Изобретение относится к способу получения энантиомеров O-деметилтрамадола и к их применению в качестве анальгетических средств.
Опиоиды применяют в течение уже многих лет в качестве анальгетиков для обезболивающего лечения, хотя они и вызывают целый ряд побочных эффектов, таких, например, как непреодолимое патологическое влечение и психическая зависимость, депрессия дыхания, ингибирующее воздействие на желудочно-кишечный тракт и запоры. По этой причине их применение допустимо только лишь при соблюдении особых мер предосторожности, таких, например, как специальные предписания при назначении препаратов на длительный период времени или в высокой дозировке, (см. Goodman, Gilman, The Pharmacological Basis of Therapeutics, издательство Pergamon Press, Нью-Йорк, 1990).
Трамадолгидрохлорид, он же гидрохлорид (1RS,2RS)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола, занимает среди основных эффективных анальгетиков особое место, поскольку это активное вещество обладает сильным обезболивающим действием, не вызывая при этом в отличие от опиоидов известных побочных явлений (см. Journ. Pharmacol. Exptl. Ther. 267, 331 (1993)). Трамадол представляет собой рацемат и состоит из равных количеств (+)- и (-)-энантиомеров. Это действующее вещество образует in vivo метаболит O-деметилтрамадол, также представленный в виде смеси энантиомеров. Проведенные исследования показали, что как оба энантиомера трамадола, так и оба энантиомера метаболитов трамадола способствуют достижению анальгетического эффекта (см. Journ. Pharmacol. Exptl. Ther. 260, 275 (1992); Arzneim. - Forschung 38, 877 (1988)).
Получение O-деметилтрамадола в виде рацемата или в форме энантиомеров известно из европейской заявки ЕР 534628 и международной заявки WO 93/04675. Согласно описанному в этих публикациях способу, осуществляемому с помощью сильного основания, как, например, гидрид натрия или гидрид калия, в присутствии тиофенола и диэтиленгликоля, хотя и можно получать O-деметилтрамадол, однако лишь с неудовлетворительным выходом.
Исходя из вышеизложенного, в основу изобретения была положена задача разработать способ, с помощью которого можно было бы получать O-деметилтрамадол с высоким выходом.
Было установлено, что благодаря применению L-(+)-винной кислоты для расщепления рацемата трамадола и последующим расщеплением метилового эфира с помощью гидрида диизобутилалюминия (ГДИБА) O-деметилтрамадол может быть получен в виде чистых энантиомеров и с высоким выходом.
В соответствии с этим предметом изобретения является способ получения энантиомеров O-деметилтрамадола, отличающийся тем, что рацемическую соль трамадола переводят в основание, с помощью L-(+)-винной кислоты путем осаждения отделяют (-)-энантиомер трамадола и после высвобождения основания с помощью ГДИБА переводят в (-)-энантиомер O-деметилтрамадола и из маточного раствора, полученного с помощью винной кислоты осадка высвобождением основания трамадола и взаимодействием с ГДИБА, получают (+)-энантиомер O-деметилтрамадола.
Для осуществления способа согласно изобретению в качестве эдукта пригоден прежде всего рацемический гидрохлорид трамадола. Этот последний в водном растворе добавлением гидроксидов щелочных металлов, предпочтительно гидроксида натрия, и экстракцией органическим растворителем, например дихлорметаном и/или диэтиловым эфиром, переводят в рацемическое основание трамадола. Затем полученное основание смешивают с L-(+)-винной кислотой, предпочтительно в присутствии органического растворителя, особенно предпочтительно в присутствии алифатического С1-С5-спирта. Образующийся тартрат (-)-энантиомера трамадола отделяют предпочтительно путем кристаллизации от образовавшегося тартрата (+)-энантиомера трамадола и после высвобождения основания трамадола в условиях, описанных выше, взаимодействием с ГДИБА переводят в (-)-энантиомер O-деметилтрамадола. Расщепление метилового эфира с помощью ГДИБА осуществляют обычно в ароматическом углеводороде, например, в толуоле при температуре от 60 до 130oС.
Растворимый в маточном растворе (+ -энантиомер трамадола в виде тартрата высвобождением основания трамадола в описанных выше условиях и последующим взаимодействием с ГДИБА в описанных выше условиях переводят в (+)-энантиомер O-деметилтрамадола.
Полученные энантиомеры O-деметилтрамадола могут быть выделены либо в виде оснований, либо в виде солей, прежде всего в виде гидрохлоридов. Эти гидрохлориды можно получать в тех же самых условиях, что и гидрохлориды трамадола.
