Изобретение относится к области неорганической химии, в частности к способу получения трифторида азота.
Трифторид азота находит широкое применение в технологиях полупроводниковых материалов, высокоэнергетических лазеров и химического газофазного осаждения. В отличие от элементного фтора его легко транспортировать в конденсированном виде при давлении до 75 кгс/см2 и плотности упаковки до 600 г/л [J. Fluor. Chem., 1991, 54, 1-3, р.37].
В основу промышленных технологий получения трифторида азота положены методы электролиза расплава гидрофторидов аммония и прямого фторирования аммиака элементным фтором.
Наиболее отработанным промышленным методом синтеза трифторида азота является электрохимический синтез NF3 из гидрофторидов аммония [Gmelin Handbook, 1986, v.4, р.172-173, Руководство по неорганическому синтезу, М., 1985, с. 220-221].
Оптимальными технологическими параметрами процесса электрохимического синтеза NF3 являются температура 100-130oС, состав расплава, соответствующий соотношению NH4F:HF=1:(1,1-1,8), плотность тока от 0,05 до 0,15 А/см2. Выход трифторида азота по току составляет 70%.
К недостаткам метода электрохимического синтеза NF3 следует отнести взрывоопасность процесса из-за образования смеси трифторида азота с водородом, использование газообразного аммиака на стадии получения гидрофторидов аммония. Кроме того, реализация этого процесса требует больших финансовых затрат из-за использования дорогостоящих никельсодержащих конструкционных материалов, наиболее устойчивых в коррозионно-активной среде расплавов гидрофторидов аммония.
Для снижения взрывоопасности смесей трифторида азота с водородом предложен способ электрохимического синтеза, предусматривающий разбавление анодного газа азотом до концентрации NF3 менее 9,5% [US 3235474, 204-63, 15.02.66] . Однако разбавление азотом продукционного газа существенно увеличивает затраты на оборудование для очистки и конденсации трифторида азота.
Из литературы известно, что в качестве исходного сырья для получения трифторида азота методом электрохимического фторирования могут быть использованы растворы мочевины и других азотсодержащих соединений - пиридина, гидразина, гуанидина, семикарбазида в безводном фтористом водороде [Z. anogr. allgem. Chem, 1969, v. 367, р.62-79]. Трифторид азота по данному способу получают с выходом от 16 до 38%.
Химизм процесса электрохимического фторирования мочевины описывается следующими уравнениями:
NH2CONH2+12F-->2NF3+COF2+4HF, (1)
2NH2CONH2+24F-->4NF3+CO2+CF4+8HF. (2)
Полученный электролизный газ после щелочной очистки от кислых примесей СО2 и COF2 содержал до 10% СF4. При фторировании пиридина содержание CF4 достигало 34% от объема получаемого трифторида азота. Это обстоятельство значительно усложняет очистку NF3 в связи с трудностью его отделения от тетрафторида углерода. Как следует из химизма процесса, отраженного уравнениями 1 и 2, на каждый моль NF3 в процессе электрохимического фторирования суммарно образуется по меньшей мере (при 100%-ном выходе NF3) 0,5 моль таких примесей как СО2, COF2 и CF4. Большое количество примесей в сырце трифторида азота и наличие водорода в газах электролиза приводит к удорожанию процесса очистки газа от примесей и необходимости принятия мер по обеспечению взрывобезопасности процесса.
Другим известным направлением получения трифторида азота является прямое фторирование аммиака элементным фтором по реакции:
4NH3+3F2-->NF3+3NH4F.
Синтез трифторида азота прямым фторированием аммиака элементным фтором в газовой фазе при мольных соотношениях NH3:F2=(1,1-2,0):1 позволяет получить целевой продукт с выходом по фтору от 10 до 25% [J. Amer. Chem. Soc, 1960, 82, 5301]. Низкий выход трифторида азота обусловлен сложностью регулирования температуры реакции, а также взаимодействием NF3 либо промежуточных продуктов его синтеза с аммиаком с образованием азота и фтористого водорода. Кроме того, возникают сложности с разделением газа от образующихся возгонов фторидов аммония.
С целью повышения эффективности охлаждения реакционной смеси и повышения выхода трифторида азота предложен способ фторирования аммиака в присутствии гексафторида серы, гексафторэтана или тетрафторметана [JP 2-255513, С 01 В 21/083, 16.10.90].
