Настоящее изобретение относится к способу расщепления рацематов трамадола.
Трамадолгидрохлорид, он же гидрохлорид (1RS,2RS)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола, занимает среди основных эффективных анальгетиков особое место, поскольку это активное вещество обладает сильным обезболивающим действием, не вызывая при этом, в отличие от опиоидов, известных побочных явлений (см. Journ. Pharmacol. Exptl. Ther. 267. 331 (1993)). Трамадол представляет собой рацемат и состоит из равных количеств (+)- и (-)-энантиомеров. Известно, что энантиомеры трамадола обладают интересным, несколько отличающимся от трамадола набором фармакологических свойств. Так, (+)-энантиомер отличается опиатоподобным анальгетическим эффектом, значительно более сильным по сравнению с трамадолом, тогда как для (-)-энантиомера характерно заметное подавление обратного захвата норадреналина.
Получение энантиомеров трамадола с использованием такого реагента расщепления рацематов, как дибензоилвинная кислота, описано в Arzneim.-Forsch. /Drug Res. 28 (I). 114 (1978). Недостаток этого способа заключается в необходимости применения очень дорогого хирального вспомогательного реагента, каковым является дибензоилвинная кислота. Повторное использование этого соединения в реакции расщепления рацематов связано со значительными трудностями, поскольку во время щелочной переработки диастереомерных солей происходит частичное отщепление дибензоильных групп. Кроме того, для выделения энантиомеров трамадола требуются оптические антиподы дибензоилвинной кислоты, чтобы получить (+)-энантиомер трамадола, необходимо проводить осаждение с помощью (-)-O, O'-дибензоил-L-винной кислоты; а чтобы получить (-)-энантиомер трамадола, необходимо проводить осаждение с помощью (+)-O,O'-дибензоил-D-винной кислоты.
В соответствии с этим в основу изобретения была положена задача разработать способ расщепления рацематов трамадола, который позволял бы избежать известных недостатков, обусловленных применением дибензоилвинной кислоты, и который обеспечивал бы получение энантиомеров трамадола с постоянно высоким выходом и высокой степенью чистоты.
Неожиданно было обнаружено, что благодаря применению L-(+)-винной кислоты, являющейся недорогим продуктом, предлагаемый способ отвечает высоким требованиям, выдвинутым при его разработке.
В соответствии с этим предметом изобретения является способ расщепления рацематов трамадола, отличающийся тем, что рацемическую соль трамадола переводят в основание, с помощью L-(+)-винной кислоты осаждением отделяют (-)-энантиомер трамадола и из маточного раствора осадка, полученного осаждением винной кислотой, высвобождением основания трамадола и последующим переводом в отличную от тартрата соль выделяют (+)-энантиомер трамадола.
Для осуществления способа согласно изобретению в качестве эдукта пригоден прежде всего рацемический гидрохлорид трамадола. Этот последний в водном растворе добавлением гидроксидов щелочных металлов, предпочтительно гидроксида натрия, и экстракцией органическим растворителем, например дихлорметаном и/или диэтиловым эфиром, переводят в рацемическое основание трамадола. Затем полученное основание смешивают с L-(+)-винной кислотой, предпочтительно в присутствии органического растворителя, особенно предпочтительно в присутствии алифатического С1-С5-спирта.
Образующийся тартрат (-)-энантиомера трамадола отделяют предпочтительно путем кристаллизации от образовавшегося тартрата (+)-энантиомера трамадола и затем при необходимости выделяют высвобождением основания трамадола в условиях, описанных выше, и переводом в отличную от тартрата соль трамадола, предпочтительно в гидрохлорид.
Растворимый в маточном растворе (+)-энантиомер трамадола в форме тартрата выделяют высвобождением основания трамадола в описанных выше условиях и последующим переводом в соль, не являющуюся тартратом, прежде всего в гидрохлорид трамадола.
Конверсию основания трамадола в гидрохлорид можно осуществлять с помощью концентрированной соляной кислоты или газообразного хлористого водорода в органическом растворителе, например в ацетоне, диоксане, диэтиловом эфире и/или диизопропиловом эфире, либо с помощью водного триметилхлорсилана в растворителе, например в 2-бутаноне.
Предлагаемый согласно изобретению способ может осуществляться экономичным и экологически безопасным путем. По сравнению с расщеплением рацематов с помощью дибензоилвинной кислоты способ согласно изобретению отличается тем, что для осуществления реакции расщепления рацематов требуется только одна энантиомерная форма винной кислоты, а именно недорогой продукт, каковым является L-(+)-винная кислота. С помощью L-(+)-винной кислоты можно получать энантиомеры с более чем 85%-ным выходом в пересчете на количество используемого рацемата и со степенью чистоты энантиомеров, превышающей 98%. Кроме того, L-(+)-винная кислота по сравнению с дибензоилвинной кислотой имеет меньшую в 2,4 раза формульную массу, следствием чего является существенное уменьшение количества отходов. Далее маточный раствор после высвобождения основания трамадола может быть возвращен в процесс расщепления рацемата.
