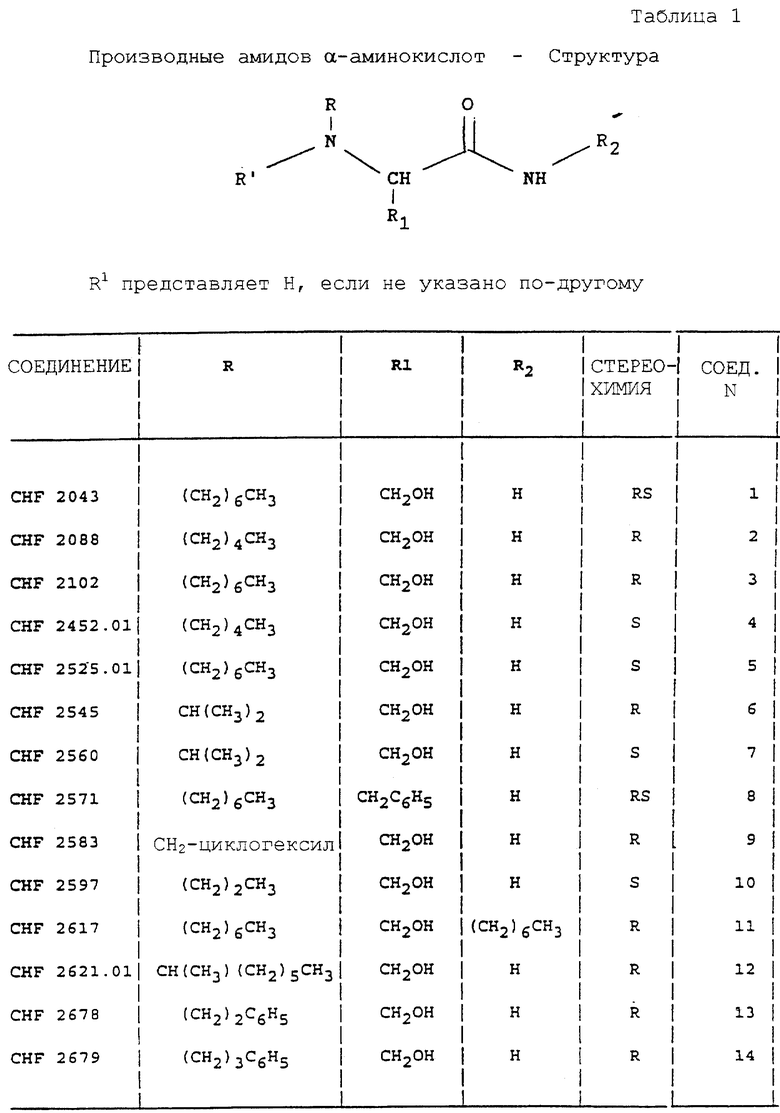
Изобретение относится к амидам α-аминокислот, способам их получения и содержащим их фармацевтическим композициям.
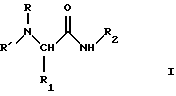
Более точно, данное изобретение относится к производным серинамида, глицинамида, аланинамида и фенилаланинамида общей формулы (I):
где R представляет линейный или разветвленный алкил; циклоалкилалкил; арилалкил или фенилалкил, необязательно замещенный в кольце алкилом, галогеном или галогеналкилом; конденсированный или неконденсированный арил, необязательно замещенный алкилом, алкокси-группой, галогеном или галогеналкилом;
R' представляет водород; алкил; фенил; фенилалкил;
R1 представляет необязательно ацилированный C1-C4 гидроксиалкил или фенилалкил, если R представляет алкил, циклоалкил, арилалкил;
R1 представляет водород, C1-C4 алкил, необязательно ацилированный C1-C4 гидроксиалкил или фенилалкил, если R представляет арил;
R2 представляет водород, алкил, фенил, фенилалкил.
Если не указано иначе, алкильная группа предпочтительно является C1-C10 алкильной группой, такой как метил, этил, н-пропил, изопропил, н-бутип, изобутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил, 2-этилпентил, 1-этилгептил, 1-метилоктил, 4-гептил.
Циклоалкилалкильная группа предпочтительно является группой, имеющей в алкильной части от 1 до 3 атомов углерода и в циклоалкильной части от 3 до 7 атомов углерода, такой как циклопропилметил, циклопентилметил, циклогексилметил, 1-(5- норборниленил)этил.
Необязательно замещенная арилалкильная или фенилалкильная группа предпочтительно является 2-нафталенилметилом, бензилом, фенетилом, фенилпропилом, фенилбутилом, 3-(4-метилфенил)пропилом, 3-(4-фторфенил)пропилом), 3-(4-хлорфенил)пропилом, 3-(4-трифторметилфенил)пропилом, 3-фенил-1-метилпропилом, 2-фенил-1-метилэтилом, 3-фенил-3-метилпропилом, 1-фенилэтилом.
Конденсированная и неконденсированная арильная группа предпочтительно является 1,2,3,4-тетрагидро-2-нафталенилом, 2-инданилом, необязательно замещенным одной (или более) алкокси-группой, галогеном или галогеналкилом.
Ацилированная C1-C4 гидроксиалкильная группа предпочтительно является ацетоксиалкильной, пропаноилоксиалкильной, 2-метилпропаноилоксиалкильной, бензоилоксиалкильной группой.
Предпочтительным классом соединений является класс, в котором:
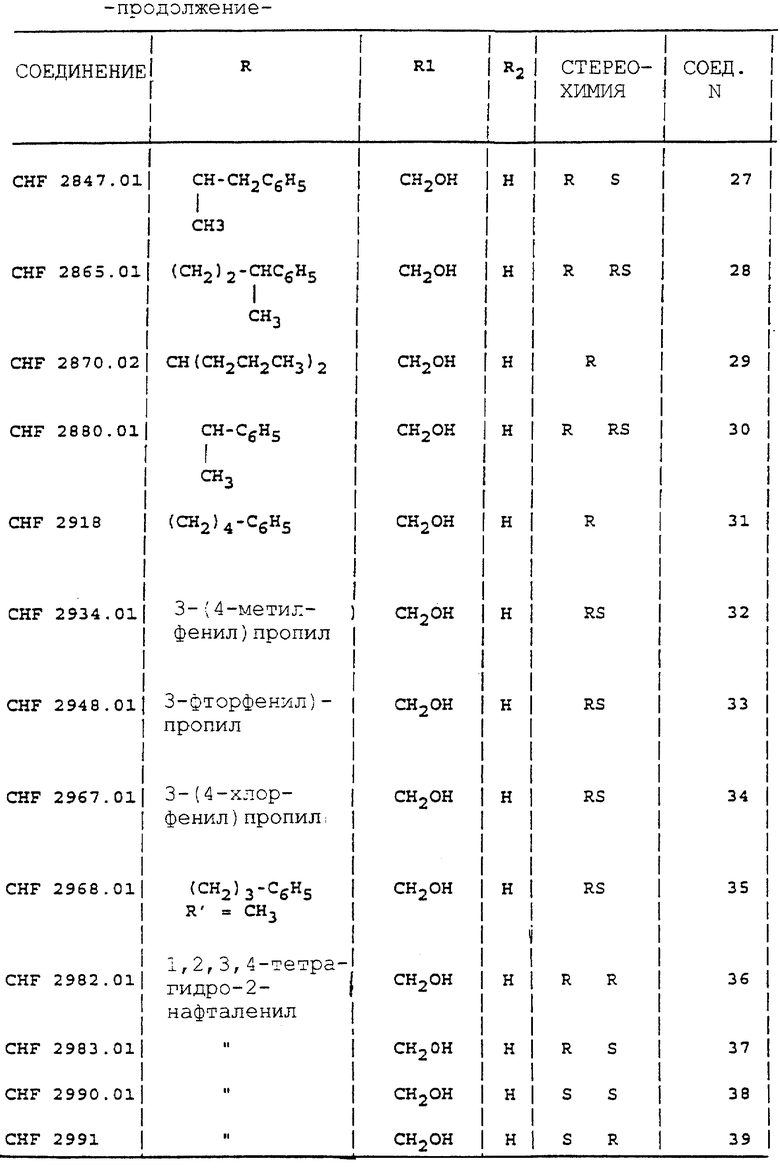
R1 представляет CH2ОН; R представляет C3-C10 алкил, C2-C4 фенилалкил, 1,2,3,4-тетрагидро-2-нафталенил или 2-инданил, необязательно замещенный алкилом, алкокси-группой, галогеном или галогеналкилом; R' представляет водород или метил, и R2 представляет водород или метил.
