Лейкотриены составляют группу гормонов местного действия, образующихся в живых системах из арахидоновой кислоты. Основные лейкотриены - это лейкотриен B4(LTB4), LTC4, LTD4 и LTE4. Биосинтез этих лейкотриенов начинается с действия фермента 5-липоксигеназы на арахидоновую кислоту с получением эпоксидного соединения, известного под названием лейкотриен A4 • (LTA4), который преобразуется в другие лейкотриены в ходе последующих стадий ферментации. Прочие особенности биосинтеза лейкотриенов, а также механизм их обмена можно найти в публикации Leukotrienes and Lipoxygenases, изд. J. Rokach, Elsevir, Амстердам (1989). Действия лейкотриенов в живых системах и их участие в различных патологических процессах также описано в указанной книге Rokach'a.
Изобретение представляет некоторые хинолинсодержащие соединения как соединения, обладающие активностью, антагонистической действию лейкотриенов. Так например, в EP 318093 C 07 D 215/18 (Мерк) описаны соединения структуры A. Структура B представлена в WO 89/12629 (Rorer).
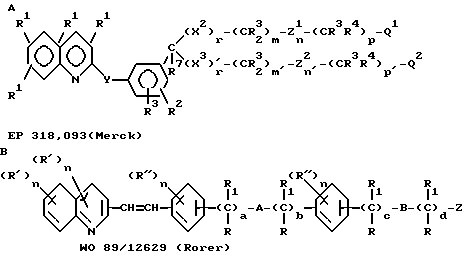
Представляемые изобретением соединения соответствуют формуле I':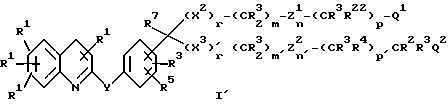
которая приведена в EP 480717, апрель 15, 1992.
Краткое описание изобретения
Настоящее изобретение касается фторированных гидроксиалкилхинолиновых кислот, обладающих активностью как антагонисты лейкотриена, способов их получения и способов, а также фармацевтических составов, пригодных для применения млекопитающим (в частности человеку).
Вследствие своей активности как антагонистов лейкотриена, представляемые изобретением соединения могут быть использованы как противоастматические, противоаллергические, противовоспалительные средства, а так же, как средства, обладающие способностью защищать клетки. Их можно применять также при лечении ангины, мозговых спазмов, глумерулярного нефрита, гепатита, эндотоксемии, увеита, реакциях отторжения аллогенного трансплантата.
Подробное описание изобретения
Представляемые изобретением соединения соответствуют формуле I,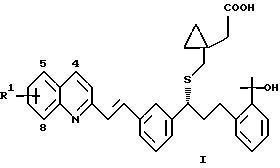
в которой R1 представляет собой 6-F или 6,7-Fe или их фармацевтически приемлемым соли.
Условные обозначения:
Приведенные ниже аббревиатуры (сокращения) имеют следующие значения:
Ac - ацетил,
DMF - диметилформамид,
Et - этил,
FAB - жесткая атомная бомбардировка,
r.t. - комнатная температура,
THF - тетрагидрофуран,
tlc - тонкослойная хроматография
Соли
Представляемые изобретением фармацевтические составы включают соединение формулы I в качестве активного ингредиента или его фармацевтически приемлемую соль и могут содержать другие фармацевтически приемлемые наполнители и необязательно другие лечебные ингредиенты.
Термин "фармацевтически приемлемые соли" касается солей, получаемых из фармацевтически приемлемых нетоксических оснований, в том числе органических и неорганических оснований. Соли-производные неорганических оснований включают в себя соли алюминия, аммония, кальция, меди, железа, железисные соли, а также соли лития, магния, марганца, марганцовистую соль, соли калия, натрия, цинка и т.д.
Особенно предпочтительными считаются соли аммония, кальция, магния, калия и натрия. Соли-производные фармацевтически приемлемых органических нетоксических оснований включают соли первичных, вторичных и третичных аминов, в том числе естественно существующих в природе, циклических аминов и основных йонообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноаминоэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, триметамин и им подобные.
Если представляемое изобретением соединение является основным, соли можно получить из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. В число таких солей входят уксусная, бензолсульфоновая, бензойная, камфорсульфоновая, глютаминовая, бромистоводородная, хлористоводородная, лимонная, этансульфоновая, фумаровая, глюконовая, глютаминовая, изетионовая, молочная, малеиновая, яблочная, миндальная, метансульфоновая, слизевая, азотная, памовая, пантотеновая, фосфорная, янтарная, серная, винная, п-толуолсульфокислота и им подобные. Особенно предпочтительными считаются лимонная, бромистоводородная, хлористоводородная, малеиновая, фосфорная, серная и винная кислоты.
Понятно, что при обсуждении способов лечения, представленных ниже, ссылки на соединение формулы I подразумевают также и фармацевтически приемлемые соли.
Использование
Способность соединений формулы I быть антагонистами действиям лейкотриенов делает их пригодными для предотвращения, торможения и возвращения к исходному состоянию симптомов, вызываемых лейкотриенами у млекопитающих, в частности у человека. Такое антагонистическое действие на лейкотриены свидетельствует о том, что соединения и содержащие их фармацевтические составы можно применять для лечения, предотвращения и возвращения к исходному состоянию симптомов ряда заболеваний млекопитающих, особенно человека, а именно:
1) легочных заболеваний, включая такие болезни, как астма, хронический бронхит, заболевания, связанные с закупоркой дыхательных путей;
2) аллергических и аллергентных расстройств, таких как аллергический ринит, контактный дерматит, аллергический конъюнктивит и им подобные;
3) воспалительных заболеваний, например артрита или воспалительных явлений кишечника;
4) болей;
5) заболеваний кожи, например таких как псориаз, диффузный нейродерматит и т.д.;
6) сердечно-сосудистых заболеваний, например ангины, миокардиальной ишемии, гипертонии, агрегации тромбоцитов и им подобных;
7) почечной инсуффляции (продувания) при ишемических явлениях, обусловленной иммунологическими или химическими (циклоспорин) причинами;
8) мигрени или "гистаминовой" головной боли;
9) глазных изменениях, таких как увеит;
10) гепатите, обусловленном химическими, иммунологическими или инфекционными раздражителями;
11) травмах или шоках, например ожогах, эндотоксемии и им подобных явлениях;
12) реакция отторжения аллогенного трансплантата;
13) предотвращении побочных явлений, связанных с введением в лечебных целях цитокинов, таких как интерлейкин 11 и некрозе опухолей;
14) хронических легочных заболеваниях, таких как фиброзно-кистозная дегенерация, хронический бронхит и других заболеваниях дыхательных путей и
15) холецистите.
