Изобретение относится к новому пептиду, проявляющему противоопухолевой активностью, более конкретно к пентапептиду, способам его получения и пептидным соединениями, проявляющим противоопухолевую активность.
В международной заявке 93/23424 описываются активные вещества на пептидной основе, которые обладают представляющими интерес противоопухолевыми активностями. Особенно хорошее действие оказывает пентапептид примера 234 указанной заявки, который отвечает следующей формуле: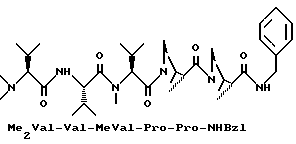
в которой Me2Val означает N,N-диметил-L-валин; MeVal означает N-метил-L-валин; Bzl означает бензильный остаток.
Пептид согласно указанной международной заявке можно получать твердофазным способом, исходя из пролина. При этом образуется активное вещество с небольшим выходом и в загрязненной форме. Требуется обязательная хроматографическая очистка. Твердофазный способ, кроме того, пригоден только для получения небольших количеств вещества. До сих пор не удалось получить вещество примера 234 международной заявки 93/23424 в кристаллической форме. Активное вещество находится в виде смолы. Вследствие этого затруднительно полное отделение остатков растворителя. Требуются дорогостоящие стадии очистки (распылительная сушка, сушка вымораживанием). Дальнейшая галеновая переработка вещества затруднительна. Для испытания и введения требуются большие количества вещества.
Задачей изобретения является получение вышеуказанного пентапептида в виде кристаллической соли, гидрохлорида.
Поставленная задача решается двумя способами получения гидрохлорида пентапептида формулы (I)
Me2Val-Val-MeVal-Pro-Pro-NHBzl • HCl, (I)
где Me означает метил, a Bzl - бензил,
первый из которых заключается в том, что соединение общей формулы (II)
Z-Val-Val-MeVal-Pro-OR1, (II)
R1 означает алкил с 1-5 атомами углерода, и Z означает бензилоксикарбонильную защитную группу, которая может быть замещена в фенильном кольце, подвергают снятию защитной группы Z на концевой аминогруппе, получаемое при этом соединение подвергают диметилированию на свободной концевой аминогруппе с последующим омылением алкоксильной группы-OR1 на концевой карбоксильной группе и получаемое при этом соединение формулы (V)
Me2Val-Val-MeVal-Pro-OH, (V)
где Me имеет вышеуказанное значение,
последовательно подвергают взаимодействию с пивалоилхлоридом и пролинбензиламидом с последующим переводом в гидрохлорид, а второй в том, что соединение общей формулы (II)
Z-Val-Val-MeVal-Pro-OR1, (II)
где R1 означает алкил с 1-5 атомами углерода, и Z означает бензилоксикарбонильную защитную группу, которая может быть замещена в фенильном кольце, подвергают омылению алкоксильной группы-OR1 на конце карбоксильной группы, получаемое при этом соединение формулы (IX)
Z-Val-Val-MeVal-Pro-OH, (IX)
где Me имеет вышеуказанное значение,
последовательно подвергают взаимодействию с пивалоилхлоридом и пролинбензиламидом и получаемое при этом соединение формулы (XI)
Z-Val-Val-MeVal-Pro-Pro-NHBzl, (XI)
где Me имеет вышеуказанное значение,
подвергают снятию защитной группы Z на концевой аминогруппе с последующим диметилированием свободной концевой аминогруппы и омылением алкоксильной группы -ОR1 на концевой карбоксильной группе и получаемое при этом соединение переводят в гидрохлорид.
Первый способ, который далее обозначается как "способ A", протекает по следующей реакционной схеме A: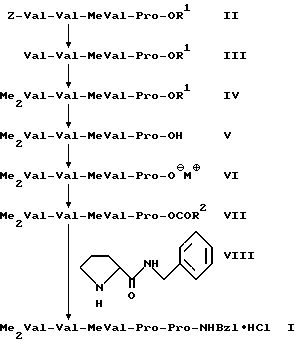
Приведенные в схеме A радикалы R1, R2, Z, Me и Bzl имеют вышеуказанные значения, M⊕ означает ион калия, ион натрия, ион лития или ион аммония, как триэтиламмоний.
Заместителями фенильного кольца бензилоксикарбонила могут быть галоген, алкил с 1-4 атомами углерода, алкоксил с 1-4 атомами углерода, ацилоксигруппа с 1-4 атомами углерода, или нитрогруппа, и в особенности хлор в положении 2, хлор в положении 3, хлор в положении 4, бром в положении 4, метоксигруппа в положении 4, ацетильная группа в положении 4, нитрогруппа в положении 2 и нитрогруппа в положении 4.
