Изобретение относится к области медицины и пригодно для лечения язвенной болезни желудка и двенадцатиперстной кишки, эрозивного и рефлюкс-эзофагита, синдрома Золлингера-Эллисона, других заболеваний, сопровождающихся желудочной гиперсекрецией, а также профилактики стрессовых и послеоперационных язв, рецидивов кровотечений из верхних отделов желудочно-кишечного тракта, аспирации желудочно-кишечного сока при операциях под наркозом.
Известно противоязвенное средство - ранитидин (или N-[2-[[[5-(диметиламино)метил] -2-фуранилметил] тио]этил]-N'-метил-2-нитро-1,1-этендиамин) [Машковский М.Д. Лекарственные средства, т. 1, изд. 12-е, М., 1993, с. 359] . Ранитидин является антагонистом H2-рецепторов и его механизм действия основан на связывании H2-рецепторов париетальных клеток слизистой оболочки желудка, в результате чего подавляется базальная и стимулированная желудочная секреция. В медицинской практике ранитидин обычно применяется в виде гидрохлорида.
В международной заявке WO 95/27486, 1995 г. описаны оральные композиции ранитидина или его солей, в том числе гидрохлорида ранитидина. Композиция на основе гидрохлорида ранитидина содержит, мас.%:
Ядро, Гидрохлорид ранитидина - 18,7
Микрокристаллическая целлюлоза - 80,6,
Стеарат магния - 0,7,
и оболочку, которая состоит из оксипропилметилцеллюлозы, двуокиси титана, триацетина и желтого оксида железа.
Однако приведенная композиция характеризуется достаточно низким содержанием действующего вещества (менее 20%), что требует приема дополнительных доз препарата или существенного увеличения веса таблетки для создания требуемой терапевтической концентрации.
Наиболее близкой к заявляемой по совокупности существенных признаков является фармацевтическая композиция, описанная в патенте России N 2131264, опубл. 10.06.99 (прототип). Она содержит ранитидин и вспомогательные вещества в следующем соотношении, маc.%:
Ранитидин - 53,3 - 59,4
Микрокристаллическая целлюлоза - 37,4 - 44,8
Поливинилпирролидон - 1,4 - 2,0
Стеарат магния - 0,5 - 1,2
Данную противоязвенную композицию получают смешиванием ранитидина и микрокристаллической целлюлозы, далее проводят влажную грануляцию при распылении водно-спиртового раствора поливинилпирролидона над псевдоожиженной массой, гранулят сушат, добавляют к нему стеарат магния и готовую смесь таблетируют.
Однако отсутствие оболочки значительно снижает стабильность указанной лекарственной формы, и, соответственно, ее срок годности, а приведенный количественный и качественный состав ингредиентов ядра таблетки не позволяет получать удовлетворительное качество препарата после нанесения оболочки.
Таким образом, по-прежнему актуальным является создание твердой лекарственной формы гидрохлорида ранитидина, которая удовлетворяет всем требованиям на фармацевтическое средство.
Техническим результатом, получаемым при реализации настоящего изобретения, является то, что новая противоязвенная композиция устойчива при хранении и при этом легко высвобождает действующее вещество, что обеспечивает его высокую биодоступность, а также последовательность операций и их технологических параметров, позволяющих производить лекарственную форму требуемого качества, в частности с удовлетворительной прочностью.
Указанный технический результат достигается тем, что предлагаемая фармацевтическая композиция, обладающая противоязвенной активностью, включает ядро, содержащее в качестве действующего вещества гидрохлорид ранитидина и в качестве вспомогательных веществ аэросил, крахмал, микрокристаллическую целлюлозу, молочный сахар, поливинилпирролидон и стеарат магния, покрытое оболочкой, которая состоит из сополимера метакриловой кислоты, полиэтиленгликоля и диоксида титана
Ингредиенты содержатся в ядре в следующем соотношении, мас.%:
Гидрохлорид ранитидина - 60,0 - 75,0
Аэросил - 0,1 - 10,0
Крахмал - 4,0 - 16,0
Микрокристаллическая целлюлоза - 8,5 - 20,3
Молочный сахар - 4,5 - 15,0
Поливинилпирролидон - 0,1 - 1,3
Стеарат магния - 0,3 - 1,0
Заявляемая пропорция ингредиентов является оптимальной, найдена экспериментально и обеспечивает необходимое качество ядра композиции.
Введение в композицию в качестве целевых добавок аэросила, крахмала и молочного сахара наряду с применением микрокристаллической целлюлозы, поливинилпирролидона и стеарата магния в указанных пределах позволило увеличить по сравнению с прототипом содержание действующего вещества (свыше 60%) и обеспечило стабильность композиции в ходе хранения при удовлетворительной прочности ядра таблетки и легкости высвобождения действующего вещества.
Несоблюдение найденных соотношений ингредиентов в ядре не позволяет получить необходимое качество и стабильность композиции при хранении.