Представляемые изобретением соединения можно использовать также для лечения или предотвращения у млекопитающих, особенно у человека, таких заболеваний, как гастрит, сопровождающийся появлением эрозии, разъедающий эзофагит (воспаление пищевода), понос, спазмы сосудов головного мозга, преждевременные роды, самопроизвольный аборт, дисменоррея, ишемия, вызванные вредными веществами поражение или некроз печени, поджелудочной железы, почек или поражение тканей миокарда, паренхиматозные поражения печени, вызванные гепатоксическими веществами, такими как CCl4 и D-галактозамин, ишемическая почечная недостаточность, заболевания, связанные с нарушением функции поджелудочной железы, нарушения деятельности поджелудочной железы и желудка, связанные с выделением желчи и солей, изменения клеток, связанные со стрессовыми явлениями и травмами, обусловленная глицерином почечная недостаточность. Соединения характеризуются также способностью защищать клетки.
Способность соединения защищать клетки можно наблюдать как в опытах на животных, так и при исследованиях на добровольцах, фиксируя повышенную устойчивость слизистой оболочки желудочно-кишечного тракта на вредное действие сильных раздражителей, например изъяpвляющее действие аспирина или индометацина.
Кроме снижения действия нестероидных противовоспалительных лекарств на желудочно-кишечный тракт, опыты на животных показывают, что подобные соединения, отличающиеся способностью оказывать защитное действие на клетки, могут предотвращать патологические изменения, вызванные оральным введением сильных кислот, сильных оснований, этанола, гипертонических солевых растворов и т.д.
Для измерения способности к защите клеток могут быть использованы два метода анализа: (A) анализ на патологические изменения, вызванные этанолом, и (B) анализ на язву, вызванную индометацином, описанные в EP 140684.
Дозы
Величина профилактической и лечебной дозы соединения I зависит, естественно, от тяжести заболевания пациента, конкретного соединения формулы I и способа введения лекарства. Кроме того, она определяется возрастом, весом тела и индивидуальными реакциями пациента в каждом конкретном случае.
В основном величина дневной дозы для противоастматического, противоаллергического и противовоспалительного действия (но не для защиты клеток) составляет от около 0,001 до около 100 мл на кг веса тела млекопитающего, предпочтительно 0,1-10 мг на кг, особенно предпочтительно 0,1 до 1 мг на кг в виде однократной или разделенной дозовой формы. С другой стороны, в ряде случаев может возникнуть необходимость использовать дозы, выходящие за пределы этих ограничений.
В тех случаях, когда необходимы составы для внутривенного введения, приемлемый диапазон доз для противоастматического, противовоспалительного или противоаллергического применения составляет от около 0,001 мг до около 25 мг (предпочтительно от 0,001 до около 1 мг) соединения формулы I на кг веса тела в день, а для целей защиты клеток соответственно от около 0,1 мг до около 100 мг (предпочтительно от около 1 мг до 100 мг и особенно предпочтительно от около 1 мг до около 10 мг) соединения формулы I на кг веса тела в день.
В случае, когда рекомендуются составы для орального введения, приемлемый диапазон доз для противоастматического, противоаллергического или противовоспалительного применения составляет от 0,01 мг до около 100 мг соединения формулы I на кг веса тела в день, предпочтительно от около 0,1 мг до около 10 мг на кг и для целей защиты клеток от около 0,1 мг до около 100 мг (предпочтительно от около 1 мг до около 100 мг и особенно предпочтительно от около 10 мг до около 100 мг) соединения формулы I на кг веса тела в день.
Для лечения заболеваний глаз используют офтальмологические составы для введения в глаза, содержащие 0,001-1% на вес раствора или суспензии соединения формулы I в приемлемых офтальмологических формах.
Точное количество соединения формулы I, которое может быть использовано как средство для защиты клеток, между прочим, зависит от того, будет ли оно вводиться для лечения пораженных клеток или же во избежание возможного поражения в будущем, от природы пораженных клеток (например, язвенных изменений желудочно-кишечного тракта, приводящих к нефротическому некрозу) и от природы этимологических факторов заболевания.
Например, использование соединения формулы I в целях защиты клеток в будущем предполагает совместное введение соединения формулы I вместе с нестероидным противовоспалительным средством, которое в противном случае может вызвать такое поражение (например, индометацином). В этом случае соединение формулы I вводят от 30 минут до и 30 минут после введения нестероидного противовоспалительного средства. Предпочтительно, введение производить до или одновременно с ним (например, в комбинированной дозовой форме).
Фармацевтические составы
Для введения млекопитающим и особенно человеку эффективной дозы представляемого изобретением соединения можно рекомендовать подходящий способ введения. Возможно, например, введение через рот, ректальное, местное, через глаза, легочное, через нос и т.д. Дозовые формы включают таблетки, пастилки, дисперсии, суспензии, растворы, капсулы, кремы, мази, аэрозоли и т.д.
