./млн в ДМСО-d6): 2,81 (2H, m), 2,98 (2H, m), 6,66 (1H, d, J = 2,2 Гц), 6,77 (1H, dd, J = 8,5, 2,5 Гц), 7,25 (2H, m), 7,53 (2H, m), 7,61 (1H, s), 7,84 (1H, d, J = 8,5 Гц), 10,41 (1H, s).
Пример 12
Синтез 6-гидрокси-2-пиперонилиден-1-тетралона
После добавления 1,0 г 6-гидрокси-1-тетралона и 1,11 г пипероналя в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение 0,5 ч, охлаждали до комнатной температуры, и выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 1,63 г целевого соединения.
FAB-MC (М+1); 295.
1H-ЯМР (част./млн в ДМСО-d6): 2,82 (2H, m), 3,01 (2H, m), 6,06 (2H, s), 6,65 (1H, d, J = 2,1 Гц), 6,75 (1H, dd, J = 8,5, 2,4 Гц), 6,98 (1H, d, J = 7,9 Гц), 7,02 (1H, dd, J = 9,4, 1,1 Гц), 7,08 (1H, d, J = 1,5 Гц), 7,55 (1H, s), 7,82 (1H, d, J = 8,5 Гц), 10,37 (1H, s).
Пример 13
Синтез 2-[(3,4-бензодиоксан)-6-метилен]-6-гидрокси-1-тетралона
После добавления 1,0 г 6-гидрокси-1-тетралона и 1,21 г 1,4-бензодиоксан-6-карбальдегида в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение одного часа, охлаждали до комнатной температуры и выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 1,57 г целевого соединения.
FAB-MC (М+1); 309.
1H-ЯМР (част./млн в ДМСО-d6): 2,81 (2H, m), 3,00 (2H, m), 4,26 (4H, m), 6,65 (1H, d, J = 2,4 Гц), 6,75 (1H, dd, J = 8,5, 2,1 Гц), 6,90 (1H, d, J = 8,2 Гц), 6,98 (1H, dd, J = 8,5, 1,8 Гц), 7,01 (1H, d, J= 2,1 Гц), 7,52 (1H, s), 7,82 (1H, d, J = 8,6 Гц), 10,36 (1H, s).
Пример 14
Синтез 2-[(3,4-дигидроксифенил)метилен]-7-гидрокси-4(4H)- бензопиранона
После добавления 0,5 г 7-гидрокси-4(4H)- бензопиранона и 0,42 г 3,4-дигидроксибензальдегида в смесь 25 мл концентрированной соляной кислоты и 35 мл метанола смесь кипятили с обратным холодильником в течение 2,5 ч, охлаждали до комнатной температуры и добавляли 250 мл воды. Смесь оставляли стоять в течение 18 ч, а выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 0,61 г целевого соединения.
FAB-MC(M+1); 285.
1H-ЯМР (част. /млн в ДМСО-d6): 5,34 (2H, d,J= 1,6 Гц), 6,29 (1H, d, J = 2,1 Гц), 6,53 (1H, dd, J = 8,6, 2,2 Гц), 6,74 (1H, dd, J = 8,2, 1,8 Гц), 6,82 (2H, m), 7,51 (1H, s), 7,71 (1H, d,J= 8,8 Гц), 9,19 (1H, s), 9,55 (1H, s), 10,57 (1H, s).
Пример 15
Синтез 7-гидрокси-3-пиперонилиден-4(4H) -бензопиранона
После добавления 1,0 г 7-гидрокси-4(4H)-бензопиранона и 1,0 г пипероналя в смесь 50 мл концентрированной соляной кислоты и 75 мл метанола смесь кипятили с обратным холодильником в течение двух часов, охлаждали до комнатной температуры и добавляли 200 мл воды. Смесь оставляли стоять в течение одного часа, а выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 0,266 г целевого соединения.
FAB-MC (М+1);297.
