Изобретение относится к способам анализа летучих органических соединений и может быть использовано в научных исследованиях и для контроля состава веществ в химической, нефтехимической, биологической и других отраслях промышленности. Преимущественно изобретение предназначено для использования в медицине для контроля чистоты медицинских препаратов.
Для разделения смесей алифатических и ароматических веществ методом газовой хроматографии обычно используют органические неподвижные жидкие фазы различной полярности, например оксидипропионитрил, полиэтиленгликоль, полиэтиленгликольсукцинат и др. (см., например, Авт. свид. СССР N 544913, кл. G 01 N 31/08, 1974). Однако такие фазы обладают недостаточно высокой селективностью к углеводородам с близкими температурами кипения, в частности к изомерам, либо, имея хорошую селективность, характеризуются большим временем удерживания. Для этих фаз селективность в гомологическом ряду полярных соединений практически отсутствует.
Известна неподвижная жидкая фаза для газовой хроматографии, которая в своем составе содержит кристаллогидраты солей различных металлов (см. авт. свид. СССР N 771542, МПК G 01 N 31/08, 1980). Способ хроматографического анализа с применением указанной неподвижной фазы основан на использовании в качестве подвижной фазы газа-носителя, содержащего пары воды (см. там же).
Известная неподвижная фаза и способ хроматографического анализа с ее применением, принятые за прототип, позволяют во многих случаях реализовать высокую селективность при анализе полярных соединений различных классов.
Однако в некоторых случаях при анализе сложных смесей веществ, содержащих, например, ароматические амины, соединения терпенового ряда и др. известная фаза проявляла недостаточную селективность и наблюдаемые пики разделяемых веществ имели несимметричный характер.
Задача изобретения состояла в создании такой неподвижной жидкой фазы и способа хроматографического анализа с ее применением, которые обеспечивают высокую селективность разделения различных классов органических соединений, включая гомологические ряды полярных соединений.
Указанная задача решается тем, что предложена неподвижная фаза для газовой хроматографии, основу которой, согласно изобретению, составляет водный раствор N-метилморфолин-N-оксида (C5H11NO2).
Задача решается также тем, что предложен способ хроматографического анализа смесей веществ с применением газа-носителя, содержащего пары воды, в котором согласно изобретению в качестве неподвижной фазы используют водный раствор N-метилморфолин-N-оксида.
Парциальное давление паров воды в газе-носителе при осуществлении предлагаемого способа может меняться в интервале от 40 до 760 мм рт. ст.
Температуру хроматографической колонки при осуществлении предлагаемого способа можно изменять в интервале от 40 до 150oC.
Используемое в качестве неподвижной фазы вещество (N-метилморфолин-N-оксид) неограниченно растворимо в воде, эфире и спирте, способно к донорно-акцепторным взаимодействиям и в настоящее время применяется как растворитель целлюлозы в процессе получения целлюлозных волокон (см. Голова Л.И. Химические волокна, 1996, N 1, с. 13 - 23).
Сущность изобретения поясняется приведенным ниже описанием способа приготовления хроматографического сорбента с применением предлагаемой неподвижной фазы и примерами хроматографического анализа различных классов соединений.
Используемый в предлагаемом способе образец N-метилморфолин-N-оксида (ММО) представляет собой кристаллическое вещество с содержанием воды 3 - 5% (по весу) и температурой плавления ≈ 150oC. Сорбент с 8% ММО готовили путем испарения при комнатной температуре летучего растворителя (воды) из суспензии, состоящей из частиц твердого носителя в растворе ММО. Твердым носителем служил белый диатомитовый носитель хроматон N-AW (фирмы "Chemapol", Чехия) фракция 0,16 - 0,20 мм. Высушенный сорбент загружали в колонку из нержавеющей стали, которую использовали при анализе различных классов химических веществ.
Пример 1.
Анализировались компоненты камфорного спирта. Условия анализа: хроматографическая колонка длиной 2,0 и внутренним диаметром 3 мм, заполненная сорбентом, приготовленным описанным выше способом. Температура колонки - 80oC, газ-носитель - азот, содержащий пары воды. Парциальное давление паров воды в газе-носителе составляло 350 мм рт. ст.