Представляемые изобретением фармацевтические составы содержат в своем составе соединение формулы I в качестве активного ингредиента или соответственно фармацевтически приемлемую соль его, в сочетании с фармацевтически приемлемым наполнителем и необязательно с другим ингредиентом, обладающим лечебным эффектом. Термин "фармацевтически приемлемые соли" относится к солям, получаемым из фармацевтически приемлемых нетоксических оснований или кислот, в том числе неорганических оснований или кислот и органических оснований или кислот.
Составы включают в себя составы, приемлемые для орального, ректального, местного, парэнтерального (в том числе подкожного, внутримышечного и внутривенного), глазного (офтальмологические), легочного (через нос или защечного) применения, а также введения через нос, хотя большинство приемлемых способов применения в ряде указанных случаев будут зависеть от природы или серьезности заболевания и состояния здоровья пациента, которого предполагается лечить, а также от природы активного ингредиента. Традиционно их используют в единой дозовой форме или же получают в соответствии с методами, хорошо известными специалистам-фармацевтам.
Для использования в виде ингаляций представляемые изобретением соединения обычно применяют в виде аэрозольных ингаляторов или емкостей, обеспечивающих распыление лекарственного средства. Соединения можно готовить также в виде порошков, приготавливаемых по специальным технологиям, или порошкообразных составов, которые можно вдыхать методом инсуффляции с помощью ингаляторов. Предпочтительная система для ингаляции представляет собой аэрозоль с отмеренной дозой, который может быть приготовлен в виде суспензии или раствора соединения I в подходящем пропелленте, например, фторированных углеродах или углеводородах.
Приемлемые составы соединения I для местного применения включают устройства для трансдермального введения, аэрозоли, кремы, мази, лосьоны, примочки, распыляемые порошки и им подобные формы.
Для практического применения соединения формулы I можно комбинировать в качестве активного ингредиента при тщательном перемешивании с фармацевтически приемлемым носителем в соответствии с традиционными фармацевтическими технологиями. С носителями и наполнителями можно получить большое разнообразие различных форм в зависимости от формы состава, который хотят получить, например оральные или парентеральные формы (включая внутривенную).
Для приготовления составов для оральной дозовой формы можно рекомендовать любую фармацевтическую среду, такую как, например, вода гликоли, масла (растительные), спирты, корригенты, консерванты, подцвечивающие средства и др. в случае, если речь идет о жидких составах для введения через рот, таких, как, например, суспензии, эликсиры и растворы; или носители (наполнители), например крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, средства грануляции, смазывающие средства, связывающие агенты, средства, обеспечивающие разложение, и им подобные в случае, когда речь идет о твердых составах для орального применения - таких, как, например, порошки, капсулы и таблетки, причем твердые составы для орального применения более предпочтительны, чем жидкие.
Вследствие простоты и легкости введения таблетки и капсулы представляют собой наиболее перспективную дозовую форму, при приготовлении которой рекомендуется применять обычные фармацевтические носители. По желанию таблетки можно покрывать, пользуясь "водными" или "безводными" методиками.
В дополнение к обычным дозовым формам, описанным выше, соединения формулы I можно вводить также, пользуясь средствами контролированного выделения вещества и/или приборами и устройствами, описанными, например, в пат. США 3845770, 3916899, 3536809, 3598123, 3630200 и 4008719, описание их дается здесь в виде ссылок.
Представляемые изобретением составы для орального применения могут представлять собой отдельные единицы, например капсулы саши или таблетки, содержащие заранее определенное количество активного ингредиента, порошки или гранулы или же растворы или суспензии в водной жидкости, неводной жидкости, эмульсии масло-в воде или жидкие эмульсии вода-в масле.
Такие составы можно получать в соответствии с известными фармацевтическими методами, однако все эти методы включают стадию соединения активного ингредиента с наполнителями, представляющими собой один или более необходимых ингредиентов. В основном, составы получают путем однородного внутреннего перемешивания активного ингредиента жидкими носителями и под конец распределяют в твердом наполнителе или производят то и другое, после чего, в случае необходимости, придают массе желаемую форму.
Например, таблетки можно получать путем прессования или формования, необязательно с одним или более дополнительными ингредиентами. Для прессования таблеток можно пользоваться специальной таблетирующей машиной, вводя активный ингредиент в форме свободного потока в виде порошка или гранул, необязательно в смеси со связывающими средствами, смазывающими агентами, инертными разбавителями, поверхностно-активными веществами и диспергирующими средствами.
Формованные таблетки можно получать в специальной формовочной машине, увлажняя смесь порошкообразных соединений инертным жидким разбавителем. Желательно, чтобы каждая таблетка содержала от около 2,5 мг до около 500 мг активного ингредиента, а каждое саше или соответственно каждая капсула - от около 2,5 до около 500 мг активного ингредиента.
Ниже приведены примеры представляемых фармацевтических дозовых форм для соединений формулы I:
Суспензия для инъекций (внутримышечно) - мг/мл:
Соединение формулы I - 10
Метилцеллюлоза - 5,0
Твин 80 - 0,5
Бензиловый спирт - 9,0
Бензальконийхлорид - 1,0
Вода для инъекций для общего объема - 1 мл
Таблетки - мг/таблетку
Соединение формулы I - 25
Микрокристаллическая целлюлоза - 415
Павидон - 14,0
Предварительно желатинизированный крахмал - 43,5
Стеарат магния - 2,5 - 500
Капсулы - мг/капсулу
Соединение формулы I - 25
Порошкообразная лактоза - 573,5
Стеарат магния - 1,5 - 600
Аэрозоли - на баллончик
Соединение формулы I - 24
Лецитин, NF, жидкий концентрированный - 1,2
Трихлорфторметан, NF, - 4,025 г
Дихлордифторметан, NF - 12,15 г
Соединение с другими лекарственными средствами
Кроме соединений формулы I, представляемые изобретением фармацевтические составы могут содержать также другие активные ингредиенты, такие как ингибиторы циклооксигеназы, нестероидные противовоспалительные средства (НСПВС), анальгетики периферического радиуса действия, такие как например, зомепирак дифлунизал и т.д.