Сложный тетрапептидный эфир формулы (II) растворяют в пригодном растворителе, например в спирте, как метанол, этанол, изопропанол, бутанол; в простом эфире, как тетрагидрофуран, диоксан, метил-трет.-бутиловый эфир; в сложном эфире, как этилацетат; или в ледяной уксусной кислоте. После добавки пригодного катализатора, как, например, палладий-на-угле или платина-на-угле, при температурах в пределах от 0oC до 50oC, предпочтительно в пределах от 10oC до 30oC, пропускают водород. Введение водорода можно осуществлять при нормальном давлении или при повышенном давлении вплоть до 10 бар. Реакцию можно ускорять, если допускать известный ток отходящего газа. По окончании поглощения водорода добавляют 2-5 эквивалентов формальдегида в виде водного раствора или также в газообразной форме или в виде параформальдегида. Затем далее вводят водород при вышеописанных условиях. После этого катализатор отфильтровывают. Соединение формулы (IV) очищают путем кристаллизации в виде гидрохлорида из пригодного растворителя или смеси растворителей. При этом оказывается пригодной смесь изопропанола с метил-трет.-бутиловым эфиром. Следовые количества сложного Z-тетрапептидного эфира формулы (II), имеющиеся в соединении формулы (IV), можно удалять также с помощью экстрактивного способа разделения.
Омыление сложного эфира формулы (IV) осуществляют в пригодном растворителе, например в спирте, как метанол, этанол, изопропанол; в простом эфире, как метил-трет.-бутиловый эфир, тетрагидрофуран, диоксан; в углеводороде, как толуол, ксилол: или в хлорированном углеводороде, как 1,2-дихлорэтан, метиленхлорид, хлороформ, с добавкой воды или без добавки воды и с помощью пригодного основания, как гидроксид натрия, гидроксид калия, гидроксид лития. Расщепление сложного эфира можно осуществлять также с помощью кислот. В случае, где R1 означает трет.-бутил, для этой цели особенно пригодны трифторуксусная кислота и раствор хлороводорода в диоксане.
Полученную тетрапептидную кислоту формулы (V) далее нужно связывать с пролинбензиламидом формулы (VIII) для получения пентапептида формулы (I). При такого рода реакциях связывания легко протекает рацемизация. Поэтому G. Pettit и др. (J. Am. Chem. Soc. 113. 6692-6693 (1991)) для аналогичного связывания с тетрапептидной кислотой формулы (V) в качестве реагента связывания применяют диэтилфосфороцианидат. Диэтилфосфороцианидата в достаточно больших количествах в продаже нет. Вследствие этого метод требует дополнительных стадий способа с использованием ядовитых фосфорных и цианидных реагентов. Цианидсодержащие отходы вызывают проблемы в отношении загрязнения окружающей среды. Поэтому способ непригоден для технологического осуществления. Методом связывания пептида, который можно особенно просто реализовать в промышленном масштабе, является метод смешанного ангидрида (см., например, J. Meienhofer в "The Peptides, Analysis, Synthesis, Biology", том 1, Академик Пресс, Орландо, 1979, с. 264 -314). При этом кислоту формулы (V) депротонируют с помощью пригодного основания, например, третичного амина, как триэтиламин, N-метилморфолин, дициклогексилэтиламин, диизопропил-этиламин, с получением соединения формулы (VI). Сложные эфиры формулы (IV) также с помощью оснований, как гидроксид натрия, гидроксид калия, гидроксид лития, можно прямо превращать в соли формулы (VI). Соединения формулы (VI) путем введения во взаимодействие с хлорангидридом кислоты ClCOR1 превращают в смешанный ангидрид формулы (VII). Наряду с пивалоилхлоридом также можно применять другие хлорангидриды кислот, как, например, хлорангидрид 2-этилгексановой кислоты, этиловый эфир хлормуравьиной кислоты, метиловый эфир хлормуравьиной кислоты и изобутиловый эфир хлормуравьиной кислоты. Смешанные ангидриды очень сильно склонны к рацемизации (см., например, J. Meienhofer в "The Peptides", том 1, Академик Пресс, Орландо, 1979, с. 276 и последующие).
В настоящее время неожиданно оказалось, что тетрапептидную кислоту формулы (V) можно превращать по методу смешанного ангидрида полностью без рацемизации. Особенно хороших результатов достигают со смешанным ангидридом, который получают из кислоты формулы (V) и пивалоилхлорида. В противоположность более новым опубликованным результатам (N.L. Benoiton и др., Can. J. Chem., 65, 619-625 (1987)) взаимодействие с пивалоилхлоридом, в том, что касается селективности и выхода, дает лучшие результаты, чем взаимодействие со сложными эфирами хлормуравьиной кислоты. Получение смешанного ангидрида формулы (VII) и последующее связывание с пролинбензиламидом осуществляют при температурах от -20oC до +5oC в пригодном растворителе, как диоксан, N-метилпирролидон, тетрагидрофуран, толуол, метиленхлорид, диметилформамид. Вместо пролинбензиламида формулы (VIII) можно также применять пригодную соль этого соединения, как, например, гидросульфат, метилсульфонат, гидрохлорид или гидробромид. При этом тогда нужно добавлять дальнейший эквивалент основания, например, триэтиламина. После осуществленного связывания пептида и обычной экстрактивной обработки сырой продукт растворяют в пригодном растворителе, например в углеводороде, как толуол, ксилол; или простом эфире, как диэтиловый эфир, тетрагидрофуран, диоксан, метил-трет.- бутиловый эфир; кетоне, как ацетон, метилэтилкетон, диэтилкетон, циклогексанон; или в хлорированных растворителях, как метиленхлорид, хлороформ, 1,2-дихлорэтан. Путем введения газообразного хлороводорода или добавления раствора хлороводорода в пригодном растворителе, как, например, тетрагидрофуран, метанол, изопропанол, н-пентанол, диизопропиловый эфир, осаждают гидрохлорид формулы (I). Особенно пригодным при этом оказывается способ, при котором пентапептид в виде свободного основания сначала растворяют в метилэтилкетоне и затем добавляют раствор хлороводорода и изопропанола.
