Изобретение относится к новым производным трет.-бутилэрголина, к способу их получения, к фармацевтической композиции, содержащей такие соединения, и к использованию их в качестве фармацевтических препаратов.
Настоящее изобретение предлагает новую группу производных эрголина, которые, как обнаружено, обладают биологической активностью, представляющей особый интерес.
Раскрытые соединения обладают селективным и высоким сродством к 5-HTIA-рецепторам и весьма отличаются от большинства других производных эрголина в том, что они показывают незначительное сродство к α1-, α2, D1-, D2- рецепторам.
Упомянутые соединения могут быть использованы для лечения различных расстройств, связанных с серотонинэргическими дисфункциями, такими как нарушение терморегуляции, нарушение функции памяти, расстройства сна, нарушение регуляции насыщения (т.е., потребления пищи и напитков), привыкание к чрезмерному употреблению лекарственных средств, регуляции синдрома отмены лекарственного препарата, гипертензия, гиперемезис, депрессия, страх и психоз, ишемический инсульт.
Конкретнее, настоящее изобретение предлагает соединения формулы (I)
в которой A представляет собой OH, NH2, COOR'3, OCONHR4, CONHR4, NHCOR4, NHCO2R4, NHC(X)NHR4, NHC(X)NHCOR4,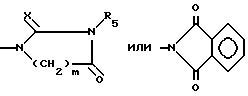
R1 представляет собой водород или линейный или разветвленный C1-4-алкил; R2 представляет собой водород, хлор, бром или S-C1-4-алкильную группу; R3 и R'3, независимо, представляют собой C1-5-алкил или водород, n равен 0, 1 или 2, m равен 1 или 2; R4 представляет собой водород, C1-7-алкил, C3-7-циклоалкил, адамантидил (трицикло-3.3.1.1.3.7)декан-1-ил, C1-5-алкилфенил, C2-алкилфенил; фенил, необязательно замещенный одной или несколькими группами, выбираемыми среди C1-4-алкила, C1-3-алкоксигруппы, метилендиоксигруппы, цианогруппы, трифторметила, гидроксигруппы, нитрогруппы и ацетила; необязательно замещенное нафтильное кольцо или фенил, сконденсированный с гетероциклической кольцевой системой, содержащей 5- или 6-членное кольцо, включающее 1 - 4 гетероатома, выбираемых среди азота, кислорода и серы; гетероциклическое 5- или 6-членное кольцо, включающее 1 или 2 гетероатома, выбираемых среди азота, кислорода и серы, которое необязательно замещено группой, выбираемой среди C1-4-алкила, фенила, необязательно замещенного так, как упоминалось выше, C1-3-алкоксигруппы и галогена; R5 представляет собой водород, C1-4-алкил или фенил, и X представляет собой NH, O или S; или их фармацевтически приемлемые соли.
Галоген, предпочтительно, представляет собой хлор или бром. Заместитель трет.бутил находится в положении 13 или 14 эрголинового скелета. Заместитель в положении 8 имеет α- или β- расположение.
Подходящие фармацевтически приемлемые кислотно-аддитивные соли включают соли как неорганических, так и органических кислот.
В настоящем описании термин "линейный или разветвленный алкил" включает метил, этил, пропил, изопропил, бутил, втор. бутил, н.бутил, пентил или гексил; термин C3-7-циклоалкил" включает циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Когда R4 представляет собой фенил, сконденсированный с гетероциклическим кольцом, его, предпочтительно, выбирают среди групп, имеющих формулы: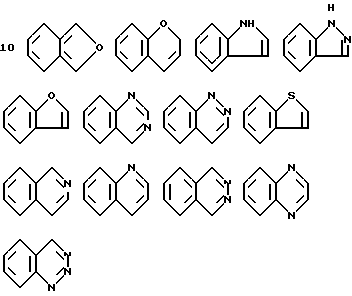
Когда R4 представляет собой гетероциклическое кольцо, его, предпочтительно, выбирают среди групп, имеющих формулы: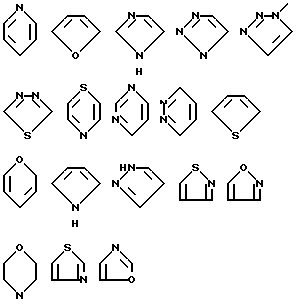
Все упомянутые выше гетероциклические системы необязательно замещенные, могут быть полностью или частично восстановлены.
Более предпочтительными соединениями формулы I являются те соединения, в которых R1 представляет собой водород или метил, R2 представляет собой водород, бром или S-C1-4-алкильную группу, R3 представляет собой метил, n равен 0, 1 или 2, и A выбирают среди OH, NH2, COOR'3, NHCO2R4, NHCONHR4, CONHR4, OCONHR4, NHCONHCOR4, NHCSNHCOR4,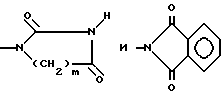
где m равен 1 или 2, R'3 представляет собой C1-4-алкил или водород, и R4 представляет собой фенил, бензил, трет.бутил, пиридил, 5-бромпиридил, этил, циклогексил, адамантил, фенилвинил, 5-бромпиридил, этил, циклогексил, адамантидил, фенилвинил, 1,5-диметил-3-пиразил, 2-метил-4-тиазолил, пиразинил, пиримидинил, тиазолил или 6-хлор-3-пиридазинил.
Предпочтительно, R3 представляет собой метильную группу.
Настоящее изобретение также предлагает способ получения соединений формулы (I) и их кислотно-аддитивных солей, который включает
(а) взаимодействие соединения формулы (II)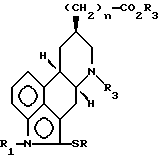
в которой n, R1 и R3 имеют установленные выше значения, и R представляет собой C1-4-алкил или фенил, с трет.бутилирующим агентом в присутствии кислоты, отделение полученного в результате 13-изомера от 14-изомера, и удаление 2-SR-группы посредством восстановителя;
(i) либо гидролиз получающегося 8-карбоксилата и конденсацию образовавшейся 8-карбоновой кислоты, необязательно после активации, с амином формулы R4-NH2, в которой R4 имеет установленные выше значения;
(ii) либо восстановление образующегося 8-карбоксилата и взаимодействие получающегося в результате 8-гидроксиметильного производного с соединением формулы R4-N= C= O или с п-нитрофенилхлоркарбонатом, и затем с соединением формулы R4-NH2, где R4 имеет установленные выше значения;
(iii) либо восстановление образующегося в результате 8-карбоксилата и восстановление получающегося в результате 8-гидроксиметильного производного трифенилфосфином, диэтилазодикарбоксилатом и фталимидом, и гидролиз полученного соединения;
(b) взаимодействие соединения, полученного на стадии a (iii), имеющего формулу (III)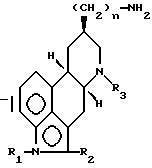
в которой n, R1, R2 и R3 имеют установленные выше значения,
(i) с соединением формулы (IV) R4 - COOH, в которой R4 имеет установленные выше значения, или с его реакционноспособным функциональным производным; или
(ii) с соединением формулы (V) R4-N=C=X, или (ii') с п-нитрофенилхлоркарбонатом, и взаимодействие полученного в любом случае соединения, с соединением формулы R4-NH2,
где R4 и X имеют установленные выше значения; или
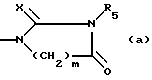
(iii) с соединением формулы (VI) Hal-(CH2)m-COOC1-2алкил, где m имеет установленные выше значения, и Hal означает галоген, и циклизацию полученного в результате соединения с соединением формулы Rs-N=C=X, где R5 и X имеют установленные выше значения; или
(iv) с соединением формулы (VII) R4OCOY, где R4 имеет установленные выше значения, и Y представляет собой хлор или п-нитрофенильную группу; или
(v) с соединением формулы (VIII) R4-CON=C=X, где R4 и X имеют установленные выше значения;
(c) если желательно, превращение соединения формулы (I), полученного таким образом, в другое соединение формулы (I) и/или, если желательно, превращение свободного соединения формулы (I) в его кислотно-аддитивную соль, и
(d) извлечение получающегося в результате соединения формулы (I) как такового или в виде его кислотно-аддитивной соли.
