Предметом настоящего изобретения являются производные амидиносульфоксида и сульфона, способы их получения, содержащие их фармацевтические составы и их применение в терапии, в частности их применение в качестве селективных ингибиторов индуцибельной синтазы оксида азота.
С начала 1980-ых годов было известно, что сосудистая релаксация, осуществляемая с помощью ацетилхолина, зависит от наличия эндотелия и данная активность приписывалась лабильному гуморальному фактору, именуемому релаксирующим фактором эндотелиального происхождения (РФЭП). Уже в течение более 100 лет известна активность глицерилтринитрата как сосудорасширяющего средства, а в настоящее время установлено, что NO является активным компонентом амилнитрита, глицерилнитрита и других нитровазодилататоров. Современная идентификация РФЭП как NO совпала с открытием биохимического пути, посредством которого происходит синтез NO из аминокислоты L-аргинина ферментом NO синтазой.
NO является эндогенным стимулятором фермента растворимой гуанилатциклазы, он задействован в серии биологических процессов, дополняющих опосредованную эндотелием релаксацию, включая цитотоксичность фагоцитарных клеток и взаимодействие клетка-клетка в центральной нервной системе [1, 2]. В настоящее время считают, что избыточное продуцирование NO может быть задействовано в ряде состояний, включающих состояния, к которым относятся и системная гипотензия, такая как септический шок, и терапия посредством некоторых цитокинов, а также многочисленные воспалительные заболевания, такие как артрит.
Синтез NO из L-аргинина может быть ингибирован аналогом L-аргинина, NG-монометил-L-аргинином (L-NMMA), и было предложено терапевтическое применение L-NMMA для лечения септического (токсического) шока и других типов системной гипотензии [3, 4]. Было предложено также терапевтическое применение некоторых других ингибиторов NO синтазы, отличных от L-NMMA, для тех же целей [3, 5].
Недавно стало очевидно, что имеются по меньшей мере три изофермента NO синтазы [6], а именно:
(I) конститутивный, Ca++ /калмодулинзависимый фермент (eNOS), который присутствует в клетках сосудистого эндотелия, который высвобождает NO в ответ на рецепторную или физическую стимуляцию,
(II) конститутивный, Ca++ /калмодулинзависимый фермент (nNOS), локализованный в мозге и отдельных периферических нервных системах, который высвобождает NO в ответ на рецепторную или физическую стимуляцию,
(III) Ca++ независимый фермент (iNOS), который индуцируется после активации сосудистого гладкого мускула, макрофагов, клеток эндотелия и ряда других клеток эндотоксином и цитокинами. Однократно экспрессированная, эта индуцибельная NO синтаза синтезирует NO в течение длительных промежутков времени.
NO, выделяемая ферментами eNOS и nNOS, действует в качестве механизма тансдукции, лежащего в основе некоторых физиологических ответов. NO, продуцируемая ферментом iNOS, действует как цитотоксическая молекула на проникающие микроорганизмы. Оказывается также, что побочные эффекты избыточного продуцирования NO, в частности патологическое расширение кровеносных сосудов, сосудистое истечение и поражение тканей, могут в значительной степени являться результатом действия NO, синтезируемого ферментом iNOS.
Ингибиторы NO синтазы, предложенные для терапевтического применения, такие как L-NMMA и нитроаргинин, являются неселективными и поэтому они ингибируют все изоферменты NO синтазы. В случае применения такого неселективного ингибитора NO синтазы необходимо большое внимание уделять тому, чтобы избежать возможных серьезных последствий сверхингибирования eNOS, включая гипертензию и возможный тромбоз и поражение тканей. В частности, в случае терапевтического применения L-NMMA для лечения септического и/или токсического шока рекомендовано, чтобы у пациента постоянно в ходе лечения контролировали кровяное давление. Таким образом, пока неселективные ингибиторы NO синтазы имеют терапевтическое применение, предполагается следование соответствующим мерам предосторожности, в то время как ингибиторы NO синтазы, которые являются селективными в том смысле, что они ингибируют iNOS в значительно большей степени, чем eNOS, могут обеспечивать даже большие терапевтические преимущества и являться намного более простыми в плане применения.
Описана [7] группа амидинопроизводных формулы (0)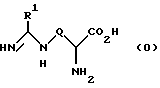
и их соли и фармацевтически приемлемые эфиры и амиды, в которых:
R1 является C1-6 алкильной группой с нормальной или разветвленной цепью, C2-6 алкенильной группой, C2-6 алкинильной группой, C3-6 циклоалкильной группой или C3-6 циклоалкил C1-6 алкильной группой;
Q является алкиленовой, алкениленовой или алкиниленовой группой, имеющей от 3 до 6 атомов углерода, которая в принципе может быть замещена одной или более C1-3 алкинильными группами; либо Q является группой формулы -(CH2)pX(CH2)q-, в которой p равно 2 или 3, q равно 1, а X является S(O)x, где x равно 0, 1 или 2, О или NR2, где R2 является H или C1-6 алкилом; либо Q является группой формулы -(CH2)rA(CH2)s-, в которой r равно 0, 1 или 2, s равно 0, 1 или 2, а A является карбоциклическим или гетероциклическим кольцом с числом членов от 3 до 6, которое может быть в принципе замещено одним или более приемлемых заместителей, таких как C1-6 алкил, C1-6 алкоксил, гидроксил, галоген, нитрогруппа, цианогруппа, трифтор C1-6 алкил, аминогруппа, C1-6 алкиламиногруппа или диC1-6 алкиламиногруппа, которые обладают ингибирующей активностью в отношении фермента NO синтазы.
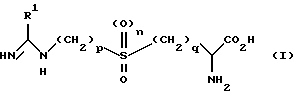
Авторы настоящего изобретения выявили частную группу соединений, которые являются селективными ингибиторами iNOS, обладающими слабым действием на eNOS или не обладающие им вообще. Согласно настоящему изобретению предложены соединения формулы (I)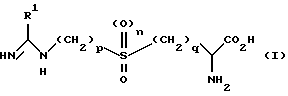
в которой R1 является C1-6 алкильной группой с нормальной или разветвленной цепью, C2-6 алкенильной группой, C2-6 алкинильной группой, C3-6 циклоалкильной группой или C3-6 циклоалкил C1-6 алкильной группой, каждая из которых возможно замещена одной-тремя группами, выбранными независимо из: галогруппы; -CN, NO2; группы -COR2, в которой R2 является водородом, C1-6 алкилом, -OR3, где R3 является водородом или C1-6 алкилом, либо NR4R5, где R4 и R5 независимо выбраны из водорода или C1-6 алкила; группы -S(O)mR6, в которой m равно 0, 1 или 2, R6 является водородом, C1-6 алкилом, гидроксилом или NR7 R8, где R7 и R8 независимо являются водородом или C1-6 алкилом; группы PO(OR9)2, в которой R9 является водородом или C1-6 алкилом; группы NR10R11, в которой R10 и R11 независимо выбраны из водорода, C1-6 алкила, -COR12,
где R12 является водородом или C1-6 алкилом, либо -S(O)m'R13, где m' равно 0, 1 или 2, а R13 является водородом или C1-6 алкилом; или группы -OR14, в которой R14 является водородом, C1-6 алкилом, возможно замещенным одним-тремя атомами галогена, C6-10 арилом или -COR15, где R15 является водородом или C1-6 алкилом;
p равно 2 или 3, q равно 1 или 2, а n равно 0 или 1, и все их соли, эфиры, амиды и физиологически приемлемые пролекарства.
