Область изобретения
Настоящее изобретение относится к способу получения производного хинолина, представленного формулой (3), которое может быть полезным промежуточным соединением для получения восстанавливающих холестерин агентов (ингибиторов HMG-CoA редуктазы).
Уровень техники
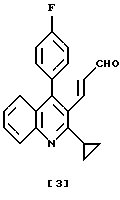
Соединение хинолина, представленное формулой (4), раскрыто в JP-A1-279866, ЕР-А-304063 и в патенте США 5011930 как полезный восстанавливающий холестерин агент (ингибитор HMG-CoA редуктазы).
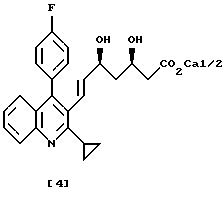
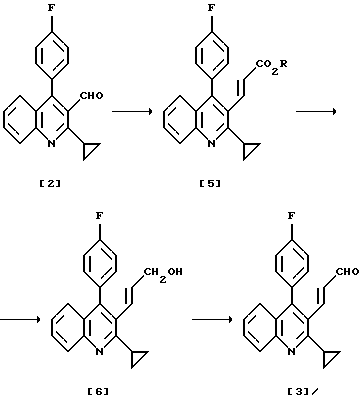
Соединение хинолина, представленное формулой (4), получают в вышеуказанных патентах как представлено далее, превращая альдегид (2) в сложный эфир α,β-ненасыщенной карбоновой кислоты (5), с последующим восстановлением до спирта (6) и окислением до целевого соединения хинолина (3). Хотя непосредственное восстановление сложного эфира α,β-ненасыщенной карбоновой кислоты до целевого соединения хинолина (3) обеспечило бы эффективность получения, проблема состоит в трудности контроля за такой схемой.
Содержание изобретения
В результате интенсивных исследований для решения вышеуказанной проблемы авторы настоящего изобретения нашли одностадийный способ получения целевого хинолина (3) через соединение нитрила (1), получаемого при взаимодействии альдегида, представленного формулой (2), с диэтилцианометилфосфонатом.
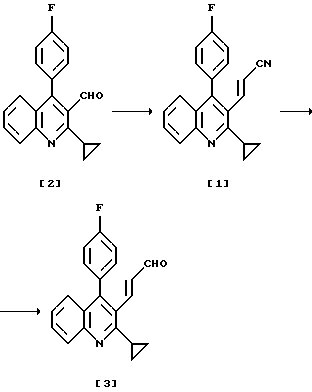
А именно, настоящее изобретение относится к способу получения производного хинолина (3) через соединение нитрила (1), получаемого при взаимодействии альдегида (2) с диэтилцианометилфосфонатом и его промежуточным соединением (1).
Одностадийное получение целевого хинолина (3) можно осуществить через соединение нитрила (формула (1)), получаемого при взаимодействии альдегида, представленного формулой (2) с диэтилцианометилфосфонатом.
Предпочтительный вариант осуществления изобретения
Далее описывается способ осуществления изобретения.
Получение соединения нитрила (1)
В качестве растворителей, которые можно использовать в этой реакции, можно указать ароматические углеводороды, такие как толуол или ксилол, эфирные растворители, такие как тетрагидрофуран или диоксан, или галогенированные растворители, такие как дихлорэтан или о-дихлорбензол.
Используют от 0,5 до 5-кратного количества молей, предпочтительно от 0,9 до 1,5-кратного количества молей диэтилцианометилфосфоната.
Можно использовать основание, такое как гидрид натрия, гидроксид натрия, гидрид калия, метоксид натрия, этоксид натрия, трет-бутоксид калия или карбонат калия, в количестве молей, кратном от 0,5 до 10, в зависимости от типа растворителя и основания. Можно, в некоторых случаях, использовать катализатор переноса фазы, такой как Aliquat 336, например, если толуол в качестве растворителя объединяют с (водным) гидроксидом натрия в качестве основания.
Температура реакции находится в интервале от -20 до 80oС, предпочтительно в интервале от 20 до 40oС.
Получение производного хинолина (3)
Использование в реакции диизобутилалюминийгидрида в качестве восстанавливающего агента и ароматического углеводорода, такого как толуол или ксилол, в качестве растворителя дает хорошие результаты. Диизобутилалюминийгидрид используют в 0,5-5-кратном количестве молей, предпочтительно в 0,9-1,5-кратном количестве молей, а температура находится в интервале от -50 до 50oС, предпочтительно в интервале от -30 до 5oС. Возможно также восстановление никелем Рэнея в муравьиной кислоте в качестве растворителя.