Второй способ, который далее обозначается как способ Б, протекает по следующей реакционной схеме Б: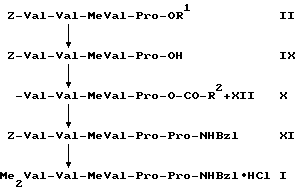
Омыление сложного эфира формулы (II), получение смешанного ангидрида формулы (X) и связывание пептида для получения соединения формулы (XI) осуществляют по аналогии с последовательностью синтеза IV---> V---> VI---> VII---> I. Отщепление защитной группы Z и диметилирование для получения соединения формулы (I) проводят по аналогии с превращением II---> III---> IV.
Также в случае варианта способа Б метод смешанного ангидрида протекает неожиданно без рацемизации. Вопреки опубликованным Беноитоном результатам, лучших выходов достигают со смешанным ангидридом, который получают из кислоты формулы (IX) и пивалоилхлорида.
Необходимое для получения пептида формулы (I) исходное соединение формулы (II) можно получать из Z-Val-O-CO-R2 (XII) и Val-MeVal-Pro-OR1 (XIII).
По предлагаемым способам A и Б активное вещество формулы (I) получают в кристаллической форме. Пептид можно очищать далее просто путем перекристаллизации. Обязательной хроматографической стадии очистки не требуется.
Изобретение относится также к следующим форпродуктам для получения соединения формулы (I):
Z-Val-Val-MeVal-Pro-OR1; (II)
Val-Val-MeVal-Pro-OR1; (III)
Me2Val- Val-MeVal-Pro-O-R1; (IV)
Z-Val-Val-MeVal-Pro-OH; (IX)
Z-Val-Val-MeVal-Pro-Pro-NHBzl, (XI)
в которых R1, R2 и Z имеют вышеуказанные значения.
Соединение формулы (I) является эффективным против твердых опухолей (опухоли легкого, груди, кишечника, мочевого и желчного пузырей, прямой кишки, матки, простаты), против лейкемии, лимфомы и других опухолевых заболеваний.
Противоопухолевая активность гидрохлорида пентапептида формулы (I) иллюстрируется следующими опытами.
Опыт 1 (ин витро)
Цитотоксичность определяли с помощью стандартной методологии, используемой при анализе приросших линий клеток, как, например, осуществляемый в микрокультурах тест с применением тетразолиевого соединения (тест МТТ). Детали этого опыта опубликованы (см. Alley, МС и др., Cancer Research 48, стр. 589-601, 1988). Экспоненциально растущие культуры клеток рака, таких как клетки НТ-29 рака толстой кишки или LX-1 рака легких использовали для приготовления культур, выращиваемых в микротитровальных пластинках. Клетки высеивали в количестве по 5000-20,000 клеток на ячейку в 96- ячейковую пластинку (в 150 мкл среды) и выращивали в течение ночи при температуре 37oC. Добавляли исследуемое соединение в 10-кратном разбавлении, причем концентрации варьировали в пределах от 10-4 М до 10-10 М. Затем клетки инкубировали на 48-72 часов. Чтобы определить количество жизнеспособных клеток в каждой ячейке, добавляли 50 мкл раствора концентрацией 3 мг/мл бромида 3-(4,5-диметилтриазол-2-ил)-2,5-дифенилтетразолия в солевом растворе. Получаемую смесь инкубировали при 37oC в течении 5 часов и затем в каждую ячейку добавляли 50 мкл 25%-го додецилсульфата натрия, pH 2. После инкубации в течение ночи поглощение при 550 нм каждой ячейки измеряли на стандартном приборе-счетчике, применяемом в случае осуществления иммуноферментного твердофазного анализа. Средние процентные значения активности исследуемого соединения по четырем ячейкам (+/-стандартное отклонение) расчитывали по следующему уравнению:
где А означает активность исследуемого соединения.
Концентрация исследуемого соединения, обеспечивающая 50%-ное торможение роста клеток, составляла 3 х 10-10.
Опыт II (ин виво)
Соединение настоящего изобретения испытывали в доклиническом опыте на активность ин виво, который весьма показателен о полезности вещества в клинической практике. Такой опыт проводили на выбритых мышах, в которые внедрена ткань опухоли, предпочтительно от человеческого организма, путем трансплантации общеизвестными приемами. Антиопухолевая активность исследуемого соединения определялась на обработанных упомянутым образом мышах. При этом человеческие опухоли груди (МХ-1), выращенные на выбритых мышах, лишенных зобной железы, трансплантировали в новый реципиент животных, используя фрагменты опухолей, размер которых около 50 мг. День трансплантации обозначали как день 0. Через 6-10 дней мышам, подразделенным на группы по 5-10 животных, давали исследуемое соединение, введенное внутривенно или внутрибрюшинным способом с помощью укола. Соединение давали в дозах 25 мг/кг веса тела три раза в неделю в течение 3 недель. Диаметры опухолей и вес тел животных измеряли дважды в неделю. Объем опухолей рассчитывали, используя значения диаметров, измеренные кронциркулем Верниер и по следующей формуле:
(длина х ширина2) 2 = мм3 объема опухоли.
Средние значения объемов опухолей рассчитывали для каждой обработанной группы и определяли величины A, определенные указанным выше образом. Данные по активности оценивают следующим образом. Значение A, равное 1,0 или более, указывает на то, что соединение не оказало воздействия на рост опухоли, тогда как значение < 1.0 указывает на некоторое понижение массы опухоли. Значения 0.15-0.49 можно рассматривать как отражение умеренного замедления активности, а < 0.01-0.14 - хорошая до превосходная активность.
При вышеуказанных условиях исследуемое соединение обеспечивает полную дегенерацию опухоли (исчезла раковая масса после терапии).
Гидрохлорид пентапептида формулы (I) относится к категории малотоксичных веществ.
Нижеследующие примеры поясняют предлагаемые способы.
Пример 1 (способ A)
A. Получение исходных соединений
a. Z-Val-Val-MeVal-Pro-OMe (II; R1 = Me)
В реактор для гидрирования емкостью 400 литров вносят 39,6 кг (83,3 моля) Z-Val-MeVal-Pro-OMe (VIII) в 320 л метанола вместе с 4 кг 5%-ного палладия-на-угле. Затем при охлаждении при 20-30oC пропускают водород до тех пор, пока в реакционном растворе более нельзя обнаружить никакого эдукта. При выпуске содержимого реактора отфильтровывают катализатор. Для обработки осуществляют концентрирование в эмалированном реакторе емкостью 400 л в вакууме водоструйного насоса вплоть до объема 50 л. Затем приливают 50 л толуола и экстрагируют с помощью 40 л 2н. соляной кислоты. Толуольную фазу еще раз дополнительно экстрагируют с помощью 40 л 1н. соляной кислоты и после этого сливают. Объединенную кислую водную фазу вносят обратно в реактор, добавляют 40 л метиленхлорида в виде нижнего слоя и затем путем приливания 50%- ного раствора гидроксида натрия, при интенсивном перемешивании и охлаждении, устанавливают pH 9. После разделения фаз метиленхлоридную фазу сливают, а водную фазу дополнительно экстрагируют еще дважды по 40 л метиленхлоридом. Объединенный метиленхлоридный раствор продукта промывают водой до нейтральной реакции. После этого метиленхлоридную фазу концентрируют до объема 90 л. Получают Val-MeVal-Pro-OMe (XIV; R1 = метил).
Выход составляет 24,2 кг, соответственно, 85,2%.
В реакторе емкостью 400 л 17,84 кг (70,88 моль) Z-валина и 4,59 кг (74,42 моля) триэтиламина растворяют в 170 л метиленхлорида. К этому раствору при температуре от -5oC до -10oC добавляют 8,58 кг (70,88 моль) хлорангидрида пивалиновой кислоты. Спустя 2 часа времени реакции при -5oC, при температуре -5oC приливают раствор 24,2 кг Val-MeVal-Pro-OMe в 86 л метиленхлорида. Спустя следующие 2 часа выдерживания при -5oC, нагревают до 20oC и при этой температуре перемешивают в течение 12 часов. Для обработки добавляют 50 л воды. После отделения водной фазы органическую фазу экстрагируют один раз с помощью 40 л 2н. соляной кислоты и два раза по 40 л каждый раз 2н. раствора гидроксида натрия. После промывки органической фазы водой до нейтральной реакции метиленхлорид отгоняют и заменяют 300 л диизопропилового эфира. Для кристаллизации продукта эмульсию находящегося в виде масла продукта нагревают до 60oC, смешивают с затравочными кристаллами и выдерживают в течение 7 часов при 60oC. Для полноты кристаллизации продолжают перемешивать последовательно в течение 5 часов при 50oC и в течение 5 часов при 40oC и потом охлаждают до 20oC. Суспензию кристаллов отфильтровывают через работающий под давлением фильтр емкостью 120 л и высушивают в токе азота.