Трет. бутилирующий агент, применяемый на стадии (а), может представлять собой, например, изобутан или трет-бутилацетат. Подходящей кислотой для этой стадии является трифторуксусная кислота. Подходящим восстановителем для стадии (a) является никель Ренея.
Когда 8-карбоновую кислоту, полученную на стадии (a)-(i), активируют, подходящим образом этот процесс осуществляют путем взаимодействия с таким соединением, как смешанный ангидрид, ацилазид или N,N'-замещенная изомочевина.
Соединение формулы (I), в котором R2 представляет собой водород, может быть хлорировано или бромировано с целью получения соединения формулы (I), в котором R2 представляет собой хлор или бром.
Соединение формулы (I), в котором R1 представляет собой водород, может быть N-алкилировано с получением соединения формулы (I), в котором R1 представляет собой C1-4-алкил.
Соединение формулы (I), в котором заместитель X в заместителе A представляет собой серу, может быть превращено в соответствующее соединение формулы (I), в котором X представляет собой кислород, путем обработки солью серебра.
Процесс стадии (b) может быть осуществлен в соответствии со стандартной методикой. Подходящие реакционноспособные функциональные производные соединений формулы (IV) включают соответствующие галоидангидриды, имидазолиды, ацилазиды. Взаимодействие с ацилгалогенидами подходящим образом осуществляют в присутствии органического основания, такого как пиридин или этиламин. Взаимодействие с имилазолидами (полученными взаимодействием соединений формулы (IV) с N,N-карбонилдиимидазолом) подходящим образом осуществляют в инертном растворителе, таком как тетрагидрофуран. Реакцию с ацилазидами (полученными взаимодействием соединения формулы (IV) с дифенилфосфороилазидом. DPPA) подходящим образом осуществляют в инертном растворителе, таком как тетрагидрофуран, при 0oC в присутствии органического основания, такого как триэтиламин.
Взаимодействие с соединениями формулы (V) или (VIII) (которые можно получить, например, исходя из соединения формулы (IV) по известным реакциям) подходящим образом осуществляют в инертном растворителе, таком как тетрагидрофуран или диоксан, при температуре в интервале от 65 до 100oC.
Взаимодействие с п-нитрофенилхлоркарбонатом может быть осуществлено в инертном растворителе, таком как тетрагидрофуран или метиленхлорид, при температуре от 0 до 30oC, в присутствии основания, такого как триэтиламин или карбонат калия.
Взаимодействие с соединением формулы R4-NH2 под индексом (ii') или взаимодействием с соединениями формулы (VI) осуществляют подходящим образом в инертном растворителе, таком как диметилформамиде или тетрагидрофуран, при температуре от 30 до 100oC.
Циклизацию по п. (iii) предпочтительно осуществляют путем нагревания в растворителе, подобном диоксану или толуолу, или путем плавления при пониженном давлении.
Взаимодействие с соединением формулы (VII) выполняют в инертном растворителе, таком как пиридин или тетрагидрофуран, при комнатной температуре в присутствии органического основания, такого как триэтиламин.
Хлорирование или бромирование соединения формулы (I), в котором R2 представляет собой водород, может быть осуществлено по известным методикам с использованием стандартных хлорирующих или бромирующих агентов, таких как N-Cl или N-Br-сукцинимид или сульфурилхлорид. Реакцию обычно осуществляют в инертном растворителе, таком как хлороформ, метиленхлорид или тетрагидрофуран.
N-Алкилирование соединения формулы (I), в котором R1 представляет собой водород, может быть осуществлено в соответствии с известными методами N-алкилирования индолов, например, при использовании соединения формулы (IX) R1-Z, в котором R1 представляет собой C1-4-алкил, и Z представляет собой отщепляющуюся группу, такую как хлор, бром, иод. Реакцию обычно осуществляют в инертном растворителе, таком как диметилсульфоксид, и в присутствии сильного основания, такого как гидроксид калия или натрия.
Конверсия заместителя X от серы в кислород может быть осуществлена в подходящем растворителе, таком как этанол или раствор метансульфоновой кислоты в воде, причем соль серебра представляет собой нитрат или сульфат серебра.
Исходные соединения формулы (II), (IV), (V), (VI), (VII), (VIII), (IX) и амин формулы R4NH2 являются известными соединениями или могут быть получены хорошо известными способами, исходя из известных соединений.
Соединения настоящего изобретения проявляют удивительные фармацевтические свойства. Испытания на связывание показывают, что соединения общей формулы (I) обладают высоким и селективным сродством к 5-HTIA-рецепторным участкам, имеющим агонистическую или антагонистическую активность на главном уровне. Соединения настоящего изобретения могут найти применение при оказании помощи при страхе, депрессии, шизофрении и боли (Pharmacology and Toxicology 1989, 64, p. 3 - 5, Drug of the future, 1988, 13 (5), p. 429 - 437, J. Neural Transm., 1988, 74, p. 195 - 198), для лечения стресса (Neuropharmac, 1989, 25 (5), p. 471 - 476), и облегчения синдрома отмены лекарственного препарата (абстинентного синдрома) - благодаря подавлению бензодиазепинов, кокаина, спирта и никотина, или для модификации всасывания пищи и сексуального поведения (J. Receptor Research, 1988, 8, p. 59 - 81), и для облегчения боли при нейронных повреждениях, следующих за церебральной ишемией, действуя как нейропротектанты (Stroke 1990, 21 (IV) p. 161; J. Cereb. Blood Flow Metabol. 1911, 11 (II), p. 426; Pharmacology of cerebral ischemia, 1990, Stuttgart, 1990, p. 493 - 497).
Следующие далее эксперименты иллюстрируют профиль связывания соединений общей формулы I.
Эксперимент 1
Сродство к серотонину IA(5-HTIA)-рецептора /испытание связывания 3H-8-гидрокси-2-дипропиламинотетралина (3H-8-OH-DPAT)/
Приготовление неочищенной синаптосомной фракции и анализ связывания проводят в соответствии со способом, о котором сообщают Hall и др. в Journal od Neurochemistry, vol. 44, page 1685, 1985. Замороженный гиппокампус, извлеченный из крыс, гомогенизируют в 40 объемах охлажденного льдом 50 мМ трис-HCl буфера (pH 7,4), и суспензию центрифугируют при 500 x g в течение 10 минут при 0oC. Супернатант центрифугируют при 40000 x g в течение 20 минут при 0oC, и образующийся после центрифугирования осадок гомогенизируют в 40 объемах вышеупомянутого буфера и инкубируют при 37oC в течение 10 минут. После завершения реакции суспензию центрифугируют при 40000 x g в течение 20 минут при 0oC. Образующийся в результате осадок дважды промывают путем повторного суспендирования в 40 объемах вышеупомянутого буфера и центрифугирования, и в заключение суспендируют в 60 объемах охлажденного льдом 50 мМ трис-HCl буфера (pH 7,4), содержащего 1 мМ хлорида марганца для использования в следующем испытании.
К аликвотам (900 мкл) раствора синаптосомных мембран добавляют 50 мкл раствора тритированного 8-OH-DPAT до конечной концентрации 0,2 нМ, и 50 мкл раствора испытуемого соединения или 50 мкл его среды и инкубируют в течение 10 минут при 37oC. Затем к смеси добавляют 5 мл охлажденного льдом 50 мМ трис-HCl буфера (pH 7,4), быстро фильтруют под вакуумом через фильтры WhatmanR GF/B и промывают дважды 5 мл того же буфера. Радиоактивность остатка на фильтрах измеряют сцинтилляционным счетчиком для жидкости. Неспецифическое связывание устанавливают по присутствию менее 10-5 М серотонина (5-HT). Концентрацию испытуемого соединения при 50% ингибирования (IC50) определяют графически. Результаты суммируют в табл. 1.
Эксперимент 2.
Сродство к серотонину 2(5-HT2)-рецептора (испытание на связывание 3H-кетонсерина)
Получение неочищенной синаптостомной фракции и анализ связывания проводят в соответствии с методом, о котором сообщают Leysen и др. в Molecular Pharmacology, vol. 21, page 301, 1981.
