Настоящее изобретение относится к новому способу получения соединения формулы

его оптически активным R или S энантиомерам, смесям этих энантиомеров и их фармацевтически приемлемым солям присоединения кислот.
Соединение формулы I известно под названием карведилол, который используется в качестве лекарства, обладающего антигипертензивным, бета-адреноблокирующим и сосудорасширяющим действием. Его химическое название: 1-(9Н-карбазол-4-илокси)-3-[/2-(2-метоксифенокси)этил/амин]-2-пропанол.
Способ получения карведилола известен из патентной публикации DE-OS № 2815926, а способ получения R и S энантиомеров описан в патентной публикации DE-OS № 3319027. Согласно известному способу, 4-(оксиранилметокси)-9Н-карбазол формулы

или его R или S энантиомеры реагируют с 2-(2-метоксифенокси)этиламином формулы
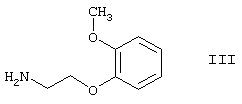
с получением соединения формулы I, выход которого составляет от 39 до 42%.
Недостаток известного способа состоит в том, что одновременно с получением карведилола также образуется бис соединение формулы
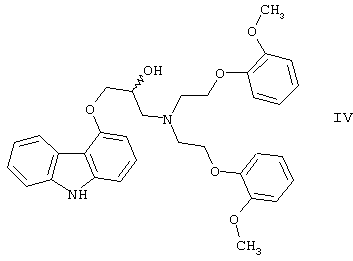
в результате реакции двух молярных эквивалентов 4-(оксиранилметокси)-9Н-карбазола формулы II и одного молярного эквивалента 2-(2-метоксифенокси)этиламина формулы III. Избежать этой побочной реакции нельзя, и бис соединение формулы IV образуется в количестве, сравнимом с количеством карведилола. Следовательно, данный способ является неэкономичным.
Другие возможности получения карведилола описаны в патентной публикации DE-OS № 2815926. Согласно этим способам, 4-(3-амино-гидроксипропокси)-9Н-карбазол может реагировать с
a) 2-(2-метоксифенокси)этилгалидом или сульфонатом;
b) 2-(2-метоксифенокси)ацетальдегидом с последующим каталитическим гидрированием;
c) 2-(2-метоксифенокси)ацетилхлоридом с последующим восстановлением полученного кислотного амида комплексным гидридом металла.
Ни один из способов от а) до с) не подходит для экономичного получения карведилола. Для каждого способа необходима стадия очистки продукта с помощью колоночной хроматографии, что делает получение исходного соединения неэкономичным. В реакции по способу а) также образуется бис соединение формулы IV. В реакции по способу b) выход составляет 41%, в то время как по способу с) - максимум 24%. В реакции по способу с) использование комплексного гидрида металла, практически гидрида лития и алюминия, также является недостатком, поскольку эта реакция сопровождается повышенной опасностью воспламенения. Данная реакция требует особых условий - даже следы влаги должны быть удалены.
В принципе, можно избежать образования бис соединения формулы IV, если использовать вместо первичного амина формулы III вторичный амин, который является производным первичного амина формулы III, содержащего защитную группу. Таким вторичным амином является, например, N-/2-(2-метоксифенокси)этил/бензиламин формулы

В Примере 5 патентной публикации DE-OS № 2815926 4-оксиранилметокси-9Н-карбазол формулы II реагирует со вторичным амином формулы V с получением бензилкарведилола формулы
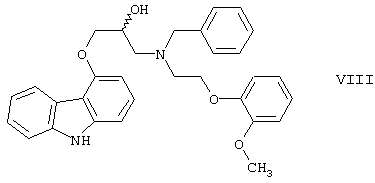
который можно выделить только благодаря очистке с помощью колоночной хроматографии. Такая процедура является не экономичной для производства в промышленном объеме. Реакцию соединений формул I и V проводили в диметиловом эфире этиленгликоля.
Пример 5 из DE OS № 2815926 был воспроизведен четыре раза. Как показано в Примере Сравнения № 1, выход продукта был 60%, причем температура плавления загрязненного продукта была 92° С, что значительно ниже значения, указанного в Примере 5 (97-99° С). Было невозможно получить продукт в кристаллическом виде без очистки с помощью колоночной хроматографии, таким образом, нельзя исключить эту стадию очистки. Это является существенным недостатком для применения в промышленности. Так как увеличение объема производства продукта по данному известному способу сопровождается снижением как выхода, так и качества продукта, указанный способ не пригоден для применения в промышленности.