Выход составляет 32,2 кг, соответственно, 79%.
Температура плавления составляет 134-135oC.
б. Гидрохлорид пролинбензиламида (XII • HCl)
К раствору 99,7 г Z-пролина и 58 мл триэтиламина в 1 л метиленхлорида при температуре от -10oC до -15oC прикапывают 48,2 г хлорангидрида пивалиновой кислоты. Перемешивают дополнительно в течение 45 минут при -10oC и затем в течение получаса при температуре -10oC добавляют 42,8 г бензиламина в 500 мл метиленхлорида. Перемешивают дополнительно в течение часа при комнатной температуре. Метиленхлоридный раствор после этого промывают с помощью 500 мл воды, дважды по 500 мл 10%-ного водного раствора гидрокарбоната натрия, дважды по 500 мл воды, дважды по 500 мл 5%-ного водного раствора лимонной кислоты и дважды по 500 мл воды, сушат над сульфатом натрия и выпаривают. Остаются 120 г остатка, который растворяют в 200 мл этилацетата. К этилацетатному раствору добавляют 1,2 л н-гептана, перемешивают в течение часа, твердое вещество отсасывают и высушивают при -50oC в вакууме.
Выход составляет 110 г, соответственно, 81,3%.
Температура плавления составляет 93-94oC.
110 г таким образом полученного Z-пролинбензиламида растворяют в 1,5 л метанола. После добавления 0,5 г 10%-ного палладия-на-угле пропускают водород. При комнатной температуре в течение полутора часов раствор поглощает 0,5 л водорода. После отфильтровывания катализатора и выпаривания остаются 4,6 г желтого цвета масла.
413 г таким образом полученного пролинбензиламида растворяют в 400 мл изопропанола. Добавляют 630 мл насыщенного раствора хлористого водорода в изопропаноле, перемешивают образовавшуюся суспензию в течение двух часов при температуре от 0oC до 5oC, подкисляют и дважды промывают с помощью 250 мл изопропанола. Остаток высушивают при 50oC в вакууме. Получают 401 г гидрохлорида пролинбензиламида; α
Б. Получение целевого продукта
а.1 Me2Val-Val-MeVal-Pro-OMe HCl (IV • HCl; R1 = Me)
В реактор для гидрирования емкостью 400 литров вносят 20 кг (34,8 моль) Z-Val-Val-MeVal-Pro-OMe (II; R1 = метил) вместе с 2 кг 5%-ного палладия-на-угле в 200 л метанола. Затем при охлаждении при температуре 20oC пропускают водород до тех пор, пока в реакционном растворе более нельзя обнаружить никакого эдукта. Затем добавляют 8,46 кг (104 моля) 37%-ного раствора формальдегида и продолжают гидрировать при 20oC вплоть до прекращения поглощения водорода. При выпуске содержимого реактора отфильтровывают катализатор. Для обработки осуществляют концентрирование в эмалированном реакторе емкостью 400 л в вакууме водоструйного насоса вплоть до объема 50 л. Затем добавляют 200 л изопропанола и снова концентрируют до объема 50 л. После этого растворяют в 135 л метил-трет.-бутилового эфира и при охлаждении при 20oC добавляют один эквивалент изопропанольного раствора хлороводорода. Образовавшуюся суспензию перемешивают далее еще 3-4 часа при 20oC и 2 часа при температуре от 0oC до 5oC и затем фильтруют через работающий под давлением фильтр емкостью 120 л. Осадок на фильтре промывают один раз с помощью 50 л свежего метил-трет.- бутилового эфира.
Выход составляет 16,2 кг, соответственно, 92,3%.
Температура плавления составляет 224oC (разложение).
а.2 Также выделяют промежуточное соединение Val-Val-MeVal- Pro-OMe (III; R1 = метил), когда после первой стадии гидрирования обработку проводят следующим образом:
Реакционный раствор отделяют от катализатора и концентрируют. Остаток растворяют в этилацетате. Этилацетатный раствор экстрагируют дважды с помощью 2н. соляной кислоты. В кислой водной фазе устанавливают pH-значение, равное 9, с помощью раствора гидроксида натрия и экстрагируют дважды метиленхлоридом. Метиленхлоридную фазу затем промывают до нейтральной реакции и выпаривают.
Высокоэффективная жидкостная хроматография (ВЭЖХ):
содержание соединения составляет 96,8%,
1H-ЯМР (400 мГц, дейтерохлороформ, тетраметилсилан в качестве внутреннего стандарта):
δ (м.д.); 0,84-1,08 (м. 18 H); 1,45-1,6 (с., уширенный, 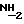 ); 1,85-2,15 (м. , 4H); 2,18-2,38 (м., 3H); 3,15 (с.,
); 1,85-2,15 (м. , 4H); 2,18-2,38 (м., 3H); 3,15 (с., 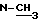 ); 3,25 (д., 1 H); 3,65-3,75 (м. , 1H); 3,73 (с.,
); 3,25 (д., 1 H); 3,65-3,75 (м. , 1H); 3,73 (с., 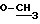 ); 3,9-4,05 (м., 1H); 4,38-4,45 (м., 1 H); 4,73-4,83 (м., 1H); 5,12 (д., 1H): 7,9 (д.,
); 3,9-4,05 (м., 1H); 4,38-4,45 (м., 1 H); 4,73-4,83 (м., 1H); 5,12 (д., 1H): 7,9 (д., 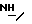 ).
).
а. 3 Me2Val-Val-MeVal-Pro-OMe • HCl (IV • HCl; R1 = Me) можно также получать по следующей методике, при которой не нужно выделять и очищать промежуточное соединение Z-Val-Val-MeVal-Pro-OMe (II; R1 = Me).
В колбе емкостью 4 л 128 г (0,51 моль) Z-валина и 55,1 г (0,54 моля) триэтиламина растворяют в 1,2 л метиленхлорида. К этому раствору при температуре от -5oC до -10oC добавляют 62,1 г (0,51 моль) хлорангидрида пивалиновой кислоты. Спустя 2 часа времени реакции при -5oC, приливают раствор 174,6 г (0,51 моль) Val-MeVal-Pro-OMe в 0,8 л метиленхлорида, перемешивают следующие 2 часа при -5oC и затем после нагревания до 20oC дальнейшие 12 часов. После этого реакционную смесь смешивают с 370 мл воды. После разделения фаз, метиленхлоридную фазу промывают один раз с помощью 290 мл 2н. соляной кислоты, дважды по 290 мл 2н. раствора гидроксида натрия и трижды по 370 мл воды. Затем метиленхлорид выпаривают и заменяют тремя литрами метанола. К этому раствору добавляют суспензию 30 г 5%-ного палладия-на-угле в 110 мл воды и гидрируют при 25oC при перемешивании за счет пропускания водорода из мерной емкости вплоть до поглощения одного эквивалента водорода. Затем добавляют 123 г (1,53 моля) 37%-ного раствора формальдегида и продолжают гидрировать вплоть до поглощения следующих двух эквивалентов водорода. После этого катализатор отделяют и реакционную смесь выпаривают на роторном испарителе. Остающееся масло растворяют в 670 мл изопропанола и 2,6 л метил-трет. -бутилового эфира. К этому раствору добавляют один эквивалент изопропанольного раствора хлороводорода. Образовавшуюся суспензию перемешивают далее в течение 12 часов при 20oC и затем отсасывают. Осадок на фильтре промывают небольшим количеством метил-трет.-бутилового эфира и после этого высушивают в сушильном вакуумном шкафу при 40oC.
Выход составляет 182,8 г, соответственно, 71%.
Температура плавления составляет 224oC (разложение).
б. Me2Val-Val-MeVal-Pro-Pro-NHBzl • HCl (I)
В реактор емкостью 400 л предварительно вносят 15,9 кг (31,5 моль) Me2Val-Val-MeVal-Pro-OMe • HCl (IV • HCl; R1 = метил) вместе со 140 л толуола и 15 л метанола. К полученной смеси добавляют 3,15 кг (76,38 моль) гидроксида натрия в таблетках. После полного омыления, то есть спустя 3 часа при 20oC, осуществляют нейтрализацию путем добавки изопропанольного раствора хлороводорода. Затем осуществляют азеотропную перегонку с толуолом при давлении 100 мбар вплоть до освобождения от спирта и воды. Отогнанный растворитель постепенно заменяют толуолом. После этого добавляют 80 л метиленхлорида и 6,44 кг (63,0 моль) триэтиламина (содержание: 99%), охлаждают до -5oC и при этой температуре добавляют 3,84 кг (31,5 моль) хлорангидрида пивалиновой кислоты. После протекания реакции в течение двух часов при температуре в пределах от -5oC до 0oC порциями добавляют 7,6 кг (31,5 моль) Pro-NHBzl • HCI. После стояния в течение двух часов при -5oC нагревают до 20oC и оставляют реагировать еще в течение следующих шести часов. Затем при давлении 500 мбар отгоняют добавленный метиленхлорид и добавляют 80 л толуола. После этого добавляют 50 л воды и в водной фазе устанавливают pH-значение, равное 9. После интенсивного перемешивания водную фазу отделяют, а органическую фазу дополнительно промывают один раз с помощью 25 л воды. Затем органическую фазу экстрагируют дважды по 50 л 2н. соляной кислоты. Продукт снова экстрагируют из кислой водной фазы, после установления pH-значения, равного 9, путем трехкратной экстракции по 50 л метиленхлорида каждый раз. После промывки метиленхлоридной фазы водой до нейтральной реакции метиленхлорид отгоняют и заменяют на 180 л метилэтилкетона. Раствор нагревают до 40oC и смешивают с одним эквивалентом (31,5 моль) изопропанольного раствора хлороводорода. Образовавшуюся суспензию нагревают до 60oC и после этого без дальнейшего нагревания перемешивают в течение 12 часов. Потом охлаждают до 20oC и перемешивают следующие 5 часов. Затем охлаждают до 5oC и фильтруют через работающий под давлением фильтр емкостью 120 л. Осадок на фильтре промывают с помощью 60 л свежего, охлажденного до 5oC, метилэтилкетона. После предварительного высушивания на фильтре продукт высушивают в вакуумном сушильном шкафу при 40oC до постоянного веса.
Выход составляет 14,36 кг, соответственно, 67%.
Температура плавления составляет 214oC (разложение). [α]
Пример 2 (способ Б)
a. Z-Val-Val-MeVal-Pro-OH (IX)
В колбе емкостью 2 л 117 г (0,2 моля)) Z-Val-Val-MeVal-Pro- OMe (II; R1 = Me, пример 1Aa) растворяют в 900 мл метанола и 47,5 мл воды. Затем добавляют 18 г (0,45 моль) гидроксида натрия в таблетках и перемешивают в течение 12 часов при 20oC. Для обработки добавляют 250 мл воды и отгоняют метанол. Затем добавляют столько этилацетата, чтобы произошло четкое разделение фаз (примерно 500 мл). Этилацетатную фазу отделяют. Водную фазу подкисляют до pH-значения, равного 1, и экстрагируют дважды по 500 мл метиленхлорида. Органическую фазу затем выпаривают досуха.
Выход составляет 105 г, соответственно, 96,4%.
1H-ЯМР (200 мГц, дейтерохлороформ, тетраметилсилан в качестве внутреннего стандарта):
δ (м. д.): 0,6-1,2 (м., 18H); 1,7-2,45 (м.,7H); 3,2 (с., 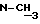 ); 3,55-3,95 (м. , 2H); 4,05-4,2 (м., 1H); 4,35-4,5 (м., 1H); 4,68-4,85 (м., 1H); 4,98-5,2 (м. , 3H); 5,93 (д., Val-1
); 3,55-3,95 (м. , 2H); 4,05-4,2 (м., 1H); 4,35-4,5 (м., 1H); 4,68-4,85 (м., 1H); 4,98-5,2 (м. , 3H); 5,93 (д., Val-1  ); 7,2-7,4 (м., 5H); 7,53-7,68 (Val-2
); 7,2-7,4 (м., 5H); 7,53-7,68 (Val-2  ); 9,6-10,2 (с., уширенный,
); 9,6-10,2 (с., уширенный, 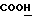 ).
).
б. Z-Val-Val-MeVal-Pro-Pro-NHBzl (XI)
5 г Z-Val-Val-MeVal-Pro-OH (8,75 ммоль) (IX) растворяют в 50 мл метиленхлорида, прикапывают 1,79 г (17,5 ммоль) триэтиламина, охлаждают до 10oC и при этой температуре прикапывают 1,08 г (8,75 ммоль) хлорангидрида пивалиновой кислоты. После перемешивания в течение двух часов при 10oC при этой температуре прикапывают раствор 2,11 г (8,75 ммоль) Pro-NHBzl • HCl в 10 мл метанола, дополнительно перемешивают в течение двух часов при 10oC и в течение ночи при комнатной температуре.
Реакционную смесь промывают три раза по 50 мл воды, один раз с помощью 50 мл воды при pH-значении, равном 9, и еще два раза по 50 мл воды. Метиленхлоридный раствор подвергают обработке на роторном испарителе. В качестве остатка получают 5,3 г (81,4%) белого кристаллического продукта (чистота: 88,7%).
Температура плавления составляет 118-122oC.
в. Me2Val-Val-MeVal-Pro-Pro-NHBzlHCl (I)
12 г Z-Val-Val-MeVal-Pro-Pro-NHBzl (XI) растворяют в 200 мл метанола. К полученному раствору добавляют 2 г 5%-ного палладия-на-угле (суспендирован в 20 мл воды) и гидрируют при 20oC вплоть до прекращения поглощения водорода. Затем добавляют 6,5 г 37%-ного раствора формальдегида и гидрируют далее вплоть до прекращения поглощения водорода. Затем катализатор отделяют и реакционный раствор выпаривают. Остаток обрабатывают толуолом, снова концентрируют, еще раз смешивают с 200 мл толуола и отфильтровывают. Толуольный раствор затем экстрагируют два раза по 50 мл 2н. соляной кислоты. Кислую водную фазу доводят до pH 9, с помощью раствора гидроксида натрия и экстрагируют трижды по 50 мл метиленхлорида. Метиленхлоридную фазу промывают водой до нейтральной реакции и концентрируют. Сырое основание растворяют в смеси 150 мл метилэтилкетона и 7,5 мл изопропанола. Из этого раствора путем добавки 4 г 25%-ного раствора хлороводорода в изопропаноле при 40oC осаждают продукт в виде соли. Суспензию дополнительно перемешивают в течение трех часов при 20oC и в течение часа при температуре от 0oC до 5oC и затем отсасывают.
Выход составляет 7,1 г.
Содержание соединения составляет 99,1% (процент площади согласно ВЭЖХ).
Температура плавления составляет 214oC (разложение). [α]
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ДОЛАСТАТИНА 15 | 1998 |
|
RU2195462C2 |
| ИНГИБИТОРЫ ТРОМБИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2172741C2 |
| ПРОТИВООПУХОЛЕВЫЕ ПЕПТИДЫ | 1996 |
|
RU2182911C2 |
| ПРОИЗВОДНЫЕ ДИПЕПТИДНЫХ П-АМИДИНО-БЕНЗИЛАМИДОВ С N-КОНЦЕВЫМИ СУЛЬФОНИЛЬНЫМИ ОСТАТКАМИ И ИХ СОЛИ С ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫМИ КИСЛОТАМИ | 1995 |
|
RU2152953C1 |
| Способ получения пентапептидов или их эфиров или их амидов или их солей | 1977 |
|
SU772481A3 |
| ПРОИЗВОДНЫЕ НИТРАТОАЛКАНОВЫХ КИСЛОТ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1991 |
|
RU2017748C1 |
| ПРОИЗВОДНЫЕ КАРБОНОВОЙ КИСЛОТЫ | 1996 |
|
RU2175315C2 |
| ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ АМИДИНЫ, ОБЛАДАЮЩИЕ СВОЙСТВАМИ ИНГИБИТОРОВ ТРОМБИНА, СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ПЯТИЧЛЕННОГО ГЕТЕРОЦИКЛИЧЕСКОГО АМИДИНА, В КАЧЕСТВЕ СОСТАВНОЙ ЧАСТИ ИНГИБИТОРОВ СЕРИНПРОТЕАЗЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2175328C2 |
| МЕТАЛЛОЦЕНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, КАТАЛИТИЧЕСКАЯ СИСТЕМА, СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВ И ПОЛИМЕРНОЕ ФОРМОВАННОЕ ИЗДЕЛИЕ | 1994 |
|
RU2147587C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАЦЕТАТА ТРИПЕПТИДА | 2014 |
|
RU2551276C1 |
Описывается гидрохлорид пентапептида формулы (I) Me2Val-Val-MeVal-Pro-Pro-NHBzl • HCl (I), где Me - метил, Bzl - бензил. Соединение обладает противоопухолевой активностью. Описываются также способы его получения. 4 с. и 1 з.п. ф-лы.
Me2Val-Val-MeVal-Pro-Pro-NHBzl•HCl,
где Me - метил;
Bzl - бензил.
Me2Val-Val-MeVal-Pro-Pro-NHBzl•HCl,
где Me - метил;
Bzl - бензил,
отличающийся тем, что соединение общей формулы II
Z-Val-Val-MeVal-Pro-OR',
где R' - алкил с 1-5 атомами углерода;
Z - бензилоксикарбонильная защитная группа, которая может быть замещена в фенильном кольце,
подвергают снятию защитной группы Z на концевой аминогруппе, получаемое при этом соединение подвергают диметилированию на свободной концевой аминогруппе с последующем омылением алкоксильной группы - OR' на концевой карбоксильной группе и получаемое при этом соединение формулы V
Me2Val-Val-MeVal-ProOH,
где Me имеет указанное значение,
последовательно подвергают взаимодействию с пивалоилхлоридом и пролинбензиламидом с последующим переводом получаемого при этом соединения в гидрохлорид.
Me2Val-Val-MeVal-Pro-Pro-NHBzl•HCl,
где Me - метил;
Bzl - бензил,
отличающийся тем, что соединение общей формулы (II)
Z-Val-Val-MeVal-Pro-OR' (II),
где R' - алкил с 1-5 атомами углерода;
Z - бензилоксикарбонильная защитная группа, которая может быть замещена в фенильном кольце,
подвергают омылению алкоксильной группы - OR' на конце карбоксильной группы, получаемое при этом соединение формулы (IX)
Z-Val-Val-MeVal-ProOH (IX)
где Me имеет указанное значение,
последовательно подвергают взаимодействию с пивалоилхлоридом и пролинбензиламидом и получаемое при этом соединение формулы (XI)
Z-Val-Val-MeVal-Pro-Pro-NHBzl (XI)
где Me имеет указанное значение,
подвергают снятию защитной группы Z на концевой аминогруппе с последующим диметилированием свободной концевой аминогруппы и омылением алкоксильной группы - OR' на конце карбоксильной группы и получаемое при этом соединение переводят в гидрохлорид.
| Способ получения глутамилсодержащих пептидов | 1977 |
|
SU715574A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |

