Изобретения относятся к биотехнологии, генной инженерии, микробиологии и медицине и могут быть использованы для производства рекомбинантной плазмиды и получения на ее основе вакцины против гепатита В.
В настоящее время известно достаточно много вакцин против гепатита В на основе рекомбинантных поверхностных антигенных детерминант этого вируса (патент США 5098704, МКИ А 61 К 39/29, НКИ 424-89, патенты Франции 2606281, МКИ А 61 К 39/29, и 2635532, МКИ С 12 N 15/51). Как правило, при создании этих вакцин используют плазмиды с природными участками генома вируса гепатита В (серотипы adr и adw), кодирующими его поверхностную антигенную детерминанту, экспрессируемыми под контролем сильных конструктивных промоторов дрожжевых генов (патент ЕПВ 0533492, МКИ А 61 К 39/29, патент США 5133961, МКИ А 61 К 39/29, НКИ 424-89). Эти конструкции обеспечивают получение высокого уровня экспрессии целевого продукта, при одновременном высоком уровне экспрессии чужеродного белка, что приводит к нестабильности штаммов, особенно при наращивании больших количеств биомассы. К тому же они требуют порой использования для роста клеток реципиентов различных, сложных и дорогостоящих питательных сред.
Наиболее близкой к заявленной рекомбинантной плазмиде ДНК, является рекомбинантная плазмида pDES20, кодирующая поверхностный антиген вируса гепатита В (HBsAg/ayw), мол. м. 4,53 MДа и размером 7067 н.п. и содержащая следующие структурные элементы: бактериальный ориджин репликации CoLEI размером 0,62 т.н.п.; бактериальный оперон β-лактомазы размером 1,05 т.н.п., фрагмент природной дрожжевой 2μ-плазмиды, обеспечивающий автономную репликацию pDES20 в Saccharomyces cerevisiae, размером 1,89 т.н.п.; дрожжевой оперон Leu2 размером 2,21 т.н.п.; дрожжевой оперон HBsAg/ayw с промотором размером 0,35 т. н. п. , терминатором размером 0,18 т.н.п. дрожжевого гена РНО5 и синтетическим геном полипептида HBsAg/ayw, первичная структура которого составлена с учетом частоты использования кодонов в Saccharomyces cerevisiae, а области стыковки РНО5-промотора и гена полипептида HBsAg/ayw, а также последнего и РНО5-терминатора имеют определенные первичные структуры (патент 2088664, Россия, МКИ А 61 К 39/29, С 12 N 15/51, 26.01.96 г., прототип).
Известную плазмиду получают путем последовательного осуществления следующих действий: химического синтеза однотяжевых фрагментов ДНК (олигодезоксирибонуклеотидов), сборки синтетического гена полипептида HBsAg/ayw с лидерной нетранслируемой областью мРНК секретируемой кислой фосфатазы, кассетой кодонов-терминаторов и 3'-концевым сайтом рестрикции BamI, получения минимального фрагмента 5'-нетранслируемой области дрожжевого оперона pho5, обеспечивающего контролируемую инициацию транскрипции, присоединения промотора к синтетическому гену мРНК полипептида HBsAg/ayw с кассетой кодонов-терминаторов и 3'-концевым сайтом рестрикции BamHI, получения минимального фрагмента 3'-нетранслируемой области дрожжевого оперона pho5, обеспечивающего эффективную терминацию транскрипции и процессинг мРНК, получения фрагмента дрожжевой ДНК с опероном leu2, получения фрагмента дрожжевой 2m-плазмиды, получения бактериального оперона β-лактамазы, получения бактериального ориджина репликации ColEl и сборкой челночной рекомбинантной плазмиды pDES20 с искусственным опероном для экспрессии в дрожжах Saccharomyces cerevisiae синтетического гена полипептида HBsAg/ayw.
Трансформируемые известной плазмидой клетки дрожжевого штамма Saccharomyces cerevisiae ВКПМ Y-2203 являются продуцентом поверхностного антигена вируса гепатита В (HBsAg/ayw).
Данная рекомбинантная плазмида, полученная способом, описанным в патенте, позволяет обеспечить получение поверхностного антигена вируса гепатита В, однако характеризуется достаточно низкой его экспрессией (около 2-5%), нерентабельной для его промышленного производства, а трансформированный ею штамм недостаточно эффективен, вследствие нестабильности известной плазмиды. Кроме того, использование различных питательных сред для синтеза HBsAg клетками штамма, сложных по своему составу (содержат большое количество разнообразных микроэлементов), приводит к повышению стоимости получения конечного продукта.
В основу заявленной группы изобретений положена задача создания стабильного, более эффективного микробиологического дрожжевого продуцента, обеспечивающего синтез рекомбинантного поверхностного антигена вируса гепатита В серотипа (HBsAg/ayw), с последующим созданием на его основе высоко иммуногенной вакцины, лишенной побочных эффектов. Упомянутая задача решена за счет достижения эффекта повышенного синтеза рекомбинантного поверхностного антигена вируса гепатита В серотипа (HBsAg/ayw), в частности за счет полимеризации участка, кодирующего дрожжевой оперон с геном HBsAg.
Технический результат, достигаемый при осуществлении заявленных изобретений, заключается в создании высоко эффективного и стабильного микробиологического дрожжевого продуцента и снижении стоимости получения конечного продукта.
Поставленная в заявленной группе изобретений задача, с достижением упомянутого выше технического результата, решена следующим образом.