Способ, описанный в Примере 5 DE OS № 2815926, был модифицирован путем использования вместо диметилового эфира этиленгликоля этилацетата или диоксана как показано в Примерах Сравнения №№2 и 3. Даже после 28 часов реакции около половины исходного соединения осталось не прореагировавшим в диоксане, а в этилацетате реакция практически не прошла совсем.
Целью настоящего изобретения было разработать экономичный способ получения карведилола формулы I.
Указанная цель была достигнута в результате создания способа по настоящему изобретению, в котором вторичный амин формулы V реагирует с эпихлоргидрином, полученное соединение формулы
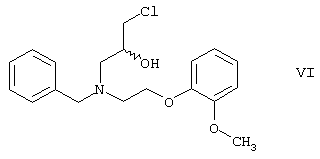
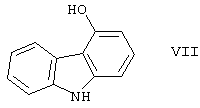
реагирует с 4-гидрокси-9Н-карбазолом формулы
и полученный бензил-карведилол формулы VIII дебензилируют путем каталитического гидрирования;
и, при необходимости, полученный продукт реагирует с неорганической или органической кислотой для получения его фармацевтически приемлемой соли присоединения кислоты.
Вторичный амин формулы V используется в виде масла, то есть в виде соединения, в котором отсутствует кристаллизационная вода, либо предпочтительно в виде соединения, в котором присутствует кристаллизационная вода.
Согласно способу настоящего изобретения реакцию вторичного амина формулы V с эпихлоргидрином проводят без растворителя или с растворителем при температуре 150° С. Растворителем может быть органический растворитель протонной, дипольной апротонной или неполярной природы. Реакцию предпочтительно проводить без растворителя; в этом случае используют необходимый избыток эпихлоргидрина, а реакцию проводят при температуре от 30 до 80° С.
Хлорсоединение формулы VI реагирует с 4-гидрокси-9Н-карбазолом формулы VII в органическом растворителе дипольной апротонной или неполярной природы при температуре 150° С. Предпочтительным растворителем является ацетонитрил, а реакцию проводят при температуре кипения реакционной смеси.
Полученный бензилкарведилол формулы VIII либо сначала выделяют, а затем дебензилируют, либо, что предпочтительно, не выделяют из реакционной смеси, в которой он был получен, перед реакцией дебензилирования. Дебензилирование проводят путем каталитического гидрирования, способом, известным для удаления бензильной группы.
Предпочтительно, в качестве катализатора используют палладий на угле. Для дебензилирования бензилкарведилола катализатор можно использовать несколько раз без регенерации. Предпочтительно, гидрирование проводят с использованием гидрата гидразина.
Карведилол очень высокой чистоты с выходом около 80% получают способом по настоящему изобретению. В данном способе не образуется бис соединения формулы IV.
Бензилкарведилол формулы VIII получается очень высокой чистоты, следовательно, не требуется его очистки и реакцию дебензилирования можно проводить в той реакционной смеси, в которой он был получен.
Приведенные выше данные свидетельствуют о том, что способ по настоящему изобретению является экономичным и простым в исполнении.
Далее настоящее изобретение поясняется следующими Примерами.
Получение исходных соединений:
Получение соединения формулы V:
Дигидрат N-/2-(2-метоксифенокси)этил/бензиламина
К 131 см3 (128,64 г, 1.2 моль) бензиламина, нагретого до 80° С, добавляют 69,33 г (0,3 моль) 1-(2-бромэтокси)-2-метоксибензола в течение 25 минут с такой скоростью, чтобы температура реакционной смеси была от 95 до 105° С. Затем реакционную смесь перемешивают при температуре от 95 до 105° С в течение 2 часов, полученную суспензию охлаждают ледяной водой до 25° С и добавляют в течение 5 минут к 1000 см3 10%-ной соляной кислоты, охлажденной ледяной водой, обращая внимание на то, чтобы температура смеси не превысила 50° С. Полученный раствор охлаждали ледяной водой. Через 5-10 минут гидрохлорид продукта осаждался в виде кристаллов. Суспензию кристаллов перемешивали при 5-10° С в течение 0,5 часа, фильтровали и промывали водой. Неочищенный гидрохлорид перекристаллизовывали из 400 см3 воды, фильтровали и промывали водой.
Таким образом было получено 71,9 г (81,6%) гидрохлорида названного соединения с температурой плавления 148-150° С.
Гидрохлорид суспендировали, суспензию нагревали до растворения кристаллов и затем к теплому раствору прибавляли по каплям 100 см3 10%-ного водного раствора гидроокиси натрия. Раствор охлаждали до температуры 5-10° С ледяной водой, кристаллы отфильтровывали, промывали 100 см3 ледяной воды и сушили на воздухе при комнатной температуре.