ПРИМЕРЫ
Пример 1
(-)-(1S,2S)-2-диметиламинометил-1-(3-метоксифенил) циклогексанол, гидрохлорид (-1)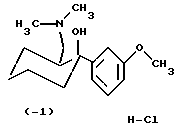
Стадия 1: Высвобождение рацемического основания
3 кг (10 молей) (1RS,2RS)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола, гидрохлорида (1), суспендировали в 4800 мл воды и смешивали с 1,6 кг истолченного льда. При перемешивании добавляли по каплям 1300 мл 36-38%-ного едкого натра (технического). Затем экстрагировали последовательно 7000 мл дихлорметана, а после разделения фаз еще 2000 мл дихлорметана. Объединенные органические фазы сушили над сульфатом натрия. После удаления перегонкой растворителя получали 2630 г (99% от теории) (1RS, 2RS)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола в виде сиропа.
Стадия 2: Осаждение с помощью L-(+)-винной кислоты
2630 г (10 молей) основания из первой стадии растворяли в 2400 мл этанола и смешивали с раствором, состоящим из 1500 г (10 молей) L-(+)-винной кислоты и 11200 мл этанола. Для кристаллизации перемешивали в течение 2 ч при комнатной температуре, после чего оставляли на 24 ч при 4oС. Выпавшие кристаллы отсасывали и промывали 6400 мл этанола, имевшего температуру 4oС. После сушки кристаллизата при комнатной температуре под вакуумом (60 мбар) получали 2050 г (49% в пересчете на общее количество использованного рацемического основания) (1S,2S)-2-диметиламинометил-1-(3-метоксифенил)циклогексанол-L-(+)-тартрата с температурой плавления 173-175oС. Удельное вращение: [α]
Стадия 3: Высвобождение основания из соли L-(+)-винной кислоты
2050 г (4,95 моля) (1S,2S)-2-диметиламинометил-1-(3-метоксифенил)циклогексанол-L-(+)-тартрата из стадии 2 растворяли в 4000 мл воды и смешивали с 900 г истолченного льда. При перемешивании добавляли по каплям 1000 мл 36-38%-ного едкого натра (технического). Затем экстрагировали последовательно 2500 мл дихлорметана, а после разделения фаз еще 500 мл дихлорметана. Объединенные органические фазы сушили над сульфатом натрия. После удаления перегонкой растворителя получали 1280 г (99% от теории) (1S,2S)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола в виде сиропа.
Стадия 4: Перевод (1S,2S)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола в гидрохлорид (-1)
1280 г (4,86 моля) полученного на стадии 3 основания растворяли в 16 л 2-бутанона и при перемешивании смешивали с 88 мл (4,9 моля) воды и 621 мл (532 г; 4,9 моля) триметилхлорсилана. Для кристаллизации перемешивали в течение 3 ч при комнатной температуре и оставляли на 24 ч при 4oС. Выпавшее в осадок твердое вещество отсасывали, промывали 5000 мл 2-бутанона, имевшего температуру 4oС, и сушили при 90oС под вакуумом (60 мбар) до получения постоянного веса. Таким путем получали 1390 г (95% от теории, в пересчете на количество использованного основания из стадии 3, и 92% в пересчете на количество энантиомера использованного рацемата из стадии 1) гидрохлорида (-1) в виде бесцветных кристаллов.
Температура плавления: 172-173oС.
Удельное вращение: [α]
Пример 2
(+)-(1R, 2R)-2-диметиламинометил-1-(3-метоксифенил) циклогексанол, гидрохлорид (+1)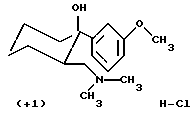
Стадия 1: Высвобождение основания из маточного раствора осадка, полученного с помощью L-(+)-винной кислоты
Этанольный маточный раствор и фазу после промывки осадка, полученного с помощью L-(+)-винной кислоты (пример 1, стадия 2), объединяли. После удаления перегонкой растворителя остаток (2080 г) растворяли в 2500 мл воды и смешивали с 900 г истолченного льда. При перемешивании добавляли по каплям 1000 мл 36-38%-ного едкого натра (технического). Затем экстрагировали последовательно 2700 мл дихлорметана, а после разделения фаз еще 600 мл дихлорметана. Объединенные органические фазы сушили над сульфатом натрия. После удаления перегонкой растворителя получали 1340 г (99% от теории) (1S,2S)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола в виде сиропа.