В подходящем случае R1 является C1-6 алкилом, C2-6 алкенилом или алкинилом либо C3-6 циклоалкилом, причем каждая из групп является возможно замещенной одной-тремя группами, независимо выбранными из -CN; -NO2; группы -COR2a, в которой R2a является водородом, C1-4 алкилом, гидроксилом или аминогруппой; группы -S(O)mR6a, в которой m соответствует приведенному выше описанию, а R6a является водородом, C1-4 алкилом, гидроксилом или аминогруппой; группы -PO(OR9a)2, в которой R9a является водородом или C1-4 алкилом; группы NR10aR11a, в которой R10a и R11a являются независимо выбранными из водорода, C1-4 алкила, -COR12a или S(O)m'R13a, где m' соответствует приведенному выше описанию, а R12a и R13a независимо являются водородом или C1-4 алкилом; группы OR14a, в которой R14a является водородом, C1-4 алкилом, возможно замещенным одним-тремя атомами галогена, фенилом, бензилом или -COR15a, где R15a является водородом или C1-4 алкилом.
Предпочтительно R1 является C1-4 алкильной группой или C2-4 алкенильной или алкинильной группой, возможно замещенной одной-тремя группами, независимо выбранными из -CN; группы -COR2, в которой R2 соответствует приведенному выше описанию, группы -S(O)mR6, в которой m и R6 имеют значения, определенные выше; группы NR10R11, в которой R10 и R11 имеют значения, определенные выше; галогена; или группы -OR14, в которой R14 имеет значение, определенное выше.
Наиболее предпочтительно, если R1 является метильной или этильной группой, возможно замещенной одним-тремя заместителями, независимо выбранными из галогена, группы -OR14, в которой R14 имеет значение, приведенное выше, или S(O)mR6, где m и R6 определены выше.
Предпочтительной группой соединений являются соединения формулы (IА)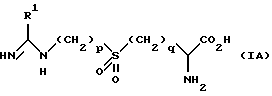
в которой R1, R2, p и q имеют значения, определенные выше.
Предпочтительно R1 является незамещенным или замещенным только одной группой.
Предпочтительные соединения включают:
2-Амино-6-(1-иминоэтиламино)-4,4-диоксо-4-тиагексановую кислоту,
2-Амино-6-(1-иминоэтиламино)-4-оксо-4-тиагексановую кислоту,
2-Амино-7-(1-иминоэтиламино)-5-оксо-5-тиагептановую кислоту,
2-Амино-7-(1-иминоэтиламино)-5,5-диоксо-5-тиагептановую кислоту,
2-Амино-6-(1-имино-2-фторэтиламино)-4,4-диоксо-4-тиагексановую кислоту,
2-Амино-6-(1-имино-2-метоксиэтиламино)-4,4-диоксо-4-тиагексановую кислоту,
2-Амино-6-(2-ацетокси-1-иминоэтиламино)-4,4-диоксо-4-тиагексановую кислоту,
2-Амино-6-(2-бензилокси-1-иминоэтиламино)-4,4-диоксо-4- тиагексановую кислоту,
2-Амино-6-(2-метилтио-1-иминоэтиламино)-4,4-диоксо-4- тиагексановую кислоту,
2-Амино-6-(2-гидрокси-1-иминоэтиламино)-4,4-диоксо-4- тиагексановую кислоту
и все их соли, эфиры, амиды и физиологически приемлемые пролекарства.
Наиболее предпочтительными являются 2-Амино-6-(1- иминоэтиламино)-4,4-диоксо-4-тиагексановая кислота и 2-Амино-6- (1-имино-2-фторэтиламино)-4,4-диоксо-4-тиагексановая кислота. Предпочтительно, чтобы соединения находились в R конфигурации.
Соединения формулы (I) могут иметь в молекуле несколько асимметричных центров в зависимости от точного значения различных групп и предполагается, что формула (I) включает все возможные изомеры. Все соединения формулы (I) содержат асимметричный центр в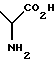
группе, и хотя предпочтительна естественная L или (S) хиральность аргинина, опять же предполагается, что формула должна включать все возможные изомеры, как индивидуальные, так и смешанные в любых соотношениях.
В одном из способов реализации настоящего изобретения предложено соединение формулы (IБ)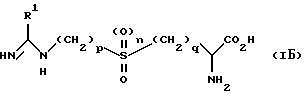
и его соли и фармацевтически приемлемые эфиры и амиды, в которых R1 является C1-6 алкильной группой с нормальной или разветвленной цепью, C2-6 алкенильной группой, C2-6 алкинильной группой, C3-6 циклоалкильной группой или C3-6 циклоалкил C1-6 алкильной группой; p равно 2 или 3, q равно 1 или 2, а n равно 0 или 1.
В другом способе реализации настоящего изобретения предложено соединение формулы (IВ)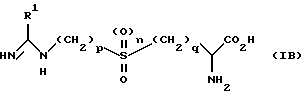
в которой n равно 0 или 1; p равно 2 или 3; q равно 1 или 2; R1 является C1-6 алкильной группой с нормальной или разветвленной цепью, C2-6 алкенильной группой, C2-6 алкинильной группой, C3-6 циклоалкильной группой или C3-6 циклоалкил C1-6 алкильной группой, каждая из которых замещена одной-тремя группами, независимо выбранными из: -CN; -NO2; группы -COR2, в которой R2 является водородом, C1-6 алкилом, -OR3, где R3 является водородом или C1-6 алкилом, или NR4R5, где R4 и R5 независимо выбраны из водорода или C1-6 алкила; группы -S(O)mR6, в которой m равно 0, 1 или 2, R6 является водородом, C1-6 алкилом, гидроксилом или NR7R8, где R7 и R8 являются независимо водородом или C1-6 алкилом; группы PO(OR9)2, где R9 является водородом или C1-6 алкилом; группы NR10R11, в которой R10 и R11 являются независимо выбранными из водорода, C1-6 алкила, -COR12, где R12 является водородом или C1-6 алкилом, либо -S(O)m'R13, где m' равно 0, 1 или 2, a R13 является водородом или C1-6 алкилом; галогеном; либо группой -OR14, в которой R14 является водородом, C1-6 алкилом, возможно замещенным одним-тремя атомами галогена, C6-10 арилом или -COR15, где R15 является водородом или C1-6 алкилом; и все его соли, эфиры, амиды и физиологически приемлемые пролекарства.
Термин "галоген" обозначает радикал фтора, хлора, брома или иода, предпочтительно фтора.
Приемлемые соли включают соли, образованные как с органическими, так и с неорганическими кислотами. Такие соли, полученные в результате добавления кислот, обычно являются фармацевтически приемлемыми, хотя соли, относящиеся к фармацевтически неприемлемым солям, могут находить применение при получении и очистке искомого соединения. Таким образом, предпочтительные соли включают соли, образованные из соляной, бромоводородной, серной, лимонной, винной, фосфорной, молочной, пировиноградной, уксусной, трифторуксусной, янтарной, щавелевой, фумаровой, малеиновой, щавелевоуксусной, метансульфоновой, этансульфоновой, n-толуолсульфоновой, бензолсульфоновой и изотионовой кислот. Соли соединений формулы (I) могут быть получены в результате реакции взаимодействия соответствующего соединения в форме свободного основания с соответствующей кислотой.
Фармацевтически приемлемые эфиры и амиды соединений формулы (I) могут иметь кислотную группу, замененную группой -CO2R3, в которой R3 является, например, C1-6 алкилом, арилом или арил C1-3 алкилом, либо -COR4, где R4 является остатком приемлемой натуральной или синтетической аминокислоты.
Термин "физиологически приемлемое пролекарство" обозначает производные соединений формулы (I), которые имеют ту же самую физиологическую функцию, что и свободное соединение формулы (I), например, в силу того, что они превращаются в него в организме. Подобные пролекарства могут обладать или не обладать активностью в силу собственной природы.