При мольном отношении газа-разбавителя к аммиаку (5-100):1 и фтора к аммиаку (3-20): 1, температуре в реакторе от 80 до 250oС выход NF3 составил от 30 до 59,5%. Максимальный выход трифторида азота был достигнут при мольном соотношении NF3: SF6, равном 4:100. Результаты, приведенные в патенте, свидетельствуют о том, что содержание целевого продукта в абгазах не должно превышать 1-2 об.%, следовательно концентрирование его до 99%-ного наряду с регенерацией газа-разбавителя потребует значительных затрат.
Кроме того, отделение возгонов (плава) гидрофторидов аммония от разбавленных газов технически сложная операция. В связи с этим возможности реализации этого способа в промышленности невысоки.
С целью преодоления вышеуказанных недостатков авторами патентов US 4091081, С 01 В 21/52, 23.05.78 и US 5637285, С 01 В 21/06, 10.06.97 были предложены способы получения трифторида азота фторированием элементным фтором аммиака, растворенного в плаве гидрофторидов аммония, при температуре процесса от 93 до 209oС и мольном соотношении NН3:HF соответственно равном 1:(2,0-2,5) и 1:(2,55-2,85).
Проведение синтеза трифторида азота в жидкой фазе расплава гидрофторидов аммония способно обеспечить эффективный отвод тепла экзотермической реакции и в основном решить проблему отделения гидрофторидов аммония от целевого продукта.
Однако промышленная реализация этих процессов потребует строжайших мер взрывобезопасности в связи с использованием в технологии аммиака и элементного фтора. Кроме того, при достаточно высоких выходах трифторида азота (65%) расход фторсодержащего сырья неоправданно велик, так как более 60% фтора, используемого в технологии, превращается в гидрофториды аммония, регенерация фтористого водорода из которых достаточно сложна.
По совокупности существующих признаков наиболее близким к предлагаемому способу является метод получения фторидов азота путем прямого фторирования элементным фтором производных аммиака, содержащих азотводородные связи, таких как амиды щелочных металлов, мочевина, биурет, сульфамид, формамид, гидразин, этилендиамин, меламин при температуре 0-300oС в присутствии катализатора - фторида металла, образующего кислую соль с HF [US 3961024, С 01 В 21/52, 01.06.76]. Так, например, при фторировании 50%-ных смесей мочевины с фторидом или гидрофторидом натрия газовой смесью, содержащей 50 об.% F2, получен газ состава: 10-17 об.% NF3 и 3-13 об.% N2F4. Наряду с фторидами азота продукционный газ содержал примеси COF2, СО2, CF4, N2O, и NO3F. Фторирование смесей биурета с фторидом лития или гидрофторидом натрия (1:1), разбавленным элементным фтором, приводит к образованию газа с содержанием NF3 6-47 об.% и N2F4 2,6-26 об.%.
Недостатками известного способа являются низкая селективность процесса получения трифторида азота из-за образования тетрафторгидразина, а также использование большого количества (100-400%) катализатора, который необходимо регенерировать.
Кроме того, при увеличении объемов реакторов в экзотермических процессах систем "газ-твердое" возникают трудности с охлаждением, а перегрев твердых смесей приводит к спеканию реагентов с последующим непрогнозируемым уменьшением скорости процесса фторирования, а также выделению избыточного аммиака, образующего взрывоопасные смеси со фтором.
Задачей изобретения является создание взрывобезопасного процесса синтеза трифторида азота и повышение селективности процесса.
Поставленная задача достигается путем прямого фторирования элементным фтором смесей (растворов или суспензий) азотсодержащих соединений с безводным фтористым водородом в условиях, обеспечивающих селективность синтеза трифторида азота.
Азотсодержащие соединения выбраны из группы, включающей мочевину и продукты ее разложения, такие как биурет, циануровая кислота, аммелид, меламин, формамид. Предпочтительно использовать для синтеза мочевину как более дешевое и доступное сырье, удобное в обращении.