Замороженную кору головного мозга, извлеченную из крыс, гомогенизируют в 30 объемах охлажденного льдом 0,32 М раствора сахарозы, и суспензию центрифугируют при 1000 x g в течение 10 минут при 0oC. Суспернатант центрифугируют при 4000 x g в течение 20 минут при 0oC, и образующийся в результате центрифугирования осадок гомогенизируют в 30 объемах охлажденного льдом 50 мМ трис-HCl буфера (pH 7,7), и инкубируют при 37oC в течение 10 минут. Суспензию снова центрифугируют при 40000 x g в течение 20 минут при 0oC. Полученный в результате центрифугирования осадок гомогенизируют в 100 объемах вышеупомянутого буфера и используют в качестве раствора синаптосомных мембран в следующем испытании.
К аликвотам (900 мкл) раствора синаптосомных мембран добавляют 50 мкл раствора 3H-кетансерина до конечной концентрации 0,2 мМ и 50 мкл испытуемого соединения или его среды и инкубируют при 37oC в течение 20 минут. После завершения реакции смесь быстро фильтруют под вакуумом через фильтры WhatmanR GF/B. Фильтры три раза промывают 5 мл упомянутого выше буфера и затем измеряют радиоактивность осадка, оставшегося на фильтрах, с помощью сцинтилляционного счетчика для жидкостей. Неспецифическое связывание устанавливают по присутствию менее 10 мкМ миансерина. Концентрацию испытуемого соединения при 50% ингибирования (IC50) определяют графически. Результаты суммируют в таблице 1.
Эксперимент 3.
Сродство к допамину 2(D2)-рецептора (испытания на связывание 3H-спиперона)
Получение неочищенной синаптосомной фракции и анализ связывания проводят в соответствии с методом, о котором сообщают 1. Creese и др. в European Journal of Pharmacology, vol. 46, page 377, 1977. Замороженное полосатое тело, извлеченное из крыс, гомогенизируют в 100 объемах охлажденного льдом 50 мМ транс-HCl буфера (pH 7,7), и суспензию центрифугируют при 500 х g в течение 10 минут при 0oC. Супернатант центрифугируют при 50000 х g в течение 15 минут при 0oC, и образующийся в результате центрифугирования осадок гомогенизируют в 100 объемах упомянутого выше буфера, и затем суспензию снова центрифугируют при 50000 х g в течение 15 минут при 0oC. Образующийся в результате центрифугирования осадок гомогенизируют в 150 объемах 50 мМ трис-HCl буфера (pH 7,1), содержащего 120 мМ хлорида калия, 2 мМ хлорида кальция, 1 мМ хлорида магния, 0,1% аскорбиновой кислоты и 10 мкМ паргилина. Суспензию инкубируют при 37oC в течение 10 минут и затем используют в качестве раствора синаптосомных мембран для следующих испытаний.
К аликвотам (900 мкл) раствора синаптосомных мембран добавляют 50 мкл раствора 3H-спиперона при конечной концентрации 0,2 нМ и 50 мкл раствора испытуемого соединения или 50 мкл и его среды, и инкубируют при 37oC в течение 20 минут. После завершения реакции смесь быстро фильтруют под вакуумом через фильтры WhatmanR GF/B. Фильтры промывают три раза 5 мл упомянутого выше буфера, и затем измеряют радиоактивность остатка на фильтрах с помощью сцинцилляционного счетчика для жидкости. Неспецифическое связывание устанавливают по присутствию менее 100 мкМ (L)-сульпирида. Концентрацию испытуемого соединения при ингибировании 50% (IC50) определяют графически. Результаты суммируют в табл. 1 (см в конце описания).
Поведенческая фармакология
Потенциальную анксиолитическую активность соединений формулы I устанавливают в скрининговой моделе страха по индуцированной стрессом гипертермии (Lecci A. , Borsini F., Volterra G. and Meli A., Pharmacological Validation of a novel animal model of anticipatory anxiety in mice. Phychopharmacology, 1990, 101, 255-261). Эта процедура основана на антагонизме возрастания ректальной температуры, наблюдаемого у посаженных в клетку мышей, когда их удаляют из их группы.
Из полученных данных, представленных в табл. 2 (см. в конце описания), очевидно, что соединения настоящего изобретения способны проявлять антагонизм к индуцированной стрессом гипертермии, обнаруживая потенциальные анксиолитические свойства.
Токсичность соединений настоящего изобретения в самом деле является незначительной, и они, следовательно, могут безопасно использоваться в качестве полезных лекарственных препаратов.
В соответствии с настоящим изобретением пациента лечат по способу, заключающемуся во введении пациенту эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Соединение формулы I или его фармацевтически приемлемая соль могут быть использованы для регуляции состояний, которые можно отнести на счет серотонинэргических дисфункций, таких как ухудшение терморегуляции или функции памяти, расстройства сна, привыкание к чрезмерному употреблению лекарственных средств, гипертензия, гиперемезис, депрессия, страх или психоз, или регуляция насыщения или синдром отмены лекарственного препарата, церебральная ишемия.
Изобретение также предлагает фармацевтическую композицию, включающую производное эрголина общей формулы I или его фармацевтически приемлемую соль в смеси с фармацевтически приемлемым разбавителем или носителем.
Соединения формулы I и их соли, описанные здесь, могут быть введены парентеральным или оральным способом, предпочтительно - оральным способом. В зависимости от способа введения композиции могут находиться в твердой, полутвердой или жидкой дозировачной форме, такой как, например, таблетки, пилюли, капсулы, порошки, жидкости, суспензии и т.п. Композиция может включать обычные фармацевтические носители, адъюванты и т.д.
Дозировка настоящих лекарственных средств изменяется в соответствии с полом, возрастом, состоянием или медицинскими картами пациентов, а также в соответствии со способом или целью введения. Вообще, лекарственные препараты могут вводиться как единичные дозы или в виде раздельных доз, чтобы обеспечить, скажем, 0,1-10 мг/кг веса тела в день эффективного ингредиента, предпочтительно - 0,1 - 5 мг/кг веса тела.
Фармацевтические композиции, содержащие соединения изобретения, готовят обычными способами с обычными ингредиентами.
Так, для орального введения фармацевтические композиции, содержащие соединения изобретения, представляют собой, предпочтительно, таблетки, пилюли или капсулы, которые содержат активное вещество вместе с разбавителями, такими как, например, лактоза, декстроза, маннит, сорбит, сахароза, целлюлоза, смазывающими веществами, например, кремнеземом, тальком, стеариновой кислотой, стеаратом кальция или магния или полиэтиленгликолями; или они также могут содержать связывающие вещества, такие как, например, крахмалы, желатин, метилцеллюлоза, аравийская камедь, трагакант, поливинилпирролидон; диспергирующие средства, такие как, например, крахмалы, альгиновая кислота, альгинаты; вспенивающую смесь, красящее вещество; подслащивающие вещества; смачиватели, такие как, например, лецитин, полисорбаты, лаурилсульфаты, и вообще нетоксичные и фармакологически неактивные вещества, применяемые при изготовлении фармацевтических препаратов.
Упомянутые фармацевтические препараты могут быть изготовлены известными способами, например, посредством процессов смещения, гранулирования, таблетирования, нанесения сахарного или пленочного покрытия.
Упомянутые фармацевтические формулировки, содержащие соединения изобретения, также могут быть изготовлены известными способами, и они могут представлять собой, например, сироп или капли для орального введения, стерильные растворы для инъекций или суппозитории.
Следующие далее примеры иллюстрируют изобретение.
Пример 1
2-Тиометил-6-метил-8 -β- метоксикарбонил-(13,14)-трет.бутилэрголин (1, R1 = H, R2 = SCH3, R3 = CH3, A = CO2CH3, n = 0)
К раствору 23 г 2-тиометил-6-метил -8β- метоксикарбонилэрголина в 230 мл трифторуксусной кислоты добавляют по каплям 24,5 мл трет.бутилацетата. Образующийся в результате раствор нагревают при 40oC в течение 5 часов. Растворитель испаряют, и темный остаток обрабатывают этилацетатом и 0,1 N гидроксидом аммония. Органическую фазу промывают соляным раствором и сушат (Na2SO4). Растворитель удаляют, и пенистый остаток растворяют в минимальном количестве метанола. Посредством охлаждения кристаллизуют 13,7 г 13-производного, т. пл. 259 - 261oC. Посредством хроматографирования маточной жидкости получают 4,2 г соответствующего 14-производного, т.пл. 280 - 283oC.