Предлагаемая фармацевтическая композиция выполняется в виде твердой лекарственной формы, имеющей оболочку, предпочтительно в форме таблеток, что обеспечивает максимальную технологичность последующей фасовки. Наличие оболочки заявленного состава, во-первых, дополнительно повышает стабильность композиции в период хранения и удлиняет срок годности препарата, во-вторых, улучшает ее внешний вид. Оптимальное соотношение указанных ингредиентов в оболочке составляет, маc.%:
Сополимер метакриловой кислоты - 15,0 - 50,0
Полиэтиленгликоль - 1,0 - 20,0
Диоксид титана - 36,0 - 84,0
В качестве основы оболочки наиболее приемлемым оказался сополимер метакриловой кислоты, и в качестве наполнителей - полиэтиленгликоль и диоксид титана. Совместное использование этих ингредиентов в приведенных соотношениях позволяет получить оболочку, имеющую необходимую прочность и защищающую ядро от внешней атмосферы. Помимо этого, применение указанной оболочки наряду с ядром заявленного состава обуславливает быстрое и целенаправленное высвобождение гидрохлорида ранитидина из лекарственной формы (в среду растворения через 30 мин переходит 94 - 97% действующего вещества, по ГФ XI - не менее 75% через 45 мин), что в свою очередь повышает биодоступность препарата и сводит к минимуму возможность побочных эффектов.
Способ получения фармацевтического состава с противоязвенной активностью, содержащего гидрохлорид ранитидина и фармацевтически приемлемые целевые добавки, включает смешение указанного действующего вещества с наполнителем, представляющим собой комбинацию крахмала, микрокристаллической целлюлозы и молочного сахара, увлажнение полученной массы водно-спиртовым раствором поливинилпирролидона при его массовом соотношении с увлажняемой массой 1: (5,2 - 9,8), соответственно, влажное гранулирование, сушку, сухое гранулирование, введение аэросила, стеарата магния и остальной части крахмала, последующее формование гранул и нанесение оболочки. Использование при получении гранулята в качестве наполнителя комбинации крахмала, микрокристаллической целлюлозы и молочного сахара при заявляемом соотношении увлажнителя и увлажняемой массы обеспечивает хорошую адгезию гидрохлорида ранитидина и вспомогательных веществ, что способствует существенному повышению прочности ядра таблетки (до 12 - 13 кг) при снижении содержания связующего вещества (поливинилпирролидона) по сравнению с прототипом. Тем самым, уменьшается отбраковка таблеток в процессе нанесения защитного покрытия и последующей фасовки. Целесообразное массовое соотношение воды и 96% этилового спирта в увлажняющем растворе составляет 1:0,75 - 1,25, однако этот интервал не является строго ограниченным. Концентрация поливинилпирролидона в растворе составляет 1,0 - 7,0 %.
Количество вводимого в композицию после сухого гранулирования крахмала составляет 5,0 - 14,0% от общей массы крахмала, содержащегося в лекарственной форме. Остальную часть крахмала применяют в качестве наполнителя. Изменение этого соотношения ухудшает текучие свойства гранулята и затрудняет нормальное проведение процесса таблетирования.
Оптимальная остаточная влажность гранул после сушки составляет 2 - 3%. Более высокое влагосодержание вызывает слипание гранул, тем самым, затрудняя получение однородной смеси и проведение таблетирования, а при уменьшении влажности возрастает отбраковка таблеток (сколы, расслоение).
Полученная фармацевтическая композиция соответствует требованиям ГФ XI (по внешнему виду, растворению и другим показателям), стабильна при хранении и имеет срок годности более 2 лет.
Изобретение иллюстрируется следующими примерами.
Пример 1. Смесь просеянных порошков ранитидина (62,4 г), молочного сахара (15,6 г), микрокристаллической целлюлозы (8,85 г) и сухого картофельного крахмала (15,8 г) увлажняют 10,4 г 1% раствора поливинилпирролидона в 50% водном этаноле, перемешивают до равномерного распределения влаги, гранулируют на установке для получения гранулята через сетку с диаметром отверстий 3 мм и сушат до остаточной влажности 2 - 3%. Сухое гранулирование проводят на установке для получения гранулята из сухих смесей. К измельченному грануляту добавляют смесь 0,8 г сухого картофельного крахмала, 0,1 г аэросила и 0,3 г стеарата магния, после чего таблетируют. На полученные ядра со средней массой 0,26 г наносят пленкообразующий состав, содержащий 6,9% сополимера метакриловой кислоты (Eudragit Е-100), 4,6% полиэтиленгликоля, 11,5% двуокиси титана в смеси ацетона и водного спирта. Наслаивание проводят до получения пленки удовлетворительной толщины. Полученные таблетки со средней массой 0,273 г удовлетворяют требованиям на фармацевтическое средство.
