Настоящее изобретение относится к рецептору глюканового активатора (здесь и далее именуемому иногда как "ER"), к молекулам ДНК, кодирующим ER, к векторам, содержащим эти молекулы ДНК и к растительным клеткам, трансформированным этими молекулами ДНК. Более конкретно настоящее изобретение относится к ER, полученным из фракции плазматической мембраны корней сои, молекулам ДНК, кодирующим ER, векторам, содержащим ДНК-молекулы и к растительным клеткам, трансформированным молекулами ДНК.
Предпосылки изобретения
Известно, что растения синтезируют и накапливают антибиотический агент, называемый фитоалексином, как реакцию на инфицирование патогеном (M.Yoshikawa, 1978, Nature 257:546). Было обнаружено, что некоторые патогены растений содержат вещества, которые заставляют их осуществлять такие реакции "сопротивления" (или устойчивости) (N.T.Keen (1975) Science 187:74), которые называют "активаторами" ("elicitors"). Считают, что биохимический процесс, начиная от инфицирования растений патогенами, до синтеза и накопления фитоалексина состоит в следующем.
Когда мицелий патогена вторгается в растительную клетку, глюканаза в растительной клетке работает таким образом, чтобы расщепить полисахариды на поверхности стенки мицелия патогена и за счет этого высвободить активатор. Если активатор связывается с рецептором в растительной клетке, продуцируется второй мессенджер, который участвует в трансдукции сигнала. Вещество трансдукции сигнала включается в ядро растительной клетки и активирует транскрипцию генов, кодирующих энзимы, синтезирующие фитоалексин, для того, чтобы индуцировать синтез фитоалексина. В то же самое время происходит ингибирование деградации фитоалексина. В результате происходит эффективное накопление фитоалексина.
Фитоалексин, играющий важную роль в устойчивости сои, называют глицеолином, и его строение известно (M.Yoshikawa et.al.(1978) Physiol.Plant.Pathol. 12: 73). Активатор является полисахаридом глюкозы и, как сообщается, представляет собой глюкан, содержащий β -1,6- и β -1,3 связи (J.K.Sharp et.al. (1984) J. Biol. Chem. 259 : 11321, M.Yoshikawa (1990) Plant Cell Technology 1,2 : 695). Специфическим рецептором для глюканового активатора, полученного из патогенов сои плесневых грибков Phytophthora megasperma f.sp.glycinea, как считают, является протеин, который играет важную роль в синтезе и накоплении антибиотического агента - глицеолина. Способ выделения ER, специфического для этого активатора, был раскрыт (E.G.Cosio et.al., (1990) FEBS 264: 235, E. G. Cosio et al. (1992) Eur.J.Biochem. 204:1115, T.Frey et al. (1993) Phytochemistry 32:543). Однако аминокислотная последовательность ER не была определена, и до сих пор не известен ген, кодирующий его.
Целью настоящего изобретения является создание рецептора глюканового активатора.
Другой целью изобретения является создание молекулы ДНК, кодирующей рецептор глюканового активатора.
Еще одной целью настоящего изобретения является создание векторов, содержащих молекулу ДНК, кодирующую рецептор глюканового активатора.
Еще одной целью изобретения является получение растительных клеток, трансформированных ДНК-молекулами, кодирующими рецептор глюканового активатора.
Описание изобретения
В результате различных исследований, проведенных для решения вышеуказанных проблем, авторам удалось выделить из корней сои ER и клонировать ген ER из кДНК библиотеки сои, что и составляет предмет настоящего изобретения. В настоящем изобретении предложен рецептор глюканового активатора с аминокислотной последовательностью, которая представлена как последовательность ID N 1. В настоящем изобретении предложены также молекулы ДНК, кодирующие рецептор глюканового активатора с аминокислотной последовательностью, представленной как последовательность ID N 1, и их фрагменты. Далее в настоящем изобретении предложены молекулы ДНК, содержащие нуклеотидные последовательности, кодирующие рецептор глюканового активатора, которые включены в плазмиду pER23-1, и их фрагменты. В настоящем изобретении предложены также векторы, содержащие ДНК-молекулы, кодирующие рецептор глюканового активатора, и растительные клетки, трансформированные ДНК-молекулами, кодирующими рецептор глюканового активатора.
Краткое описание чертежей
Фиг. 1 представляет картины трех стадий очистки за счет электрофореза на SDS-полиакриламидном геле.
На фиг. 2 представлены карты плазмид pER23-1 и pER23-2.
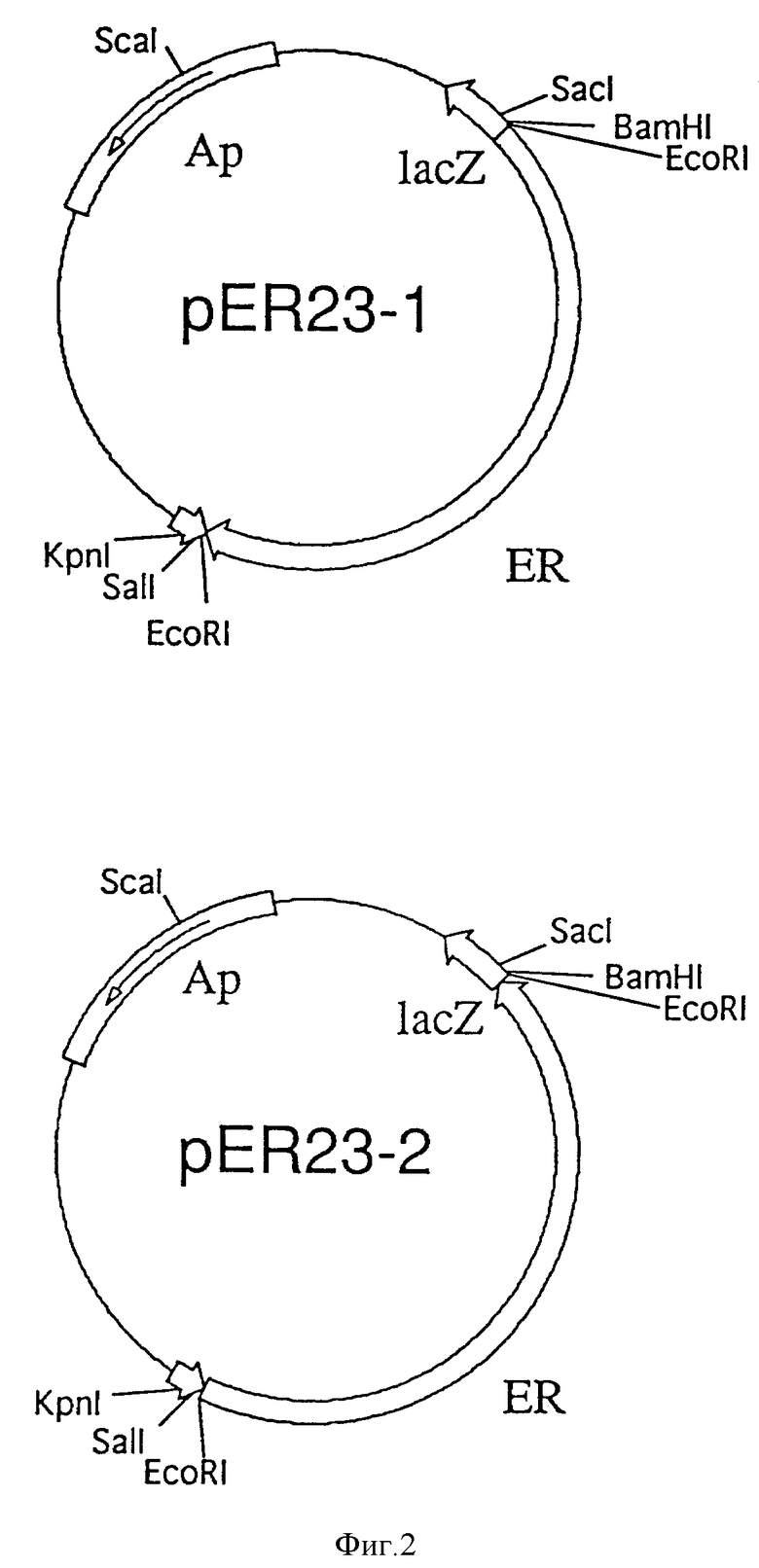
На фиг. 3 представлена схема конструирования плазмиды pK V1-ER23.
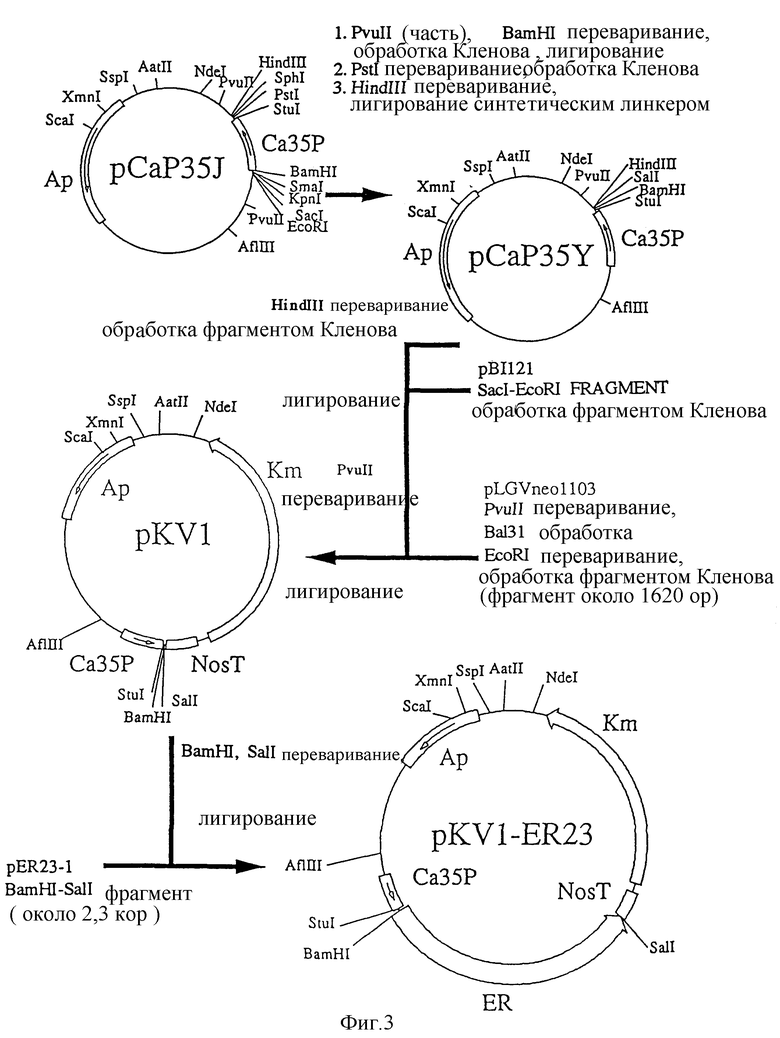
На фиг. 4 представлено кратковременное повышение концентрации внутриклеточного Ca2+ после добавления активатора к культивируемым клеткам сои.
