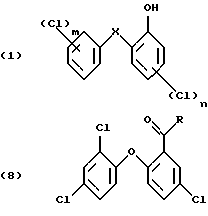
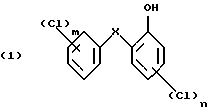
Изобретение относится к получению галоген-о-гидроксидифениловых соединений формулы: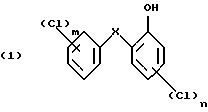
в которой Х- -О- или -СН2-;
m - от 1 до 3; и
n - 1 или 2;
и к применению этих соединений для защиты органических материалов от микроорганизмов или, например, в косметических композициях.
Получение галоген-о-гидроксидифениловых соединений, в частности 2-гидрокси-2', 4,4'-трихлордифенилового эфира (Triclosan; соединения формулы (3) ниже), обычно проводят диазотированием и последующим гидролизом 2-амино-2', 4,4'-трихлордифенилового эфира (TADE; соединения формулы (2) ниже):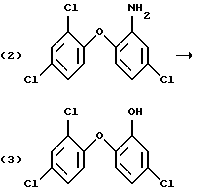
Однако выход в этом способе получения неудовлетворителен, поскольку могут иметь место различные конкурентные химические реакции.
Поэтому цель настоящего изобретения - найти экономичный способ получения галоген-о-гидроксидифениловых соединений, в котором подавляются нежелательные побочные реакции.
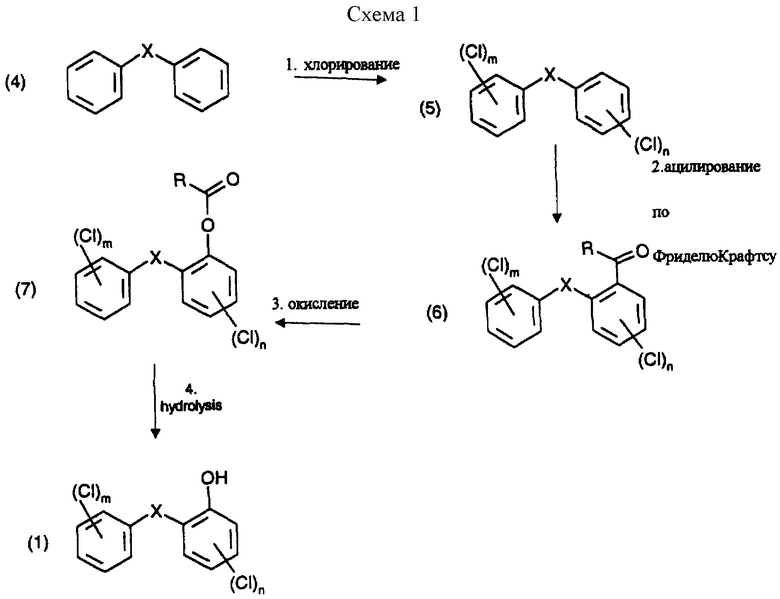
Настоящее изобретение предлагает способ получения галоген-о-гидроксидифениловых соединений, включающий четыре стадии, в котором в первой стадии дифениловое соединение хлорируют; во второй стадии хлорированное соединение ацилируют реакцией Фриделя-Крафтса и его можно вновь хлорировать после ацилирования; в третьей стадии ацильное соединение окисляют; и в четвертой окисленное соединение подвергают гидролизу по схеме 1 реакции, приведенной в конце описания.
В схеме1:
R - незамещенный С1-С8алкил или С1-С8алкил, замещенный 1-3 атомами галогена или гидрокси; или незамещенный С6-С12арил или С6-С12арил, замещенный 1-3 атомами галогена, С1-С5алкилом или C1-С8алкокси, или их комбинация;
Х- -О- или -СН2-;
m - от 1 до 3; и
n - 1 или 2.
С1-С8алкил означает разветвленный или неразветвленный алкил, такой как метил, этил, пропил, изопропил, н-бутил, втор-бутил, изобутил, т-бутил, 2-этилбутил, н-пентил, изопентил, 1-метилпентил, 1,3-диметилбутил, н-гексил, 1-метилгексил, н-гептил, изогептил, 1,1,3,3-тетраметилбутил, 1-метилгептил, 3-метилгептил, 2-этилгексил или н-октил.
С1-С8алкокси представляет собой остатки с прямой или разветвленной цепью, такие как метокси, этокси, пропокси, бутокси, пентокси, гексилокси, гептилокси или октилокси.
Галоген означает фтор, бром или предпочтительно хлор.
В вышеприведенной схеме реакции в формулах (6) и (7) R предпочтительно С1-С4алкил, конкретно метил.
В первой стадии реакции в качестве хлорирующего реагента можно применить хлористый сульфурил или лучше газообразный хлор. Реакцию лучше проводить в присутствии катализатора, такого как дибензотиофен, сернистый метил, сернистый пропил, сернистый фенил, кислота по Льюису, например хлорид алюминия, или в смеси таких соединений.
