Изобретение относится к области биотехнологии, в частности к генной инженерии растений, и может быть использовано для получения трансгенных микроводорослей рода хлорелла, устойчивых к глифосату.
Глифосат [N-(фосфонометил)-глицин] - наиболее распространенный гербицид, применяемый в сельском хозяйстве по всему миру. Причинами его популярности являются высокая эффективность, широкий спектр действия, крайне низкая токсичность для человека и безопасность для окружающей среды.
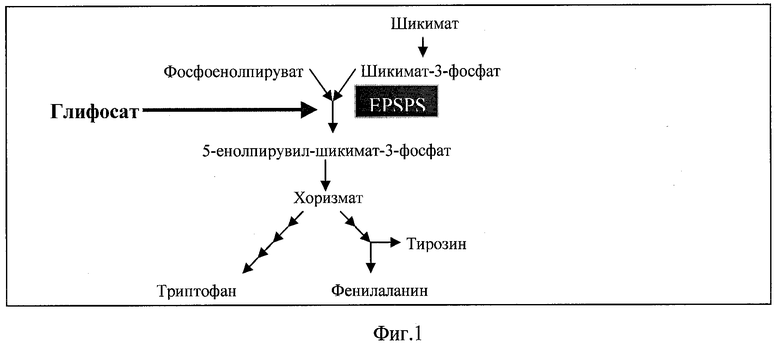
Глифосат был синтезирован и запатентован в качестве гербицида Джоном Е. Францем из компании «Монсанто» в 1970 году (1). Механизм действия глифосата уникален, поскольку он является единственным ингибитором фермента 5-енолпирувил-шикимат-3-фосфат-синтетазы (EPSPS), участвующего в пути метаболизма шикимата. Путь метаболизма шикимата и его ингибирование глифосатом представлен на фиг.1.
Фермент EPSPS катализирует реакцию, в которой из шикимат-3-фосфата (S3P) и фосфоенолпирувата образуется 5-енолпирувил-шикимат-3-фосфат (ESP), который далее превращается в хоризмат (хоризмовую кислоту) - растительный предшественник ароматических аминокислот (фенилаланина, тирозина и триптофана).
Глифосат по своей структуре является аналогом переходного состояния фосфоенолпирувата, одного из субстратов EPSPS. Ингибирование EPSPS приводит к подавлению обратной связи данного пути, вследствие чего накапливается шикимат-3-фосфат, который превращается в избыток шикимата (2). Большое количество шикимата накапливается в тканях растений, обработанных глифосатом (3).
В конце 1980-х годов были предприняты многочисленные попытки создания трансгенных растений, устойчивых к глифосату. В результате был найден ген CP4 из Agrobacterium sp., кодирующий устойчивую к глифосату форму фермента EPSPS (4). После помещения гена с соответствующими регуляторными элементами в геном растений растения стали проявлять устойчивость к высокому уровню глифосата. Кроме гена СР4 был найден ген глифосат-оксидоредуктазы (GOX) из бактерий Ochrobactrum anthropi, этот ген содержится в трансгенных растениях рапса (5). Для кукурузы ген EPSPS был изменен методом сайт-направленного мутагенеза, в результате была получена форма фермента EPSPS, устойчивая к глифосату, которая встречается в некоторых разновидностях трансгенной кукурузы. В 1986 году была запатентована технология получения таких устойчивых к глифосату трансгенных растений, как соя, хлопок, люцерна, рапс, лен, томат, сахарная свекла, подсолнух, картофель, табак, кукуруза, пшеница, рис и салат-латук (6). В 1996 году на рынке появилась устойчивая к глифосату соя, в 1996 году - рапс, в 1997 году - хлопчатник, в 1998 - кукуруза, на настоящий момент почти 90% всех трансгенных зерновых растений составляют культуры, устойчивые к глифосату (6).
Одноклеточные зеленые микроводоросли рода хлорелла (Chlorella) являются потенциальной альтернативной системой экспрессии гетерологичных белков. В отличие от системы экспрессии E.coli хлорелла имеет эукариотическую организацию клеток, что делает возможным посттрансляционную модификацию белков. В отличие от дорогостоящих культур клеток млекопитающих культивирование клеток хлореллы достаточно дешево, поскольку хлорелла является автотрофом и в качестве источника энергии может использовать только свет и диоксид углерода. Микроводоросли рода Chlorella обладают устойчивостью к большинству антибиотиков, используемых в качестве селективных маркеров, что затрудняет отбор трансформированных растительных клеток. Антибиотики, которые сейчас используют при работе с клетками хлореллы, имеют высокую стоимость и токсичность (ярким примером служит гигромицин). Поэтому актуальной задачей является поиск новых селективных маркеров и создание соответствующих генно-инженерных конструкций, несущих гены устойчивости, для последующей трансформации клеток микроводоросли Chlorella.