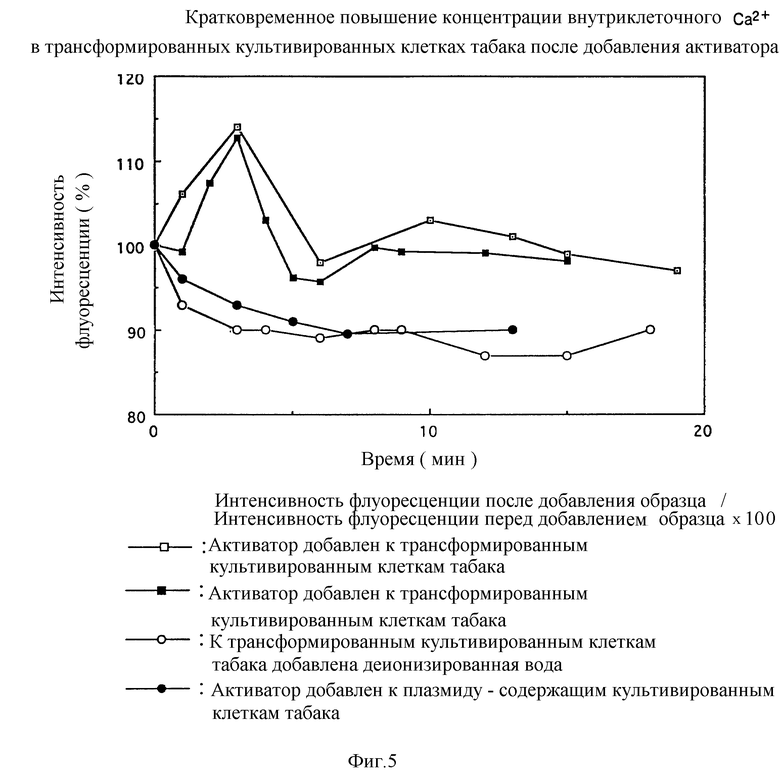
Фиг. 5 представляет кратковременное повышение концентрации внутриклеточного Ca2+ после добавления активатора к культивируемым трансформированным клеткам табака.
На фиг.6 представлены активатор-связывающие активности полной длины или частичной длины ER, экспрессированных в E.coli.
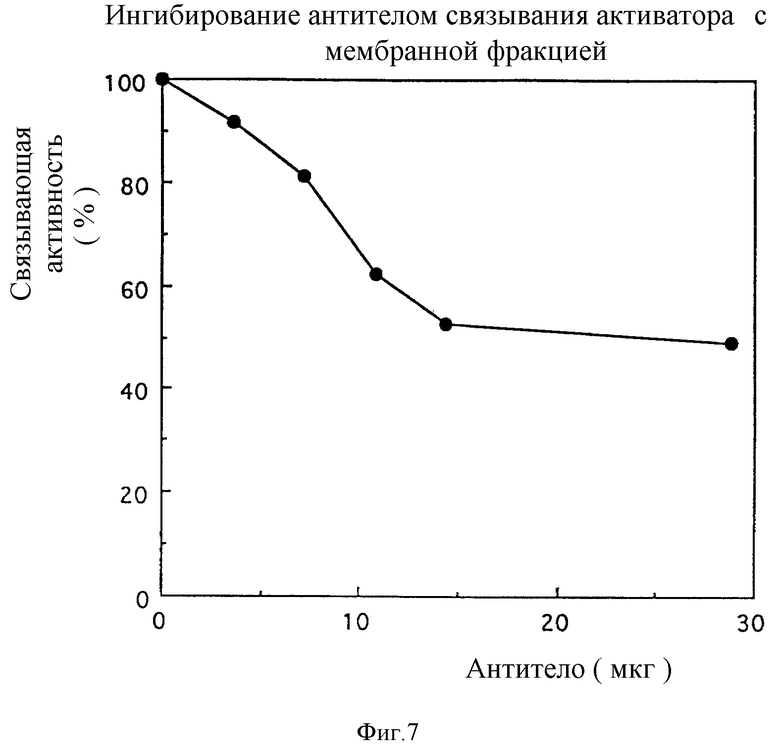
На фиг. 7 представлено ингибирование связывания активатора со связывающим активатор протеином в мембранной фракции семядолей сои за счет антитела против активатор-связывающего домена.
На фиг. 8 представлено ингибирование накопления фитоалексина, индуцированное активатором, в семядолях сои за счет антитела против активатор-связывающего домена.
Предпочтительный вариант осуществления изобретения
Рецептор глюканового активатора настоящего изобретения представляет собой протеин, обладающий функцией рецептора для глюкановых активаторов, полученный из патогенов растений, в частности Phytophthora. Более конкретно, он представляет собой протеин, который связывается с глюкановым активатором, полученным при расщеплении частей стенки мицелия патогена за счет β - 1,3-глюканазы в растительных клетках во время внедрения растительных патогенов, особенно микроорганизмов, принадлежащих к роду Phytophthora, и который затем заставляет микросомы и ядра продуцировать повышенное количество фитоалексина в растительных клетках. Рецептор глюканового активатора настоящего изобретения содержит аминокислотную последовательность, представленную как последовательность ID N 1. "Аминокислотная последовательность практически в соответствии с последовательностью ID N 1'' включает аминокислотные последовательности, как представлено последовательностью ID N 1, в которой могут быть делеции, замены или добавления аминокислоты (аминокислот), при условии, что они сохраняют функции рецептора глюканового активатора.
Рецептор глюканового активатора настоящего изобретения можно получить, например, частично модифицированным способом Козио (E.J.B.Cosio (1992), 204: 1115). Короче, корни, листья и стебли сои, предпочтительно сорта green homer, гомогенизируют, и мембранную часть собирают из полученной суспензии, очищают с помощью ионообменной хроматографии, и далее очищают с помощью афинной хроматографии, используя в качестве лиганда активатор. Активатор, который используют в афинной хроматографии, предпочтительно получают из Phytophthora megasperma f.sp.glycinea расы 1 (АТСС 34566), так как она демонстрирует несовместимость с green homer (т.е. устойчивость к патогену).
Полученную таким образом аминокислотную последовательность рецептора глюканового активатора можно определить следующим образом.
Очищенный ER переносят на PVDF в мембрану (Миллипор Ко) с помощью электроблоттинга и переваривают лизилендопептидазой (АР-1). Фрагментированные пептиды выделяют из PVDF мембраны и фракционируют с помощью ВЭЖХ с обращенной фазой (μ-Bondasphere 5 мк C8). Пиковые фракции анализируют с помощью газофазного протеинового секвенатора (Applied Biosystems Co) ER настоящего изобретения пригодны для выяснения механизма устойчивости растений к грибкам и для создания производных активатора, способных вызывать устойчивость к грибкам, и их можно использовать в качестве антигена для получения антител против ER.
Настоящее изобретение охватывает ДНК-молекулы, содержащие нуклеотидные последовательности, кодирующие рецептор глюканового активатора, и их фрагменты. ДНК-молекулы настоящего изобретения содержат, предпочтительно, по крайней мере один стоп кодон (например, TAG), соседний c 3' концом.
Более конкретно, настоящее изобретение охватывает ДНК-молекулы, содержащие нуклеотидные последовательности, кодирующие рецептор глюканового активатора, содержащего аминокислотные последовательности, практически в соответствии с представленной последовательность ID N 1, и их фрагменты. "ДНК-молекулы, содержащие нуклеотидные последовательности, кодирующие рецептор глюканового активатора" включает все дегенеративные изомеры. Термин "дегенеративные изомеры" относится к ДНК-молекулам, кодирующим один и тот же полипептид с различной вырожденностью кода. Если в качестве примера взять ДНК-молекулу, имеющую нуклеотидную последовательность SEQ ID N 2, ДНК-молекула, в которой кодон для любой аминокислоты, например, ААС для Asn изменен на дегенеративный кодон AAT, будет называться дегенеративным изомером. Примеры таких дегенеративных изомеров включают ДНК-молекулы, содержащие нуклеотидную последовательность, представленную как последовательность ID N 2 (SEQ ID N 2).
В другом аспекте в настоящем изобретении предложены ДНК-молекулы, содержащие нуклеотидные последовательности, кодирующие рецепторы глюканового активатора, которые включены в плазмиду pER23-1, и их фрагменты. E.coli ДН5-α EKB633, трансформированная плазмидой pER23-1, была депонирована в Национальном институте Bioscience and Human-Technology, Agency of Industrial Sience and Technology 15 июня 1994 г. под регистрационным номером FERM BP-4699.
Фрагменты ДНК-молекул, содержащие нуклеотидные последовательности, кодирующие рецептор глюканового активатора настоящего изобретения, могут содержать нуклеотидные последовательности, кодирующие аминокислотную последовательность ID N 1 (SEQ ID N 1).
ДНК-молекулы настоящего изобретения, которые содержат нуклеотидные последовательности, кодирующие рецептор глюканового активатора и их фрагменты, могут необязательно связываться с ATG кодоном для инициирования метионина вместе с трансляционной рамкой в участке в обратном направлении от 5' конца и также связываться с другими ДНК-молекулами, имеющими соответствующие длины в качестве нетранслируемых районов в участке в обратном направлении от 5' конца и в участке в прямом направлении от 3' конца.
ДНК-молекулы настоящего изобретения, которые содержат нуклеотидные последовательности, кодирующие рецептор глюканового активатора и их фрагменты, можно представить в форме частей, составляющих плазмидные или фаговые ДНК-молекулы или в форме частей, составляющих плазмидные, фаговые или геномные ДНК-молекулы, которые введены в микроорганизмы (в частности, бактерии, включая E.coli и Agrobacterium), фаговые частицы или растения.
Для стабильной экспрессии ДНК-молекул, кодирующих рецептор глюканового активатора и их фрагментов в растениях, промотор, ДНК-молекулу (ATG), кодирующую кодон инициирования и терминатор, можно добавить к ДНК-молекулам настоящего изобретения и их фрагментам в соответствующих комбинациях. Примеры промоторов включают промотор генов, кодирующих малую субъединицу рибулозо-1,5-бифосфаткарбоксилазы (Fluhr et al. , Proc. Natl. Acad. Sci. USA (1986); 83:2358), промотор гена нопалинсинтазы (Langridge et al., Plant Cell Rep. (1985) 4:355), промотор продуцирования вируса мозаики цветной капусты 19S-RNA (Guilley et al., Cell (1982) 30:763), промотор продуцирования вируса мозаики цветной капусты 35S-RNAI Odell et al., Nature (1985) 313:810) и т.п. Примеры терминаторов включают терминатор гена нопалинсинтазы (Depicker et al. , J. Mol. Appl. Gen. (1982) 1:561) и терминатор гена октопинсинтазы (Gielen et al., EMBO J. (1984) 3:835).
ДНК-молекулу, содержащую нуклеотидную последовательность, кодирующую рецептор глюканового активатора, можно получить способом, который включает стадии химического синтеза по крайней мере части ДНК-молекулы, в соответствии с обычными способами синтеза нуклеиновых кислот, и получение целевой ДНК-молекулы из соответствующей кДНК-библиотеки с использованием синтезированных ДНК-молекул в качестве зондов обычными способами, например иммунологически или способом гибридизации.
Некоторые плазмиды, различные типы рестрикционных энзимов, Т4ДНК-лигаза и другие энзимы для использования в этом способе, коммерчески доступны. Клонирование ДНК, конструирование плазмид, трансфекцию хозяев, культивирование трансфектантов, выделение ДНК-молекул из культуры и другие стадии можно осуществить в соответствии со способами, описанными в книге Самбрука (Molecular Cloning, J.Sambrook et al., CSH Laboratory (1989), Current Protocols in Molecular Biology, F.M.Ausubel et al., John Wiley and Sons (1987)) и др.
Более конкретно, ДНК-молекулы настоящего изобретения, которые содержат нуклеотидные последовательности, кодирующие рецептор глюканового активатора, можно получить следующим образом.