На фиг. 1 представлена полученная при указанных условиях хроматограмма разделения 10%-ного камфорного спирта (фармакопейный препарат). На хроматограмме видны отчетливо разделенные пики камфоры (1) и этанола (2). Следует отметить, что хроматографическое разделение этого же состава в аналогичных условиях (tk = 80oC, 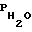 = 350 мм рт. ст.) с использованием водно-солевой неподвижной фазы (прототипа) не обеспечивало разделение пиков камфоры и этанола.
= 350 мм рт. ст.) с использованием водно-солевой неподвижной фазы (прототипа) не обеспечивало разделение пиков камфоры и этанола.
Пример 2.
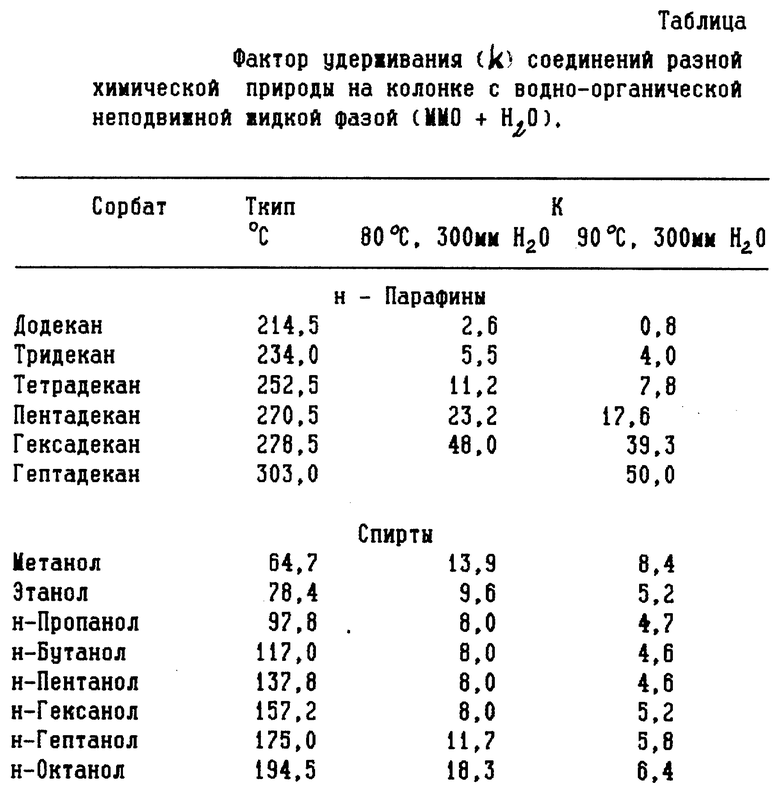
Анализу подвергали аэрозольный препарат "Каметон". Условия хроматографирования аналогичны условиям в примере 1, за исключением температуры хроматографической колонки, которая в данном примере составляла 90oC.
На фиг. 2 представлена полученная в этих условиях хроматограмма разделения, на которой: 1 - компоненты эвкалиптового масла; 2 - 1,8 - цинеол; 33 - камфора; 4 - нафталин; 5 - хлорбутанолгидрат; 6 - ментол.
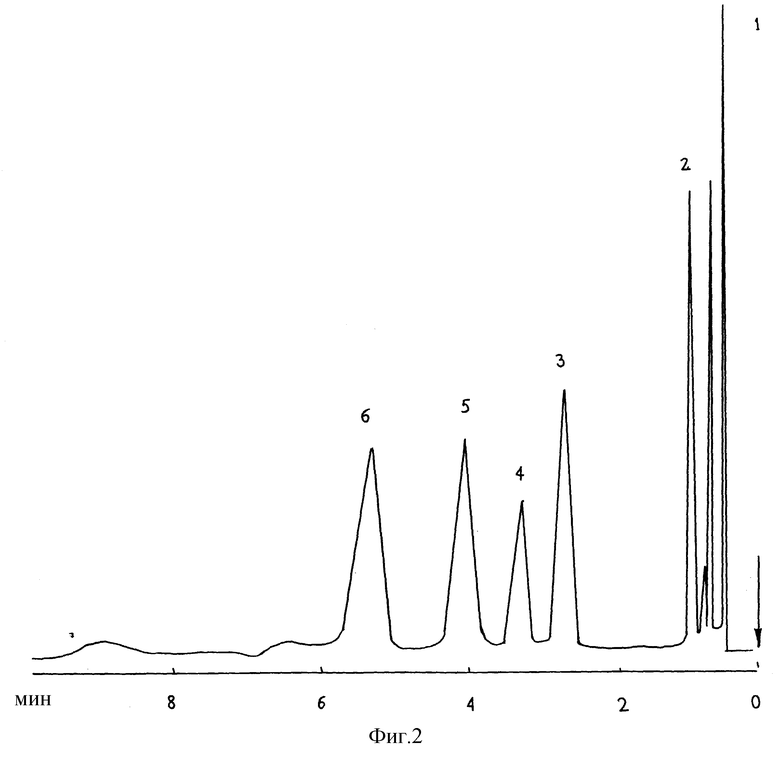
Пример 3.
Анализу подвергали бензин АИ-95. Условия анализа аналогичны условиям в примере 1. На фиг.3 представлена полученная при этих условиях хроматограмма разделения, на которой идентифицированы основные компоненты: 1 - н-ундекан; 2 - н-додекан; 3 - н-тридекан; 4 - н-тетрадекан; 5 - метанол.
Пример 4.
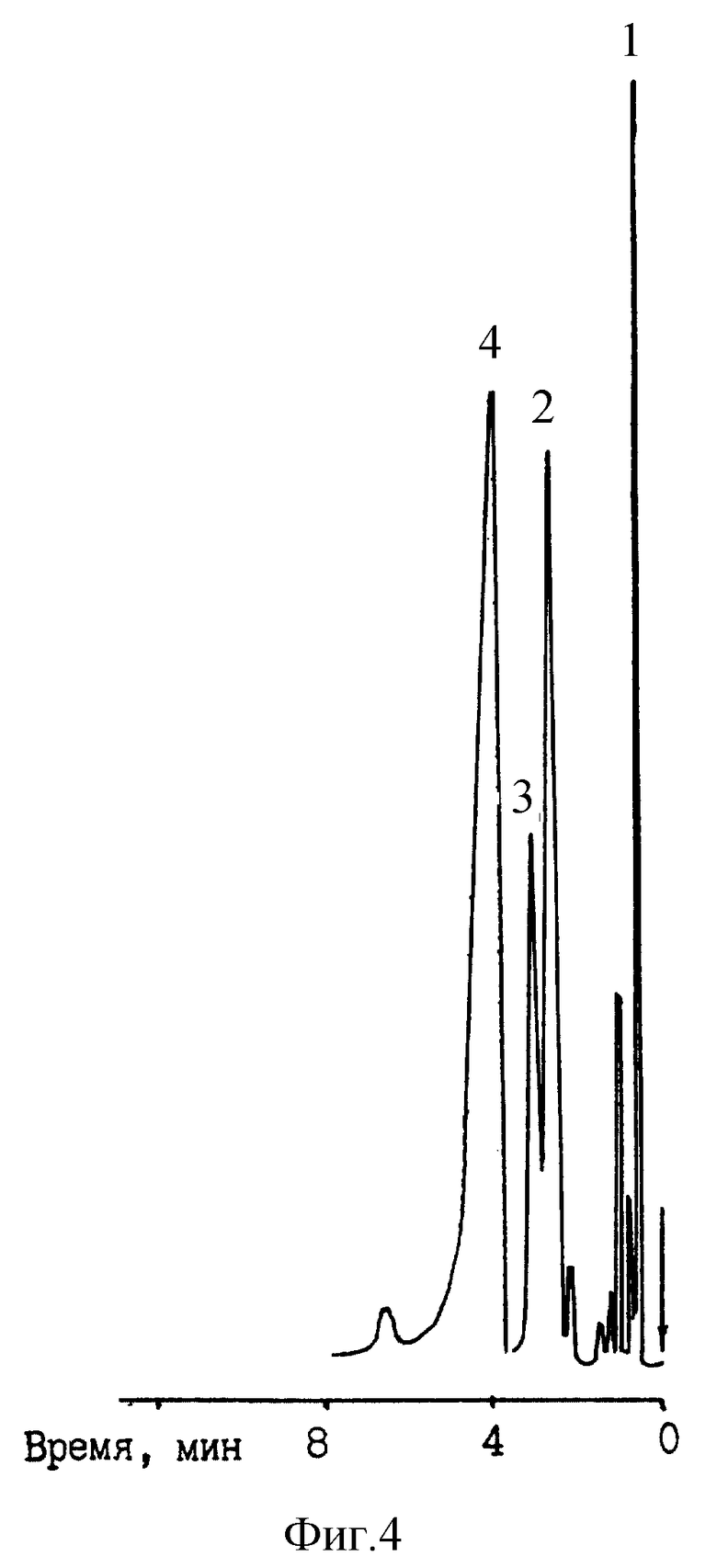
Анализу подвергалась смесь алифатических аминов. Температура колонки составляла 90oC. Упругость паров воды в газе-носителе (N2) составляла 335 мм рт. ст. Температура испарителя - 190oC. Температура детектора (ПИД) - 250oC.
На фиг. 4 представлена полученная при этих условиях хроматограмма разделения, на которой: 1 - триэтиламин; 2 - н-бутиламин; 3 - н-амиламин; 4 - диэтиламин.
Пример 5.
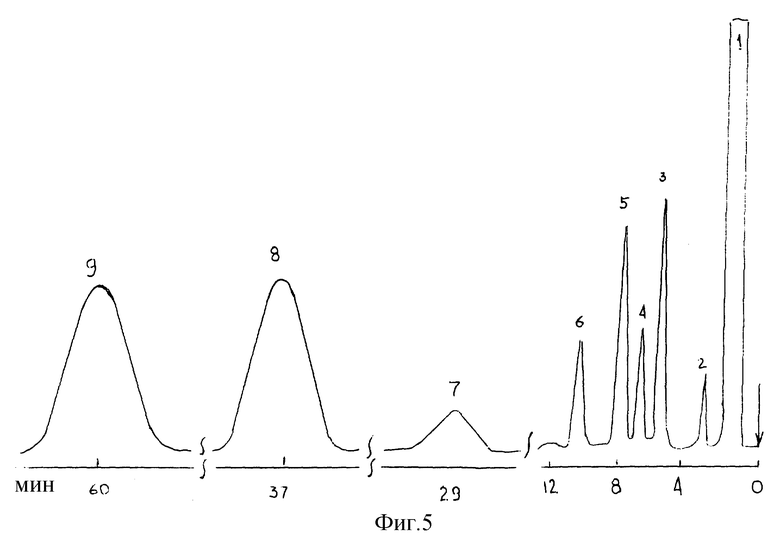
На фиг. 5 представлена хроматограмма сложной смеси соединений разной химической природы, включая близкокипящие компоненты, например, октанол-1 и 2,6-диметиланилин (Tкип - 214,5oC). Температура разделения - 90oC, парциальное давление паров воды в азоте - 400 мм рт. ст., температура испарителя - 300oC, детектора - 250oC. Основные идентифицированные компоненты: 1-бензол; 2 - н-тридекан; 3 - нафталин; 4 - пиридин; 5 - метанол; 6 - октанол-1; 7 - 2,6-диметиланилин; 8 - анилин; 9 - хинолин.
Пример 6.
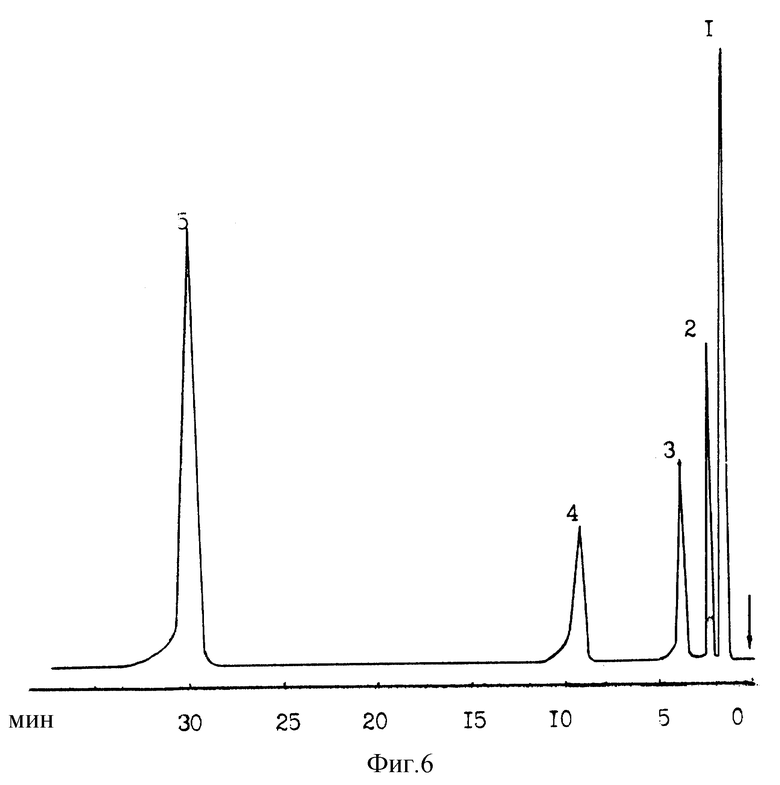
Анализу подвергалась смесь высококипящих соединений. Температура колонки составляла 125oC, температура испарителя - 300oC. Температура детектора - 350oC. Подвижная фаза - 100%-ный водяной пар.
На фиг. 6 представлена хроматограмма анализируемой смеси веществ, полученная при указанных условиях.
Идентифицированные компоненты смеси: 1 - пиридин; 2 - нафталин; 3 - бифинил; 4 - анилин; 5 - n-толуидин.
Пример 7.
Анализу подвергалась смесь парафинов C5-C11. Температура колонки 40oC, температура испарителя - 150oC, температура детектора (ПИД) - 200oC. Упругость паров воды в газе-носителе (N2) - 55 мм рт. ст.
На хроматограмме (не показана) парафины выходили узкими симметричными пиками, в то время как при анализе их с использованием известной неподвижной фазы, принятой за прототип, пики парафинов имели асимметричную форму.
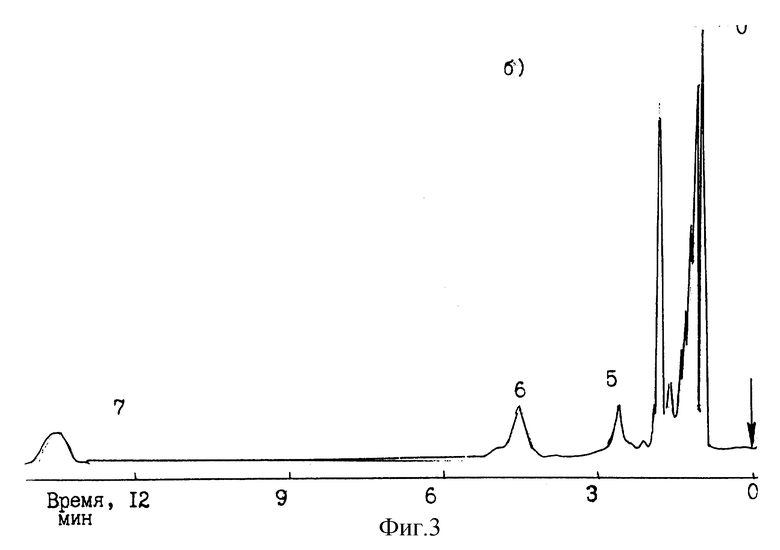
В таблице приведены полученные с применением предлагаемой неподвижной фазы данные по удерживанию (фактор удерживания - К) соединений различной химической природы при двух различных температурах хроматографической колонки (80 и 90oC) и разных парциальных давлениях паров воды в газе-носителе (300 мм рт. ст. и 350 мм рт. ст.).