В следующем аспекте настоящего изобретения предложены также соединения формулы (I), описанные выше, и все их соли, эфиры, амиды и физиологически приемлемые пролекарства для применения в медицине, в частности для лечения состояний, при которых благоприятно ингибирование продуцирования NO из L-аргинина под действием NO синтазы, и более специфично, состояний, при которых благоприятно ингибирование продуцирования NO в результате действия iNOS изофермента в большей степени, чем продуцирования NO под действием eNOS.
Согласно следующему аспекту в настоящем изобретении предложено применение соединения формулы (I) и всех его солей, эфиров, амидов и физиологически приемлемых пролекарств для изготовления лекарственного препарата для лечения состояния, при котором благоприятно ингибирование NO продуцирования из аргинина под действием NO синтазы и, более конкретно, под действием iNOS.
В следующем аспекте предложено применение соединения формулы (I) или его соли, эфира, амида либо физиологически приемлемого пролекарства для изготовления лекарственного препарата для лечения шоковых состояний, проистекающих из перепроизводства NO под действием iNOS, таких как септический шок или шок, вызванный внезапной печеночной недостаточностью, либо терапией цитокинами, такими как ТНФ, ИЛ-1 и ИЛ-2, или терапией цитокин-индуцирующими агентами, например 5,6-диметилксантенонуксусной кислотой.
В еще одном аспекте настоящего изобретения предложено применение соединения формулы (I) или его соли, эфира, амида или физиологически приемлемого пролекарства для изготовления лекарственного препарата для лечения воспалительного состояния, например артрита.
С дугой стороны, предложен способ лечения состояния, при котором благоприятно ингибирование NO продуцирования из L-аргинина под действием NO синтазы, и в частности под действием iNOS, включающий в себя воздействие на млекопитающего, нуждающегося в таком лечении, фармацевтически эффективным количеством соединения формулы (I), описанного выше, или его соли, эфира, амида или физиологически приемлемого пролекарства.
Другие состояния, при которых благоприятно ингибирование iNOS, включают широкий круг ауто-иммунных и/или воспалительных заболеваний, таких как болезни суставов (например, ревматоидный артрит, остеоартрит), заболевания желудочно-кишечного тракта (например, язвенный колит и другие воспалительные заболевания желудочно-кишечного тракта, гастрит и воспаление слизистой оболочки, являющееся результатом инфекции, поражение кишечника, вызванное нестероидными потивовоспалительными лекарствами), легких (респираторный дистресс-синдром взрослых, астма), сердца (миокардит), нервной ткани (например, рассеянный склероз), поджелудочной железы (например, сахарный диабет), почек (например, гломерулонефрит), кожи (например, дерматит, псориаз, крапивница), а также трансплантированных органов (отторжение) и множественные заболевания органов (например, системная волчанка). Кроме того, очевидным является перепроизводство NO ферментом iNOS при атеросклерозе.
Далее, в еще одном аспекте настоящего изобретения предложено применение соединения формулы (I) или его соли, эфира, амида или физиологически приемлемого пролекарства для изготовления лекарственного препарата, предназначенного для лечения указанных выше состояний.
Ингибирование nNOS и/или iNOS благоприятно при лечении заболеваний нервной системы, обусловленных сверхпродуцированием NO этим изоферментом, особенно при лечении церебральной ишемии. Другие заболевания включают повреждение ЦНС, эпилепсию, слабоумие, связанное со СПИДом, хроническое нейродегенеративное заболевание и хроническую боль, а также состояния, в которые может быть вовлечен неадренергический нехолинергический нерв, например приапизм, ожирение и гиперфагия. Согласно настоящему изобретению предложено также применение соединения формулы (I) или его соли, эфира, амида или физиологически приемлемого пролекарства для изготовления лекарственного средства, предназначенного для применения при лечении указанных выше состояний.
Кроме того, ингибирование NO синтазы может быть благоприятно для профилактики потери лимфоцитов, ассоциированной с ВИЧ инфекцией, для повышения радиочувствительности опухолей в ходе радиотерапии и для уменьшения роста опухолей и метастаз.
Ингибирование как iNOS, так и nNOS может быть благоприятным при лечении некоторых состояний, в которых играют определенную роль оба фермента, например таких состояний ЦНС, как церебральная ишемия.
Подразумевается, что используемая в тексте ссылка на "лечение" пациента включает профилактику; подразумевают, что термин "млекопитающее" включает человека или животное.
Активность соединений формулы (I) и всех их солей, эфиров, амидов и физиологически приемлемых пролекарств как ингибиторов NO синтазы может быть определена с помощью выделенных ферментов человека или грызунов, крысиного кольца аорты или in vivo на мышах согласно нижеописанным способам.
Хотя для соединений формулы (I) и всех их солей, эфиров, амидов и физиологически приемлемых пролекарств может быть допустимым введение в виде неочищенных химических веществ, предпочтительно применять их в виде фармацевтических препаратов. Согласно следующему аспекту в настоящем изобретении предложен фармацевтический препарат, содержащий соединение формулы (I) и все его соли, эфиры, амиды и физиологически приемлемые пролекарства совместно с одним или несколькими фармацевтически приемлемыми носителями и возможно одним или несколькими другими терапевтическими ингредиентами. Носитель(и) должны быть "приемлемыми" в том смысле, что они являются совместимыми с другими ингредиентами препарата и не являются опасными для самого реципиента.
Препараты включают препараты, пригодные для перорального, парентерального (включая подкожное, внутрикожное, внутримышечное, внутривенное и внутриартериальное), ректального и местного (включая кожное, трансбуккальное, подъязычное и внутриглазное) введения, хотя наиболее приемлемый путь может зависеть, например, от состояния и заболевания пациента.
Препараты могут быть изготовлены, как правило, в виде унифицированных доз и могут быть изготовлены любым из способов, известных из уровня состояния фармакопеи. Все способы включают стадию смешивания соединения формулы (I) и всех его солей, эфиров, амидов и физиологически приемлемых пролекарств ("активный ингредиент") с носителем, который включает один или несколько вспомогательных ингредиентов. В целом препараты готовят путем гомогенного и тщательного смешивания активного ингредиента с жидкими носителями или тонкоизмельченными твердыми носителями или с обоими сразу, а затем, если это необходимо, формования продукта в желаемый препарат.
Препараты по настоящему изобретению, приемлемые для перорального введения, могут быть изготовлены в виде отдельных единиц, таких как капсулы, лепешки или таблетки, каждая из которых содержит определенное количество активного ингредиента; в виде порошка или гранул; в виде раствора или суспензии в водной жидкости или неводной жидкости; либо в виде жидкой эмульсии масло в воде или жидкой эмульсии вода в масле. Активный ингредиент также может быть представлен в виде шарика, лекарственной кашки или пасты.
Таблетка может быть изготовлена путем прессования или формования возможно с одним или несколькими вспомогательными ингредиентами. Спрессованные таблетки могут быть изготовлены путем прессования в соответствующем механизме активного ингредиента в свободно-текучей форме, такой как порошок или гранулы, возможно смешанного со связующим веществом, лубрикантом, инертным разбавителем, поверхностно-активным или диспергирующим агентом. Формованные таблетки могут быть изготовлены путем формования в соответствующем механизме смеси порошкообразного соединения, смоченного инертным жидким разбавителем. Таблетки в случае необходимости могут быть покрыты оболочкой или на них могут быть нанесены бороздки, они могут быть изготовлены таким образом, чтобы обеспечивать медленное и контроллируемое выделение из них активного ингредиента.
Препараты для парентерального введения включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатики и растворы, которые делают препарат изотоническим с кровью предположительного реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Препараты могут быть изготовлены в виде унифицированных разовых доз или много-дозовых контейнеров, например герметичных ампул или флаконов, и могут храниться в высушенном замораживанием (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например солевого раствора или воды для инъекций, непосредственно перед применением. Приготовленные для немедленного приема растворы для инъекций и суспензии могут быть приготовлены из стерильных порошков, гранул и таблеток вышеописанного типа.