1H-ЯМР (част. /млн в ДМСО-d6): 5,33 (2H, d, J = 1,6 Гц), 6,09 (1H, s), 6,31 (1H, d, J = 2,1 Гц), 6,53 (1H, dd, J = 8,8, 2,5 Гц), 6,93 (1H, dd, J = 7,9, 1,2 Гц), 7,02 (2H, m), 7,59 (1H, s), 7,72 (1H, d, J = 8,8 Гц), 10,64 (1H, s).
Пример 16
Синтез 3-[(1,4-бензодиоксан)-6-метилен]-7-гидрокси-4(4H)-бензопиранона
После добавления 1,0 г 7-гидрокси-4(4H)-бензопиранона и 1,2 г 1,4-бензодиоксан-6-карбоксальдегида в смесь 50 мл концентрированной соляной кислоты и 40 мл метанола смесь кипятили с обратным холодильником в течение 1,5 ч, охлаждали до комнатной температуры и выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 1,066 г целевого соединения.
FAB-MC (М+1); 311.
1H-ЯМР (част. /млн в ДМСО-d6): 4,29 (4H, m), 5,33 (2H, d, J= 1,6 Гц), 6,31 (1H, d, J = 2,1 Гц), 6,53 (1H, dd, J = 8,8, 2,4 Гц), 6,89-6,96 (3H, m), 7,56 (1H, s), 7,72 (1H, d,J = 8,5 Гц), 10,62 (1H, s).
Пример 17
Синтез 3-[(3,4-диметоксифенил)метилен]-7-гидрокси-4(4H)-бензопиранона
После добавления 1,0 г 7-гидрокси-4(4H)бензопиранона и 1,22 г 3,4- диметоксибензальдегида в смесь 50 мл концентрированной соляной кислоты и 40 мл метанола смесь кипятили с обратным холодильником в течение 1,5 ч, охлаждали до комнатной температуры, добавляли 100 мл воды, и смесь оставляли стоять в течение 30 мин. Выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение семи часов под пониженным давлением с получением 0,255 г целевого соединения.
FAB-MC (М+1); 313.
1H-ЯМР (част./млн в ДМСО-d6): 3,80 (3H, s), 3,81 (3H, s), 5,39 (2H, d), 6,32 (1H, d, J= 2,1 Гц), 6,54 (1H, dd, J = 8,8, 2,2 Гц), 6,97- 7,05 (3H, m), 7,63 (1H, s), 7,73 (1H, d, J = 8,8 Гц).
Пример 18
Синтез 3-[(3-этокси-4-метоксифенил)метилен] -7-гидрокси-4(4H)-бензопиранона
После добавления 15 мл насыщенного метанольного раствора хлорида водорода к 1,0 г 7-гидрокси-4(4H)- бензопиранона и 1,30 г 3-этокси-4-метоксибензальдегида смесь перемешивали в течение 22,5 ч, добавляли 100 мл воды и выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы добавляли в 30 мл метанола при 55oC и смесь фильтровали. Образовывавшиеся кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 0,415 г целевого соединения.
FAB-MC (М+1); 327.
1H-ЯМР (част./млн в ДМСО-d6): 1,33 (3H, t), 3,81 (3H, s), 4,05 (2H, q), 5,38 (2H, d), 6,31 (1H, d, J= 2,5 Гц), 6,53 (1H, dd, J = 8,5, 2,1 Гц), 6,96-7,05 (3H, m), 7,62 (1H, s), 7,73 (1H, d, J = 8,5 Гц).
Пример 19
Синтез 3-[(3-метокси-4-этоксифенил)метилен] -7-гидрокси-4(4H)-бензопиранона
После добавления 20 мл насыщенного метанольного раствора хлорида водорода к 1,0 г 7-гидрокси-4(4H)-бензопиранона и 1,30 г 3-метокси-4-этоксибензальдегида смесь перемешивали в течение 18 ч, добавляли 100 мл воды, и выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы добавляли в 100 мл метанола при 55oC, смесь перемешивали в течение 15 мин и фильтровали. Образовывавшиеся кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 0,216 г целевого соединения.