Таким образом было получено 43,1 г (48,9%) названного соединения с температурой плавления 53-55° С. Продукт содержал кристаллизационную воду (C16H19NO2·2H2O).
Получение соединения формулы VI:
1-[бензил-/2-(2-метоксифенокси)этил/амин]-3-хлор-2-пропанола
10,3 г (40 ммоль) N-/2-(2-метоксифенокси)этил/бензиламина растворяли в 16 см3 эпихлоргидрина, полученный раствор перемешивали при 55-60° С в течение 3 часов. Реакционную смесь упаривали при пониженном давлении, добавляли 20 см3 толуола, упаривали и еще раз повторяли данную процедуру.
Таким образом было получено 13,8 г (99%) названного соединения в виде маслянистого осадка.
Пример 1
1-/N-бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанол
3,5 г (10 ммоль) 1-[бензил-/2-(2-метоксифенокси)этил/амин]-3-хлор-2-пропанола и 0,92 г (5 ммоль) 4-гидрокси-9Н-карбазола растворяли в 25 см3 диоксана. К полученному раствору добавляли 0,69 г (5,0 ммоль) карбоната калия, реакционную смесь кипятили 28 часов, затем охлаждали до 25° С. Неорганическую соль отфильтровывали, промывали диоксаном, фильтрат упаривали при пониженном давлении. К 4,6 г кубового остатка добавляли 25 см3 изопропанола. Смесь нагревали до 60° С и охлаждали при перемешивании. Через 24 часа кристаллы отфильтровывали и промывали диизопропиловым эфиром.
Таким образом было получено 1,79 г (71,7%) названного соединения в виде белых кристаллов с температурой плавления 93-95° С.
Пример 2
1-(9Н-карбазол-4-илокси)-3-/2-(2-метоксифенокси)этиламин/-2-пропанол
В колбу на 100 см3 помещали 1,99 г (4 ммоль) 1-/N-бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанола, 40 см3 этанола, 4 см3 воды и 1.0 г катализатора - увлажненного палладия на угле с содержанием влаги - 50,6% и содержанием палладия - 16.2% в расчете на сухой катализатор. К смеси добавляли по каплям 4 см3 (80 ммоль) 98%-ного гидрата гидразина при температуре от 5 до 10° С в течение 10 минут, затем реакционную смесь перемешивали при температуре 25° С в течение 2 часов. После этого катализатор отфильтровывали, промывали дважды по 20 см3 этанола, фильтрат упаривали при пониженном давлении. К кубовому остатку прибавляли 50 см3 воды и 50 см3 1,2-дихлорэтана, смесь перемешивали до растворения осадка, фазы разделяли, водную фазу экстрагировали еще 2 раза по 50 см3 1,2-дихлорэтана. Органическую фазу сушили и упаривали при пониженном давлении. Кубовый остаток нагревали с 9 см3 этилацетата до кипения, горячую смесь фильтровали через бумажный складчатый фильтр, фильтрат охлаждали при перемешивании. После выпадения кристаллов смесь перемешивали еще один час при 25° С, кристаллы отфильтровывали и дважды промывали по 1 см3 холодного этилацетата. Полученный продукт сушили под лампой инфракрасного излучения.
Таким образом было получено 1,33 г (81,6%) названного соединения в виде белых кристаллов с температурой плавления 114-116° С.
Пример 3
1-(9Н-карбазол-4-илокси)-3-/2-(2-метоксифенокси)этиламин/-2-пропанол
К 60 см3 этанола прибавляли 1,1 г (2,2 ммоль) 1-/N-бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанола и 0,5 г катализатора - увлажненного палладия на угле с содержанием влаги - 50,6% и содержанием палладия - 16.2% в расчете на сухой катализатор. Смесь тщательно перемешивали в атмосфере водорода с давлением 5 бар при комнатной температуре. После этого катализатор отфильтровывали, промывали дважды по 20 см3 этанола, фильтрат упаривали при пониженном давлении. К кубовому остатку прибавляли 5 см3 этилацетата, смесь перемешивали 2 часа при 25° С, полученные кристаллы отфильтровывали и дважды промывали по 1 см3 холодного этилацетата. Полученный продукт сушили под лампой инфракрасного излучения.
Таким образом было получено 0,69 г (76,6%) названного соединения в виде белых кристаллов с температурой плавления 114-116° С.