Два типа частичных аминокислотных последовательностей выбирают из аминокислотных последовательностей рецептора глюканового активатора. Приготавливают праймеры, состоящие из комбинаций всех оснований, которые могут кодировать C-конец выбранной частичной последовательности, и праймеры, состоящие из комбинаций всех оснований, которые могут кодировать N-конец выбранной частичной последовательности. Эти синтезированные праймеры используют в качестве смешанных праймеров для осуществления двух PCR с использованием ДНК-молекул подходящей кДНК-библиотеки сои в качестве матрицы. Затем два амплифицированных фрагмента заданной длины, амплификацию которых ожидают (эти фрагменты соответствуют ДНК-молекулам, кодирующим вышеуказанные две частичные аминокислотные последовательности), выделяют и определяют их нуклеотидные последовательности. На основании определения нуклеотидных последовательностей синтезируют праймер, содержащий нуклеотидную последовательность, кодирующую C-конец аминокислотной частичной последовательности, расположенной у C-конца рецептора глюканового активатора, и праймер, содержащий аминокислотные последовательности, кодирующие N-конец аминокислотной частичной последовательности, расположенной у N-конца рецептора глюканового активатора. Эти два синтезированных праймера используют для осуществления PCR, с использованием ДНК-молекул вышеуказанной соевой кДНК-библиотеки в качестве матрицы. Полученные амплифицированные фрагменты используют в качестве зондов для гибридизации вышеуказанной соевой кДНК- библиотеки, в результате чего получают ДНК-молекулы, содержащие нуклеотидные последовательности, кодирующие рецептор глюканового активатора.
Полученные ДНК-молекулы, содержащие нуклеотидные последовательности, кодирующие рецептор глюканового активатора, можно секвенировать любым известным способом, например способом Максам-Джилберта (Maxam-Gilbert Method Enzymol. , 65:499, 1980), методом терминации дидеоксинуклеотидной цепи, используя М13 фаг (J.Messing et al., Gene, 19:269, 1982) и т.п.
Так как результаты различных исследований глюканового активатора предполагают, что рецептор глюканового активатора играет важную роль в устойчивости растений по отношению к грибкам, ожидается, что ДНК-молекулы, кодирующие рецептор глюканового активатора настоящего изобретения, и их фрагменты могут придавать растениям устойчивость к грибкам, если их ввести и экспрессировать в растительных клетках (особенно в клетки высших растений), которые не содержат рецептора глюканового активатора, в соответствии с обычной процедурой. Было выдвинуто предположение, что грибки, способные инфицировать растения, обычно имеют супрессоры, за счет чего обладают способностью подавлять устойчивость растений по отношению к грибкам. Ожидается, что новые растения, обладающие устойчивостью к грибкам, можно будет получить за счет введения и экспрессии ДНК-молекул, кодирующих рецептор глюканового активатора настоящего изобретения и их фрагментов так, чтобы ER работал, или за счет модификации ДНК-молекул и их фрагментов, или за счет регулирования их экспрессируемых количеств. Более того, если ДНК-молекулы настоящего изобретения и их фрагменты вводят и экспрессируют в растительных клетках, особенно в клетках высших растений, наряду с генами, повышающими устойчивость к грибкам, или такие свойства, как те, которыми обладает ген глюканазы, который придает растениям устойчивость к грибкам, ожидается, что в таком случае растениям можно придать более высокую устойчивость к грибкам нежели в том случае, когда вводят ген глюканазы.
Векторы, которые используют для введения молекул ДНК, кодирующих рецептор глюканового активатора, и их фрагментов можно сконструировать так, чтобы рецептор глюканового активатора мог быть стабильно экспрессирован в растения. Более конкретно, промотор, ДНК-молекула, кодирующая кодон инициирования (ATG) и терминатор можно вводить в ДНК-молекулы, кодирующие рецептор глюканового активатора, и их фрагменты, в соответствующих комбинациях. Примеры таких промоторов включают промотор генов, кодирующих малую субъединицу рибозо-1,5-бис-фосфаткарбоксилазы (Fluhr et al., Proc. Natl. Acad. Sci. USA (1986) 83:2358), промотор гена нопалин-синтазы (Langridge et al. , Plant Cell Rep. (1985) 4:355), промотор для продуцирования вируса мозаики цветной капусты 19S-RNA (Guilley et al., Cell. (1982) 30:763), промотор для продуцирования вируса мозаики цветной капусты 35S-RNA (Odell et al., Nature (1985) 313:810) и т.п. Примеры терминаторов включают терминатор гена нопалин-синтазы (Depicker et al., J.Mol. Appl. Gen. (1982) 1:561) и терминатор гена октопин-синтазы (Gielen et al., EMBO J. (1984) 3:835).
ДНК-молекулы, кодирующие рецептор глюканового активатора и их фрагменты можно ввести в растительные клетки обычными хорошо известными способами, например способом, указанным в "Plant genetic transformation and gene expression; a laboratory manual", J. Draper et al. eds., Blackwell Scientific Publications, 1988. Примеры таких способов включают такие биологические способы, как те, которые используют вирусы или Agrobacteria, и такие физико-химические способы, как электропорация, способ с использованием полиэтиленгликоля, микроинъекции и т.п.
Далее настоящее изобретение будет раскрыто более подробно со ссылками на следующие примеры, которые никоим образом не должны ограничивать объем настоящего изобретения.
Пример 1
Очистка ER, полученного из корней сои.
1) Определение активности связывания глюканового активатора ER.
Комплекс активатора (средний молекулярный вес :10000) и тирамина (TOKYO KAS EI KOGYO CO., LTD) синтезируют способом Jong-Joo Cheong (The Plant Cell (1991) 3:127). Комплекс активатор-тирамин метят иодом, используя хлорамин T.
Образец (количество протеина менее 500 мкг) суспендируют в 500 мкл аналитического буфера (50 мМ Tris - HCl pH 7,4, 0,1 М сахароза, 5 мМ MgCl2, 1 мМ PMSF и 5 мМ ЕДТА) и инкубируют при 0oC в течение 2 часов. Меченый иодом комплекс активатор-тирамин в количестве 7,0 нМ (70 Кюри/ммоль, число молей рассчитывают в предположении, что молекулярный вес активатора составляет 10000, и это значение используют во всем описании) добавляют к суспензии, и полученную смесь инкубируют при 4oC в течение 2 часов. Реакционный раствор фильтруют через Watman GF/B и обрабатывают 0,3% раствором полиэтиленимина в течение по крайней мере 1 часа. Остаток трижды промывают 5 мл буфера, охлажденного льдом (10 мМ Tris - HCl pH 7,0, 1 М NaCl, 10 мМ MgCl2). Определяют радиоактивность, сохранившуюся на фильтре, с помощью гамма-счетчика (отсчет A). Для того чтобы исключить эффект неспецифического связывания, осуществляют ту же процедуру, что и раньше, за исключением того, что к каждому образцу добавляют 17 мкМ рецептора, полученную смесь суспендируют в аналитическом буфере, и полученную суспензию инкубируют при 0oC в течение 2 часов. Полученную величину вычитают из отсчета A, что дает величину ( Δ срм=число импульсов в минуту). Эту полученную величину ( Δ срм) делят на полное количество импульсов, а затем умножают на полное количество активаторов, использованных в эксперименте, для того, чтобы рассчитать количество протеина, связывающего активатора (в молях).
Степень чистоты ER проверяют вышеуказанным способом.
2) Выделение ER, полученных из корней сои
Семена сои (Glycine max. CV. Green Homer) (Takayama Seed Co.) культивируют на вермикулите в течение 1 недели, а затем ведут аквакультуру на вермикулите в течение 15 дней для сбора корней (около 40 кг влажного веса). Собранные корни хранят при -80oC до тех пор, пока их не используют для выделения ER. К корням (2 кг влажн. вес) добавляют охлажденный льдом буфер (25 мМ Tris - HCl pH 7,0, 30 мМ PMSF) в количестве 1,25 л, и полученную смесь гомогенизируют в смесителе Waring в течение 2 минут.
Полученную суспензию фильтруют через Miracloth (Calbiochem Co.), и полученный фильтрат центрифугируют со скоростью 9000 об/мин при 4oC в течение 15 минут. Надосадочную жидкость ультрацентрифугируют со скоростью 37000 об/мин при 4oC в течение 20 минут. Осадок суспендируют в 160 мл охлажденного льдом буфера (25 мМ Tris - HCl pH 7,4, 0,1 М сахарозы, 5 мМ MgCl2, 1 мМ PMSF и 5 мМ EDTA) до получения мембранной фракции. К мембранной фракции добавляют амфолитический детергент ZW3-12 (Boehringer Co.) до получения финальной концентрации 0,25% для солюбилизации ER из мембранной фракции, и полученную смесь перемешивают при 8oC в течение 30 минут. Полученную смесь ультрацентрифугируют со скоростью 37000 об/мин при 4oC в течение 20 минут для того, чтобы собрать надосадочную жидкость, содержащую солюбилизированные ER (растворимая фракция). 165 мл растворимой фракции диализуют против 2 л буфера (50 мМ Tris - HCl pH 8,0, 0,2% ZW3-12, 4oC) 4 раза. К образцу добавляют 5 мл Protrap (TAKARA SHUZO Co., ТД), и полученную смесь перемешивают при 8oC в течение 30 минут для удаления протеаз из образца и для стабилизации ER. Полученную смесь центрифугируют при 2800 об/мин при 4oC в течение 2 минут для того, чтобы собрать надосадочную жидкость. Полученную надосадочную жидкость (160 мл) концентрируют до примерно 50 мл, используя ультрафильтрационную мембрану VM-10 (Amicon Co. ), и концентрат диализуют против A-буфера (50 мМ Tris - HCl pH 8,0, 0,1 М сахарозы, 5 мМ MgCl2, 1 мМ PMSF, 5 мМ EDTA и 0,2% ZW3-12, 4oC).
Полученный диализат наносят на Q-Sepharose HP 26/10 (Pharmacia Co.), и ER элюируют с линейным градиентом 0-1 М NaCl (Q-Sepharose активная фракция). ER элюируют при концентрации NaCl около 0,45 М. Q-Sepharose активную фракцию разбавляют в 3 раза буфером A, и разбавленную фракцию наносят на Mono Q 10/10 (Pharmacia Co.). ER элюируют с линейным градиентом 0-1 М NaCl (Mono Q активная фракция, 8 мл). ER элюируют при концентрации NaCl около 0,25 М.
ER очищают с помощью афинного геля, используя активатор в качестве лиганда, следующим образом.
Активатор приготавливают в соответствии со способом N.T.Keen с некоторыми модификациями (Plant Physiol. (1983) 71:460, Plant Physiol. (1993) 71: 466). Короче, стенки мицелия патогенной Phytophthora megasperma f.sp. glycinea paca 1 (ATCC 34566) обрабатывают зимолиазой 100T (KJRJN BREWERY CO. , LTD) для высвобождения активатора. После обработки зимолиазу (Zymolyase 100T) удаляют с помощью СМ-целлюлозы, набитой в колонку. Полученную после пропускания через колонку фракцию очищают с помощью гель-проникающей хроматографии G75 (Pharmacia Co.) для отбора той фракции активатора, средний молекулярный вес которой составляет 10000 Да. Индуцирующую глицеолин активность активатора собранной фракции определяют по способу M.Yoshikawa (Nature (1978) 257:546). Добавление 8 мкг активатора к семядолям сои вызывает индуцирование около 550 мкг глицеоллина после 24 часов инкубирования.