До проведения взаимодействия с ГДИБА целесообразно высвобожденное из тартрата соответствующего энантиомера трамадола основание переводить в отличную от тартрата соль трамадола, предпочтительно в гидрохлорид, и затем из этого последнего в условиях, описанных выше, высвобождать основание трамадола.
Конверсию основания трамадола в гидрохлорид можно осуществлять с помощью концентрированной соляной кислоты или газообразного хлористого водорода в органическом растворителе, например в ацетоне, диоксане, диэтиловом эфире и/или диизопропиловом эфире, либо с помощью водного триметилхлорсилана в растворителе, например в 2-бутаноне.
С помощью предлагаемого согласно изобретению способа можно получать энантиомеры O-деметилтрамадола экономичным, экологически безопасным путем и с высоким выходом. Для расщепления рацематов соли трамадола необходима только лишь энантиомерная форма винной кислоты, а именно не требующая больших затрат L-(+)-винная кислота. С помощью L-(+) -винной кислоты можно получать энантиомеры трамадола с выходом более 85% в пересчете на используемый рацемат и со степенью чистоты энантиомеров выше 98%. После высвобождения основания трамадола маточный раствор может быть возвращен в процесс расщепления рацематов. В результате расщепления метилового эфира получают энантиомеры 0-деметилтрамадола с выходом более 95%.
В европейской заявке ЕР 534628 и международной заявке WO 93/04675 описано применение O-деметилтрамадола в комбинации с кодеином, оксикодоном, гидрокодоном или ацетаминофеном для лечения болезненных состояний. Было установлено, что уже сам по себе O-деметилтрамадол или в сочетании с трамадолом обладает высоким анальгетическим эффектом.
Другим предметом изобретения в соответствии с этим является применение O-деметилтрамадола в виде основания и/или соли в форме рацемата либо энантиомера индивидуально или в комбинации с трамадолом в виде основания и/или соли в форме рацемата либо энантиомера в качестве анальгетического действующего вещества в лекарственном средстве.
Предпочтительно применяют (+)-энантиомер O-деметилтрамадола.
Анальгетики согласно изобретению наряду с основанием и/или по крайней мере одной из солей O-деметилтрамадола индивидуально либо в комбинации с основанием трамадола и/или по крайней мере одной из солей трамадола содержат носители, наполнители, растворители, разбавители, красители и/или связующие вещества. Выбор этих вспомогательных веществ, равно как и их применяемые количества, зависят от того, назначено ли лекарственное средство для орального, внутривенного, щечного, интраперитонеального, интрадермального, внутримышечного, назального или локального введения, например, на коже, слизистых оболочках или глазах. Для орального приема пригодны композиции в виде таблеток, драже, капсул, гранулятов, капель, микстур и сиропов; для парентерального, местного и ингаляционного введения могут использоваться растворы, суспензии, легко реконструируемые сухие композиции, а также аэрозоли. В качестве композиций, пригодных для чрескожного назначения, могут служить применяемые согласно изобретению соединения в депо в растворенном виде или в пластыре, при необходимости с добавками средств, способствующих кожной пенетрации. Высвобождение применяемых согласно изобретению соединений из композиционных форм, назначаемых для орального или чрескожного введения, может происходить постепенно.
Количество действующих веществ, назначаемых пациенту, варьируют в зависимости от веса пациента, метода введения, показания и степени серьезности заболевания. Обычно применяют от 5 до 500 мг/кг по крайней мере одного из 10 вышеуказанных соединений.
Пример 1. (-)-(1S,2S)-2-Диметиламинометил-1-(3-метоксифенил)циклогексанол, гидрохлорид (-1)
Стадия 1: Высвобождение рацемического основания.
3 кг (10 моль) (1RS,2RS)-2-Диметиламинометил-1-(3-метоксифенил)циклогексанола, гидрохлорида (1), суспендировали в 4800 мл воды и смешивали с 1,6 кг истолченного льда. При перемешивании добавляли по каплям 1300 мл 36-38%-ного едкого натра (технического). Затем экстрагировали последовательно 7000 мл дихлорметана, а после разделения фаз еще 2000 мл дихлорметана. Объединенные органические дозы сушили над сульфатом натрия. После удаления перегонкой растворителя получали 2630 г (99% от теории) (1RS, 2RS)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола в виде сиропа.
Стадия 2: Осаждение с помощью L-(+)-винной кислоты.