FAB-MC (М+1); 327.
1H-ЯМР (част. /млн в ДМСО-d6: 1,33 (3H, t), 3,80 (3H, s), 4,05 (2H, q), 5,38 (2H, d), 6,31 (1H, d, J = 2,1 Гц), 6,53 (1H, dd, J = 8,5, 2,1 Гц), 6,93 - 7,03 (3H, m), 7,63 (1H, s), 7,73 (1H, d, J = 8,8 Гц).
Пример 20
Синтез 3-[(3,4-диэтоксифенил)метилен] -7-гидрокси-4(4H)-бензопиранона
После добавления 20 мл насыщенного метанольного раствора хлорида водорода к 1,0 г 7-гидрокси-4(4H)- бензопиранона и 1,58 мл 3,4-диэтоксибензальдегида смесь перемешивали в течение 72 ч, добавляли 100 мл воды, и выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы добавляли в 60 мл метанола при 55oC, смесь перемешивали в течение 15 мин и фильтровали. Образовывавшиеся кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 0,762 г целевого соединения.
FAB-MC (М+1); 341.
1H-ЯМР (част./млн в ДМСО-d6): 1,33 (6H, t), 4,07 (4H, q), 5,37 (2H, d), 6,31 (1H, d, J = 2,4 Гц), 6,53 (1H, dd, J = 8,8, 2,4 Гц), 6,93 - 7,02 (3H, m), 7,62 (1H, s), 7,73 (1H, d, J = 8,6 Гц).
Пример 21
Синтез 3-[(3-метил-4-метоксифенил)метилен] -7-гидрокси-4(4Н)-бензопиранона
После добавления 20 мл насыщенного метанольного раствора хлорида водорода к 1,0 г 7-гидрокси-4(4H)-бензопиранона и 0,87 мл 3-метил-4-метоксибензальдегида смесь перемешивали в течение 72 ч, добавляли 100 воды и выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы добавляли в 50 мл метанола при 55oC, смесь перемешивали в течение 15 мин и фильтровали. Образовывавшиеся кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 0,447 г целевого соединения.
FAB-MC (М+1); 297.
1H-ЯМР (част./млн в ДМСО-d6): 2,17 (3H, s), 3,83 (3H, s), 5,35 (2H, d), 6,31 (1H, d, J = 2,2 Гц), 6,53 (1H, dd, J = 8,6, 2,2 Гц), 7,02 (1H, d, J = 8,5 Гц), 7,24 (2H, m), 7,59 (1H, s), 7,73 (1H, d, J = 8,6 Гц).
Пример 22
Синтез 2-метокси [(3-метокси-4- этоксифенил)метилен]-6-метокси-1-тетралона
После добавления 1,0 г 6-метокси-1-тетралона и 1,27 г 3-метокси-4-этоксибензальдегида в смесь 60 мл концентрированной соляной кислоты и 50 мл метанола смесь кипятили с обратным холодильником в течение 2,5 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли 100
100 мл насыщенного солевого раствора. Этилацетатный раствор обезвоживали сульфатом магния и при 40oC под пониженным давлением концентрировали до остаточного объема 20 мл. Выпадавшие в осадок кристаллы отфильтровывали. Эти кристаллы сушили над пентоксидом фосфора в течение четырех часов под пониженным давлением с получением 0,89 г целевого соединения.
FAB-MC (М+1); 339.
1H-ЯМР (част. /млн в CDCl3): 1,46 (3H, t), 2,89 (2H, m), 3,10 (2H, m), 3,83 (3H, s), 3,86 (3H, s), 4,22 (2H, q, 4 Гц), 6,67 (1H, d, J= 8,2 Гц), 6,83 (1H, dd, J = 8,3, 1,9 Гц), 6,85 (1H, d, J = 8,2 Гц), 6,65 (1H, d, J= 1,8 Гц), 7,00 (1H, dd, J = 8,5, 2,1 Гц), 7,65 (1H, s), 8,07 (1H, d, J = 8,8 Гц).
