Техническая область
Данное изобретение относится к способу получения фторспирта общей формулы I
H(CFR1CF2)nCH2OH (I)
где R1 представляет F или СF3, когда n=1, и R1 представляет собой F, когда n= 2, который является по существу свободным от примесей, и к применению указанного фторспирта для производства носителя записи информации, включающего субстрат и создаваемый на нем записывающий (регистрирующий) слой, приспособленный для лазерной записи и/или считывания.
Предшествующий уровень
Относительно технологии получения H(CF2CF2)nCH2OH (n=1, 2) следует отметить, что в Японской не прошедшей экспертизу патентной публикации 154707/1979 и патенте США 2559628, описывается, что смесь теломеров, т.е. Н(СF2СF2)nСН2OН (максимальное значение n= 12) может быть получена взаимодействием метанола с тетрафторэтиленом в присутствии трет-бутилоктилпероксида.
Однако даже если теломерную смесь, полученную с помощью данного способа, очищают с помощью перегонки, образующийся после выпаривания остаток порядка нескольких сотен ч. /млн. не может быть удален, и в результате, когда эта смесь используется в качестве растворителя при изготовлении носителя записи информации, включающего субстрат и создаваемый на нем записывающий слой, приспособленный для лазерной записи и/или считывания, такого как, например, CD-R и DVD-R, неизбежным недостатком является недостаточно высокое качество носителя для записи информации вследствие влияния указанного остатка, образующегося после выпаривания.
Предметом данного изобретения является предоставление фторспирта общей формулы I
H(CFR1CF2)nCH2OH (I)
где n и R1 имеют определенные выше значения,
который является по существу свободным от примесей, таких как остаток после выпаривания и вещества, абсорбирующие УФ-излучение, способа получения указанного фторспирта, применение указанного фторспирта в качестве растворителя для изготовления носителя записи информации, включающего субстрат и создаваемый на нем записывающий слой, приспособленный для лазерной записи и/или считывания.
Описание изобретения
Изобретение относится к приведенным далее пунктам 1-20.
1. Способ получения фторспирта следующей формулы I
H(CFR1CF2)nCH2OH (I)
где R1 представляет собой F или СF3, когда n=1, и R1 представляет собой F, когда n=2,
включающий взаимодействие метанола с тетрафторэтиленом или гексафторпропиленом в присутствии источника свободных радикалов, в котором реакционная смесь подвергается перегонке либо в присутствии основания, либо после контактирования указанной реакционной смеси с основанием.
2. Способ получения фторспирта по п.1, в котором основание представляет собой вещество, имеющее значение рКb не более 2.
3. Способ получения фторспирта по п.1, в котором основание представляет собой алкоксид щелочного металла или гидроксид щелочного металла.
4. Способ получения фторспирта по п.1, в котором основание представляет собой, по меньшей мере, одно вещество, выбранное из группы, включающей алкоксиды натрия, гидроксид натрия и гидроксид калия.
5. Способ получения фторспирта по п.1, в котором фторспирт формулы I
H(CFR1CF2)nCH2OH (I)
где R1 и n имеют определенные выше значения,
который получен с помощью перегонки, имеет остаток после выпаривания не более 50 ч./млн.
6. Способ получения фторспирта по п.5, в котором фторспирт формулы I
H(CFR1CF2)nCH2OH (I)
где R1 и n имеют определенные выше значения,
который получен с помощью перегонки, имеет остаток после выпаривания не более 25 ч./млн.
7. Способ получения фторспирта по п.5, в котором фторспирт формулы I
H(CFR1CF2)nCH2OH (I)
где R1 и n имеют определенные выше значения,
который получен с помощью перегонки, имеет остаток после выпаривания не более 10 ч./млн.
8. Способ получения фторспирта по п.1, в котором источник свободных радикалов представляет собой, по меньшей мере, один представитель, выбранный из группы включающей инициатор реакции. УФ-излучение и нагрев.
9. Способ получения фторспирта по п.8, в котором источник свободных радикалов представляет собой инициатор реакции, имеющий период полураспада при температуре реакции около 10 часов.
10. Способ получения фторспирта по п.8, в котором источник свободных радикалов представляет собой пероксид.
11. Способ получения фторспирта по п.8, в котором источник свободных радикалов представляет собой ди-трет-бутилпероксид, трет-бутилпероксиизопропилкарбонат или трет-бутилперокси-2-этилгексаноат.
12. Способ получения фторспирта по п.1, в котором вместе с источником свободных радикалов используется акцептор кислоты.
13. Фторспирт формулы I
H(CFR1CF2)nCH2OH (I)
где R1 представляет собой F или СF3, когда n=1, R1 представляет собой F, когда n=2,
который имеет остаток от выпаривания не более 50 ч./млн.
14. Фторспирт по п.13, остаток от выпаривания которого составляет не более 25 ч./млн.
15. Фторспирт по п.13, остаток от выпаривания которого составляет не более 10 ч./млн.
16. Фторспирт по п.13, абсорбция (190-300 нм) которого в метаноле составляет не более 0,2 абс.
17. Фторспирт по п.13, абсорбция (205 нм) которого в метаноле составляет не более 0,1 абс.
18. Фторспирт по п.17, абсорбция (205 нм) которого в метаноле составляет не более -0,2 абс.
19. Применение фторспирта по п.13 для изготовления носителя записи информации, включающего субстрат и создаваемый на нем записывающий слой, приспособленный для лазерной записи и/или считывания.
20. Носитель записи информации, включающий субстрат и создаваемый на нем записывающий слой, приспособленный для лазерной записи и/или считывания и изготовленный с использованием фторспирта формулы I
H(CFR1CF2)nCH2OH (I)
где R1 представляет собой F или CF3, когда n=1, R1 представляет собой F, когда n=2,
полученного по способу по п.1, или фторспирта формулы I
H(CFR1CF2)nCH2OH (I)
где R1 представляет собой F или СF3, когда n=1, R1 представляет собой F, когда n=2, по п.13.
В способе получения согласно данному изобретению метанол используется в избытке относительно тетрафторэтилена или гексафторпропилена. Температура реакции равна около 40-140oС, время реакции составляет около 3-12 часов, давление реакции равно около 0,2-1,2 МПа. Данная реакция может проводиться в реакторе высокого давления, таком как, например, автоклав. Реакционная система предпочтительно продувается инертным газом с использованием азота, аргона или подобного газа.
После завершения реакции избыток метанола необязательно отгоняют и остаток дополнительно подвергают перегонке в присутствии основания. Кроме того, в том случае, когда реакционная смесь содержит в качестве примеси Н(СF2СF2)nСН2ОН (n≥3) или H(CF(CF3)CF2)nCH2OH (n≥2), примесь предпочтительно удаляют заранее с помощью перегонки. Реакционная смесь, содержащая фторспирт формулы I
H(CFR1CF2)nCH2OH (I)
где n и R1 имеют значения, определенные выше,
подвергается перегонке либо в присутствии основания, либо после контактирования реакционной смеси с основанием.
Основание, добавляемое к указанной реакционной смеси или контактирующее с ней, предпочтительно представляет собой основание со значением рКb не более 2, и включает таким образом алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия, пропоксид натрия, трет-бутоксид калия, этоксид лития и др. , гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия, гидроксид лития и др., гидроксид кальция, гидроксид алюминия, гидроксид бария, гидроксид магния и натриевую известь. Количество основания составляет около 0,05-1,0 моль, предпочтительно приблизительно 0,1-0,5 моль на 1 кг реакционной смеси, из которой удаляют метанол.