Как видно из представленной таблицы, предлагаемая НЖФ позволяет разделять с высокой степенью селективности самые разные классы химических соединений. При этом температура колонки оказывает очень сильное влияние на удерживание компонентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА СМЕСЕЙ ВЕЩЕСТВ И ГАЗОВЫЙ ХРОМАТОГРАФ | 1991 |
|
RU2018821C1 |
| Неподвижная жидкая фаза длягАзОВОй ХРОМАТОгРАфии | 1979 |
|
SU840733A1 |
| Способ хроматографического анализа | 1978 |
|
SU771542A1 |
| НЕПОДВИЖНАЯ ФАЗА ДЛЯ ГАЗОВОЙ ХРОМАТОГРАФИИ | 2014 |
|
RU2570705C1 |
| Неподвижная фаза для газовой хроматографии | 1978 |
|
SU771543A1 |
| Способ получения модифицированных адсорбентов и твердых носителей для хроматографии | 1976 |
|
SU612170A1 |
| СПОСОБ ПРОИЗВОДСТВА ЭКСПЕРТИЗЫ АЛКОГОЛЬНОГО ОПЬЯНЕНИЯ ПРИ СУДЕБНО-ХИМИЧЕСКОМ ИССЛЕДОВАНИИ БИОЖИДКОСТЕЙ ТРУПОВ | 1998 |
|
RU2130754C1 |
| Неподвижная фаза для газовой хроматографии | 1987 |
|
SU1476373A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ИЗОТЕРМ СОРБЦИИ ГАЗОВ И ПАРОВ В МЕМБРАННЫХ МАТЕРИАЛАХ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2013 |
|
RU2567402C2 |
| СПОСОБ ХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА В ЗАКРЫТОМ ТОНКОМ СЛОЕ СОРБЕНТА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2011 |
|
RU2494391C2 |


Предложены способ анализа летучих органических соединений методом парофазной хроматографии и неподвижная фаза. Последняя в своем составе содержит водный раствор N-метилморфолин-N-оксида. Парциальное давление паров воды в газе-носителе может составлять 40-760 мм рт.ст., а температура хроматографической колонки - выбираться в интервале 40-150oС. Изобретение обеспечивает высокую селективность разделения различных классов органических соединений, включая гомологические ряды полярных соединений. 2 с. и 2 з.п. ф-лы, 1 табл., 6 ил.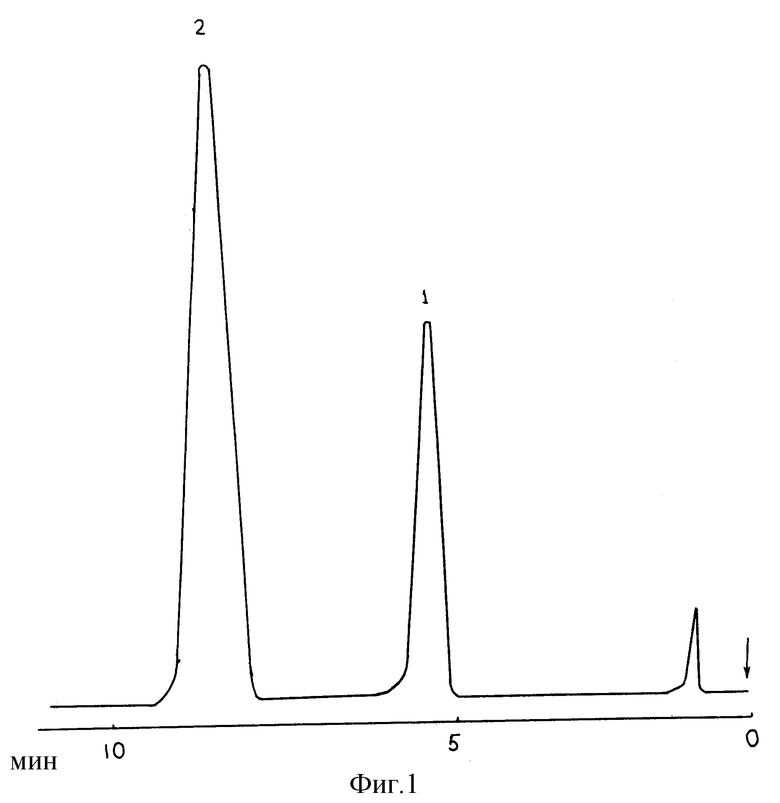
| Неподвижная фаза для газохроматографического разделения смесей углеводородов | 1974 |
|
SU544913A1 |
| Прибор для изучения контракции цементов | 1979 |
|
SU771552A1 |
| RU 96122605 A, 20.01.1999 | |||
| US 5503749 A, 02.04.1996 | |||
| АНАЛИТИЧЕСКИЙ КОМПЛЕКС ДЛЯ ОПРЕДЕЛЕНИЯ СПИРТОВ В БИОЛОГИЧЕСКИХ ЖИДКОСТЯХ | 1993 |
|
RU2110795C1 |
| Способ определения глюкозы и/или фруктозы в жидких средах | 1987 |
|
SU1434368A1 |