Наиболее ближайшей к заявляемой рекомбинантной плазмидной ДНК - прототипом, является рекомбинантная плазмидная ДНК pMON546, кодирующая синтез EPSPS под 35S промотором вируса табачной мозаики с сигнальным пептидом (7). В патенте описан вектор, устойчивый к канамицину, в составе которого имеется ген EPSPS. Полипептид синтезируется с сигнальным пептидом, обеспечивающим его перенос из цитоплазмы в хлоропласты растительных клеток. Трансформацию растительных клеток табака, томата, подсолнечника, картофеля, риса, кукурузы и сахарной свеклы проводили с хелперной плазмидой pGV3111SE, кодирующей определенные ферменты, необходимые для успешной трансформации рекомбинантной плазмидной ДНК pMON546. Полученные таким образом трансгенные растения были устойчивы к глифосату.
Недостатком известной плазмиды является то, что она не обеспечивает экспрессию гена EPSPS в трансгенных микроводорослях рода хлорелла.
Поиск по источникам научно-технической и патентной информации не выявил технических решений, раскрывающих генетические конструкции, обеспечивающие экспрессию гена EPSPS в трансгенных микроводорослях рода хлорелла, а также технологии, касающиеся использования микроводорослей рода хлорелла для получения устойчивых к глифосату форм.
Технической задачей изобретения является создание рекомбинантной плазмидной ДНК, содержащей ген агробактериальной 5-енолпирувил шикимат-3-фосфат-синтетазы, для получения трансгенных микроводорослей рода хлорелла, устойчивых к глифосату.
Поставленная техническая задача достигается предлагаемой рекомбинантной плазмидной ДНК pBinPLUS-ARS-EPSPS, содержащей ген агробактериальной 5-енолпирувил-шикимат-3-фосфат-синтетазы под промотором гена альфа-тубулина хлореллы, для получения трансгенных микроводорослей рода хлорелла, устойчивых к глифосату.
Конструирование рекомбинантной плазмиды осуществляют в несколько этапов. Сначала проводят сборку из олигонуклеотидов фрагмента ДНК, кодирующего транзиторный пептид (СР4), и клонирование гена агробактериальной 5-енолпирувил-шикимат-3-фосфат-синтетазы (EPSPS). Фрагмент ДНК, кодирующий транзиторный пептид (СР4), собирают из частично перекрывающихся между собой олигонуклеотидов, структуры которых приведены на фиг.2.
Фрагмент ДНК, кодирующий ген EPSPS, амплифицируют с помощью олигонуклеотидных праймеров EPSPS-U и EPSPS-R на матрице геномной ДНК Agrobacterium tumefaciens str. С58. Фрагменты гена EPSPS и гена, кодирующего транзиторный пептид, объединяют с помощью ПЦР. Далее проводят клонирование объединенного фрагмента ДНК, кодирующего CP4-EPSPS, в плазмиду pMTL22 по сайтам гидролиза эндонуклеазами рестрикции EcoRI и BglII. Затем к кодирующей последовательности добавляют регуляторные элементы: промотор и терминатор для экспрессии в растениях. На 5'-конец кодирующей последовательности гена EPSPS добавляют 35S терминатор вируса мозаики цветной капусты из плазмиды pAVA389 по сайтам гидролиза XbaI и HindIII, получают плазмиду pMTL22-EPSPS-35S term. Фрагмент ДНК, содержащий промотор гена альфа-тубулина хлореллы, получают с помощью ПЦР с праймеров CLV1 и CLV2, в качестве матрицы используют геномную ДНК хлореллы, затем клонируют в вектор pBluescriptIISK+ по сайтам гидролиза FauNDI и HindIII. После этого проводят ПЦР с праймерами Т7 и tua-pr-BglII (второй праймер содержит сайт узнавания и гидролиза рестриктазы BglII) и переносят промотор в плазмиду pMTL22-EPSPS-35S term по сайтам гидролиза Sfr274I и BglII. Таким образом, получают плазмиду pMTL22-casEPSPS, содержащую функциональные элементы, необходимые для экспрессии целевого гена в растении.