В качестве катализатора для реакции хлорирования по изобретению наиболее приемлема смесь сернистого пропила и эквимолярное количество хлорида алюминия. Для реакции в первой стадии температуру можно выбирать в пределах большого диапазона, например от -10 до 50oС. Предпочтительно реакцию проводят при температуре от 0 до 40oС. Время реакции имеет большой диапазон. Обычно реакцию проводят в пределах от 1 до 48, лучше от 2.5 до 10 часов.
Реакцию ацилирования (2 стадию) обычно проводят в присутствии кислоты Льюиса, например хлорида алюминия. Кислоту Льюиса можно применять в молярных количествах от 1 до 3, предпочтительно 1.25-2 от количества хлорированного соединения формулы (5). Подходящим ацилирующим реагентом в этой реакции является ацилгалоид, лучше хлористый ацетил. Другими приемлемыми ацилирующими реагентами являются, например,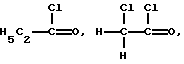
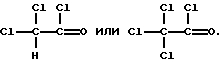
Кислоту Льюиса и ацилирующие реагенты применяют предпочтительно в эквимолярных количествах. Реакцию можно проводить в растворителях, которые обычно применяют для реакций Фриделя-Крафтса, таких как галоидированные растворители типа метиленхлорида или этиленхлорида. Время реакции в этой стадии не имеет особой важности и может составлять достаточно большой диапазон, например от 1 до 18 часов.
После реакции ацилирования реакционную смесь можно подвергнуть последующей реакции хлорирования, аналогичной первой, особенно, если в первой стадии реакции получены смеси хлорированных различным образом дифениловых соединений, такие как смеси соединений 4,4'-дихлородифенила и 2,4,4'-трихлородифенила. При последующем хлорировании получают однородно хлорированные ацильные соединения.
Реакцию хлорирования (первую стадию) и реакцию ацилирования (вторую стадию) и при необходимости последующую реакцию хлорирования лучше проводить в одном реакторе.
Окисление ацильного соединения формулы (6) в соединение формулы (7) (окисление по Байеру-Виллигеру) можно проводить с применением различных окислительных агентов. Подходящие окислительные агенты представляют собой, например:
- эквимолярную смесь разбавленной надуксусной кислоты и уксусного ангидрида в присутствии каталитического количества хлорной кислоты;
- избыток 3-хлор-надбензойной кислоты в воде;
- динаддодекандикислоту (DPDDA);
- смесь разбавленной надуксусной кислоты и уксусного ангидрида и серной кислоты;
- смесь м-хлорнадбензойной кислоты (МСРВА), трифторуксусной кислоты и дихлорметана;
- смесь бората натрия и трифторуксусной кислоты;
- смесь муравьиной кислоты, перекиси водорода, уксусного ангидрида, фосфорного ангидрида и уксусной кислоты;
- смесь уксусной кислоты, перекиси водорода, уксусного ангидрида и фосфорного ангидрида;
- смесь K2S2O8, серной кислоты и смеси вода/метанол 1:1;
- смесь уксусной кислоты и калиевой соли мононадмалеиновой кислоты;
- смесь трихлорметилена, калиевой соли мононадмалеиновой кислоты и кислого сульфата натрия;
- смесь малеинового ангидрида, уксусного ангидрида, перекиси водорода и трихлорметана;
- смесь малеинового ангидрида, комплекса мочевина-перекись водорода и уксусной кислоты; и
- мононадфталат магния.
Для окисления предпочтительно применяют смесь малеинового ангидрида, комплекса мочевина-перекись водорода и уксусной кислоты в качестве растворителя. При желании к окислительному агенту можно дополнительно добавить увлажняющий агент. Время реакции имеет большой диапазон от 1 часа до около недели, наиболее предпочтителен срок от 4 до 6 дней.
Температура реакции также имеет большой диапазон от -20 до около 80oС, предпочтительно реакцию проводят при комнатной температуре.
И, наконец, гидролиз в нужные галогено-гидроксидифениловые эфиры формулы (1) протекает количественно.
Способ по изобретению относится в основном к получению галоген-о-гидроксидифениловых соединений формулы (1), в которой
Х - кислород, и особенно тех соединений, в которых
m- 2 и
n - 1.
Наиболее предпочтительно соединение формулы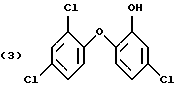
Некоторые ацильные соединения, полученные во второй стадии реакции (ацилирование по Фриделю-Крафтсу), являются новыми соединениями. Это соединения формулы
в которой R - незамещенный С1-С8алкил или С1-С8алкил, замещенный 1-3 атомами галогена или гидрокси; или незамещенный С6-С12арил или С6-С12арил, замещенный 1-3 атомами галогена, С1-С5алкилом или C1-С8алкокси, или их комбинация.
В формуле (8) R предпочтительно С1-С4алкил, в частности метил.
Эти новые соединения представляют собой еще один аспект изобретения.