Препараты для ректального введения могут быть изготовлены в виде суппозитория с обычными носителями, такими как масло какао или полиэтиленгликоль.
Препараты для локального введения в рот, например трансбуккально или под язык, включают таблетки, содержащие активный ингредиент в ароматизированной основе, такой как сахароза и гуммиарабик или трагакант, и пастилки, содержащие активный ингредиент в такой основе, как желатин и глицерин или сахароза и гуммиарабик.
Предпочтительными препаративными формами в виде унифицированных доз являются формы, содержащие эффективную дозу активного ингредиента, как изложено ниже, или ее соответствующую долю.
Следует иметь в виду, что в дополнение к перечисленным конкретно выше ингредиентам препараты по данному изобретению могут включать другие агенты, известные из уровня техники, соответствующие типу искомого препарата, например препараты, пригодные для перорального введения, могут включать ароматизирующие агенты.
Соединения по изобретению могут быть введены перорально или путем инъекции в дозе от 0,1 до 1500 мг/кг в день, предпочтительно от 0,1 до 500 мг/кг в день. Интервал доз для взрослых людей лежит в целом от 5 мг до 35 г/день и предпочтительно от 5 мг до 2 г/день. Таблетки или другие формы, предназначенные для введения в виде отдельных единиц, как правило, могут содержать количество соединения по изобретению, эффективное в такой дозировке, или применяться в виде многократных доз, например единицы, содержащие от 5 мг до 500 мг, обычно приблизительно от 10 мг до 200 мг.
Соединения по настоящему изобретению могут быть введены в сочетании с одним или более других активных ингредиентов. Подобные дополнительные активные ингредиенты, пригодные для совместного введения, очевидны для врача и могут, например, представлять собой противовоспалительное средство, такое как кортикостероид, например метилпреднизолон. Соединения формулы (I) являются конкурентными ингибиторами по отношению к L-аргинину и поэтому может быть нецелесообразным одновременно лечить пациентов препаратами (например, общее парентеральное питание), которые имеют высокое содержание L-аргинина.
Соединения формулы (I) и все их соли, эфиры, амиды и физиологически приемлемые пролекарства предпочтительно вводить перорально или путем инъекции (внутривенно или подкожно). Точное количество соединения, вводимого пациенту, оставляется на усмотрение лечащего врача. Однако применяемая доза будет зависеть от ряда факторов, включая возраст и пол пациента, конкретное нарушение, подлежащее лечению, и его тяжесть. Также способ введения может изменяться в зависимости от состояния и его тяжести.
Соединения формулы (I) являются новыми, и согласно следующему аспекту настоящего изобретения предложен способ их получения.
Соединения формулы (I) могут быть получены:
а) путем окисления соединения формулы (II):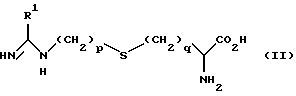
Окисление может быть осуществлено способами, известными из уровня техники, например посредством обработки обогащенным кислородом соединением, таким как 30%-ная перекись водорода и 0,5 M молибдат аммония в перхлорной кислоте.
Соединения формулы (II) могут быть получены в результате реакции взаимодействия аминокислоты формулы (III)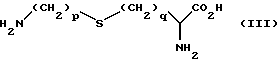
или ее защищенного производного с соединением формулы (IV)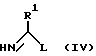
в которой L является уходящей группой, a R1, p и q имеют значения, определенные выше, с последующим удалением любых присутствующих защитных групп, с образованием соединения формулы (II).
Приемлемые уходящие группы L включают -OR5 и -SR5, где R5 является низшей алкильной группой, например C1-4 алкилом, предпочтительно метилом или этилом.
Соединения формулы (III) в общем следует применять в такой форме, в которой функциональная группа аминокислоты защищена приемлемыми защитными группами, в этом отношении можно сослаться на [8]. Защитные группы могут быть затем удалены соответствующим способом в качестве заключительной стадии процесса, в результате чего получают соединение формулы (II). Например, аминокислотная функциональная группа может быть защищена в виде соли меди с последующим снятием защиты, происходящим на ионообменной колонке, предназначенной для удаления неорганических ионов из реакционной смеси. Соединения формулы (IV) могут применяться в форме свободного основания либо в виде соли, полученной при добавлении кислоты, например соли соляной или иодоводородной кислоты. Реакцию обычно проводят в соответствующем растворителе в присутствии основания, например гидроксида щелочного металла, предпочтительно при pH примерно от 9 до 11 и, как правило, при температуре от 0oC до температуры дефлегмации растворителя, предпочтительно от 0oC до 50oC. Предпочтительным растворителем является вода, хотя реакция также может быть проведена в таком полярном растворителе, как низший спирт, например метанол или этанол, либо амид, например диметилформамид, чистом или в смеси с водой, что может являться преимуществом в определенных условиях.
Соединения формулы (III) являются, в целом, известными соединениями, которые могут быть известными способами превращены в соответствующие защищенные производные. Имидаты и тиоимидаты формулы (IV) (L является -OR5 и -SR5 соответственно) также являются в общем известными соединениями, и реакция взаимодействия таких соединений с первичным амином известна [9].
(б) путем снятия защиты с соединения формулы (V)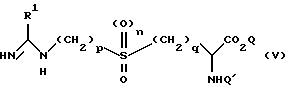
в которой R1, n, p и q определены выше, Q является водородом или карбоксильной защитной группой, а Q' является защитной группой, возможно с последующим превращением в другое соединение формулы (I). Примеры приемлемых защитных групп включают третбутоксикарбонил, бензилоксикарбонил, алкил, трет-бутил, бензил и т.д. Другие приемлемые группы, которые могут быть применены, будут очевидны для специалиста. Если защитная группа является кислотно лабильной, например в том случае, когда Q является алкилом, реакция может быть осуществлена посредством кислотного гидролиза, например в результате реакции с хлороводородом в диоксане, HBr/уксусная кислота в ледяной уксусной кислоте или трифторуксусной кислоте в дихлорметане, при неэкстремальной температуре от -5oC до 100oC, предпочтительно при комнатной температуре. Когда защитная группа отщепляется посредством гидрогенолиза, например бензилоксикарбонил, снятие защиты может быть осуществлено с помощью водорода над катализатором, например палладий/уголь.
Соединения формулы (V) могут быть получены в результате реакции взаимодействия соединения формулы (VI)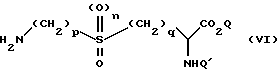
в которой n, p, q, Q и Q' определены выше, с соединением формулы (VII)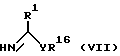
в которой R1 имеет значения, определенные ранее, Y является О или S, а R16 является C1-6 алкилом, фенил C1-6 алкилом или нафтил C1-6 алкилом, например бензилом. Реакция может быть проведена в полярном растворителе, например C1-6 спирте, таком как метанол или этанол, при неэкстремальной температуре от -50oC до 150oC, например от -5oC до 50oC, в частности при комнатной температуре.
Промежуточные соединения формулы (VI) и их производные, у которых свободная аминогруппа является защищенной, являются новыми и составляют еще один аспект настоящего изобретения. Наиболее предпочтительным промежуточным соединением является т-бутил-6- бензилоксикарбониламино-2-т-бутоксикарбониламино-4,4-диоксо-4- тиагексаноат.
Определенные соединения формулы (VII), в которой Y является S, являются новыми и могут быть получены, как описано ниже.