Акцептор кислоты включает, но не ограничивается ими, карбонаты и гидрокарбонаты щелочных металлов или щелочноземельных металлов, такие как карбонат кальция, карбонат магния, карбонат бария, карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия и т.п., оксид кальция, гидроксид кальция и натриевую известь. Предпочтительным акцептором кислоты является вещество, способное захватывать кислоту, образуемую в течение реакции, например HF, не придавая реакционной системе высокой основности.
Количество акцептора кислоты конкретно не ограничивается, и может быть около 0,001-0,1 моля из расчета на 1 моль тетрафторэтилена или гексафторпропилена.
В качестве источника или генератора свободных радикалов может использоваться, по меньшей мере, один представитель группы, включающей инициатор реакции, УФ-излучение и нагрев. Когда источником свободных радикалов является УФ-излучение, может использоваться УФ-свет от ртутной лампы, например, среднего давления или высокого давления. Когда источником свободных радикалов является нагрев, температура может выбираться в интервале, например, между 250 и 300oС. Инициатор реакции включает, но не ограничивается ими, пероксиды, и предпочтительно использовать инициатор с периодом полураспада при температуре реакции около 10 часов.
Предпочтительный источник свободных радикалов включает пербутил D (ди-трет-бутилпероксид), пербутил О (трет-бутилперокси-2-этилгексаноат) и пербутил I (трет-бутилпероксиизопропилкарбонат). Количество инициатора реакции должно обычно составлять около 0,005-0,1 моля в расчете на 1 моль тетрафторфэтилена или гексафторпропилена.
Количество остатка после выпаривания фторспирта, получаемого согласно данному изобретению, составляет 50 ч./млн. (м.д.) или менее, предпочтительно 25 м.д. или менее, предпочтительнее 10 м.д. или менее.
Количество остатка после выпаривания может определяться следующим образом. Фторспирт выпаривают при 40oС /5 мм рт. ст., остаток взвешивают и выражают в массовых долях "частях на миллион" (м.д.) из расчета на фторспирт, такой как НСF2СF2СН2ОН.
УФ-абсорбция фторспирта формулы I, полученного согласно данному изобретению, в метаноле при 205 нм составляет не более 0,1 абс, предпочтительно -0,1 абс или менее, предпочтительнее -0,2 абс или менее. УФ-абсорбция в метаноле может быть измерена с использованием смеси 1 мл фторспирта общей формулы I и 3 мл метанола в качестве образца и метанола в качестве контроля.
То, что фторспирт данного изобретения является "по существу свободным от примесей" означает, что (i) остаток после выпаривания фторспирта составляет не более 50 м. д. , предпочтительно не более 25 м.д., предпочтительнее не более 10 м.д., и/или (ii) его УФ-абсорбция (205 нм) в метаноле составляет не более 0,1 абс, предпочтительно не более -0,1 абс, предпочтительнее не более -0,2 абс.
Носитель записи информации, включающий субстрат и создаваемый на нем записывающий слой, приспособленный для лазерной записи и/или считывания, может быть изготовлен путем растворения красителя в растворителе, содержащем фторспирт общей формулы I согласно изобретению, предпочтительно в растворителе фтористого ряда, содержащем указанный фторспирт, с последующим осуществлением стандартного ряда операций с использованием полученного раствора красителя, включая покрытие субстрата этим раствором и сушку покрытого субстрата для обеспечения записывающего слоя, содержащего краситель. Указанный выше краситель включает цианиновые красители, фталоцианиновые красители, пирилиевые красители, тиопирилиевые красители, скварилиевые красители, азулениевые красители, индофенольные красители, индоанилиновые красители, трифенилметановые красители, хиноновые красители, аминиевые красители, диаммониевые красители и красители на основе комплексов металлов. Сырьевой материал для субстрата включает пластики, такие как поликарбонаты, поли(метилметакрилат), эпоксисмола, аморфные полиолефины, сложные полиэфиры, поли(винилхлорид) и т.д., стекло и керамику. С целью улучшения гладкости поверхности и адгезии или предотвращения разложения записывающего слоя между записывающим слоем и субстратом может быть предусмотрено грунтовочное покрытие или прокладка, и/или на записывающем слое может быть образован защитный слой.
Согласно изобретению можно легко получить по существу свободные от примесей НСF2СF2СН2ОН, Н(СF2СF2)2СН2OН и НСF(СF3)СF2СН2OН, которые годятся для использования в производстве носителя записи информации, включающего субстрат и создаваемый на нем записывающий слой, приспособленный для лазерной записи и/или считывания (оптические диски, такие как CD-R, DVD-R и т.д.) или фоточувствительного материала для пленки.
Наилучший способ осуществления изобретения
Следующие примеры иллюстрируют изобретение более подробно.
ПРИМЕР 1
В автоклав добавляют метанол (2 л), ди-трет-бутилпероксид (45 г) и карбонат кальция (30 г). После продувки азотом в автоклав вводят тетрафторэтилен с начальной скоростью 600 г/час. Реакцию проводят в течение 6 часов при регулировании температуры и давления на уровне соответственно 125oС и 0,8 МПа.
После охлаждения реакционную смесь перегоняют для удаления метанола и затем H(CF2CF2)nCH2OH (n - целое число 2 или более), получая фракцию НСF2СF2СН2ОН (1,2 л). Остаток после выпаривания фракции HCF2CF2CH2OH составляет приблизительно 600 м.д., и абсорбция (205 нм) составляет 2,0 абс. Анализ капиллярной ГХ/МС показывает наличие в качестве примеси различных альдегидов, таких как НСНО, HCF2CF2CHO, HCF2CHFCHO, HCF2CF2CF2CF2CHO, HCF2COOCH2CH=CHCHO, HCF2CH2COOCH=CHCHO, HCF2CF2CH(ОН)ОСН2СНО.
Повторная перегонка указанной фракции почти не вызывает каких-либо изменений любого из количеств указанной примеси, остатка от выпаривания и абсорбции (205 нм).
К полученной выше фракции HCF2CF2CH2OH (1 л) добавляют 28% метоксид натрия в метаноле (30 г) и смесь перегоняют при нагревании, получая HCF2CF2CH2OH, который по существу свободен от примеси. Остаток после перегонки полученного таким образом HCF2CF2CH2OH составлял не более 10 м.д., его абсорбция (205 нм) составляла не более -0,2 абс. Количество упомянутых выше альдегидов было ниже предела обнаружения для ГХ/МС.
Условия анализа ГХ/МС:
1) Колонка: жидкая фаза DB-1301
Толщина пленки 1,00 мкм
Размер колонки 60 м х 0,247 мм
2) Условия анализа:
Носитель Нe 200 кПа
Воздух 40 кПа
Н2 50 кПа
Температура 50oС в течение 5 минут - 220oС в течение 15 минут (температура повышается со скоростью 15oС/мин).
Инжекция 200oС.
ПРИМЕР 2
Фракцию H(CF2CF2)nCH2OH (n≥2) подвергают фракционной перегонке для выделения фракции Н(СF2СF2)2СН2ОН. К этой фракции добавляют метоксид натрия, как показано в примере 1, и смесь перегоняют, получая фракцию H(CF2CF2)2CH2OH, показывающую остаток от выпаривания не более 25 м.д.
ПРИМЕР 3
Реакцию и процедуру очистки с помощью перегонки осуществляли по методике примера 1, за исключением того, что использовали гексафторпропилен вместо тетрафторэтилена. В результате получен НСF(СF3)СF2СН2ОН, имеющий остаток от выпаривания не более 25 м.д., УФ-абсорбция (205 нм) не более 0,1 абс.
ПРИМЕР 4
Фракцию HCF2CF2CH2OH до перегонки в присутствии основания, полученную в примере 1, пропускали через колонку с натриевой известью для удаления HF. В результате газохроматографическая чистота падала с 99,974% до 99,5368%.
Когда данную фракцию HCF2CF2CH2OH пониженной чистоты перегоняли, получали HCF2CF2CH2OH, дающий остаток от выпаривания не более 50 м.д., значение УФ-абсорбции (205 нм) не более 0,1 абс.
СРАВНИТЕЛЬНЫЙ ТЕСТ
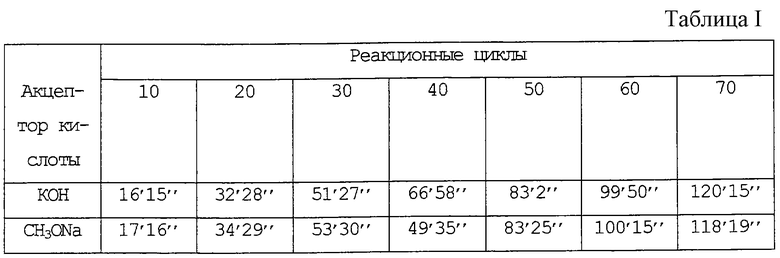
Тест проводили в тех же условиях, что и в примерах 19 и 20 патента США 4346250.
В стеклянный автоклав (внутренний объем 2000 мл), снабженный мешалкой из нержавеющей стали, загружали метанол (800 г, 25 моль) и перекись ди-трет-бутила (пербутил D, 20 г), после закрытия автоклава снижали внутреннее давление при перемешивании до тех пор, пока метанол не закипал. Затем добавляли тетрафторэтилен (ТФЭ) для достижения атмосферного давления и при нагревании повышали температуру до 125oС. Реакцию инициировали введением в автоклав ТФЭ до давления 0,8 МПа (около 8 кг/см2). Когда давление снижалось до 0,7 МПа (около 7 кг/см2), немедленно добавляли ТФЭ для повышения давления до 0,8 МПа (около 8 кг/см2). Данный цикл реакции повторяли, при этом на каждом цикле вводили одно и то же количество ТФЭ (около 3 г) и в то же время получали такое же количество теломеров. На 30-м цикле после инициирования реакции в автоклав под давлением добавляли раствор метанола (10 мл), содержащий КОН (0,56 г) или СН3ОNа (0,54 г) (1/100 моль в виде КОН или СН3ОNа) и реакцию продолжали далее. Время реакции через каждые 10 циклов после инициирования реакции приведено в таблице I в конце описания.
Как показано в таблице I, скорость реакции восстанавливается при добавлении КОН или СН3ОNа.
По окончании реакции автоклав охлаждали до комнатной температуры. ТФЭ выпускали для сбора реакционной смеси. Метанол удаляли из реакционных смесей перегонкой, получая 210 г и 215 г смеси теломеров соответственно. рН и содержание HF обеих смесей теломеров составили: рН 3,5 и HF 0,95 г (0,048 моль). Следовательно, весь КОН и СН3ОNа, добавленный в ходе реакции, расходуется в реакции нейтрализации.
Впоследствии Н(CH2CF2)nСН2OН, в котором n представляет целое число, равное 2 или более, отделяли, получая фракции HCH2CF2CH2OH (150 г в обоих случаях). Остаток от выпаривания и поглощения (205 нм) фракции HCH2CF2CH2OH приведены в таблице II.
Анализ фракций с помощью капиллярной ГХ/масс-спектрометрии показал, что в обеих фракциях содержится большое число альдегидов, таких как формальдегид, HCF2CF2CHO, HCF2CHFCHO, HCF2CF2CF2CF2CHO, HCF2COOCH2CH=CHCHO, HCF2CH2COOCH=CHCHO и HCF2CF2CH(OH)OCH2CHO.
Повторная перегонка указанной фракции вызывала лишь незначительные изменения в любом из показателей количества указанной примеси, остатка от выпаривания и поглощения (205 нм).
Причины, по которым остаток от выпаривания нельзя удалить из фторспирта повторными перегонками
Реакционная смесь содержит большое число органических соединений, таких как формальдегид (НСНО), остаточный инициатор реакции и продукты разложения иные, чем исходный материал (метанол), и продукты реакции [Н (CFRlCF2)nCH2ОH] , где R1 представляет F, когда n=1, R1 представляет F или CF3, когда n=2.
Альдегидные соединения, включающие (3) HCF2CHFCHO, (5) HCF2COOCH2CH= CHCHO, (6) HCF2CH2COOCH=CHCHO и (7) HCF2CF2CH(OH)OCH2CHO, образуются в реакционной смеси под действием радикалов или в окислительных условиях. Хотя высококипящие альдегидные соединения (имеющие более длительное время удерживания в ГХ) можно удалить, альдегидные соединения (3), (5) и (6) удалять трудно из-за близости их температур кипения к HCF2CF2CH2OH и большого числа азеотропов.
Похоже, альдегидные компоненты (3), (5) и (6) не вызывают проблем при использовании для производства CD-R, поскольку альдегидные компоненты (3), (5) и (6) выпариваются вместе с HCF2CF2CH2ОH и не остаются в CD-R. Однако компоненты (3), (5) и (6), содержащиеся в фторспирте, постепенно превращаются в высококипящие соединения, так что следовое количество альдегидных компонентов остается в CD-R в виде высококипящих соединений и влияет на записывающие свойства CD-R дисков.
При проведении перегонки после превращения компонентов (3), (5) и (6) в высококипящие соединения при термическом разложении HCF2CF2CH2ОH во время перегонки образуются неотделимые компоненты (3), (5) и (6). Поэтому содержание альдегидных компонентов (3), (5) и (6) в дистилляте нельзя уменьшить (смотри таблицу III). В заключение, с помощью перегонки в отсутствие основания практически невозможно уменьшить остаток от выпаривания до 50 м.д.
Примеси во фторспирте:
(I) HCF2CF2CHO
(2) СН3ОН
(3) HCF2CHFCHO
(4) HCF2CF2CF2CF2CHO
(5) HCF2COOCH2CH=CHCHO
(6) HCF2CH2COOCH=CHCHO
(7) HCF2CF2CH(ОН)ОСН2СНО
(8) HCF2CF2CH2OCH2OCH2CF2CF2H
(9) другие, такие как высококипящие соединения.
Экспериментальные данные (1)
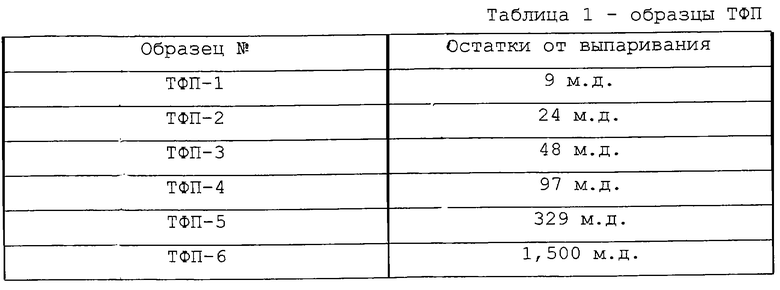
Сравнительные данные с использованием фторспирта (HCF2CF2CH2OH: ТФП) с различными количествами остатка от выпаривания
[Образец фторспирта]
Как показано в таблице 1, использовались образцы HCF2CF2CH2OH (ТФП 1-6) с шестью уровнями остатка от выпаривания.
[Условия производства CD-R]
Сто CD-R дисков приготавливали в следующих условиях с использованием образцов HCF2CF2CH2ОH (ТФП 1-ТФП 6):
Матрица
Высота бороздки ~200 нм
Ширина бороздки ~450 нм
Состав образцов растворов цианинового красителя:
Концентрация: содержание твердого вещества 40 г/(ТФП)
Содержание твердого вещества: краситель: S04/L04 (70/30) (продукт NIPPON KANKO SHIKISO).
Процедура производства:
Температура 22±0,5oС
Влажность 45±2,5%
Обращение: стандартное (изменяемое со шкалой на пятом уровне)
Сушка 40oС x 1 мин
Отражающий слой: серебро, 80 нм толщиной
Защитный слой: тип DSM 5100 (лак)
Производство CD-R дисков:
аппарат тщательно промывают и сушат перед направлением образцов,
раствор красителя используют в течение дня, когда этот раствор приготовлен,
после того, как процесс производства CD-R становится стабильным, производство CD-R продолжают до тех пор, пока не получают 100 пластинок CD-R дисков в хорошем состоянии.
Диски хранят в помещении.
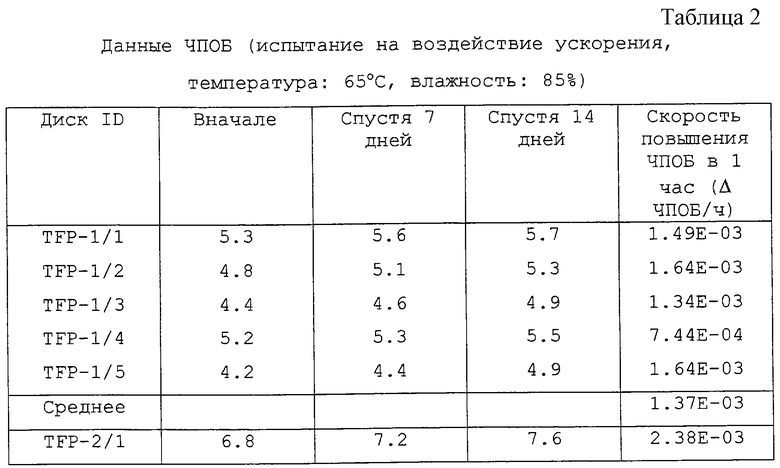
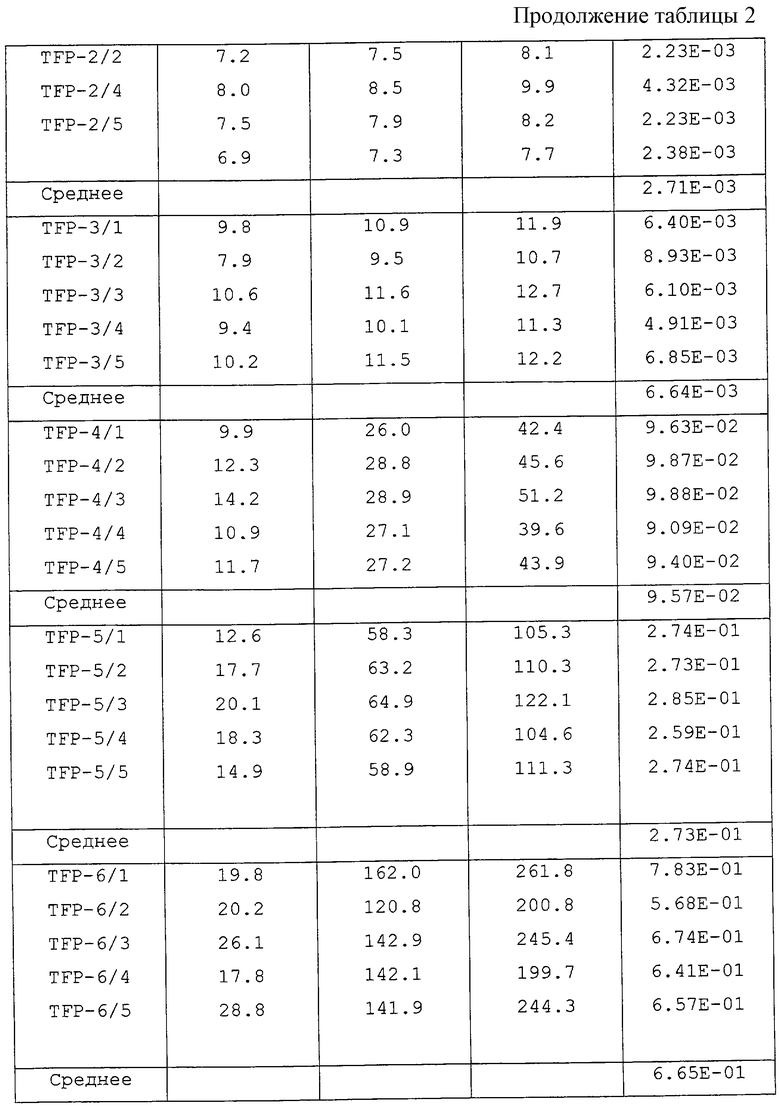
[Тест записанного CD-R диска]
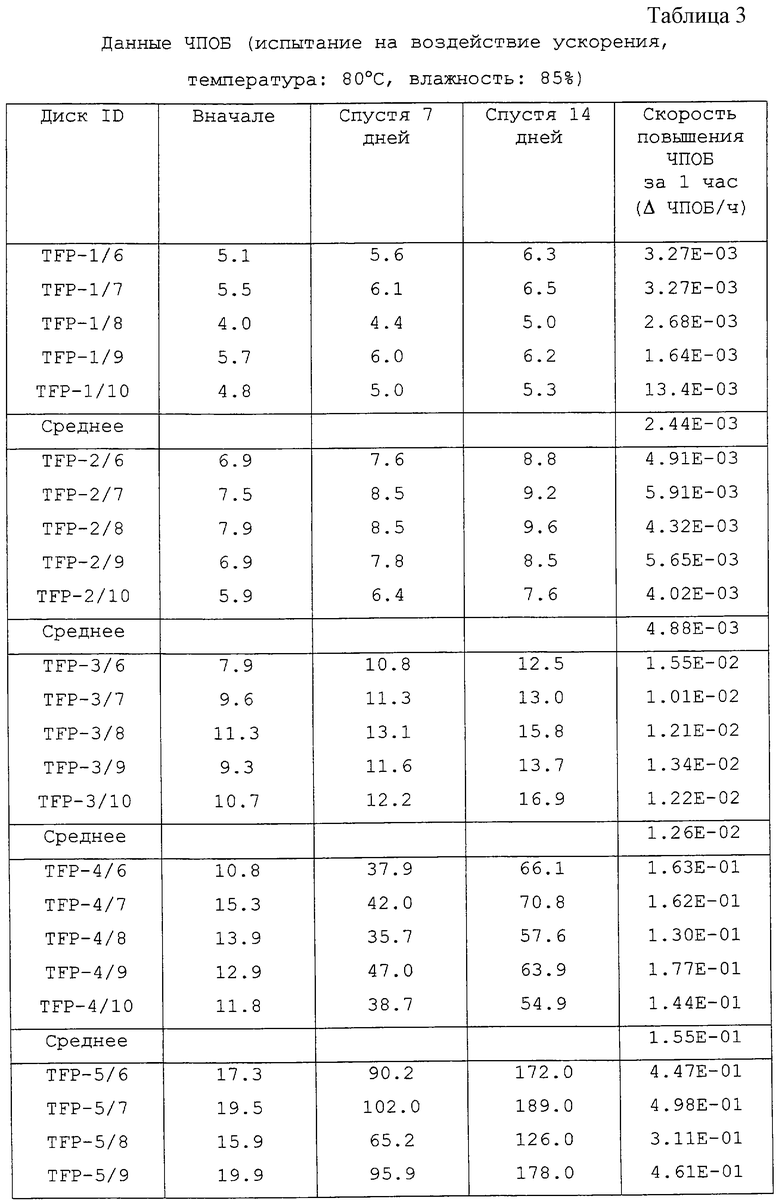
Три комплекта дисков, состоящих каждый из 5 дисков, выбрали произвольно из изготовленных дисков с использованием образцов ТФП. На выбранные диски произвели запись в следующих условиях. Что касается каждого диска, ЧПОБ (частоту появления ошибочных блоков) определяли (i) сразу же после произведения записи, (ii) после испытания на воздействие ускорения при температуре 65oС и влажности 85% в течение 7 дней и 14 дней, (iii) после испытания на воздействие ускорения при температуре 80oС и влажности 85% в течение 7 дней и 14 дней. Результаты приведены в таблицах 2, 3 и 4, при этом каждая таблица соответствует испытаниям на воздействие ускорения (ii), (iii) и (iv).
Условия произведения записи:
устройство записи: Plextor Plexwriter 6/28
скорость записи: четырехкратная скорость
продолжительность записи 72 мин
Остатки от выпаривания каждого образца ТФП суммированы в таблице 5. Скорости повышения ЧПОБ в час для CD-R, произведенных с использованием каждого из образцов ТФП, приведены в таблице 6.
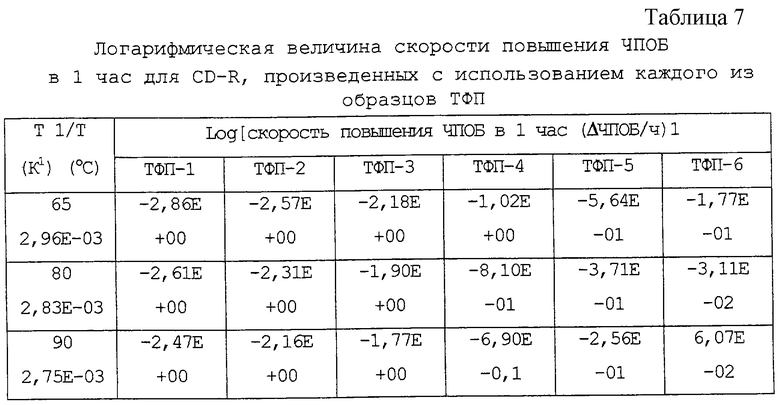
Скорость повышения ЧПОБ в 1 час в таблице 6 переводят в логарифмическую величину, она приведена в таблице 7.
Данные таблицы 7 нанесены на график согласно уравнению Аррениуса, где на оси абсцисс представлена величина, обратная абсолютной температуре (1/Т), а на оси ординат представлено логарифмическое значение скорости повышения ЧПОБ в 1 час (фиг.1).
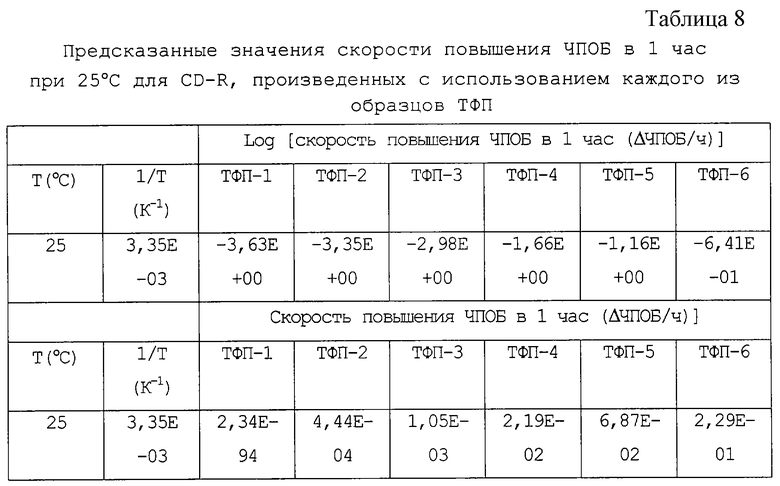
На фиг.1 скорость повышения ЧПОБ в ЧПОБ в 1 час при 25oС (1/Т=3/35К-03) экстраполируют путем расчета приблизительной прямой методом наименьших квадратов с использованием данных при трех различных температурах. Предсказанные значения при 25oС приведены в таблице 8.
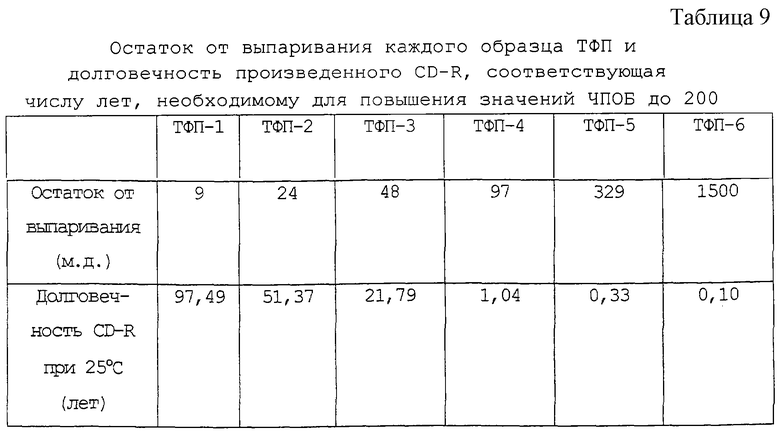
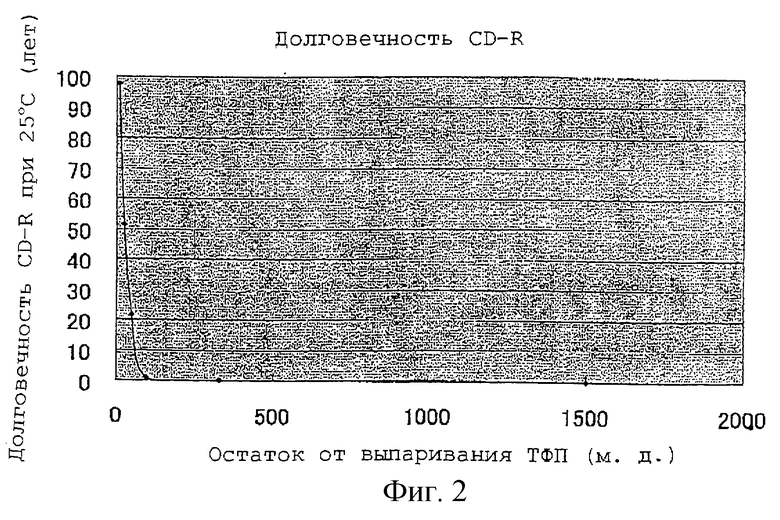
Число лет, необходимое для повышения значений ЧПОБ для каждого CD-R до 200, рассчитано исходя из скорости повышения ЧПОБ в 1 час, как показано в таблице 8, и представлено как долговечность CD-R при 25oС в таблице 9. Фиг.2 представляет собой график, на котором на оси абсцисс представлен остаток от выпаривания ТФП, а на оси ординат представлена долговечность CD-R при 25oС.
Данные результаты, приведенные на фиг.2, показывают, что для получения CD-R с практической долговечностью подходящим является предпочтительно не более чем 25 м.д. остатка от выпаривания ТФП и что более чем 50 м.д. остатка от выпаривания резко сокращают долговечность CD-R.
Дополнительные примеры 5-41
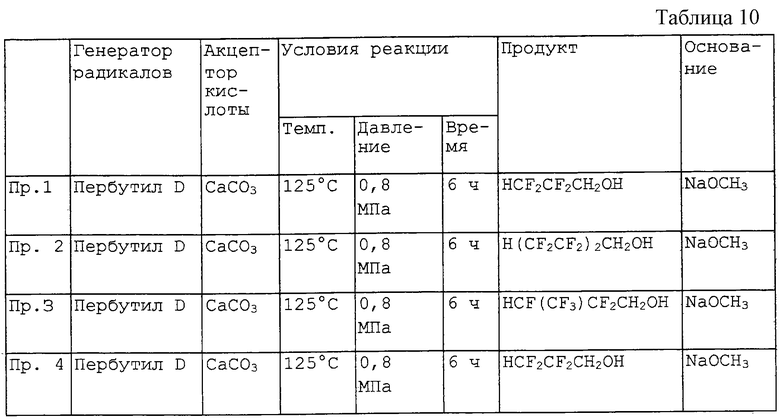
Сводка примеров 1-4 описания (см. табл.10).
ПРИМЕР 5
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2OH (1 литр) добавляли 95%-ный этилат натрия (11 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученного при этом HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (абсорбция) (205 нм) составляло не более -0,2 абс.
ПРИМЕР 6
Фракцию HCF2CF2CH2ОH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2ОH (1 литр) добавляли 25%-ный пропилат натрия в растворе пропанола (51 г, около 0,16 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученного при этом HCF2CF2CH2ОH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 7
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2OH (1 литр) добавляли 97%-ный трет-бутилат калия (18 г, около 0,16 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученного при этом HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не боле -0,2 абс.
ПРИМЕР 8
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2OH (1 литр) добавляли 10%-ный этилат лития в растворе этанола (81 г, около 0,16 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученного при этом HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 9
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2OH (1 литр) добавляли 25%-ный водный раствор гидроксида натрия (25 г, около 0,16 моль) и смесь перегоняли при нагревании. Остаток после выпаривания полученного при этом HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 10
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2OH (1 литр) добавляли 25%-ный водный раствор гидроксида калия (35 г, около 0,16 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученного при этом HCF2CF2CH2ОH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 11
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2OH (1 литр) добавляли 25%-ный водный раствор гидроксида лития (15 г, около 0,16 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученного при этом HCF2CF2CH2ОH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 12
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. Фракцию HCF2CF2CH2OH (1 литр) пропускали через трубку, которую заполняли гидроксидом кальция (50 г, около 0,68 моль) для удаления HF. Чистота фракции HCF2CF2CH2OH, определенная методом газовой хроматографии, снижалась с 99,968% до 99,523% при обработке в трубке с Ca(OH)2. Фракцию HCF2CF2CH2OH пониженной чистоты перегоняли при нагревании. Остаток от выпаривания полученного при этом HCF2CF2CH2OH составлял не более 25 м.д., а поглощение (205 нм) составляло не более 0,1 абс.
ПРИМЕР 13
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. Фракцию HCF2CF2CH2OH (1 литр) пропускали через трубку, заполненную гидроксидом алюминия (50 г, около 0,64 моль), для удаления HF. Чистота фракции HCF2CF2CH2ОH, определенная методом газовой хроматографии, снижалась с 99,968% до 99,523% при обработке в трубке с А1(ОН)3. Фракцию HCF2CF2CH2OH пониженной чистоты перегоняли при нагревании. Остаток от выпаривания полученного при этом HCF2CF2CH2OH составлял не более 25 м.д., а поглощение (205 нм) составляло не более 0,1 абс.
ПРИМЕР 14
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. Фракцию HCF2CF2CH2OH (1 литр) пропускали через трубку, заполненную октагидратом гидроксида бария (150 г, около 0,48 моль), для удаления HF. Чистота фракции HCF2CF2CH2OH, определенная методом газовой хроматографии, снижалась с 99,972% до 99,511% при обработке в трубке с Ва(ОН)3•6Н2О. Фракцию HCF2CF2CH2OH пониженной чистоты перегоняли при нагревании. Остаток от выпаривания полученного при этом HCF2CF2CH2OH составлял не более 25 м.д., а поглощение (205 нм) составляло не более 0,1 абс.
ПРИМЕР 15
Фракцию HCF2CF2CH2ОH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. Фракцию HCF2CF2CH2OH (1 литр) пропускали через трубку, заполненную гидроксидом магния (50 г, около 0,86 моль), для удаления HF. Чистота фракции HCF2CF2CH2OH, определенная методом газовой хроматографии, снижалась с 99,975% до 99,535% при обработке в трубке с Мg(ОН)2. Фракцию HCF2CF2CH2OH пониженной чистоты перегоняли при нагревании. Остаток от выпаривания полученного при этом HCF2CF2CH2OH составлял не более 25 м.д., а поглощение (205 нм) составляло не более 0,1 абс.
ПРИМЕР 16
В автоклав, снабженный ртутной лампой высокого давления, загружали метанол (2 литра) и карбонат кальция (30 г). После замещения азотом загружали ТФЭ с начальной скоростью 600 г/ч. Реакцию проводили при облучении УФ-светом при помощи ртутной лампы высокого давления в течение 48 часов при температуре 125oС и давлении 0,8 МПа. Реакционную смесь охлаждали и перегоняли для удаления метанола, а затем перегоняли дополнительно для отделения фракции HCF2CF2CH2OH от Н(CF2CF2)nСН2ОН (n представляет целое число, равное 2 или более). Полученная при этом фракция HCF2CF2CH2OH имела остаток от выпаривания около 400 ч./млн. и поглощение (205 нм) 1,5 абс.
Анализ данной фракции методом капиллярной ГХ/масс-спектрометрии показал, что во фракции содержится большое число альдегидов. Повторная перегонка указанной фракции привела к небольшому изменению остатка от выпаривания и поглощения (205 нм).
К полученной таким образом фракции HCF2CF2CH2OH (0,2 литра) добавляли 28%-ный NаОСН3 (6 г, 0,03 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 17
Реакцию и перегонку проводили таким же образом, что и в примере 16, за исключением того, что вместо тетрафторэтилена использовали гексафторпропилен, получая 0,22 литра фракции HCF(CF3)CF2CH2OH. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2ОH составлял 450 м.д., а поглощение (205 нм) составляло 1,6 абс.
К полученной таким образом фракции HCF2CF2CH2OH (0,2 литра) добавляли 28%-ный NаОСН3 (6 г, 0,03 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 25 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 18
Реакцию проводили в течение 72 часов таким же образом, что и в примере 16, за исключением того, что вместо ртутной лампы высокого давления использовали ртутную лампу низкого давления. Реакционную смесь перегоняли, получая фракцию HCF2CF2CH2ОH (0,23 литра). Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял 400 м.д., а поглощение (205 нм) составляло 1,5 абс.
К полученной таким образом фракции HCF2CF2CH2OH (0,2 литра) добавляли 28%-ный NaOCH3 (6 г, около 0,03 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 19
В автоклав добавляли метанол (2 литра) и карбонат кальция (30 г). После замещения азотом загружали ТФЭ с начальной скоростью 600 г/ч. Реакцию проводили в течение 48 часов при температуре 280oС и давлении 1,0 МПа. Реакционную смесь охлаждали и перегоняли для удаления метанола, а затем дополнительно перегоняли для отделения фракции HCF2CF2CH2OH (0,35 литра) от H(CF2CF2)nCH20H (n представляет целое число, равное 2 или более). Полученная при этом фракция HCF2CF2CH2OH имела остаток от выпаривания около 500 м.д. и поглощение (205 нм) 2,0 абс. Анализ данной фракции методом капиллярной масс-спектрометрии-ГХ показал, что фракция содержит большое количество альдегидов. Повторная перегонка привела к небольшому изменению остатка от выпаривания и поглощения (205 нм).
К полученной таким образом фракции HCF2CF2CH2OH (0,2 литра) добавляли 28%-ный NаОСН3 (6 г, около 0,03 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 20
Реакцию и перегонку проводили таким же образом, что и в примере 19, за исключением того, что вместо тетрафторэтилена использовали гексафторпропилен, получая 0,3 литра фракции HCF2CF2CH2OH. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял 550 м.д., а поглощение (205 нм) составляло 2,1 абс.
К полученной таким образом фракции HCF2CF2CH2OH (0,2 литра) добавляли 28%-ный NаОСН3 (6 г, около 0,03 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2ОH составлял не более 25 м.д., а поглощение (205 нм) составляло не более -0,1 абс.
ПРИМЕР 21
В автоклав добавляли метанол (2 литра), пербензойную кислоту (75 г) и карбонат кальция (30 г). После замещения азотом загружали ТФЭ с начальной скоростью 600 г/ч. Реакцию проводили в течение 6 часов при температуре 75oС и давлении 0,8 МПа. Реакционную смесь охлаждали и перегоняли для удаления метанола, а затем перегоняли далее для отделения фракции HCF2CF2CH2OH (0,6 литра) от H(CF2CF2)nСН2ОН (n представляет целое число, равное 2 или более). Полученная при этом фракция HCF2CF2CH2OH имела остаток от выпаривания около 600 м.д. и поглощение (205 нм) 2,0 абс. Анализ данной фракции методом капиллярной масс-спектрометрии-ГХ показал, что фракция содержит большое количество альдегидов. Повторная перегонка привела к небольшому изменению остатка от выпаривания и поглощения (205 нм).
К полученной таким образом фракции HCF2CF2CH2ОH (0,5 литра) добавляли 28%-ный NаОСН3 (15 г, около 0,08 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH20H составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 22
Реакцию и перегонку проводили таким же образом, что и в примере 21, за исключением того, что вместо пербензойной кислоты использовали АИБН (50 г), получая 0,6 литра фракции HCF2CF2CH2ОH. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2ОH составлял 600 м.д., а поглощение (205 нм) составляло 1,8 абс.
К полученной таким образом фракции HCF2CF2CH2ОH (0,5 литра) добавляли 28%-ный NаОСН3 (15 г, около 0,08 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 23
Реакцию проводили в течение 72 часов таким же образом, что и в примере 16, за исключением того, что вместо ртутной лампы высокого давления и тетрафторэтилена использовали лампу инфракрасного излучения и смесь тетрафторэтилен/озон (200/1 в молярном соотношении) соответственно. Реакционную смесь перегоняли, получая фракцию HCF2CF2CH2ОH (0,21 литра). Остаток от выпаривания полученной при этом фракции HCF2CF2CH2ОH составлял 400 м.д., а поглощение (205 нм) составляло 1,5 абс.
К полученной таким образом фракции HCF2CF2CH2OH (0,2 литра) добавляли 28%-ный NаОСН3 (6 г, около 0,03 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2ОH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 24
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2ОH (1 литр) постепенно добавляли NaH (3,6 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 25
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2OH (1 литр) постепенно добавляли NaH 15%-ный раствор бутиллития в гексане (64 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2ОH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 26
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2ОH (1 литр) постепенно добавляли NaH, 97%-ный диизопропиламид лития (16,5 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 27
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2OH (1 литр) постепенно добавляли амид натрия (5,9 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 28
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2OH (1 литр) постепенно добавляли (С6Н5)3СNа (40 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2ОH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 29
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2OH (1 литр) постепенно добавляли (С6Н5)3СОNа (14,4 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 30
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2OH (1 литр) постепенно добавляли Na (3,5 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 31
Фракцию HCF2CF2CH2OH перед перегонкой в присутствии основания получали таким же образом, что и в примере 1. К фракции HCF2CF2CH2OH (1 литр) постепенно добавляли Мg (3,6 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 32
Реакцию и перегонку проводили таким же образом, что и в примере 1, за исключением того, что вместо СаСО3 в качестве акцептора кислоты использовали МgСО3 (30 г), получая 1,2 литра фракции HCF2CF2CH2OH. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял 600 м.д., а поглощение (205 нм) составляло 2,0 абс.
К полученной таким образом фракции HCF2CF2CH2OH (1 литр) добавляли 28%-ный NаОСН3 (30 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 33
Реакцию и перегонку проводили таким же образом, что и в примере 1, за исключением того, что вместо СаСО3 в качестве акцептора кислоты использовали Nа2СО3 (30 г), получая 1,2 литра фракции HCF2CF2CH2ОH. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял 600 м.д., а поглощение (205 нм) составляло 2,0 абс.
К полученной таким образом фракции НСF2СF2СН2ОН (1 литр) добавляли 28%-ный NaOCH3 (30 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 34
Реакцию и перегонку проводили таким же образом, что и в примере 1, за исключением того, что вместо СаСО3 в качестве акцептора кислоты использовали К2СО3 (30 г), получая 1,2 литра фракции HCF2CF2CH2OH. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял 600 м.д., а поглощение (205 нм) составляло 2,0 абс.
К полученной таким образом фракции HCF2CF2CH2OH (1 литр) добавляли 28%-ный NаОСН3 (30 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции НСF2СF2СН2ОН составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 35
Реакцию и перегонку проводили таким же образом, что и в примере 1, за исключением того, что вместо СаСО3 в качестве акцептора кислоты использовали NаНСО3 (30 г), получая 1,2 литра фракции HCF2CF2CH2OH. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2ОH составлял 600 м.д., а поглощение (205 нм) составляло 2,0 абс.
К полученной таким образом фракции HCF2CF2CH2OH (1 литр) добавляли 28%-ный NаОСН3 (30 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 36
Реакцию и перегонку проводили таким же образом, что и в примере 1, за исключением того, что вместо СаСО3 в качестве акцептора кислоты использовали КНСО3 (30 г), получая 1,2 литра фракции HCF2CF2CH2OH. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял 600 м.д., а поглощение (205 нм) составляло 2,0 абс.
К полученной таким образом фракции HCF2CF2CH2OH (1 литр) добавляли 28%-ный NaOCH3 (30 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 37
Реакцию и перегонку проводили таким же образом, что и в примере 1, за исключением того, что вместо СаСО3 в качестве акцептора кислоты использовали СаО (30 г), получая 1,2 литра фракции HCF2CF2CH2OH. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял 600 м.д., а поглощение (205 нм) составляло 2,0 абс.
К полученной таким образом фракции HCF2CF2CH2OH (1 литр) добавляли 28%-ный NаОСН3 (30 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 38
Реакцию и перегонку проводили таким же образом, что и в примере 1, за исключением того, что вместо СаСО3 в качестве акцептора кислоты использовали ВаСО3 (30 г), получая 1,2 литра фракции HCF2CF2CH2OH. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял 600 м.д., а поглощение (205 нм) составляло 2,0 абс.
К полученной таким образом фракции HCF2CF2CH2OH (1 литр) добавляли 28%-ный NаОСН3 (30 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 39
Реакцию и перегонку проводили таким же образом, что и в примере 1, за исключением того, что вместо СаСО3 в качестве акцептора кислоты использовали KYOWORD 200В (А12O3• хН2О, 30 г), получая 1,2 литра фракции HCF2CF2CH2ОH. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2ОH составлял 600 м.д., а поглощение (205 нм) составляло 2,0 абс.
К полученной таким образом фракции HCF2CF2CH2OH (1 литр) добавляли 28%-ный NаОСН3 (30 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 40
Реакцию и перегонку проводили таким же образом, что и в примере 1, за исключением того, что вместо СаСО3 в качестве акцептора кислоты использовали KYOWORD 500 (Mg6Al2(ОН)16СО3• 4Н2О, 30 г), получая 1,2 литра фракции HCF2CF2CH2OH. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял 600 м.д., а поглощение (205 нм) составляло 2,0 абс.
К полученной таким образом фракции HCF2CF2CH2OH (1 литр) добавляли 28%-ный NаОСН3 (30 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
ПРИМЕР 41
Реакцию и перегонку проводили таким же образом, что и в примере 1, за исключением того, что вместо СаСО3 в качестве акцептора кислоты использовали SEGARD (Al2O3• mSiO2• nH2O+А1(ОН)3, 30 г), получая 1,2 литра фракции HCF2CF2CH2OH. Остаток от выпаривания полученной при этом фракции HCF2CF2CH2OH составлял 600 м.д., а поглощение (205 нм) составляло 2,0 абс.
К полученной таким образом фракции HCF2CF2CH2OH (1 литр) добавляли 28%-ный NаОСН3 (30 г, около 0,15 моль) и смесь перегоняли при нагревании. Остаток от выпаривания полученной при этом фракции HCF2 CF2CH2OH составлял не более 10 м.д., а поглощение (205 нм) составляло не более -0,2 абс.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ФТОРСПИРТА (ВАРИАНТЫ) | 1999 |
|
RU2166495C1 |
| СПОСОБ ВЫДЕЛЕНИЯ ПЕНТАФТОРЭТАНА ИЗ СМЕСИ С ХЛОРПЕНТАФТОРЭТАНОМ | 1995 |
|
RU2121993C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРАМИДА И ФТОРНИТРИЛА | 2008 |
|
RU2440975C2 |
| Способ получения линейных простых галогенсодержащих полиэфиров | 1984 |
|
SU1806149A3 |
| ГАЛОГЕНСОДЕРЖАЩИЙ ПРОСТОЙ ПОЛИЭФИР | 1992 |
|
RU2107074C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРСОДЕРЖАЩИХ ПРОСТЫХ ЭФИРОВ | 2009 |
|
RU2463286C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРСОДЕРЖАЩЕГО ПРОСТОГО ЭФИРА ВЫСОКОЙ ЧИСТОТЫ | 2010 |
|
RU2486170C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГАЛОГЕНСОДЕРЖАЩЕГО ПРОСТОГО ПОЛИЭФИРА | 1991 |
|
RU2073692C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1,1,1-ТРИФТОРХЛОРЭТАНА И 1,1,1,2-ТЕТРАФТОРЭТАНА | 1991 |
|
RU2072975C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1,1,1,2,2-ПЕНТАФТОР-3,3-ДИХЛОРПРОПАНА И 1,1,2,2,3-ПЕНТАФТОР-1,3-ДИХЛОРПРОПАНА | 1991 |
|
RU2041193C1 |






Изобретение относится к способу получения фторспирта формулы H(CFR1CF2)nCH2OH (I), где R1 представляет F или CF3, когда n=1, и R1 представляет F, когда n=2, включающий взаимодействие метанола с тетрафторэтиленом или гексафторпропиленом в присутствии источника свободных радикалов. Полученную реакционную смесь подвергают перегонке или в присутствии основания, или после контакта указанной смеси с основанием. Фторспирт формулы I, полученный с помощью перегонки, имеет остаток от выпаривания не более 50 м.д. В качестве источника свободных радикалов используют пероксидный инициатор реакции, УФ-излучение либо нагрев. Состав представляет собой фторспирт формулы I с остатком от выпаривания не более 50 м.д. Состав применяют в качестве растворителя красителя в производстве носителя записи информации для лазерной записи и/или считывания. С использованием фторспирта формулы I создают оптический диск для лазерной записи и/или считывания. Технический результат - получение фторспирта, пригодного для изготовления носителя лазерной записи информации и/или считывания. 4 с. и 16 з.п. ф-лы, 13 табл., 2 ил.
H(CFR1CF2)nCH2OH (I),
где R1 представляет F или CF3, когда n=1, и R1 представляет F, когда n= 2,
включающий взаимодействие метанола с тетрафторэтиленом или гексафторпропиленом в присутствии источника свободных радикалов, по которому реакционную смесь подвергают перегонке или в присутствии основания, или после контакта указанной реакционной смеси с основанием.
H(CFR1CF2)nCH2OH (I),
где R1 и n имеют значения, определенные выше,
полученный с помощью перегонки, имеет остаток от выпаривания не более 50 м.д.
H(CFR1CF2)nCH2OH (I),
где R1 и n имеют значения, определенные выше,
полученный с помощью перегонки, имеет остаток от выпаривания не более 25 м.д.
H(CFR1CF2)nCH2OH (I),
где R1 и n имеют значения, определенные выше,
полученный с помощью перегонки, имеет остаток от выпаривания не более 10 м.д.
H(CFR1CF2)nCH2OH (I),
в которой R1 представляет F или CF3 когда n=1, R1 представляет F, когда n=2,
в котором остаток от выпаривания не более 50 м.д.
H(CFR1CF2)nCH2OH (I),
где R1 представляет F или CF3, когда n=1, R1 представляет собой F, когда n=2,
полученного по способу по п.1, или состав по п.13.
Приоритеты по пунктам:
28.12.1998 по пп.1-12;
25.02.1999 по пп.13-20.
| US 4346250 A, 24.08.1982 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИФТОРИРОВАННЫХ СПИРТОВ | 1999 |
|
RU2150459C1 |
| RU 95106316 A1, 27.01.1997 | |||
| ТОНЕР, ПРОЯВИТЕЛЬ, ИСПОЛЬЗУЮЩИЙ ДАННЫЙ ТОНЕР, УСТРОЙСТВО ФОРМИРОВАНИЯ ИЗОБРАЖЕНИЯ | 2012 |
|
RU2559628C1 |