2630 г (10 моль) Основания из первой стадии растворяли в 2400 мл этанола и смешивали с раствором, состоящим из 1500 г (10 моль) L-(+)-винной кислоты и 11200 мл этанола. Для кристаллизации перемешивали в течение 2 ч при комнатной температуре, после чего оставляли на 24 ч при 4oС. Выпавшие кристаллы отсасывали и промывали 6400 мл этанола, имевшего температуру 4oС. После сушки кристаллизата при комнатной температуре под вакуумом (60 мбар) получали 2050 г (49% в пересчете на общее количество использованного рацемического основания) (1S, 2S)-2-диметиламинометил-1-(3-метоксифенил)циклогексанол-L-(+)-тартрата с температурой плавления 173-175oС. Удельное вращение: [α]
Стадия 3: Высвобождение основания из соли L-(+)-винной кислоты.
2050 г (4,95 моля) (1S,2S)-2-диметиламинометил-1-(3-метоксифенил)циклогексанол-L-(+)-тартрата из стадии 2 растворяли в 4000 мл воды и смешивали с 900 г истолченного льда. При перемешивании добавляли по каплям 1000 мл 36-38%-ного едкого натра (технического). Затем экстрагировали последовательно 2500 мл дихлорметана, а после разделения фаз еще 500 мл дихлорметана. Объединенные органические фазы сушили над сульфатом натрия. После удаления перегонкой растворителя получали 1280 г (99% от теории) (1S,2S)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола в виде сиропа.
Стадия 4: Перевод (1S,2S)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола в гидрохлорид (-1)
1280 г (4,86 моля) полученного на стадии 3 основания растворяли в 16 л 2-бутанона и при перемешивании смешивали с 88 мл (4,9 моль) воды и 621 мл (532 г; 4,9 моль) триметилхлорсилана. Для кристаллизации перемешивали в течение 3 ч при комнатной температуре и оставляли на 24 ч при 4oС. Выпавшее в осадок твердое вещество отсасывали, промывали 5000 мл 2-бутанона, имевшего температуру 4oС, и сушили при 90oС под вакуумом (60 мбар) до получения постоянного веса. Таким путем получали 1390 г (95% от теории в пересчете на количество использованного основания из стадии 3 и 92% в пересчете на количество энантиомера использованного рацемата из стадии 1) гидрохлорида (-1) в виде бесцветных кристаллов.
Температура плавления: 172-173oС.
Удельное вращение: [α]
Стадия 5: Перевод гидрохлорида (-1) в (-)-(1S,2S)-3-(2-диметил-аминометил-1- гидроксициклогексил) фенол, гидрохлорид
Из гидрохлорида (-1) с помощью раствора дихлорметан-гидроксид натрия в условиях, описанных на стадии 1, высвобождали основание. После сушки раствора дихлорметан отгоняли под вакуумом. 208,1 г (0,79 моль) полученного основания, растворенного в 360 мл толуола, добавляли по каплям при комнатной температуре к 1,6 л 20%-ного раствора гидрида диизобутилалюминия (1,58 моль) в толуоле. Затем в течение 11 ч нагревали с обратным холодильником и после охлаждения до комнатной температуры продолжали дальнейшее охлаждение смесью лед-поваренная соль до приблизительно 0oС. Далее по каплям добавляли 450 мл этанола таким образом, чтобы внутренняя температура не превышала 15oС. После завершения добавления продолжали перемешивать в течение 15 мин и затем разбавляли 1000 мл толуола. При охлаждении смесью лед-поваренная соль по каплям добавляли 450 мл водно-этанольной смеси (1:1) и после завершения добавления перемешивали в течение 1 ч при комнатной температуре. Выпавший в осадок гидроксид алюминия отсасывали и для проведения последующей экстракции смешивали при 60oС с пятью объемными частями этилового эфира уксусной кислоты. После повторного отсасывания объединенные органические фазы сушили над сульфатом натрия и концентрировали с помощью ротационного испарителя при 60oС. Таким путем получали 193 г (98% от теории) основания, причем это основание кристаллизовалось в виде твердого вещества с температурой плавления 139-142oС.
Полученный неочищенный продукт растворяли в 1,93 л ацетона и смешивали с 65 мл концентрированной соляной кислоты. После начавшейся кристаллизации перемешивали в течение 1 ч при охлаждении ледяной баней и затем осадок отсасывали. Далее дважды промывали ацетоном и диэтиловым эфиром, после чего кристаллизат сушили под вакуумом, образуемым масляным насосом, при 70oС до получения постоянного веса. Таким путем получали 216,8 г (96% от теории) бесцветных кристаллов.
Температура плавления: 247-248oС (разложение).