Соединения формулы (VI) могут быть получены путем окисления соединения формулы (VIII)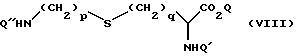
в которой p, q, Q и Q' имеют значения, определенные выше, а Q'' является подходящей защитной группой, например бензилоксикарбонилом, с последующим удалением защитной группы Q''. Реакция окисления может быть осуществлена стандартными способами, известными из уровня техники, например согласно способу [10] , или посредством реакции взаимодействия с м-надхлорбензойной кислотой с образованием сульфоксидного продукта, с последующей реакцией с "Oxone", если необходим сульфоновый продукт. Реакцию удобно проводить в полярном растворителе, например воде или низшем спирте, таком как этанол, или их смеси.
Удаление защитной группы может быть осуществлено стандартными способами, известными специалистам, например с катализатором переноса состояний гидрогенизации, применяя, например, муравьиную кислоту в спирте, таком как метанол, в присутствии подходящего катализатора, такого как 10%-ный палладий на угле.
Соединения формулы (VIII) могут быть получены в результате реакции взаимодействия
(i) соединения формулы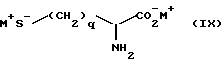
в которой q определено ранее, а M+ является подходящим катионом, например Na+, с соединением формулы (X)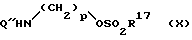
в которой p и Q'' определены ранее, a R17 является C1-6 алкильной группой или арильной группой, с последующей защитой карбоксильной группы и аминогруппы защитными группами Q и Q' либо наоборот.
Соединения формулы (IX) не выделяются обычным способом, поэтому они могут быть получены путем восстановительного отщепления соединения формулы (XI):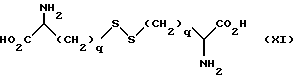
в которой q определено ранее.
Отщепление может быть осуществлено способами, известными из уровня техники, например посредством применения натрия в жидком аммонии при температуре от -78oC до 0oC, предпочтительно около -40oC.
Соединения формулы (IX) также могут быть образованы путем обработки соединения формулы (XII)
приемлемым неорганическим основанием, например гидрокарбонатом натрия. Реакцию проводят в подходящем растворителе, например ДМФ.
(ii) Посредством реакции взаимодействия соединения формулы (XII), описанной выше, с подходящим органическим основанием, например ДБУ. Реакцию проводят в соответствующем растворителе, например толуоле, при не экстремальной температуре от 0 до 100oC, предпочтительно при комнатной температуре.
Соединения формул (X), (XI) и (XII) являются коммерчески выпускаемыми препаратами либо могут быть получены способами, хорошо известными специалистам.
Большинство промежуточных соединений формулы (VII), в которой Y представляет собой S, являются новыми. Согласно настоящему изобретению далее предложены промежуточное соединение формулы (VII')
в которой R1 и R16 определены выше, и способ их получения при условии, что:
(а) соединение формулы (VII') не является бензиловым эфиром 2,2-дихлортиопропионимидина; а
(б) R1 не является C1-5 алкилом, циклопропилом или циклогексилом.
Предпочтительные промежуточные соединения включают:
S-бензил-2-метокситиоацетимидат
S-бензил-2-фтортиоацетимидат
S-(2-нафтилметил)-2,2-дифтортиоацетимидат
S-(2-нафтилметил)-2-тиометилтиоацетимидат
Промежуточные соединения формулы (VII') могут быть получены посредством реакции взаимодействия соединения формулы (XIII)
с реагентом формулы R16L', приемлемо PhCH2L', в которой R16 определено ранее, а L' является подходящей концевой группой, например атомом галогена, таким как хлор.
Соединения формулы (XIII) являются коммерчески доступными либо могут быть получены способами, известными специалистам.
Настоящее изобретение будет далее проиллюстрировано следующими примерами.
Промежуточное соединение А
Получение т-Бутил-6-амино-2-т-бутоксикарбониламино-4, 4-диоксо-4-тиагексаноата формиатной соли
Т-бутил-6-бензилоксикарбониламино-2-т-бутоксикарбониламино-4,4- диоксо-4-тиагексаноат (152 мг), растворенный в 5%-ной смеси муравьиная кислота/метанол (8 мл), по каплям добавляли к перемешивавшейся суспензии 10%-ного Pd/C в 5%-ной смеси муравьиная кислота/метанол (2 мл) при 0oC. Реакционную смесь перемешивали при 0oC в течение 1 часа, а затем при комнатной температуре в течение 1-2 часов. Реакционную смесь фильтровали через hyflo, а катализатор промывали метанолом (25 мл) и водой (25 мл). Фильтрат концентрировали до одной четвертой объема под вакуумом и разбавляли водой (25 мл). Этот процесс повторяли дважды, а затем упаривали до сухого состояния под вакуумом. Остаток растворяли в воде и лиофилизировали, получая в результате беловатое вещество (111 мг).
Промежуточное соединение 1Б
Получение бромистоводородного S-Бензил-2-фтортиоацетимидата
Фторацетамид (3,39 г) и бензилбромид (6,23 г) подвергали дефлегмации в хлороформе (40 мл) в атмосфере азота в течение 16 часов. После охлаждения смесь разбавляли эфиром (200 мл), а полученное оранжевое вещество отфильтровывали. Твердую фазу промывали дополнительным количеством эфира и сушили над P2O5 под вакуумом, получая в результате 5,19 г искомого продукта.
Аналогичным способом получали следующие промежуточные соединения:
Наименование промежуточного соединения - Т. пл./oC
бромистоводородный S-бензил-2-метокситиоацетимидат из 2-метокситиоацетамида - 140
бромистоводородный S-бензил-2-бензилокситиоацетимидат из 2-бензилокситиоацетамида - 148-149 (разл)
бромистоводородный S-бензил-2-метилтиотиоацетимидат из 2-метилтиотиоацетамида - 163-165 (разл)
Промежуточное соединение В
Получение гидрохлорида S-бензилтиоацетимидата (промежуточное соединение)
Смесь тиоацетамида (15,0 г, 0,20 моль) и бензилхлорида (25,3 г, 0,20 моль) в хлороформе (75 мл) нагревали с дефлегмацией в течение 90 минут (для тиоацетамида требовалось около 40 минут для перехода в раствор). Затем реакционную смесь оставляли охлаждаться до комнатной температуры, после чего она стояла ночь при 0oC, и при этом образовывались бесцветные кристаллы в виде слоя на поверхности. Продукт отфильтровывали, промывали холодной 10%-ной смесью эфир-хлороформ и отсасывали досуха, получая в результате соединение, указанное в заголовке, (25,55 г) в виде бесцветных призм. Т.пл.=161-163oC.
Соль бромистоводородной кислоты получали аналогичным способом с выходом 85%; т.пл.=184-186oC dec.
Пример 1
Получение дигидробромида 2-амино-6-(1-имино-2-фторэтиламино)- 4,4-диоксо-4-тиагексановой кислоты
(а) Гидробромид т-бутил-2-т-бутоксикарбониламино-6-(1- имино-2-фторэтиламино)-4,4-диоксо-4-тиагексановой кислоты
К промежуточному соединению A (609 мг) в этаноле (10 мл) при 0oC добавляли Промежуточное соединение 1Б (407 мг) в виде одной порции. Смесь перемешивали при 0oC в течение 1 часа, затем при комнатной температуре в течение 2 часов. Растворитель удаляли под вакуумом, а остаток фракционировали между водой и эфиром. Водный слой промывали еще дважды эфиром, а затем концентрировали под вакуумом. Оставшуюся смолу очищали посредством колоночной хроматографии на двуокиси кремния, элюируя смесью дихлорметан-метанол (8:1), получая в результате бесцветную пену (317 мг).