Для исключения неспецифической адсорбции на геле-носителе, Mono Q активную фракцию собирают и перемешивают с примерно 33 мг соединенного с мальтозой стеклянного геля (объем слоя около 100 мкл) при 8oC в течение 1 часа. Этот гель осаждают центрифугированием (1000 об/мин, 4oC, 2 минуты) для сбора надосадочной жидкости (фракция, пропущенная через соединенный мальтозой стеклянный гель). Соединенный мальтозой стеклянный гель получают по способу А. М. Jeffrey et al. (Biochem. Biophys. Res. Commun. (1975) 62:608). Короче, 120 мг мальтозы и 6 г Glass Aminopropyl (Sigma Co.) суспендируют в 36 мл H2O, и полученную суспензию перемешивают при комнатной температуре в течение ночи. К полученной суспензии добавляют 36 мл этанола. Сразу после этого к смеси добавляют раствор боргидрида натрия (864 мг) в 72 мл этанола. Полученную смесь обрабатывают ультразвуком в течение 2 минут и перемешивают при комнатной температуре в течение 5 часов. Добавляют 288 мл воды к реакционной смеси, и полученную смесь охлаждают льдом и доводят до pH 5,6 уксусной кислотой. Этот гель промывают примерно 1,8 л H2O для удаления свободной мальтозы. Мальтозу, содержащуюся в промывочном растворе определяют количественно по способу J.H.Roe (J.Biol. Chem. (1955) 212:335), используя реагент антрон. Количество соединенной в геле мальтозы определяют, учитывая количество мальтозы, содержащееся в промывочном растворе. В результате оказывается, что 60 мг мальтозы соединилось с 6 г Glass Aminopropyl.
Около 17 мг соединенного с активатором стеклянного геля (объем слоя около 50 мкл) добавляют к 8 мл фракции, пропущенной через соединенный с мальтозой стеклянный гель, и полученную смесь осторожно перемешивают при 8oC в течение ночи. Гель собирают центрифугированием (1000 об/мин, 4oC, 2 минуты) и промывают 2 объемами слоя буфера A дважды. Этот гель промывают дополнительно 4 объемами слоя 0,1% SDS трижды для сбора соединенных с гелем ER (фракции, элюированные из соединенного с активатором стеклянного геля). Объединенный с активатором стеклянный гель получают по способу A.M.Jeffrey et al. (Biochem. Biophys. Res. Commun. (1975) 62:608). Короче, активатор (37 мг) и Glass Aminopropyl. (490 мг) суспендируют в 6 мл H2O и перемешивают при комнатной температуре в течение ночи. К суспензии добавляют 6 мл этанола, и немедленно после этого добавляют раствор боргидрида натрия (144 мг) в 12 мл этанола. Полученную смесь обрабатывают ультразвуком в течение 2 минут и перемешивают при комнатной температуре в течение 5 часов. К полученной смеси добавляют 48 мл воды. Эту смесь охлаждают льдом и pH устанавливают 5,6 за счет уксусной кислоты. Свободный активатор определяют количественно реагентом антроном. Количество объединенного с гелем активатора оценивают из количества свободного активатора, содержащегося в промывочном растворе. В результате оказывается, что 34 мг активатора соединилось с 490 мг Glass Aminopropyl.
Количества протеина и ER на вышеуказанных стадиях очистки представлены в таблице 1.
Mono Q активная фракция, фракция, пропущенная через соединенный с мальтозой стеклянный гель, и фракция, элюированная из соединенного с активатором стеклянного геля (по 10 мкл каждая), были обработаны электрофоретически на электрофоретическом градиентном геле, SDS-PAGE пластине 10/20 (Daiich Kagaku Jakuhin Co.) и окрашены серебром (Daiich Kagaku Jakuhin Co.). Картины электрофореза представлены на фиг. 1. На фиг. 1 полоса 1 представляет Mono Q активную фракцию; полоса 2 - пропущенную через соединенный с мальтозой стеклянный гель; полоса 3 - фракцию, элюированную из соединенного с активатором стеклянного геля.
Из фиг. 1 видно, что ER полосы детектируются с молекулярным весом около 70000 Да.
Протеин с молекулярным весом около 70000 метят иодом-125, используя при этом 125I-меченый комплекс фотоафинного реагента SASD (Pierce Co.) и активатор. SDS-PAGE полосу мембранной фракции переносят на PVDF мембрану с помощью Вестернблоттинга и инкубируют с тем же самым 125I-меченым активатором, который используют при определении активатор-связывающей активности ER на PVDF мембране так, чтобы протеин с молекулярным весом около 70000 Да был помечен иодом-125. Эти факты показывают, что протеин с молекулярным весом около 70000 Да обладает активатор-связывающей активностью.
Около 4 мкг ER выделяют из примерно 40 мг (влажный вес) соевых корней вышеуказанным способом.
3) Анализ ER-фрагментированных пептидов
ER фрагментируют за счет переваривания протеазой до пептидов. Аминокислотные последовательности фрагментированных пептидов определяют по способу Jwamatsu (Akihiro Jwamatsu, Seikagaku (1991) 63:139, A.Jwamatsu, Electrophoresis (1992) 13:142). Раствор ER, очищенных вышеуказанным способом, концентрируют до около 100 мкл на Centricon-30 (Amicon Co.) и обрабатывают электрофоретически на 10 - 20% SDS полиакриламидном геле. Полученные протеиновые полосы переносят на PVDF мембрану (Millipore Co.) с помощью аппарата электроблоттинга (Sartrius Co.). Перенесенные на PVDF мембрану полосы окрашивают 0,1% Ponceau S (Sigma Co.)/ 1% уксусной кислотой. Основную полосу с молекулярным весом 70000 Да вырезают и обесцвечивают 0,5 мМ NaOH. Эту полосу восстановительно S-карбоксиметилируют. К полученной полосе добавляют лизилендопептидазу (AP-1) при отношении энзим:субстрат (мол:мол) 1:100, и полученную смесь подвергают взаимодействию при 30oC в течение 16 часов. Полученные фрагментированные пептиды вводят в мк-Bondasphere 5 мкм C8-300 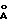 (2,1 х 150 мм Waters) колонку, уравновешенную 98% растворителя A и 2% растворителя B, и элюируют с 2 - 50% линейным градиентом растворителя B в течение 30 минут при скорости потока 0,25 мл/минуту (растворитель A: 0,05% TFA раствор, растворитель B: 0,02% TFA в 2-пропанол/ацетонитриле = 7:3 (объем/объем)). Элюированные пептиды определяют по поглощению на 214 нМ, фракции каждого пика собирают вручную. Полученные пиковые фракции анализируют на газофазном секвенаторе протеинов (Модель 470 A Applied Biosystem). В результате анализа всех полученных пиковых фракций точно определяют следующие аминокислотные последовательности фрагментированных пептидов:
(2,1 х 150 мм Waters) колонку, уравновешенную 98% растворителя A и 2% растворителя B, и элюируют с 2 - 50% линейным градиентом растворителя B в течение 30 минут при скорости потока 0,25 мл/минуту (растворитель A: 0,05% TFA раствор, растворитель B: 0,02% TFA в 2-пропанол/ацетонитриле = 7:3 (объем/объем)). Элюированные пептиды определяют по поглощению на 214 нМ, фракции каждого пика собирают вручную. Полученные пиковые фракции анализируют на газофазном секвенаторе протеинов (Модель 470 A Applied Biosystem). В результате анализа всех полученных пиковых фракций точно определяют следующие аминокислотные последовательности фрагментированных пептидов:
N 1: Val Asn Ile Gln Thr Asn Thr Ser Asn Ile Ser Pro Gln (N-конец)
N 5: Lys Ser Ile Asp Gly Asp Leu Val Gly Val Val Gly Asp Ser
N 6: Lys Tyr Lys Pro Gln Ala Tyr Ser Ile Val Gln Asp Phe Leu Asn Leu Asp
N 7: Lys Thr Asp Pro Leu Phe Val Thr Trp His Ser Ile Lys (смешанная последовательность).
Пример 2
Клонирование ER гена сои
Семена сои (Glycine max cv. Green Homer) (Takayama Seed Co.) культивируют на вермикулите в течение 1 недели, а затем ведут водную культуру в течение 15 дней для сбора корней (около 40 кг, влажный вес). Часть собранных корней хранят при -80oC до использования. Полную РНК получают по способу Ishida (Cell Technology Laboratory Manipulation Manual, Kodansha Scientific). Короче, хранившиеся корни (28,5 г влажный вес) измельчают в ступке, добавляя жидкий азот. К полученному порошку добавляют 35,6 мл GTC раствора, хранившегося при 65oC, и полученную смесь гомогенизируют в смесителе Waring. Полученную суспензию центрифугируют со скоростью 6000 об/мин при комнатной температуре в течение 15 минут до сбора 40 мл надосадочной жидкости. Надосадочную жидкость осторожно наносят на слой раствора цезия в центрифужной ампуле и центрифугируют со скоростью 35000 об/мин при 25oC в течение 20 часов. Полученный осадок растворяют в 9 мл TE/ 0,2% SDS. После того, как дважды проводят экстракцию смесью фенол/хлороформ, полную РНК выделяют осаждением этанолом (4,37 мг).
Полученную полную РНК (2,2 мг) очищают с помощью олиготекса (Oligotex dT30, Japan Roche Co.) в соответствии с указаниями изготовителей, до получения 60 мкг poly (A) + RNK.
2) Получение библиотеки кДНК сои
Молекулы кДНК синтезируют из 5 мкг poly (A) + RНК с помощью набора для синтеза кДНК (Pharmacia Co.) в соответствии с указаниями изготовителей. Синтезированные фрагменты кДНК лигируют с лямбда-фаговым вектором λ gt10 (Stratagene Co. ) с помощью T4 лигазы (TAKARA SHUZO CO., LTD). Для упаковки смеси ДНК в фаговые частицы используют Gigopack (Stratagene Co.) и получают кДНК библиотеку сои - примерно 1,5 • 106 б.о.е. Эту библиотеку амплифицируют до 160 мл библиотеки кДНК сои 1,6 • 1011 б.о.е./мл.
Полную ДНК в библиотеке кДНК получают следующим образом: смесь хлороформа и изоамилового спирта (24:1) добавляют к 500 мкл раствора фага (1,6 • 1011 б.о.е./мл) в равном количестве. Полученную смесь встряхивают в течение 30 секунд и центрифугируют для сбора водного слоя. Водный слой снова экстрагируют смесью хлороформа и изоамилового спирта (24:1). К полученному водному слою добавляют 5 мкл 3 М раствора ацетата натрия (pH 5,4) и 125 мкл этанола, и полученную смесь центрифугируют для сбора осадка. Полученный осадок промывают 70% этанольным раствором и растворяют в 10 мМ Tris -HCl раствора (pH 8), содержащего 1 мкг/мл RN-ase A (RNA e A, Sigma Co.). Полученный раствор используют в качестве PCR матрицы.