Пример 4
1-/N-бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанол
41,2 г (160 ммоль) N-/2-(2-метоксифенокси)этил/бензиламина растворяли в 64 см3 эпихлоргидрина, полученный раствор перемешивали при 55-60° С в течение 3,5 часов. Затем реакционную смесь упаривали при пониженном давлении, к кубовому остатку добавляли 20 см3 толуола, упаривали и еще раз повторяли данную процедуру. К 56,0 г маслянистого кубового остатка добавляли 300 см3 ацетонитрила и 14,66 г (80 ммоль) 4-гидрокси-9Н-карбазола. К полученному раствору прибавляли 13,45 г (160 ммоль) бикарбоната натрия и перемешивали при кипячении в течение 20 часов. Смесь охлаждали до 25° С, неорганическую соль отфильтровывали и промывали ацетонитрилом. Фильтрат упаривали при пониженном давлении. К 68,6 г кубового остатка добавляли 160 см3 изопропанола. Смесь нагревали до 60° С и охлаждали при перемешивании. Через 3 часа охлажденную ледяной водой густую суспензию фильтровали и промывали 100 см3 холодного изопропанола и 100 см3 диизопропилового эфира. Продукт сушили под лампой инфракрасного излучения.
Таким образом было получено 31,80 г (80,0%) названного соединения в виде белых кристаллов с температурой плавления 94-96° С.
Пример 5
1-/N-бензил-2-(2-метоксифенокси)этиламин/-3-(9Н-карбазол-4-илокси)-2-пропанол
К 3,5 г (10 ммоль) 1-[бензил-/2-(2-метоксифенокси)этил/амин]-3-хлор-2-пропанола добавляли 25 см3 тетрагидрофурана и 0,92 г (5 ммоль) 4-гидрокси-9Н-карбазола. К полученному раствору добавляли 0,69 г (5,0 ммоль) карбоната калия, смесь перемешивали при кипячении в течение 30 часов, охлаждали до 25° С, неорганическую соль отфильтровывали и промывали тетрагидрофураном. Фильтрат упаривали при пониженном давлении. К 4,5 г кубового остатка добавляли 20 см3 изопропанола. Смесь нагревали до 60° С и охлаждали при перемешивании. Через 24 часа выпавшие кристаллы отфильтровывали, промывали диизопропиловым эфиром и сушили под лампой инфракрасного излучения.
Таким образом было получено 1,3 г (52,1%) названного соединения в виде белых кристаллов с температурой плавления 93-95° С.
Пример Сравнения №1
(Воспроизведение Примера 5 патентной публикации DE-OS No. 2815926)
Смесь 60,4 г 4-оксиранилметокси-9Н-карбазола, 64,8 г N-/2-(2-метоксифенокси)этил/бензиламина и 200 мл диметилового эфира этиленгликоля кипятили в течение 24 часов. При охлаждении смеси продукт не выпал в осадок. Смесь упаривали при пониженном давлении, кубовый остаток не могли выделить в виде кристаллов из этанола, изопропанола и ацетона, следовательно, его перенесли на колонку, заполненную силикагелем. При хроматографии на колонке в качестве растворителя использовали 1,2-дихлорметан; смесь, содержащую 9 объемов 1,2-дихлорметана и 1 объем этилацетата; смесь, содержащую 7 объемов 1,2-дихлорметана и 3 объема этилацетата; этилацетат. Содержащие продукт фракции собирали и упаривали. В результате получали 75 г (60%) 1-(9Н-карбазол-4-илокси)-3-/2-(2-метоксифенокси)этиламин/-2-пропанола с температурой плавления 92° С.
