Изобретение относится к способам получения галоидзамещенных соединений гидроксидифенила формулы
где m - от 1 до 2.
Получение галозамещенных соединений гидроксидифенила, в частности 2-гидрокси-2', 4,4'-трихлородифенилового эфира (Triclosan; соединение формулы (3)) обычно проводят диазотированием и последующим гидролизом 2-амино-2', 4,4'-трихлородифенилового эфира (TADE; соединения формулы (2)).
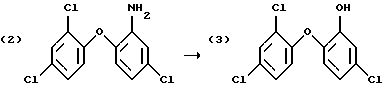
Однако выход в этом способе получения неудовлетворителен, поскольку могут иметь место различные конкурентные химические реакции.
Поэтому цель настоящего изобретения - найти экономичный способ получения галоидзамещенных соединений гидроксидифенила, в котором подавляются нежелательные побочные реакции.
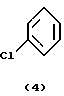
Поставленную задачу изобретение реализует реакцией, включающей четыре стадии, в первой стадии ацетилируют галоидзамещенное соединение бензола в присутствии кислоты Льюиса, на второй стадии соответствующее ацетилированное соединение этерифицируют соответствующим галоидзамещенным соединением фенола, на третьей стадии этерифицированное соединение окисляют и на четвертой стадии полученное окисленное соединение подвергают гидролизу по следующей реакционной схеме 1, приведенной в конце описания.
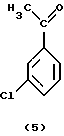
На первой стадии процесса (реакция ацетилирования) получают соединения формулы (5). Обычно эту реакцию проводят в присутствии кислоты Льюиса, например галида алюминия, в частности хлорида алюминия. В этом случае кислоту Льюиса используют в молярном количестве 1-3, лучше 1,25-2 по отношению к галоидзамещенному соединению формулы (5). Возможным ацилирующим реагентом в этой реакции является ацилгалоид, в частности хлористый ацетил.
Кислоту Льюиса и ацилирующий реагент лучше применять в эквимолярных количествах. Реакцию проводят в растворителях, обычно используемых в реакциях Фриделя-Крафтса, например метиленхлориде или этиленхлориде. Время реакции для этой стадии не играет большой роли и имеет большой диапазон, например от 1 до 18 часов.
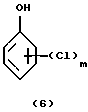
Во второй стадии процесса получают соединения формулы (7). Этерификацию свободной ОН группы галоидзамещенного соединения фенола формулы (6) проводят обычно в щелочной среде с применением сильного органического или лучше неорганического основания, такого как NaOH или КОН, и в присутствии медного катализатора и инертного органического растворителя, например, изомерной смеси толуола или ксилола. Время реакции на этой стадии составляет обычно 1-24 часа, предпочтительно 2-10 часов; температура колеблется от 80 до 250oС, точнее от 100 до 170oС.
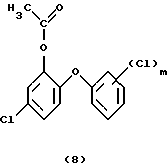
В третьей стадии процесса (реакция окисления) получают соединения формулы (8). Окисление ацетилированного соединения формулы (7) в соединение формулы (8) (окисление по Байеру-Виллигеру) можно проводить с применением различных окислительных агентов. Подходящие окислительные агенты представляют собой, например:
- смесь разбавленной надуксусной кислоты и уксусного ангидрида в присутствии каталитического количества хлорной кислоты;
- м-хлорнадбензойную кислоту (МСРВА) в воде;
- динаддодекандикислоту (DPDDA);
- смесь разбавленной надуксусной кислоты и уксусного ангидрида и серной кислоты;
- надбензойную кислоту (РВА);
- смесь бората натрия и трифторуксусной кислоты;
- смесь муравьиной кислоты, перекиси водорода, уксусного ангидрида, фосфорного ангидрида и уксусной кислоты;
- смесь уксусной кислоты, перекиси водорода, уксусного ангидрида и фосфорного ангидрида;
- смесь K2S2O8, серной кислоты и смеси вода/метанол 1:1;
- смесь уксусной кислоты и калиевой соли мононадмалеиновой кислоты;
- смесь трихлорметилена, калиевой соли мононадмалеиновой кислоты и кислого сульфата натрия;
- смесь малеинового ангидрида, уксусного ангидрида, перекиси водорода и трихлорметана;
- смесь малеинового ангидрида, комплекса мочевина-перекись водорода и уксусной кислоты;
- монопернадфталат магния;
- смесь уксусного ангидрида, серной кислоты и H2O2;
- смесь дихлоруксусной кислоты и Н2O2.