Удельное вращение: [α]
Пример 2. (+)-(1R,2R)-2-Диметиламинометил-1-(3-метоксифенил)циклогексанол, гидрохлорид (+1)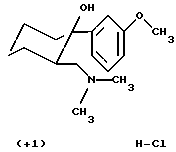
Стадия 1: Высвобождение основания из маточного раствора осадка, полученного с помощью L-(+)-винной кислоты.
Этанольный маточный раствор и фазу после промывки осадка, полученного с помощью L-(+)-винной кислоты (пример 1, стадия 2), объединяли. После удаления перегонкой растворителя остаток (2080 г) растворяли в 2500 мл воды и смешивали с 900 г истолченного льда. При перемешивании добавляли по каплям 1000 мл 36-38%-ного едкого натра (технического). Затем экстрагировали последовательно 2700 мл дихлорметана, а после разделения фаз еще 600 мл дихлорметана. Объединенные органические фазы сушили над сульфатом натрия. После удаления перегонкой растворителя получали 1340 г (99% от теории) (1R,2R)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола в виде сиропа.
Стадия 2: Перевод (1R,2R-2-диметиламинометил-1-(3-метокси-фенил)циклогексанола в гидрохлорид (+1).
1340 г (5,09 моль) полученного на стадии 1 основания растворяли в 17,5 л 2-бутанона и при перемешивании смешивали с 105 мл (5,8 моль) воды и 670 мл (573 г; 5,3 моль) триметилхлорсилана. Для кристаллизации перемешивали в течение 3 ч при комнатной температуре и на 24 ч оставляли при этой температуре. Выпавшее в осадок твердое вещество отсасывали, промывали 5000 мл 2-бутанона и при 90oС сушили под вакуумом (60 мбар) до получения постоянного веса. Таким путем получали 1350 г (88% от теории в пересчете на количество использованного основания из стадии 1 и 89% в пересчете на количество энантиомера использованного рацемата из примера 1, стадия 1) гидрохлорида (+1) в виде бесцветных кристаллов.
Температура плавления: 171-172oС.
Удельное вращение: [α]
Стадия 3: Перевод гидрохлорида (+1) в (+)-(1R,2R)-3-(2-диметиламинометил-1-гидроксициклогексил)фенол, гидрохлорид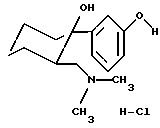
Исходя из гидрохлорида (+1), в условиях, описанных в примере 1, стадия 5, получали (+)-энантиомер O-деметилтрамадола в виде гидрохлорида с выходом 96%.
Температура плавления: 247-248oС (разложение).
Угол вращения: [α]
Фармакологические исследования
Испытания анальгезии на мышах в тесте на болевой синдром
Исследования анальгетической эффективности проводили на мышах в тесте на болевой синдром, индуцированный фенилхиноном (модифицированный метод I.C. Hendershot, J. Forsaith, Journ. Pharmacol. Exp. Ther. 125, 237-240 (1959)). В этих целях использовали мужских особей мышей линии NMRI весом 25-30 г. Группам по 10 животных в каждой, предназначенным для испытания одной дозы субстанции, через 10 мин после внутривенной инъекции применяемых согласно изобретению соединений в дозировке 0,3 мл/мышь вводили внутрибрюшинно 0,02%-ный водный раствор фенилхинона (фенилбензохинон, фирма Sigma, Дейзенхофен; приготовление раствора с добавками 5% этанола и выдержкой в водяной бане при 45oС). Подопытных животных помещали поодиночке в специальные клетки для наблюдения и с помощью кнопочного счетчика в интервалы времени от 5 до 20 мин, после введения фенилхинона подсчитывали число индуцированных болевых разгибательных движений (так называемые реакции на болевой синдром, т. е. прогибание тела с вытягиванием задних конечностей). Исходя из зависимого от дозировки уменьшения числа реакций на болевой синдром по сравнению с таковыми у тестируемых одновременно контрольных групп, которых обрабатывали только фенилхиноном посредством регрессионного анализа (обрабатывающая программа Martens EDV Service, Эккенталь) рассчитывали значения ED50 с 95%-ной доверительной областью касательно реакций на болевой синдром.