3) Амплификация и клонирование кДНК-фрагментов ER сои за счет PCR
Нижеследующие четыре олигодеоксинуклеотида (смешанные праймеры U5, U7, U10 и U12) синтезируют с помощью автоматического синтезатора нуклеиновых кислот (модель 394 Applied Biosystems Co.) на основании аминокислотных последовательностей фрагментированных пептидов, полученных в примере 1 (N 5 и N 6):
Праймер 5 5'-AARAGYATHGAYGGNGA-3'
Праймер 7 5'-WRTCNCCNACNAC-3'
Праймер 10 5'-GTNAAYAARATNCARAC-3'
Праймер 12 5'-ARRTTNAGRAARTCYTC-3'
(R:A/G, Y:C/T, W:A/T, H:A/C/T, N:A/G/T/C)
Полную ДНК в 0,5 мкг кДНК библиотеки растворяют в 79 мкл дистиллированной воды. К 8 мкл 2,5 мМ dNTP в 10 мкл 10хPCR буфера (присоединенного к Tag ДНК полимеразе TAKARA SHUZO CO., LTD) добавляют либо комбинацию праймеров U5 и U7, либо комбинацию праймеров U10 и U12 (по 100 пмолей каждого) и 0,5 мкл Tag ДНК полимеразы (TAKARA SHUZO CO., LTD) до конечного объема 100 мкл. PCR реакцию ведут с помощью Gene Amp. PCR System 9600 (Perkin - Elmer Co.) в 50 циклов 1) денатурирование при 94oC х 30 секунд, 2) ренатурирование при 47oC х 30 секунд и 3) удлинение при 72oC х 1 минуту. После реакции 15 мкл реакционного раствора обрабатывают электрофоретически на 15% полиакриламидном геле. Этот гель подкрашивают 0,5 мкг/мл этидиумбромидного раствора в течение 10 минут. Полосы, демонстрирующие специфические амплифицированные фрагменты 40bp и 47bp, амплификацию которых ожидали, вырезают, наблюдая под УФ-светом. Кусочки геля снимают пластиковым стержнем и элюируют элюирующим буфером (0,5 М ацетат аммония, 10 мМ ацетат магния, 1 мМ ЕДТА и 0,10 SDS) в течение ночи для того, чтобы собрать ДНК-содержащий раствор.
Собранные ДНК-фрагменты клонируют в плазмиду pT7Blue (R) с помощью pT7Blue T-Vector набора (Novagene Co.). Полученные плазмиды pN 5 - 1, 2 и pN 6 - 1, 2, 3, 4, 5, 6, 7, 8 и 9 секвенируют на флуоресцентном автоматическом ДНК секвенаторе (модель 373A Applied Biosystem Co.). Полученные результаты показывают, что полученные амплифицированные ДНК-фрагменты, отличные от праймеров, также кодируют аминокислотные последовательности фрагментированных пептидов N 5 и N 6.
Два нижеследующих олигодеоксинуклеотида (смешанные праймеры U18 и U19) были синтезированы с помощью автоматического синтезатора нуклеиновых кислот на основании результатов ДНК секвенирования.
Праймер U18 5'-AAGTAYAAGCCRCAAGCCTATTCA-3'
Праймер U19 5'-ATCGCCRACAACMCCAA-3'
(Y и R имеют указанные ранее значения, M:A/C).
Полную ДНК в 0,5 мкг кДНК-библиотеки растворяют в 79 мкл дистиллированной воды. Комбинацию праймеров U18 и U19 (100 пмолей каждого) и 0,5 мкл Tag ДНК полимеразы добавляют к 8 мкл 2,5 мМ dNTP в 10 мкл 10хPCR буфера до получения конечного количества 100 мкл. PCR реакцию осуществляют в 40 циклов 1) денатурирование при 94oC х 30 секунд, 2) ренатурация при 52oC х 30 секунд и 3) удлинение при 72oC х 1 минуту. Пятнадцать микролитров реакционного раствора обрабатывают электрофоретически на 1% агарозном геле.
Гель окрашивают 0,5 мкг/мл этидиумбромидного раствора в течение 15 минут. Полосу, демонстрирующую специфический амплифицированный фрагмент около 540bp вырезают, наблюдая под УФ-светом. Кусочки геля обрабатывают Gene Clean 11 (Bio 101 Co.) для сбора ДНК-содержащего раствора.
Собранный ДНК- фрагмент клонируют в плазмиду pT7Blue (R) с помощью pT7Blue T-Vector набора. Полученную плазмиду pN 5 - pN 6 секвенируют флуоресцентным секвенатором. Полученные результаты показывают, что амплифицированный ДНК-фрагмент состоит из 539 bp и кодирует не только аминокислотные последовательности фрагментированных пептидов N 5 и N 6 с обеих сторон, но также и пептида N 7 в амплифицированной части.
4) Скринирование и клонирование библиотеки за счет гибридизации.
Плазмиду N 5 - N 6, в которую был клонирован ER кДНК- фрагмент, переваривают рестрикционными энзимами Bam H1 и Pst 1. Выделяют ДНК фрагмент около 540bp и используют в качестве зонда. Выделенный ДНК-фрагмент метят [α-32P] dCTP с использованием системы введения метки в ДНК Megaprime (Amersham Co.) в соответствии с инструкциями изготовителей, и реакционный раствор используют в экспериментах по гибридизации.
Фаг кДНК-библиотеки инфицируют E.coli C600 hfl (Invitrogen Co.) и инокулируют в 10 мг/мл MgCl2-содержащей Z-среды на пластинах диаметром около 15 см, до получения всего около 1 • 106 бляшек. Осуществляют блоттинг бляшек на нейлоновой мембране (Hybond-N; Amersham Co.). Мембрану подвергают взаимодействию с 32P-dCTP меченым ER кДНК-фрагментом, и положительные фаги определяют ауторадиографически, затем их снова скринируют тем же способом до получения около 30 фаговых клонов, обладающих различными интенсивностями сигналов. Клон λ ER 23 (PA23), содержащий самый длинный вставленный ДНК-фрагмент, отбирают.
ДНК-молекулу λ-фага выделяют с помощью Lambdasoro (Promega Co.) из раствора позитивного клона λ ER23, выделенного в эксперименте по гибридизации. 10 мкл 10 х EcoR1 расщепленного буфера (рестрикционный энзим EcoR1 10 Ед.) добавляют к 5 мкг ДНК раствора до получения полного количества 100 мкл, и полученную смесь подвергают взаимодействию при 37oC в течение ночи. Реакционный раствор обрабатывают электрофоретически на 1% агарозном геле. Вырезают полосу около 2,3 кв и обрабатывают Gene Clean II (Bio 101 Co) для сбора ДНК-содержащего раствора. Вектор p Blue Script II KS - (0,02 мкг) (Stratagene Co) расщепляют рестрикционным энзимом EcoRI.
После того как смешивают два ДНК раствора, 2 мкл 10 х лигазного буфера и 0,2 мкл Т4 ДНК лигазы (TAKARA SHUZO CO., LTD.), добавляют до полного объема 20 мкл. Полученную смесь подвергают взаимодействию при 16oC в течение 4 часов, и реакционный раствор используют для трансформации E.coli DH5α (Gibco BRL Co. ). Приготавливают пластину 2% агарной среды с 25 мл Z-среды, содержащей 50 мкг/мл ампициллина, 40 мкг/мл YPTG и 40 мкг/мл X-gal. Трансформированную E. coli инокулируют на пластину агарной среды и культивируют при 37oC в течение ночи. Из образовавшихся колоний отбирают белые колонии, и их культивируют в 3 мл Z-среды, содержащей 50 мкг/мл ампициллина при 37oC в течение 8 часов. Из этих бактериальных клеток выделяют плазмиды за счет щелочного способа, и определяют, являются ли они клонами, в которые был клонирован целевой фрагмент с рестрикционным энзимом, получая при этом плазмиды pER23-1 и pER23-2 (5225 bp), которые ориентированы в сторону, противоположную вектору. На фиг. 2 представлены карты плазмид pER23-1 и pER23-2.
5) Определение нуклеотидной последовательности ER-кодирующего клона.
ДНК-нуклеотидные последовательности плазмид pER23-1 и pER23-2 были определены в обеих ориентациях с помощью флуоресцентного секвенатора 1) используя плазмиды pER23-1 и pER23-2, переваренные соответствующими рестрикционными энзимами, 2) используя соответствующие праймеры, синтезированные на основе информации об уже определенных нуклеотидных последовательностях, или 3) расщепляя pER23-1 рестрикционными энзимами Kpn 1 и Xhol, и pER23-2-рестрикционными энзимами Kpn 1 и Clal, а затем используя набор Kilosequence deletion (TAKARA SHUZO CO. , LTD) для получения плазмид, с делецией с интервалами около 200 - 300 bp. ДНК-нуклеотидная последовательность представлена в последовательности ID N 2 в разделе "Описание последовательностей". Полученные результаты показывают, что ДНК-фрагмент содержит 667 аминокислот, кодирующих открытую рамку считывания из 2001 bp, начиная у нуклеотидной последовательности, соответствующей N-концевой последовательности (фрагментированный пептид N 1), секвенированной с помощью аминокислотного секвенатора. Аминокислотная последовательность представлена в последовательности ID N 1 в разделе "Описание последовательностей". Аминокислотная последовательность, построенная на основе полученных ДНК-нуклеотидных последовательностей, соответствует определенной ранее аминокислотной последовательности ER сои.
Кроме того, были проведены поиски в высокой степени гомологичных аминокислотных последовательностей с помощью блока программного обеспечения для анализа последовательностей нуклеиновых кислот и аминокислот (Macvector:Kodak Co.), используя базу данных последовательностей аминокислот и нуклеиновых кислот (Entrez:NCBI). Однако было обнаружено, что ни одна из аминокислотных последовательностей не была в высокой степени гомологична известным к этому моменту последовательностям. Следовательно, становится очевидным, что полученный ER является новым протеином.
Пример 3
Экспрессия ER сои в растениях табака
1) Конструирование растительной экспрессионной плазмиды pKV1-ER23.
Как представлено на фиг. 3, вектор экспрессии в растения pKV1, который используют в данном примере, получают из плазмиды pCaP35J, содержащей промотор вируса 35S мозаики цветной капусты (J. Yamaya et al., (1988) Mol. Gen. Cenet. 211:520) следующим образом.
Плазмиду pCaP35J полностью переваривают рестрикционным энзимом BamHI для того, чтобы удалить сайт мультиклонирования, представленный в обратном направлении от 35S промотора. После частичного переваривания Pvull, проводят обработку фрагментами Кленова (TAKARA SHUZO CO., LTD) для получения тупых концов. Полученную плазмидную ДНК замыкают лигированием и вводят в E.coli DH5α. Целевую плазмиду отбирают из полученных клонов. Отобранную плазмиду переваривают рестрикционным энзимом Pst1 для встраивания сайта мультиклонирования, присутствующего в прямом направлении от 35S промотора. Проводят обработку фрагментами Кленова для получения тупых концов. Полученную плазмидную ДНК переваривают Hind III. С помощью автоматического синтезатора нуклеиновых кислот синтезируют следующие синтетические линкерные ДНК, отжигают и лигируют с Hind III переваренной плазмидой. Полученную плазмидную ДНК вводят в E.coli DH5α. Целевую плазмидную pCaP35Y (2837 bp) отбирают из полученных клонов.
5'-GGAATTCGAGCTCGGTACCCGGGGGATCCTCTAGAGTCGACCTGCAGGCATGCA-3' (последовательность ID N 3)
5'-CCTTAAGCTCGAGCCATGGGCCCCCTAGGAGATCTCAGCTGGACGTCCGTACGTTCGA-3' (последовательно ID N 4).
Для введения терминатора нопалин-синтазы в pCaP35Y, плазмиду pBI121 (Clontech Co.) переваривают Sacl и EcoR1, и Sacl-EcoRI фрагмент обрабатывают фрагментами Кленова для затупления концов; затем полученный фрагмент pB1121 лигируют с плазмидой pCaP35Y, в которой тупые концы образованы по Hind III сайту в прямом направлении от 35S промотора. Полученную плазмидную ДНК вводят в E.coli ДН5 α. Из полученных клонов отбирают целевую плазмиду. Для введения кассеты устойчивости к канамицину в отобранную плазмиду, последнюю переваривают PvuII и лигируют с фрагментом (около 1620bp) pLGVneo 1103 (R. Hain et al. (1985) Mol. Gen. Genet. 199:161), которую получают в стадиях расщепления по PvuII сайту, расположенному в прямом направлении от терминатора октопии-синтазы, обрабатывают Ba131 (TAKARA SHUZO CO., LTD) до получения делеции, расщепляют по EcoRI сайту в обратном направлении от промотора нопалин-синтазы и создают тупые концы с обеих сторон. Полученную плазмидную ДНК вводят в E.colii ДН5α. Целевую плазмиду или вектор экспрессии в растения pKV1 (4828 bp) отбирают из полученных клонов.
Полученную pKV1 переваривают по уникальному сайту рестрикционными энзимами BamHI и SaII, и лигируют в содержащий ER ген фрагмент (то есть BamHI, SaII фрагмент pEB23-1, около 2,3 kbp). Полученную плазмидную ДНК вводят в E. coli ДН5α. Целевую ER-экспрессионную плазмиду pKV1-ER23 (около 7,1 kbp) отбирают из полученных клонов.
2) Кратковременная экспрессия ER в культивируемых клетках табака.
ER-ген вводят в культивируемые клетки табака за счет электропорации для кратковременной ER-экспрессии с помощью частично модифицированного метода Ватанабэ (J. Watanabe (1987) FEBS 219:65). Молекулы ДНК плазмиды pKV1-ER23 очищают щелочным методом. Культивируемые клетки табака получают по способу Hirai et al. (Plant Cell Cultivation Manual, Gakkai Shuppan Center 1982) для использования в кратковременной экспрессии ER. Семена табака (сорт Bright Yellow, полученные от профессора Hirofumi Uchimiya из университета в Токио) стерилизуют 1% раствором гипохлорита, а затем проращивают. Ювенильные ткани табака сразу после прорастания переносят в культуральную агарную среду табака (Murashige - Skoog среда (Flow Laboratories Co.) дополненную 2 ч. на млн. 2,4-дихлорфеноксиуксусной кислоты, 3% сахарозы и 8% агара) для стимуляции образования каллюсов в течение 3 недель. Около 1 г каллюсной массы суспендируют в 50 мл культуральной среды табака (Murashige - Skoog среда / Flow Laboratories Co.), дополненной 2 ч. на млн. 2,4-дихлорфеноксиуксусной кислоты и 3% сахарозы) для подготовки культивируемых клеток. Эти клетки табака культивируют до тех пор, пока они не вступают в логарифмическую фазу развития. Культивируемые клетки собирают центрифугированием (600 об/мин, 3 минуты) и суспендируют в растворе, состоящем из 1% целлюлазы Ohozuka (Yakult Co.), 1% Дрицелазы (Dricelase Kyowa Hakko Co. LTD), 0,1% Пектриазы (Pectriase, Seishin Seiyaku Co.) и 0,4 М D-маннитом (Wako Pure Chemicals Co. , LTD), и pH доводят до 5,7 за счет HCl. Реакцию ведут при 30oC в течение 90 минут до получения протопластов. Реакционный раствор промывают 0,4 М D-маннитом при 4oC тремя циклами центрифугирования
для удаления энзимного раствора. Операция электропорации состоит в суспендировании 1•106 клеток в 0,8 мл электропорационного раствора (70 мМ KCl, 5 мМ MES и 0,3 М маннита), смешивания суспензии с 10 мкг молекул ДНК pKV1-ER23 и обработке смеси в genepluser (Biorad Co.) в условиях 125 мкф и 300 B в электропорационной кювете (Biorad Co., расстояние между электродами 0,4 см). После обработки полученный раствор собирают пастеровской пипеткой и оставляют выстаиваться на льду в течение 30 минут. Реакцию ведут при 30oC в течение 5 минут, и реакционный раствор повторно суспендируют в протопластовой среде (среда Murashige - Skoog/ Flow Laboratories Co.), дополненной 0,2 ч. на млн. 2,4-дихлорфеноксиуксусной кислоты, 1% сахарозы и 0,4 М маннита и pH установлена 5,7). Клетки оставляют выстаиваться в темноте при 25oC в течение ночи и собирают центрифугированием (8000 об/мин, 3 минуты). 60 микролитров суспензионного буфера (25 мМ Tris - HCl pH 7,0, 30 мМ MgCl2, 2 мМ дитиотреитола, 2,5 мМ метабисульфита калия и 1 мМ PMSF) добавляют к клеткам, и полученную смесь перемешивают на Vortex в течение 3 минут. Полученный образец хранят при -80oC до проведения экспериментов по связыванию активатора.
Для контроля повторяют вышеуказанную процедуру, за исключением того, что ДНК-молекулу pKV1 вместо pKVI-ER23 вводят в клетки табака.
3) Стабильная экспрессия ER в культивируемых клетках суспензии табака
Трансформированные культивированные клетки табака, способные постоянно удерживать ген ER, отбирают из культивируемых клеток табака, способных к кратковременной ER экспрессии, следующим образом.
Протопласты, полученные при получении культивируемым клеток табака, способных к кратковременной ER-экспрессии, суспендируют в 1% агарозе, содержащей протопластную среду (Murashige - Skoog/ Flom Laboratories Co.), дополненную 0,2 ч. на млн. 2,4-дихлорфеноксиуксусной кислотой, 1% сахарозы и 0,4 М маннита, и доведенной до pH 5,7). Суспензию накапывают на пластину с помощью капельной пипетки перед тем, как агароза отверждается, в результате чего протопласты фиксируются в шарикоподобной твердой среде. После отверждения агарозы, не содержащую агарозы протопластную среду добавляют на пластину, за счет чего происходит погружение протопластфиксирующей агарозной среды в жидкую среду. После того как протопласты культивируют в темноте в течение 1 недели, добавляют канамицин до конечной концентрации 100 мкг/мл и культивирование продолжают. Трансформанты, отобранные из выросших колоний, переносят в канамицинсодержащую жидкую среду и культивируют.
Получают два клона (I1 и I2) культивированных клеток табака, стабильно трансформированных pKV1-ER23 и два клона (C2-1 и C2-4) культивированных клеток табака, стабильно трансформированных pKV1.
4) Эксперимент определения активности связывания активатора
Активность связывания активатора определяют следующим образом.
Комплекс активатора и тирамина (TOKYO KAS E1 KOGYO CO., LTD) был синтезирован по способу Jong-Joo Cheong (The Plant Cell (1991) 3:127). Комплекс активатор-тирамин метят иодом-125, используя хлорамин T. Полученный образец (количество протеина менее 500 мкг) суспендируют в 500 мкл аналитического буфера (50 мМ Tris - HCl pH 7,4, 0,1М сахарозы, 5 мМ MgCl2, 1 мМ PMSF и 5 мМ EDTA) и инкубируют при 0oC в течение 2 часов. Меченый иодом комплекс активатор-тирамин в количестве 100 нМ (70 кюри/ммоль) добавляют к суспензии, и полученную смесь инкубируют при 4oC в течение 2 часов. Реакционный раствор фильтруют через Watman GF/B (обработанный 0,3% водным раствором полиэтиленимина в течение по крайней мере 1 часа), промывают трижды 5 мл буфера, охлажденного льдом (10 мМ Tris - HCl pH 7,0 1М NaCl, 10 мМ MgCl2). Радиоактивность, удержанную на фильтре-мембране определяют с помощью гамма-счетчика (отсчет A). Для того чтобы исключить эффект неспецифического связывания, указанную ранее процедуру повторяют за исключением того, что к тому же самому образцу добавляют 17 мкМ активатора, полученную смесь суспендируют в аналитическом буфере, и полученную суспензию инкубируют при 0oC в течение 2 часов. Полученную величину вычитают из отсчета A до получения величины (дельта импульсов в минуту), характеризующей активатор-специфическое связывание. Полученную величину ( Δ срм) делят на полное количество импульсов, а затем умножают на полное количество активатора, использованного в эксперименте, чтобы рассчитать количество активатор-связывающего протеина (в молях).
В результате специфическое связывание с активатором наблюдают в клетках табака, трансформированных молекулой ДНК pKV1-ER23, тогда как никакого специфического связывания с активатором не наблюдается в контрольных клетках табака, в которые была введена молекула ДНК pKV1 (таблица 2). Этот факт позволил выявить, что полученный таким образом ген кодирует протеин, обладающий активатор-связывающей активностью.
5) Кратковременное повышение внутриклеточной Ca2+ концентрации в трансформированных культивированных клетках табака за счет добавления активатора - глюкана.
Растения распознают активатор у рецептора, который ему специфичен, и затем промотируют накопление фитоалексина или индуцируют гиперчувствительную реакцию для предотвращения инвазии грибков. Сообщалось для некоторых растений, что поступление ионов кальция в клетки на ранней фазе таких реакций сопротивления является важным (U.Conrath et al., (1991) FEBS LETTERS 279: 141, M.N. Zook et al. (1987) Plant Physiol 84:520, F.Kurosaki et al., (1987) Phytochemistry 26: 1919; C.L. Preisig and R.A. Moreau (1994) Phytochemistry 36: 857). Сообщалось также о предположении, что поступление ионов кальция в клетки стимулирует промотирование накопления фитоалексина в сое, что используют авторы изобретения для получения ER (M.R. Stab and Ebel (1987) Archi Biochem. Biophys 257: 416). Следовательно, если трансформированные культивируемые клетки табака получают, вводя ER-ген в не содержащие ER культивируемые клетки табака для экспрессии ER и если изменяют внутриклеточную концентрацию ионов кальция за счет добавления глюканового активатора, такое изменение препятствует стимулированию реакции сопротивления за счет глюканового активатора у растений, отличающихся от сои (например, у табака), что позволяет им тем самым продемонстрировать устойчивость к широкому кругу грибков, которые используют глюкан в качестве компоненты стенок мицелия.
Исследовали изменение концентрации внутриклеточного Ca2+ в трансформированных культивируемых клетках табака за счет добавления активатора.
В этом эксперименте используют трансформированные культивируемые клетки табака (I6), полученные с помощью селекции канамицином, и плазмидусодержащие культивируемые клетки табака (C2 - 4).