Галоген-о-гидроксидифениловые соединения, полученные в соответствии с изобретением, нерастворимы в воде, но растворимы в разбавленных растворах гидроксида натрия и гидроксида калия, и практически во всех органических растворителях. Благодаря таким свойствам в отношении растворимости их применение для борьбы с микроорганизмами, в частности бактериями, и для защиты органических материалов от микроорганизмов очень обширно. Итак, их можно применять, например, вместе с увлажняющими или диспергирующими агентами, как мыла или как синтетические моющие растворы для дезинфекции и мытья тела и рук, а в разбавленном или неразбавленном виде их можно хранить во флаконах или других жестких контейнерах.
Следующие примеры более подробно иллюстрируют изобретение, никоим образом не ограничивая его.
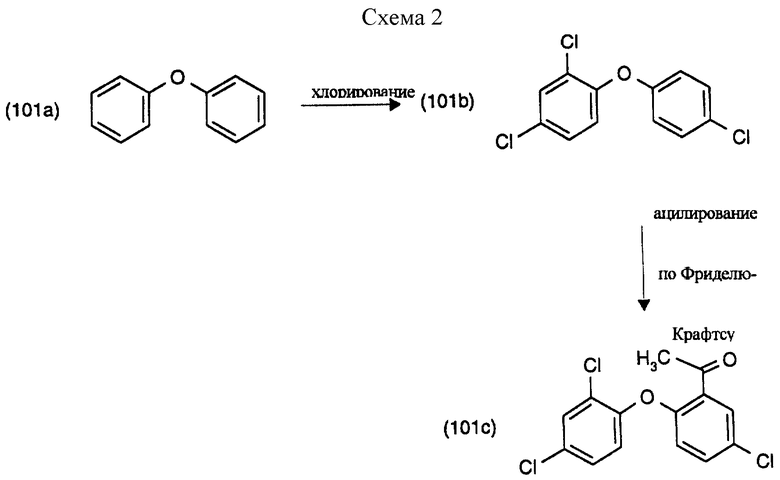
Пример 1а): Хлорирование дифенилового эфира и непосредственное применение продукта реакции для реакции с хлористым ацетилом: (см. схему 2).
Смесь из 265 г (1.56 М) дифенилового эфира (формула 101а), 7.36 г (0.06 М) дипропилсульфида и 7.46 г (0.06 М) АlСl3 помещают в реактор и расплавляют перемешиванием и подогревом до 30oС. Хлорирование проводят введением газообразного хлора с такой скоростью, чтобы температуру реакционной смеси можно было поддерживать с помощью внешнего охлаждения ниже 40oС. Реакцию контролируют газовой или жидкостной хромагографией. Хлорирование прекращают, когда содержание 2,4,4'-трихлордифенилового эфира (соединения формулы 101b) достигает 80% выхода (около 6 часов).
Для ацилирования 265 г (3.37 М) хлористого ацетила по каплям добавляют на 450 г (3.37 М) АlСl3 в 1100 мл 1,2-дихлорэтана при 20-40oС. Реакционную смесь перемешивают 15 минут при 40oС. Наконец, раствор по каплям добавляют к хлорированной реакционной смеси в 800 мл дихлорэтана при температуре 40oС на протяжении времени свыше 1 часа. Далее реакционную смесь перемешивают при около 40oС в течение 10 часов. Потом реакционную смесь обрабатывают 4 кг льда и 550 мл концентрированной НСl и на короткое время экстрагируют ее. Полученные водную и органическую фазы разделяют. После отгонки растворителя из органической фазы остается темный вязкий остаток, который при выстаивании кристаллизуется.
Выход: около 490 г реакционной смеси; содержание основного компонента составляет около 340 г, что соответствует теоретическому выходу 70% от применяемого дифенилового эфира (формула 101а).
Основной компонент: 2-ацетил-4,2,4'-трихлордифениловый эфир, соответствующий формуле (101с).
Состав реакционной смеси (% поверхности ГХ или ЖХ): около 70% основного компонента, около 15% 2,2',4,4'-тетрахлордифенилового эфира, остальное количество представлено неизвестными соединениями.
Реакционную смесь можно непосредственно применять для последующего окисления по Байеру-Виллигеру (пример 1b).
Пример 1b): Окисление по Байеру-Виллигеру (см. схему 3).
6.32 г 2-ацетил-4,2,4'-трихлордифенилового эфира, соответствующего формуле (101с), полученного в примере 1а), и 6.88 г м-хлорнадбензойной кислоты (МСРВА) диспергируют увлажняющим агентом в 40 мл воды при 20-25oС. Суспензию нагревают до 80oС и выдерживают при этой температуре с энергичным перемешиванием 3 часа. С добавлением 30 мл тетрахлорэтилена образуются две прозрачные фазы. Избыток надкислоты разлагают добавлением 0.5 г кислого сульфита натрия, смесь доводят до величины рН около 8 добавлением NaOH и водную фазу (содержащую м-хлорнадбензойную кислоту) отделяют.
Феноловый эфир формулы (101d) регенерируют в виде белого порошка с температурой плавления 48-49oС кристаллизацией из органической фазы.