Весовое соотношение соединения формулы I и вторичного активного ингредиента может варьироваться и будет зависеть от эффективной дозы каждого ингредиента. Обычно используют эффективную дозу каждого. Так, например, если соединение формулы I соединяют с нестероидным противовоспалительным средством, то соотношение соединения (весовое) формулы I и нестероидного противовоспалительного средства будет колебаться в пределах от 1000:1 до около 1: 1000, предпочтительно от около 200:1 до около 1:200.
Соединение формулы I и другие активные ингредиенты обычно комбинируют в указанных выше соотношениях, но в каждом случае должна быть использована эффективная доза каждого активного ингредиента.
Нестероидные противовоспалительные средства можно разделить на пять групп:
(1) производные пропионовой кислоты,
(2) производные уксусной кислоты,
(3) производные фенаминовой кислоты,
(4) оксикамы и
(5) производные бифенилкарбоновой кислоты или их фармацевтически приемлемые соли.
Производные пропионовой кислоты, которые можно использовать, включают в себя: альминопрофен, беноксапрофен, буклоксиновую кислоту, карпрофен, фенбуфен, фенопрофен, флупрофен, флубипрофен, ибупрофен, индопрофен, кетопрофен, миропрофен, напроксен, оксапрозин, пирпрофен, пронопрофен, супрофен, тиапрофеновую кислоту и тиоксапрофен. Структурно родственные производные пропионовой кислоты, обладающие аналогичными анальгетическими (обезболивающими) и противовоспалительными свойствами, также предполагается включить в эту группу.
Таким образом "производные пропионовой кислоты" в указанном здесь значении представляют собой лекарственные средства смешанного ненаркотического обезболивающего и нестероидного противовоспалительного действия, которые содержат свободную -CH(CH3)COOH или -CH2CH2COOH (которые в свою очередь необязательно могут существовать в форме фармацевтически приемлемой солевой группы, например -CH(CH3)COO-Na+ или -CH2CH2COO-Na+), как правило, соединенной непосредственно или через карбонильную группу с циклической системой, предпочтительно с системой ароматического кольца.
Производные уксусной кислоты, пригодные для использования в указанных целях, включают в себя: индометацин, предпочтительно представляющий собой нестероидное противовоспалительное средство, ацеметацин, альклофенак, клиданак, диклофенак, фенклофенак, фенклозиновую кислоту, фентиазак, фурофенак, ибуфенак, изоксепак, окспинак, сулиндак, тиопинак, тольметин, зидометацин и зомепирак. Структурно родственные производные уксусной кислоты, обладающие аналогичными обезболивающими противовоспалительными свойствами, также могут быть включены в указанную группу.
Таким образом, "производные уксусной кислоты" в указанном здесь значении представляют собой лекарственные средства смешанного ненаркотического обезболивающего и нестероидного противовоспалительного действия, имеющие свободную -CH2COOH-группу (которая необязательно может существовать в форме фармацевтически приемлемой солевой группы, например -CH2COO-Na+), как правило, соединенную непосредственно с циклической системой, предпочтительно с системой ароматического или гетероароматического кольца.
Производные фенаминовой кислоты, пригодные для использования в указанных целях, включают в себя: флуфенаминовую кислоту, меклофенаминовую, мефенаминовую, нифиминовую и тольфенаминовую кислоту. Структурно родственные производные фенаминовой кислоты, обладающие аналогичными анальгетическими и противовоспалительными свойствами, также могут быть включены в указанную группу.
Таким образом, "производные фенаминовой кислоты" в указанном здесь значении представляют собой лекарственные вещества смешанного ненаркотического обезболивающего и нестероидного противовоспалительного действия, содержащие основную структуру: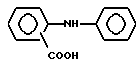
которая может иметь целый ряд заместителей и в которой свободная -COOH-группа может существовать в форме фармацевтически приемлемой солевой группы, например -COO-Na+.
Производные "бифенилкарбоновой кислоты", которые можно использовать, включают в себя дифлунизал и флуфенизал. Структурно родственные производные бифенилкарбоновой кислоты, обладающие аналогичными обезболивающими и противовоспалительными свойствами, также могут быть включены в указанную группу.
Таким образом, "производные бифенилкарбоновой кислоты" в указанном здесь значении представляют собой лекарственные средства смешанного ненаркотического обезболивающего и нестероидного противовоспалительного действия, содержащие основную структуру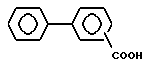
которая может иметь ряд заместителей и в которой свободная -COOH-группа может существовать в форме фармацевтически приемлемой солевой группы, например, -COO-Na+.
Оксикамы, которые можно использовать в представляемом изобретении, включают в себя: изоксикам, пироксикам, судоксикам и теноксикам. Структурно родственные оксикамы, обладающие аналогичными обезболивающими и противовоспалительными свойствами, также могут быть включены в указанную группу.
Таким образом, "оксикамы" в указанном здесь значении представляют собой лекарственные средства смешанного ненаркотиеского обезболивающего и нестеродиного противовоспалительного действия с общей формулой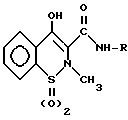
в которой R представляет собой арильную или гетероарильную циклическую структуру.