(б) Дигидробромид 2-амино-6-(1-имино-2-фторэтиламино)-4,4- диоксо-4-тиагексановой кислоты
Гидробромид т-бутил-2-т-бутоксикарбониламино-6-(1-имино- 2-фторэтиламино)-4,4-диоксо-4-тиагексановой кислоты (300 мг) растворяли в ледяной уксусной кислоте (4,5 мл) и охлаждали во время добавления HBr в уксусной кислоте (45% в/об, 1,5 мл). Затем смесь перемешивали при комнатной температуре в течение 2 часов. Летучие вещества выпаривали под вакуумом, а остаток растворяли в воде. Раствор упаривали до сухого состояния под вакуумом и повторяли эту процедуру дважды. К остатку добавляли этанол и концентрировали смесь под вакуумом до беловатой пены. Материал может быть подвергнут дальнейшей очистке путем растворения в минимальном объеме теплого этанола и осаждения продукта эфиром с образованием гигроскопичного вещества (220 мг) в виде дигидрата.
Аналогичным способом могут быть получены соединения, приведенные в табл. А (см. в конце описания).
Пример 4
Получение 0,33 гидробромида 1,67 гидрохлорида 2- амино-6-(2-бензилокси-1-иминоэтиламино)-4,4-диоксо-4- тиагексановой кислоты
Гидробромид т-бутил-6-(2-бензилокси-1-иминоэтиламино)-2- т-бутоксикарбониламино-4,4-диоксо-4-тиагексаноата (0,97 г) (полученный способом, аналогичным способу, описанному в Примере 1А из Промежуточных соединений А и 3Б) перемешивали в 4 М смеси HCl/диоксан (12 мл) при комнатной температуре в течение 6 часов. Растворитель упаривали, а оставшуюся смолу растирали в порошок с сухим эфиром (20 мл). При стоянии медленно образовывалось белое вещество. Эфир декантировали, а очень гигроскопичное вещество промывали свежим эфиром и сушили под вакуумом при нагревании до 65oC.
Аналогичным способом может быть получено следующее соединение:
Пример 5
Наименование соединения: 2-Амино-6-(2-метилтио-1-иминоэтиламино)-4,4-диоксо-4-тиагексановая кислота, полученная из Промежуточных соединений А и 5Б.
Пример 6
Получение 0,33 гидробромида 1,67 гидрохлорида 2- амино-6-(2-гидрокси-1-иминоэтиламино)-4,4-диоксо-4-тиагексановой кислоты
Соединение из Примера 4 (350 мг, 0,81 ммоль) в смеси этанола (15 мл) и воды (5 мл) гидрогенизировали при комнатной температуре и атмосферном давлении над 5%-ным палладием на угле (Degussa type Е101 NO/W, 80 мг) в течение 18 часов. Катализатор отфильтровывали и промывали водой. Затем гидрогенизацию повторяли, используя свежий катализатор (100 мг), еще в течение 18 часов. Катализатор отфильтровывали и промывали водой, а растворители упаривали. Оставшуюся смолу истирали в порошок с небольшим количеством этанола, в результате чего происходило медленное образование бледно-желтого вещества. Этанол удаляли, а очень гигроскопичное вещество сушили в вакууме при комнатной температуре. Выход 240 мг.
Пример 7
Получение 2-амино-6-(1-иминоэтиламино)-4,4-диоксо- 4-тиагексановой кислоты
Способ А
(a) S-[2-(1-иминоэтиламино)этил]цистеин
Гидробромид S-(2-аминоэтил)цистеина (12,25 г, 50 ммоль) растворяли в теплой воде (50 мл) и растирали в порошок с основным карбонатом меди (5,85 г). Раствор охлаждали и фильтровали через Hyflo, а остаток промывали водой. Голубой раствор защищенного медью производного цистеина перемешивали и охлаждали до 10oC. Посредством добавления 2 н. гидроксида натрия pH доводили до значения от 10 до 11, в процессе чего порциями добавляли гидрохлорид этилацетимидата (9,375 г, 75 ммоль). Температуре позволяли достичь комнатного значения, а раствор доводили до pH 3 путем добавления 2 н. соляной кислоты. Раствор наносили на Dowex AG50WX8 колонку (объем слоя 100 мл), промывали водой и элюировали 0,2 н. раствором аммиака. Нингидринположительные фракции собирали, а раствор аммиака удаляли посредством выпаривания под вакуумом. Оставшийся раствор доводили до pH 4 посредством добавления 2 н. соляной кислоты. Раствор упаривали до сухого состояния на роторном испарителе, получая в результате 3 г гидрохлорида S-[2-(1-иминоэтиламино)этил]цистеина, который сушили в вакуумном эксикаторе.
(б) 2-Амино-6-(1-иминоэтиламино)-4,4-диоксо-4- тиагексановая кислота
Раствор S-[2-(1-иминоэтиламино)этил] цистеина (3,25 г, 10 ммоль) в 1 М хлорной кислоте (20 мл) обрабатывали 30%-ным раствором перекиси водорода (6,8 мл, 60 ммоль) и 0,5 М молибдатом аммония (1 мл). Температуру результирующей реакции поддерживали на 30oC посредством водяного охлаждения. Реакционную смесь перемешивали при 25oC в течение 2 часов, спустя это время ее наносили на AG 1X8 (ацетатную) ионообменную колонку (50 мл, 60 ммоль). Аминокислоту элюировали из колонки водой, а растворитель упаривали из нингидринположительных фракций до получения масла. Масло обрабатывали этанолом и снова упаривали под вакуумом. Оставшееся масло (приблизительно 4 г) очищали посредством колоночной хроматографии быстрого разделения, применяя в качестве элюанта смесь метанол/аммиак (9:1) и получая в результате 0,8 г 2-амино- 6-(1-иминоэтиламино)-4,4-диоксо-4-тиагексановой кислоты.
Способ Б
(а) Сульфат S-(2-бензилоксикарбониламиноэтил)цистеина
Жидкий аммиак (6 л) перемешивали в реакторе и осторожно добавляли L-цистеин (300 г, 1,25 моль). Реакционную смесь перемешивали в течение 1 часа. Через 2 часа добавляли кусковой натрий (115 г, 5 моль). Образовывался серый раствор. В течение 15 минут порциями добавляли 2-(бензилоксикарбониламино) этанфенилсульфонат (836 г, 2,5 моль). Реакционную смесь перемешивали с аммиаком при дефлегмации в течение ночи. Аммиаку позволяли испаряться посредством отключения циркуляции. Реакционную смесь оставляли на ночь, затем добавляли 9 литров воды и нагревали реакционную смесь до 40oC при перемешивании. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат нейтрализовали разбавленной серной кислотой. Белое/желтое вещество оделяли фильтрованием и сушили в вакуумной печи при 80oC. Получали 715 г соединения, указанного в заголовке. Т.пл. = 220-221,5oC.
(б) S-(2-бензилоксикарбонил-аминоэтил)-N-бутилоксикарбонилцистеин
К перемешивавшейся и охлаждавшейся на льду суспензии тщательно размолотого сульфата S-(2-бензилоксикарбониламиноэтил) цистеина (39,5 г, 0,1 моль) в диоксане (400 мл) и воде (200 мл) добавляли тремя порциями в течение 1,5 часов ангидрид бутилоксикарбонила (24 г, 0,11 моль), доводя pH до 9,5 посредством добавления приблизительно 200 мл 1 М гидроксида натрия. Диоксан удаляли под вакуумом, а к водному раствору добавляли этилацетат. Водный слой доводили до pH 2,8 путем добавления водной серной кислоты и фильтровали. Фильтрат экстрагировали этилацетатом. Этилацетатные экстракты объединяли, промывали водой и рассолом, сушили над сульфатом натрия и фильтровали. Растворитель упаривали под вакуумом, получая в результате 42,95 г соединения, указанного в заголовке.
(в) Трет-бутиловый эфир S-(2-бензилоксикарбониламино)-N- бутилоксикарбонилцистеина
Раствор S-(2-бензилоксикарбонил-аминоэтил)-N- бутилоксикарбонилцистеина (3,98 г, 10 ммоль) в сухом бензоле (15 мл) нагревали с дефлегмацией и отгоняли небольшое количество бензола для удаления последних следов влаги. К указанному раствору в состоянии дефлегмации по каплям добавляли в течение 20 минут ди- третбутилацеталь N,N-диметилформамида (8,2 г, 40 ммоль). Реакционную смесь затем нагревали с дефлегмацией в течение 30 минут, до тех пор, пока не переставали регистрироваться изменения посредством ТСХ. Реакционную смесь охлаждали до комнатной температуры, обрабатывали водой (15 мл) и перемешивали. Фазы разделяли и промывали органический слой 10%-ным KHCO3 (3х10 мл) и рассолом (2х10 мл). Водные фазы промывали свежей смесью эфир-бензол (1: 1) (2х15 мл) и 4 объединенных экстракта сушили над Na2SO4, обрабатывали углем, фильтровали и упаривали до сухого состояния, получая бледное таннированное масло. Сырой материал очищали посредством хроматографии быстрого разделения на SiO2, применяя 65%-ную смесь циклогексан-этилацетат в качестве элюанта. В результате получали 2,56 г продукта, указанного в заголовке, в виде практически бесцветного масла.
(г) Трет-бутиловый эфир 2(бутилоксикабониламино)-6- (бензилоксикабониламино)-4,4-диоксо-4-тиагексановой кислоты
К трет-бутиловому эфиру S-(2-бензилоксикабониламино)-N- бутилоксикарбонилцистеина (454 мг, 1,0 ммоль) в метаноле (5 мл) при 0oC по каплям добавляли раствор "Oxone" (925 мг, 3,0 ммоль) в воде (5 мл) в течение 10 минут. В ходе процедуры добавления наблюдали экзотермический эффект, а температуру поддерживали ниже 2oC посредством внешнего охлаждения льдом. Реакционную смесь перемешивали при 0oC в течение 4 часов, а затем позволяли ей нагреться до 15oC в течение 10 часов. Из-за высокой вязкости реакционной смеси для облегчения перемешивания к ней добавляли дополнительные аликвоты метанола (4 мл) и воды (2 мл). Добавляли еще одну порцию "Oxone" (308 мг, 1,0 ммоль) в воде (1,5 мл) и перемешивали смесь еще в течение 14 часов при комнатной температуре. Затем реакционную смесь обрабатывали эфиром (3х25 мл) и каждый раз отгоняли эфирный раствор от твердого остатка. Объединенные эфирные экстракты сушили над Na2SO4, фильтровали и упаривали до сухого состояния, получая 0,428 г соединения, указанного в заголовке, в виде бесцветного масла, которое быстро превращалось в белое вещество. Т.пл.=116-118oC.
(д) Трет-бутиловый эфир 2-(бутилоксикарбониламино)- 6-амино-4,4-диоксо-4-тиагексановой кислоты
К раствору трет-бутилового эфира 2-бутилоксикарбониламино)-6- (бензилоксикарбониламино)-4,4-диоксо-4-тиагексановой кислоты (200 мг) в метаноле (5 мл) добавляли Pd/C (50%-ный) (Degussa тип) (200 мг) и гидрогенизировали смесь при комнатной температуре до тех пор, пока не переставали наблюдаться изменения. Затем реакционную смесь фильтровали через слой Hyflo, а остаток промывали свежим метанолом. Объединенные фильтраты упаривали до сухого состояния, получая 119 мг соединения, указанного в заголовке, в виде практически бесцветного стекла.
(е) Гидрохлорид трет-бутилового эфира 2-(бутилоксикарбониламино) -6-(1-иминоэтиламино)-4,4-диоксо-4-тиагексановой кислоты
К раствору трет-бутилового эфира 2-(бутилоксикарбониламино)-6- амино-4,4-диоксо-4-тиагексановой кислоты (340 мг) в метаноле (10 мл) порциями добавляли Промежуточное соединение В (202 мг). После перемешивания в течение 30 минут
при 0oC реакционной смеси позволяли нагреваться до комнатной температуры, а затем упаривали до сухого состояния. Масляный остаток обрабатывали водой (2 мл) и экстрагировали эфиром (4х4 мл) для удаления бензилмеркаптана. Органические экстракты промывали свежей водой (2 мл), а объединенные экстракты упаривали под вакуумом при 40oC, получая в результате 396 мг соединения, указанного в заголовке, в виде бесцветного стекла.
(ж) Дигидрохлорид 2-Амино-6-(1-иминоэтиламино)-4,4-диоксо-4- тиагексановой кислоты
К раствору гидрохлорида трет-бутилового эфира 2- (бутилоксикарбониламино)-6-(1-иминоэтиламино)-4,4-диоксо-4- тиагексановой кислоты (300 мг) в диоксане (15 мл) добавляли 4 н. HCl/диоксан (10 мл) и оставляли смесь при комнатной температуре в течение 24 часов. Реакционную смесь затем упаривали до сухого состояния, а полутвердый остаток обрабатывали водой (2 мл) и упаривали, получая в результате бесцветное стекло. Сырой продукт очищали посредством хроматографии быстрого разделения на SiO2, применяя в качестве элюанта смесь CH3CN:CH3CO2H:H2O (5:2:2), получая в результате 172,5 мг соединения, указанного в заголовке.
Пример 8
Получение 2-Амино-6-(1-иминоэтиламино)-4-оксо-4- тиагексановой кислоты
Способ А
(a) S-[2-(Иминоэтиламино)этил]цистеин
S-[2-(Иминоэтиламино)этил] цистеин (3 г, 0,0113 моль) получали согласно способу, описанному в Примере 7, способ А, описанная выше стадия (а).
(б) 2-Амино-6-(1-иминоэтиламино)-4-оксо-4-тиагексановая кислота
Раствор S-[2(Иминоэтиламино)этил] цистеина (3 г, 0,0113 моль) в 1 М соляной кислоте (30 мл) перемешивали, а затем обрабатывали 30%-ным раствором перекиси водорода (1,5 мл, 11,6 ммоль). Реакционную смесь оставляли на ночь, после чего проверяли окончание реакции посредством тонкослойной хроматографии. Реакционную смесь наносили на AG50WX8 колонку, промывали водой и элюировали 0,5 М раствором аммиака. Нингидринположительные фракции собирали, а растворитель упаривали под вакуумом. Сырой продукт очищали посредством колоночной хроматографии быстрого разделения, применяя в качестве элюанта смесь метанол/аммиак (9:1) и получая 200 мг 2-Амино-6-(1-иминоэтиламино)-4-оксо-4-тиагексановой кислоты.
Способ Б
(а) Трет-бутиловый эфир 2-(бутилоксикарбониламино)-6- (бензилоксикарбониламино)-4-оксо-4-тиагексановой кислоты
К раствору трет-бутилового эфира S-(2- бензилоксикарбониламино)- N-бутилоксикарбонилцистеина (454 мг, 1,0 ммоль) (полученного, как описано в Примере 7, Способ Б, стадии от (а) до (в)) в дихлорметилене (25 мл) при -2oC добавляли м-хлорпербензойную кислоту (426 мг @ 85%=2,10 ммоль). Наблюдали слабый экзотермический эффект, приводивший к повышению температуры до +2oC. Затем реакционную смесь перемешивали при 0oC в течение 1,5 часов. Спустя 1 час реакция практически завершалась. Затем реакционную смесь экстрагировали 5%-ным KHCO3 (3х25 мл), органический слой сушили над Na2SO4, фильтровали и упаривали до сухого состояния, получая очень светлое таннированное масло. Сырое соединение очищали посредством хроматографии быстрого разделения на SiO2, применяя в качестве элюанта 1,25%-ную смесь МеОН-ЭтОАц, получая в результате 395 г соединения, указанного в заголовке.