Для гидролиза часть воды добавляют к органической фазе и гидроксидом натрия величину рН доводят до 12. Конечный продукт формулы (101) получают из промежуточного продукта формулы (101d). Хлористоводородной кислотой рН доводят до около 1, водную фазу отделяют и фазу тетрахлорэтилена концентрируют.
Получают 5.7 г желтоватого масла, которое содержит 80% общего выхода соединения формулы (101). После рекристаллизации из петролейного эфира продукт получают в виде белого порошка с температурой плавления 55-56oС. Данные совпадают с данными первоначального соединения.
Пример 1с): Альтернативное окисление по Байеру-Виллигеру в безводной среде.
К раствору 3 г (10 мМ) 2-ацетил-4,2,4'-трихлордифенилового эфира формулы (101с) в 20 мл безводного дихлорметана добавляют 4.5 г (13 мМ) м-хлорнадбензойной кислоты. Смесь охлаждают до 0oС и туда добавляют 0.77 мл (10 мМ) трифторуксусной кислоты. Реакционной смеси позволяют медленно согреться до комнатной температуры. После времени реакции на протяжении 8 часов при комнатной температуре реакционную смесь охлаждают раствором сульфита натрия и промывают насыщенным раствором бикарбоната натрия. Слой дихлорметана промывают несколько раз водой, высушивают над безводным сульфатом натрия и концентрируют до получения масляного остатка. Этот остаток гидролизуют кипячением его в течение 15 часов с флегмацией в 10 мл 1N раствора NaOH. Получают 2 г сырого продукта реакции, который после подкисления очищают колонной хроматографией. Таким образом получают 1.5 г (54%теретического выхода) соединения формулы (101) в виде белого кристаллического порошка.
Альтернативный гидролиз
0.9 г сырого продукта, полученного в реакции окисления по Байеру-Виллигеру, кипятят с флегмацией 4 часа в 5 мл этанола, содержащего несколько капель концентрированной НСl. Реакцию контролируют тонкослойной хроматографией. После завершения реакции спирт отгоняют в вакууме. Масляный остаток растворяют в 10 мл дихлорэтана и раствор повторно промывают водой. Органическую фазу высушивают над безводным Na2SO4 и концентрируют. Получают 0.8 г сырого соединения формулы (101). Рекристаллизация из петролейного эфира дает 0.64 г (70% теоретического выхода) кристаллического порошка.
Пример 2: Повторяют процедуру примера 1а), за исключением того, что хлорирование проводят примерно в 30% растворе дифенилового эфира в 1,2-дихлорэтане.
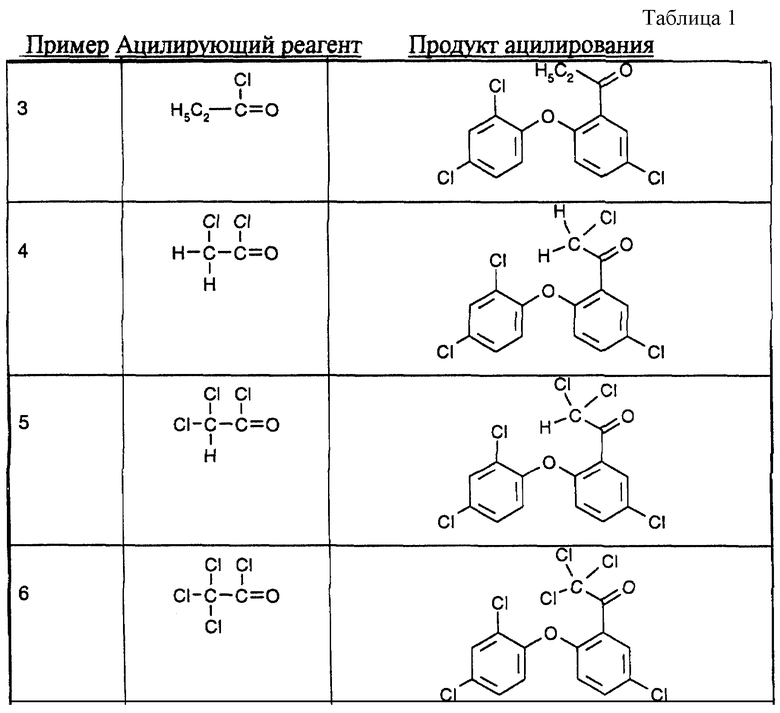
Примеры 3-6: В реакции ацилирования, описанной в примере 1а), в дополнение к хлористому ацетилу можно также применять ацилирующие реагенты, указанные в табл.1 в конце описания.
Пример 7: Ацилирование смеси 2-ацетил-4,2,4'-трихлордифенилового эфира и 4,4'-дихлордифенилового эфира и последующая реакция хлорирования.