В части плазмиды поставленная задача решается тем, что заявляемая плазмида содержит следующие существенные для ее функционирования структурные элементы:
- искусственный дрожжевой оперон HBsAg, включающий:
промоторную область АОХ1 оперона (7-933 н.п.), обеспечивающую эффективную транскрипцию контролируемой мРНК, а также интеграцию по гомологичному участку в области промотора АОХ1 гена; ген HBsAg вируса гепатита В, обеспечивающий экспрессию целевого белка (HBsAg/ayw) (972-1652 н.п.); нетранслируемую область терминации транскрипции дрожжевого оперона АОХ1, обеспечивающую эффективное окончание транскрипции и процессинг мРНК (1655-2013 н. п. ); нуклеотидная последовательность дрожжевого оперона HBsAg представляет собой отдельный структурный модуль, который может быть повторен в плазмиде контролируемое число раз при помощи разработанной генно-инженерной процедуры;
- дрожжевой оперон оротидин-5'-фосфатдекарбоксилазы - Ura3-селективный маркер, придающий штамму Hansenula polymorpha способность расти на селективной среде без урацила (селективный маркер для трансформации штамма Hansenula polymorpha) 2019-3188 н.п., а сама кодирующая область гена Ura3 со стоп-кодоном - 2290-3093 н.п.);
- 3'-концевую нетранслируемую область АОХ1 оперона, обеспечивающую эффективную интеграцию по гомологичному участку в дистальную область АОХ1 гена (3193-3640 н.п.);
- бактериальный оперон bla, кодирующий белок бета-лактамазу, являющийся селективным маркером для трансформации штаммов Е. coli (кодирующая область 4880-5740 н.п. в комплементарной цепи) и бактериальный участок инициации репликации типа ColEl, обеспечивающий репликацию плазмиды в штаммах Е. coli.
Таким образом создана рекомбинантная плазмида pHPS1, кодирующая поверхностный антиген (HBsAg) вируса гепатита В, размером 5945 н.п., содержащая следующие структурные элементы: искусственный дрожжевой оперон HBsAg, включающий промоторную область АОХ1 оперона (927 н.п.), ген HBsAg вируса гепатита В (HBsAg/ayw) (680 н.п.) и терминатор транскрипции (358 н.п.); дрожжевой оперон оротидин-5'-фосфатдекарбоксилазы - Ura3; 3'-область АОХ1 оперона, (447 н.п.); бактериальный оперон бета-лактамазы, (860 н.п.) и бактериальный участок инициации репликации типа Col El (90 н.п.). Кроме того, создана рекомбинантная плазмида дополнительно содержащая одну или несколько копий искусственного дрожжевого оперона HBsAg.
В части способа получения плазмиды поставленная задача, в соответствии с настоящим изобретением, решается тем, что осуществляют химический синтез олигодезоксирибонуклеотидов, после чего клонируют 3'-концевую нетранслируемую область ДНК гена АОХ, клонируют ген Ura3 с промоторной областью, получают плазмиду, содержащую терминатор АОХ гена, ген Ura3 с промоторной областью, получают плазмиду, содержащую терминатор АОХ гена, ген Ura3 с промоторной областью и 3'-концевую нетранслируемую область ДНК гена АОХ, клонируют 5'-концевую нетранслируемую область ДНК гена АОХ, содержащую промотор этого гена, и собирают вектор для экспрессии в дрожжах Hansenula polymorpha, в который клонируют ген HBsAg. Таким образом создают дрожжевой оперон гена HBsAg, включающий все необходимые элементы для экспрессии его в дрожжах Hansenula polymorpha.
Поставленная задача решается также тем, что осуществляют полимеризацию участка, кодирующего искусственный дрожжевой оперон HBsAg, состоящего из промоторной области АОХ1 оперона, гена HBsAg вируса гепатита В и участка терминации транскрипции дрожжевого оперона АОХ1 в плазмиде pHPS2. Плазмиды pHPS1 и pHPS2, в соответствии с настоящим изобретением, включают искусственный дрожжевой оперон HBsAg/ayw под контролем промотора алкогольоксидазы (ген АОХ1) в одном, двух или более повторах. Для полимеризации искусственного дрожжевого оперона HBsAg используют генно-инженерный подход, позволяющий проводить контролируемое увеличение количества копий участка ДНК в плазмиде in vitro.
Таким образом создан способ получения рекомбинантных плазмид, обеспечивающих эффективную экспрессию белков в штаммах дрожжей Hansenula polymorpha, включающий химический синтез фрагментов ДНК, клонирование и сборку вектора для экспрессии HBsAg гена, с последующим созданием дрожжевого оперона с HBsAg геном, после чего проводят операцию полимеризации оперона с HBsAg геном.
Заявляемый штамм - продуцент поверхностного антигена вируса гепатита В получают трансформацией клеток дрожжевого штамма A Hansenula polymorpha плазмидой pHPS2. Полимеризованный оперон HBsAg обеспечивает повышенный выход рекомбинантного поверхностного антигена вируса гепатита В в штамме, содержащем плазмиду pHPS2, по сравнению с опероном в одной копии (штамм с плазмидой pHPS1) и ранее описанными штаммами.
В качестве штамма-продуцента HBsAg гепатита В был использован штамм A Hansenula polymorpha. Штамм A Hansenula polymorpha несет мутацию в URA3 гене, что позволяет селективно отбирать трансформанты, несущие плазмиды pHPS1 и pHPS2.
Морфологические признаки. Клетки округлой, слегка овальной формы, размером 5-10 мкм, часть клеток имеет на поверхности почки или соединена с дочерними клетками.
Культуральные признаки. Клетки штаммов дрожжей Hansenula polymorpha A (pHPSl) и A (pHPS2) хорошо растут на полной органической среде YEPD: 2% пептона, 1% дрожжевого экстракта, 2% глюкозы или 1% глицерина; минеральной среде SC: 1,34% Yeast Nitrogen Base ("Difco", США), 2% глюкозы; 1% глицерина, 0,5% метанола, а также на других синтетических средах для дрожжей. При росте на твердых средах клетки образуют гладкие круглые колонии с матовой поверхностью светло-кремового цвета, край неровный. При росте в жидких средах образуют интенсивную ровную суспензию. Культура имеет характерный запах метилотрофных дрожжей.
Физиолого-биохимические признаки. Клетки растут в пределах широкого диапазона температур от 4 до 37oС и рН 4-8. Оптимальной температурой выращивания является 35oС, при росте в анаэробных условиях клетки незначительно защелачивают среду. Оптимум рН для роста составляет 5,0-6,5. В качестве источника углерода могут быть использованы различные простые органические соединения, такие, как глюкоза, глицерин, метанол. В качестве источника азота клетки могут использовать минеральные соли в аммонийной форме, мочевину, аминокислоты. Клетки способны как к аэробному, так и к анаэробному росту.
Существенным при использовании данного штамма является отсутствие потребности в урациле.
