Камптотецин представляет собой природное соединение, которое было впервые выделено из листьев и коры китайского растения, называемого camptotheca acuminata (смотри Wall et collaborateurs, J.Amer.Chem.Soc., 88: 3888 (1966)). Камптотецин представляет собой пентациклическое соединение, образованное индолизино[1,2-b]хинолиновым фрагментом, соединенным с шестизвенным α-гидроксилактоном. Углерод в положении 20, который несет α-гидроксигруппу, является асимметричным и придает молекуле вращающую способность. Природная форма камптотецина имеет абсолютную "S" конфигурацию на углероде в положении 20 и отвечает следующей формуле:
Камптотецин обладает антипролиферативной активностью по отношению к нескольким линиям раковых клеток, включающим в себя линии клеток человеческих опухолей ободочной кишки, легкого и молочной железы (Suffness, M, et collaborateurs: The Alkaloids Chemistry and Pharmacology, Bross, A., ed., Vol.25, p.73 (Academic Press, 1985)). Предполагают, что антипролиферирующая активность камптотецина связана с его ингибирующей активностью по отношению к топоизомеразе I ДНК.
Указывалось, что α-гидроксилактон абсолютно необходим одновременно для активности камптотецина in vivo и in vitro (Camptothecines: New Anticancer Agents, M.Putmesil ef collaborateurs, ed., p.27 (CRC Press, 1995); M.Wall et collaborateurs. Cancer Res., 55: 753 (1995); Hertzberg et collaborateurs, J.Med.Chem., 32: 715 (1982) и Crow et collaborateurs, J.Med.Chem., 35: 4160 (1992)). Позднее заявитель разработал новый класс аналогов камптотецина, в которых β-гидроксилактон замещает природный α-гидроксилактон камптотецина (смотри международную заявку на патент 97/00876).
Предметом изобретения является новый способ получения энантиометрически чистого промежуточного продукта синтеза, а также новых энантиометрически чистых аналогов камптотецина.
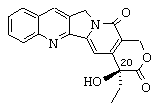
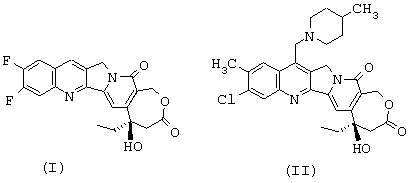
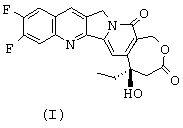
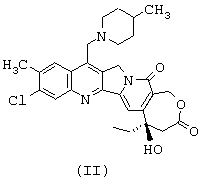
Итак, прежде всего предметом изобретения являются новые аналоги камптотецина, которые отличаются от любого известного соединения, отличающиеся тем, что они имеют соответствующие формулы (I) и (II), представленные ниже,
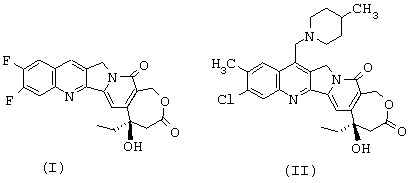
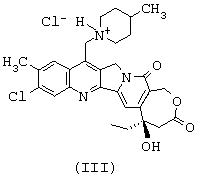
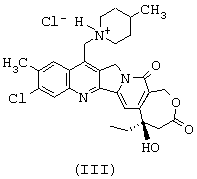
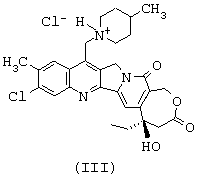
или тем, что он представляет собой одну из солей соединения формулы (II), такую, например, как соль формулы (III), представленной ниже
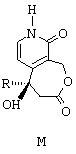
Ключевым промежуточным продуктом в синтезе оптически чистых соединений этого типа является продукт общей формулы М, представленной ниже,
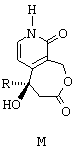
в которой R обозначает линейный или разветвленный алкильный радикал, насчитывающий от 1 до 10 атомов углерода. Предпочтительно, R представляет собой этильный радикал.
Соединения формул (I) и (II) могут быть получены следующим образом:
- соединение формулы
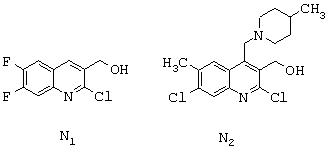
связывают с одним или с другим из соединений формул N1 или N2, представленных ниже:
с получением соответственно соединения формулы О1 или соединения формулы O2:
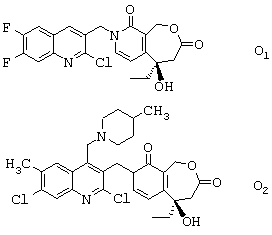
- затем соединение O1 циклизуют и получают соединение формулы (I); циклизация соединения О2 дает соединение формулы (II), которое после солеобразования может давать соединение формулы (III).
Образование соединений O1 или О2 из соединения общей формулы М, в которой R обозначает этильный радикал и N1 и N2, осуществляется при помощи обработки, известной специалистам под названием реакция Мицунобу (смотри O.Mitsunobu et coll., Synthesis, p.1 (1981)). Речь идет о замещении гидроксильной группы соединения N нуклеофилом, таким как соединение М, или депротонированным производным этого последнего, путем обработки фосфином, например трифенилфосфином, и азодикарбоксилатным производным, например диэтилазодикарбоксилатом или диизопропилазодикарбоксилатом, в апротонном растворителе, таком как, например, тетрагидрофуран или N,N-диметилформамид. Циклизация соединений O1 и О2 с образованием соединений формул (I) и (II) осуществляется, предпочтительно, в присутствии палладийсодержащего катализатора (например, диацетата палладия) в щелочных условиях (создаваемых, например, ацетатом щелочного металла, в случае необходимости в сочетании с веществом, улучшающим межфазный перенос, таким как, например, тетрабутиламмонийбромид) в апротонном растворителе, таком как ацетонитрил или N,N-диметилформамид, при температуре, составляющей от 50°С до 120°С (R.Grigg et coll., Tetrahedron 46, page 4003 (1990)).
Изобретение предлагает также в качестве нового промышленного продукта соединение общей формулы М, такое как определенное перед этим. Этот продукт может быть использован для изготовления лекарственных средств.
Соединение формулы М синтезируют согласно новому способу, составляющему часть изобретения и состоящему из следующих последовательных стадий:
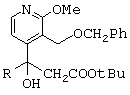
- рацемический сложный трет-бутиловый эфир, представленный ниже,
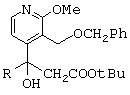
(по поводу его получения см., в частности, международную заявку на патент 97/00876) обрабатывают при комнатной температуре в течение 18 часов трифторуксусной кислотой, в результате чего получают соответствующую карбоновую кислоту;
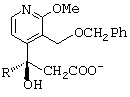
- затем соль хинидина кислоты, полученной перед этим, нагревают в изопропиловом спирте до температуры выше 30 (с, предпочтительно приблизительно 50 (с, после чего дают реакционной среде охладиться до комнатной температуры таким образом, что соль одного из оптических изомеров вышеупомянутой кислоты кристаллизуется, в то время как соль другого оптического изомера, анион которого представлен ниже, остается в растворе;
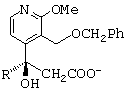
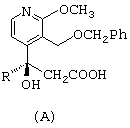
- раствор незакристаллизовавшейся соли энантиомера в изопропиловом спирте концентрируют, обрабатывают соляной кислотой и перемешивают, получая соединение общей формулы А; представленной ниже;
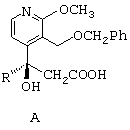
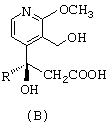
- затем соединение общей формулы А приводят в контакт с палладием на влажном угле, потом к смеси добавляют формиат аммония или муравьиную кислоту, в результате чего получают дебензилированный продукт общей формулы В, представленной ниже;
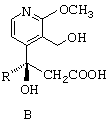
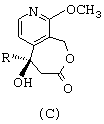
- затем циклизуют соединение общей формулы В путем воздействия дициклогексилкарбодиимидом с получением лактонного соединения общей формулы С, представленной ниже;
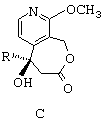
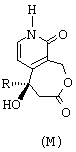
- наконец, путем воздействия иодидом натрия и триметилсилилхлоридом превращают группу -ОСН3 лактонного соединения общей формулы С в карбонильную группу с получением соединения общей формулы М, представленной ниже.
В способе, описанном выше, реакцию, приводящую от соединения общей формулы А к соединению общей формулы В, осуществляют предпочтительно в метаноле и, предпочтительно нагревая реакционную среду после добавления формиата аммония, приблизительно до 40°С. Циклизация соединения общей формулы В с получением соединения С может осуществляться в ТГФ, предпочтительно при температуре приблизительно 50 (с, тогда как реакцию, приводящую от соединения общей формулы С к соединению общей формулы М, осуществляют предпочтительно при комнатной температуре, используя в качестве растворителя ацетонитрил.
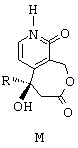
В особом случае, когда R представляет собой этил, соединение формулы М синтезируют согласно способу, состоящему из следующих последовательных стадий:
- рацемический сложный трет-бутиловый эфир, представленный ниже,
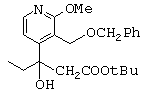
(по поводу его получения см., в частности, международную заявку на патент 97/00876) обрабатывают при комнатной температуре в течение 18 часов трифторуксусной кислотой, в результате чего получают соответствующую карбоновую кислоту;
- затем соль хинидина 3-(3-бензилоксиметил-2-метокси-4-пиридил)-3-гидроксипентановой кислоты нагревают в изопропиловом спирте до температуры выше 30 (с, предпочтительно приблизительно до 50°С, после чего дают реакционной среде охладиться до комнатной температуры таким образом, что соль оптического изомера (+)-3-(3-бензилоксиметил-2-метокси-4-пиридил)-3-гидроксипентановой кислоты кристаллизуется, в то время как соль изомера (-), анион которого представлен ниже, остается в растворе;
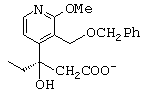
- раствор соли энантиомера (-)-3-(3-бензилоксиметил-2-метокси-4-пиридил)-3-гидроксипентановой кислоты в изопропиловом спирте концентрируют, обрабатывают соляной кислотой и перемешивают, получая соединение формулы А', представленной ниже;
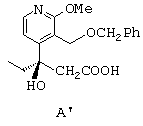
- затем соединение А' приводят в контакт с палладием на влажном угле, потом к смеси добавляют формиат аммония или муравьиную кислоту, в результате чего получают дебензилированный продукт В', представленный ниже;
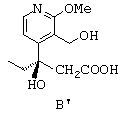
- затем циклизуют соединение формулы В' путем воздействия дициклогексилкарбодиимидом с получением лактонного соединения формулы С', представленной ниже:
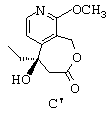
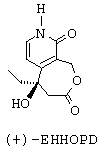
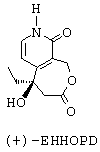
- наконец, путем воздействия иодидом натрия и триметилсилилхлоридом превращают группу -ОСН3 лактонного соединения формулы С' в карбонильную группу с получением (+)-5-этил-5-гидрокси-1,3,4,5,8,9-гексагидрооксепино[3,4-с]пиридин-3,9-диона (или (+)-ЭГГОПД), представленного ниже.
Соединение формулы N1 может быть получено из анилина формулы P1, представленной ниже,
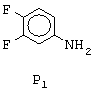
согласно следующему способу. Анилин формулы P1 N-ацетилируют путем обработки ацетилирующим агентом, таким как, например, уксусный ангидрид. Полученный таким образом ацетанилид обрабатывают при температуре, составляющей от 50°С до 100 (с, предпочтительно 75 (с, реагентом, известным специалисту под названием реактив Вильцмейера (полученным действием фосфорилоксихлорида на N,N-диметилформамид при температуре, составляющей от 0°С до 10°С), в результате чего получают соответствующий 2-хлор-3-хинолинкарбальдегид (смотри, например, Meth-Cohn et coll., J.Chem.Soc., Perkin Trans.I, p.1520 (1981); Meth-Cohn et coll., J.Chem.Soc., Perkin Trans.I р.2509 (1981); Nakasimhan et coll., J.Am.Chem.Soc., 112, p.4431 (1990)). 2-Хлор-6,7-дифтор-3-хинолинкарбальдегид легко восстанавливается в соответствующий 2-хлор-6,7-дифтор-3-хинолинметанол формулы N1 в обычных условиях, известных специалисту, таких как обработка в спиртовом растворителе (например, метаноле) боргидридом натрия при температуре, составляющей от 0°С до 40°С.
Соединение формулы N2 может быть получено согласно следующему способу: анилин формулы Р2, представленной ниже,
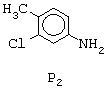
ацилируют в орто-положении реакцией с хлорацетонитрилом в присутствии трихлорида бора и другой кислоты Льюиса, такой как трихлорид алюминия, тетрахлорид титана или диэтилалюминийхлорид, в апротонном растворителе или в смеси апротонных растворителей с последующим гидролизом (смотри, например, Т. Sugasawa et coll., J.Am.Chem.Soc., 100, р.4842 (1978)). Полученный таким образом промежуточный продукт затем обрабатывают этилмалонилхлоридом в апротонном растворителе, таком как ацетонитрил, в присутствии основания, такого как триэтиламин, затем обрабатывают алкоголятом щелочного металла, например метилатом натрия, в метаноле, в результате чего получают этил-7-хлор-4-хлорметил-6-метил-2-оксо-1,2- дигидро-3-хинолинкарбоксилат. Этот последний превращают в этил-2,7-дихлор-4-хлорметил-6-метил-3-хинолинкарбоксилат путем обработки фосфорилоксихлоридом. Затем, путем обработки 4-метилпиперидином осуществляют нуклеофильное замещение. Функциональную группу этилкарбоксилата затем восстанавливают диизобутилалюминийгидридом в апротонном растворителе, таком как дихлорметан, в результате чего получают соединение формулы N2. В случае необходимости можно менять порядок проведения двух последних стадий.
Аналоги промежуточных соединений типа N1 или N2 были описаны в литературе, в частности в заявке РСТ 95/05427.
Соединение формулы (II) может быть превращено в фармацевтически приемлемую соль согласно обычным способам. Приемлемые соли включают, в качестве примера, не носящего ограничительного характера, соли присоединения с неорганическими кислотами, такие как гидрохлорид, сульфат, фосфат, дифосфат, гидробромид и нитрат, или органическими кислотами, такие как ацетат, малеат, фумарат, тартрат, сукцинат, цитрат, лактат, метансульфонат, п-толуолсульфонат, памоат, салицилат, оксалат и стеарат. По поводу других примеров фармацевтически приемлемых солей можно сослаться на "Pharmaceutical Salts", J.Pharm.Sci., 66: 1 (1977).
Соединения по изобретению обладают интересными фармакологическими свойствами. Так например, соединения по изобретению обладают ингибирующей активностью по отношению к топоизомеразе I и/или II и противоопухолевой активностью. Уровень техники дает основание полагать, что соединения согласно изобретению обладают противопаразитарной и/или противовирусной активностью. Соединения согласно настоящему изобретению могут также быть использованы в других терапевтических применениях.
Ниже, в экспериментальной части, найдет место иллюстрация фармакологических свойств соединений согласно изобретению.
Соединения могут ингибировать топоизомеразу, например типа I и/или II, у пациента, например млекопитающего, такого как человек, при введении этому пациенту терапевтически эффективного количества соединения формулы (I) или формулы (II), или фармацевтически приемлемой соли соединения формулы (II), или любой смеси этих последних.
Соединения согласно изобретению обладают противоопухолевой активностью. Они могут быть использованы для лечения опухолей, например опухолей, выделяющих топоизомеразу, у пациента путем введения вышеупомянутому пациенту терапевтически эффективного количества соединения формулы (I) или формулы (II), или фармацевтически приемлемой соли соединения формулы (II), или любой смеси этих последних. Примеры опухолей или раковых опухолей содержат раковые опухоли пищевода, желудка, кишечника, прямой кишки, полости рта, глотки, гортани, легкого, ободочной кишки, молочной железы, шейки матки, тела эндометрия, яичников, предстательной железы, яичек, мочевого пузыря, почек, печени, поджелудочной железы, костей, соединительных тканей, кожи, глаз, головного мозга и центральной нервной системы, а также рак щитовидной железы, лейкемия, боллезнь Ходжкина, лимфомы, отличные от лимфом Ходжкина, множественные миеломы и другие.
Равным образом они могут быть использованы для лечения паразитарных инфекций путем ингибирования жгутиковых, паразитирующих в крови (например, при трипаносомозе или инфекциях, возбуждаемых лейшманией), или путем ингибирования малярийного плазмодия (как например, при малярии), но также для лечения вирусных инфекций или заболеваний.
Эти свойства делают продукты формул (I) и (II) пригодными для фармацевтического применения. Предметом настоящей заявки являются также, в качестве лекарственных средств, продукты формул (I) и (II), такие как определенные выше, или фармацевтически приемлемые соли присоединения продукта формулы (II) с минеральными или органическими кислотами, такая как, например, соль формулы (III), описанная перед этим, или любая смесь этих последних. Изобретение касается также фармацевтических композиций, содержащих в качестве действующего начала по меньшей мере одно из лекарственных средств, таких, как определенные выше.
Изобретение касается также фармацевтических композиций, содержащих соединение согласно изобретению, или его соль присоединения с фармацевтически приемлемой кислотой, в сочетании с фармацевтически приемлемым носителем, сообразно выбранному способу введения (например, перорально, внутривенно, внутрибрюшинно, внутримышечно, чрескожно или подкожно). Фармацевтическая композиция (например, терапевтическая) может быть в форме твердого вещества, жидкости, липосом или липидных мицелл.
Фармацевтическая композиция может быть в форме твердого вещества, например порошков, пилюль, гранул, таблеток, липосом, желатиновых капсул или суппозиториев. Пилюля, таблетка или желатиновая капсула могут быть покрыты веществом, способным защитить композицию от воздействия желудочного сока или ферментов в желудке пациента в течение периода времени, достаточного для того, чтобы позволить этой композиции пройти непереваренной в тонкую кишку. Соединение может быть также введено локально, например, в само место нахождения опухоли. Соединение может быть также введено согласно способу пролонгированного высвобождения (например, композиция с пролонгированным высвобождением или насос для вливания). Подходящими твердыми носителями могут быть, например, фосфат кальция, стеарат магния, карбонат магния, тальк, сахара, лактоза, декстрин, крахмал, желатин, целлюлоза, метилцеллюлоза, Na-карбоксиметилцеллюлоза, поливинилпирролидин и воск. Фармацевтические композиции, содержащие соединение согласно изобретению, могут также находиться в форме жидкости, как например растворов, эмульсий, суспензий или составов с пролонгированным высвобождением. Подходящими жидкими носителями могут быть, например, вода, органические растворители, такие как глицерин или гликоли, такие как полиэтиленгликоль, а также их смеси в различных пропорциях в воде.
Равным образом, предметом изобретения является применение продуктов формул (I) и (II), таких как определенные выше, или фармацевтически приемлемых солей присоединения продукта формулы (II) с минеральными или органическими кислотами, таких, как например соль формулы (III), описанная перед этим, или смеси этих последних, для получения лекарственных средств, предназначенных для ингибирования топоизомераз, и, более конкретно, топоизомераз типа I и II, лекарственных средств, предназначенных для лечения опухолей, лекарственных средств, предназначенных для лечения паразитарных инфекций, а также лекарственных средств, предназначенных для лечения вирусных инфекций или заболеваний.
Доза соединения согласно настоящему изобретению, предусматриваемая для лечения вышеупомянутых заболеваний или расстройств, зависит от способа введения, возраста и массы тела пациента, а также от его состояния, и, в конце концов, она будет определена лечащим врачом или ветеринаром. Такое количество, определенное лечащим врачом или ветеринаром, называется здесь "терапевтически эффективное количество".
Если только не будет определено иначе, все используемые здесь технические и научные термины имеют то же самое значение, что и значение, обычно понимаемое рядовым специалистом в области, к которой относится изобретение. Также, все публикации, заявки на патенты, все патенты и все другие упоминавшиеся здесь ссылки включены через ссылку.
Следующие примеры приведены для иллюстрации вышеупомянутых процедур и ни в коем случае не должны рассматриваться в качестве ограничивающих объем патентной охраны изобретения.
Экспериментальная часть
ПРИМЕР 1: (+)-5-Этил-5-гидрокси-1,3,4,5,8,9-гексагидрооксепино[3,4-с]пиридин-3,9-дион[(+)-ЭГГОПД]
1.а. Соль хинидина 3-(3-бензилоксиметил-2-метокси-4-пиридил)-3-гидроксипентановой кислоты
Трет-бутил-3-(3-бензилоксиметил-2-метокси-4-пиридил)-3-гидроксипентаноат (40 г, 100 ммоль) обрабатывают трифторуксусной кислотой (150 мл) и реакционную среду перемешивают в течение 18 часов при 20°С. После выпаривания трифторуксусной кислоты вливают метиленхлорид (200 мл) и вводят насыщенный раствор бикарбоната натрия до получения рН=7,5-8. После декантации водную фазу промывают 100 мл метиленхлорида. Величину рН водной фазы доводят затем до 1 путем добавления раствора 6н соляной кислоты. Затем продукт экстрагируют из водной фазы метиленхлоридом (2 раза по 200 мл). Раствор сушат над сульфатом магния и концентрируют. Полученную таким образом 3-(3-бензилоксиметил-2-метокси-4-пиридил)-3-гидроксипентановую кислоту (31,1 г, 90 ммоль), взятую в изопропиловый спирт (30 мл), обрабатывают раствором хинидина (29,2 г, 90 ммоль) в изопропиловом спирте (30 мл) при 50°С при перемешивании до полного растворения. После этого позволяют температуре опуститься до 40 (с, перемешивание прекращают и позволяют температуре снизиться до 20°С. Среду доводят до 0°С без перемешивания, затем выдерживают при этой температуре в течение 16 ч. Затем позволяют температуре подняться до 20°С и перемешивают до кристаллизации. Среду разбавляют изопропиловым спиртом, затем фильтруют. Осадок промывают изопропиловым спиртом. Соль энантомера (+) осаждается (m=26,6 г), тогда как соль оптического изомера (-) остается в растворе в изопропиловом спирте. Собирают таким образом фильтрат, который концентрируют с получением масла (34 г), которое без дополнительной очистки вводят в следующую стадию.
Продукты анализируют методом ВЭЖХ на колонке с CHIRAL AGP 5 мкм (10 см × 4 мм), элюируемой смесью изопропиловый спирт/вода/фосфатный буфер рН=6,5: 30/920/50; с расходом 1,2 мл/мин; УФ-детектирование на 280 нм. Полученное время удерживания составляет 6,4 мин для оптического изомера (-) и 2,8 мин для оптического изомера (+). Отношение оптический изомер (-)/оптический изомер (+) равно 83/17.
1.б. (-)-3-(3-Бензилоксиметил-2-метокси-4-пиридил)-3-гидроксипентановая кислота
Раствор соли хинидина энантиомера (-)-3-(3-бензилоксиметил-2-метокси-4-пиридил)-3-гидроксипентановой кислоты (стадия 1.а) концентрируют. Концентрат извлекают в 270 мл метиленхлорида и 270 мл раствора 1н соляной кислоты. Реакционную среду перемешивают в течение 16 ч при 20°С. После декантации органическую фазу концентрируют, концентрат забирают метанолом для введения в следующую стадию.
Получают 13,5 г продукта (выход 87%) и отношение энантиомер (-)/энантиомер(+) равное 85/15.
Время удерживания при ВЭЖХ (тот же самый протокол, что в пункте 1.а) составляет:
- энантиомер (-): 6,4 мин;
- энантиомер (+): 2,8 мин.
1.в. (+)-5-Этил-5-гидрокси-1,3,4,5,8,9-гексагидрооксепино[3,4-с]пиридин-3,9-дион:
(-)-3-(3-Бензилоксиметил-2-метокси-4-пиридил)-3-гидроксипентановую кислоту (13,5 г, 39 ммоль; стадия 1.б) растворяют в 87 мл метанола. Этот раствор под азотом выливают на палладированный уголь с влажностью 50%, содержащий 10% палладия (27,7 г, 13 ммоль). Реакционную среду перемешивают в течение 5 мин, затем приливают раствор формиата аммония (11,5 г, 183 ммоль) в 135 мл метанола. Реакционную среду перемешивают в течение 30 мин, позволяя температуре эволюционировать, затем ее нагревают при 40°С в течение 30 мин. Тогда среду фильтруют через слой кларсила и концентрируют. Приливают 40 мл толуола, который выпаривают; эту операцию повторяют для того, чтобы удалить метанол. Осадок, полученный таким образом, извлекают в 45 мл ТГФ. Затем приливают раствор дициклогексилкарбодиимида (7,180 г, 34,5 ммоль) в 20 мл ТГФ. Реакционную среду греют при 50°С в течение 1 ч. Смесь возвращают к 20 (с, затем отфильтровывают дициклогексилкарбамид. Фильтрат концентрируют досуха. Осадок растворяют в 46 мл ацетонитрила, добавляют к нему 6,0 г (40,5 ммоль) иодида натрия, затем 5,13 мл (40,5 ммоль) триметилсилилхлорида. Реакционную смесь выдерживают при перемешивании при комнатной температуре в течение 5 ч. Тогда вводят 28 мл ацетонитрила и 5,6 мл воды. Полученный осадок фильтруют, затем забирают в 1 мл воды и доводят рН до 7,5 путем добавления раствора гидроксида аммония. Полученное твердое вещество отфильтровывают и сушат. Получают m=4,2 г конечного продукта с выходом 34% и отношением энантиомер (+)/энантиомер (-), равным 88,4/11,6.
Анализ методом ВЭЖХ осуществляют на колонке с Chiracel OD 25 см × 4,6 мм, используемыми элюентами являются гептан 600 и этанол 400, расход составляет 1 мл/мин, регистрация на длине волны 210 нм. Полученное время удерживания составляет:
- энантиомер (-): 7,1 минуты;
- энантиомер (+): 9 минут.
Продукт забирают в ацетон (40 мл), затем добавляют воду (150 мл). Дают выпасть осадку и получают 3 г продукта с отношением энантиомер (+)/энантиомер (-), равным 99,4/0,6.
1Н ЯМР (250 МГц, ДМСО D6): 0,8 (т, 3Н, СН3-СН2); 1,65 (м, 2Н, СН2-СН3); 3,00-3,35 (к, 1Н+1Н,-СН2С=O); 5,3 (к, 2Н, СН2-O); 5,7 (с, -ОН); 6,35 (д, 1Н ароматический): 7,3 (д, 1Н ароматический): 11,7 (с, N-H).