Для окисления предпочтительно применяют м-хлорнадбензойную кислоту (МСРВА), смесь бората натрия и трифторуксусной кислоты или смесь уксусного ангидрида и H2O2. При желании к окислительному агенту можно дополнительно добавить увлажняющий агент. Время реакции имеет большой диапазон от 0,5 до около 15 часов и конкретно составляет от 1 до 8 часов. Температура реакции также имеет большой диапазон от -20 до около 100oС, предпочтительно от 0 до около 85oС.
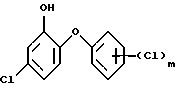
Последующий гидролиз с получением нужного галоидзамещенного гидроксидифенилового эфира формулы (1) количественно протекает в кислой или щелочной среде.
Способ по изобретению относится к получению галоидзамещенных соединений гидроксидифенила формулы (1), в которой m=1.
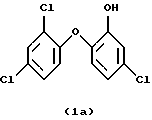
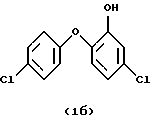
Особенно предпочтительны соединения формулы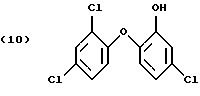
или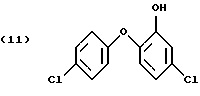
Галоидзамещенные соединения гидроксидифенила, полученные в соответствии с изобретением, нерастворимы в воде, но растворимы в разбавленных растворах гидроксида натрия и гидроксида калия, а также во всех органических растворителях. Учитывая эти требования, предъявляемые к растворимости, их применение для борьбы с микроорганизмами, в частности бактериями, и в качестве дезинфекционных средств для защиты органических материалов и изделий от микроорганизмов очень обширно. Итак, их можно применять в разбавленном или неразбавленном виде, например, вместе с увлажняющими или диспергирующими агентами, такими как синтетические мыльные или моющие растворы для дезинфекции и мытья тела и рук, изделий, в стоматологических гигиенических композициях.
Следующие примеры иллюстрируют изобретение, никоим образом не ограничивая его.
Примеры получения
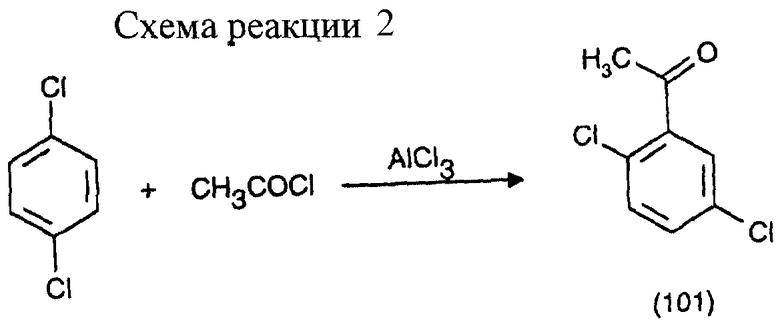
Пример 1: Получение 2,5-дихлороацетофенона (первая стадия) (см. схему реакции 2 в конце описания).
147 г (1,0 М) п-дихлоробензола полностью расплавляют при 60oС в реакторе, снабженном капельной воронкой, мешалкой и дефлегматором. К расплаву добавляют 120 г (0,9 М) безводного AlCl3. К перемешиваемой суспензии в течение 1 часа при 60oС по каплям добавляют 39,3 г (0,5 М) хлористого ацетила, при этом суспензия медленно превращается в чистый раствор. После нагрева до 110oС смесь перемешивают при этой температуре в течение 7 часов. После охлаждения до комнатной температуры коричневую реакционную массу подвергают гидролизу осторожным декантированием на смеси из 200 мл воды и 200 г льда. Посредством внешнего охлаждения температуру смеси во время гидролиза поддерживают между 30 и 40oС. После разделения фаз нижнюю органическую фазу промывают 400 мл воды и после еще одного разделения фаз подвергают фракционной перегонке. Водные фазы выливают.
Выход: 66 г 2,5-дихлорацетофенона (70% от теоретического на основе ацетилхлорида).
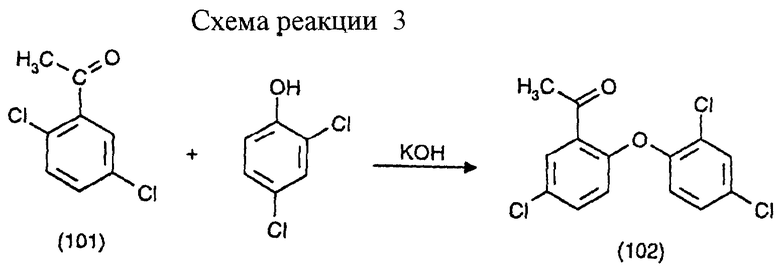
Пример 2: Получение 1-(5-хлор-2-(2,4-дихлорфенокси)фенилэтанона (вторая стадия) (см. схему реакции 3 в конце описания).
163 г 2,4-дихлорофенола вначале вводят вместе с 22,0 г 85% КОН и 167 мл смеси изомеров ксилола. Все нагревают до флегмации и воду удаляют азеотропной перегонкой. Красно-коричневый прозрачный раствор охлаждают до 100oС, обрабатывают 189 г 2,5-дихлорацетофенона (соединение формулы 101) и 0,8 г основного карбоната меди, нагревают до 140oС и перемешивают при этой температуре 8 часов. В ходе этого процесса реакционный раствор становится темно-красно-коричневым и в осадок выпадает белое вещество, которое отфильтровывают. После отгонки большей части ксилола в мягком впрыскивающем вакууме получают фракцию в 208 г, которая в дополнение к исходным продуктам 2,4-дихлорфенола и 2,5-дихлорацетофенона содержит какую-то часть ксилола (т. кип. 30-85oС/0,5-1 мм рт.ст.). Получают 82 г второй слегка желтоватой фракции (т. кип. 165-175oС/1 мм рт. ст. ) соединения формулы (102), что соответствует теоретическому выходу в 82% от количества используемого КОН (т.пл.=91oС).
Анализ для C14H9Cl3О2 (ММ=315.58):
Рассчитано, %: C 53,28; H 2,87; Cl 33,7; O 10,14;
Получено, %: C 53,31; H 2,85; Cl 33,37; O 10,31;
Пример 3: Получение 1-(5-хлор-2-(4-хлорфенокси)фенил)этанона (вторая стадия)
Процедура та же, что описана в Примере 2, но вместо 163 г дихлорфенола применяют 128,6 г 4-хлорфенола. После времени реакции на протяжении 2 часов при 140oС и последующих процедур, описанных в Примере 2, получают основную фракцию 91 г (т. кип. 112-173oС/1 мм рт.ст.) после предваряющей ее фракции в 223 г (т. кип. 30-112oС/1 мм рт.ст.), которая в дополнение к исходному материалу 2,5-дихлороацетофенона содержит более 80% реакционного продукта формулы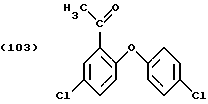
который при охлаждении отверждается и представляет собой порошок белого цвета (т.пл.=64oС).
Анализ для C14H10Cl2O2 (MM=281,14):
Рассчитано, %: C 59,81; H 3,59; Cl 25,22; O 11,38;
Получено, %: C 59,74; H 3,48; Cl 25,49; O 11,29;
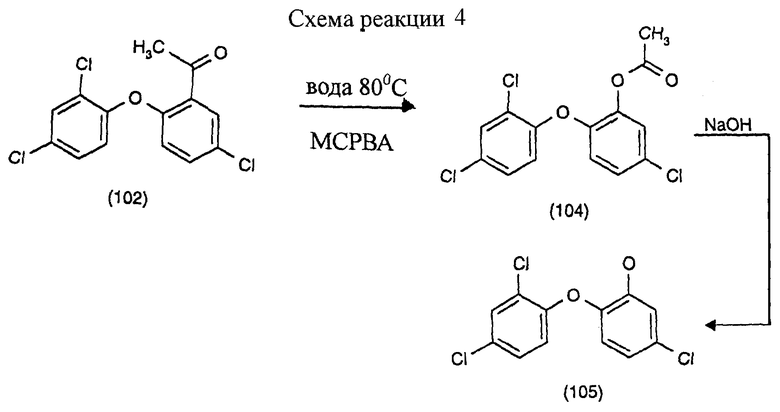
Пример 4: Получение 5-хлор-2-(2,4-дихлорфенокси)фенола окислением по Байеру-Виллигеру м-хлорнадбензойной кислотой (третья и четвертая стадии) (см. схему реакции в конце описания).
6,3 г ацетилированного соединения формулы (102) суспендируют в 40 мл деионизированной воды при 20oС. Туда высыпают 9,8 г м-хлорнадбензойной кислоты (МСРВА) и смесь нагревают с перемешиванием. Начиная с 52oС, образуется фаза смола/вода; смесь нагревают примерно до 80oС и эту температуру поддерживают в течение 7 часов.
Смесь обрабатывают 0,5 г бисульфита натрия с целью удаления избытка перекиси. С добавлением 50 мл этиленхлорида и 4 г 10N NaOH получают две прозрачные фазы. Водную фазу с рН около 12 отделяют; фазу с растворителем промывают водой до нейтральной. После перегонки растворителя остается 5,5 г кристаллизованного соединения формулы (104) (промежуточного соединения эфира фенола).
Для гидролиза эфир фенола растворяют в 50 мл этиленхлорида и 10 мл 5N раствора гидроксида натрия. Раствор нагревают до 70-73oС, эту температуру поддерживают в течение 15 минут, затем уксусной кислотой доводят рН до около 4 и фазы разделяют. После удаления растворителей получают 4,9 г сырого продукта формулы (105) бежевого цвета. После очищающей фильтрации и рекристаллизации из петролейного эфира 80/110 получают чистый продукт в виде бесцветных кристаллов с температурой плавления 56-57oС.
Пример 5: Получение 5-хлор-2-(2,4-дихлорфенокси)фенола окислением по Байеру-Виллигеру NaBO3 (третья и четвертая стадии)
6,3 г ацетилированного соединения формулы (102) суспендируют в 20 мл трифторуксусной кислоты, и суспензию нагревают при 20oС с 9,3 г тетрагидрата надборнокислого натрия. Ее нагревают до 40oС и эту температуру поддерживают 90 минут с хорошим перемешиванием. После гидролиза полученного фенолового эфира и последующей обработки аналогично Примеру 4 получают 5,4 г сырого продукта формулы (105).
Пример 6: Получение 5-хлор-2-(2,4-дихлорфенокси)фенола окислением по Байеру-Виллигеру уксусным ангидридом/Н2О2
18 мл уксусного ангидрида смешивают при -5oС с 4,5 мл 98% серной кислоты. С энергичным перемешиванием при температуре от -5 до -3oС в течение 25 минут по каплям добавляют 4,2 мл 30% перекиси водорода. Эмульсию молочного цвета обрабатывают при -5oС 12,5 мл метиленхлорида с получением прозрачного раствора. Его в течение 3 минут с очень энергичным перемешиванием добавляют к смеси 7,9 г ацетилового соединения формулы (102), 15 мл метиленхлорида, 18 мл 100% уксусной кислоты и 13,5 мл 98% серной кислоты при температуре от 0 до -5oС.
Реакционная смесь становится двухфазной и темной. Реакцию поддерживают при 0-5oС один час, затем при 10oС 4 часа и при 15oС 1 час.
Реакция заканчивается при 20oС в течение 20 часов, при этом полученный промежуточный продукт фенолового эфира формулы (104) также частично гидролизуется до производного фенола формулы (105). После разложения избыточной перекиси водорода отгоняют метиленхлорид. Для полного гидролиза температуру поддерживают на 100oС в течение 4 часов. Продукт регенерируют экстракцией метиленхлорида. После перегонки растворителя остается масляный остаток 7 г сырого продукта формулы (105), который после традиционной очистки дает продукт с температурой плавления 56-57oС.
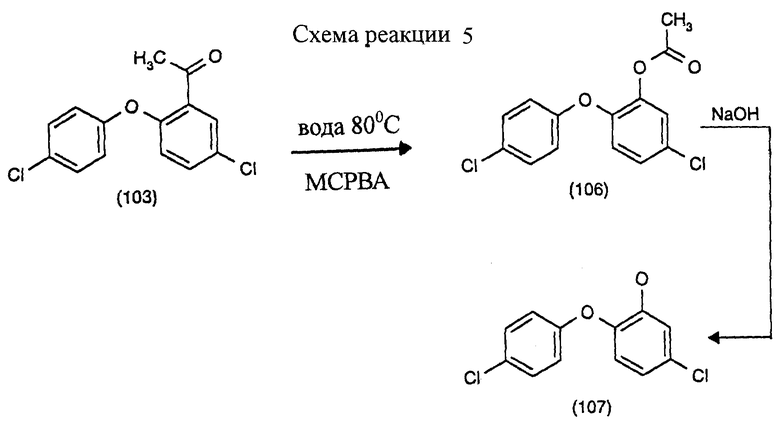
Пример 7: Получение 5-хлор-2-(4-хлорфенокси)фенола (третья и четвертая стадии) (см. схему реакции в конце описания).
14 г ацетилированного соединения формулы (103) суспендируют в 100 мл деионизированной воды при 20oС с применением увлажняющего агента. Туда высыпают 29 г 70% 3-хлорнадбензойной кислоты (МСРВА) и смесь нагревают с перемешиванием. Начиная с 52oС, образуется фаза смола/вода; смесь нагревают примерно до 80oС и выдерживают при этой температуре 7 часов.
Смесь обрабатывают 0,5 г бисульфита натрия с целью удаления избытка перекиси. С добавлением 50 мл смеси изомеров ксилола и 9 г 10N NaOH получают две прозрачные фазы. Водную фазу с рН около 12 отделяют; фазу растворителя, содержащую соединение формулы (106), промывают водой до нейтральной.
Для гидролиза эфира фазу ксилола обрабатывают 24 г 10% NaOH и перемешивают в условиях флегмации (около 95oС) 5 часов. Затем фазу ксилола отделяют и рН бледно-коричневой водной фазы доводят до около 3, используя 4 г 34% хлористоводородной кислоты при 25oС. В ходе этого продукт осаждается кристаллами бежевого цвета и после фильтрации его можно тщательно промыть водой на фильтре, работающем под разрежением. После высушивания получают 5 г сырого продукта формулы (107) с температурой плавления от 73 до 74oС.
После рекристаллизации из петролейного эфира 80/110 чистое вещество получают в виде бесцветных кристаллов с т.пл. 74-74,5oС.
Примеры 8-14
Соединения, представленные в таблице, получают аналогично способу, описанному выше.


Изобретение относится к новому способу получения галоидзамещенных соединений гидроксидифенила, которые применяются для борьбы с микроорганизмами. Способ получения галоидзамещенных соединений гидроксидифенила общей формулы (1), в которых m - от 1 до 2, например, соединения (1а) или (1б), включает ацетилирование соответствующего галоидзамещенного соединения бензола формулы (4), преимущественно ацетилхлоридом, в присутствии кислоты Льюиса на первой стадии, на второй стадии - этерификацию полученного соответствующего ацетилированного соединения формулы (5) соответствующим галоидзамещенным соединением фенола формулы (6), обычно в щелочной среде с применением сильного основания, такого как NaOH или КОН. На третьей стадии проводят окисление полученного этерифицированного соединения формулы (7), желательно м-хлорнадбензойной кислотой, возможно, в присутствии смеси бората натрия и трифторуксусной кислоты. На четвертой стадии проводят гидролиз полученного окисленного соединения формулы (8). Предлагаемый способ является экономичным, позволяет подавить нежелательные побочные реакции и получить продукты с хорошим выходом. 10 з.п.ф-лы, 1 табл.
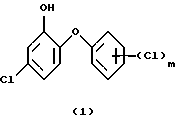

где m - от 1 до 2,
ацетилированием соответствующего галоидзамещенного соединения бензола формулы (4)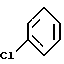
в присутствии кислоты Льюиса на первой стадии, на второй стадии этерификацией полученного соответствующего ацетилированного соединения формулы (5)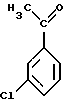
соответствующим галоидзамещенным соединением фенола формулы (6)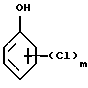
на третьей стадии окислением полученного этерифицированного соединения формулы (7)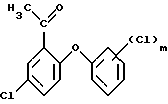
и на четвертой стадии гидролизом полученного окисленного соединения формулы (8)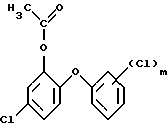
2. Способ по п. 1, отличающийся тем, что в реакции ацетилирования на первой стадии получают соединение формулы (5).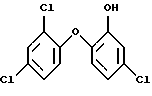
11. Способ по любому из пп. 1-10, относящийся к получению соединения формулы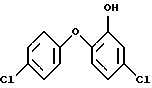
| СТЕНД ДЛЯ СБОРКИ И РАЗБОРКИ КАБИН ТРАКТОРОВ | 0 |
|
SU384043A1 |
| ATKINSON D.C | |||
| et al., Substituted (2-Phenoxyphenyl)acetic Acids with Antiinflammatory Activity | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US 4065503 A, 27.12.1977 | |||
| Способ определения границы руда-закладочный бетон | 1987 |
|
SU1493776A1 |
| Пломба | 1984 |
|
SU1288747A1 |
| DE 3723994 A1, 04.02.1988 | |||
| The Merck Index, Thelfth Edition, 1996, MERCK & CO, WHITEHOUSE STATION | |||
| Зеркальная камера с моноклем | 1924 |
|
SU1646A1 |
| Способ получения дихлорпроизводных дифенилового эфира | 1972 |
|
SU455936A1 |
| Способ получения м-феноксифенола | 1990 |
|
SU1740365A1 |