Изобретение проиллюстрировано графическими материалами, на которых изображены: на фиг.1 - схема плазмиды pHPS1, на фиг.2 - схема плазмиды pHPS2, на фиг. 3 - нуклеотидная последовательность плазмиды pHPS2, и следующими примерами, приведенными ниже. Все стандартные генно-инженерные и микробиологические манипуляции, а также амплификацию и секвенирование ДНК проводили по известным методикам (Маниатис Т., Фрич Э., Сэмбрук Дж., Молекулярное клонирование. М. : Мир, 1984; Клонирование ДНК. Методы. Под ред. Д. Гловера. Пер. с англ., М., Мир, 1988; Saiki R.K., Gelfand D.H., Stoffel S., Sharf SJ. , Higuchi R. , Horn G.T., Mullis K.B., Erlich H.A. Science, 1988, v.239, 4839, p. 487-491; Sanger F., Nicklen S., Coulson A.R. Proc. Nat. Acad. Sci. USA, 1977, v.74, p. 5463-5467).
Пример 1. Получения плазмиды pHPS1.
а) Химический синтез олигодезоксирибонуклеотидов.
Олигодезоксирибонуклеотиды были синтезированы твердофазным аминофосфитным методом с помощью синтезатора АСМ 100-2 (Новосибирск) и очищены электрофорезом в 12%-ном ПААГ геле.
б) Получение участка 3'-концевой нетранслируемой области ДНК гена АОХ с последующим ее клонированием.
Копию 3'-концевой нетранслируемой области гена АОХ получали методом ПЦР, для чего были выбраны праймеры, соответствующие началу и концу данной области ДНК. Используя в качестве ДНК-матрицы хромосомную ДНК дрожжей Pichina pastoris, методом полимеразной цепной реакции с использованием олигонуклеотидных праймеров состава (подчеркнуты сайты рестриктаз ВаmНI и Xbal и PstI)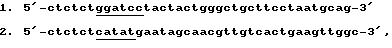
соответствующих начальной и концевой части ДНК, амплифицировали последовательность нуклеотидов. Реакцию проводили в 25 мкл реакционной смеси, содержащей 2 мкг ДНК, 10 пМ каждого праймера, 67 мМ трис-НСl (рН 8,8 при 25oС), 15 мМ сульфата аммония, 2,5 мМ хлористого магния, 0,01% Твин-20, смесь дезоксинуклеотидтрифосфатов (дАТФ, дЦТФ, дТТФ, дГТФ по 2,5 мМ) и 1 ед. Тaq-ДНК-полимеразы. Реакцию амплификации проводили под вазелиновым маслом: 5 циклов: 93oС-2 мин, 57oС-5 мин и 72oС-30 сек; 30 циклов: 93oС-1 мин, 63oС-1 мин и 72oС-30 сек; затем 63oС-5 мин и 72oС-10 мин. Продукт амплификации размером 478 н. п. обрабатывали хлороформом, переосаждали этиловым спиртом и растворяли в воде. Для клонирования данного продукта полученную ДНК рСRАОХ3', а также ДНК плазмиды pQE16 гидролизовали рестрикционными эндонуклеазами BamHI и XbaI при 37oС в буфере, содержащем 66 мМ Трис-ацетат (рН 7,9 при 37oС), 20 мМ ацетата магния, 132 мМ ацетата калия и 0,2 мг/мл BSA в течение 1,5 часов. Выделенные из геля рестрикционные фрагменты: фрагмент ДНК, включающий 3'-концевую область гена АОХ размером 460 н.п. и фрагмент плазмиды pQE16 размером 2419 н.п. лигировали с помощью ДНК-лигазы фага Т4. Полученной лигированной смесью трансформировали клетки Е. coli M15[REP4] методом электропорации. Трансформированные клетки отбирали на агаризованной среде LB с антибиотиками Ар и Km. Плазмидную ДНК выделяли методом щелочного лизиса, анализировали с помощью рестриктаз ВаmHI и ХbаI, а также рестриктаз, находящихся внутри клонируемых фрагментов, и отбирали клоны pUY1 (размером 2879 н. п. ), плазмидная ДНК которых содержит 3'-концевую нетранслируемую область гена АОХ. Первичную структуру полученной плазмиды подтверждали секвенированием дидезоксиметодом Сэнгера.
в) Получение гена Ura3 с промотором с последующим его клонированием.
Копию гена Ura3 с собственным промотором получали методом ПЦР, для чего были выбраны праймеры, соответствующие участку ДНК перед промоторной областью гена и концу кодирующей части гена со стоп-кодоном. Используя в качестве ДНК-матрицы хромосомную ДНК дрожжей Saccharomyces сerevisiae, методом полимеразной цепной реакции с олигонуклеотидными праймерами состава (подчеркнуты сайты рестриктаз ВаmHI, XbaI и BglII):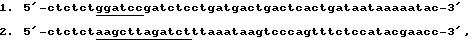
соответствующих начальной и концевой части ДНК, амплифицировали последовательность нуклеотидов. Реакцию проводили в 25 мкл реакционной смеси, содержащей 2 мкг ДНК, 10 пМ каждого праймера, 67 мМ трис-НСl (рН 8,8 при 25oС), 15 мМ сульфата аммония, 2,5 мМ хлористого магния, 0,01% Твин-20, смесь дезоксинуклеотидтрифосфатов (дАТФ, дЦТФ, дТТФ, дГТФ по 2,5 мМ) и 1 ед. Тaq-ДНК-полимеразы. Реакцию амплификации проводили под вазелиновым маслом: 5 циклов: 93oС-2 мин, 58oС-5 мин и 72oС-2 мин; 30 циклов: 93oС-1 мин, 63oС-1 мин и 72oС-2 мин, а затем 63oС-5 мин и 72oС-10 мин. Продукт амплификации размером 2647 н.п. обрабатывали хлороформом, переосаждали этииловым спиртом и растворяли в воде. Для клонирования данного продукта полученную ДНК pCRUra3, а также ДНК плазмиды pQE16 гидролизовали рестрикционными эндонуклеазами BamHI и XbaI при 37oС в буфере, содержащем 66 мМ Трис-ацетат (рН 7,9 при 37oС), 20 мМ ацетата магния, 132 мМ ацетата калия и 0,2 мг/мл BSA в течение 1,5 часов. Выделенные из геля рестрикционные фрагменты: фрагмент ДНК, соответствующий гену Ura3 с промоторной областью размером 1177 н.п и фрагмент плазмиды pQE16 размером 2419 н.п. лигировали с помощью ДНК-лигазы фага Т4. Полученной лигированной смесью трансформировали клетки Е. coli M15[REP4] методом электропорации. Трансформированные клетки отбирали на агаризованной среде LB с антибиотиками Ар и Km. Плазмидную ДНК выделяли методом щелочного лизиса, анализировали с помощью рестриктаз ВаmНI и XbaI, а также рестриктаз, находящихся внутри клонируемых фрагментов, и отбирали клоны pUY2 (размером 3596 н.п.), плазмидная ДНК которых содержит ген Ura3 с промоторной областью. Первичную структуру полученной плазмиды подтверждали секвенированием дидезоксиметодом Сэнгера.