В этом случае селективность синтеза трифторида азота в заявляемом способе обеспечивается условиями преимущественного протекания реакций фторирования, описываемых следующими химическими уравнениями:
2CO(NH2)2+3F2-->NF3+NH2CONHCONH2+3HF, (3)
3CO(NH2)2+9F2-->3NF3+C3N3O3H3+9HF, (4)
4CO(NH2)2+12F2-->4NF3+C3N4O2H4+12HF. (5)
при незначительном протекании реакций 1 и 2, приведенных ранее.
Процесс фторирования осуществляют в начальном периоде при температуре от -20 до -10oС и расходе элементного фтора, соответствующего мольному отношению к исходным соединениям не более 0,5. При этом создаются условия для протекания следующих реакций:
CO(NH2)2+2F2-->CO(NH2)(NF2)+2HF, (6)
CO(NH2)(NF2)+H2O+HF-->CO2+NF2H+NH4F, (7)
обеспечивающих практически полное удаление следов воды, а также значительной части таких примесей, как NO, CO2, CF4, N2O.
При температуре фторирования ниже -20oС начинается кристаллизация исходных соединений, а при температуре выше -10oС возрастают потери фторидов азота с абгазами. Увеличение мольного отношения фтора к исходным соединениям более 0,5 в начальный период приводит к потерям фторидов азота с абгазами и снижению выхода готового продукта.
Процесс фторирования в описанных выше условиях проводят до появления в абгазах следов трифторида азота. Затем, после образования трифторида азота, дальнейшее фторирование осуществляют при температуре от -15 до 0oС и расходе фтора, соответствующем мольному отношению к исходным соединениям не более 3,0.
При увеличении расхода фтора более 3 моль на 1 моль исходных соединений резко возрастает в готовом продукте концентрация таких примесей, как СО2, COF2 и CF4. Повышение температуры фторирования выше 0oС приводит к преимущественному протеканию реакций 1 и 2 и снижению селективности процесса.
Для фторирования используется элементный фтор с концентрацией не менее 50 мас. %, наиболее целесообразно использовать элементный фтор с концентрацией 90-98 мас.%.
Содержание исходного продукта в смеси с безводным фтористым водородом составляет от 20 до 50 мас.%. При содержании исходных соединений в смеси менее 20% значительно снижается скорость фторирования. В то же время при использовании смесей исходных соединений с безводным фтористым водородом с концентрацией более 50% растворы начинают кристаллизоваться и создают технологические трудности в процессе синтеза.
Процесс фторирования протекает при давлении от минус 0,7 до 1,7 кгс/см2. При большем давлении скорость фторирования снижается.
Синтез трифторида азота может быть осуществлен как в насадочной колонне, так и в колонне с распылением жидкости либо в реакторе барботажного типа. Наиболее простое аппаратурное оформление процесса фторирования включает емкостной реактор, снабженный охлаждающими элементами. В реактор загружают раствор мочевины или другого исходного соединения в безводном фтористом водороде и по сифону подают элементный фтор. Регулирование температуры раствора и отвод тепла реакции осуществляют с помощью рассола -40oС, подаваемого в охлаждающие элементы.
Продукты реакции фторирования не образуют устойчивых соединений с безводным фтористым водородом, поэтому регенерация последнего сводится к отгонке при температуре до 150oС с выходом до 90% по HF.
Полученную газовую смесь, содержащую в качестве основных примесей N2, F2, NO, N2F2, СО2, COF2, N2O и CF4, пропускают через обратный холодильник, охлаждаемый рассолом -40oС, и подвергают щелочной очистке растворами KOH или K2CO3 от избыточного фтора и других окислителей. Дальнейшая очистка газа осуществляется сорбционными методами до получения конденсированного трифторида азота с содержанием 98,0-99,0 мас.% NF3.
Определение состава газообразных продуктов фторирования проводят с использованием химического, потенциометрического, хроматографического и спектрального методов анализа.
Таким образом, разработан взрывобезопасный способ получения трифторида азота прямым фторированием мочевины или продуктов ее разложения. Способ позволяет получать продукт с максимальным содержанием трифторида азота и минимальной концентрацией примесей с выходом до 90%. Мягкие условия фторирования дают возможность упростить аппаратурное оформление процесса и достичь высокой степени использования фторсодержащего сырья.
Нижеследующие примеры иллюстрируют предлагаемое изобретение, но не ограничивают его.
Пример 1.
Для синтеза используют металлический реактор емкостью 60 л, снабженный рубашкой охлаждения, термопарой, барботажным устройством в виде сифона для подачи газообразного фтора, обратным холодильником, охлаждаемым рассолом с температурой -35oС для исключения выноса и потерь фтористого водорода из реактора, технологическими штуцерами для загрузки и выгрузки продуктов, датчиком давления.
В охлажденный реактор последовательно загружают 20 кг мочевины и 32 кг безводного фтористого водорода. Полученный раствор мочевины в безводном фтористом водороде охлаждают до температуры -20oС и подают газообразный фтор (98 об.%) со скоростью, обеспечивающей содержание окислителей в перерасчете на F2 в абгазах фторирования не более 0,5%. Содержание окислителей определяют йодометрическим методом. Первоначально объем пропущенного фтора, при котором в абгазах появляется NF3, составляет 3 м3, что соответствует мольному отношению F2:CO(NH2)2, равному 0,4. Выходящую из реактора газовую смесь, которая содержит в основном примеси (CF4 5,6 об.%, СО2 12,1 об.%, СОF2 10,3 об. %, N2F2 и NF2H 0,2 об.%, N2O 11,2 об.%, окислители 0,4 об.%) и 0,7 об.% NF3 охлаждают в обратном холодильнике и сдувают через колонну с известковым поглотителем.
После появления в абгазах трифторида азота фторирование осуществляют при температуре -5oС, подавая газообразный фтор со скоростью, обеспечивающей содержание окислителей в пересчете на F2 в абгазах фторирования не более 3,0%.
Выходящую из реактора газовую смесь охлаждают в обратном холодильнике и с помощью компрессора закачивают в ресивер емкостью 1 м3. Полученная газовая смесь имеет следующий состав, об.%: NF3 86,2; CF4 0,7; СO2 и COF2 4,3; N2F2 0,2; N2O 2,7, сумма окислителей 1,5.
Количество израсходованного фтора составляет 3,8 м3, что соответствует мольному отношению F2:CO(NH2)2, равному 0,5. Дальнейшую очистку NF3 осуществляют сорбционными методами для получения конденсированного трифторида азота с содержанием основного вещества 99,0 об.%.
Выход трифторида азота, определенный по соотношению объемного количества полученного в ресивере трифторида азота к одной трети объема, затраченного в процессе газообразного фтора, составляет 94,9%.