Примеры
Далее настоящее изобретение будет раскрыто более подробно со ссылкой на примеры. Однако эти конкретные примеры ни коим образом не ограничивают настоящее изобретение.
Получение нитрила (1)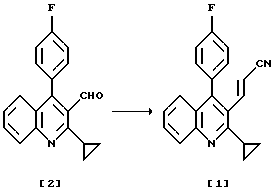
К раствору 199 г (683 ммоль) 2-циклопропил-4-(4-фторфенил)хинолин-3-карбоальдегида в 960 г толуола добавляют 136 г (765 ммоль, 1,1 экв) диэтилцианометилфосфоната и 5,5 г (13,6 ммоль, 0,02 экв) Aliquat 336.
По каплям добавляют 400 г 20%-ного водного гидроксида натрия в течение 0,5-1 часа, причем температуру внутри реактора поддерживают при 25-35oС, и реакционный раствор перемешивают при этой же температуре в течение 1 часа.
После завершения реакции добавляют 200 г воды и полученную смесь перемешивают в течение 30 минут и оставляют для разделения. Полученный органический слой промывают 400 мл 10%-ного насыщенного водного гидроксида натрия, объединяют с 400 мл насыщенного водного хлорида натрия, рН доводят до рН 7 с помощью 1 н. водной соляной кислоты и оставляют для разделения. После добавления 50 г сульфата натрия полученный органический слой перемешивают в течение 1 часа, затем перемешивают еще в течение 30 минут вместе с 5 г активированного угля и 20 г силикагеля и фильтруют через воронку со слоем целита.
Растворитель отгоняют из фильтрата при пониженном давлении до тех пор, пока не останется около 400 г, и выпавшие в осадок кристаллы плавят in situ, нагревая и кипятя с обратным холодильником вместе с 580 г гексана в течение 30 минут, затем охлаждают до 5oС и перемешивают при этой температуре в течение 2 часов. Выпавшие в осадок кристаллы собирают фильтрованием, промывают смесью толуол-гексан (1: 5, вес/вес) и гексаном и сушат, получая 189 г 3-(2-циклопропил-4-(4-фторфенил)-3-хинолил)-проп-2-ененитрила с выходом 88%. Т. плавления 176-178oС.
Получение производного хинолина (3)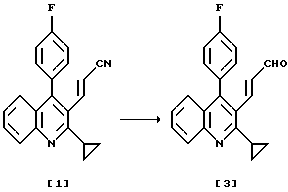
Раствор 181 г (576 ммоль) 3-(2-циклопропил-4-(4-фторфенил)-3-хинолил)проп-2-ененитрита в 1812 мл толуола охлаждают до температуры внутри реактора -10oС. По каплям добавляют 650 мл 1,02 М раствора диизобутилалюминийгидрида (663 ммоль, 1,15 экв) в толуоле в течение 1 часа, при этом температуру внутри реактора поддерживают в интервале от -10 до -5oС, и полученную смесь перемешивают при той же самой температуре в течение 1 часа.
После этой реакции добавляют по каплям 30,5 г этанола, при этом температуру поддерживают в интервале от -10 до -5oС, и полученную смесь перемешивают при той же самой температуре в течение 30 минут. По каплям добавляют 155 мл 1 н. соляной кислоты, поддерживая температуру при 10oС или ниже, и полученную смесь перемешивают при этой же температуре в течение 1 часа. Затем по каплям добавляют 9,06 мл 35% соляной кислоты, поддерживая ту же самую температуру, и полученную смесь перемешивают при температуре внутри реактора 25-30oС; полученную смесь фильтруют через воронку со слоем целита.
После добавления 725 мл 1 н. соляной кислоты фильтрат перемешивают в течение 30 минут и оставляют для разделения. Органический слой промывают 360 мл 1 н. соляной кислоты и 545 мл насыщенного водного хлорида натрия. Все водные слои объединяют и снова экстрагируют 725 мл этилацетата, экстракты промывают 360 мл насыщенного водного хлорида натрия и объединяют с вышеуказанным органическим слоем. После добавления 1090 мл воды рН смеси доводят до рН 7 с помощью насыщенного водного бикарбоната натрия и промывают 1090 мл воды и 1090 мл насыщенного водного хлорида натрия.
Растворитель отгоняют из полученного раствора при пониженном давлении и добавляют 360 г циклогексана и 720 г н-гексана. Полученную смесь кипятят с обратным холодильником в течение 30 минут, затем охлаждают до 0-5oС и перемешивают при той же самой температуре в течение 2 часов. Выпавшие в осадок кристаллы собирают фильтрованием, промывают смесью циклогексан-н-гексан (1: 2, вес/вес) и н-гексаном и сушат, получая 3-(2-циклопропил-4-(4-фторфенил)-3-хинолил)проп-2-енала с выходом 93%. Т. плавления: 146-147oС.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ХИНОЛИЛАКРИЛОНИТРИЛА И СООТВЕТСТВУЮЩИХ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ | 2001 |
|
RU2260000C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХИНОЛИНКАРБАЛЬДЕГИДА | 1999 |
|
RU2217423C2 |
| ОПТИЧЕСКИ-АКТИВНЫЙ β-АМИНОАЛКОКСИБОРАНОВЫЙ КОМПЛЕКС, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ОПТИЧЕСКИ АКТИВНОЕ β-АМИНОСПИРТОВОЕ ПРОИЗВОДНОЕ ДЛЯ ЕГО ПОЛУЧЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНЫХ СПИРТОВ С УЧАСТИЕМ КОМПЛЕКСА | 1994 |
|
RU2126412C1 |
| СПОСОБ ПОЛУЧЕНИЯ 7-ХИНОЛИНИЛ-3,5-ДИГИДРОКСИГЕПТ-6-ЕНОАТА | 2002 |
|
RU2260001C2 |
| ИНГИБИТОР АТЕРОСКЛЕРОТИЧЕСКОГО УТОЛЩЕНИЯ ВНУТРЕННЕЙ ОБОЛОЧКИ СОСУДОВ | 1992 |
|
RU2114620C1 |
| БЕНЗОПИРАНОВОЕ СОЕДИНЕНИЕ | 2005 |
|
RU2366658C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОПИРАНА В КАЧЕСТВЕ ПРОТИВОАРИТМИЧЕСКИХ АГЕНТОВ | 2005 |
|
RU2380370C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ХИНОЛИНКАРБОКСАЛЬДЕГИДА И ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2002 |
|
RU2264391C2 |
| ПРОИЗВОДНЫЕ 4-АМИНОБЕНЗОПИРАНА | 2002 |
|
RU2288226C2 |
| Способ получения производных 3(2Н)-пиридазинона | 1988 |
|
SU1584750A3 |
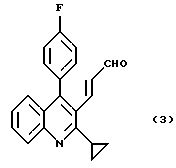
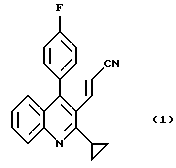
Изобретение относится к способу получения 3-((2-циклопропил-4-(4-фторфенил)-3-хинолил)проп-2-еналя формулы (3), которое является промежуточным соединением в синтезе восстанавливающих холестерин агентов. Указанный способ включает взаимодействие 2-циклопропил-4-(4-фторфенил)хинолин-3-карбоальдегида с диэтилцианометилфосфонатом в присутствии основания при температуре 20 - 40oС с получением 3-(2-циклопропил-4-(4-фторфенил)-3-хинолил)-проп-2-ененитрила и взаимодействие 3-(2-циклопропил-4-(4-фторфенил)-3-хинолил)-проп-2-ененитрила с диизобутилалюминийгидридом при температуре (-5) - (-10)oС с получением целевого соединения. Также описано промежуточное соединение 3-(2-циклопропил-4-(4-фторфенил)-3-хинолил)-проп-2-ененитрил. Технический результат: упрощение процесса. 2 с.п. ф-лы.
2. Способ получения производного хинолина формулы (3)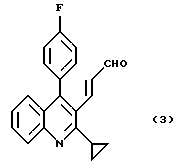
который заключается во взаимодействии соединения формулы (2)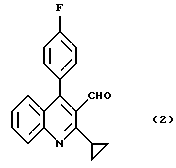
с диэтилцианометилфосфонатом в присутствии основания при температуре 20-40oС с получением соединения формулы (I)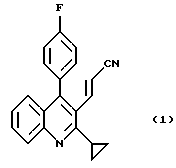
и далее во взаимодействии соединения (1) с диизобутилалюминийгидридом при температуре (-5)-(-10)oC с получением соединения формулы (3).
| ИНГИБИТОР АТЕРОСКЛЕРОТИЧЕСКОГО УТОЛЩЕНИЯ ВНУТРЕННЕЙ ОБОЛОЧКИ СОСУДОВ | 1992 |
|
RU2114620C1 |
| DE 3905908 A1, 06.09.1990 | |||
| Система единого времени для управления вторичными часами | 1974 |
|
SU535548A1 |
| US 5102888 А, 07.04.1992. | |||