г) Получение терминатора АОХ гена с последующим его клонированием.
Копию терминатора АОХ гена получали методом ПЦР, для чего были спланированы праймеры, соответствующие началу и концу выбранной области ДНК. Используя в качестве ДНК-матрицы хромосомную ДНК дрожжей Pichia pastoris, методом полимеразной цепной реакции с олигонуклеотидными праймерами состава (подчеркнуты сайты рестриктаз EcoRI, BglII и ВаmНI)
соответствующих начальной и концевой части ДНК, амплифицировали последовательность нуклеотидов. Реакцию проводили в 25 мкл реакционной смеси, содержащей 2 мкг ДНК, 10 пМ каждого праймера, 67 мМ трис-НСl (рН 8,8 при 25oС), 15 мМ сульфата аммония, 2,5 мМ хлористого магния, 0,01% Твин-20, смесь дезоксинуклеотидтрифосфатов (дАТФ, дЦТФ, дТТФ, дГТФ по 2,5 мМ) и 1 ед. Тaq-ДНК-полимеразы. Реакцию амплификации проводили под вазелиновым маслом: 5 циклов: 93oС-2 мин, 60oС-5 мин и 72oС-30 сек; 30 циклов: 93oС-1 мин, 65oС-1 мин и 72oС-30 сек, а затем 65oС-5 мин и 72oС-10 мин. Продукт амплификации pCRAOXTer размером 384 н.п. обрабатывали хлороформом, переосаждали этиловым спиртом и растворяли в воде. Для клонирования данного продукта полученную ДНК pCRAOXTer, а также ДНК плазмиды pQE16 гидролизовали рестрикционными эндонуклеазами ЕсоRI и BglII при 37oС в буфере, содержащем 66 мМ Трис-ацетат (рН 7,9 при 37oС), 20 мМ ацетата магния, 132 мМ ацетата калия и 0,2 мг/мл BSA в течение 1,5 часов. Выделенные из геля рестрикционные фрагменты: фрагмент ДНК, включающий терминатор АОХ гена размером 366 н.п. и фрагмент плазмиды pQE16 размером 3388 н.п. лигировали с помощью ДНК-лигазы фага Т4. Полученной лигированной смесью трансформировали клетки Е. Coli М15[REP4] методом электропорации. Трансформированные клетки отбирали на агаризованной среде LB с антибиотиками Ар и Km. Плазмидную ДНК выделяли методом щелочного лизиса, анализировали с помощью рестриктаз EcoRI и BglII, а также рестриктаз, находящихся внутри клонируемых фрагментов, и отбирали клоны pY3 (размером 3745 н.п.), плазмидная ДНК которых держит терминатор АОХ гена. Первичную структуру полученной плазмиды подтверждали секвенированием дидезоксиметодом Сэнгера.
д) Получение плазмиды, содержащей терминатор АОХ гена и ген Ura3 с промоторной областью.
Для получения плазмиды, содержащей терминатор АОХ гена и ген Ura3 с промоторной областью, ДНК плазмиды pY2 гидролизуют рестрикционными эндонуклеазами BamHI и Pνul при 37oС в буфере, содержащем 66 мМ Трис-ацетат (рН 7,9 при 37oС), 20 мМ ацетата магния, 132 мМ ацетата калия в течение 1 часа, а плазмиду pY3 гидролизовали рестрикционными эндонуклеазами BglII и Pνul при 17oС в буфере, содержащем 50 мМ Трис-НСI (рН 7,5 при 37oС), 10 мМ хлорида магния, 100 мМ хлорида натрия и 0,1 мг/мл BSA в течение 1 часа.
Выделенные из геля рестрикционные фрагменты: фрагмент ДНК плазмиды pUY2, включающий ген Ura3 с промоторной областью размером 2853 н.п., а также фрагмент ДНК плазмиды pY3, включающий терминатор АОХ гена размером 1076 н.п. лигировали с помощью ДНК-лигазы фага Т4. Полученной лигированной смесью трансформировали клетки Е. coli M15[REP4] методом электропорации. Трансформированные клетки отбирали на агаризованной среде LB с антибиотиками Ар и Km. Плазмидную ДНК выделяли методом щелочного лизиса, анализировали с помощью рестриктаз PνuI, BamHI и BglII, а также рестриктаз, находящихся внутри клонируемых фрагментов, и отбирали клоны pUY4 (м.в. 3929 н.п.), плазмидная ДНК которых содержит терминатор АОХ гена и ген Ura3 с промоторной областью. Первичную структуру полученной плазмиды подтверждали секвенированием дидезоксиметодом Сэнгера.
е) Получение плазмиды, содержащей терминатор АОХ гена, ген His4 с промоторной областью и 3'-концевую нетранслируемую область ДНК после гена АОХ.
Для получения плазмиды, содержащей терминатор АОХ гена, ген Ura3 с промоторной областью и 3'-концевую нетранслируемую область ДНК после гена АОХ, ДНК плазмиды pY1 гидролизовали рестрикционными эндонуклеазами ВаmHI и PνuI при 37oС в буфере 66 мМ Трис-ацетат (рН 7,9 при 37oС), 20 мМ ацетат магния, 132 мМ ацетат калия и 0,2 мг/мл BSA в течение 1 часа, а плазмиду pUY4 гидролизовали рестрикционными эндонуклеазами BglII и PνuI при 37oС в буфере, содержащем 50 мМ Трис-НСI (рН 7,5 при 37oС), 10 мМ хлорида магния, 100 мМ хлорида натрия и 0,1 мг/мл BSA, в течение 1 часа. Выделенные из геля рестрикционные фрагменты: фрагмент ДНК плазмиды pUYl, включающий 3'-концевую нетранслируемую область ДНК после гена АОХ размером 2136 н.п., а также фрагмент ДНК плазмиды pUY4, включающий терминатор АОХ гена и ген Ura3 с промоторной областью размером 2250 н.п. лигировали с помощью ДНК-лигазы фага Т4. Полученной лигированной смесью трансформировали клетки Е. coli M15[REP4] методом электропорации. Трансформированные клетки отбирали на агаризованной среде LB с антибиотиками Ар и Km. Плазмидную ДНК выделяли методом щелочного лизиса [2] , анализировали с помощью рестриктаз PνuI, EcoRI и XbaI, а также рестриктаз, находящихся внутри клонируемых фрагментов, и отбирают клоны pUY5 (размером 4386 н.п.), плазмидная ДНК которых содержит терминатор АОХ гена, ген Ura3 с промоторной областью и 3'-концевую нетранслируемую область гена АОХ. Первичную структуру полученной плазмиды подтверждали секвенированием дидезоксиметодом Сэнгера.
ж) Получение 5'-концевой нетранслируемой области ДНК гена АОХ, содержащей промотор этого гена, с последующим ее клонированием.
Копию 5'-концевой нетранслируемой области ДНК перед геном АОХ, содержащей промотор этого гена, получали методом ПЦР, для чего были выбраны праймеры, соответствующие началу участка 5'-концевой нетранслируемой области ДНК и концу этого участка после промотора гена АОХ. Используя в качестве ДНК-матрицы хромосомную ДНК дрожжей Pichia pastoris, методом полимеразной цепной реакции с использованием олигонуклеотидных праймеров состава (подчеркнуты сайты рестриктаз XhoI, Kpn2I и EcoRI)
соответствующих начальной и концевой части ДНК, амплифицировали последовательность нуклеотидов. Реакцию проводили в 25 мкл реакционной смеси, содержащей 2 мкг ДНК, 10 пМ каждого праймера, 67 мМ трис-НСI (рН 8,8 при 25oС), 15 мМ сульфата аммония, 2,5 мМ хлористого магния, 0,01% Твин-20, смесь дезоксинуклеотидтрифосфатов (дАТФ, дЦТФ, дТТФ, дГТФ по 2,5 мМ) и 1 ед. Тaq-ДНК-полимеразы. Реакцию амплификации проводили под вазелиновым маслом: 5 циклов: 93oС-2 мин, 55oС-5 мин и 72oС-1 мин; 30 циклов: 93oС-1 мин, 60oС-1 мин и 72oС- 1 мин; затем 60oС-5 мин и 72oС-10 мин. Продукт амплификации размером 987 н. п. обрабатывали хлороформом, переосаждали этиловым спиртом и растворяли в воде. Для клонирования данного продукта полученную ДНК pCRAOXPr, а также ДНК плазмиды pQE16 гидролизовали рестрикционными эндонуклеазами XhoI и Крn2I при 37oС в буфере, содержащем 66 мМ Трис-ацетат (рН 7,9 при 37oС), 20 мМ ацетата магния, 132 мМ ацетата калия и 0,2 мг/мл BSA в течение 1,5 часов. Выделенные из геля рестрикционные фрагменты: фрагмент ДНК, включающий 5'-концевую область ДНК перед геном АОХ, и промотор этого гена размером 969 н.п., и фрагмент плазмиды pQE16 размером 2908 н.п. лигировали с омощью ДНК-лигазы фага Т4. Полученной лигированной смесью трансформировали клетки Е. coli M15[REP4] методом электропорации. Трансформированные клетки отбирали на агаризованной среде LB с антибиотиками Ар и Km. Плазмидную ДНК выделяли методом щелочного лизиса, анализировали с помощью рестриктаз XhoI и Крn2I, а также рестриктаз, находящихся внутри клонируемых фрагментов, и отбирали клоны pY6 (размером 3877 н.п.), плазмидная ДНК которых содержит 5'-концевую область перед геном АОХ, содержащую промотор этого гена. Первичную структуру полученной плазмиды подтверждали секвенированием дидезоксиметодом Сэнгера.
з) Сборка вектора для экспрессии в дрожжах Hansenula polymorpha.
Для получения плазмиды, содержащей все элементы, необходимые для экспрессии в дрожжах: терминатор АОХ гена, ген Urа3 с промоторной областью, 3'-концевую нетранслируемую область ДНК гена АОХ, а также 5'-концевую нетранслируемую область ДНК перед геном АОХ, содержащую промотор этого гена, ДНК плазмиды pUY5 и pY6 гидролизовали рестрикционными эндонуклеазами EcoRI и Pνul при 37oС в буфере, 66 мМ Трис-ацетат (pН 7,9 при 37oС), 20 мМ ацетата магния, 132 мМ ацетата калия и 0,2 мг/мл BSA в течение 1 часа. Выделенные из геля рестрикционные фрагменты: фрагмент ДНК плазмиды pUY5, включающий 3'-концевую нетранслируемую область гена АОХ, терминатор АОХ гена и ген Ura3 с промоторной областью размером 3673 н.п., а также фрагмент ДНК плазмиды pY6, включающий 5'-концевую нетранслируемую область гена АОХ, содержащую промотор этого гена, размером 1591 н.п., лигировали с помощью ДНК-лигазы фага Т4. Полученной лигированной смесью трансформировали клетки Е. coli M15[REP4] методом электропорации. Трансформированные клетки отбирали на агаризованной среде LB с антибиотиками Ар и Кm. Плазмидную ДНК выделяли методом щелочного лизиса, анализировали с помощью рестриктаз, ЕсоRI и Pνul, а также рестриктаз, находящихся внутри клонируемых фрагментов, и отбирали клоны pUY7 (размером 5264 н. п.), плазмидная ДНК которых содержит в своем составе все элементы, необходимые для экспрессии в дрожжах: 5'-концевую нетранслируемую область перед геном АОХ, включающую промотор этого гена, терминатор АОХ гена, ген Ura3 с промоторной областью, а также 3'-концевую нетранслируемую область гена АОХ. Первичную структуру полученной плазмиды подтверждали секвенированием по методу Сэнгера.
и) Получение HBsAg в дрожжевом векторе с последующим ее клонированием.
Копию гена HBsAg получали методом ПЦР, для чего были выбраны праймеры, соответствующие началу гена, включающему ATG-кодон и концу кодирующей области гена после стоп-кодона. Используя в качестве ДНК-матрицы ДНК вируса HBV субтипа ayw, методом полимеразной цепной реакции с олигонуклеотидными праймерами состава (подчеркнуты сайты рестриктаз EcoRI и NotI)
соответствующих начальной и концевой части гена, амплифицировали последовательность нуклеотидов. Реакцию проводили в 25 мкл реакционной смеси, содержащей 2 мкг ДНК, 10 пМ каждого праймера, 67 мМ трис-НСl буфера (рН 8,8 при 25oС), 15 мМ сульфата аммония, 2,5 мМ хлористого магния, 0,01% Твин-20, смесь дезоксинуклеотидтрифосфатов (дАТФ, дЦТФ, дТТФ, дГТФ по 2,5 мМ) и 1 ед. Тaq-ДНК-полимеразы. Реакцию амплификации проводили под вазелиновым маслом: 5 циклов: 93oС-2 мин, 55oС-5 мин и 72oС-40 сек; 30 циклов: 93oС-1 мин, 60oС-1 мин и 72oС-40 сек; а затем 60oС-5 мин и 72oС-10 мин. Продукт амплификации pCRHBsAg размером 707 н.п. обрабатывали хлороформом, переосаждали этиловым спиртом и растворяли в воде. Для клонирования данного продукта полученную ДНК pCRHBsAg, a также ДНК плазмиды pUY7 гидролизовали рестрикционными эндонуклеазами ЕсоRI и NotI при 37oС в буфере, содержащем 10 мМ Трис-HCI (рН 8,5 при 37oС), 10 мМ хлорида магния, 100 мМ хлорида калия и 0,1 мг/мл BSA в течение 1,5 часов. Выделенные из геля рестрикционные фрагменты: фрагмент дрожжевого вектора размером 5258 н.п. и фрагмент, содержащий кодирующую область HBsAg размером 687 н.п., лигировали с помощью ДНК-лигазы фага Т4. Полученной лигированной смесью трансформировали клетки Е. coli M15[REP4] методом электропорации. Трансформированные клетки отбирали на агаризованной среде LB с антибиотиками Ар и Кm. Плазмидную ДНК выделяли методом щелочного лизиса, анализировали с помощью рестриктаз EcoRI и NotI, а также рестриктаз, находящихся внутри клонируемых фрагментов, и отбирали клоны pHPS1 (размером 5945 н.п.), плазмидная ДНК которых содержит в своем составе ген HBsAg и все элементы, необходимые для его экспрессии в дрожжах Hansenula polymorpha. Первичную структуру полученной плазмиды подтверждали секвенированием дидезоксиметодом Сэнгера. Схема плазмиды pHPS1 приведена на фиг. 1.
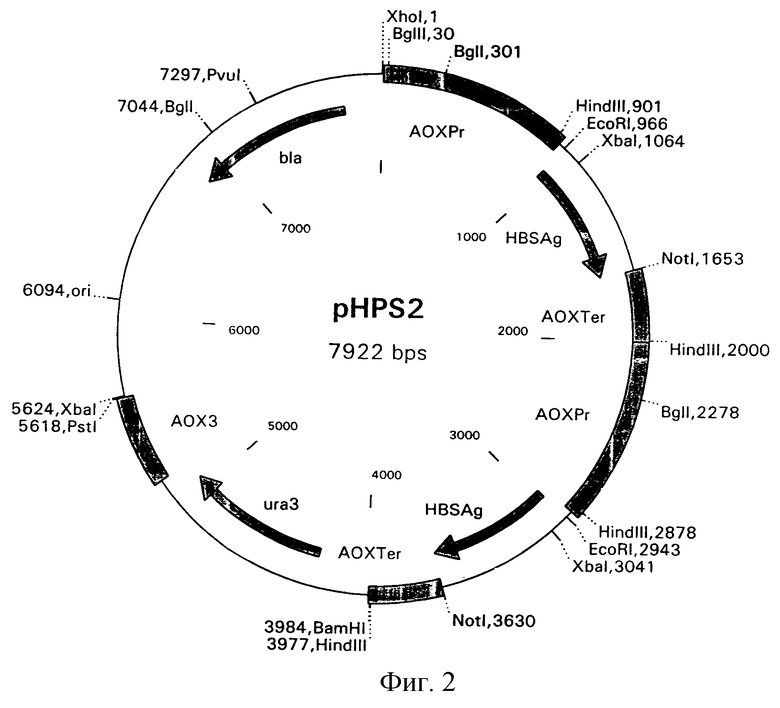
Пример 2. Полимеризация искусственного дрожжевого оперона гена HBsAg.
Для получения плазмиды, содержащей 2 копии дрожжевого оперона гена HBsAg, ДНК плазмиды pHPS1 гидролизовали рестрикционными эндонуклеазами ВаmHI и PνuI при 37oС в буфере 66 мМ Трис-ацетат (рН 7,9 при 37oС), 20 мМ ацетата магния, 132 мМ ацетата калия и 0,2 мг/мл BSA в течение 1 часа, и туже плазмиду pHPS1 гидролизовали рестрикционными эндонуклеазами BglII и PνuI при 37oС в буфере, содержащем 50 мМ Трис-НСI (рН 7,5 при 37oС), 10 мМ хлорида магния, 100 мМ хлорида натрия и 0,1 мг/мл BSA в течение 1 часа. Выделенные из геля рестрикционные фрагменты: фрагмент, полученный гидролизом BamHI и PνuI размером 2632 н. п., а также фрагмент, полученный гидролизом ВglII и PvuI размером 5290 н.п., лигировали с помощью ДНК-лигазы фага Т4. Полученной лигированной смесью трансформировали клетки Е. coli M15[REP4] методом электропорации. Трансформированные клетки отбирали на агаризованной среде LB с антибиотиками Ар и Km. Плазмидную ДНК выделяли методом щелочного лизиса, анализировали с помощью рестриктаз PνuI, ВаmHI и BglII, а также рестриктаз, находящихся внутри полимеризованных фрагментов HCBsAg, и отбирали клоны pHPS2 (размером 7922 н.п.), плазмидная ДНК которых содержит повторяющиеся модули дрожжевого оперона гена HBsAg. Схематичное изображение плазмиды pHPS2 представлено на фиг. 2. Первичная структура полученной плазмиды pHPS2 представлена на фиг.3.
Пример 3. Получение штамма Hansenula polymorpha - продуцента рекомбинантного поверхностного антигена вируса гепатита В (HBsAg).
Для получения штаммов дрожжей Hansenula polymorpha - продуцентов HBsAg клетки дрожжей штамма А трансформировали плазмидами pHPS1 и pHPS2.
Для трансформации использовали ДНК плазмиды pHPS1 и pHPS2, линеаризованные рестрикционными эндонуклеазами BglII и PstI] при 37oС в буфере, содержащем 50 мМ Трис-НСI (рН 7,5 при 37oС), 10 мМ хлорида магния, 100 мМ хлорида натрия и 0,1 мг/мл BSA в течение 1,5 часов.
Клетки дрожжей выращивали в 100 мл среды YEPD при 35oС до достижения культурой оптической плотности, соответствующей 2-4 ед. поглощения при длине волны 600 нм. Клетки дважды промывали стерильной водой, после чего суспендировали в 0,3 мл 100 мМ раствора ацетата лития и инкубировали при 35oС в течение 30 мин. К 50 мкл полученной суспензии клеток добавляли 0,1-1 мкг плазмидной ДНК, 50 мкг ДНК спермы лосося, предварительно денатурированной нагреванием (10 мин при 100oС) и 0,3 мл раствора 100 мМ ацетата лития, содержащего 40% полиэтиленгликоля 4000. Далее пробу инкубировали 30 мин при 30oС и 20 мин при 42oС, помещали на 15 сек в ледяную баню и центрифугировали 10 сек при 10000 об/мин. Клетки суспендировали в 1 мл стерильной воды и высевали на твердую среду SC. Клоны трансформантов вырастали через 2-3 суток. Выросшие клоны пересевали на чашки со средой SC, содержащей 2% глюкозу, отдельными колониями, затем перепечатывали на среду ММ (1,34% Yeast Nitrogen Base ("Difco", QUA), 0,5% метанола, 2% агара ("Difco", США)) для отбора трансформантов, не растущих на среде с метанолом, что свидетельствовало об интеграции чужеродного гена в локус AOXI (фенотип Met").
Плазмиды pHPS1 и pHPS2 не претерпевали структурных перестроек в Hansenula polymorpha как при хранении штаммов, так и в процессе их культивирования. Это происходило вследствие устойчивой интеграции данных плазмид в геном штамма, что обеспечивало им необходимую стабильность в условиях митотического деления. В проведенных экспериментах в условиях длительного культивирования на богатой неселективной среде не было выявлено клеток штамма Hansenula polymorpha, утративших плазмиды pHPS1 или pHPS2 (не способных расти на селективной среде в отсутствие гистидина).
Для анализа продукции HBsAg белка клетками трансформантов фенотипа Met" их выращивали при 30oС в 100 мл жидкой среды BMGY (2% пептона, 1% дрожжевого экстракта, 1% глицерина, 10 мл 1М калий-фосфатного буфера, рН 6,0, 1,34% Yeas Nitrogen Base ("Difco", США) до стационарной фазы роста в течение 2 суток. Клетки собирали центрифугированием при 5000 об/мин в течение 10 мин, супернатант сливали и переносили всю биомассу в 20 мл жидкой среды ВMMY (2% пептона, 1% дрожжевого экстракта, 0,5% метанола, 10 мл 1 М калий-фосфатного буфера, рН 6,0, 1,34% Yeast Nitrogen Base ("Difco", США) для индукции экспрессии HBsAg. Индукцию проводили при 35oС в течение 4 суток.
По окончании индукции клетки дрожжей собирали центрифугированием, суспендировали в равном объеме буфера для вскрытия (50 мМ Na-фосфатый буфер, рН 8,0, 5 мМ ЭДТА, 4 М мочевина, 2 мМ PМSF, 0,5% Твин-20), добавляли стеклянные шарики (0-0,1 мм) и разрушали на дезинтеграторе "Dyno-Mill" в течение 10 мин, далее центрифугировали 10 мин при 3000 об/мин для удаления неразрушенных клеток. В супернатанте определяли присутствие HBsAg иммуноферментным методом и при помощи электрофореза в полиакриламидном геле.
Для определения содержания HBsAg методом иммуноферментного анализа использовали тест-систему Вектогеп B-HBs-антиген-стрип (ЗАО "Вектор-Бест"). Тестируемые пробы разводили в соотношениях от 1:10 до 1:100 10 мМ К-фосфатным буфером, рН 6,7, и помещали в лунки планшета для иммуноферментного анализа. Далее пробы инкубировали при 37oС в течение часа, лунки промывали 5 раз буфером ФСБ-Т (10 мМ К-фосфатый буфер, рН 6,7, содержащий 137 мМ NaCI, 2,7 мМ КCl, 0,05% Твин-20) и вносили в лунки раствор моноклональных антител к HBsAg, меченных пероксидазой хрена. Затем пробы инкубировали при 37oС в течение часа, промывали буфером ФСБ-Т, добавляли по 100 мкл субстрата ТМБ (тетраметилбензидин), выдерживали в темноте в течение 15-20 мин при комнатной температуре. Реакцию останавливали 50 мкл 1 М H2SO4 и измеряли оптическую плoтность при 450 нм.
Согласно полученным таким образом данным, клетки дрожжей штамма A(pHPS1) и A(pHPS2) синтезируют HBsAg, дающий положительную реакцию с моноклональными антителами к нативному HBsAg. В то же время, в клетках дрожжей штамма, трансформированных плазмидой UY7, отсутствует белок, иммунологически родственный нативному HBsAg.
Для контроля продукции HBsAg клетками дрожжей штаммов А (pHPS1) и A(pHPS2) использовали также метод электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия. Разделение белков проводили в 15% полиакриламидном геле в стандартной системе буферов (электродный буфер: 25 мМ Tris-HCl, 192 мМ глицин, 0,1% додецилсульфат натрия, рН 8,3; буфер для геля: 375 мМ трис-хлоридный буфер, рН 8,8). Параллельно проводили разделение белков контрольного штамма А, трансформированного плазмидой pY7), выращенного в идентичных условиях. В качестве стандартов молекулярной массы использовали карбоангидразу, ингибитор трипсина, миоглобин, лизоцим. По окончании электрофореза гели окрашивали 0,15% раствором кумасси G250 в 25% изопропаноле и 10% уксусной кислоте и отмывали в 10% уксусной кислоте. При сравнении спектра белков трех штаммов у штаммов A(pHPS1) и A(pHPS2) обнаруживали появление двуx дополнительных белковых полос с молекулярной массой 24 кДа и 45 кДа, что соответствует молекулярной массе белка HBsAg и его димера. Уровень синтеза HBsAg белка определяли, сравнивая интенсивность окрашивания полосы рекомбинантного белка с полосой соответствующего белка - стандарта молекулярной массы.
Согласно полученным данным клетки дрожжей штамма A(pHPS2) синтезируют около 150-180 мг иммунореактвного HBsAg белка на литр культуры дрожжей (а штамма A (pHPS1) около 80-90 мг HBsAg белка.
Проведенные эксперименты показали, что плазмиды pHPS1 и pHPS2 не претерпевают структурных перестроек в Hansenula polymorpha в процессе культивирования. Это обеспечивается интеграцией данных плазмид в геном штамма, что приводит к их стабильности в условиях методического выделения, которая является одним из важнейших условий промышленного культивирования Hansenula polymorpha. Проведенные эксперименты в условиях длительного культивирования на богатой неселлективной среде подтвердили отсутствие клеток штамма Hansenula polymorpha, утративших плазмиды pPBS1 или pHPS2, то есть не способных расти на селективной среде, в отсутствие урацила, даже через 200-300 циклов деления клеток.
Созданный челночный вектор с двойной копией гена оказался генетически стабильным при культивировании в Е. coli.


Изобретение относится к биотехнологии и медицине и касается рекомбинантной плазмиды, кодирующей поверхностный антиген (HBsAg) вируса гепатита В, ее получения и штамма дрожжей Hansenula polymorpha - продуцента поверхностного антигена вируса гепатита В. Сущность изобретения состоит в создании рекомбинатной плазмиды pHPS2, имеющей размер 7922 н.п. и состоящей из следующих элементов: димера искусственного дрожжевого оперона HBsAg/ayw, состоящего из промоторной области АОХ1 оперона, гена HBsAg вируса гепатита В и участка терминации транскрипции, дрожжевого оперона оротидин-5-фосфатдикарбоксилазы Ura3, области АОХ1 оперона, бактериального оперона бета-лактамазы и бактериального участка инициации репликации типа Col El. Изобретение также включает штамм Hansenula polymorpha - продуцента поверхностного антигена (HBsAg) вируса гепатита В. Преимущество заключается в повышении экспрессии HBsAg. 4 с. и 2 з.п.ф-лы, 3 ил.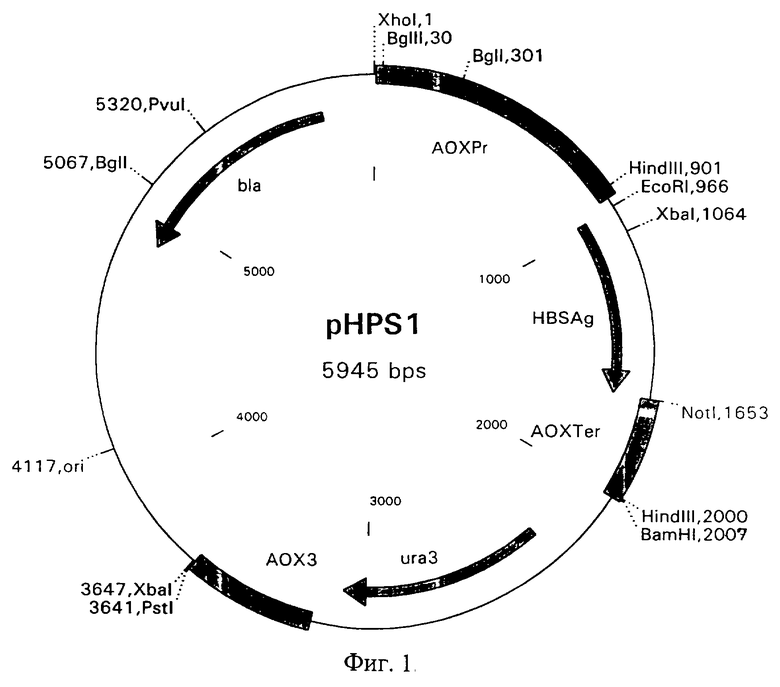
| US 6103519 А, 15.08.2000 | |||
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК PDES20, КОДИРУЮЩАЯ ПОВЕРХНОСТНЫЙ АНТИГЕН ВИРУСА ГЕПАТИТА В (HBSAG/AYW), ШТАММ ДРОЖЖЕЙ SACCHAROMYCES CEREVISIAE, СОДЕРЖАЩИЙ РЕКОМБИНАНТНУЮ ПЛАЗМИДНУЮ ДНК PDES20 - ПРОДУЦЕНТ ПОВЕРХНОСТНОГО АНТИГЕНА ВИРУСА ГЕПАТИТА В (HBSAG/AYW) | 1996 |
|
RU2088664C1 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДА PFS 19, ОПРЕДЕЛЯЮЩАЯ СИНТЕЗ ПОВЕРХНОСТНОГО АНТИГЕНА S ВИРУСА ГЕПАТИТА B ЧЕЛОВЕКА | 1992 |
|
RU2115730C1 |
| US 5935789 А, 10.08.1999 | |||
| SOHN J.H | |||
| et al | |||
| Novel autonomously replicating sequence (APS) for muetiple integration in the yeast Hansenula polymopha DL, J | |||
| of Bacteriology, 1996, v.178, №15, p.4420-4428 | |||
| DIMINSKY D | |||
| et al | |||
| Comparison be tween hepatitis B surface antigen (HBsAg) particles derived from mammalian cells (CHO) and yeast all (Hansenula polymorpha) : composition, structure and ummunogenicity, Vaccine, 1997, v.15, №6-7, p.637-647. | |||