В представляемом изобретении могут быть использованы также: амфенакнатрий, аминопрофен, анитразафен, антрафенин, ауранофин, бендазат лизинат, бензиданин, бепрозин, броперамол, буфезолак, синметацин, ципроквазон, клоксимат, дазидамин, дебоксамет, дельметацин, детомидин, дексиндопрофен, диацерин, ди-физаламин, дифенпирамид, эморфазон, энфенаминовая кислота, эноликам, эпиризол, этерсалат, этодолак, этофенамат, фанетизол мезилат, фенклорак, фендозал, фенфлумизол, фенпразон, флоктафенин, флуниксин, флуноксапрофен, флупроквазон, фопиртолин, фосфозал, фурклопрофен, глукаметацин, кваймезоал, ибупроксам, изофезолак, изониксим, изопрофен, изоксинам, лефетамин HCl, лефлуномид, лофемизол, лоназолак кальция, лотифазол, локсопрофен, лизин клониксинат, меклофенамат натрия, мезеклазон, набуметон, никтиндол, нимесулид, орпаноксин, оксаметацин, оксападол, перизоксал цитрат, пимепрофен, пиметацин, пипрофен, пирозолак, пирфенидон, проглуметацин малеат, проквазон, пиридоксипрофен, судоксикам, талметацин, талнифлумат, теноксикам, тиазолинбутазон, тиелавин B, тиарамид HCl, тифламизол, тимегадин, толпадол, триптамид и уфенамат.
Могут быть использованы также следующие нестероидные противовоспалительные средства, обозначенные компанией кодовыми номерами (см. напр. Pharmacoprojects), а именно:
480156, AA861, AD1590, AFP802, AFP860, A177B, AP504, AU8001, BPPC, BW540C, CHIN01N 127, CN100, EB3821, EL 508, FI044, GV3658, ITF182, KCNTE16090, KME4, LA2851, MR714, MP897, MY309, ON03144, PR823, PV108, PV102, R830, RS2131, SCR152, SH440, SIR133, SPA 510, SQ27239, ST281, SY6001, TA60, TAI-901 (4-бензоил-1-инданкаробоновая кислота), TVX2706, U60257, UR2301 WY41770.
И, наконец, пригодные для указанной цели соединения включают в себя салицилаты, особенно ацетилсалициловую кислоту и фенилбутазоны, и их фармацевтически приемлемые соли.
Кроме индометацина, предпочтительными нестероидными противовоспалительными средствами считают ацетилсалициловую кислоту, диклофенак, фенбуфен, фенопрофен, флубипрофен, ибупрофен, кетопрофен, напроксен, фенилбутазон, пироксикам, сулиндак и толметин.
Фармацевтические составы, имеющие в своем составе соединения формулы I, могут содержать также ингибиторы биосистеза лейкотриенов, описанных, например, в EP 138481 (апрель 24, 1985), EP 1156394 (август 8, 1984), EP 136893 (апрель 10, 1985) и EP 140709 (май 8, 1985), и представленных здесь в виде ссылок.
Соединения формулы I можно использовать также в сочетании с антагонистами лейкотриена, описанными, например, в EP 106565 (апрель 25, 1984) и EP 104885 (апрель 4, 1984), приведенными здесь в виде ссылок и другими известными соединениями такого типа, описанными в приложении к EP Nos 56172 (июль 21, 1982) и 61800 (июнь 10, 1982) и спецификации к пат. Великобритании N 2058785 (апрель 15, 1981), представленными здесь в виде ссылок.
Содержащие соединения формулы I фармацевтические составы могут иметь в своем составе в качестве второго активного ингредиента антагонисты простагландина, такие как описаны например, в EP 11067 (май 28, 1980) или антагонисты тромбоксана, представленные в пат. США 4 237160. Они могут содержать также ингибиторы гисдитин декарбоксилазы, такие как α -фторметилгистидин, описанный в пат. США 4325961.
Перспективным может оказаться также соединение веществ формулы I с антагонистом H1 или H2-рецепторами, например, ацетамазолом, аминотиадиазолами, описанными в EP 40696 (декабрь 2, 1981), а также веществами типа бенадрил, циметидин, фамотидин, фрамамин, гистадил, фенерган, ранитидин, терфенадин и им подобные, описанные, например, в пат. США 4283408; 4362736 и 4394508. Фармацевтические составы могут содержать также ингибитор K+/H+АТФ-азы, например, омепразол, описанный в пат. США 4255431 и им подобные.
Другие фармацевтические составы, включающие соединения формулы I в сочетании с антагонистами серотонина, такими как метисергин, антагонистами серотонина, описанными, например, в Nature, том 316, стр. 126-131, 1985 и им подобные. Каждая из ссылок, касающаяся подобного использования, приводится в виде ссылки.
Другие перспективные фармацевтические составы включают соединения формулы I в комбинации с антихолинергетиками, например, ипратропий бромидом, бронхолитическими средствами, такими как бета агонист сольбутамол, метапротеренол, тербуталин, фенотерол и аналогичными им веществами, а также противоастматическими лекарственными средствами теофиллином, холин теофиллинатом и энпрофиллином, антагонистами кальция нифедипином, дильтиаземом, нитрендипином, верапамилом, нимодипином, фелодипином и т.д., а также кортикостероидами, гидрокортизоном, метилпреднизалоном, бетазоном, дексаметазоном, беклометазоном и им подобными.
Таблица иллюстрирует представляемые изобретением соединения.
Способы синтеза.
Представляемые изобретением соединения можно получать в соответствии с методами, изложенными в EP 480717.
Анализ на определение биологической активности:
Соединение формулы I можно испытывать при использовании следующих методов анализа на определение их активности как антагонистов лейкотриена и их способности ингибировать биосинтез лейкотриена у млекопитающих.
Изучение связывания LTD4-рецептора на оболочке легкого морской свинки, трахеях морской свинки и опытах in vivo на морских свинках, находящихся под наркозом.
Подробное описание этих трех тестов представлено в публикации T.R. Jones и сотр., Can. J. Physiol. Pharmacol. 67, 17-28 (1989).
Используя нижеследующие методы анализа соединения формулы I, можно использовать для определения их активности в отношении ингибирования биосинтеза лейкотриенов у млекопитающих. Анализы подробно описаны в EP 480717, апрель 15, 1992.
Определение способности ингибирования 5-липогеназы LTB4-анализ на определение полиморфноядерных нейтрофилов у человека.