Примеры 2, 3 выполняют аналогично. Полученные таблетки удовлетворяют требованиям на фармацевтическое средство. Результаты представлены в таблице.
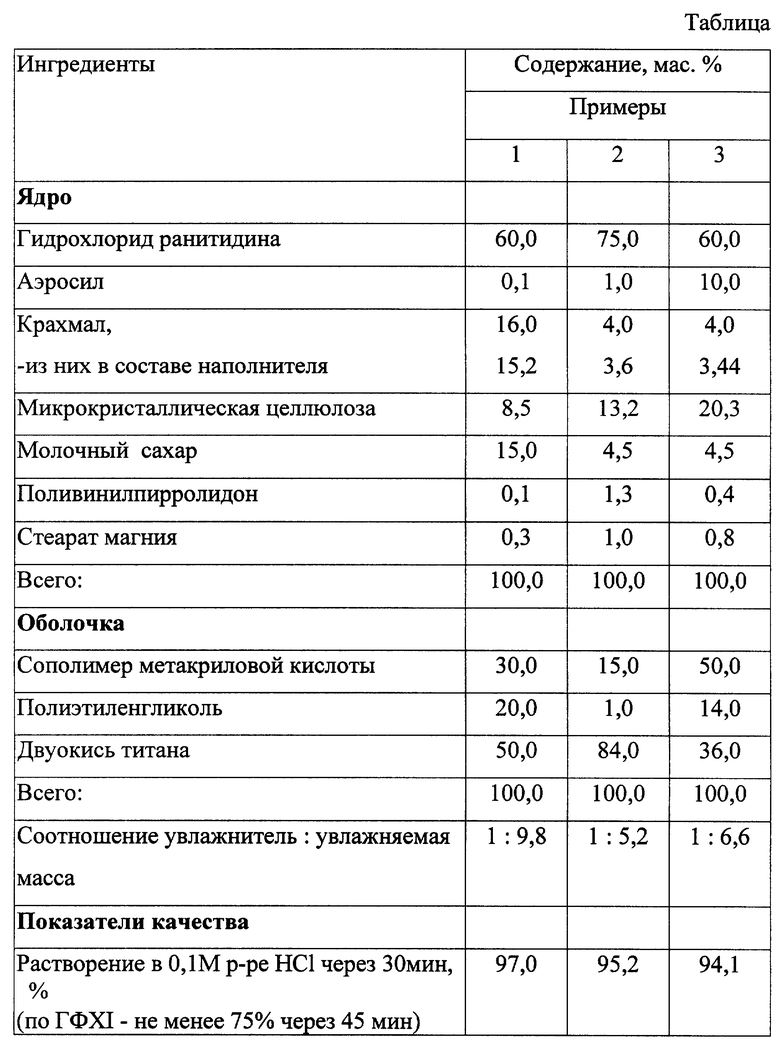
Фармацевтическая композиция выполнена в виде твердой дозированной формы, преимущественно в виде таблетки. Ядро твердой дозированной формы содержит, мас.%: гидрохлорид ранитидина 60,0 - 75,0, аэросил 0,1 - 10,0, крахмал 4,0 - 16,0, микрокристаллическую целлюлозу 8,5 - 20,3, молочный сахар 4,5 - 15,0, поливинилпирролидон 0,1 - 1,3 и стеарат магния 0,3 - 1,0. Оболочка ядра состоит из, мас.%, сополимера метакриловой кислоты 15,0 - 50,0, полиэтиленгликоля 1,0 - 20,0 и диоксида титана 36,0 - 84,0. Способ получения фармацевтической композиции включает смешение гидрохлорида ранитидина с наполнителем, влажное гранулирование, сушку, сухое гранулирование, формование гранул и нанесение оболочки. Новая фармацевтическая композиция устойчива при хранении, имеет удовлетворительную прочность и обеспечивает высокую биодоступность действующего вещества - гидрохлорида ранитидина. 2 с. и 5 з.п. ф-лы, 1 табл.
Гидрохлорид ранитидина - 60,0 - 75,0
Аэросил - 0,1 - 10,0
Крахмал - 4,0 - 16,0
Микрокристаллическая целлюлоза - 8,5 - 20,3
Молочный сахар - 4,5 - 15,0
Поливинилпирролидон - 0,1 - 1,3
Стеарат магния - 0,3 - 1,0
а оболочка состоит из сополимера метакриловой кислоты, полиэтиленгликоля и диоксида титана.
Сополимер метакриловой кислоты - 15,0 - 50,0
Полиэтиленгликоль - 1,0 - 20,0
Диоксид титана - 36,0 - 84,0
3. Фармацевтическая композиция по п.1 или 2, отличающаяся тем, что она выполнена в форме таблетки.
| US 4880636 A, 14.11.1989 | |||
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| ПРОТИВОЯЗВЕННОЕ СРЕДСТВО В ВИДЕ ТАБЛЕТОК И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1996 |
|
RU2131264C1 |