ПРИМЕР 2: (+)-5-Этил-9,10-дифтор-5-гидрокси-4,5,13,15-тетрагидро-1Н,3Н-оксепино[3',4':6,7]индолизино[1,2-b]хинолин-3,15-дион
2.a. N-(3,4-Дифторфенил)ацетамид:
Смесь 3,4-дифторанилина (50 мл, 0,5 моль) и триэтиламина (70 мл, 0,5 моль) в дихлорметане (1,5 л) охлаждают при помощи ледяной бани. По каплям добавляют уксусный ангидрид (71,5 мл, 0,75 моль) и затем реакционную смесь перемешивают в течение 1 ч при комнатной температуре. Полученную смесь затем последовательно промывают водой, 10%-ным раствором бикарбоната натрия и насыщенным рассолом. Органическую фракцию сушат над сульфатом натрия и концентрируют при пониженном давлении. Остаток суспендируют в пентане, фильтруют и сушат при пониженном давлении с получением титульного продукта (78 г, выход 91%) в форме твердого белесого вещества (Тпл 126-127,5°С).
1Н ЯМР (ДМСО): 2,15 (с, 3Н); 7,10-7,65 (м, 2Н); 7,65-8,10 (м, 1Н); 10,30 (широкий пик, 1Н).
2.б. 2-Хлор-6,7-дифтор-3-хинолин-3-карбальдегид
Используют обычную процедуру, описанную Meth-Cohn et al., J.Chem.Soc., Perkin Trans.I, (1981), 1520 и 2509. К реактиву Вильцмейера, полученному добавлением по каплям в атмосфере аргона оксихлорида фосфора (103 мл, 1,1 моль) к безводному ДМФ (34 мл, 440 ммоль), охлажденному на ледяной бане и перемешанному в течение получаса, после чего позволяют температуре снова подняться до комнатной температуры, добавляют продукт стадии 2.а (32 г, 220 ммоль). Смесь, полученную таким образом, перемешивают при 70°С в течение 16 ч. После возвращения к комнатной температуре реакционную смесь по каплям добавляют к смеси воды со льдом (400 мл) и перемешивают в течение 2 ч. Полученный осадок отфильтровывают и промывают водой, затем сушат с получением титульного продукта (9 г, выход 18%) в форме желтого твердого вещества (Тпл 226,5-229°С).
1Н ЯМР (ДМСО): 8,17 (дд, 1Н); 8,39 (дд, 1Н); 8,97 (д, 1Н); 10,34 (д, 1Н).
ИК-спектр (КВr): 888, 1061, 1262, 1507, 1691 см-1.
2.в. 2-Хлор-6,7-дифтор-3-хинолилметанол
Суспензию продукта стадии 2.б (9 г, 39 ммоль) в метаноле (400 мл) обрабатывают боргидридом натрия (2 г, 53 ммоль) при комнатной температуре в течение получаса. Избыток боргидрида разлагают уксусной кислотой (2 мл). Летучие вещества удаляют при пониженном давлении. Остаток растворяют в этилацетате (500 мл), полученную смесь затем последовательно промывают разбавленным раствором бикарбоната натрия, водой и насыщенным рассолом. Затем сушат над сульфатом магния и концентрируют при пониженном давлении. Остаток перекристаллизовывают из 1,2-дихлорэтана с получением титульного продукта (8 г, выход 80%) в форме бежевого твердого вещества (Тпл 166,5-167°С).
1Н ЯМР (ДМСО): 4,67 (д, 2Н); 5,80 (т, 1Н); 8,01 (дд, 1Н); 8,22 (дд, 1Н); 8,48 (с, 1Н).
ИК-спектр (КВr): 871, 1038, 1253, 1513 см-1.
2.г. (+)-8-(2-Хлор-6,7-дифтор-3-хинолинметанол)-5-этил-5-гидрокси-1,3,4,5,8,9-гексагидрооксепино[3,4-с]-пиридин-3,9-дион
К раствору (+)-ЭГГОПД (1,58 г, 7,08 ммоль; стадия 1.в), продукта стадии 2.в (1,62 г, 7,06 ммоль) и трибутилфосфина (1,91 мл, 7,87 ммоль) в безводном ДМФ (30 мл) при комнатной температуре в атмосфере аргона по каплям добавляют диэтилазодикарбоксилат (1,24 мл, 7,87 ммоль). Смесь, полученную таким образом, затем перемешивают в течение 16 ч. Затем реакционную среду выпаривают досуха при пониженном давлении. Остаток очищают хроматографией на колонке с диоксидом кремния (элюент: этилацетат). Полученное твердое вещество забирают в диэтиловый эфир, фильтруют и сушат, в результате чего получают титульный продукт (1,56 г, выход 51%) в форме белесого твердого вещества (Тпл 196°С).
1Н ЯМР (ДМСО): 0,84 (т, 3Н); 1,74 (м, 2Н); 3,02 (д, 1Н); 3,34 (д, 1Н): 5,29 (с, 2Н); 5,31 (дд, 2Н); 5,75 (с, 1Н); 6,51 (д, 1Н); 7,80 (д, 1Н); 8,03 (дд, 1Н); 8,07 (с, 1Н); 8,17 (дд, 1Н).
ИК-спектр (КВr): 875, 1057, 1360, 1507, 1574, 1647, 1749 см-1.
2.д. (+)-5-этил-9,10-дифтор-5-гидрокси-4,5,13,15-тетрагидро-1H,3Н-оксепино[3',4':6,7]индолизино[1,2-b]-хинолин-3,15-дион
Смесь продукта стадии 2.г (1,53 т, 3,52 ммоль, стадия 2.г), тетрабутиламмонийбромида (1,25 г, 3,87 ммоль), ацетата калия (520 мг, 5,28 ммоль), трифенилфосфина (180 мг, 0,70 ммоль) и ацетата палладия (II) (79 мг, 0,35 ммоль) перемешивают в атмосфере аргона в безводном ацетонитриле, нагреваемом с обратным холодильником в течение 22 ч. После возвращения реакционной среды к комнатной температуре ее концентрируют при пониженном давлении, после чего проводят хроматографию на колонке с диоксидом кремния (элюент: смесь CH2Cl2/MeOH 98/2). Получают титульный продукт (960 мг, выход 68%; степень чистоты, определенная методом ВЭЖХ: 97,1%). Этот продукт забирают в безводный CH2Cl2 (100 мл) и перемешивают 24 ч, затем фильтруют и сушат, в результате чего получают очищенный титульный продукт (850 мг, выход 61%; степень чистоты, определенная методом ВЭЖХ: 99,6%) в форме белого твердого вещества.