На заключительном этапе экспрессионную кассету извлекают по сайтам гидролиза Sfr274I и BamHI и переносят в бинарный вектор pBinPLUS-ARS, гидролизованный по Sail и BamHI (поскольку эндонуклеазы Sfr274I и SalI дают совместимые «липкие» концы).
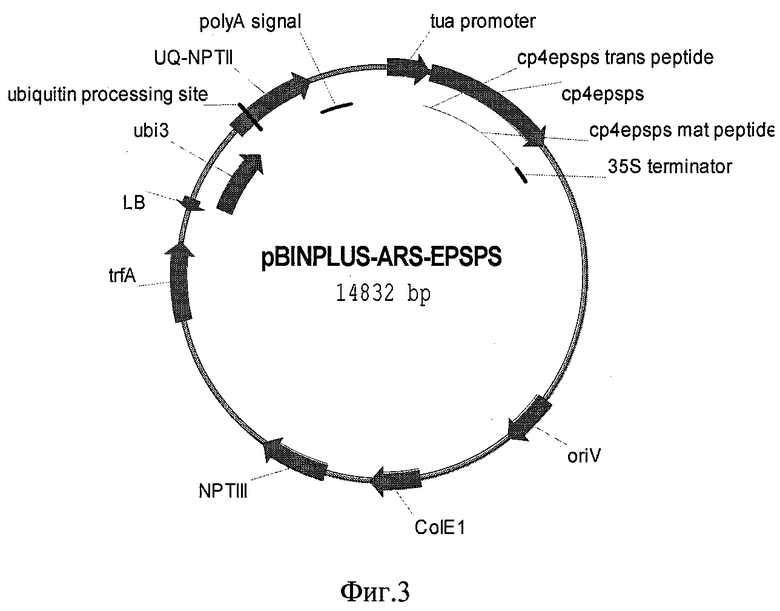
В результате получают рекомбинантную плазмидную ДНК pBinPLUS-ARS-EPSPS, несущую ген агробактериальной 5-енолпирувил-шикимат-3-фосфат-синтетазы под управлением промотора гена альфа-тубулина хлореллы размером 14,8 т.п.н., которая позволяет осуществить агробактериальный перенос целевой последовательности ДНК в геном растения и таким образом получить устойчивую к глифосату форму одноклеточной микроводоросли рода хлорелла.
Плазмида состоит из следующих элементов:
- ДНК векторной плазмиды pBinPLUS-ARS размером 12440 п.н.
- HindIII-BglII фрагмент ДНК размером 500 п.н., содержащий промотор гена альфа-тубулина хлореллы
- BglII-XbaI фрагмент ДНК размером 1610 п.н., содержащий кодирующую последовательность гена EPSPS с сигнальным пептидом
- XbaI-HindIII фрагмент ДНК размером 230 п.н., содержащий 35S терминатор вируса мозаики цветной капусты
Физическая карта плазмиды pBinPLUS-ARS-EPSPS с указанием генетических маркеров приведена на фиг.3.
Отличие полученной рекомбинантной плазмидной ДНК от прототипа состоит в использовании промотора гена альфа-тубулина хлореллы для экспрессии гена EPSPS, что позволяет получать трансгенные микроводоросли рода хлорелла, устойчивые к глифосату.
Сущность изобретения поясняется следующими примерами.
Пример 1. Конструирование плазмиды pBinPLUS-ARS-EPSPS
1.1 Получение плазмиды pMTL22-EPSPS
Для сборки фрагмента смесь, содержащую 1 мкМ олигонуклеотиды Transit 1-4, прогревали при 95°C в течение 5 мин, после чего добавляли буфер для ПЦР С18 (65 Мм Tris-HCl (pH 8,9); 16 Мм (NH4)2SO4; 0,05% Tween 20; 1,8 Мм MgCl2) и Tag-полимеразу. Олигонуклеотиды для сборки гена, кодирующего транзиторный пептид, представлены в таблице 1.
Проводили 10 циклов денатурации при 95°C и элонгации-отжига при 62°C. Реакционную смесь в дальнейшем использовали в качестве матрицы для ПЦР с праймерами U-transit и R-transit. ПЦР проводили в буфере С18, содержащем 200 мкМ dNTP, 3 мкМ каждого из праймеров, 1 ед. акт. Taq-полимеразы и матрицу. Фрагмент ДНК, кодирующий ген EPSPS, амплифицировали с помощью олигонуклеотидных праймеров EPSPS-U и EPSPS-R на матрице геномной ДНК Agrobacterium tumefaciens str. С58. Структуры используемых олигонуклеотидных праймеров представлены в таблице 2.
Фрагменты гена EPSPS и гена, кодирующего транзиторный пептид, объединяли с помощью ПЦР в буфере С18, содержащем 200 мкМ dNTP, по 3 мкМ каждого из праймеров U-transit и EPSPS-R и 1 ед. акт. Taq-полимеразы.
5 мкг ДНК плазмиды pMTL22 подвергали гидролизу эндонуклеазами рестрикции BglII (50 е.а.) и EcoRI (50 е.а.) в течение 2 часов при 37°C в 50 мкл буфера Orange, содержащего 50 mM Tris-HCl pH 7.6; 10 мМ MgCl2; 100 mМ NaCl; 1 мМ DTT. Рестрикционную смесь подвергали электрофорезу в 1.2%-ном агарозном геле, гель окрашивали раствором бромистого этидия в воде (1 мкг/мл), фрагмент размером 2500 п.н. вырезали и выделяли из геля методом сорбции ДНК на силикагеле.
10 мкг амплификационного фрагмента ДНК CP4-EPSPS, кодирующего EPSPS с транзиторным пептидом, подвергали гидролизу эндонуклеазами рестрикции BglII (100 е.а.) и EcoRI (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера Orange, содержащего 50 мМ Tris-HCl pH 7.6, 10 мМ MgCl2, 100 мМ NaCl, 1 мМ DTT. Рестрикционную смесь подвергали электрофорезу в 1.5%-ном агарозном геле, гель окрашивали раствором бромистого этидия в воде (1 мкг/мл), фрагмент размером 1600 п.н. вырезали и выделяли из геля методом сорбции ДНК на силикагеле.
Далее проводили лигирование полученных вектора и фрагмента, для чего смешивали 0,05 мкг BglII-EcoRI фрагмента плазмиды pMTL22 и 0,05 мкг BglII-EcoRI амплификационного фрагмента ДНК CP4-EPSPS в 20 мкл лигазного буфера, содержащего 50 мМ Tris-HCl pH 7.8; 10 мМ MgCl2; 10 мМ DTT; 1 мМ АТР; 25 мкг/мл БСА. К смеси добавляли 10 е.а. ДНК-лигазы фага Т4 и инкубировали 6 часов при 4°C.
10 мкл лигазной смеси использовали для трансформации клеток E.coli штамма XL1-Blue. Клетки высевали на чашку с агаризованным LB-агаром с ампициллином (100 мкг/мл), IPTG (0,5 мкМ/мл) и X-gal (100 мкг/мл). Отбор колоний со вставкой проводили методом ПНР с праймерами pMTLl, pMTL2 среди колоний белого цвета. Продукты ПЦР анализировали при помощи электрофореза в 1,5%-ном агарозном геле: при наличии вставки длина ПЦР-фрагмента составляла 1830 п.н., при отсутствии - 265 п.н. Было отобрано 16 клонов. ДНК этих клонов дополнительно проверяли с помощью гидролиза эндонуклеазой рестрикции PvuII с последующим разделением продуктов гидролиза электрофорезом в 1,5%-ном агарозном геле. Наличие вставки подтвердилось для 15 клонов. Было проведено секвенирование последовательностей полученных клонов pMTL22-EPSPS с праймеров pMTLl, pMTL2.
1.2 Получение плазмиды pMTL22-EPSPS-35S term
10 мкг ДНК плазмиды pMTL22-EPSPS подвергали гидролизу эндонуклеазой рестрикции HindIII (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера White, содержащего 10 мМ Tris-HCl, pH 8.5, 10 мМ MgCl2, 100 мМ NaCl, 1 мМ DTT, инактивировали фермент прогреванием при 65°С в течение 20 мин, осаждали ДНК из рестрикционной смеси изопропанолом и растворяли в воде. Затем ДНК гидролизовали эндонуклеазой рестрикции XbaI (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера Orange, содержащего 50 мM Tris-HCl pH 7.6; 10 mМ MgCl2; 100 mМ NaCl; 1 mМ DTT. Рестрикционную смесь подвергали электрофорезу в 1.0%-ном агарозном геле, гель окрашивали раствором бромистого этидия в воде (1 мкг/мл), фрагмент размером 4070 п.н. вырезали и выделяли из геля методом сорбции ДНК на силикагеле.
10 мкг ДНК плазмиды pAVA389, содержащей 35S терминатор вируса мозаики цветной капусты, подвергали гидролизу эндонуклеазой рестрикции HindIII (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера White, содержащего 10 мМ Tris-HCl, pH 8.5, 10 мМ MgCl2, 100 мМ NaCl, 1 мМ DTT, инактивировали фермент прогреванием при 65°C в течение 20 мин, осаждали ДНК из рестрикционной смеси изопропанолом и растворяли в воде. Затем ДНК гидролизовали эндонуклеазой рестрикции XbaI (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера Orange, содержащего 50 mМ Tris-HCl pH 7.6; 10 mM MgCl2; 100 mМ NaCl; 1 mМ DTT. Рестрикционную смесь подвергали электрофорезу в 1.5%-ном агарозном геле, гель окрашивали раствором бромистого этидия в воде (1 мкг/мл), фрагмент размером 230 п.н. вырезали и выделяли из геля методом сорбции ДНК на силикагеле.
Далее проводили лигирование полученных вектора и фрагмента, для чего смешивали 0,05 мкг HindIII-XbaI фрагмента плазмиды pMTL22-EPSPS и 0,05 мкг HindIII-XbaI фрагмента плазмиды pAVA389 в 20 мкл лигазного буфера, содержащего 50 мМ Tris-HCl pH 7.8; 10 мМ MgCl2; 10 мМ DTT; 1 мМ ATP; 25 мкг/мл БСА. К смеси добавляли 10 е.а. ДНК-лигазы фага Т4 и инкубировали 6 часов при 4°C.
10 мкл лигазной смеси использовали для трансформации клеток E.coli штамма XL1-Blue. Клетки высевали на чашку с агаризованным LB-агаром с ампициллином (100 мкг/мл). Отбор колоний со вставкой проводили методом ПЦР с праймерами pMTLl, pMTL2. Продукты ПЦР анализировали при помощи электрофореза в 1,5%-ном агарозном геле. Наличие вставки подтверждали при помощи рестрикционного анализа эндонуклеазами рестрикции HindIII и XbaI с последующим разделением продуктов гидролиза электрофорезом в 1,5%-ном агарозном геле.
1.3 Получение плазмиды pBlu CLV1-2
Фрагмент ДНК, содержащий промотор гена альфа-тубулина хлореллы, получали с помощью ПЦР с праймеров CLV1 и CLV2 в буфере С18, содержащем 200 мкМ dNTP, по 3 мкМ каждого из праймеров и 1 ед. акт. Taq-полимеразы, в качестве матрицы использовали геномную ДНК хлореллы. 10 мкг амплификационного фрагмента ДНК CLV1-2 гидролизовали эндонуклеазой рестрикции HindIII (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера White, содержащего 10 мМ Tris-HCl, pH 8.5, 10 мМ MgCl2, 100 мМ NaCl, 1 мМ DTT, инактивировали фермент прогреванием при 65°C в течение 20 мин, осаждали ДНК из рестрикционной смеси изопропанолом и растворяли в воде. Затем ДНК гидролизовали эндонуклеазой рестрикции FauNDI (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера Yellow, содержащего 33 мМ Tris-ацетат, pH 7.9, 10 мМ магния ацетат, 66 мМ калия ацетат, 1 мМ DTT. Рестрикционную смесь подвергали электрофорезу в 1.5%-ном агарозном геле, гель окрашивали раствором бромистого этидия в воде (1 мкг/мл), фрагмент размером 740 п.н. вырезали и выделяли из геля методом сорбции ДНК на силикагеле.
10 мкг ДНК плазмиды pBluescriptSK(+) подвергали гидролизу эндонуклеазами рестрикции HindIII (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера White, содержащего 10 мМ Tris-HCl, pH 8.5, 10 мМ MgCl2, 100 мМ NaCl, 1 мМ DTT, инактивировали фермент прогреванием при 65°C в течение 20 мин, осаждали ДНК из рестрикционной смеси изопропанолом и растворяли в воде. Затем ДНК гидролизовали эндонуклеазой рестрикции FauNDI (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера Yellow, содержащего 33 мМ Tris-ацетат, pH 7.9, 10 мМ магния ацетат, 66 мМ калия ацетат, 1 мМ DTT. Рестрикционную смесь подвергали электрофорезу в 1.0%-ном агарозном геле, гель окрашивали раствором бромистого этидия в воде (1 мкг/мл), фрагмент размером 2930 п.н. вырезали и выделяли из геля методом сорбции ДНК на силикагеле.
Далее смешивали 0,05 мкг FauNDI-HindIII фрагмента плазмиды pBluescriptSK(+) и 0,05 мкг FauNDI-HindIII амплификационного фрагмента ДНК CLV1-2, содержащего промотор гена альфа-тубулина хлореллы в 20 мкл лигазного буфера, содержащего 50 мМ Tris-HCl pH 7.8; 10 мМ MgCl2; 10 мМ DTT; 1 мМ АТР; 25 мкг/мл БСА. К смеси добавляли 10 е.а. ДНК-лигазы фага Т4 и инкубировали 6 часов при 4°C.
10 мкл лигазной смеси использовали для трансформации клеток E.coli штамма XL1-Blue. Клетки высевали на чашку с агаризованным LB-агаром с ампициллином (100 мкг/мл). Отбор колоний со вставкой проводили методом ПЦР с праймерами CLV1, CLV2. Продукты ПЦР анализировали при помощи электрофореза в 1,5%-ном агарозном геле. Наличие вставки подтверждали при помощи рестрикционного анализа эндонуклеазами рестрикции HindIII и FauNDI с последующим разделением продуктов гидролиза электрофорезом в 1,5%-ном агарозном геле.
1.4 Получение плазмиды pMTL22-casEPSPS
Фрагмент, содержащий промотор гена альфа-тубулина хлореллы, получали методом ПЦР с праймеров Т7 и tua-pr-BglII (5'-CCTAAAGATCTGGTTGCAGATTTAGAAAGAA - 3') в буфере С18, содержащем 200 мкМ dNTP, по 3 мкМ каждого из праймеров и 1 ед. акт. Taq-полимеразы, в качестве матрицы использовали плазмиду pBlu CLV1-2. С помощью праймера tua-pr-BglII вводили сайт гидролиза эндонуклеазы рестрикции BglII.
10 мкг амплификационного фрагмента ДНК tua prom, кодирующего промотор гена альфа-тубулина хлореллы, подвергали гидролизу эндонуклеазой рестрикции BglII (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера Orange, содержащего 50 мМ Tris-HCl pH 7.6, 10 мМ MgCl2, 100 мМ NaCl, 1 мМ DTT, осаждали ДНК из рестрикционной смеси изопропанолом и растворяли в воде. Затем ДНК гидролизовали эндонуклеазой рестрикции Sfr274I (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера Blue, содержащего 10 мМ Tris-HCl, pH 7.6, 10 мМ MgCl2, 1 мМ DTT. Рестрикционную смесь подвергали электрофорезу в 1.5%-ном агарозном геле, гель окрашивали раствором бромистого этидия в воде (1 мкг/мл), фрагмент размером 520 п.н. вырезали и выделяли из геля методом сорбции ДНК на силикагеле.
10 мкг ДНК плазмиды pMTL22-EPSPS-35S term подвергали гидролизу эндонуклеазой рестрикции BglII (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера Orange, содержащего 50 мМ Tris-HCl pH 7.6, 10 мМ MgCl2, 100 мМ NaCl, 1 мМ DTT, осаждали ДНК из рестрикционной смеси изопропанолом и растворяли в воде. Затем ДНК гидролизовали эндонуклеазой рестрикции Sfr274I (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера Blue, содержащего 10 мМ Tris-HCl, pH 7.6, 10 мМ MgCl2, 1 мМ DTT. Рестрикционную смесь подвергали электрофорезу в 1.0%-ном агарозном геле, гель окрашивали раствором бромистого этидия в воде (1 мкг/мл), фрагмент размером 4290 п.н. вырезали и выделяли из геля методом сорбции ДНК на силикагеле.
Далее смешивали 0,05 мкг Sfr274I-BglII фрагмента плазмиды pMTL22-EPSPS-35S term и 0,05 мкг Sfr274I-BglII амплификационного фрагмента ДНК tua prom, кодирующего промотор гена альфа-тубулина хлореллы, в 20 мкл лигазного буфера, содержащего 50 мМ Tris-HCl рН 7.8; 10 мМ MgCl2; 10 мМ DTT; 1 мМ ATP; 25 мкг/мл БСА. К смеси добавляли 10 е.а. ДНК-лигазы фага Т4 и инкубировали 6 часов при 4°С.
10 мкл лигазной смеси использовали для трансформации клеток E.coli штамма XL1-Blue. Клетки высевали на чашку с агаризованным LB-агаром с ампициллином (100 мкг/мл). Отбор колоний со вставкой проводили методом ПЦР с праймерами pMTLl, tua-pr-BglII. Продукты ПЦР анализировали при помощи электрофореза в 1,5%-ном агарозном геле. Наличие вставки подтверждали при помощи рестрикционного анализа эндонуклеазами рестрикции Sfr274I и BglII с последующим разделением продуктов гидролиза электрофорезом в 1,5%-ном агарозном геле.
1.5 Получение плазмиды pBinPLUS-ARS-EPSPS
10 мкг ДНК плазмиды pMTL22-casEPSPS, содержащей экспрессионную кассету гена EPSPS, подвергали гидролизу эндонуклеазами рестрикции Sfr274I (100 е.а.) и BamHI (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера Green, содержащего 10 мМ Tris-HCl, pH 7.6, 10 мМ MgCl2, 50 мМ NaCl, 1 мМ DTT. Рестрикционную смесь подвергали электрофорезу в 1.0%-ном агарозном геле, гель окрашивали раствором бромистого этидия в воде (1 мкг/мл), фрагмент размером 2380 п.н. вырезали и выделяли из геля методом сорбции ДНК на силикагеле.
10 мкг ДНК бинарного вектора pBinPLUS-ARS подвергали гидролизу эндонуклеазой рестрикции Sfr274I (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера Blue, содержащего 10 мМ Tris-HCl, pH 7.6, 10 мМ MgCl2, 1 мМ DTT, осаждали ДНК из рестрикционной смеси изопропанолом и растворяли в воде. Затем ДНК гидролизовали эндонуклеазой рестрикции Sail (100 е.а.) в течение 2 часов при 37°C в 50 мкл буфера Orange, содержащего 50 мМ Tris-HCl pH 7.6, 10 мМ MgCl2, 100 мМ NaCl, 1 мМ DTT. Рестрикционную смесь подвергали электрофорезу в 1.0%-ном агарозном геле, гель окрашивали раствором бромистого этидия в воде (1 мкг/мл), фрагмент размером 12450 п.н. вырезали и выделяли из геля методом сорбции ДНК на силикагеле.
Далее смешивали 0,05 мкг Sfr274I-BamHI фрагмента плазмиды pMTL22-casEPSPS и 0,05 мкг Sfr274I-SalI фрагмента ДНК бинарного вектора pBinPLUS-ARS в 20 мкл лигазного буфера, содержащего 50 мМ Tris-HCl рН 7.8; 10 мМ MgCl2; 10 мМ DTT; 1 мМ АТР; 25 мкг/мл БСА. К смеси добавляли 10 е.а. ДНК-лигазы фага Т4 и инкубировали 6 часов при 4°C.
10 мкл лигазной смеси использовали для трансформации клеток E.coli штамма XL1-Blue, клетки высевали на чашку с агаризованным LB-агаром с ампициллином (100 мкг/мл). Отбор колоний со вставкой проводили методом ПЦР с праймерами CaMV PrU3 и NOST2. Продукты ПЦР анализировали при помощи электрофореза в 1,5%-ном агарозном геле. Наличие вставки подтверждали при помощи рестрикционного анализа эндонуклеазой рестрикции BamHI с последующим разделением продуктов гидролиза электрофорезом в 1,5%-ном агарозном геле.
Пример 2.
Получение трансгенной микроводоросли рода хлорелла, устойчивой к глифосфату
Одноклеточную микроводоросль Chlorella vulgaris выращивали при 25°C в следующем режиме: 18 часов свет интенсивностью около 3000 Люм, 6 часов без света в среде Ф/2 медиум, содержащей антибиотики хлорамфеникол (50 мкг/мл) и стрептомицин (50 мкг/мл) в течение 8 дней. Клеточную суспензию центрифугировали 5 мин при 1500 g, один раз промывали 25 мМ фосфатным буфером pH=6 и ресуспендировали в фосфатном буфере, содержащем 0,6 М сорбитол, 0,6 М маннитол, 4% целлюлазу, 2% мацеразы и 50 единиц пектиназы. Клетки инкубировали в течение 16 часов в темноте при 25°C с качанием. Таким образом пролучили хлоропласты для последующей трансформации.
Трансформация
Полученные хлоропласты центрифугировали 5 мин при 400 g, осадок один раз промыли, затем ресуспендировали в среде Ф/2 медиум, содержащей 0,6 М сорбитол, 0,6 М маннитол.
0,4 мл клеточной суспензии, содержащей 107-108 протопластов, переносили в пробирку с 5 мкг плазмиды pBinPLUS-ARS-EPSPS и 25 мкг тимусной ДНК теленка и в пробирку с 25 мкг тимусной ДНК теленка. После 15 мин инкубирования при комнатной температуре добавляли 200 мкл PNC (40% ПЭГ 4 000, 0,8 хлорида натрия, 50 мМ хлорида кальция) и плавно перемешивали в течение 30 мин. Затем добавляли 0,6 мл среды Ф/2 медиум, содержащей 0,6 М сорбитол, 0,6 М маннитол, 1% глюкозы, 1% дрожжевого экстракта. Инкубировали при 25°C в темноте для регенерации клеток. Затем трансформированные клетки выращивали, как описано выше, в среде Ф/2 медиум, содержащей антибиотики хлорамфеникол (50 мкг/мл), стрептомицин (50 мкг/мл) и глифосат (50 мкг/мл) в течение 8 дней.
Интенсивность роста определяли спектрофотометрически при длине волны 550 нм. Одноклеточная микроводоросль Chlorella vulgaris, содержащая плазмиду pBinPLUS-ARS-EPSPS показала оптическую плотность клеточной суспензии 1,6-1,8 ОЕ, тогда как клеточная суспензия не трансгенной хлореллы показала оптическую плотность 0,1-0,2 ОЕ.
Использование рекомбинантной плазмидной ДНК pBinPLUS-ARS-EPSPS позволит получать трансгенные микроводоросли рода хлорелла, устойчивые к глифосату.



Изобретение относится к области биотехнологии и представляет собой рекомбинантную плазмидную ДНК pBinPLUS-ARS-EPSPS размером 14,8 т.п.н., обеспечивающую экспрессию гена агробактериальной 5-енолпирувил-шикимат-3-фосфат-синтетазы под управлением промотора гена альфа-тубулина хлореллы в трансгенных микроводорослях рода хлорелла, состоящую из следующих элементов: ДНК векторной плазмиды pBinPLUS-ARS размером 12440 п.н., HindIII-BglII фрагмента ДНК размером 500 п.н., содержащего промотор гена альфа-тубулина хлореллы, BglII-XbaI фрагмента ДНК размером 1610 п.н., содержащего кодирующую последовательность гена EPSPS с сигнальным пептидом, XbaI-HindIII фрагмента ДНК размером 230 п.н., содержащего 35S терминатор вируса мозаики цветной капусты. Использование рекомбинантной плазмидной ДНК pBinPLUS-ARS-EPSPS позволит получать трансгенные микроводоросли рода хлорелла, устойчивые к глифосату. 3 ил., 2 табл.
Рекомбинантная плазмидная ДНК pBinPLUS-ARS-EPSPS, обеспечивающая экспрессию гена агробактериальной 5-енолпирувил-шикимат-3-фосфат-синтетазы под управлением промотора гена альфа-тубулина хлореллы в трансгенных микроводорослях рода хлорелла, размером 14,8 т.п.н., состоящая из следующих элементов:
ДНК векторной плазмиды pBinPLUS-ARS размером 12440 п.н.,
HindIII-BglII фрагмента ДНК размером 500 п.н., содержащего промотор гена альфа-тубулина хлореллы,
BglII-XbaI фрагмента ДНК размером 1610 п.н., содержащего кодирующую последовательность гена EPSPS с сигнальным пептидом,
XbaI-HindIII фрагмента ДНК размером 230 п.н., содержащего 35S терминатор вируса мозаики цветной капусты.
| US 4940835, 10.07.1990 | |||
| BELKNAP W, et.al | |||
| pBINPLUS/ARS: an improved plant transformation vector based on pBINPLUS | |||
| Biotechniques | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| XIE L.X | |||
| et.al | |||
| Construction of a vector conferring herbicide and pest resistance in tobacco plant, Sheng Wu Gong Cheng Xue Bao | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| УНИВЕРСАЛЬНЫЙ ВЕКТОР ДЛЯ СТАБИЛЬНОЙ ТРАНСФОРМАЦИИ ХЛОРОПЛАСТНОГО ГЕНОМА, СПОСОБ СТАБИЛЬНОЙ ТРАНСФОРМАЦИИ РАСТЕНИЯ-МИШЕНИ, СПОСОБ ПРИДАНИЯ УСТОЙЧИВОСТИ К ГЕРБИЦИДУ РАСТЕНИЮ-МИШЕНИ, СПОСОБ ОПРЕДЕЛЕНИЯ ТРАНСФОРМАЦИИ ХЛОРОПЛАСТА И СТАБИЛЬНО ТРАНСФОРМИРОВАННЫЙ ХЛОРОПЛАСТНЫЙ ГЕНОМ | 1998 |
|
RU2238324C2 |