Пример Сравнения №2
Смесь 1,92 г (8 ммоль) 4-оксиранилметокси-9Н-карбазола, 3,08 г (10,5 ммоль) N-/2-(2-метоксифенокси)этил/бензиламина и 20 см3 этилацетата кипятили. После этого продукт выделяли с помощью ВЭЖХ. После 28 часов реакции реакционная смесь содержала только 3% бензилкарведилола, практически весь исходный 4-оксиранилметокси-9Н-карбазол остался непрореагировавшим. Характеристики ВЭЖХ:
Колонка: LiChrospher 100RP-18 (5 um), λ =254 нм,
Ацетонитрил: буферный раствор = 1:1,
Скорость потока: 0,5 см3/мин,
Буферный раствор: 7,74 г ацетата аммония и 10,5 см3 уксусной кислоты на 1000 см3 водного раствора,
Время удерживания:
N-/2-(2-метоксифенокси)этил/бензиламин 2,0 мин
4-оксиранилметокси-9Н-карбазол 4,6 мин
Бензилкарведилол 14,1 мин
Пример Сравнения №3
Повторяли процедуру, описанную в Примере Сравнения №2, которая отличалась тем, что в качестве растворителя использовали диоксан. Через 28 часов реакции в реакционной смеси присутствовало около 50% исходного 4-оксиранилметокси-9Н-карбазола, что было определено с помощью ВЭЖХ.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОГО ПРОДУКТА - КАРВЕДИЛОЛА И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1998 |
|
RU2216539C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-ГИДРОКСИ-9H-КАРБАЗОЛА | 1998 |
|
RU2204551C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКО ОПТИЧЕСКИ ЧИСТОГО КАРВЕДИЛОЛА | 2006 |
|
RU2415130C2 |
| Способ получения производныхКАРбАзОлил (4)-ОКСи-пРОпАНОлАМиНАили иХ СОлЕй (ЕгО ВАРиАНТы) | 1979 |
|
SU810079A3 |
| СПОСОБ ПОЛУЧЕНИЯ РЕНТГЕНОАМОРФНОЙ МОДИФИКАЦИИ КАРВЕДИЛОЛА | 2006 |
|
RU2366653C2 |
| Способ получения производных триазолопиримидина или их смеси, или их фармацевтически приемлемых солей с металлами | 1990 |
|
SU1776261A3 |
| НОВЫЕ ПРОИЗВОДНЫЕ N-(ИМИНОМЕТИЛ)АМИНОВ, ИХ ПОЛУЧЕНИЕ, ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1999 |
|
RU2230742C2 |
| АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1996 |
|
RU2198878C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ И АГОНИСТОВ БЕТА-2-АДРЕНЕРГИЧЕСКИХ РЕЦЕПТОРОВ | 2013 |
|
RU2661877C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
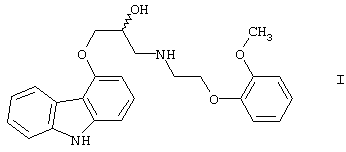
Изобретение относится к новому способу получения 1-(9Н-карбазол-4-илокси)-3-[/2-(2-метоксифенокси)этил/амин]-2-пропанола формулы

и его фармацевтически приемлемым солям присоединения кислот. Способ заключается в том, что вторичный амин формулы V 
реагирует с эпихлоргидрином, полученное хлор-соединение формулы 
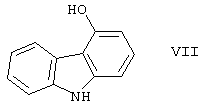
реагирует с 4-гидрокси-9Н-карбазолом формулы
и полученный бензил-карведилол формулы 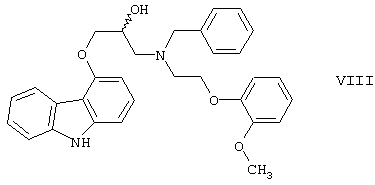
дебензилируют путем каталитического гидрирования; и, при необходимости, полученный продукт подвергают реакции с неорганической или органической кислотой для получения его фармацевтически приемлемой соли присоединения кислоты.
Способ получения 1-(9Н-карбазол-4-илокси)-3-[/2-(2-метоксифенокси)этил/амин]-2-пропанола формулы
и его фармацевтически приемлемых солей присоединения кислот, отличающийся тем, что вторичный амин формулы V
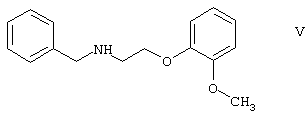
реагирует с эпихлоргидрином, полученное хлор-соединение формулы
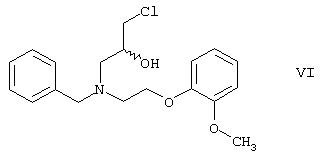
реагирует с 4-гидрокси-9Н-карбазолом формулы

и полученный бензил-карведилол формулы VIII
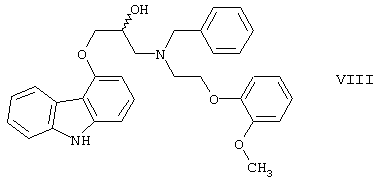
дебензилируют путем каталитического гидрирования и при необходимости полученный продукт подвергают реакции с неорганической или органической кислотой для получения его фармацевтически приемлемой соли присоединения кислоты.
| АНТИТЕЛА ПРОТИВ КЛАУДИНА И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2815926C2 |
| DE 3319027 A1, 29.11.1984 | |||
| Способ получения 1-( @ -карбазолил)-пропандиола-2,3 | 1981 |
|
SU1068429A1 |