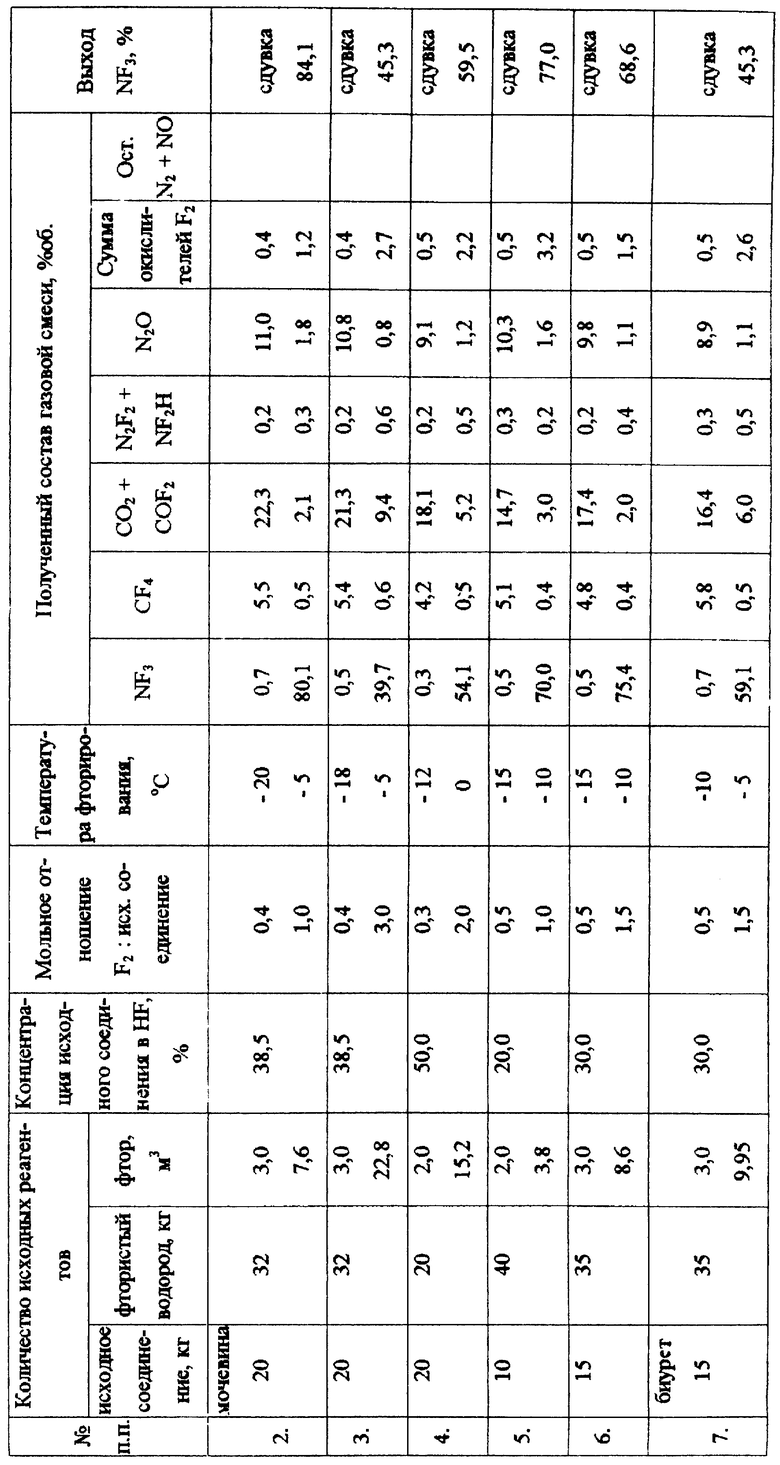
Последующие синтезы (примеры 2-7) проведены аналогично примеру 1. Условия проведения и полученные результаты представлены в таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ переработки полифторида аммония и способ получения трифторида азота, применяемый в нем | 2024 |
|
RU2829882C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИФТОРИДА АЗОТА | 2006 |
|
RU2317251C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРИДОВ АЗОТА | 2001 |
|
RU2178384C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИФТОРИДА АЗОТА | 2004 |
|
RU2256605C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИФТОРИРОВАННЫХ ЭТАНОВ | 1993 |
|
RU2043327C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИФТОРИДА АЗОТА | 2001 |
|
RU2182556C1 |
| ЭКОЛОГИЧЕСКИ-БЕЗОПАСНЫЙ СПОСОБ ПОЛУЧЕНИЯ УГЛЕРОДНОГО СОРБЕНТА | 2015 |
|
RU2601762C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИФТОРИДА АЗОТА | 2001 |
|
RU2182555C1 |
| СПОСОБ ОЧИСТКИ ГАЗООБРАЗНОГО ТРИФТОРИДА АЗОТА | 2002 |
|
RU2206499C1 |
| НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ 2-ФТОРСУЛЬФОНИЛТЕТРАФТОРЭТИЛТРИФТОРВИНИЛОВОГО ЭФИРА | 2011 |
|
RU2475477C1 |
Изобретение относится к новому способу получения трифторида азота, который находит широкое применение в технологиях полупроводниковых материалов, высокоэнергетических лазеров и химического газофазного осаждения. Трифторид азота получают фторированием мочевины или продуктов ее разложения элементным фтором в безводном фтористом водороде при температуре от -20 до 0oС и мольном отношении фтора к исходным соединениям не более 3. Концентрация исходных соединений в безводном фтористом водороде составляет 20-50 мас.%. Разработанный способ взрывобезопасен и позволяет получать продукт с максимальным содержанием трифторида азота и минимальной концентрацией примесей с выходом до 90%. 4 з.п. ф-лы, 1 табл.
| US 3961024 А, 01.06.1976 | |||
| СПОСОБ ПОЛУЧЕНИЯ ТРЕХФТОРИСТОГО АЗОТА | 0 |
|
SU196734A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US 4543242 А, 24.09.1985 | |||
| Устройство для передвижения конвейера на крутопадающих пластах | 1979 |
|
SU787684A1 |