Соединения формулы I можно испытывать при использовании следующих методов анализа на определение их активности в качестве антагонистов лейкотриена и ингибиторов биосинтеза лейкотриена. Методы анализа описаны в EP 480717, апрель 15, 1992.
Анализ на крысах, страдающих астмой.
Динамика легких у прирученных белок, находящихся в сознании. Обезьяны.
Профилактика вызванного бронхостеноза у овец, страдающих аллергией.
Далее предмет изобретения иллюстрируется приведенными ниже примерами, описанием которых он не ограничивается. Все температурные данные даются в градусах Цельсия.
Пример 1.
Натрий-1-((R)-(3)(2))6,7-дифтор-2-хинолинил(-этенил/фенил)-3-(2-(2-гидрокси-2-пропил(фенил)-пропил)тио(метил)циклопропанацетат
Стадия 1: 6,7-дифтор-2-метилхинолин
Кротоновый альдегид (226,34 г, 3,23 моля) в 100 мл 2-бутанола добавляют по каплям к кипящему с обратным холодильником раствору 3,4-дифторанилина (417,27 г, 3,23 моля) п-хлоранила (794,65 г, 3,23 моля) и концентрированной соляной кислоты (808 мл) в 5,4 л 2-бутанола. 2 часового нагревания 2,7 л растворителя отгоняют при вакууме при температуре около 60o.
Затем к реакционной смеси добавляют 2 л толуола с последующим удалением 2,5-3 растворителя до тех пор, пока не образуется тестообразное твердое вещество. Добавляют тетрагидрофуран (2 л), и нагревают смесь в течение 30 минут, после чего производят охлаждение до 0oC. Твердый продукт собирают и промывают тетрагидрофураном, очищая методом тонкослойной хроматографии. Затем твердый продукт растворяют в эквивалентном количестве K2CO3 (этилацетата) и отделяют органическую фазу. Водную фазу экстрагируют этилацетатом (2х), органические фазы соединяют, сушат над сульфатом магния, и растворитель отгоняют. Продукт кристаллизуют в минимальном количестве этилацетата с получением 328,08 г (57%) указанного в заголовке соединения.
1H ЯМР (CDCOCD3): δ 8,19 (1H, д), 7,75 (2H, м), 7,4 (1H, д), 2,64 (3H, с).
Стадия 2: 3-(2)6,7-дифтор-2-хинолинил (этенил)-бензальдегид
В соответствии с методикой, представленной в примере 24, стадия 1 пат. США 4851409, но при использовании дифторметилхинолина со стадии 1 получают указанное в заголовке соединение.
1H ЯМР (CDCOCO3): δ 10,12 (1H, с), 8,4 (1H, д), 8,29 (1H, с), 8,1 - 7,85 (6H, м), 7,7 - 7,55 (2H, м).
Стадия 3: 1-(3-/2-)6,7-дифтор-2-хинолинил(этенил)-фенил-2-пропен-1-ол.
В соответствии с методикой, представленной в примере 80, стадия 1 EP 480717, но при использовании 3-(2-)6,7-дифтор-2-хинолинил (этенил)бензальдегида со стадии 2 получают указанное в заголовке соединение.
1H ЯМР (CD3COCO3): δ 8,32 (1H, д), 7,92 - 7,8 (4H, м), 7,75 (1H, шир.с), 7,6 (1H, м), 7,5 - 7,25 (3H, м), 6,05 (1H, ддд), 5,37 (1H, ддд), 5,25 (1H, м), 5,1 (1H, ддд), 4,61 (1H, д).
Стадия 4: Этил 2-(3-)3-(2-)6,7-дифтор-2-хинолинил(-этенил(фенил)-3-оксопропил)бензоат.
В соответствии с методикой, представленной в примере 146, стадия 1 EP 480717, но при использовании дифторированного спирта со cтадии 3 получают указанное в заголовке соединение.
1H ЯМР (CD3COCO3): δ 8,35 (2H, м), 8,0 - 7,8 (7H, м), 7,6 - 7,3 (5H, м), 4,33 (2H, кв.), 3,5 - 3,3 (4H, м), 1,32 (3H, т).
Стадия 5: Этил 2-(3)(-)3-(2-)6,7-дифтор-2-хинолинил(этенил)фенил-3-гидроксипропил/бензоат.
В соответствии с методикой, представленной в примере 146, стадия 2 EP 480,717, но при использовании дифторкетона со стадии 4 получают указанное в заголовке соединение.
1H ЯМР (CD3COCO3): δ 8,31 (1H, д), 7,92 - 7,75 (6H, м), 7,6 - 7,25 (7H, м), 4,78 (1H, м), 4,47 (1H, д), 4,3 (2H, кв.), 3,2 - 2,95 (2H, м), 2,05 (2H, м), 1,32 (3H, т).
Стадия 6: 2-(2)3-(S)-(3-)2-/6,7-дифтор-2-хинолинил(этенил(фенил)-3-гидроксипропил(фенил)-2-пропанол
Смесь безводного CeCl3 (40,5 г, 164 ммоля) в тетрагидрофуране (500 мл) кипятят с обратным холодильником в течение ночи, используя ловушку Дина Старка, заполненную активированным 3  молекулярным ситом. К суспензии CeCl3 добавляют по каплям в течение 30 минут при 0oC метилмагнийхлорид (263 мл, 3.0М в тетрагидрофуране, 790 ммолей). После 2-х-часового перемешивания при 0oC смесь охлаждают до -5oC и добавляют по каплям раствор гидроксиэфира (71,8 г, 152 ммоля) в толуоле (600 мл) со стадии 5.
молекулярным ситом. К суспензии CeCl3 добавляют по каплям в течение 30 минут при 0oC метилмагнийхлорид (263 мл, 3.0М в тетрагидрофуране, 790 ммолей). После 2-х-часового перемешивания при 0oC смесь охлаждают до -5oC и добавляют по каплям раствор гидроксиэфира (71,8 г, 152 ммоля) в толуоле (600 мл) со стадии 5.
Реакционную смесь перемешивают еще в течение часа, прежде чем добавить 2М (HOAc) (600 мл) и толуол (600 мл). Органический слой промывают насыщенным водным раствором NaHCO3 и соляным раствором. После концентрирования в вакууме и очистки остатка методом флеш-хроматографии (30% этилацетата в толуоле) получают 63,48 г (91%) указанного в заголовке соединения.
1H ЯМР (CD3COCD3): δ 8,4 (1H, д), 8,0 - 7,8 (5H, м), 7,65 (1H, м), 7,5 (3H, м), 7,35 - 7,1 (4H, м), 4,88 (1H, м), 4,58 (1H, д), 4,19 (1H, с), 3,22 (2H, м), 2,15 (2H, м), 1,70 (3H, с), 1,68 (3H, с).
Стадия 7: Циклический сульфит 1,1-циклопропандиметанола
К раствору комплексного соединения BH3:ТГФ (1 моль в тетрагидрофуране, 262 мл) добавляют диэтил 1,1-циклопропандикарбоксилат (25 г, 134 ммоля) при 25oC в атмосфере азота. Раствор кипятят с обратным холодильником в течение 6 часов, охлаждают до комнатной температуры и осторожно добавляют MeOH (30 мл). Раствор перемешивают в течение 1 часа, после чего концентрируют с получением маслянистого продукта.
Неочищенный диол растворяют в CH2Cl2 (234 мл) и (15,9 г, 134 ммоля) и добавляют по каплям в течение 15 минут при 25oC SOCl, (15,9 г, 134 ммоля). После перемешивания еще в течение 15 минут смесь промывают водным NaHCO3. Органический экстракт сушат над сульфатом натрия, фильтруют и концентрируют с получением количественно указанного в заголовке соединения в виде твердого вещества белого цвета.
Стадия 8: 1-(гидроксиметил)циклопропанацетонитрил
К раствору циклического сульфитного продукта со стадии 7 (14,7 г, 99 ммолей) в диметилформамиде (83 мл) добавляют NaCH (9,74 г, 199 ммолей). Смесь нагревают при 90oC в течение 20 часов. После охлаждения добавляют этилацетат (400 мл) и промывают раствор насыщенным NaHCO3-раствором (55 мл), H2O (4 х 55 мл), насыщенным раствором NaCl и сушат над сульфатом натрия. Раствор концентрируют с получением 7,1 г (65%) указанного в заголовке соединения.
Стадия 9: 1-(ацетилтиометил)циклопропанацетонитрил
К раствору спирта со стадии 8 (42 г, 378 ммолей) в сухом CH2Cl (450 мл) при -30oC добавляют по каплям EF3N (103,7 мл, 741 ммолей) при -30oC, а затем CH3SO2Cl (43,3 мл, 562 ммолей). Смесь нагревают до 25oC, промывают NaHCO3, сушат над сульфатом натрия и концентрируют в вакууме с получением соответствующего мезилата. Затем мезилат растворяют в диметилформамиде (450 мл) и охлаждают до 0oC. Добавляют тиоацетат калия (55,4 г, 485 ммолей), и перемешивают смесь при 25oC в течение 18 часов. Добавляют этилацетат (1,5 л), раствор промывают NaHCO3, сушат над сульфатом натрия и концентрируют в вакууме с получением 45 г (70%) указанного в заголовке соединения.
Стадия 10: Метил 1-(тиометил)циклопропанацетат
К раствору нитрила со стадии 9 (45 г, 266 ммолей) в MeOH (1,36 л) добавляют воду (84 мл) и концентрированную серную кислоту (168 мл). Смесь кипятят с обратным холодильником в течение 20 часов, охлаждают до 25oC, добавляют воду (1 л) и экстрагируют продукт CH2Cl2 (2 х 1,5 л). Органический экстракт промывают водой и сушат над сульфатом натрия. После концентрирования органического раствора получают 36 г (93%) указанного в заголовке соединения.
Стадия 11: Метил 1-(((R)-(3-)2-)6,7-дифтор-2-хинолинил(этенил)-фенил)-3(2-)2-гидрокси-2-пропил фенил)пропил(тио)метил)циклопропанацетат
Диол со стадии 6 (1,0 г, 2,1 ммоля) растворяют в тетрагидрофуране (1 мл) и диметилформамиде (1 мл) и охлаждают до -40oC. Добавляют диизопропилэтиламин (383 мкл, 2,2 ммоля), а затем метансульфонилхлорид (170 мкл, 2,2 ммолей). Смесь перемешивают в течение 2 часов при медленном нагревании до -30oC. К мутной реакционной смеси добавляют тиол (370 мг, 2,3 ммоля) со стадии 10, а затем по каплям вводят раствор трет.-бутоксида калия в тетрагидрофуране (2,52 мл, 1,75 М, 4,4 ммоля).
Реакционную смесь перемешивают при -30oC в течение 3,5 часов, прежде чем погасить ее 25%-ным водным раствором NH4OAc. После экстрагирования этилацетатом (3х), промывания органического слоя соляным раствором и выпаривания растворителя получают остаток, который после очистки методом флеш-хроматографии (5%-10% этилацетат в толуоле) дает 658 мг (53% указанного в заголовке соединения).
1H ЯМР (CD3COD3): δ 8,21 (1H, д), 7,9 - 7,7 (5H, м), 7,57 (1H, м), 7,4 (3H, м), 7,25 - 7,05 (4H, м), 4,07 (1H, т), 3,95 (1H, с), 3,58 (3H, с), 3,2 (1H, ддд), 2,93 (1H, ддд), 2,58 (2H, с), 2,41 (2H, дд), 2,25 (2H, м), 1,58 (6H, с), 0,55 - 0,35 (4H, м).
Стадия 12: Натрий 1-(((1R)-(3-)2-)6,7-дифтор-2-хинолинил(этенил)- фенил(3-)2-)2-гидрокси-2-пропил(фенил)пропил(тио)метил) циклопропанацетат
В соответствии с методикой гидролиза из примера 146, стадия 11 EP 480717, но при использовании дифторированного эфира со стадии 1, получают кислоту указанного в заголовке соединения.
1H ЯМР (CD3COD3): δ 8,32 (1H, д), 7,95 - 7,77 (5H, м), 7,65 - 7,38 (5H, м), 7,2 - 7,0 (3H, м), 4,07 (1H, т), 3,18 (1H, ддд), 2,9 (1H, ддд), 2,8 (1H, шир. с) 2,6 (2H, с), 2,42 (2H, с), 2,2 (2H, м), 1,53 (6H, с), 0,55 - 0,35 (4H, м).
В соответствии с методикой примера 146, стадия 12 EP 480,717 для вышеупомянутой кислоты получают указанное в заголовке соединение.
1H ЯМР (CD3COD3): δ 8,2 (1H, д), 7,85 - 7,7 (5H, м), 7,52 - 7,25 (5H, м), 7,1 - 7,0 (3H, м), 4,04 (1H, т), 3,2 (1H, м), 3,8 - 2,5 (4H, м), 2,3 - 2,05 (4H, м), 1,54 (3H, с), 150 (3H, с), 0,45 (2H, м), 0,25 (2H, м).
Масс-спектр FAB: MH+ при 610, (M+23)+ при 632. Расчетные данные микроанализа для C35H34NO3SF2Na • 3H2O:
C 63,99, H 5,97, N 2,07.
Найдено: C 64,52, H 5,95, N 2,07.
Пример 2
Натрий 1-(((R)-(3)(2-)6-фтор-2-хинолинил)этенил)-фенил)-3-(2-(2-гидрокси-2-пропил(фенил)пропил (тио)метил)циклопропанацетат
В соответствии с методикой примера 1 стадия 2, но при использовании в качестве исходного соединения 6-фтор-2-метил-хинолин (см. C.M. Leir, J. Org. Chem, т. 42, стр. 911 - 913, 1977) получают указанное в заголовке соединение.
Кислота:
1H ЯМР (CD3COD3): δ 8,15 (1H, д), 8,02 (1H, дд), 7,9 (1H, д), 7,87 (1H, д), 7,8 (1H, с), 7,55 - 7,35 (6H, м), 7,25 - 7,0 (4H, м), 4,05 (1H, т), 3,18 (1H, ддд), 2,9 (1H, ддд), 2,59 (2H, с), 2,43 (2H, д), 2,2 (2H, м), 1,52 (6H, с), 0,55 - 0,35 (4H, м).
Натриевая соль
1H ЯМР (CD3COD3): δ 8,35 (1H, д), 8,09 (1H, дд), 8,0 - 7,35 (10H, м), 7,1 (3H, м), 4,09 (1H, т), 3,2 (2H, м), 2,85 - 2,55 (3H, м), 2,35 - 2,0 (4H, м), 1,52 (3H, с), 1,49 (3H, с), 0,45 (2H, м), 0,25 (2H, м).
Расчетные данные микроанализа для C35H35FNNaO3S • H2O: C 68,93, H 6,13, N 2,30.
Найдено: C 68,42, H 5,99, N 2,29.
Масс-спектр (FAB): MH+ при 592, (M + 23)+ при 614 (30%).
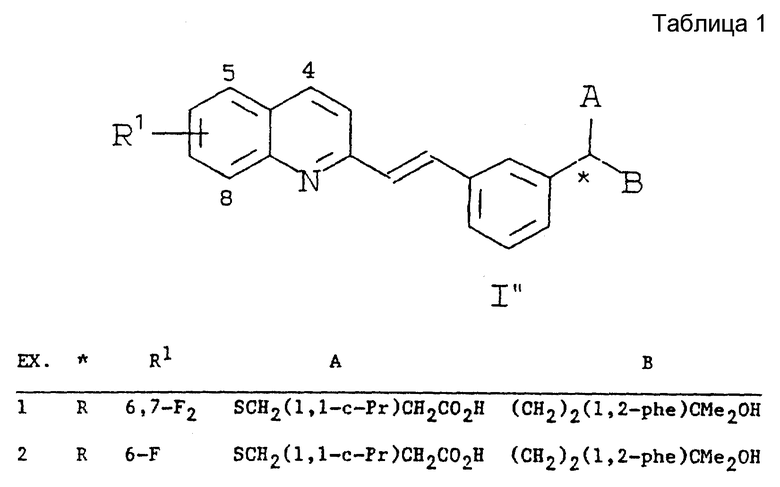
Хинолин-производные формулы I, в которой R1 представляет собой 6-F или 6,7-F2, или их фармацевтически приемлемая соль, проявляет активность антагониста лейкотриена. 2 с. и 4 з.п. ф-лы. 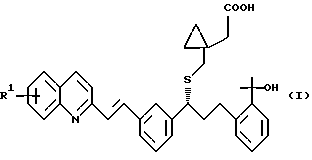
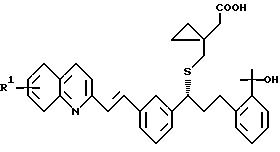
в которой R' представляет собой 6-F или 6,7-F2,
или их фармацевтически приемлемая соль.
| Способ получения производных хинолинкарбоновых кислот или их фармацевтически приемлемых солей или эфиров | 1984 |
|
SU1393314A3 |
| Способ получения диалкилфосфатов | 1974 |
|
SU480716A1 |
| Способ получения диалкил - -имидометилдитиофосфатов | 1973 |
|
SU480717A1 |