Пример 2
6-Метил -8β- метоксикарбонил-13-трет.бутилэрголин (1: R1 = R2 = H, R3 = CH3, A = CO2CH3, n = 0)
К кипящему с обратным холодильником раствору 5,75 г 2-тиометил-6-метил-13-трет. бутилэрголина в 300 мл метанола в атмосфере азота добавляют частями 10 г никеля Ренея.
После 10-минутного кипячения кипящую смесь фильтруют, и Ni Ренея тщательно промывают метанолом.
Растворитель удаляют, и остаток кристаллизуют из этилацетата, и получают 4,3 г названного в заголовке соединения, т.пл. 175 - 177oC.
Пример 3
6-Метил -8β- гидроксиметил-13-трет-бутилэрголин (1; R1 = R2 = H R3 = CH3, A = OH, n = 0)
К раствору 4,5 г борогидрида натрия в 50 мл метанола добавляют по каплям раствор 4,5 г 6-метил -8β- метоксикарбонил-13-трет.бутилэрголина в 30 мл метанола. Получающуюся в результате суспензию при перемешивании нагревают при 50oC в течение 1 часа. Образующийся в результате мутный раствор разбавляют 200 мл воды, и затем экстрагируют хлороформом. Органическую фазу промывают соляным раствором и сушат. Упаривание растворителя и кристаллизация из этанола дают 3,3 г названного в заголовке соединения, т.пл. 240 - 243oC.
Пример 4
6-Метил -8β- аминометил-13-трет-бутилэрголин (1: R1 = R2 = H, R3 = CH3, A = NH2, n = 1)
К суспензии 3 г 6-метил -8β- гидроксиметил-13-бутилэрголина, 3 г трифенилфосфина и 2 г фталимида в 30 мл тетрагидрофурана при перемешивании добавляют по каплям раствор 2,35 г диэтилазодикарбоксилата в 20 мл тетрагидрофурана.
Перемешивание продолжают в течение 2 часов; затем образовавшийся в результате оранжевый раствор разбавляют 200 мл 0,1 метансульфоновой кислоты и экстрагируют этилацетатом. Водную фазу подщелачивают гидроксидом аммония и затем экстрагируют этилацетатом. После промывания соляным раствором и высушивания раствор концентрируют при пониженном давлении, и получают 4 г 6 метил -8β- фталимидометил-13-трет-бутилэрголина, т.пл. 132-137oC. К раствору, содержащему это фталимидное производное в 100 мл этанола, добавляют 10 мл 98% гидразингидрата. После перемешивания в течение 3 часов суспензию фильтруют, и фильтрат после упаривания растворителя растворяют в этилацетате, и раствор тщательно промывают 0,1N гидроксидом натрия. Органическую фазу промывают соляным раствором и сушат. Концентрацией раствора получают 2,4 г названного в заголовке соединения, т.пл. 321 - 233oC.
Пример 5
6-Метил -8β- бензоиламинометил-13-трет.-бутилэрголин (1: R1 = R2 = H, R3 = CH3, A = NHCOPh, n = 1)
К раствору 2 г 6-метил -8β- аминометил-13-трет.бутилэрголина в 50 мл пиридина добавляют 0,95 г бензоилхлорида. После перемешивания при комнатной температуре в течение 3 часов образующийся в результате раствор разбавляют этилацетатом и промывают 0,1 N раствором гидроксида натрия и затем соляным раствором. После высушивания растворитель удаляют, и остаток растворяют в обработанном углем метаноле, затем дважды кристаллизуют из ацетона, и получают 2,4 г названного в заголовке соединения, т.пл. 190 - 193oC.
Пример 6
6-Метил- -8β, фенилацетиламинометил-13-трет. бутилэрголин (I; R1=R2=H, R3=CH3, A = NHCOCH2Ph, n = 1)
Действуя как в примере 5, но используя вместо бензоилхлорида фенилацетилхлорида получают названное в заголовке соединение с выходом 74%, т.пл. 165-167oC.
Пример 7
6-Метил- -8β- пивалоиламинометил-13-трет.бутилэрголин (I; R1= R2 = H, R3 = CH3, A = NHCO+, n = 1)
Действуя как в примере 5, но используя вместо бензоилхлорида пиловалоилхлорид, получают названное в заголовке соединение с выходом 60%, т.пл. 235-238oC.
Пример 8
6-Метил- -8β- изоникотиноиламинометил-13-трет.бутилэрголин (I: R1 = R2 = H, R3 = CH3, A = NHCOC5H4N, n = 1)
Действуя как в примере 5, но используя вместо бензоилхлорида гидрохлорид изоникотиноилхлорида, получают названное в заголовке соединение с выходом 45%, т.пл. 241-243oC.
Пример 9
6-Метил- -8β-(5-бромникотиноил)аминометил-13-трет.бутилэрголин (I, R1 = R2 = H, R3 = CH3, A = NHCOC5H3NBr, n = 1)
Действуя как в примере 5, но используя вместо бензоилхлорида гидрохлорид 5-бромникотиноилхлорида, получают названное в заголовке соединения с выходом 75%, т.пл. 285-287oC.
Пример 10
6-Метил- -8β- бензилоксикарбониламинометил-13-трет.бутилэрголин (I: R1 = R2 = I, R3 = CH3, A = NHCOOPh, n = 1)
Действуя как в примере 4, но используя вместо бензоилхлорида бензилоксикарбонилхлорид, получают названное в заголовке соединение с выходом 55%, т.пл. 139-142oC.
Пример 11
6-Метил-8-β- этоксикарбониламинометил-13-трет.бутилэрголин (I: R1 = R2 = H, R3 = CH3, A = NHCO2C2H5, n = 1)
Действуя как в примере 5, но используя вместо бензоилхлорида этоксикарбонилхлорид, получают названное в заголовке соединение с выходом 80%, т. пл. 235-237oC.
Пример 12
6-Метил- -8β- фениламинокарбониламинометил-13-трет.бутилэрголин (I: R1 = R2 = H, R3 = CH3, A = NHCONHPh, n = 1)
К раствору 3,2 г 6-Метил- -8β- аминометил-13-трет.бутилэрголина в 50 мл диоксана добавляют 1,5 г фенилизоцианата. Образующийся в результате раствор кипятят с обратным холодильником в течение 1 часа. Растворитель удаляют, и остаток хроматографируют на прокладке из силикагеля, элюируя хлороформом. После упаривания растворителя и кристаллизации из этанола получают названное в заголовке соединение с выходом 70%, т.пл. 238 - 240oC.
Пример 13
6-Метил- -8β- циклогексилкарбониламинометил-13-трет.бутилэрголин (I: R1 = R2 = H, R3 = CH3, A = NHCOC6H11, n = 1)
К раствору 1,92 циклогексанкарбоновой кислоты в 30 мл тетрагидрофурана добавляют 1,7 г карбонилдиимидазола. Полученный в результате раствор нагревают при 50oC в течение 10 минут. К образующемуся в результате прозрачному раствору добавляют 3,1 г 6-метил -8β- аминометил-13-трет.бутилэрголина, и нагревание продолжают в течение 3 часов. Растворитель удаляют испарением, остаток обрабатывают этилацетатом и промывают насыщенным раствором гидрокарбоната натрия. После промывания соляным раствором и сушки получившийся в результате раствор концентрируют, и получают 3,8 г названного в заголовке соединения, т.пл. 151- 154oC.
Пример 14
6-Метил -8β- (1-адамантил)карбониламинометил-13-трет.-бутилэрголин (I, R1 = R2 = H, R3 = CH3, A = NHCOAd, n = 1)
Действуя как в примере 13, но используя вместо циклогексанкарбоновой кислоты адамантан-1-карбоновую кислоту, получают названное в заголовке соединение с выходом 60%, т.пл. 240 - 243oC.