Испытание ингибирующего действия на 17β-ГСД in vitro
Оценивали ингибирующее действие на 17β-ГСД соединений, полученных в примерах 1-22 (в дальнейшем сокращенно называемых испытываемым материалом). Конкретно каждый испытываемый материал растворяли в этаноле с получением раствора конечной концентрации 260 нМ, который помещали в пробирку и упаривали досуха в газообразном азоте. К этому материалу добавляли 590 мкл буферного раствора 10 мМ фосфата (при pH 7,5), содержавшего 100 мМ хлорида калия, 1 мМ этилендиаминтетрауксусной кислоты, 0,5 мМ никотинамидадениндинуклеотидфосфата восстанавливающего типа (все химикаты приобретены у фирмы Wako Junyaku Company) и 1 мкМ [4- 14С]эстрона (фирма NEN Research Products Company), и 10 мкл микросомовой фракции, полученной из человеческой плаценты в соответствии с методом Е.А. Thompson и др. [J. Biol. Chem., том 249, 5364-5372 (1974)] , и при температуре 37oC при встряхивании в течение 30 мин в этой смеси проводили реакцию.
Сразу же по завершении реакции добавляли 2 мл дихлорметана. Смесь тщательно перемешивали и центрифугировали в течение пяти минут при скорости вращения 3000 об/мин. Образовавшийся нижний слой (дихлорметановый слой) удаляли в другую пробирку и упаривали досуха. В эту пробирку добавляли 100 мкл этанола, содержавшего 20 мкг эстрона и 20 мкг эстрадиола, и 20 мкл этой смеси в качестве пробы наносили на пластинку для тонкослойной жидкостной хроматографии (силикагель 60 F254, фирма Merck Company). После проявления пластинки для тонкослойной жидкостной хроматографии смесью бензол/ацетон (4:1) под ультрафиолетовым облучением отсекали пятна, которые соответствовали эстрону и эстрадиолу, для определения с помощью жидкостного сцинцилляционного счетчика количества остаточного [4-14С]эстрона и количества полученного [4- 14С] эстрадиола по активности фермента 17β-ГСД добавляли жидкий сцинцилляционный коктейль [Filter count (товарный знак), фирма Hewlett Packard Company] . Более того, в качестве контрольного эксперимента аналогичный процесс проводили без добавления испытываемых материалов. Степень ингибирования активности фермента 17β-ГСД в контрольном эксперименте принимали равной за 0% и в процентах определяли степень ингибирования фермента 17β-ГСД испытываемыми материалами. Полученные результаты представлены на фиг. 1-3.
Промышленная применимость
Вышеприведенные результаты подтверждают, что производные по настоящему изобретению (испытываемые материалы) обладают превосходным ингибирующим действием на 17β-ГСД. Таким образом, по настоящему изобретению предлагаются новые тетралоновые и бензопираноновые производные и способ получения этих производных. Производные по настоящему изобретению обладают ингибирующим действием на 17β-ГСД,, поэтому действие этих производных может быть использовано при создании терапевтического средства для профилактики и/или лечения андроген- и эстрогензависимых заболеваний, в частности рака предстательной железы, доброкачественной гиперплазии предстательной железы, вирилизма, рака молочной железы, мастопатии, рака матки, эндометриоза, рака яичника и т.п.
Более того, эти соединения по настоящему изобретению могут быть использованы в составе композиции фоторезиста позитивного типа.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗОФУРАНОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ И ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО | 1998 |
|
RU2152391C1 |
| ГЛИКОПРОТЕИН TCF-II И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭФФЕКТИВНОЕ КОЛИЧЕСТВО ГЛИКОПРОТЕИНА TCF-II | 1991 |
|
RU2097432C1 |
| ФРАГМЕНТ К ДНК, КОДИРУЮЩИЙ ГЛИКОПРОТЕИН TCF-II | 1990 |
|
RU2113480C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1994 |
|
RU2176640C2 |
| ГЕТЕРОБИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2170737C2 |
| ЗАЩИТНОЕ СРЕДСТВО ДЛЯ НЕЙРОННЫХ КЛЕТОК СЕТЧАТКИ, СОДЕРЖАЩЕЕ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА ПРОИЗВОДНЫЕ ИНДАЗОЛА | 2006 |
|
RU2392938C2 |
| ПРОИЗВОДНЫЕ КОНДЕНСИРОВАННЫХ ПОЛИЦИКЛИЧЕСКИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2167877C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА | 1995 |
|
RU2137770C1 |
| ЦИКЛОПЕПТИД ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2108342C1 |
| ПРОИЗВОДНЫЕ ЭТАНОЛАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1993 |
|
RU2125983C1 |
Описываются новые тетралоновые или бензопираноновые производные и способ получения этих производных, которые могут быть использованы в составе терапевтического лекарственного средства для профилактики и/или лечения гормонзависимых заболеваний. Новые тетралоновые или бензопираноновые производные имеют общую формулу I в которой R1 и R2 каждый обозначает соответственно водород, гидроксильную группу, алкилоксигруппу; R3-R7 каждый обозначает водород, гидроксильную группу, прямую или разветвленную алкильную, алкилокси-группу, содержащую 1-6 углеродных атомов, галоген, аминогруппу или алкилендиоксигруппу, присоединенную соответственно по месту положения R3 и R4, R4 и R5, R5 и R6 или R6 и R7, и А обозначает метилен или кислород. Соединения обладают ингибирующим действием в отношении активности 17β- гидроксистероиддегидрогеназы, поэтому такие производные могут быть использованы при приготовлении терапевтического средства для профилактики и/или лечения андроген- и эстрогензависимых заболеваний, в частности рака предстательной железы, доброкачественной гиперплазии предстательной железы, вирилизма, рака молочной железы, мастопатии, рака матки, эндометриоза, рака яичника и т.п. 3 с.п. ф-лы, 3 ил.
в которой R1 и R2 каждый обозначает соответственно водород, гидроксильную группу или алкилоксигруппу;
R3 - R7 каждый обозначает водород, гидроксильную группу, прямую или разветвленную алкильную, алкилоксигруппу, содержащую 1 - 6 углеродных атомов, галоген, аминогруппу или алкилендиоксигруппу, присоединенную соответственно по месту положения R3 и R4, R4 и R5, R5 и R6 или R6 и R7;
А обозначает метилен или кислород.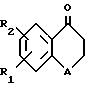
в которой R1 и R2 каждый обозначает водород, гидроксильную группу или алкилоксигруппу;
А обозначает метилен или кислород,
и бензальдегидного соединения общей формулы III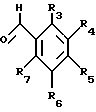
в которой R3 - R7 каждый обозначает водород, гидроксильную группу, прямую или разветвленную алкильную, алкилоксигруппу, содержащую 1 - 6 углеродных атомов, галоген, аминогруппу или алкилендиоксигруппу, присоединенную соответственно по месту положения R3 и R4, R4 и R5, R5 и R6 или R6 и R7, и кипячение с обратным холодильником в кислых условиях с последующей очисткой целевого продукта от реакционной смеси.
Приоритет по пунктам:
29.01.97 - по пп.1 - 3;
08.08.97 - по пп.1 - 3 - разновидности радикалов.
| 2-МЕТИЛ-5-МЕТОКСИ-3-МЕТОКСАЛИЛАЦЕТОБЕНЗОФУРАН, ПРОЯВЛЯЮЩИЙ ПРОТИВОВИРУСНУЮ АКТИВНОСТЬ В ОТНОШЕНИИ ВИРУСА ГРИППА ТИПА А И В | 1991 |
|
RU2032683C1 |
| SU 11388027 A, 30.01.1985 | |||
| EP 0313295 A2, 26.04.1989.EP 0557075 A1, 25.08.1993 | |||
| US 3975380 A, 17.08.1976 | |||
| Способ получения пери-инденонов | 1973 |
|
SU509212A3 |