(б) Трет-бутиловый эфир 2-(бутилоксикабониламино)-6- (бензилоксикарбониламино)-4,4-диоксо-4-тиагексановой кислоты
Соединение, указанное в заголовке, получали способом, аналогичным описанному в Примере 7, Способ Б, стадии от (д) до (ж).
Пример 9
Биологическая активность
Ингибирование очищенных человеческих NO синтаз определяли после получения человеческой nNOS [11], iNOS [12] и eNOS [13] согласно описанию. Их активность регистрировали по превращению [14C]-L-аргинина в цитруллин, как описано в [14] , в реакционных смесях (100 мкл), содержавших 50 мМ HEPES pH 7,0, 8 мкМ тетрагидробиоптерина, 1 мМ НАДФН и 0,5 мкМ [14C]-L-аргинина (30000 импульсов в минуту) при 30oC. Результаты представлены в таблице 1.
Ингибирование рекомбинантной человеческой iNOS определяли по экспрессии в клеточной системе бакуловирус/насекомое [15], тестируя цитозоль насекомого на NO синтазу посредством микротитрационного планшетного анализа, основанного на спектрофотометрическом анализе, описанном ранее [16]. Анализ проводили при 37oC в реакционных смесях, содержавших HEPES (100 мМ), ДТТ (0,1 мМ), тетрагидробиоптерин (5 мкМ), НАДФН (100 мкМ), гемоглобин (5 мкМ), L-аргинин (30 мкМ) и ингибитор (0-300 мкМ), измеряя изменение поглощения при 405-420 нм и определяя ингибирование в устойчивом состоянии между 15 и 30 минутами инкубации. Результаты представлены в таблице 2.
Ингибирование eNOS и iNOS in situ в кольцах аорт крыс определяли путем измерения увеличения кольцевого напряжения, вызываемого ингибированием NO синтазы. Для изучения базального тонуса (отражающего eNOS) готовили кольца грудной аорты с интактным эндотелием, как описано ранее [17], и получали кривые кумулятивных концентраций для ингибиторов в присутствии трехкратной концентрации фенилфрина (ЭД10 = 10 нМ). Для исследований индуцированного тонуса гладкого мускула (отражающего iNOS) кольца с обнаженным эндотелием подвергали действию ЛПС (0,1 мкг/мл из S.typhosa) в присутствии фенилфрина в концентрации, приблизительно равной ЭД90, в течение 6 часов, как описано ранее [18] . В течение этого времени наблюдали прогрессивную потерю тонуса вследствие iNOS индукции. Затем для ингибиторов были получены кривые кумулятивных концентраций. Результаты представлены в табл. 2.
Ингибирование eNOS и iNOS in vivo определяли по действию ингибиторов на кровяное давление либо у нормальных (eNOS) мышей, либо у мышей в состоянии шока из-за действия эндотоксина (iNOS), находящихся в сознании. Самок CD-1 мышей (25-35 г) анастезировали на короткий срок изофтораном (2%-ным). В бедренную вену имплантировали трубки канюли, проводили их подкожно таким образом, чтобы они выходили в верхней части спины и сообщались с шарнирно закрепленной системой для продолжительной регистрации кровяного давления и для введения ингибитора соответственно. После восстановления после хирургического вмешательства животных со средним кровяным давлением в нормальном диапазоне (90-110 мм Hg) использовали для снятия кумулятивных концентрационных кривых для ингибиторов, действующих на кровяное давление, либо без дальнейшей обработки ("нормальные мыши"), либо 7 часов спустя после введения липополисахарида (12,5 мг/кг ЛПС из E.coli 026:B6 внутривенно в течение 30 секунд) для индукции шока ("шоковые мыши"). У нормальных мышей соединение из Примера 7 не оказывало действия на кровяное давление в пределах дозового интервала 1-1000 мг/кг. Однако у шоковых мышей соединение из Примера 7 было способно полностью восстанавливать кровяное давление до нормального уровня.
Действие соединения из Примера 7 на индуцированное эндотоксином сосудистое истекание исследовали на крысах, как описано [19]. Соединение из Примера 7 в дозах до 5 мг/кг при введении конкурентно с эндотоксином не вызывало ухудшения эндотоксин-индуцированного плазменного истекания в отличие от неселективных ингибиторов, таких как L-NMMA. Однако соединение из Примера 7 не устраняло iNOS-зависимое замедленное плазменное истекание с ЭД50, равной 1 мг/кг.
ЛИТЕРАТУРА
1. Moncada et al. Biochemical Pharmacology, 38, 1709-1715 (1989).
2. Moncada et al., Pharmacological Reviews, 43, 109-142 (1991).
3. WO 91/04024.
4. GB-A-2240041.
5. EP-A-0446699.
6. Knowles and Moncada, Biochem. J., 298, 249-258 (1994).
7. PCT/GB 9202387.
8. T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2nd Edition, John Wiley and Sons Inc., 1991.
9. The Chemistry of Amidines and Imidates, Vol. 2, Eds. Saul Patai and Zvi Rappoport, John Wiley and Sons Inc., 1991.
10. Tet. Lett., 22 (14), 1287 (1981).
11. Furfine et al., Biochem. 32, 8512-8517 (1993).
12. Sherman et al., Biochem. 32, 11600-11605 (1993).
13. Garvey et al. Arch. Bioch. Biophys. (1994), in press.
14. Schmidt et al., Proc. Natl. Acad. Sci. USA, 88, 365-369 (1991).
15. Charles et al. In: The Biology of Nitric Oxide 4, Moncada S., Feelisch M. , Busse R. and Higgs E.A., eds., Portland Press, London, pp. 316-320 (1994).
16. Knowles et al. , Biochem. Biophys. Res. Commun., 172, 1042-1048 (1990).
17. Rees et al., Br. 3. Pharmacol. 96, 418-424 (1989).
18. Rees et al., Biochem. Biophys. Res. Commun. 173, 541- 547 (1990).
19. Laszlo et al., Brit. J. Pharmacol. 111, 1309-1315 (1994).


Описываются новые производные амидиносульфона формулы I, где R1 является метилом или фторметилом, или его соль, эфир или амид. Соединения селективно ингибируют изоферментом NO синтазы, продуцирующим оксид азота, и могут найти применение для лечения септического шока или шока, вызванного внезапной печеночной недостаточностью. Описываются также способы получения соединений формулы I, фармацевтическая композиция и способ лечения. 7 с. и 2 з.п. ф-лы, 3 табл.
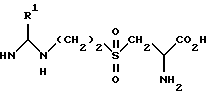
где R1 является метилом или фторметилом,
или его соль, эфир или амид.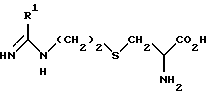
где R1 такой, как определено выше,
возможно с последующим превращением в другое соединение формулы I.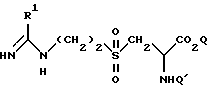
где R1 такой, как определено выше;
Q является водородом или карбоксилзащитной группой;
Q' является защитной группой,
возможно с последующим превращением в другое соединение формулы I.
Приоритеты по признакам и пунктам:
15.05.1995 при R1 - фторметил в п.1, второе соединение в п.2;
15.06.1994 - все признаки в п.1 за исключением R1 - фторметил, первое соединение в п.2, пп.3 - 9.
| WO 9313055 A, 08.07.1993 | |||
| Способ получения производных бензодиоксола | 1977 |
|
SU686615A3 |
| DE 4310202 A, 07.10.1993. | |||