Стадия 2: Перевод (1R,2R)-2-диметиламинометил-1-(3-метокси-фенил)циклогексанола в гидрохлорид (+1)
1340 г (5,09 молей) полученного на стадии 1 основания растворяли в 17,5 л 2-бутанона и при перемешивании смешивали с 105 мл (5,8 молей) воды и 670 мл (573 г; 5,3 молей) триметилхлорсилана. Для кристаллизации перемешивали в течение 3 ч при комнатной температуре и на 24 ч оставляли при этой температуре. Выпавшее в осадок твердое вещество отсасывали, промывали 5000 мл 2-бутанона и при 90oС сушили под вакуумом (60 мбар) до получения постоянного веса. Таким путем получали 1350 г (88% от теории, в пересчете на количество использованного основания из стадии 1, и 89% в пересчете на количество энантиомера использованного рацемата из примера 1, стадия 1) гидрохлорида (+1) в виде бесцветных кристаллов.
Температура плавления: 171-172oС.
Удельное вращение: [α]
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРОВ O-ДЕМЕТИЛТРАМАДОЛА | 1997 |
|
RU2185371C2 |
| ДИМЕТИЛ(3-АРИЛБУТ-3-ЕНИЛ)АМИНОСОЕДИНЕНИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1997 |
|
RU2167146C2 |
| 6-ДИМЕТИЛАМИНОМЕТИЛ-1-ФЕНИЛЦИКЛОГЕКСАНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИ ДЕЙСТВУЮЩИХ ВЕЩЕСТВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1996 |
|
RU2178409C2 |
| 1-ФЕНИЛ-2-ДИМЕТИЛАМИНОМЕТИЛЦИКЛОГЕКСАН-1-ОЛОВЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1996 |
|
RU2167148C2 |
| О-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 6-МЕТИЛТРАМАДОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2286334C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЛИ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2002 |
|
RU2309942C2 |
| КОМБИНАЦИЯ ОПРЕДЕЛЕННЫХ ОПИОИДОВ С МУСКАРИНОВЫМИ АНТАГОНИСТАМИ ДЛЯ ТЕРАПИИ НЕДЕРЖАНИЯ МОЧИ | 2002 |
|
RU2305562C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 1-АМИНОБУТАН-3-ОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2001 |
|
RU2288219C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ С-ЦИКЛОГЕКСИЛМЕТИЛАМИНА, ЛЕКАРСТВЕННОЕ СРЕДСТВО И ПРИМЕНЕНИЕ | 2001 |
|
RU2295515C2 |
| ПРИМЕНЕНИЕ ЗАМЕЩЕННЫХ 6-ДИМЕТИЛАМИНОМЕТИЛ-1-ФЕНИЛЦИКЛОГЕКСАНОВЫХ СОЕДИНЕНИЙ ДЛЯ ТЕРАПИИ НЕДЕРЖАНИЯ МОЧИ | 2001 |
|
RU2279875C2 |
Изобретение относится к способу расщепления рацематов трамадола. Гидрохлорид трамадола-гидрохлорид (1RS,2RS)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола является сильным анальгетиком, не вызывающим при этом, в отличие от опиоидов, известных побочных эффектов. Энантиомеры трамадола обладают интересным, несколько отличающимся от рацемата трамадола, набором фармакологических свойств. Способ заключается в том, что рацемическую соль трамадола, преимущественно гидрохлорид, переводят в основание, с помощью L-винной кислоты в среде подходящего растворителя, обычно органического растворителя, предпочтительно в присутствии алифатического С1-С5-спирта, осаждением отделяют (-)энантиомер трамадола, как правило, кристаллизацией. Из маточного раствора осадка, полученного осаждением винной кислотой, высвобождением основания и последующим переводом в отличную от тартрата соль подходящей кислоты выделяют (+)энантиомер трамадола. Предпочтительно выделять (-) и (+)-энантиомеры трамадола в виде гидрохлорида. Предлагаемый способ позволяет получить энантиомеры с постоянно высоким выходом и высокой степенью чистоты. 4 з.п.ф-лы.
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ циклоАЛКЕНОВ С ОСНОВНЫМИ ЗАМЕСТИТЕЛЯМИ | 0 |
|
SU234951A1 |
| US 3652589 A, 28.03.1972 | |||
| Муфельная печь | 1975 |
|
SU534628A1 |
| Способ выделения аминоспиртов или их солей | 1974 |
|
SU546607A1 |
| Устройство для контроля углов гибки и пружинения к трубогибочному станку | 1975 |
|
SU517356A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |