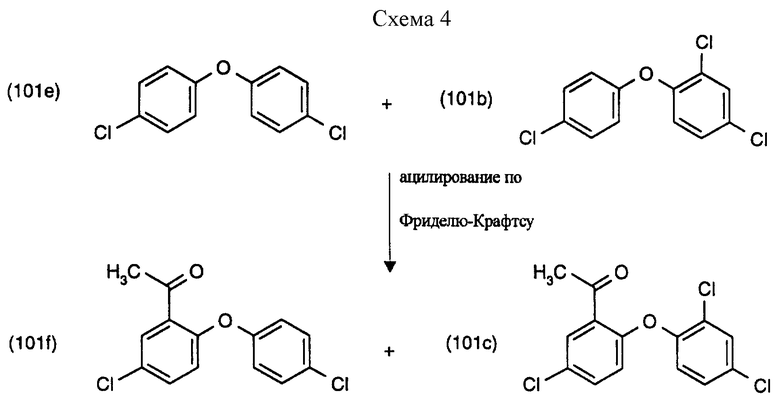
Пример 7а: Ацилирование (см. схему 4).
В трехгорлый реактор для сульфирования, снабженный капельной воронкой, уравновешивающей давление, входной трубкой для азота, мешалкой и предохранительной трубкой, помещают 480 мл безводного 1,2-дихлорэтана и 221.8 г (11.456 М) 88% хлорида алюминия. Смесь перемешивают и охлаждают в ледяной бане в атмосфере азота. К этой смеси добавляют 104 мл хлористого ацетила свежей перегонки (114.4 г, 1.456 М) в течение 15-20 минут. Экзотермической реакции позволяют остыть до комнатной температуры и смесь перемешивают 30 минут. Образуется гомогенная темно-коричневая смесь, к которой с перемешиванием при комнатной температуре на протяжении 15-30 минут по каплям добавляют 251.9 г смеси, содержащей 2,4,4'-трихлордифениловый эфир (79%) (= соединение формулы (101b)) и 4,4'-дихлордифениловый эфир (9%) (соединение формулы (101b)), растворенной в 480 мл безводного 1,2-дихлорэтана. Реакцию контролируют газовой хроматографией. После перемешивания примерно в течение 15 часов при комнатной температуре смесь добавляют к 500 мл ледяной воды, содержащей 50 мл концентрированной НСl. После перемешивания на протяжении 15 минут органическую фазу отделяют от водной фазы. Водную фазу дважды экстрагируют 1,2-дихлорэтаном, каждый раз 100 мл 1,2-дихлорэтана. Соединенные органические фазы 6 раз промывают водой по 500 мл каждый раз и высушивают над безводным сульфатом натрия. После удаления растворителя получают 244 г смеси, содержащей соединение формулы (101с) и соединение формулы (101f).
Эту реакционную смесь применяют для дальнейшей реакции хлорирования.
Пример 7b: Дальнейшее хлорирование (см. схему 5).
В трехгорлый реактор для сульфирования, снабженный капельной воронкой, входной трубкой для газообразного хлора, мешалкой, предохранительной трубкой, а также системой очистки для кислотных паров, помещают 0.077 г (0.65 мМ) пропилсульфида и 88% хлорид алюминия в 120 мл безводного 1,2-дихлорэтана. В эту смесь с перемешиванием вводят на протяжении 15 минут газообразный хлор. После прерывания подачи газа туда добавляют по каплям в течение 1.5-2 часов 244 г смеси, содержащей 2-ацетил-2,4,4'-трихлордифениловый эфир (84.4%) и 2-ацетил-4,4'-дихлордифениловый эфир (1.9%), растворенной в 120 мл безводного 1,2-дихлорэтана.
В течение 1 часа вводят с перемешиванием газообразный хлор. Реакцию контролируют газовой хроматографией. После завершения реакции смесь добавляют к 500 мл ледяной воды, содержащей около 15% НСl. Органическую фазу отделяют и водную фазу дважды промывают 1,2-дихлорэтаном, каждый раз 100 мл 1,2-дихлорэтана. Соединенные органические фазы пять раз промывают насыщенным раствором бикарбоната натрия, 200 мл раствора каждый раз, затем пять раз промывают водой, используя 200 мл воды каждый раз и высушивают над сульфатом натрия. Наконец, растворитель удаляют в вакууме. После этого получают 240 г сырого продукта, содержащего соединения формул (101с) и (101g).
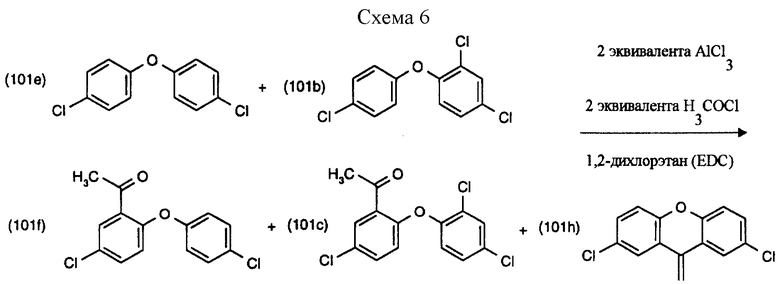
Пример 8: Ацилирование (см. схему 6).
В трехгорлую колбу с плоским круглым дном объемом 500 мл, снабженную капельной воронкой, уравновешивающей давление, верхней мешалкой и предохранительной трубкой, в атмосфере азота вводят 80 мл 1,2-дихлорэтана (EDC) и 9.85 г (0.0664 М) хлорида алюминия. Смесь охлаждают в водяной бане до 15oС и с перемешиванием в течение 30-45 минут по каплям добавляют 4.7 мл хлористого ацетила (5.18 г, 0.0660 М). К этому комплексу на протяжении 20 минут при комнатной температуре с перемешиванием добавляют по каплям 10 г смеси соединения формулы (101е) (4,4'-дихлордифенилового эфира, DCDPE), соединения формулы (101b) (2,4,4'-трихлордифенилового эфира, TCDPE) и 2,2',4,4'-тетрахлордифенилового эфира (0.0333 М DCDPE и TCDPE вместе, TetCDPE), растворенной в 20 мл EDC. При добавлении хлорирующей смеси не отмечалось значительного подъема температуры. Реакция начинала флегмировать и была под контролем ГХ (с применением FID и нормализации поверхности) через определенные промежутки времени. Потребовался 1 час, чтобы завершить преобразование TCDPE в соединение формулы (101с). Во время преобразования образовалось также 2.3% соединения формулы (101h) (= ксантена).
В табл. 2а показан ход реакции.
В другой реакции количество EDC снижено с 10 объемов до 2 объемов, хотя реакцию проводят в тех же условиях, что выше (табл. 2б).
Пример 9: Ацилирование (см. схему 7).
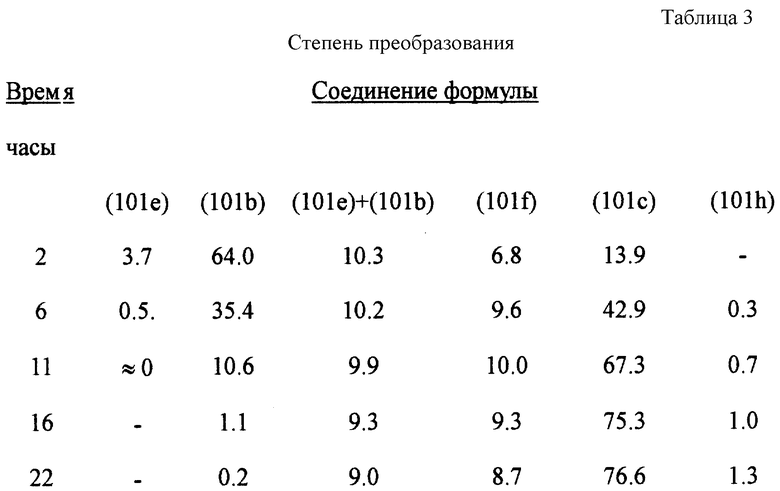
В трехгорлую колбу с плоским круглым дном объемом 20 л, снабженную капельной воронкой, уравновешивающей давление, верхней мешалкой и предохранительной трубкой, в атмосфере азота вводят 7 л дихлорметана (DCM) и 1088.2 г (7.328 М) хлорида алюминия. Смесь охлаждают в водяной бане до 15oС и с перемешиванием в течение 20 минут добавляют 522.9 мл хлористого ацетила (575.2 г, 7.328 М). В течение этого времени внутренняя температура реакции поднимается до около 20oС. Раствор перемешивают еще 10 минут, пока он не станет прозрачным. К этому комплексу на протяжении 20 минут с одновременным нагреванием реакционной смеси с перемешиванием добавляют 1100 г смеси соединений формул (101e) (DCDPE), (101b) (TCDPE) и 2,2',4,4'-тетрахлордифенилового эфира (3.664 М DCDPE и TCDPE вместе), растворенной в 4000 мл DCM. При добавлении хлорирующей смеси не отмечалось значительного подъема температуры. Реакция начинает флегмировать приблизительно спустя 1.5 часа. Через равные промежутки времени брались пробы на определение продолжительности реакции под контролем ГХ (с применением FID и нормализации поверхности). Требуется приблизительно 22-24 часа для преобразования TCDPE в соединение формулы (101с) (TCADPE). Во время конверсии образовалось также 1.3% соединения формулы (101h) (= ксантена).
Данные ГХ, приведенные в табл. 3, показывают преобразование как функцию времени.
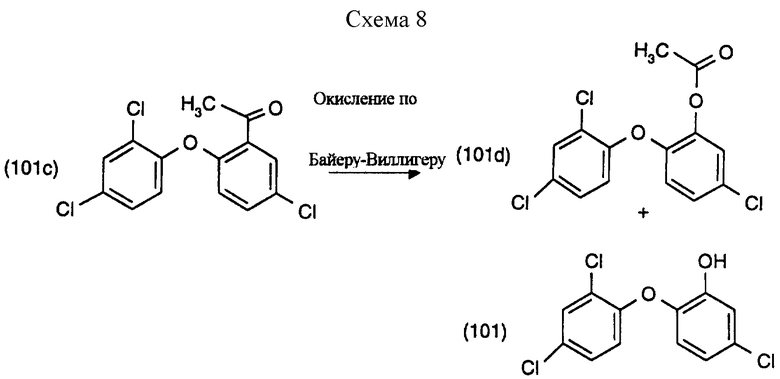
Пример 10: Окисление по Байеру-Виллигеру (см. схему 8).
5 мл ацетонитрила помещают в колбу с плоским круглым дном объемом 50 мл и туда добавляют 0.75 г (0.0079 М) комплекса мочевина-перекись водорода (UHP) и 92 г (0.0008 М) малеиновой кислоты. К этой перемешанной гетерогенной смеси при комнатной температуре в течение 10 минут порциями добавляют 0.75 г (0.0076 М) малеинового ангидрида. К раствору добавляют 0.25 г (0.0008 М) соединения формулы (101с). Реакцию продолжают перемешиванием при комнатной температуре. Реакционная смесь становится прозрачной примерно после 45 минут и ее контролируют ГХ (FID детектор и нормализация поверхности). Спустя 19 часов преобразование в реакции достигло около 40%. См. табл. 4.
Пример 11: Окисление по Байеру-Виллигеру (схема реакции соответствует схеме примера 10).
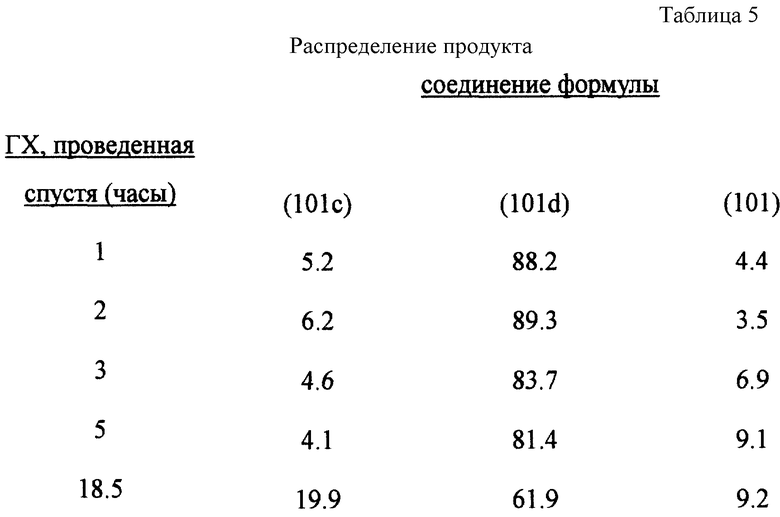
25 мл трифторуксусной кислоты и 5 мл (0.0441 М) 30%раствора перекиси водорода помещают в колбу с плоским круглым дном объемом 100 мл. Смесь перемешивают 15 минут и с перемешиванием при комнатной температуре добавляют 5.0 г (0.0158 М) соединения формулы (101с). Дальше реакцию продолжают перемешиванием ее при комнатной температуре. Спустя 15 минут раствор становится желтовато-оранжевым, его подвергают анализу ГХ (FID детектор и нормализация поверхности).
Распределение продукта было следующим (табл. 5):
Пример 12: Окисление по Байеру-Виллигеру (схема реакции соответствует примеру 10).
15 мл уксусной кислоты помещают в двухгорлую колбу с круглым плоским дном с конденсатором и капельной воронкой. Туда добавляют 4 мл (0.0280 М) 70% раствора хлорной кислоты и 2.0 г (0.0063 М) соединения формулы (101с). Гомогенную смесь нагревают до 70-75oС. Через капельную воронку в течение 30 минут по каплям добавляют 4.4 мл (0.0647 М) 50% раствора перекиси водорода. После завершения реакции смесь подвергают анализу ГХ (FID детектор и нормализация поверхности).
Распределение продукта было следующим (табл. 6).
Пример 13: Окисление по Байеру-Виллигеру (схема реакции соответствует примеру 10).
10 мл воды помещают в двухгорлую колбу с круглым плоским дном и капельной воронкой. Туда медленно добавляют 10 мл H2SO4, добавляют 2.0 г (0.0063 М) соединения формулы (101с) (2-ацетил-2',4,4'-трихлорацетилдифенилового эфира) и 50% раствор H2SO4 нагревают до 80oС. Затем температуру поднимают до около 130oС. К этому раствору на протяжении 15-20 минут по каплям добавляют 3.6 мл (0.318 М) 30% раствора перекиси водорода. После завершения реакции смесь подвергают анализу ГХ (FID детектор и нормализация поверхности).
Распределение продукта было следующим (табл. 7).








Изобретение относится к новому способу получения галоген-о-гидроксидифениловых соединений формулы (1), в которых Х- -О- или -СН2-, m = от 1 до 3, n = 1 или 2, которые применяются для защиты органических материалов от микроорганизмов, и к новым ацильным соединениям формулы (8), которые являются промежуточными продуктами, в которых R - незамещенный C1-С8алкил, замещенный 1-3 атомами галогена или гидрокси; или незамещенный С6-С12арил или С6-С12арил, замещенный 1-3 атомами галогена, С1-С5алкилом или C1-С8алкокси. Способ получения соединений формулы (1) включает четыре стадии. На первой стадии хлорируют соответствующее дифениловое соединение формулы (4), преимущественно газообразным хлором, желательно в присутствии пропил-сульфида и эквимолярного количества хлорида алюминия. На второй стадии полученное соответствующее хлорированное соединение формулы (5) ацилируют преимущественно хлористым ацетилом по реакции Фриделя-Крафтса в присутствии кислоты Льюиса, например хлористого алюминия, при эквимолярном соотношении хлористого ацетила и хлорида алюминия, обычно в присутствии галоидированного растворителя, с последующим возможным хлорированием полученного продукта после ацилирования. При этом возможно проведение ацилирования и возможного хлорирования в одном реакторе. На третьей стадии полученное ацильное соединение формулы (6), в котором значения заместителя R указаны выше, окисляют, как правило, смесью малеинового ангидрида, комплекса мочевина-перекись водорода и уксусной кислоты в качестве растворителя. На четвертой стадии гидролизуют полученное окисленное соединение формулы (7), в котором значения заместителя R указаны выше. Способ является экономичным и позволяет подавить нежелательные побочные реакции. 2 с. и 13 з.п. ф-лы, 8 табл.
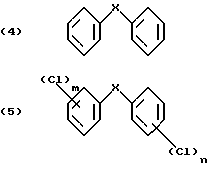
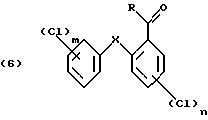
включающий четыре стадии, в котором в первой стадии хлорируют соответствующее дифениловое соединение формулы (4)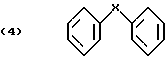
на второй стадии ацилируют полученное соответствующее хлорированное соединение формулы (5)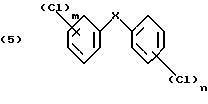
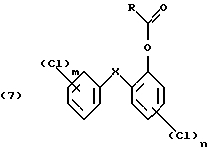
реакцией Фриделя-Крафтса в присутствии кислоты Льюиса с последующим возможным хлорированием полученного продукта после ацилирования, в третьей стадий окисляют полученное ацильное соединение формулы (6)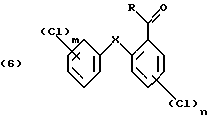
и на четвертой стадии гидролизуют полученное окисленное соединение формулы (7)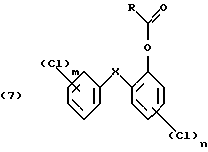
причем указанные в структурных формулах радикалы имеют следующие значения:
R - незамещенный С1-С8алкил или С1-С8алкил, замещенный 1-3 атомами галогена или гидрокси; или незамещенный С6-С12арил или С6-С12арил, замещенный 1-3 атомами галогена, С1-С5алкилом или C1-С8алкокси;
Х- -О- или -СН2-;
m = от 1 до 3; и
n = 1 или 2.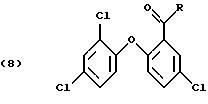
где R - незамещенный С1-С8алкил или С1-С8алкил, замещенный 1-3 атомами галогена или гидрокси; или незамещенный С6-С12арил или С6-С12арил, замещенный 1-3 атомами галогена, С1-С5алкилом или C1-С8алкокси.
| СТЕНД ДЛЯ СБОРКИ И РАЗБОРКИ КАБИН ТРАКТОРОВ | 0 |
|
SU384043A1 |
| W.D.WATSON ET.AL | |||
| Noncatalytic Chlorination of Diphenyl Ether, J.Org.Chem., 1979, vol.44, №7, pp.1155-1158 | |||
| G.A | |||
| OLAH ET | |||
| AL | |||
| Способ получения морфия из опия | 1922 |
|
SU127A1 |
| Regioselective para Halogenation of Phenols, Phenol Ethers and Anilines with Halodimethylsulfonium Halides, SYNTHESIS, 1986, №10, pp | |||
| Запорный к лапан для тушения горящих нефтяных фонтанов | 1914 |
|
SU868A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| D.C | |||
| ATKINSON ET.AL | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ определения границы руда-закладочный бетон | 1987 |
|
SU1493776A1 |
| Пломба | 1984 |
|
SU1288747A1 |
| DE 3723994 A1, 04.02.1988 | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Гибочный станок | 1976 |
|
SU642044A1 |
| "The Merck Index, Thelfth Edition" | |||
| Предохранительное устройство для паровых котлов, работающих на нефти | 1922 |
|
SU1996A1 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРСОДЕРЖАЩИХ ДИФЕНОЛОВ | 0 |
|
SU232980A1 |
| Способ получения м-феноксифенола | 1990 |
|
SU1740365A1 |