1Н ЯМР (ДМСО): 0,87 (т, 3Н); 1,85 (м, 2Н); 3,08 (д, 1Н); 3,44 (д, 1Н); 5,26 (с, 2Н); 5,39 (д, 2Н); 5,52 (д, 2Н); 5,99 (широкий пик, 1Н); 7, 39 (с, 1Н); 8,15 (дд, 1Н); 8,23 (дд, 1Н); 8,68 (с, 1Н).
ИК-спектр (KBr): 871, 1261, 1512, 1579, 1654, 1746 см-1.
ПРИМЕР 3: (+)-1-[9-хлор-5-этил-5-гидрокси-10-метил-3,15-диоксо-4,5,13,15-тетрагидро-1Н,3Н-оксепино[3',4':6,7] индолизино[1,2-b]хинолин-12-илметил]-4-метил-гексагидропиридинийхлорид
3.а. 1-(2-амино-4-хлор-5-метилфенил)-2-хлорэтанон
3-Хлор-4-метиланилин (44,4 мл, 0,366 моль) в 1,2-дихлорэтане (440 мл) в атмосфере аргона охлаждают на ледяной бане. К этой смеси по порядку по каплям добавляют трихлорид бора (1М в гептане, 400 мл, 0,4 моль), хлорацетонитрил (28 мл, 0,44 моль) и диэтилалюминийхлорид (1М в гептане, 400 мл, 0,4 моль). В процессе добавления температуру поддерживают ниже 20°С. Полученную смесь затем нагревают с обратным холодильником в течение 3 ч, затем охлаждают до 10°С. Тогда с осторожностью приступают к гидролизу реакционной среды при помощи 2н соляной кислоты (240 мл) и нагревают ее с обратным холодильником в течение 1 ч. Добавляют воду (1л) и этилацетат (1 л), перемешивают полученную смесь в течение 15 мин перед разделением фаз. Водную фазу вновь экстрагируют этилацетатом (200 мл), а объединенные органические фазы промывают водой (500 мл). Органическую фазу сушат над сульфатом магния и концентрируют. Остаток забирают в петролейный эфир (фракция с температурой кипения от 45 до 60 (с, 150 мл) и смеси, полученной таким образом, дают отстояться в течение 16 ч при 4°С. Образовавшийся осадок выделяют путем фильтрования, промывают петролейным эфиром и сушат при пониженном давлении с получением титульного продукта (25 г, выход 31%). Тпл=129-130°С.
1Н ЯМР (ДМСО): 2,20 (с, 3Н); 4,98 (с, 2Н); 6,90 (с, 1Н); 7,15 (широкий пик, 2Н); 7,70 (с, 1Н).
ИК-спектр (КВr): 871, 1018, 1183, 1225, 1270, 1533, 1577, 1619, 1662 см-1.
3.б. Этил-7-хлор-4-хлорметил-6-метил-2-оксо-1,2-дигидро-3-хинолинкарбоксилат
Продукт стадии 3.а (25 г, 0,11 моль) и триэтиламин (30,6 мл, 0,22 моль) смешивают в ацетонитриле (520 мл). При комнатной температуре в атмосфере аргона добавляют этилмалонилхлорид (28,1 мл, 0,22 моль). Полученную смесь перемешивают в течение 3 ч. Затем по каплям добавляют этилат натрия (полученный растворением 3 г или 0,13 моль натрия в 140 мл абсолютного этанола) и полученную смесь перемешивают при комнатной температуре в течение 16 ч. Осадок выделяют путем фильтрования и последовательно промывают этанолом, водой, этанолом и простым эфиром. Затем его сушат при пониженном давлении при 70°С над пятиокисью фосфора, в результате чего получают титульный продукт (28,6 г, выход 83%) в форме белесого порошка.
1Н ЯМР (ДМСО): 1,30 (т, 3Н); 2,40 (с, 3Н); 4,35(к, 2Н); 4,85 (с, 2Н); 7,41 (с, 1Н); 7,91 (с, 1Н); 12,15 (широкий пик, 1Н).
ИК-спектр (КВr): 879, 1108, 1250, 1288, 1483, 1664, 1721 см-1.
3.в. Этил-2,7-дихлор-4-хлорметил-6-метил-3-хинолинкарбоксилат
Продукт стадии 3.б (28,4 г, 90 ммоль) в течение 4 ч нагревают с обратным холодильником в оксихлориде фосфора (400 мл). Полученную смесь концентрируют при пониженном давлении (20 мм рт.ст.) при 80°С. Остаток забирают в диизопропиловый эфир (400 мл). Образовавшийся осадок выделяют путем фильтрования, промывают простым эфиром и петролейным эфиром, затем сушат при пониженном давлении, в результате чего получают титульный продукт (25,4 г, выход 85%) в форме белесого порошка (Тпл=126-127°С).
1Н ЯМР (ДМСО): 1,37 (т, 3Н); 2,58 (с, 3Н); 4,49 (к, 2Н); 5,14 (с, 2Н); 8,16 (с, 1Н); 8,35 (с, 1Н).
ИК-спектр (КВr): 874, 1006, 1163, 1243, 1278, 1577, 1723 см-1.
3.г. 2,7-Дихлор-4-хлорметил-6-метил-3-хинолилметанол
Продукт стадии 3.в (25,2 г, 76,5 ммоль) смешивают в атмосфере аргона с дихлорэтаном (630 мл). По каплям добавляют диизобутилалюминийгидрид (1М в дихлорметане, 307 мл, 307 ммоль), в то время как реакционную смесь перемешивают и поддерживают температуру ниже 20°С. Затем реакционную смесь в течение 3 ч перемешивают при комнатной температуре, затем вливают в водный раствор тартрата калия (концентрация 20 мас.%, 1,5 л). Эмульсию, полученную таким образом, тщательно перемешивают в течение 1 ч, фильтруют через целит и тогда две фазы оказываются разделенными. Водную фазу экстрагируют (200 мл), а объединенные органические фазы промывают водным раствором хлорида натрия (концентрация 20 мас.%, 500 мл). Полученную органическую фазу сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Остаток забирают в диэтиловый эфир (50 мл) и образовавшийся осадок выделяют путем фильтрования. Сушкой при пониженном давлении получают титульный продукт (18,3 г, выход 93%) в форме белесого порошка (Тпл=169-170°С).
1Н ЯМР (ДМСО): 2,57 (т, 3Н); 4,84 (с, 2Н); 5,36 (с, 2Н); 8,06 (с, 1Н); 8,27 (с, 1Н).
ИК-спектр (КВr): 870, 1022, 1102, 1304, 1482, 1567 см-1.
3.д. 2,7-Дихлор-6-метил-4-(4-метилпиперидинометил)-3-хинолилметанол
Раствор продукта стадии 3.г (16,2 г, 55,7 ммоль) в ТГФ (70 мл) обрабатывают раствором 4-метилпиперидина (23 мл, 195 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 2 ч. Добавляют воду (200 мл) и дихлорметан (200 мл). Органическую фазу промывают водным раствором хлорида натрия (концентрация 20 мас.%, 100 мл), сушат над сульфатом магния и концентрируют при пониженном давлении. Кристаллизацией остатка в диэтиловом эфире получают титульный продукт (18,3 г, выход 93%) в форме кристаллического белого твердого вещества (Tпл=170-171,5°С).