Внутриклеточные концентрации Ca2+ в культивируемых клетках определяют следующим образом с помощью ацетоксиметильного производного (Fura-2 AM) флюоресцентного хелатора (Fura - 2) для измерения концентрации Ca2+:
Клетки собирают с примерно 2 мл культуры трансформированных клеток табака (что соответствует объему клеток около 250 мл после выстаивания в течение 10 минут) за счет центрифугирования (600 об/мин, 30 секунд), и образующуюся надосадочную жидкость удаляют. К этим клеткам добавляют 2 мл культуральной среды табака, и полученную смесь перемешивают осторожно и центрифугируют (600 об/мин, 30 сек) для удаления надосадочной жидкости. Те же самые операции повторяют для того, чтобы промыть культивируемые клетки. Промытые культивируемые клетки гомогенно суспендируют в 2 мл среды. К 1 мл суспензии культивируемых клеток в среде добавляют 1 мл среды и 4 мкл 1 мМ Fura-2 AM (конечная концентрация: 2 мкМ, Dojin Chemical Co.), и полученную смесь инкубируют в темноте в течение 30 минут, иногда перемешивая ее. Затем клетки промывают 2 раза 2 мл среды с помощью центрифугирования (600 об/мин, 30 сек) для исключения свободного Fura-2 AM, который не включился в клетки. Промытые культивируемые клетки гомогенно суспендируют в 2 мл среды, и 2 мл этой среды переносят в ячейку для измерения флуоресценции. Включенные Fura-2 AM должны быть изменены на Fura-2 за счет гидролиза внутриклеточной эстеразой. Флуоресценцию за счет связывания Fura-2 с внутриклеточным Ca2+ определяют на длине волны флуоресценции 505 нм, при длине волны возбуждающего света 335 нм, причем культивируемые клетки перемешивают, чтобы предотвратить осаждение культивируемых клеток. Излучают изменение концентрации внутриклеточного Ca2+, измеряя интенсивность флуоресценции через определенные промежутки времени после добавления 50 мкл глюканового активатора (1 мг/мл) или деионизированной воды к культивируемым клеткам. Для контроля определяют изменения концентрации внутриклеточного Ca2+ для плазмидусодержащих культивируемых клеток табака описанным ранее способом. В качестве другого контроля исследуют изменение концентрации внутриклеточного Ca2+ для культивируемых клеток сои тем же способом, что и ранее, за исключением того, что культивируемые клетки промывают средой для клеток сои следующего состава:
NaH2PO4•H2O 75 мг/мл, KH2PO4 170 мг/мл, KNO3 2200 мг/мл, NH4NO3 60 мг/мл, (NH4)2SO4 67 мг/мл, MgSO4•7H2O 310 мг/мл, CaCl2•2H2O 295 мг/мл, FeSO4•7H2O 28 мг/мл, EDTA•Na2 37,3 мг/мл, KI 0,75 мг/мл, MnSO4•4H2O 10,0 мг/мл, H3BO3 3,0 мг/мл, ZnSO4•7H2O 2 мг/мл, Na2MoO4• 2H2O 0,25 мг/мл, CuSO4•5H2O 0,025 мг/мл, CoCl2•6H2O 0,025 мг/мл. Инозитол 100 мг/мл, никотиновая кислота 1,0 мг/мл, пиридоксин•HCl 1,0 мг/мл, тиамин•HCl 10,0 мг/мл, глюкоза 5 г/мл, сахароза 25 г/мл, ксилоза 250 мг/мл, пируват натрия 5,0 мг/мл, лимонная кислота 10,0 мг/мл, малеиновая кислота 10,0 мг/мл, фумаровая кислота 10,0 мг/мл, N-Z-амин 500,0 мг/мл, 2,4-дихлорфеноксиуксусная кислота 1,0 мг/мл и зеатинрибозид (Zeatine riboside) 0,1 мг/мл, pH доводят до 5,7 за счет КОН.
В результате этого эксперимента наблюдается около 7% кратковременного возрастания интенсивности флуоресценции в культивируемых клетках сои через 3 минуты после добавления активатора, тогда как вовсе никакого изменения не наблюдается после добавления деионизированной воды (фиг. 4). Полученные результаты дают возможность предположить, что явление, в котором можно наблюдать связывание ER с активатором-глюканом, вызвано кратковременным поступлением Ca2+ в клетки, что подтверждает сообщение о том, что ионы кальция играют важную роль в реакции устойчивости, вызываемой активатором в культивируемых клетках сои. В трансформированных культивируемых клетках табака наблюдается около 10% возрастания интенсивности флуоресценции через 3 минуты после добавления активатора, тогда как никакого изменения не наблюдается после добавления деионизированной воды.
В случае плазмидусодержащих культивируемых клеток табака, не наблюдается никаких изменений интенсивности флуоресценции после добавления активатора (фиг. 5).
Эти результаты показывают, что растения, отличные от сои (например, табак), которые не являются реакционноспособными по отношению к глюкановому активатору, приобретают реакционную способность за счет введения гена, полученного из сои рецептора глюканового активатора для экспрессии ER. Хотя характер сигнальной трансдукции для каждого из растений не совсем ясен, ожидается, что растения, отличающиеся от табака, приобретут реакционную способность по отношению к глюкановому активатору (т.е. кратковременное возрастание концентрации внутриклеточного Ca2+) за счет введения гена настоящего ER для ER-экспрессии, тем самым обеспечивая возможность получения растений, обладающих устойчивостью к широкому кругу грибков, которые используют глюкан в качестве компоненты стенок мицелия.
Пример 4
Экспрессия ER сои в E.coli и определение активатор-связывающего домена.
1) Экспрессия активатор-связывающего домена в E.coli.
Протеин слияния частичного фрагмента ER сои с протеином, связывающим мальтозу (MBP), получают с помощью системы для слияния и очистки протеинов (Protein Fusion and Purification System, New England Biolabs Co.) для экспрессии частичного фрагмента ER сои в E.coli. PCR проводят, используя pER23 в качестве матрицы для получения фрагментов ДНК различной длины. Создают праймеры для получения MBP и протеина слияния в клонировании в плазмиду pMAL-c2 (New England Biolabs Co.), добавляя BamHl сайт с 5' стороны и Sall сайт с 3' стороны, внешней по отношению к молекуле ДНК, кодирующей полной длины участок и фрагмента ER сои, представленные на фиг. 6. Эти праймеры синтезируют с помощью автоматического синтезатора нуклеиновых кислот (модель 394 Applied Biosystems Co.). Для амплификации цепи ДНК используют следующие праймеры.
Праймер 35 5'-ATGGATCCATGGTTAACATCCAAACC-3',
Праймер 36 5'-ATGGATCCGAATATAACTGGGAGAAG-3',
Праймер 37 5'-ATGGATCCCCAGCATGGGGTAGGAAG-3',
Праймер 38 5'-TAGTCGACTACTTCTCCCAGTTATATTC-3'
Праймер 39 5'-TAGTCGACTACTTCCTACCCCATGCTGG-3',
Праймер 40 5'-TAGTCGACTATTCATCACTTCTGCTATG-3',
Праймер 41 5'-ATGGATCCGCCCCACAAGGTCCCAAA-3' и
Праймер 42 5'-ATGGATCCAATGACTCCAACACCAAG-3'.
Молекулы ДНК pER23-1 (0,01 мкг) растворяют в 79 мкл дистиллированной воды. К 8 мл 2,5 мМ dNTP в 10 мкл 10хPCR буфера (присоединенного к Tag ДНК полимеразе TAKARA SHUZO CO.,ZTD) добавляют либо комбинацию праймеров U5 и U7, либо комбинацию праймеров U10 и U12 (по 100 пмолей каждого) и 0,5 мкл Tag ДНК полимеразы (TAKARA SHUZO CO., LTD) до получения конечного объема 100 мкл. PCR реакцию ведут с помощью системы Gene Amp PCR System 9600 (Perkin - Elmer Co. , ) в 30 циклов 1) денатурирования при 94oC 30 секунд, 2) ренатурирования при 55oC х 30 секунд и 3) удлинения при 72oC х 1 минуту. После реакции 15 мкл реакционного раствора переваривают рестрикционными энзимами BamHl и Sall и обрабатывают электрофоретически на 1% агарозном геле.
Этот гель окрашивают 0,5 мкг/мл раствора этидиумбромида в течение 15 минут. Полосу, которая демонстрирует ожидаемую специфическую амплификацию, вырезают, наблюдая под УФ-светом. Этот кусочек геля обрабатывают Gene Clean II (Biol 01 Co. ) для сбора ДНК-содержащего раствора. Собранные ДНК фрагменты клонируют в BamHl-Sall сайт плазмиды pMAL-c2, и полученные клоны вводят в E. coli DH5α.
2) Получение растворимой фракции протеинов из E.coli
E. coli клетки, в которые были введены плазмиды, предварительно культивируют в экспрессионную среду (10 г/л тритона (Gibco Co.), 5 г/л дрожжевого экстракта (Gibco Co.), 5 г/л NaCl, 2 г/л глюкозы и 100 мкг/мл ампициллина). Предварительно культивированный раствор (0,4 мл) добавляют к 40 мл экспрессионной среды и культивируют при 37oC при встряхивании до тех пор, пока не достигают значения оптической плотности ОД600 = 0,55. К культуральному раствору добавляют изопропилтиогалактозид до конечной концентрации 0,3 мМ и продолжают культивирование при встряхивании в течение еще 4 часов для того, чтобы вызвать экспрессию. E. coli клетки собирают центрифугированием, и клетки E.coli промывают промывочным буфером (20 мМ Tris-HCl, pH 7,4, 200 мМ NaCl и 1 мМ EDTA). Клетки обрабатывают ультразвуком в течение всего 2 минут (5 сек • 8). К обработанным ультразвуком клеткам добавляют ZW3-12 до конечной концентрации 0,25%, и полученную смесь инкубируют при 4oC в течение 30 минут. Надосадочную жидкость собирают центрифугированием (10000 об/мин, 5 минут) до получения фракции растворимого протеина E.coli. Экспрессию протеина слияния подтверждают с помощью иммуноблоттинга, используя антитело к анти-мальтозо-связывающему протеину (New England Biolabs Co.).
3) Эксперимент по связыванию активатора
Активность связывания активатора определяют следующим образом.
Комплекс активатора и тирамина (TAKYO KASEI KOGYO CO, LTD) синтезируют по способу Jong-Joo Cheong (The Plant Cell (1991) 3:127). Комплекс активатора и тирамина метят иодом-125, используя хлорамин T. Полученный образец (количество протеина менее 800 мкг) суспендируют в 500 мкл аналитического буфера (50 мМ Tris - HCl pH 7,4, 0,1 М сахарозы, 5 мМ MgCl2, 1 мМ PMSF и 5 мМ EDTA) и инкубируют при 0oC в течение 2 часов. Меченый иодом комплекс активатор-тирамин в количестве 100 нМ (70 Кюри/ммоль) добавляют к суспензии, и полученную смесь инкубируют при 4oC в течение 2 часов. Реакционный раствор фильтруют через Watman GF (обработанный 0,3% водным раствором полиэтиленимина в течение по крайней мере 1 ч) и трижды промывают 5 мл охлажденного льдом буфера (10 мМ Tris - HCl pH 7,0 1 М NaCl, 10 мМ MgCl2). Радиоактивность, удержанную на фильтровальной мембране, подсчитывают с помощью гамма-счетчика (отсчет A). Для того чтобы исключить эффект неспецифического связывания, ту же процедуру, что и ранее осуществляют, за исключением того, что к тому же самому образцу добавляют 17 мкМ активатора, полученную смесь суспендируют в аналитическом буфере, и полученную суспензию инкубируют при 0oC в течение 2 часов. Полученную величину вычитают из отсчета A для получения величины ( Δ срм = количество импульсов в минуту) специфического связывания активатора. Полученную величину ( Δ срм) делят на полное количество импульсов, а затем умножают на полное количество активаторов, использованных в эксперименте, для подсчета количества протеина, связывающего активатор (в молях).
В результате специфическое связывание с активатором наблюдают для E. coli, трансформированных молекулами ДНК, кодирующими ER (фиг. 6). Следовательно, это является новым подтверждением того, что полученный ген кодирует протеин, обладающий активатор-связывающей активностью, и было показано, что существует активатор- связывающий домен в последовательности 239-442 аминокислот, последовательности ID N 1.