Пример 15
6-Метил -8β- (3-фенил)акрилоиламинометил-13-трет.-бутилэрголин (I, R1 = R2 = H, R3 = CH3, A = NHCOC2H2Ph, n = 1)
Действуя, как в примере 13, но используя вместо циклогексанкарбоновой кислоты 3-(фенил)акриловую кислоту, получают названное в заголовке соединение с выходом 35%, с т.пл. 190 - 191oC.
Пример 16
6-Метил -8β- (1,5-диметил-3-пиразоил/аминометил-13-трет.бутилэрголин (1; R1 = R2 = H, R3 = CH3, A = NHCOC3HN2(CH3)2, n = 1)
К раствору 2,2 г 1,5-диметилпиразол-3-карбоновой кислоты и 1,5 мл триэтиламина в 20 мл диметилформамида при 0oC добавляют по каплям 1,1 г этилкарбоната.
После перемешивания в течение 5 минут добавляют по каплям раствор 3,1 г 6-метил -8β- аминометил-13-трет. бутилэрголина в 20 мл диметилформамида, и образующийся в результате раствор перемешивают в течение ночи при комнатной температуре.
После удаления растворителя остаток обрабатывают метиленхлоридом, и раствор промывают раствором 0,1N гидроксида натрия. После промывания соляным раствором и сушки растворитель удаляют, и остаток дважды кристаллизуют из ацетона, получают 2,7 г названного в заголовке соединения, т.пл. 262 - 265oC.
Пример 17
6-Метил -8β- (2-метил-4-тиазоил)аминометил-13-трет.бутилэрголин (1; R1 = R2 = H, R3 = CH3, A = NHCOC3H2NS(CH3), n = 1)
Действуя как в примере 16, но используя вместо 1,5-диметилпиразол-3-карбоновой кислоты 2-метил-4-тиазолкарбоновую кислоту, получают названное в заголовке соединение с выходом 45%, т.пл. 265 - 268oC.
Пример 18
6-Метил -8β- бензоиламинометил-13-трет.бутилэрголин (I, R1= R2 = H, R3 = CH3, A = NHCOPh, n = 2)
Действуя как в примерах 1-5, но используя в качестве исходного вещества 2-тиометил-6-метил -8β- метоксикарбонилметилэрголин вместо 2-тиометил-6-метил -8β- метоксикарбонилэрголина, получают названное в заголовке соединение с выходом 55%, т.пл. 187 - 189oC.
Пример 19
2-Бром-6-метил -8β- бензоиламинометил-13-трет.бутилэрголин (I, R1 = H, R2 = Br, R3 = CH3, A = NHCOPh, n = 1)
К раствору 2 г 6-метил -8β- бензоиламинометил-13-трет.бутилэрголина в 75 мл диоксана добавляют порциями 0,9 г N-бромсукцинимида. После перемешивания при 40oC в течение 2 часов растворитель удаляют, и остаток хроматографируют на силикагеле, элюируя этилацетатом.
После кристаллизации из изопропанола получают 1,3 г названного в заголовке соединения, т.пл. 151 - 155oC.
Пример 20
6-Метил -8β- аминометил-14-трет.бутилэрголин (I; R1 = R2 = H, R3 = CH3, A = NH2, n = 1, 14-изомер)
Действуя как в примерах 2-4, но используя в качестве исходного вещества 2-тиометил-6-метил -8β- метоксикарбонил-14-трет. бутилэрголин, получают названное в заголовке соединение, т. пл. 215 - 217oC.
Пример 21
6-Метил -8β- бензоиламинометил-14-трет.бутилэрголин (I, R1 = R2 = H, R3 = CH3, A = NHCOPh, n = 1, 14-изомер)
Действуя как в примере 5, но используя 6-метил -8β- аминометил-14-трет. бутилэрголин, получают названное в заголовке соединение с выходом 80%, т.пл. 173 - 175oC.
Пример 22
6-Метил -8β- карбокси-13-трет.бутилэрголин (I, R1 = R2 = H, R3 = CH3, A = CO2H, n = 0)
К раствору 6,2 г 6-метил -8β- метоксикарбонил-13-трет-бутилэрголина в 50 мл метанола при перемешивании добавляют по каплям 20 мл IM NaOH.
После выдерживания при комнатной температуре в течение 2 часов растворитель удаляют и остаток разбавляют 100 мл H2O и обрабатывают 20 мл 1 M HCl. Образующийся в результате осадок отфильтровывают, промывают водой, затем кристаллизуют из кипящего метанола, и получают 5,1 г названного в заголовке соединения, т.пл. 237 - 239oC.
Пример 23
6-Метил -8β- (2-пиразинил)-аминокарбонил-13-трет.бутилэрголин (I, R1 = R2 = H, R3 = CH3, A = CONCH4H3N2, n = 0)
К раствору 4 г 6-метил -8β- карбокси-13-трет.бутилэрголина и 1,7 г N-гидроксибензотриазола в 50 мл диметилформамида добавляют 2,5 г дициклогексилкарбодиимида, и образующийся в результате раствор перемешивают при 0oC в течение получаса, затем оставляют нагреваться до комнатной температуры и обрабатывают 1,2 г 2 аминопиразина. Образующийся в результате мутный раствор греют при 80oC в течение 5 часов и затем удаляют растворитель. Остаток обрабатывают этилацетатом и промывают насыщенным раствором гидрокарбоната натрия. После промывания соляным раствором и сушки удаляют растворитель и сырую реакционную смесь хроматографируют на силикагеле, элюируя метиленхлоридом. Кристаллизация из ацетона дает 3,7 г названного в заголовке соединения, т.пл. 271 - 273oC.
Пример 24
6-Метил -8β- (2,6-диметил-4-пиримидинил)аминокарбонил-14-трет.бутилэроголин (1,R1=R2=H,R3=CH3, A=CONHC4N2(CH3)2, n=O, 14-изомер)
Действуя как в примерах 22 и 23, но используя вместо 6-метил -8β- метоксикарбонил-13-трет.бутилэрголина 6-метил -8β- метоксикарбонил-14-трет.бутилэрголин, а вместо 2-аминопиразина-2,6- диметил-4-аминопиримидин, получают названное в заголовке соединение с выходом 45%, т.пл. 284 - 287oC.
Пример 25
6-Метил -8β- (2-тиазолидинил)аминокарбонил-13-трет.бутилэрголин (1; R1= R2=H, R3=CH3, A=CONHC3H2NS, n=O)
Действуя как в примере 23, но используя вместо 2-аминопиразина 2-аминотиазол, получают названное в заголовке соединение с выходом 70%, т.пл. 230 - 234oC.
Пример 26
6-Метил -8β- (6-хлор-3-пиридазинил)аминокарбонил-13-трет.бутилэрголина (1; R1=R2=H; R3=CH3, A=CONHC4H2N2Cl, n=O)
Действуя как в примере 23, но используя вместо 2-аминопиразина 6-хлор-3-аминопиридазин, получают названное в заголовке соединение с выходом 35%, т.пл. 257 - 259oC.
Пример 27
6-Метил -8β- (1H,3H)-2,4-диоксодигидро-1-пиримидинилметил/-13-трет.бутилэрголин (1; R1=R2-1, R3=CH3, A=C4H5N2O2, n=1)
Смесь 5,1 г 6-метил -8β- аминометил-13-трет.бутилэрголина и 1,8 мл метилакрилата в 100 мл метанола кипятят с обратным холодильником в течение 4 часов. Растворитель удаляют испарением, и остаток кристаллизуют из этилацетата, получают 6 г 6-метил -8β- N-(2-метоксикарбонилэтил)-аминометил-13-трет. бутилэрголина, плавящегося при 153 - 157oC (1; R1=R2=H, R3=CH3, A= NCH2H4CO2CH3, n=1).
К раствору 2,9 г цианата калия в 30 мл добавляют по каплям раствор 6 г 6-метил -8β- N-(2-метоксикарбонилэтил)-аминометил-13-трет. бутилэрголина в 120 мл воды и 35 мл 1N HCl. Реакционную смесь нагревают при 80oC в течение 4 часов, и затем оставляют стоять при комнатной температуре в течение ночи. Твердое вещество отделяют, промывают водой и кристаллизуют из этанола, получают 4,3 г названного в заголовке соединения, т.пл. 297 - 302oC.