Испытание анальгезии на крысах в тесте на вздрагивание хвоста
Применяемые согласно изобретению соединения испытывали на анальгетическую эффективность на крысах в тесте на вздрагивание хвоста с использованием теплового излучения по методу D'Amour и Smith (см. Journ. Pharm. Exp. Ther. 72, 74-79 (1941)). Для этой цели использовали женских особей крыс Sprague Dawley весом 120-160 г. Подопытных животных помещали поодиночке в специальные клетки и на основание хвоста воздействовали сфокусированным тепловым лучом от электрической лампы (анальгезиметр типа Rhema для крыс). При этом интенсивность излучения лампы устанавливали таким образом, что промежуток времени с момента включения лампы до внезапного, судорожного вздрагивания хвоста (болевая латентность) у необработанных животных составлял 3-6 с. До введения применяемых согласно изобретению соединений животных в течение пяти минут дважды подвергали тестированию и по среднему значению полученных результатов измерений рассчитывали исходное среднее значение перед тестированием. Проявление болевых ощущений фиксировали соответственно через 20, 40 и 60 мин после внутривенного введения соединений по изобретению. При повышении болевой латентности максимальное время облучения ограничивали 12 с и увеличение латентного периода до > 150% от исходного среднего значения перед тестированием принимали за показатель анальгетического действия. Для определения зависимости от величины дозировки соединения вводили 3-5 дозами в логарифмически возрастающем количестве, включавшем соответственно пороговую и максимально эффективную дозу, и по числу анальгезированных животных по методу Литхфилда и Вилкоксона (см. Journ. Pharm. Exp. Ther. 96. 99-113 (1949)) определяли значения ED50. Определение ED50 осуществляли в максимальном диапазоне действия по истечении 20 мин после внутривенного введения субстанций.
Применяемые согласно изобретению энантиомеры O-деметилтрамадола проявили в тесте на болевой синдром у мышей, равно как и в тесте на вздрагивание хвоста у крыс, ярко выраженный анальгетический эффект. Полученные результаты представлены в нижеследующей таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ РАСЩЕПЛЕНИЯ РАЦЕМАТОВ ТРАМАДОЛА | 1997 |
|
RU2192407C2 |
| ДИМЕТИЛ(3-АРИЛБУТ-3-ЕНИЛ)АМИНОСОЕДИНЕНИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1997 |
|
RU2167146C2 |
| 1-ФЕНИЛ-2-ДИМЕТИЛАМИНОМЕТИЛЦИКЛОГЕКСАН-1-ОЛОВЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1996 |
|
RU2167148C2 |
| 6-ДИМЕТИЛАМИНОМЕТИЛ-1-ФЕНИЛЦИКЛОГЕКСАНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИ ДЕЙСТВУЮЩИХ ВЕЩЕСТВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1996 |
|
RU2178409C2 |
| О-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 6-МЕТИЛТРАМАДОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2286334C2 |
| ЗАМЕЩЕННЫЕ ЦИКЛОГЕПТЕНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1999 |
|
RU2233268C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИХ БЕНЗОЦИКЛОАЛКЕНОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2197484C2 |
| ЗАМЕЩЕННЫЕ АМИНОСОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2197474C2 |
| КОМБИНАЦИЯ ОПРЕДЕЛЕННЫХ ОПИОИДОВ С МУСКАРИНОВЫМИ АНТАГОНИСТАМИ ДЛЯ ТЕРАПИИ НЕДЕРЖАНИЯ МОЧИ | 2002 |
|
RU2305562C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 1-АМИНОБУТАН-3-ОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2001 |
|
RU2288219C2 |

Изобретение относится к усовершенствованному способу получения энантиомеров O-деметилтрамадола, обладающему сильным обезболивающим действием, которое в отличие от опиоидных анальгетиков не имеет известных побочных эффектов. Способ заключается в том, что рацемическую соль трамадола переводят в основание, с помощью L(+)винной кислоты в среде подходящего растворителя отделяют (-)-энантиомер трамадола, после высвобождения основания трамадола взаимодействием с гидридом диизобутилалюминия переводят в (-)-энантиомер O-деметилтрамадола и из маточного раствора осадка, полученного с помощью винной кислоты, высвобождением основания трамадола и взаимодействием с гидридом диизобутилалюминия получают (+)-энантиомер O-деметилтрамадола. В качестве исходной рацемической соли преимущественно используют рацемический гидрохлорид трамадола, L(+)винную кислоту применяют преимущественно в присутствии органического растворителя, такого, как алифатический С1-5-спирт. Обычно (-)энантиомер трамадола отделяют путем кристаллизации. Перед проведением взаимодействия с гидридом диизобутилалюминия можно соответствующий энантиомер основания трамадола перевести в соль, отличную от тартрата, например в гидрохлорид, из которого затем высвобождают основание. 5 з.п.ф-лы, 1 табл.
| Способ получения W-аминоалкоксиалканов или их солей | 1977 |
|
SU906367A3 |
| US 3652289 А, 28.03.1972 | |||
| Муфельная печь | 1975 |
|
SU534628A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Устройство для контроля углов гибки и пружинения к трубогибочному станку | 1975 |
|
SU517356A1 |