Особо предпочтительным подклассом соединений является тот, где R1 представляет CH2OH; R представляет фенил-(C2-C3)-алкил, или 1,2,3,4-тетрагидро-2-нафталенил, или 2-инданил, необязательно замещенный одной (или более) алкокси-группой, галогеном, галогеналкилом; R' и R2 представляют водород.
Вторым классом предпочтительных соединений является класс, где R1 представляет водород или метил; R представляет 1,2,3,4-тетрагидро-2-нафталенил или 2-инданил, необязательно замещенный одной (или более) алкокси-группой, галогеном, галогеналкилом; R' и R2 представляют водород.
Соединения данного изобретения могут находиться в форме солей присоединения органических или неорганических кислот.
Кроме того, они могут иметь один или более асимметрических атомов углерода, следовательно, их можно применять как в виде смеси, содержащей несколько диастереоизомеров в любом соотношении, так и в виде рацемической смеси, содержащей пары энантиомеров в равных или различных соотношениях, и в виде оптически чистых соединений.
Соединения данного изобретения можно применять при лечении хронических нейродегенеративных заболеваний, таких как болезнь Альцгеймера, разные формы слабоумия, болезнь Паркинсона, болезнь Хантингдона (Huntingdon's disease), или острой нейродегенеративной недостаточности, такой как удар или повреждение головы; при лечении эпилепсии и депрессии.
Уровень техники
В патенте Великобритании 2048852 (Continental Pharma S.A.) раскрыты производные 2-аминоацетамида (в общем обозначенных как глицинамид), которые можно применять при лечении эпилепсии, дискинезии, такой как болезнь Паркинсона, при лечении нарушений памяти и, возможно, при лечении депрессии.
Некоторые из раскрытых соединений, вводимых перорально в дозе 10-100 мг/кг, демонстрируют противосудорожную активность у мышей против индуцированных бикукуллином тонических судорог.
Детально исследованы 2-н-пентиламиноацетамид (далее дано тривиальное название милацемид) и его гидрохлорид.
Милацемид применяют в качестве сравнительного соединения для тестирования фармакологической активности соединений настоящего изобретения.
В Европейском Патенте EP-B1-0400495 (Famitalia Carlo Erba) раскрыты N-фенилалкил-замещенные производные α-аминокарбоксамидов.
Примерами особо предпочтительных соединении являются N-фенилалкил-замещенные производные аминопропионамида (в особенности, аланинамида и серинамида) и аминоацетамида (глицинамида). Указанные соединения действуют на центральную нервную систему и могут быть использованы в качестве агентов против эпилепсии, против болезни Паркинсона, антинейродегенеративных агентов, антидепрессантов, гипнотических агентов и антиспастических агентов.
Активность соединений оценивают на мышах как противосудорожное действие против судорог, индуцированных бикукуллином или 3-меркаптопропионовой кислотой.
Соединения, описанные в Европейском Патенте EP-B1-0400495, являются также ингибиторами моноаминооксидазы (МАО).
В WO 94/22808 и WO 94/22809 (Pharmacia/Farmitalia Carlo Erba) раскрыты другие производные аминопропионамидов, действующие на центральную нервную систему, соответственно арилалкоксибензил- и арилалкиламинобензилзамещенные.
Одним из наиболее типичных соединений изобретения WO 94/22808 является FCE28245 с химическим названием 2-{4-[3-фенилпропил] оксибензил}-амино-3-гидроксипропанамид метансульфонат, заявленный как обладающий активностью в тесте на мышах с судорогами, вызванными электрошоком.
В PCT n. WO 95/17617 (Teva-Technion) и PCT n. WO 96/21640 (Teva-Lemmon) описаны производные 1- аминобензоциклоалканов, такие как 1-аминоинданы и 1- аминотетралины, которые можно применять при лечении болезни Паркинсона, слабоумия, эпилепсии и посттравматических заболеваний.
Однако некоторые из описанных соединений, такие как рацемический N-(2-ацетамидо)-1-аминоиндан и его оптически активные формы, N-(2- ацетамидо)-6-фтор-1-аминоиндан, N-(2-ацетамидо)-1-аминотетралин, N, N-ди-(2-ацетамидо)-1-аминоиндан и N-(2-пропионамидо)-1- аминоиндан, не очень эффективны в тесте на противосудорожную активность и, повидимому, не демонстрируют особо подходящий терапевтический индекс.
В настоящее время обнаружено, что амиды аминокислот общей формулы (I) отличаются более высокой эффективностью и/или лучшим фармакологическим профилем по сравнению с соединениями прототипов.
Соединения данного изобретения можно получить реакцией эфиров или амидов аминокислот формулы II
где R1 такой, как определено выше, и X представляет алкоксигруппу или группу NHR2, где R2 такой, как определено выше, с соединениями формулы III
где Y представляет атом кислорода или группу NH, тогда как R3 и R4, одинаковые или разные, представляют водород или вместе с атомом углерода, к которому они присоединены, образуют одну из определенных выше групп R или R', за исключением конденсированного арила
с получением соединений формулы IV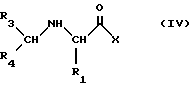
которые затем можно трансформировать в соединения формулы I с помощью одной (или более) из следующих реакций:
- если X представляет алкокси-группу реакция с амином формулы R2-NH2;
- N-алкилирование;
- ацилирование любой из гидроксильных групп, присутствующих в R1;
- образование соли и/или оптическое разделение;
- удаление всех защитных групп.
Первый вариант описанного выше процесса включает восстановительное аминирование соединения формулы II или его соли (обычно гидрохлорида), где X является алкокси-группой, например метокси-группой, соединением общей формулы III, где Y является кислородом, и последующую реакцию с амином формулы R2-NH2.
Восстановительное аминирование проводят в соответствии с обычными способами, используя стехиометрические количества или небольшой избыток реагентов, при температурах в диапазоне от 0 до 40oC и в органических растворителях, таких как спирты или ацетонитрил. В качестве восстановителя можно использовать гидрид, такой как цианборгидрид натрия, или водород в присутствии катализатора, такого как Pd на углероде.
Следующую реакцию аминирования проводят, используя избыток амина, в воде или органическом растворителе, в частности в метаноле или этаноле, при комнатной температуре или нагревании в химическом реакторе.
Второй вариант включает восстановительное аминирование соединения II, где X является группой R2-NH, в соответствии с уже описанной методикой.
И в заключение, третий вариант включает переиминирование соединения II, где X является группой R2-NH-, иминным соединением III (обычно фенилимином), где Y является NH. Эту реакцию проводят в органическом растворителе, например спирте, метиленхлориде или ацетонитриле при температуре от 0 до 40oC. Последующее восстановление полученного соединения проводят в органическом растворителе, обычно в спирте, таком как этанол или метанол, используя в качестве восстановителя гидрид, такой как боргидрид натрия, при температуре от 0 до 40oC.
Альтернативно, соединение 1 можно также получить конденсацией альфа-галогенэфира формулы V
где R1 такой, как определено выше, и предпочтительно H; W представляет атом галогена (обычно хлор или бром), и Rs представляет алкильную группу, с амином формулы VI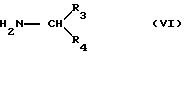
где R3 и R4 такие, как определено выше, и последующее аминирование амином R2-NH2. Конденсацию соединения V с соединением VI проводят в органическом растворителе, например ацетонитриле, спирте, диметилформамиде, при температуре от 40 до 140oC в присутствии агента, связывающего кислоту, например карбоната калия, и предпочтительно в присутствии каталитических количеств иодида калия. Затем проводят аминирование, как описано выше.
Можно также конденсировать альфа-галогенамид с амином VI.
Для рассмотренных терапевтических применений соединение 1 готовят в виде подходящих фармацевтических композиций, которые также являются предметом данного изобретения.
Указанные композиции обычно содержат от 1 до 1000 мг активного ингредиента, в частности от 10 до 100 мг, и их вводят один (или более) раз в день в зависимости от заболевания, фармакокинетики выбранного активного ингредиента и данных пациента (вес, пол, возраст).
Композиции получают, используя общеизвестные методики и наполнители, как описано, например, в книге Remington's Pharmaceutical Sciences Handbook, Mack.Pub., - N.Y., U.S.А., и вводят перорально, парентерально или ректально. Примеры препаратов включают таблетки, капсулы, сиропы, гранулы, стерильные растворы для инъекций или суспензии, суппозитории и подобное.
Приведенные далее примеры дополнительно иллюстрируют данное изобретение.
ПРИМЕРЫ
Пример 1
а) Получение гидрохлорида N-(3-фенилпропил)-L-серин метилового эфира
Гидрохлорид L-серин метилового эфира (0,9 моль, 14 г), триэтиламин (0,9 моль, 9,1 г) и 3-фенилпропиоальдегид (0,9 моль, 12,1 г) растворяют в сухом метаноле (370 мл) в сосуде Парра и гидрируют под давлением 3,164 кг/см2 (45 psi) в присутствии 10% Pd/C до тех пор, пока не прекращается абсорбция водорода.
Катализатор отфильтровывают, а фильтрат выпаривают в вакууме досуха. Полученное масло помещают в метиленхлорид (500 мл), органический раствор промывают водой и выпаривают в вакууме досуха, получая светло-желтое масло.
Продукт выделяют в виде гидрохлорида растворением в этиловом эфире (800 мл) и подкислением метанольным раствором соляной кислоты. Осадок фильтруют и сушат в вакууме при 45oC. Выход: 17,5 г (71%), Т.пл. = 126-129oC.
b) Получение гидрохлорида (-)-(S)-3-гидрокси-2-(3- фенилпропиламино)пропанамида (CHF 2803.01)
Продукт, полученный в а) (0,06 моль, 17 г), растворяют в воде (500 мл) и подщелачивают 10% водным карбонатом калия до pH=8. Свободное основание экстрагируют метиленхлоридом и выпаривают в вакууме досуха. Полученное светло-желтое масло (16,3 г) растворяют в метаноле (150 мл). Через раствор, охлажденный до -5oC, барботируют аммиак до концентрации ~15 М. Герметично закупоренная система реагирует в течение 5 дней при комнатной температуре (к. т. ), затем ее выпаривают в вакууме досуха. Продукт выделяют в виде гидрохлорида растворением в этаноле (40 мл), подкислением эфирным раствором HCl и осаждением этиловым эфиром (500 мл).
Отфильтровывают белое твердое вещество и сушат его в вакууме при 40oC. Выход: 7,6 г (46,5%), Т.пл.=153-155oC.
[α]589 (с=1, метанол) = -13,5
Пример 2
а) Получение N-(2-тетралил)-D-серин метилового эфира
Цианборгидрид натрия (0,07 моль, 4,5 г) добавляют к раствору β-тетралона (0,068 моль, 10,5 г) и гидрохлорида D-серин метилового эфира (0,07 моль, 11 г) в смеси 10/1 этанол/метанол (550 мл). Смесь реагирует при комнатной температуре в течение 24 часов, смесь выпаривают в вакууме досуха, растворяют в воде и экстрагируют этилацетатом (2х500 мл). Объединенные органические фазы экстрагируют IN HCl (2х300 мл). Водные фазы подщелачивают бикарбонатом натрия и экстрагируют этилацетатом (3х200 мл). Объединенные органические фазы выпаривают в вакууме досуха, получая продукт в виде желтого масла. Выход 12 г (72%).
b) Получение (R)-3-гидрокси-2-(1,2,3,4-тетрагидронафтален-2-(R,S)-иламино)пропанамида (CHF 2818)
Через раствор N-(2-тетралил)-D-серин метилового эфира (0,048 моль, 12 г) в метаноле (150 мл) барботируют аммиак при 0oC до концентрации ~15 М. Герметично закупоренная система реагирует в течение 120 часов при комнатной температуре. Раствор выпаривают в вакууме досуха, а оставшееся масло отверждают растиранием в петролейном эфире.
Выход: 9 г (80%), Т.пл. = 104-115oC.
с) Разделение диастереомеров CHF 2818
Получение 3-гидрокси-2-(R)-(1,2,3,4-тетрагидронафтален-2-(S)-иламино)пропанамида (CHF 2983)
2-(R)-(1,2,3,4-тетрагидро-2-(R,S)-нафталениламино)-3- гидроксипропанамид (0,038 моль, 8,8 г) кристаллизуют в этилацетате (200 мл) и полученное твердое вещество дважды перекристаллизовывают из этилацетата (200 мл) и окончательно из этанола (50 мл). Белое кристаллическое вещество сушат в вакууме при 45oC. Выход: 1,9 г (43%, Т.пл. = 142-145oC.
[α]589 (с = 1, метанол) = -105,1
Пример 3
а) Получение 3-(4-метилфенил)пропаноилхлорида
3-(4-Метилфенил)пропановую кислоту (0,055 моль, 9 г) растворяют в тионилхлориде (1,008 моль, 120 г). Смесь перемешивают в течение 30 минут при комнатной температуре, затем кипятят с обратным холодильником в течение 30 минут, выпаривают в вакууме до масла и извлекают толуолом и гексаном, выпаривая каждый раз досуха.
Выход: 12,4 г.
b) Получение 3-(4-метилфенил)пропаналя
К раствору трифенилфосфина (0,117 моль, 30,8 г) в ацетоне (200 мл) добавляют под током азота при комнатной температуре бис-(трифенилфосфин)-тетрагидроборат Cu (1) (0,067 моль, 40,69 г), затем добавляют по каплям за 45 минут 3-(4-метилфенил)пропаноилхлорид (0,055 моль, 10 г), растворенный в ацетоне (85 мл). Смесь перемешивают при комнатной температуре в атмосфере азота в течение 1 часа. Осажденное твердое вещество отфильтровывают, промывают ацетоном, а фильтрат выпаривают в вакууме. Остаток растворяют в хлороформе (340 мл), добавляют хлорид меди (I) (0,135 моль, 13,38 г) и перемешивают под током азота в течение 1 часа при комнатной температуре. Смесь фильтруют через целит, фильтрат выпаривают досуха, а полученный остаток извлекают этиловым эфиром и петролейным эфиром, фильтруют и выпаривают в вакууме, получая масло.
Выход: 6,7 г (83%).
с) Получение гидрохлорида метилового эфира 3-гидрокси-2-(3-(4-метилфенил)пропиламино)пропановой кислоты
2% Натрий в метаноле (0,045 моль, 51,7 мл) добавляют к гидрохлориду метилового эфира D,L-серина (0,045 моль, 7 г), растворяют в метаноле (70 мл), получая свободное основание.
Образовавшийся хлорид натрия осаждают этиловым эфиром (150 мл) и отфильтровывают. Фильтрат выпаривают в вакууме досуха. Полученный остаток растворяют в метаноле (450 мл), добавляют 3-(4-метилфенилпропаналь) (0,045 моль, 6,7 г) и доводят pH до 6 при помощи уксусной кислоты, добавляют цианборгидрид натрия (0,048 моль, 3 г) и смесь оставляют реагировать при комнатной температуре в течение 24 часов. Смесь подкисляют HCl в метаноле, выпаривают в вакууме досуха, извлекают метиленхлоридом (600 мл), затем подщелачивают триэтиламином и промывают водой (3 х 500 мл). Органическую фазу выпаривают в вакууме досуха, а продукт выделяют в виде гидрохлорида, извлекая его этиловым эфиром (400 мл) и подкисляя эфирным раствором HCl. Осажденное белое твердое вещество сушат в вакууме при 30oC. Выход: 8,8 г (68%).
d) Получение гидрохлорида 3-гидрокси-2-(3-(4-метилфенил)- пропиламино)пропанамида (CHF 2934.01)
Гидрохлорид метилового эфира 3-гидрокси-2-(3-(4-метилфенил)пропиламино)пропановой кислоты (0,03 моль, 8,6 г) растворяют в воде (500 мл), подщелачивают 10% водным карбонатом калия до pH = 8. Свободное основание экстрагируют метиленхлоридом и выпаривают в вакууме досуха. Полученное светло-желтое масло (16,3 г) растворяют в метаноле (150 мл). Через этот раствор, охлажденный до -5oC, барботируют аммиак до концентрации ~15 М. Герметически закупоренная система реагирует в течение 5 дней при комнатной температуре, затем ее выпаривают в вакууме досуха. Продукт выделяют в виде гидрохлорида растворением в этаноле (40 мл), подкислением раствором HCl в эфире и осаждением этиловым эфиром (500 мл). Белое твердое вещество отфильтровывают и сушат в вакууме при 40oC.
Выход: 4,9 г (60%), Т.пл.=173-176oC.
Пример 4
а) Получение 4-фенилбутаноилхлорида
4-Фенил-масляную кислоту (0,83 моль, 13,57 г) добавляют к тионилхлориду (0,114 моль, 8,27 мл) и нагревают для растворения твердого вещества. Смесь перемешивают в течение 30 минут при комнатной температуре, затем кипятят с обратным холодильником в течение 10 минут, окончательно выделяют продукт, как в примере За).
Получают 100% выход (0,083 моль, 15,09 г).
b) Получение 4-фенилбутаналя
Получают по методике примера 3b), используя в качестве исходного реагента 4-фенилбутаноилхлорид (0,083 моль, 15,09 г) и получая 11 г продукта.
с) Получение гидрохлорида метилового эфира (R)-3-гидрокси-2-(4-фенилбутиламино)пропановой кислоты
2% Натрий в метаноле (0,052 моль, 59,8 мл) добавляют к гидрохлориду метилового эфира D-серина (0,052 моль, 8,16 г), растворяют в метаноле (81,6 мл) для выделения основания. Образовавшийся хлорид натрия осаждают этиловым эфиром (163 мл) и отфильтровывают. Фильтрат выпаривают в вакууме досуха. Полученный остаток растворяют в метаноле (150 мл), добавляют 4-фенилбутаналь (0,051 моль, 10,69 г) и доводят pH до 6 уксусной кислотой, добавляют цианборгидрид натрия (0,055 моль, 3,62 г) и оставляют реагировать при комнатной температуре в течение 24 часов. Смесь подкисляют раствором HCl в метаноле, выпаривают в вакууме досуха, извлекают метиленхлоридом (600 мл) и 1 М раствором бикарбоната натрия. Фазы разделяют, водную фазу снова экстрагируют метиленхлоридом (3х200 мл), затем объединенные органические фазы промывают водой и выпаривают в вакууме досуха. Продукт выделяют в виде гидрохлорида, извлекая его этиловым эфиром (180 мл) и подкисляя раствором HCl в эфире. Осажденное белое твердое вещество сушат в вакууме при 30oC.
Выход: 5,83 г (40%).
d) Получение (R)-3-гидрокси-2-(4-фенилбутиламино)пропанамида
Гидрохлорид метилового эфира (R)-3-гидрокси-2-(4-фенилбутиламино)пропановой кислоты (0,02 моль, 5,83 г) растворяют в воде (250 мл) и подщелачивают 10% водным карбонатом натрия. Свободное основание экстрагируют метиленхлоридом (3х200 мл) и выпаривают в вакууме досуха. Полученное светло-желтое масло растворяют в метаноле (150 мл). Через этот раствор, охлажденный до -5oC, барботируют аммиак до концентрации ~15 М. Герметично закупоренная система реагирует в течение 5 дней при комнатной температуре, затем ее выпаривают в вакууме досуха, получая вязкое масло. Твердый продукт получают, извлекая его этиловым эфиром (25 мл) и осаждая гексаном (400 мл). Белое твердое вещество отфильтровывают и сушат в вакууме при 35oC.
Выход: 4,43 г (92%), Т.пл. = 59-61oC.
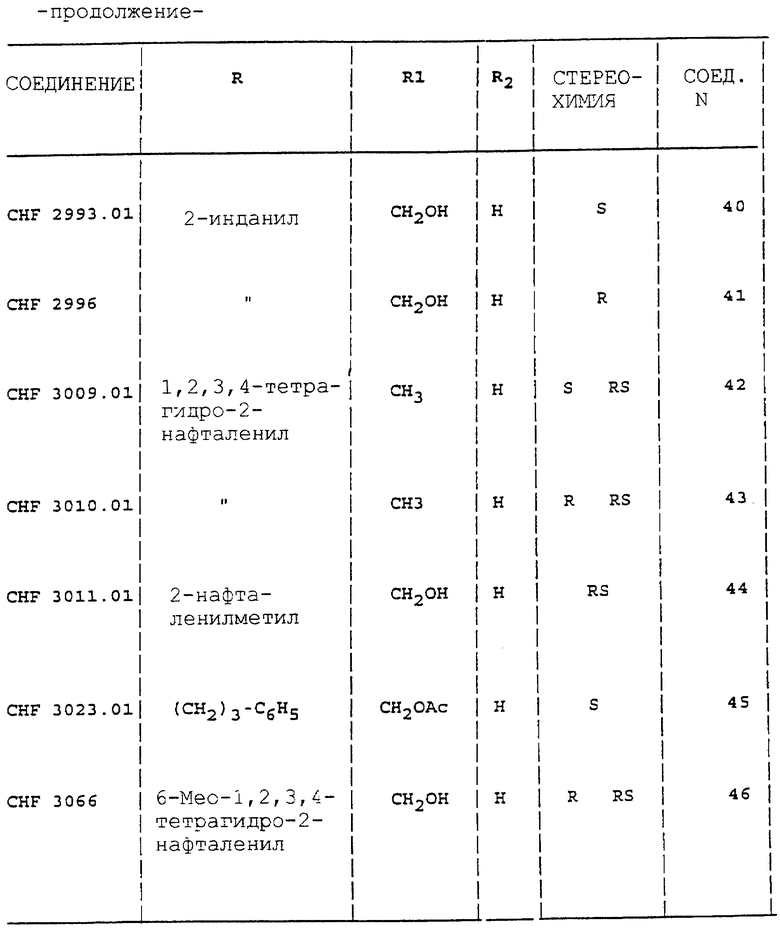
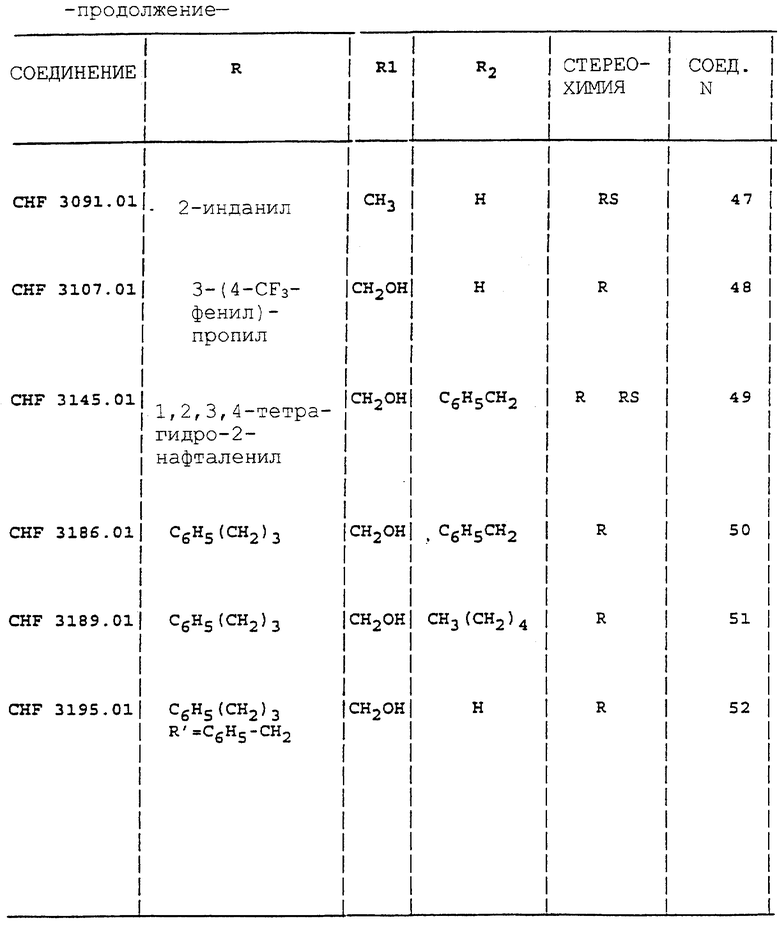
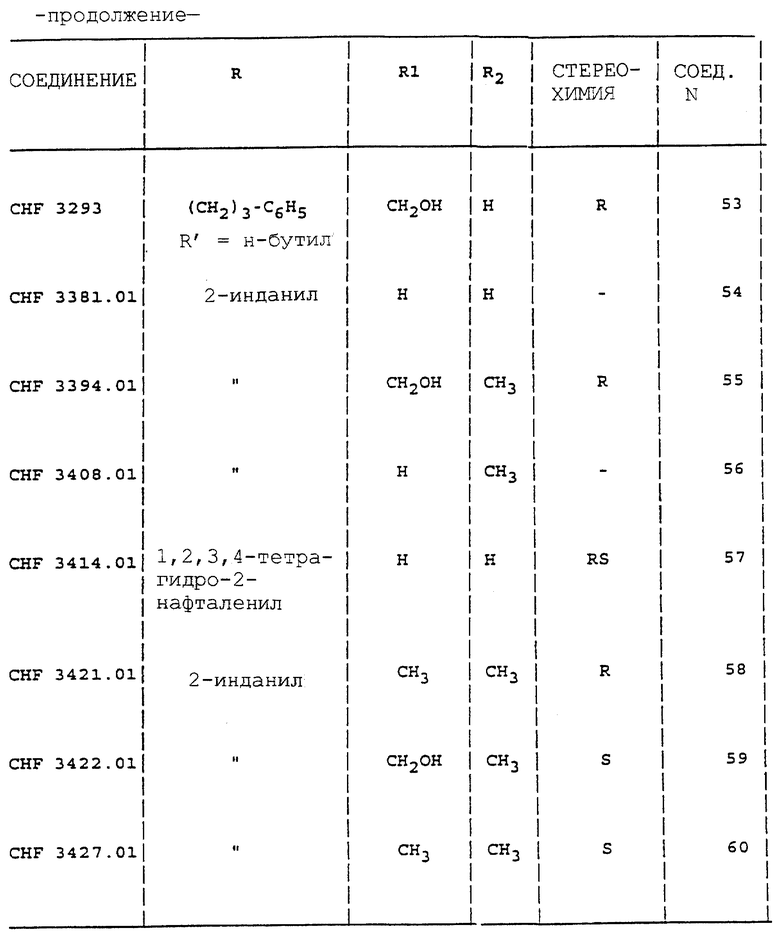
Аналогично способу, описанному в примерах с 1 по 4, получают соединения 1-5, 27, 28, 30, 33, 34, 38, 39, 42-44, 46-48, 52 и 53 таблицы 1.
Пример 5
Получение (R)-3-гидрокси-2-(3-фенилпропиламино)пропанамида (СРА 2679).
D-Серинамид (0,038 моль, 4 г) и 3-фенилпропаналь (0,038 моль, 5,1 г) растворяют в метаноле (400 мл) в сосуде Парра и гидрируют в присутствии 10% Pd/C (3 г) при 2,812 кг/см2 (40 psi) до тех пор, пока не прекращается абсорбция водорода. Катализатор отфильтровывают, а раствор выпаривают в вакууме досуха, извлекают остаток этилацетатом (300 мл) и промывают водой (2х200 мл). Органическую фазу сушат и растворяют в теплом этиловом эфире (300 мл), осаждая продукт в виде белого твердого вещества при медленном испарении растворителя при комнатной температуре.
Выход; 3,8 г (45%), Т. пл. = 76-78oC.
[α]589 (c=1, метанол) = +13,2
Аналогично способу, описанному в примере 5, получают соединения 6-13, 15-22, 24, 26, 49-51 таблицы 1.
Пример 6
Получение фосфата (R)-2-(4-гептиламино)-3-гидроксипропанамида (CHF 2870.03).
К раствору D-серинамида (0,01 моль, 1 г) и 4-гептанона (0,01 моль, 1,1 г) в метаноле (50 мл) добавляют 4 М раствор HCl в метаноле (0,0033 моль, 0,85 мл) и цианборгидрид натрия (0,005 моль, 0,33 г). Смесь реагирует при комнатной температуре в течение 10 дней, ее подкисляют раствором HCl в метаноле до pH 2 и выпаривают в вакууме досуха. Остаток извлекают водой (100 мл), промывают этиловым эфиром (100 мл), подщелачивают карбонатом натрия и экстрагируют хлороформом (3х100 мл). Полученное масло (1,3 г) растворяют в метаноле (50 мл) и обрабатывают 85% фосфорной кислотой (0,0065 моль, 0,75 г), затем выпаривают в вакууме досуха, получая продукт в виде очень легкой твердой пены.
Выход: 2 г (77%), Т.пл. = 150-156oC.
[α]589 (с=1, вода) = +1,9.
Пример 7
а) Получение метилового эфира 3-гидрокси-2-(N-метил-(3-фенилпропил)амино)пропановой кислоты
К раствору N-(3-фенилпропил)серин метилового эфира (6,4 г, 0,027 моль) в метаноле (150 мл) добавляют 10% Pd на углероде (0,7 г) и 40% муравьиный альдегид (3,0 мл, 0,04 моль), растворенный в метаноле (50 мл). Смесь перемешивают при комнатной температуре в атмосфере водорода при небольшом давлении (2,812 кг/см2 = 40 psi) до тех пор, пока не прекращается абсорбция. Смесь фильтруют, а фильтрат выпаривают в вакууме. Остаток извлекают этиловым эфиром (300 мл), промывают водой (2х200 мл), сушат над сульфатом натрия и выпаривают в вакууме.
b) Получение гидрохлорида 3-гидрокси-2-(N-метил-3- фенилпропил)аминопропанамида (CHF 2968.01)
Повторяют методику примера 3b.
Через раствор метилового эфира 3-гидрокси-2-(N-метил-(3- фенилпропиламино)пропановой кислоты (0,026 моль, 6,6 г) в метаноле барботируют аммиак при 0oC до концентрации ~15 М. Система реагирует в закрытом реакторе при температуре ~ 80oC в течение 120 часов. Раствор выпаривают в вакууме досуха, получая масло, из которого выделяют продукт способом жидкостной хроматографии при пониженном давлении. Полученное масло извлекают абсолютным этанолом и этилацетатом, затем подкисляют раствором HCl в эфире. Смесь перемешивают, добавляя этиловый эфир, затем осадок отфильтровывают и сушат его в вакууме при 40oC.
Выход: 3,5 г. Т.пл. = 112-114oC.
Аналогично способу, описанному в примере 7, получают следующие соединения:
гидрохлорид (R)-3-гидрокси-2-(N-метил-2-инданиламино) -пропанамида (CHF 3440.01; соединение N 63)
гидрохлорид (S)-3-гидрокси-2-(N-метил-2-инданиламино)- пропанамида (CHF 3462.01; соединение n.67).
Пример 8
а) Получение метил 2-(инданиламино)ацетата
Метиловый эфир глицина (0,053 моль, 6,64 г) растворяют в абсолютном этаноле (420 мл) и метаноле (42 мл), добавляют 2-инданон (0,053 моль, 7 г) и цианборгидрид натрия (0,058 моль, 3,7 г) при перемешивании в течение 40 минут. Эту смесь оставляют перемешиваться при комнатной температуре в течение ночи, выпаривают в вакууме досуха и остаток извлекают водой (500 мл) и этилацетатом (500 мл). Фазы разделяют и органическую фазу экстрагируют 0,1N раствором HCl (3х 200 мл). Доводят pH кислого раствора до 8,5-9 насыщенным раствором бикарбоната натрия и экстрагируют этилацетатом (3х250 мл). Органический раствор отделяют, сушат над сульфатом натрия и выпаривают в вакууме. Полученный продукт сушат в вакууме при комнатной температуре.
Выход: 5,8 г (53,4%).
b) Получение гидрохлорида 2-(2-инданиламино)ацетамида (CHF 3381.01)
Метил 2-(инданиламино) ацетат (0,028 моль, 5,8 г) растворяют в ~15 М растворе аммиака в метаноле (150 мл) и хранят в закрытой пробирке при комнатной температуре в течение нескольких дней. Раствор выпаривают в вакууме досуха, извлекают абсолютным этанолом (2х200 мл) и каждый раз выпаривают. Масло извлекают метанолом (30 мл) и подкисляют при перемешивании до кислого pH раствором HCl в сухом метаноле. Продукт осаждают, добавляя этиловый эфир, фильтруют и сушат в вакууме при 45oC.
Выход: 6,05г (94,6%), Т.пл. = 212-213oC.
Пример 9
Получение 2-(N-метил-2-инданиламино)ацетамида (CHF 3488)
2-(2-Инданиламино)ацетамид (0,016 моль, 3,00 г) растворяют в метаноле (60 мл) и добавляют при перемешивании карбонат калия (0,016 моль, 2,18 г). При комнатной температуре за 15 мин добавляют по каплям метилйодид (0,028 моль, 4,14 г). Эту смесь оставляют реагировать в течение 4 часов при комнатной температуре, затем выпаривают в вакууме. Полученное твердое вещество извлекают водой (100 мл), водный раствор экстрагируют этилацетатом (3х150 мл). Органический раствор сушат над сульфатом натрия, затем выпаривают в вакууме, получая масло, которое хроматографируют при среднем давлении на силикагеле (элюент: хлороформ/метанол = 90/10).
Выход: 1,31 г (40%), Т.пл. = 122-123oC
Пример 10
а) Получение метил (S)-3-гидрокси-2-(2-инданиламино)пропаноата
Гидрохлорид метилового эфира L-серина (0,05 моль, 7,78 г) растворяют в абсолютном этаноле (400 мл) и метаноле (40 мл), добавляют 2-инданон (0,05 моль, 6,74 г) и цианборгидрид натрия (0,55 моль, 3,64 г) при перемешивании в течение 30 минут. Эту смесь перемешивают при комнатной температуре в течение 5 часов, затем выпаривают в вакууме досуха, а остаток извлекают водой (150 мл) и этилацетатом (150 мл). Фазы разделяют и затем водную фазу экстрагируют этилацетатом (200 мл). Объединенные органические фазы экстрагируют 0,2 н. раствором HCl (2х200 мл). Водный раствор отделяют, доводят pH до 8 бикарбонатом натрия и экстрагируют этилацетатом (2х300 мл). Органический раствор отделяют, сушат над сульфатом натрия и выпаривают в вакууме. Полученный продукт сушат в вакууме при комнатной температуре.
Выход: 8,4 г (71,4%).
b) Получение гидрохлорида (S)-3-гидрокси-2- (2-инданиламино)-пропанамида (CHF 2993.01)
Метил (S)-3-гидрокси-2-(2-инданиламино)пропаноат (0,0357 моль, 8,4 г) растворяют в ~12 М метанольном растворе аммиака (150 мл) и хранят в закрытой пробирке при комнатной температуре в течение нескольких дней. Раствор выпаривают в вакууме досуха, получая масло, которое извлекают метанолом (2х250 мл), каждый раз выпаривая досуха. Полученный продукт (основание) извлекают этилацетатом (170 мл) и подкисляют при перемешивании до кислого pH 4,75 н. раствором HCl в сухом этилацетате. Продукт фильтруют, кристаллизуют из абсолютного этанола и сушат в вакууме при 40oC.
Выход: 6,7 г (72,8%), Т.пл. = 186-187oC,
[α]589 (c=1, метанол) = +16,6 (гидрохлорид)
[α]589 (с=1, метанол) = -24,6 (основание)
Известными в практике способами получают соль мезилат (CHF 2993.02).
Т.пл. = 180-183oC
[α]589 (с-1, метанол) = +13,4.
Аналогично способу, описанному в примере 10, получают соединения 41, 57, 61, 62, 69, 72, 75 и 76 таблицы 1.
Пример 11
Получение гидрохлорида (S)-2-(2-инданиламино)-3-(2- метилпропаноилокси)пропанамида (CHF 3542.01)
Гидрохлорид (S)-3-гидрокси-2-(2-инданиламино)пропанамида (CHF 2993.01, 0,006 моль, 1,6 г) растворяют в трифторуксусной кислоте (3,75 мл) и добавляют по каплям 2-метилпропаноилхлорид (0,022 моль, 2,3 г). Через 2 часа хранения при комнатной температуре раствор выпаривают в вакууме и полученное масло извлекают этиловым эфиром (100 мл). Продукт фильтруют, помещают в этиловый эфир (50 мл) и выделяют фильтрованием. Твердое вещество сушат в вакууме при комнатной температуре.
Выход: 1,8 г (90%), Т.пл. = 171-174oC.
Аналогично способу, описанному в примере 11, получают следующие соединения:
гидрохлорид (S)-3-ацетилокси-2-(3-фенилпропиламино)пропанамида (CHF 3023.01; соединение n. 45)
гидрохлорид (S)-3-ацетилокси-2-(2-инданиламино)пропанамида (CHF 3519.01; соединение n. 74)
[α]589 (с=1, метанол) = +29,8
гидрохлорид (S)-3-бензоилокси-2-(2-инданиламино)пропанамида (CHF 3548.01; соединение n. 78)
Пример 12
Получение гидрохлорида (S)-3-гидрокси-2-(2-инданиламино)- N-метилпропанамида (CHF 3422.01)
N-(2-инданил)-(S)-серин метиловый эфир (0,025 моль, 5,95 г) растворяют в закрытой пробирке в 8,03 М растворе метиламина в этаноле (155 мл). Через день раствор выпаривают в вакууме, извлекают полученное масло метанолом (3х100 мл), петролейным эфиром (фракция 40-70, 100 мл), каждый раз выпаривая. Полученное масло извлекают петролейным эфиром (250 мл), отверждают при перемешивании. Отфильтрованное твердое вещество (~5,42 г) растворяют в слегка теплом этилацетате (250 мл) и добавляют при перемешивании HCl в сухом этилацетате (3,5М) до заметно кислого pH. Продукт фильтруют, несколько раз промывают этиловым эфиром (150 мл) и сушат в вакуумной печи.
Выход: 5,33 г (77,9%), Т.пл. = 190,5-192oC.
[α]589 (с=1, ДМСО) = +18
[α]589 (c=1, метанол) = - 2
Аналогично способу, описанному в примере 12, получают соединения 55, 56, 58, 60, 64-66.
Пример 13
а) Получение 5,6-диметокси-2-гидроксиимино-инданона
5,6-Диметокси-1-инданон (0,078 моль, 15 г) растворяют в абсолютном этаноле (550 мл), термостатируют при 50oC и добавляют изоамилнитрит (0,086 моль, 11,9 мл) и концентрированную HCl (11,9 мл). Через несколько минут осаждается продукт. Реакцию оставляют еще на 3 часа при 50oC, затем охлаждают до комнатной температуры и отфильтровывают твердое вещество, промывают его абсолютным этанолом (50 мл) и этиловым эфиром (100 мл). Продукт сушат в вакуумной печи при комнатной температуре.
Выход: 16,3 г (94,4%).
b) Получение 5,6-диметокси-2-инданиламина
К раствору 5,6-диметокси-2-гидроксиимино-1-инданона (0,27 моль, 6 г) в ледяной уксусной кислоте (500 мл) добавляют 96% серную кислоту (3,3 мл) и 10% Pd/C (1,5 г). Смесь гидрируют в аппарате Парра (комнатная температура, 2,461 кг/см2=35 psi). Когда прекращается абсорбция водорода, отфильтровывают через целит катализатор, промывают метанолом (70 мл). Раствор выпаривают досуха, получая белое твердое вещество, которое растворяют в воде, затем экстрагируют этилацетатом (2х70 мл). Водный раствор подщелачивают 1М раствором гидроксида натрия до pH = 8-8,5. Продукт экстрагируют метиленхлоридом (2х70 мл). Органический раствор сушат над сульфатом натрия и выпаривают в вакууме, получая твердый продукт.
Выход: 4,6 г (88,5%).
с) Получение 2-(5,6-диметокси-2-инданиламино)ацетамида (CHF 3511)
5,6-Диметокси-2-инданиламин (0,024 моль, 4,6 г) и бикарбонат натрия (0,026 моль, 2,2 г) добавляют к раствору хлорацетамида) (0,024 моль, 2,2 г) в абсолютном этаноле (100 мл) и кипятят с обратным холодильником в течение 10 часов. Смесь фильтруют при комнатной температуре и выпаривают раствор досуха. Полученное масло хроматографируют на силикагеле при среднем давлении (элюент; метиленхлорид/метанол = 90/10).
Выход; 1,95 г (32,7%), Т. пл. = 135-138oC.
Аналогично способу, описанному в примере 13, получают следующие соединения:
гидрохлорид 2-(5-фтор-2-инданиламино)ацетамида (CHF 3480.01);
гидрохлорид 2-(5,6-дифтор-2-инданиламино)ацетамида (CHF 3480.01).
В приведенной далее таблице 1 сокращения и структурные формулы соединений данных примеров такие же, как используемые для других соединений, полученных способами, аналогичными описанным выше.
Противосудорожная активность
Соединения данного изобретения оценивали с помощью нескольких фармакологических тестов для определения их потенциальной противосудорожной активности. С этой целью соединения тестировали в тесте с максимальным электрошоком (maximal electrochock, MES). Эту модель широко используют для оценки эффективности противоэпилептических агентов против распространенных и отдельных припадков. Это исследование проводили на крысах и мышах, применяя экспериментальную методику, описанную в работе W.Losher и др. Epilepsy Res., 2 (1988), 145-181. Говоря кратко, при помощи электрического стимулятора через роговичные электроды подавали переменный ток 60 Гц (мышам 50 мА, крысам 150 мА) в течение 0,2 с. Противосудорожную активность соединения определяли через 60 минут и до 180 минут после его введения (перорально), рассчитывая ЕД50 для подавления тонического распрямления задних конечностей. Каждую дозу проверяли на группе из 10 животных и рассчитывали ЕД50 кривой доза-эффект согласно работе Litchfield и Wilcoxon (J. Pharmacol. Exp.Ther., 96 (1949), 99-113).
Только на крысиной модели MES оценивали период действия противосудорожной активности. Группы из 10 крыс обрабатывали дозами с равной активностью за 30, 60, 120, 240, 360 и 480 мин до электрошока. Затем оценивали максимум противосудорожной активности и продолжительность действия.
В другой серии экспериментов, проводимых на мышах, оценивали нейротоксичность данных соединений как меру ослабления двигательной функции, применяя тест с горизонтальным экраном (L.L. Coughenour и др., Pharmacol. Bioch. and Behav. , 6 (1977), 351-353). В этой модели мышей помещают индивидуально на край квадратного проволочного экрана, который закреплен горизонтально на металлическом стержне, который затем поворачивают на 180o так, что мышь оказывается под экраном. Наблюдают ослабление двигательной функции у ряда животных, которые падают с экрана или не могут вскарабкаться наверх. Затем рассчитывают среднюю неврологическую токсическую дозу (ТД50), как указано выше. Соотношение ТД50 и ЕД50 называется терапевтическим индексом (Т.И.) каждого соединения. Т.И. используют для того, чтобы показать полезное различие нейротоксичности и противоэпилептической активности. Больший Т. И. указывает на большее различие указанных выше активностей и хороший профиль соединения как противосудорожного агента.
На мышиной модели MES все исследуемые соединения показали значительную противосудорожную активность, выраженную в молях, чем активность милацемида (milacemide) и/или вальпроата натрия. Значения ЕД50 находятся в интервале от 1,2 до 0,5 ммоль/кг при соотношении активностей в интервале от 4,5 до 35 относительно милацемида и от 1 до 6,7 относительно вальпроата натрия.
Оценка периода действия противосудорожной активности показала, что некоторые соединения быстро абсорбируются с максимальным эффектом через 30 минут после введения, тогда как другие соединения оказывают замедленное действие с максимальным эффектом даже через три часа после введения.
Общей характеристикой для многих соединений является хорошая продолжительность действия при подавлении судорог, равном 50% или более, статистически значима в течение более 3 часов после введения.
Указанная продолжительность действия больше, чем у милацемида, продолжительность действия которого около одного часа.
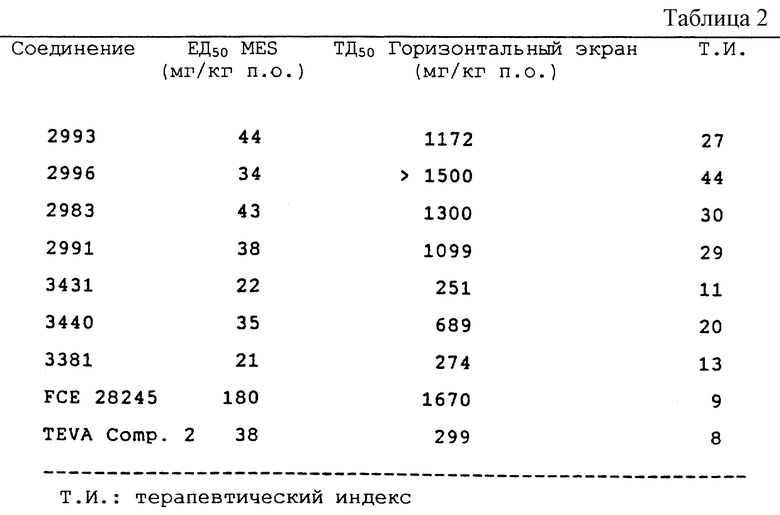
В приведенной далее таблице 2 показаны результаты MES теста на мышах для наиболее типичных соединений данного изобретения через 60 минут после введения.
Активность данных соединений сравнивали с активностью соединений двух прототипов: FCE 28245, прототипа новой серии производных серина, обладающих противосудорожной активностью, и соединения TEVA compound 2 (производное 1-аминоиндана), описанных в WO 94/22808 и в WO 95/18617, соответственно.
Все соединения вводят в виде солей HCl. Индивидуальные энантиомеры производных 2-аминоиндана и аминотетралина проявляют существенную противосудорожную активность. Производное (3)-гидроксил-2-аминоиндана (CHF 2993) и его энантиомер (R) CHF 2996 обладают одинаковой эффективностью в MES тесте. Последнее соединение дает также больший Т.И. (44) вследствие низкого значения нейротоксичности ( > 500 мг/кг перорально). Введение метильной группы в часть CHF 2993 дает в результате соединение CHF 3440 с такой же противосудорожной активностью, но с более высокой нейротоксичностью (ТД50 = 689). Наоборот, как производные (S) 2-(2-инданиламино)пропанамида, так и 2-(2- инданиламино)ацетамида CHF 3431 и CHF 3381 более активны в ингибировании MES, но более нейротоксичны, чем ранее упомянутые соединения. Хороший профиль противосудорожной активности наблюдают у производных 3-гидрокси-2-тетралинамина CHF 2983 (R,S энантиомер) и CHF 2991 (S,R энантиомер).
Все исследованные соединения примерно в 3-4 раза более активны, чем FCE 28245, химически 3-гидрокси-2-(4-(3-фенилпропилокси) бензиламино)пропанамид метансульфонат.
Также стоит отметить, что производное 2-аминоиндана CHF 3381 имеет более высокий Т. И., чем соединение 2 прототипа, известное из WO 95/18617, производное 1-аминоиндана, химическое название которого (S)-2-(инданиламино)ацетамид. Действительно, соединение 2-аминоиндана данного изобретения является более активным противосудорожным агентом, чем 1-аминоиндан прототипа. Из совместного рассмотрения этих результатов ясно, что класс соединений 2-аминоиндана данного изобретения демонстрирует меньшее снижение двигательной координации, чем соединение прототипа. В дополнение к MES тесту на мышах некоторые соединения оценивали также в MES тесте на крысах и на химической модели тонических судорог, индуцированных бикукуллином, согласно методике, описанной в работе Swinyard E.D. и др. Antiepileptic Drugs, 3-е изд. Raven Press, New York (1989). В этой модели мышей наблюдают в течение 30 минут после введения дозы бикукуллина (подкожно), которая вызывает тонические судороги у 97% животных. У животных, обработанных соединениями, в качестве конечной точки берут устранение компонента тонического вытягивания задних конечностей, полагая, таким образом, что исследуемое вещество обладает способностью предупреждать распространение приступов.
В качестве соединения сравнения используют FCE 26743 (S)-2-(4- (3-фторбензилокси)бензиламино)пропанамид, раскрытый в прототипе EP-B1-0400495.
Результаты представлены в таблице 3.
В MBS тесте на крысах соединения CHF 2993, CHF 2996 и FCE 26743 демонстрируют противосудорожную активность, близкую к активности, обнаруженной в аналогичном тесте на мышах. В этом тесте CHF 2983 является более активным и имеет ЕД50 19 мг/кг (перорально), ниже значения, указанного для MES-теста на мышах. Анализ кинетики фармакологических эффектов показывает, что CHF 2993 имеет большую продолжительность действия (6 часов). В любом случае продолжительность действия всех исследованных соединений больше, чем для соединения сравнения FCE 26743 (1 час). В случае бикукуллин-индуцированных судорог у мышей ЕД50 для CHF 2993, CHF 2983 и FCE 26743 составляет 65, 62 и 20 мг/кг (перорально) соответственно. Хотя эти значения больше, чем значения, полученные в MES тесте на мышах, но они все еще одного порядка с MES-противосудорожной активностью.
Рассматривая эти результаты вместе, можно сделать вывод, что описанные здесь соединения показывают существенную противосудорожную активность в MES-модели на мышах и на крысах, а также на модели бикукуллин-индуцированных судорог у мышей. Активность, показанная этими соединениями на мышах, почти такая же, как активность, обнаруженная для некоторых стандартных противоэпилептических лекарств, включая фенитоин, карбамазепин, и по крайней мере в четыре раза выше активности вальпроата натрия, соединений, для которых литературные данные показывают меньшие терапевтические индексы по сравнению с величинами, найденными в данном исследовании.
Соединения данного изобретения можно вводить в виде различных дозированных форм, например перорально в виде таблеток, капсул, таблеток с сахарным или пленочным покрытием, жидких растворов; ректально в виде суппозиториев; парентерально, например внутримышечно, или внутривенно, или путем инъекций или вливаний. Схему лечения различных клинических синдромов следует адаптировать к типу патологии, как обычно, принимая во внимание способ приема, форму, в которой вводят данное соединение, и возраст, вес и состояние пациента.
На практике терапевтически эффективное количество составляет примерно от 1 до 1000 мг, предпочтительно примерно от 10 до 300 мг.
Конечно, схемы приема лекарственных средств можно регулировать для обеспечения оптимальной терапевтической реакции.
Природа фармацевтических композиций, содержащих соединения данного изобретения в сочетании с фармацевтически приемлемыми носителями или разбавителями, конечно, будет зависеть от необходимого способа введения.
Композиции можно приготовлять общеизвестным способом с обычными ингредиентами. Например, соединения данного изобретения можно вводить в виде водных или масляных растворов или суспензий, таблеток, пилюль, желатиновых капсул, сиропов, капель или суппозиториев.
Таким образом, предпочтительные фармацевтические композиции для перорального введения, содержащие соединения данного изобретения, представляют собой таблетки, пилюли или желатиновые капсулы, включающие активное вещество вместе с разбавителями, такими как лактоза, декстроза, сахароза, маннит, сорбит, целлюлоза; смазывающими агентами, например диоксидом кремния, тальком, стеариновой кислотой, стеаратом магния или кальция и/или полиэтиленгликолями; или они могут также содержать связующие агенты, такие как крахмалы, желатин, метилцеллюлоза, карбоксиметилцеллюлоза, гуммиарабик, трагакант, поливинилпирролидон; разрыхлители, такие как крахмалы, альгиновая кислота, альгинаты, крахмал гликолат натрия; шипучие смеси; красители; подслащающие агенты; увлажняющие агенты, такие как лецитин, полисорбаты, лаурилсульфаты; и вообще нетоксические и фармакологически неактивные вещества, используемые в фармацевтических препаратах. Указанные фармацевтические препараты можно получать известными способами, например, смешиванием, гранулированием, таблетированием, покрытием сахаром или пленкой.
Жидкие дисперсии для перорального введения могут быть, например, сиропами, эмульсиями и суспензиями.
Сиропы могут содержать в качестве носителя, например, сахарозу или сахарозу с глицерином, и/или маннитом, и/или сорбитом. Суспензии и эмульсии могут содержать в качестве носителя, например, натуральную смолу, агар, альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт. Суспензии или растворы для внутримышечных инъекций могут содержать вместе с активным веществом фармацевтически пригодный носитель, например стерильную воду, оливковое масло, этилолеат, гликоли, например пропиленгликоль, и, если требуется, подходящее количество гидрохлорида лидокаина.
Растворы для внутривенных инъекций или вливаний могут содержать в качестве носителя, например, стерильную воду, или предпочтительно они могут быть в виде стерильных изотонических солевых растворов.
Суппозитории могут содержать вместе с активным соединением фармацевтически пригодный носитель, например масло какао, полиэтиленгликоль, поверхностно-активный жирнокислотный эфир полиоксиэтиленсорбитана или лецитин.


Изобретение относится к производным серинамида, глицинамида, аланинамида и фенилаланинамида формулы I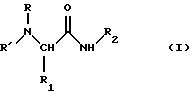
где R - 1,2,3,4-тетрагидро-2-нафталенил или 2-инданил, необязательно замещенные низшим алкилом, алкокси или галогеном; R' - водород, низший алкил, фенил (низший) алкил; R1 - водород, C1 - C4алкил, необязательно ацилированный C1 - C4 гидроксиалкил или фенил (низший) алкил; R2 - водород, C1 - C7 алкил, фенил (низший)алкил. Описан способ их получения из соответствующих эфиров или амидов аминокислот, также фармацевтическая композиция, включающая в качестве активного ингредиента соединения формулы I. Соединения I полезны для лечения неврологических заболеваний. 4 с. и 3 з.п.ф-лы, 3 табл.
где R представляет 1,2,3,4-тетрагидро-2-нафталенил или 2-инданил, необязательно замещенные низшим алкилом, алкокси или галогеном;
R' представляет водород, низший алкил, фенил(низший)алкил;
R1 представляет водород, С1-С4алкил, необязательно ацилированный С1-С4гидроксиалкил или фенил(низший)алкил;
R2 представляет водород, С1-С7алкил, фенил(низший)алкил.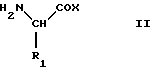
где R1 такой, как определено выше;
Х представляет алкоксигруппу или группу NHR2, где R2 такой, как определено выше,
с соединениями формулы III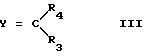
где Y представляет атом кислорода или группу NH, тогда как R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют одну из определенных выше групп R или R' с получением соединений формулы IV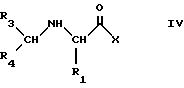
которые затем можно трансформировать в соединения формулы I с помощью одной или более из следующих реакций: если Х представляет алкоксигруппу, с помощью реакции с амином формулы R2-NH2; если Х представляет аминогруппу, с помощью реакции N-алкилирования; ацилирования любой из гидроксильных групп, присутствующих в R1, с последующим удалением всех защитных групп и выделением целевого продукта в виде соли и/или оптического разделения.
| Способ получения @ -аминоамидов | 1979 |
|
SU1220568A3 |
| Производные N-(3-пропаналкиламидо)- @ -аланина в качестве пенообразователей | 1985 |
|
SU1346636A1 |
| Пожарный двухцилиндровый насос | 0 |
|
SU90A1 |
| СПОСОБ АКУСТИЧЕСКОГО КАРОТАЖА ПРИСКВАЖИННОЙЗОНЫ | 0 |
|
SU330247A1 |
| Экономайзер | 0 |
|
SU94A1 |
| Экономайзер | 0 |
|
SU94A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| УСТРОЙСТВО для АВТОМАТИЧЕСКОГО РЕГУЛИРОВАНИЯ ГЛУБИНЫ ЗАСЫПКИ НА КОЛЕНОРЫЧАЖНЫХ ПРЕССАХ | 0 |
|
SU400495A1 |