Пример 28
6-Метил -8β- (2-тиазолидинил)аминокарбонил-14-трет.бутилэрголин (1; R1= R2=H, R3=CH3, A=CONCH3H2NS, n=0)
Действуя как в примере 25, но используя вместо 6 метил -8β- карбокси-13-трет. бутилэрголлина 6-метил -8β- карбокси-14-трет.бутилэрголин, получают названное в заголовке соединение с выходом 609; т.пл. 243 - 247oC.
Пример 29
N-/(6-Метилэрголин-14-трет.бутил-8-ил)метил-N'-ацетилтиомочевина (1, R1= R2=H, R3=CH3, A=NH-CS-NHCOCH3, n=1)
К раствору 3,2 г 6-метил -8β- аминометил-14-трет.бутилэрголина в 30 мл тетрагидрофурана добавляют 1,5 г свежеприготовленного ацетилизотиоцианата.
После перемешивания в течение 3 часов получающийся в результате желтый раствор упаривают досуха, и остаток фильтруют через тонкий слой силикагеля, элюируя дихлорметаном. После кристаллизации из ацетона получают названное в заголовке соединение с выходом 40%, т.пл. 220 - 223oC.
Пример 30
N-/(6-Метилэрголин-14-трет. бутил -8β- ил)метил/-N'-бензоилтиомочевина (1; R1=R2=1; R3=CH3, A=NHCS-NHCOC6H6, n=1).
Действуя как в примере 29, но используя вместо ацетилизотиоцианата бензоилизотиоцианат, получают названное в заголовке соединение с выходом 75%, т.пл. 287 - 289oC.
Пример 31
N-/(6-Метилэрголин-14-трет. бутил -8β- ил)метил/-N'-бензоилмочевина (1; R1=R2=H, R3=CH3, A=NHCON HCOC6H5, n=1)
К раствору 4,4 г N-/(6-метилэрголин-14-трет.бутил -8β- ил)-метил/-N'-бензоилтиомочевины в 50 мл этанола добавляют по каплям раствор 3,5 г нитрата серебра в 20 мл воды. Получающийся в результате черный раствор кипятят с обратным холодильником в течение 10 минут и затем фильтруют через цеитную прокладку.
Растворитель удаляют и остаток обрабатывают этилацетатом. После промывания соляным раствором и сушки (MgSO4) растворитель испаряют и сырой продукт дважды перекристаллизовывают из ацетона, получают 2,8 г названного в заголовке соединения, т.пл. 293 - 297oC.
Пример 32
6-Метил -8β- бензоиламина-13-трет.бутилэрголин (1; R1=R2=H, R3=CH3, A= NHCOPh, n=0).
Действуя как в примере 5, но используя вместо 6-метил -8β- аминометил-13-трет. бутилэрголина 6-метил -8β- амино-13-трет.бутилэрголин, получают в заголовке соединение с выходом 60%, т.пл. 143 - 145oC.
Пример 33
6-Метил -8β- (2-фурил)акрилоиламинометил-13-трет.бутилэрголин (1; R1=R2= H, R3=CH3, A=NHCOC2H2C2H3O, n=1)
Действуя как в примере 13, но используя вместо циклогексанкарбоновой кислоты 3-(2-фурил)акриловую кислоту (E), получают названное в заголовке соединение с выходом 25%, т.пл. 173 - 178oC.
Пример 34
6-Метил -8β- (3,4-диметоксибензоил)аминометил-13-трет.бутилэрголин (1; R1=R2=H; R3=CH3, A=NHCOC8H9O2, n=1).
Действуя как в примере 5, но используя вместо бензоилхохлорида 3,5-диметоксибензоилхлорид, получают названное в заголовке соединение с выходом 75%, т.пл. 153 - 157oC.
Пример 35
6-Метил -8β- (1-фенил-2-пирролил)аминометил-13-трет.бутилэрголин (1; R1= R2=H, R3=CH3, A=NHCOC4H3NC6H5, n=1)
Действуя как в примере 16, но используя вместо 1,5-диметилпиразол-3-карбоновой кислоты 1-фенилпиррол-2-карбоновую кислоту, получают названное в заголовке соединение с выходом 35%, т.пл. 135 - 137oC.
Пример 36
Приготовление таблеток
Соединение примера 5 (FCE 23892) - 5 мг
лактоза - 200 мг
кукурузный крахмал - 50 мг
стеарат магния - 5 мг
Способ приготовления
FСЕ 23892, лактозу и кукурузный крахмал смешивают и равномерно увлажняют водой. После просеивания увлажненной массы и сушки на полочной сушилке смесь снова пропускают сквозь сито и добавляют стеарат магния. Получающуюся в результате смесь прессуют в таблетки весом в 260 мг каждая.
Пример 37
Приготовление капсул
Соединение примера 9 (FСЕ-27823) - 5 мг
лактоза - 200 мг
стеарат магния - 5 мг
Способ приготовления
FCE 27823 смешивают с остальными добавочными продуктами, образующуюся в результате смесь пропускают через сито и перемешивают до однородного состояния в подходящем аппарате. Образующейся в результате смесью заполняют твердые желатиновые капсулы (по 210 мг на капсулу).
Пример 38
1,6-диметил -8-β- бензоиламинометил-13-трет-бутилэрголин (1, R1=R2=H3; R3=H, A=NHCOPh, n=1).
К раствору 2 г гидроксида натрия в 50 мл диметилсульфоксида добавляют 2 г 6-метил -8β- бензоиламинометил-13-трет-бутил эрголина. После перемешивания в течение 20 мин добавляют раствор 3 г метилиодида в 5 мл диметилсульфоксида и перемешивание продолжают в течение 30 мин. Реакционную смесь разбавляют водой и экстрагируют этилацетатом. После высушивания растворитель удаляют и остаток хроматографируют на силикагеле, элюируя смесью этилацетат/циклогексан 1/1.
Фракции, содержащие продукт, объединяют. Выпаривание растворителя и кристаллизация из этилацетата Δaст 1,3 г целевого соединения. Т.пл. 172 - 175oC.
Пример 39
6-метил -8β- аминокарбонилоксиметил-13-третбутилэрголин (1, R1=R2=H, R3= CH3, A=OCONH2, n=1).
К раствору 3,1 г 6-метил -8β- гидроксиметил-13-третбутил эрголина в 30 мл пиридина порциями добавляют 3 г n-нитрофенилхлоркарбоната при комнатной температуре.
После перемешивания в течение 1 часа получают желтоватый раствор, обрабатывают 10 мл концентрированного раствора гидроксида аммония и полученный желтый раствор выставляют в течение 1 часа. Растворитель удаляют и остаток разделяют между этилацетатом и разбавленным раствором карбоната натрия. После высушивания, растворитель удаляют и остаток, растворенный в ацетоне, обрабатывают древесным углем.
Концентрирование чистого раствора дает 2,1 г целевого соединения. Т.пл. 196-200oC.
Пример 40.
6-метил -8β- аминокарбонилоксиметил-14-трет-бутил-эрголин (1, R1=R2=H, R3=CH3, A = OCONH2; n=1).
Соединение получают по методике примера 39, используя 6-метил -8β- гидроксиметил-14-третбутил эрголин вместо 6-метил -8β- гидроксиметил-13-третбутил эрголина, выход 70%, Т.пл. 171-173oC.
Пример 41
6-метил -8β- (4-метоксибензоил)аминометил-13-третбутил эрголин (1, R1= R2=H, R3=CH3; A = NHCOC6OCH3, n=1).
Соединение получают по методике примера 5, используя 4-метоксибензоилхлорид вместо бензоилхлорида, выход 80%, Т. пл. 162-167oC.
Пример 42
6-метил -8β- (4-трифторметилбензоил)-аминометил-14-третбутил эрголин (1, R1=R2=H; R3=CH3, A = NHCOC6H4CF3, n=1).
Соединение получают по методике примера 5, используя 6-метил -8β- аминометил-14-третбутил эрголин вместо 6-метил 8β- аминометил-13-третбутил эрголина и 4-трифторметилбензоилхлорид вместо бензоихлорида, выход 65%, т.пл. 183-185oC.
Пример 43
6-метил -8β- (2-нафтил)карбониламинометил-13-третбутил эрголин (1, R1= R2=H; R3=CH3; A=NHCOC10H7, n=1).
Соединение получают по методике примера 13, используя β- нафтойную кислоту вместо циклогексанкарбоновой кислоты, выход 45%, т.пл. = 212-215oC.
Пример 44
6-метил -8β- (2-бензтиофен)карбониламинометил)-13-третбутил эрголин (1, R1=R2=H, R3=CH3, A=NHCOC8H6S, n=1).
Соединение получают по методике примера 13, используя 2-бензтиофенкарбоновую кислоту вместо циклогексанкарбоновой кислоты, выход 50%, Т.пл. 215-219oC.
Пример 45
6-метил -8β- гуанидинметил-13-третбутил эрголин (1, R1=R2=H, R3=CH3, A= NHC(NH)-NH2, n=1).
К раствору 3 г 6-метил -8β- аминометил-13-трет-бутил эрголина в 50 мл этанола добавляют порциями 2 г нитрат 3,5-диметил-1-гуанилпиразола. Раствор кипятят с обратным холодильником в течение 3 часов, после охлаждения твердое кристаллическое вещество дважды перекристаллизовывают из кипящего метанола, получая 1,3 г целевого соединения в виде нитрата, Т.пл. 260-270oC.
Пример 46
6-метил -8β- (5-фенил-2-перроил)аминометил-13-трет-бутил эрголин (1, R1= R2=H, R3=CH3, A=COC4H3N(C6H5), n=1).
Соединение получают по методике примера 13, используя 5-фенилпирроил-2-карбоновую кислоту вместо циклогексанкарбоновой кислоты, выход 35%, Т.пл. 142-149oC.
Пример 47
6-метил -8β- амино-13-третбутил эрголин (1, R1=R2=H, R3=CH3, A=NH2, n= 0).
Раствор 25 г 6-метил -8β- метоксикарбонил-13-третбутил эрголина в смеси 200 мл этанола и 50 мл гидразингидроксида 98% кипятят с обратным холодильником в течение 4 ч.
Растворитель удаляют и осадок перекристаллизовывают дважды из этанола, получая 18 г 6-метил -8-β- гидразинкарбонил-13-трет-бутил эрголина, т.пл. 187-192oC.
К перемешиваемому раствору 16 г 6-метил -8β- гидразинкарбонил-13-третбутил эрголина в 100 мл соляной кислоты 1 М добавляют по каплям раствор 3,8 г нитрата натрия в 10 мл воды при 5oC.
Через 15 минут полученный раствор обрабатывают 5 мл соляной кислоты 12 М и быстро нагревают до кипения с обратным холодильником в течение 10 мин.
После охлаждения раствор подщелачивают концентрированным раствором гидроксида аммония и по частям этилацетатом.
После высушивания остаток, растворенный в этаноле, обрабатывают древесным углем. Концентрирование раствора дает 10,4 г 6-метил- -8-β- амино-13-третбутил эрголина, Т. пл. = 145-149oC.
Пример 48
6-метил -8β- гидрокси-13-третбутил эрголин (1, R1=R2=H, R3=CH3, A=OH, n= 0).
К перемешиваемому раствору 10 г 6-метил -8β- амино-13-третбутил эрголина в 50 мл уксусной кислоты добавляют 4,5 г нитрита натрия в 10 мл воды при 5oC.
После перемешивания в течение 1 часа полученный желтый раствор гидрируют при давлении водорода 1 атм, используя в качестве катализатора 10% Pd/c.
После удаления растворителя полученную реакционную смесь разбавляют хлороформом, подщелачивают концентрированным раствором гидроксид аммония и по частям добавляют солевой раствор.
После высушивания и удаления растворителя остаток пропускают через колонку с маленьким слоем силикагеля, элюируя смесью этилацетат /циклогексан 2/1.
Фракции, содержащие продукт, объединяют, концентрируют раствор с получением 7,4 г 6-метил -8β- ацетилокси-13-трет-бутил эрголина, Т.пл. = 165-168oC.
К раствору 5 г 6-метил -8β- ацетилокси-13-третбутил эрголина в 30 мл этанола добавляют 3,5 мл гидроксида натрия 5 м.
По истечении 30 мин осадок фильтруют, промывая этанолом, и сушат, получая 3,9 г целевого соединения, Т.пл. = 187-193oC.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ЭРГОЛИНА | 1993 |
|
RU2118323C1 |
| ФТОРИРОВАННЫЕ ПРОИЗВОДНЫЕ 17β-ЗАМЕЩЕННОГО -4-АЗА-5α-АНДРОСТАН-3-ОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2125061C1 |
| ПРОИЗВОДНЫЕ АЗА-АНТРАЦИКЛИНОНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2159245C2 |
| АНТРАЦИКЛИНОВЫЙ ГЛИКОЗИД, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2118328C1 |
| ПРОИЗВОДНЫЕ АНТРАЦИКЛИНОНА И ИХ ИСПОЛЬЗОВАНИЕ ПРИ АМИЛОИДОЗЕ | 1995 |
|
RU2167661C2 |
| БИОЛОГИЧЕСКИ АКТИВНЫЕ ПОЛИМЕРСВЯЗАННЫЕ АНТРАЦИКЛИНЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2145965C1 |
| ПРОИЗВОДНЫЕ 3'-АЗИРИДИНО-АНТРАЦИКЛИНА, СПОСОБЫ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2149163C1 |
| ПРОИЗВОДНЫЕ АНТРАЦИКЛИНА | 1995 |
|
RU2159619C2 |
| ПОЛИМЕРНЫЙ КОНЪЮГАТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2149646C1 |
| СВЯЗУЮЩИЙ АГЕНТ ДЛЯ БИОАКТИВНЫХ ПРЕПАРАТОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2116087C1 |
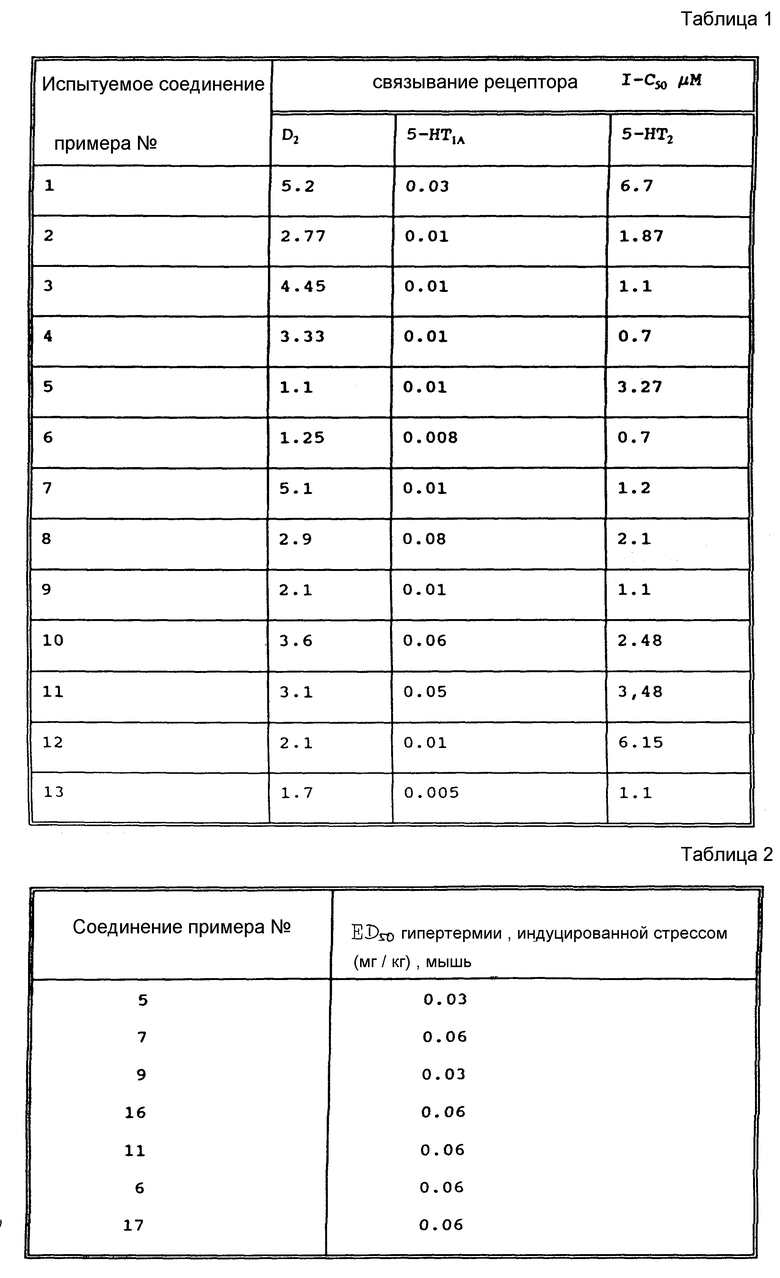
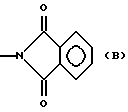
Производные эрголина общей формулы 1, где R1-C1-4алкил; A-OH. NH2, 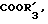 CONHR4, NHCOOR4, NHC(X)NHR4, NHC(X)NHCOR4, CONHR4 или остаток формулы (а-в); R2 - водород, бром, -S-C1-4алкил;
CONHR4, NHCOOR4, NHC(X)NHR4, NHC(X)NHCOR4, CONHR4 или остаток формулы (а-в); R2 - водород, бром, -S-C1-4алкил; 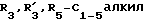 или водород, n= 0,1 или 2, m=1 или 2; R4 - водород, C1-7алкил; C3-7циклоалкил, адамантил, C1-5алкилфенил, C2алкенил-фенил, фенил, замещенный CF3 или одной или двумя алкоксигруппами, нафтил, бензотиофен, алкенилфурил, 5-6 членное гетероциклическое кольцо, с 1-2 гетероатомами, выбираемыми из N, S, возможно замещенное алкилом, фенилом или галогеном, X-O или S, или их фармацевтически приемлемые соли могут быть использованы для регуляции состояний, которые можно отнести за счет серотонинэргических дисфункций. 3 с. и 2 з.п.ф-лы, 2 табл.
или водород, n= 0,1 или 2, m=1 или 2; R4 - водород, C1-7алкил; C3-7циклоалкил, адамантил, C1-5алкилфенил, C2алкенил-фенил, фенил, замещенный CF3 или одной или двумя алкоксигруппами, нафтил, бензотиофен, алкенилфурил, 5-6 членное гетероциклическое кольцо, с 1-2 гетероатомами, выбираемыми из N, S, возможно замещенное алкилом, фенилом или галогеном, X-O или S, или их фармацевтически приемлемые соли могут быть использованы для регуляции состояний, которые можно отнести за счет серотонинэргических дисфункций. 3 с. и 2 з.п.ф-лы, 2 табл.
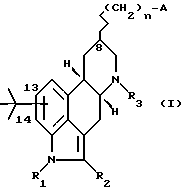

где R1 представляет собой C1-C4 алкил;
A-OH, NH2, COOR'3, CONHR4, NHCOR4, NHCOOR4, NHC(X)NHR4, NHC(X)NHCOR4, CONHR4,

или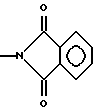
R2 - водород, бром, -S-C1-C4-алкил;
R3 и R'3 независимо C1-C5 алкил или водород;
n = 0,1 или 2;
m = 1 или 2;
R4 представляет собой водород, C1-C7 алкил, C3-C7-циклоалкил, адамантил, C1-C5 алкилфенил, C2-алкенил-фенил, фенил, замещенный трифторметилом или одной или двумя C1-C4 алкоксигруппами, нафтил, бензотиофен, C2-алкенилфурил; 5-6 членное гетероциклическое кольцо, включающее 1 - 2 гетероатома, выбираемых из азота, серы, которое может быть замещено C1-C4 алкилом, фенилом или атомом галогена;
R5 - водород, C1-C4 алкил;
X-O, NH или S,
или их фармацевтически приемлемые соли.
R3-метил; n = 0,1 или 2; A - OH, NH2, COOR'3 NHCOOR4, NHCONHR4, CONHR4, OCONHR4, NHCSNHCOR4,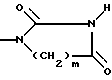
и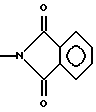
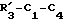 алкил или водород;
алкил или водород;
m = 1 или 2;
R4 - фенил, бензил, пиридил, 5-бромпиридил, этил, циклогексил, адамантил, фенилвинил, 1,5-диметил-3-пиразил, 2-метил-4-тиазолил, пиразинил, пиримидинил, тиазолил или 6-хлор-3-пиридазинил.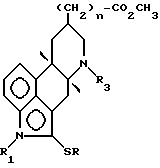
в которой n, R1 и R3 имеют значения, установленные в п.1 или 2, и R представляет собой C1-4 алкил или фенил,
с трет. бутилирующим агентом в присутствии трифторуксусной кислоты, отделение полученного в результате 13-изомера от 14-изомера и необязательное восстановление 2-тиопроизводного в присутствии никеля Ренея; (i) либо гидролиз образующегося в результате 8-карбоксилата и конденсирования образовавшейся 8-карбоновой кислоты, необязательно после активации, с амином формулы R4-NH2, где R4 имеет значения, установленные в п.1 или 2 с получением соединения, где A представляет CONНR4; (ii) либо восстановление образующегося в результате 8-карбоксилата и взаимодействие образующегося в результате 8-гидроксиметильного производного с соединением формулы R4 -N = C = O или с п-нитрофенилхлоркарбонатом и затем - с соединением формулы R4 -NH2, где R4 имеет установленные выше значения с получением соединения, где A представляет OH или OCONHR4; (iii) либо восстановление образующегося в результате 8-карбоксилата и взаимодействие образующего в результате 8-гидроксиметильного производного с трифенилфосфином, диэтилазодикарбоксилатом и фталимидом и затем - гидролиз полученного соединения с получением соединения, где A - представляет NH2; (b) если необходимо, взаимодействие соединения, полученного на стадии а (iii) формулы III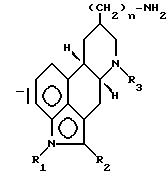
в которой n, R1 и R3 имеют установленные выше значения и R2 имеет значения, установленные в п.1 или 2; (i) с соединением формулы (IY) R4 - COOH, где R4 имеет установленные выше значения, или с его реакционноспособным функциональным производным или (ii) с соединением формулы (Y) R4 - N = C = X или (ii') с п-нитрофенилхлоркарбонатом и взаимодействие полученного соединения, в любом случае, с соединением формулы R4 - NH2, или R4 и X имеют установленные выше значения; (с) если желательно, (i) галогенирование соединения, где R2 = H с получением соединения, где R2-галоген; (ii) обработка соединения, где X = S нитратом серебра с получением соединения, где X = 0; (iii) метилирование соединения, где R1 = H с получением соединения, где X = CH3; (iv) обработка соединения, где A = COOR'3, и n = 0 гидразингидроксидом, нитритом натрия и соляной кислотой с получением соединения где A - NH2 и n = 0; (v) обработка соединения, где A - NH2 и n = 0 нитритом натрия и последующие гидрирование и гидролиз с получением соединения, где A - OH и n = 0; (d) извлечение полученного соединения формулы I как такового или виде его кислотно-аддитивной соли.
Приоритет по пунктам:
24.12.92 - по пп.1 - 3;
19.03.93 - по пп.4 и 5
| Способ металлотермического восстановления магния | 1946 |
|
SU70562A1 |
| СПОСОБ НАЗЕМНОЙ ПРОВЕРКИ РАБОТОСПОСОБНОСТИ ГИДРОСИСТЕМЫ ВОЗДУШНОГО ВИНТА ЛЕТАТЕЛЬНОГОАППАРАТА | 0 |
|
SU197241A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИФТОРАРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 0 |
|
SU296748A1 |
| SU 1178324 C1, 1998 | |||
| US 4500712 A, 1985. | |||