Пример 5
Ингибирование связывания глюканового активатора с активатор-связывающим протеином в мембранной фракции семядолей сои и ингибирование накопления фитоалексина в семядолях сои за счет антител против активатор-связывающего домена
1) Экспрессия активатор-связывающего домена в E.coli
Протеин слияния активатор-связывающего домена, полученного из ER, с мальтозо-связывающим протеином (MBP) получают с помощью системы слияния и очистки протеинов (Protein Fusion and Purification System, New England Biolabs Co. ) для экспрессии большого количества активатор-связывающего домена в E. coli. PCR ведут для получения молекулы ДНК, кодирующей активатор-связывающий домен. Нижеследующие праймеры синтезируют с помощью автоматического синтезатора нуклеиновых кислот (модель 394 Applied Biosystems Co.):
Праймер 36 5'-ATGGATCCGAATATAACTGGGAGAAG-3' и
Праймер 39 5'-TAGTCGACTACTTCCTACCCCATGCTGG-3'
Молекулы ДНК pER23-1 (0,01 мкг) растворяют в 79 мкл дистиллированной воды. К 8 мкл 2,5 мМ dNTP в 10 мкл 10хPCR буфера (присоединенного к Tag ДНК полимеразе TAKARA SHUZO CO., LTD) добавляют либо комбинацию праймеров U5 и U7, либо комбинацию праймеров U10 и U12 (по 100 пмолей каждого) и 0,5 мкл Tag ДНК полимеразы (TAKARA SHUZO CО., LTD) до получения конечного количества 100 мкл. PCR ведут с помощью системы Gene Amp PCR System 9600 (Perkin-Elmer Co.) в 30 циклов 1) денатурирования при 94oC х 30 секунд, 2) ренатурирования при 55oC х 30 секунд и 3) удлинения при 72oC х 1 минуту. После реакции 15 мкл реакционного раствора переваривают рестрикционными энзимами BamHl и Sall и обрабатывают электрофоретически на 1% агарозном геле.
Этот гель окрашивают 0,5 мкг/мл раствора этилиумбромида в течение 15 минут. Полосу, которая демонстрирует специфическую амплификацию, вырезают, наблюдая под УФ- светом. Кусочек геля обрабатывают Gene Clean II (Biol 01 Co.) для сбора ДНК-содержащего раствора. Собранные ДНК-фрагменты клонируют в BamHl-Sall сайт плазмиды pMAL-c2, и полученные клоны вводят в E.coli DH5α.
2) Выделение протеина слияния, экспрессированного в E.coli, и получение антитела
E. coli клетки, трансформированные плазмидами, предварительно культивируют в экспрессионной среде (10 г/л триптона (Gibco Co.), 5 г/л дрожжевого экстракта (Gibco Co. ), 5 г/л NaCl, 2 г/л глюкозы и 100 мл мкл/мл ампициллина) в течение ночи. Предварительно культивированный раствор (150 мл) добавляют к 1,5 л экспрессионной среды и культивируют в склянке Sakaguchi при 37oC при встряхивании до тех пор, пока не достигают значения плотности ОД600 = 0,55. К культуральному раствору добавляют изопропилтиогалактозид до конечной концентрации 0,3 мМ и культивируют со встряхиванием в течение еще 4 часов для индуцирования экспрессии. E. coli собирают центрифугированием, и клетки E. coli промывают промывочным буфером (20 мМ Tris-HCl pH 7,4, 200 мМ NaCl и 1 мМ EDTA). Полученные клетки обрабатывают ультразвуком в течение 2 минут (15 сек • 8). Фракцию растворимого протеина получают центрифугированием. Из этой фракции выделяют MBP-слитый протеин с помощью амилозной смолы. MBP- и активатор-связывающий домен расщепляют фактором Xa, и активатор-связывающий домен выделяют с помощью гель-фильтрации на хроматографической колонке. Очищенный протеин дважды вводят мышам в абдоминальную полость для иммунизации по способу E. Harlow and D. Lane (Antibody (1988), Cold Spring Harbor Co. , pp. 53 - 137). После того как увеличение титра подтверждается с помощью ELISA, получают асциты и подвергают осаждению за счет 50% насыщенного сульфата аммония и обрабатывают Protein A Sepharose (Pharmacia Co.) для получения очищенного антитела. При обработке Protein A Sepharose антитело связывается с Protein A Sepharose за счет 0,1 М натрийфосфата (pH 8,0) и его элюируют 0,1 М лимонной кислотой (pH 3,5). С помощью иммуноблоттинга подтверждают, что полученное антитело распознает только ER протеин сои.
3) Получение мембранной фракции семядолей сои
Мембранную фракцию семядолей сои получают следующим образом.
Семена сои выращивают в почве в течение 9 дней и собирают семядоли (36 г, влажный вес). Эти семядоли суспендируют в 47 мл охлажденного льдом буфера (25 мМ Tris - HCl pH 7,0, 30 мМ MgCl2, 2 мМ дитиотреитола, 2,5 мМ метабисульфита натрия и 1 мМ PMSF). Мембранную фракцию семядолей получают тем же способом, что и получали мембранную фракцию корней сои. Полученную мембранную фракцию семядолей суспендируют в 1 мл охлажденного льдом буфера (10 мМ Tris - HCl pH 7,4, 0,1 М сахарозы, 5 мМ MgCl2, 1 мМ PMSF и 5 мМ EDTA), и хранят при температуре -80oC.
4) Определение ингибирования связывания глюканового активатора с активатор-связывающим протеином мембранной фракции семядолей сои
Активатор-связывающую активность определяют следующим образом.
Комплекс активатора и тирамина (TOKYO KASEI KOGYO, CO. LTD) синтезируют по способу Jono-Joo Cheong (The Plant Cell (1991) 3:127). Комплекс активатор-тирамин метят иодом-125, используя хлорамин T. Мембранную фракцию семядолей соли (100 мкл, 820 мкг) суспендируют в 500 мкл аналитического буфера (50 мМ Tris - HCl pH 7,4, 0,1 М сахарозы, 5 мМ MgCl2, 1 мМ PMSF и 5 мМ EDTA) и инкубируют при 0oC в течение 2 часов. Меченый иодом комплекс активатор-тирамин в количестве 714 нг (143 мМ; Кюри/ммоль) добавляют к суспензии, и полученную смесь инкубируют при 4oC в течение 2 часов. Реакционный раствор фильтруют через Watman GF/B (обработанный 0,3% водным раствором полиэтиленимина в течение по крайней мере 1 часа) и трижды промывают 5 мл охлажденного льдом буфера (10 мМ Tris - HCl pH 7,0, 1 М NaCl, 20 мМ MgCl2). Радиоактивность, удержанную на фильтровальной мембране, подсчитывают с помощью гамма-счетчика (отсчет A). Для того чтобы исключить эффект неспецифического связывания, осуществляют ту же самую процедуру, что и раньше, за исключением того, что 10- кратное количество молей (75 мкг, 15 мкМ) холодного активатора добавляют к образцу, полученную смесь суспендируют в аналитическом буфере, и полученную суспензию инкубируют при 0oC в течение 2 часов. Полученную величину вычитают из отсчета A для получения величины ( Δ срм = количество импульсов в минуту) активатор-специфического связывания. Величину связывания, полученную при добавлении 3,6, 7,1, 10,8, 14,4 и 28,8 мкг очищенного антитела, а не холодного активатора, вычитают из отсчета A. Полученное значение сравнивают со значением, полученным для холодного активатора, и выражают как процент, причем величину ( Δ срм) для активатор-специфического связывания принимают за 100% (фиг. 7). Добавление 28,8 мкг антител приводит к ингибированию связывания активатора примерно на 51%. Эти результаты подтверждают тот факт, что антитела против активатор-связывающего домена ингибируют связывание активатора с активатор-связывающим протеином.
5) Ингибирование накопления фитоалексина за счет антител против активатор-связывающего домена.
Количество фитоалексина, накапливаемое за счет действия глюканового активатора, определяют с помощью семядолей сои по способу M.G. Hahn et al. ((1992) Molecular Plant Pathology, Volume II A Practical Approach, IRL Press, pp. 118 - 120).
Очищенные антитела против активатор-связывающего домена (0,1, 2,3, 4, 10 и 20 мкг/25 мкл семядолей) или очищенное антитело против полученной из дрожжей ds RNASe, pac 1 (4, 10 и 20 мкг/25 мкл/на семядолю) в качестве контроля, добавляют к семядолям сои, и полученную смесь инкубируют в течение 1 часа. Глюкановый активатор (200 нг) (25 мкл семядолей) добавляют к семядолям сои, и полученную смесь инкубируют в течение 20 часов для определения того, ингибируется ли антителом накопление фитоалексина под действием глюканового активатора. Количество накопленного фитоалексина, вызванное добавлением активатора после добавления антител, выражают в процентах, причем количество накопленного фитоалексина под действием добавления только активатора принимают за 100% (фиг. 8). Когда антитела против активатор-связывающего домена добавляют в количестве 20,0 мкг на семядолю сои, количество накопленного фитоалексина снижается примерно на 53%. В контрольном случае, количество накопленного фитоалексина изменяется мало, даже если антитела против рас 1 добавляют в количестве 20,0 мкг на семядолю сои. Полученные результаты показывают, что полученный ген кодирует не просто активатор-связывающий протеин, но кодирует ER, индуцирующий реакцию устойчивости в растениях сои.
Промышленное применение
В соответствии с настоящим изобретением предложены рецептор глюканового активатора, молекулы ДНК, кодирующие рецептор глюканового активатора, и их фрагменты, векторы, содержащие молекулы ДНК и их фрагменты, и клетки растений, трансформированные молекулами ДНК или их фрагментами.
ER настоящего изобретения можно использовать для выработки устойчивости к грибкам и для создания производных активатора, способных индуцировать устойчивость к грибкам, и его можно использовать в качестве антитела для получения антител против ER.
ДНК-молекулы настоящего изобретения, которые содержат нуклеотидные последовательности, кодирующие рецептор глюканового активатора и их фрагменты, можно использовать в качестве материала, обеспечивающего создание методик получения растений, устойчивых к грибкам. Другими словами, молекулы ДНК настоящего изобретения и их фрагменты можно вводить и экспрессировать в различные растения для повышения их устойчивости к грибковым заболеваниям.
Антитела против рецептора глюканового активатора настоящего изобретения, молекулы ДНК настоящего изобретения, которые содержат нуклеотидные последовательности, кодирующие рецептор глюканового активатора их мутанты и анти-смысловые ДНК можно использовать в исследованиях активатор-связывающего сайта ER и сигнальной трансдукции.
Кроме того, информацию об аминокислотной последовательности ER и о нуклеотидной последовательности его кодирующей можно использовать в исследованиях активатор-связывающего сайта ER и сигнальной трансдукции, в которой ER участвует.














Изобретение может быть использовано в биотехнологии растений, в частности для получения растений, устойчивых к грибковым заболеваниям. Определены аминокислотная последовательность рецептора глюканового активатора; нуклеотидная последовательность молекулы ДНК, кодирующей рецептор глюканового активатора; молекула ДНК, кодирующая рецептор глюканового активатора, включенная в плазмиду р ER 23-1; вектор, содержащий молекулу ДНК, кодирующую рецептор глюканового активатора. Молекулы ДНК настоящего изобретения, введенные и экспрессированные в растения, приводят к повышению устойчивости растений к грибкам. 4 с. и 1 з.п. ф-лы, 8 ил., 2 табл.
| Дементьева М.И | |||
| Фитопатология.-М.: Колос, 1977, с.109-114 | |||
| Frey T | |||
| et al | |||
| Affinity purification and characterization of a finding protein for a hepta-beta glucoside phytoalexin elicitor in soybean | |||
| Phytochemistry, 1993, v.32, N3, p.543-550. |