1Н ЯМР (СDСl3): 0,88 (д, 3Н); 1,17 (м, 2Н); 1,42 (м, 1Н); 1,60 (м, 2Н); 2,19 (т, 2Н); 2,56 (с, 3Н); 2,82 (д, 2Н); 4,02 (с, 2Н); 4,93 (с, 2Н); 6,36(широкий пик, 1Н); 1, 95 (с, 1Н); 8,02 (с, 1Н).
ИК-спектр (KBr): 971, 1013, 1105, 1293, 1479, 1559 см-1.
3.е. (+)-8-[2-7-Дихлор-6-метил-4-(4-метилпиперидинометил)-3-хинолилметил]-5-этил-5-гидрокси-1,3,4,5,8,9-гексагидрооксепино [3,4-с]пиридин-3,9-дион
Суспензию (+)-ЭГГОПД (получен на стадии 1.в, 1,56 г, 7,0 ммоль) в безводном диоксане (70 мл) в атмосфере аргона последовательно обрабатывают продуктом стадии 3.д (2,47 г, 7,0 ммоль), трифенилфосфином (2,02 г, 7,7 ммоль) и диизопропилазодикарбоксилатом (1,07 мл, 10,5 ммоль). Смесь перемешивают при комнатной температуре в течение 16 ч. Летучие вещества затем выпаривают при пониженном давлении. Остаток очищают хроматографией на колонке с диоксидом кремния (элюент: этилацетат). Полученное твердое вещество забирают в диэтиловый эфир, фильтруют и сушат с получением титульного продукта (1,96 г, выход 50%) в форме белесого твердого вещества (Тпл=182°С).
1Н ЯМР (ДМСО): 0,89 (м, 8Н); 1,23 (м, 1Н); 1,41 (т, 2Н); 1,64 (м, 2Н); 2,09(к, 2Н); 2,59 (м, 5Н); 3,15 (дд, 2Н), 4,06 (дд, 2Н); 5,31 (дд, 2Н); 5,35 (дд, 2Н); 5,75 (с, 1Н); 6,29 (д, 1Н); 7,17 (д, 1Н); 8,06 (с, 1Н); 8,46 (с, 1Н):
ИК-спектр (KBr): 878, 1053, 1275, 1474, 1572, 1648, 1747 см-1.
3.ж. (+)-9-Хлор-5-этил-5-гидрокси-10-метил-12-((4-метилпиперидинометил)-4,5,13,15-тетрагидро-1Н,3Н-оксепино[3',4':6,7]-индолизино[1,2-с]хинолин-3,15-дион
Смесь продукта стадии 3.е (3,80 г, 6,80 ммоль), тетрабутиламмонийбромида (2,42 г, 7,5 ммоль), ацетата калия (1,00 г, 10,2 ммоль), трифенилфосфина (890 мг, 3,4 ммоль) и ацетата палладия (II) (220 мг, 0,68 ммоль) перемешивают в атмосфере аргона в безводном ацетонитриле (85 мг) при нагревании с обратным холодильником в течение 24 ч. После охлаждения до комнатной температуры образовавшийся осадок выделяют путем фильтрования и последовательно промывают ацетонитрилом, водой, ацетоном и диэтиловым эфиром с получением после сушки при пониженном давлении титульного продукта (2,5 г, выход 70%) в форме белесого порошка.
1Н ЯМР (ДМСО): 0,86 (м, 6Н); 1,12(к, 2Н); 1,36 (м, 1Н); 1,56 (д, 2Н); 1,84(к, 2Н); 2,12 (т, 2Н); 2,56 (с, 3Н), 2,83 (дд, 2Н), 3,26 (дд, 2Н); 4,03 (дд, 2Н); 5,28 (дд, 2Н); 5,45 (дд, 2Н); 6,04 (с, 1Н), 7,34 (с, 1Н); 8,14 (с, 1Н); 8,38 (с, 1Н).
ИК-спектр (КВr): 870, 1058, 1208, 1280, 1477, 1593, 1655, 1749 см-1.
3.з. (+)-1[(5R)-9-хлор-5-этил-5-гидрокси-10-метил-3,15-диоксо-4,5,13,15-тетрагидро-1Н,3Н-оксепино[3',4':6,7]индолизино[1,2-с]хинолин-12-илметил]-4-метил-гексагидропиридинийхлорид
Смесь продукта стадии 3.ж (2,3 г, 7,7 ммоль) и абсолютного этанола (300 мл) подвергают в течение 2 мин ультразвуковой обработке. Полученную молочную суспензию перемешивают и обрабатывают соляной кислотой (1 н. раствор, 13,2 мл, 13,2 ммоль) с получением прозрачного желтого раствора, который в состоянии покоя образовывает осадок типа геля. Осадок выделяют путем фильтрования с использованием воронки Бюхнера и последовательно промывают этанолом и простым эфиром, затем сушат при пониженном давлении, в результате чего получают титульный продукт (2,1 г, выход 85%).
1Н ЯМР (ДМСО): 0,87 (м, 6Н); 1,59 (м, 5Н); 1,84(к, 2Н); 2,64 (с, 3Н); 3,28 (дд, 2Н); 3,45 (с, 2Н); 4,93 (с, 2Н); 5,47 (дд, 2Н); 5, 61 (с, 2Н); 6, 04 (широкий пик, 1Н); 7,41 (с, 1Н); 8,28 (с, 1Н); 8,63 (с, 1Н); 10,30(широкий пик, 1Н).
ИК-спектр (КВr): 1043, 1212, 1479, 1585, 1655, 1751 см-1.
Фармакологическое исследование продуктов согласно изобретению
Тест на пролиферацию клеток.
В этом исследовании используют пять линий опухолевых клеток: SW620 (аденокарцинома ободочной кишки человека), OVCAR-5 (аденокарцинома яичника человека), РС-3 и DU-145 (линия клеток предстательной железы человека) и NCI-H69 (аденокарцинома легкого человека). Эти линии происходят из NCI/Frederick Cancer Research and Development Center (Frederick, MD). Их культивируют в полной среде, содержащей среду RMPI-1640, обогащенную 10% зародышевой телячьей сыворотки и 2 мМ L-глутамина. Их инкубируют при 37°С во влажной атмосфере, содержащей 5% СО2. Прилипающие клетки отслаивают путем обработки раствором, содержащим 0,25% трипсина и 0,2% ЭДТА (Worthington Biochemical Corp., Freehold, NJ), в течение 5 мин при 37°С. Подсчет клеток осуществляют при помощи счетчика Coulter Z1 (Coulter Corp., Hialeah, FL). Жизнеспособность оценивают путем окрашивания клеток пропидийиодидом и последующего их пересчета при помощи проточного цитометра EPICS Elite (Coulter).
Тестируемое соединение примеров 2 и 3 в количестве 5 мМ растворяют в N,N-диметилацетамиде (ДМА, Aldrich). Последующие разведения осуществляют при помощи культуральной среды. Тестируемые конечные молярные концентрации составляют: 1·10-6, 2·10-7, 4·10-8, 8·10-9, 1,6·10-9, 3,2·10-10, 6,4·10-11, 1,28·10-11, 2,56·10-12 и 5,12·10-13. Каждую концентрацию тестируют в восьми лунках. На всех клеточных линиях проведены контрольные эксперименты по влиянию ДМА. Из этих контрольных экспериментов следует, что при максимальной используемой концентрации (0,02%) ДМА не оказывает воздействия. В качестве положительного контрольного вещества используют доксорубицин в концентрациях 1·10-7 М и 2·10-7 М.
Клетки высевают по 5·103 клеток на лунку в 96-лунковую микроплашку (Costar Corporation, Cambridge, MA). Клетки инкубируют 24 ч при 37°С с целью обеспечения возобновления размножения клеток. Затем, в указанных выше концентрациях, добавляют тестируемое соединение примеров 2 и 3 и инкубируют клетки при 37°С во влажной атмосфере, содержащей 5% СО2, в течение 3 дней для прилипающих клеток (SW620, OVCAR-5, РС-3 и DU 145) и в течение 5 дней для суспендированных клеток (NCI-H69).
Прилипающие клетки тестируют методом SRB (описанным в работе L.V. Rubenstein, R.H. Shoemaker, K.D. Paull, R.M. Simon, S. Tosini, P. Skehan, D.A. Scudiero, A. Monks and M.R. Boyd "Comparison of in vitro anticancer-drug-screening data generated with tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines", J.Nat. Cancer Inst., 82: 1113-1118, 1990). После трех дней инкубирования удаляют супернатант и добавляют 200 мкл RMPI-1640, не содержащей зародышевую телячью сыворотку. Клетки фиксируют путем добавления 50 мкл 50%-ной трифторуксусной кислоты (конечная концентрация трифторуксусной кислоты равна 10%) и инкубируют при 4°С в течение 1 ч. Лунки 5 раз промывают водой, затем окрашивают 50 мкл 0,4%-ного раствора сульфородамина В (СРВ, Sigma) в 1%-ной уксусной кислоте при комнатной температуре в течение 10 мин. Красящий раствор солюбилизируют 100 мкл буферного раствора Трис с концентрацией 10 мМ, рН 10, в течение приблизительно 5 мин при перемешивании, считывание микроплашек осуществляют спектрофотометрически при длине волны 570 нм.
Суспендированные клетки тестируют методом ХТТ (описанным в работе D.A. Scudiero, R.H Shoemaker, K.D. Paull, A. Monks, S. Tierney, Т.Н. Nofziger, M.J. Currens, D. Seniff et M.R. Boyd: "Evaluation of a soluble tetrazolium/forroazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines'", Cancer Research, 48: 4827-4833, 1988). После инкубации в присутствии тестируемого соединения примеров 2 и 3 к культурам добавляют ХТТ [натриевая соль 2,3-бис(2-метокси-4-нитро-5-сульфофенил)-2Н-тетразолий-5-карбоксанилида (Sigma)] и феназинметосульфат (ФМС, Sigma) в растворе в солевом фосфатном буфере и клетки инкубируют в течение 4 ч при 37°С в атмосфере, содержащей 5% СО2. Конечные концентрации ХТТ и ФМС составляют 50 и 0,38 мкг/лунка соответственно. Продуцирование формазана останавливают путем добавления 10 мкл 10%-ного раствора додецилсульфата натрия (Sigma) и поглощение считывают спектрофотометрически при 450 нм с эталонным фильтром на 600-650 нм.
Результаты
Молярные концентрации соединений примеров 2 и 3, ингибирующие пролиферацию клеток на 50%, компилированы в следующей таблице.

Примеры композиций
Жидкая композиция 10 мг соединения (I, II или III) по изобретению: 3,3 г диметиламина, qsq 200 мг физиологического раствора.
Твердая композиция (таблетки): 1 мг соединения (I, II или III) по изобретению: 75 мг порошкообразной лактозы, 20 мг микрокристаллической целлюлозы, 3 мг безводной коллоидной двуокиси кремния, 1 мг стеарата.
Изобретение относится к соединениям, представленным формулами (I), (II), (III)
Предложено соединение формулы М, в качестве промежуточного продукта,
где R обозначает этильный радикал. Предложен способ получения продукта формулы М. Предложено лекарственное средство, обладающее противоопухолевой активностью, которое содержит соединения формулы I, II, III или их соли с фармацевтически приемлемыми минеральными или органическими солями. Также предложена фармацевтическая композиция, обладающая противоопухолевой активностью, содержащая действующее начало и фармацевтически приемлемый носитель. В качестве действующего начала фармацевтическая композиция содержит соединения формулы I, II, III. Технический результат - аналоги камптотецина, обладающие антипролиферативной активностью по отношению к раковым клетками. 5 н. и 4 з.п.ф-лы, 1 табл.
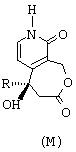
где R обозначает этильный радикал.
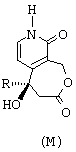
в которой R обозначает этильный радикал,
отличающийся тем, что он состоит из следующих последовательных стадий: рацемический сложный трет-бутиловый эфир формулы
обрабатывают при комнатной температуре в течение 18 ч трифторуксусной кислотой, в результате чего получают соответствующую карбоновую кислоту; затем соль хинидина кислоты, полученной перед этим, нагревают в изопропиловом спирте до температуры выше 30°С, предпочтительно приблизительно 50°С, после чего дают реакционной среде охладиться до комнатной температуры таким образом, что соль одного из энантиомеров указанной кислоты кристаллизуется, в то время как соль другого энантиомера, анион которого представлен формулой
остается в растворе; раствор незакристаллизовавшейся соли энантиомера в изопропиловом спирте концентрируют, обрабатывают соляной кислотой и перемешивают, получая соединение общей формулы (А)
затем соединение общей формулы А приводят в контакт с палладием на влажном угле, потом к смеси добавляют формиат аммония или муравьиную кислоту, в результате чего получают дебензилированный продукт общей формулы (В)
затем циклизуют соединение общей формулы (В) путем воздействия дициклогексилкарбодиимидом с получением лактонного соединения общей формулы (С)
наконец, путем воздействия иодидом натрия и триметилсилилхлоридом превращают группу -ОСН3 лактонного соединения общей формулы (С) в карбонильную группу с получением соединения общей формулы (М)
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |

