Предметом настоящего изобретения является способ получения простых азоиминоэфиров (в форме гидрохлоридов) и их гидролиза с образованием эфиров азокарбоновых кислот, которые, будучи полученными таким способом, могут быть использованы в качестве инициаторов свободных радикалов в реакциях полимеризации. Предметом настоящего изобретения являются также полученные таким образом смешанные простые азоиминоэфиры и образуемые из них смешанные эфиры азокарбоновых кислот.
Получение эфиров азокарбоновых кислот традиционно производят с помощью двухстадийного способа, первой стадией которого является превращение азонитрила взаимодействием со спиртом в присутствии НСl по реакции Пиннера с образованием гидрохлорида соответствующего простого азоиминоэфира и второй стадией является гидролиз полученного таким образом гидрохлорида.
Названный способ обладает рядом недостатков, которые делают его совершенно непригодным для производства в промышленном масштабе. Действительно, способ является слишком дорогостоящим и трудно регулируемым, а очистка конечного продукта затруднительна и требует большого количества спирта. Кроме того, выход продукта в этом способе недостаточен, а чистота продукта неудовлетворительна. Такой способ описан, например, G.A.Mortimer в Journal de chimie organique, стр. 1632-33 (1965) для получения диметилазо-бис-изобутирата, используемого в качестве полупродукта в синтезе инициатора полимеризации - ди-ацетата азобисизобутанола.
Для разрешения некоторых из названных проблем и создания возможности реализации способа получения эфиров азокарбоновых кислот в промышленном масштабе было предложено несколько решений.
Описанный ниже способ, приводящий к повышению выхода и одновременно к значительному уменьшению времени реакции, а также к значительно лучшему и значительно более быстрому отделению фазы эфира азодиизомасляной кислоты от водной фазы, описан в DE 2254572. Согласно этому патенту усовершенствования такого рода могут быть получены в результате проведения превращения Пиннера в присутствии водорастворимого циклического простого эфира, и/или водорастворимого низкомолекулярного диола, и/или простого эфирдиола, или линейного поли(простого эфирдиола) с низкой молекулярной массой (до 1800) в количестве от 0,001 до 11,0% мас. в алифатический спирт C1-С6.
В патенте ЕР 80275 сообщается, что 2,2'-азо-бис(2-метилпропионитрил), а также родственные ему соединения могут быть превращены с отличным выходом при использовании реакции Пиннера с не превышающим стехиометрическое количеством спирта, если проводить реакцию в присутствии соединения, содержащего простую эфирную группу. Описывается, что более быстрое превращение (но не более высокие выход и/или селективность) может быть достигнуто при повышении в реакционной смеси концентрации НСl. Однако присутствие простого эфира выдвигает новые проблемы, в частности проблемы разделения и последующей обработки.
В ЕР 230586 показывается, что получение простых азоиминоэфиров может быть осуществлено в одну стадию в одной емкости с использованием очень легко регулируемой реакции, в процессе которой протекают окислительное галогенирование гидразонитрила и иминоэтерификация азонитрила. Способ получения по ЕР 230586 включает таким образом взаимодействие гидразонитрила с хлором в присутствии спирта, способного превратить цианогруппу в простую иминоэфирную группу в присутствии НСl (образующегося in situ). Это взаимодействие проводят в неводной системе в присутствии растворителя, используемого для реакций окислительного галогенирования и иминоэтерификации, такого как ароматические углеводороды, галогенсодержащие углеводородные и некоторые другие растворители. Количество используемого спирта варьирует от теоретически необходимого количества до количества, превышающего теоретически необходимое в 1,2 раза, а количество хлора варьирует от теоретического количества до его небольшого избытка. Конечный продукт реакции, представляющий собой гидрохлорид простого азоиминоэфира, может быть превращен в сложный азоэфир путем гидролиза.
Однако ни в одном из приведенных выше документов не предложен способ, который мог бы быть использован в промышленном масштабе с повышенным выходом и чистотой продукта, достаточной для того, чтобы освободиться от громоздкого процесса разделения.
Заявитель неожиданным образом разработал способ получения солей азоиминоэфиров и эфиров азокарбоновых кислот, который достигает поставленных выше целей. Таким образом, настоящее изобретение относится к способу получения солей азоиминоэфиров превращением азонитрила по реакции Пиннера, в которой взаимодействие осуществляют в ароматическом растворителе в присутствии большого избытка НСl.
Предметом настоящего изобретения является таким образом способ получения гидрохлоридов простых азоиминоэфиров, включающий взаимодействие азонитрилов со спиртами и хлористоводородной кислотой в ароматическом растворителе при молярном отношении R=HCl/азонитрил, большем 2, если спиртом является метанол, и большем 3, если спиртом является этанол или высший спирт.
Согласно одному из вариантов осуществления изобретения азонитрил образуется in situ при взаимодействии соответствующего гидразонитрила с хлором.
Согласно другому варианту осуществления изобретения растворитель выбирают из группы, в которую входят толуол, хлорбензол, ксилол и бензол, из которых в том случае, когда азонитрил образуется in situ, преимущественно используют хлорбензол.
Согласно еще одном варианту осуществления изобретения спиртом является этанол.
Согласно еще одном варианту осуществления изобретения используемый спирт представляет собой смесь спиртов, в частности смесь, содержащую метанол или метанол и этанол.
Согласно одному из вариантов осуществления способа получения по изобретению гидрохлорид простого азоиминоэфира соответствует формуле II:
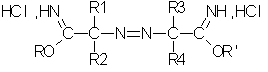
в которой:
R1, R2, R3 и R4, одинаковые или разные, независимо выбирают из группы, в которую входят следующие заместители: нормальные или разветвленные C1-С9-алкилы (преимущественно C1-C4), незамещенные или замещенные одним или несколькими заместителями, выбранными из гидроксила, C1-С6-алкокси, галогена;
С3-С6-циклоалкилы, незамещенные или замещенные одним или несколькими заместителями, выбранными из C1-С6-алкила, C1-С6-алкокси, гидрокси, галогена;
С7-С12-арилалкилы, незамещенные или замещенные одним или несколькими заместителями, выбранными из C1-С6-алкила, C1-С6-алкокси, гидрокси, галогена;
С7-С12-арилы, незамещенные или замещенные одним или несколькими заместителями, выбранными из C1-С6-алкила, C1-С6-алкокси, гидрокси, галогена;
причем по крайней мере одна из комбинаций R1-R2 и R3-R4 может образовывать алифатический цикл;
R и R', одинаковые или разные, независимо выбирают из группы, в которую входят нормальные или разветвленные алифатические C1-С10-радикалы (преимущественно C1-C4).
Согласно другому варианту осуществления изобретения R и R' в формуле II отличны один от другого и выбираются из нормальных алифатических С1-С4-радикалов.
Согласно особому варианту осуществления изобретения R1, R2, R3 и R4 являются С1-С4-алкильными группами.
Предметом изобретения является также способ получения сложного эфира азокарбоновой кислоты, включающий синтез гидрохлорида простого азоиминоэфира описанным выше способом и гидролиз полученного таким образом гидрохлорида простого азоиминоэфира в присутствии воды.
Согласно одному из вариантов осуществления изобретения гидролиз осуществляют путем постепенного добавления воды к реакционной смеси или приливанием реакционной смеси к воде при температуре от 15 до 50°С, преимущественно при приблизительно 30°С.
Согласно особому варианту осуществления способа получения эфира азокарбоновой кислоты после стадии синтеза гидрохлорид простого азоиминоэфира отфильтровывают, промывают органическим растворителем, после чего осуществляют гидролиз постепенным прибавлением отфильтрованного осадка к воде при температуре от 15 до 50°С, преимущественно от 25 до 35°С.
Предметом изобретения является также способ получения жидкой смеси сложных эфиров азокарбоновых кислот, включающий синтез гидрохлоридов простых азоиминоэфиров описанным выше способом, гидролиз полученных выше солей в присутствии воды и выделение органической фазы, содержащей сложные эфиры.
Согласно особому варианту осуществления этого способа получения, в первой стадии реакции вводят во взаимодействие более тяжелый спирт и затем, во второй стадии реакции, вводят менее тяжелый спирт.
Изобретение распространяется также на жидкую смесь сложных эфиров азокарбоновых кислот, которая может быть получена описанным выше способом, и, в частности, на смесь, которая является жидкой при температуре от -5 до +20°С.
Согласно одному из вариантов осуществления изобретения жидкая смесь содержит первый симметричный сложный эфир первого спирта, второй симметричный сложный эфир второго спирта и смешанный сложный эфир этих первого и второго спиртов. Согласно этому варианту осуществления названный выше первый симметричный сложный эфир является метиловым симметричным сложным эфиром, названный второй симметричный сложный эфир является этиловым симметричным сложным эфиром, а названный смешанный сложный эфир является метилэтиловым сложным эфиром.
Предметом изобретения является также способ получения инициаторов полимеризации, включающий синтез сложного эфира азокарбоновой кислоты с использованием описанного выше способа получения сложных эфиров азокарбоновых кислот и, в случае необходимости, превращение полученного сложного эфира в инициаторы обычными методами.
Согласно другому аспекту изобретения предметом настоящего изобретения являются соли смешанных простых азоиминоэфиров, соответствующие формуле II':
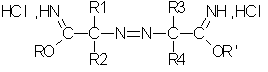
в которой:
R1, R2, R3 и R4, одинаковые или разные, независимо выбирают из группы, в которую входят следующие заместители: нормальные или разветвленные C1-С9-алкилы (преимущественно C1-C4), незамещенные или замещенные одним или несколькими заместителями, выбранными из гидроксила, C1-С6-алкокси, галогена;
С3-С6-циклоалкилы, незамещенные или замещенные одним или несколькими заместителями, выбранными из C1-С6-алкила, C1-C6-алкокси, гидрокси, галогена;
С7-С12-арилалкилы, незамещенные или замещенные одним или несколькими заместителями, выбранными из C1-C6-алкила, C1-C6-алкокси, гидрокси, галогена;
С7-С12-арилы, незамещенные или замещенные одним или несколькими заместителями, выбранными из C1-C6-алкила, C1-C6-алкокси, гидрокси, галогена;
причем по крайней мере одна из комбинаций R1-R2 и R3-R4 может образовывать алифатический цикл;
R и R' отличны один от другого и независимо выбираются из группы, в которую входят нормальные или разветвленные алифатические C1-С10-радикалы, преимущественно С1-С4-радикалы. Изобретение преимущественно касается солей азоиминоэфиров, в которых R обозначает метил и R' обозначает этил и в которых R1, R2, R3 и R4 преимущественно являются С1-С4-алкильными группами.
Предметом настоящего изобретения также являются эфиры азокарбоновых кислот, полученные из солей смешанных простых азоиминоэфиров, определенных выше.
Предметом настоящего изобретения является также способ получения азогуанильных производных, включающий синтез соответствующих гидрохлоридов азоиминоэфиров описанным выше способом и реакцию последних с аммиаком или амином в присутствии спирта с использованием любого подходящего для этой цели способа.
Более детально изобретение описано в следующем ниже описании.
В способе получения гидрохлоридов азоиминоэфиров по изобретению исходный азонитрил, используемый в реакции превращения Пиннера, может быть симметричным или асимметричным. В качестве примера таких азонитрилов можно назвать азонитрилы, отвечающие формуле I:
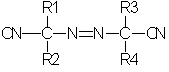
в которой:
R1, R2, R3 и R4, одинаковые или разные, независимо выбирают из группы, в которую входят следующие заместители: нормальные или разветвленные C1-C9-алкилы (преимущественно C1-C4), незамещенные или замещенные одним или несколькими заместителями, выбранными из гидроксила, C1-С6-алкокси, галогена;
С3-С6-циклоалкилы, незамещенные или замещенные одним или несколькими заместителями, выбранными из C1-C6-алкила, C1-C6-алкокси, гидрокси, галогена;
С7-С12-арилалкилы, незамещенные или замещенные одним или несколькими заместителями, выбранными из C1-С6-алкила, C1-C6-алкокси, гидрокси, галогена;
С7-С12-арилы, незамещенные или замещенные одним или несколькими заместителями, выбранными из C1-С6-алкила, C1-C6-алкокси, гидрокси, галогена;
причем по крайней мере одна из комбинаций R1-R2 и R3-R4 может образовывать алифатический цикл.
Конкретными примерами таких азонитрилов являются: 2,2'-азобисизобутиронитрил, 2,2'-азо-бис(2-метилбутиронитрил), 2,2'-азо-бис(2,4-диметилвалеронитрил) и 1,1'-азо-бис(1-цианоциклогексан).
В том, что касается спирта, используемого для реакции превращения Пиннера, следует уточнить, что используют нормальные или разветвленные алифатические C1-С10-спирты, преимущественно нормальные и преимущественно C1-C4. Особенно предпочтительны этанол и/или метанол. Спирт используют в количестве, равном требуемому стехиометрическому количеству, или в небольшом избытке по отношению к стехиометрическому количеству, т.е. до 1,5-кратного избытка по отношению к теоретическому значению. Под спиртом, в рамках настоящего изобретения, подразумевают также смеси указанных выше спиртов, преимущественно смеси, содержащие по крайней мере метанол. В этом случае получают смеси сложных эфиров. Например, если в качестве реагента используют смесь метанол/этанол, то получают сложные метиловый, этиловый и этилметиловый эфиры. Смесями спиртов могут быть также смеси с более чем двумя спиртами, например смесь метанол/этанол/пропанол.
Хлористоводородную кислоту используют в большом избытке по отношению к требуемому стехиометрическому количеству. Заявитель неожиданным образом установил, что, с одной стороны, избыток НСl должен быть значительным (например, до 3 раз превышающим стехиометрическое значение) и, с другой стороны, количество НСl, которое должно быть добавлено, зависит также от природы спирта, используемого на стадии превращения Пиннера. Молярное отношение R=HCl/азонитрил больше 2 в случае метанола и больше 3 в случае этанола или высшего спирта. В том случае, когда используют смеси спиртов, значение R является средневесовым значением для каждого индивидуального спирта. Например, в случае смеси метанол/этанол 50/50 моль/моль значение R для отношения HCl/азонитрил смеси обычно выполняется условие R≥2+3/2=2,5. Значение R лежит в пределах от 2 до 6 в случае метанола и от 3 до 6, как правило, от 4 до 6, для высших спиртов.
Что касается ароматического растворителя, следует уточнить, что может быть использован любой ароматический растворитель, галогенированный или негалогенированный, который достаточно летуч для того, чтобы он мог быть удален в конце реакции упариванием при сравнительно низкой температуре и пониженном давлении. К числу особо подходящих растворителей относятся хлорбензол, толуол, ксилол и бензол.
Реакцию превращения Пиннера осуществляют, как правило, при температуре от 10 до 40°С, преимущественно от 15 до 25°С, в течение времени, зависящего от природы азонитрила и температуры реакции и имеющего порядок от 8 до 24 час. Реакцию превращения по изобретению, как правило, проводят следующим образом: смешивают растворитель со спиртом и к полученной таким образом смеси добавляют азонитрил. После этого в реакционную смесь вводят известным способом требуемое количество безводной хлористоводородной кислоты, поддерживая температуру в пределах от 10 до 40°С, преимущественно от 15 до 25°С. Способ может быть осуществлен как без давления, так и под давлением.
Согласно особому варианту осуществления изобретения азонитрил приготовляют in situ из соответствующего гидразонитрила взаимодействием с хлором и спиртом, способным превратить цианогруппу в простую иминоэфирную группу в присутствии НСl, как это описано в ЕР 230586. Однако в отличие от того, что указано в этом документе, реакцию следует проводить в присутствии большого избытка НСl, как это определено выше в отношении реакции превращения Пиннера. В этом случае R обозначает (НСl, образующаяся in situ, +добавленная НСl)/гидразонитрил.
Взаимодействие по изобретению может быть проведено с сухим гидразонитрилом, но заявитель установил, что его возможно проводить и с влажным гидразонитрилом. В этом случае вода удаляется при растворении влажного гидразонитрила в реакционном растворителе и отделении водной фазы. Растворенные в растворителе следы воды могут быть удалены азеотропной отгонкой перед добавлением спирта и других реагентов.
Заявитель также установил, что реакция in situ, когда ее проводят в присутствии галогенсодержащего, в частности хлорсодержащего, растворителя, преимущественно хлорбензола, при получении солей простых азоиминоэфиров не дает токсичных хлорсодержащих производных.
Полученные таким образом гидрохлориды азоиминоэфиров соответствуют общей формуле II:
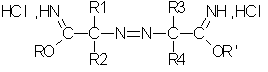
в которой:
R1, R2, R3 и R4 определены выше и
R и R', одинаковые или разные, независимо выбирают из группы, в которую входят нормальные или разветвленные алифитические C1-С10-радикалы (преимущественно C1-C4), причем R и R' преимущественно являются разными.
Особенно предпочтительны соли простых азоиминоэфиров, у которых R1, R2, R3 и R4 и R и R' являются С1-С4-алкилами.
Полученный таким образом гидрохлорид простого азоиминоэфира может быть использован для получения соединений азогуанильного типа взаимодействием газообразного аммиака или первичного или вторичного амина в присутствии спирта с использованием любого подходящего для этой цели известного способа.
Полученный таким образом гидрохлорид простого азоиминоэфира может быть также использован для получения эфира азокарбоновой кислоты формулы III:
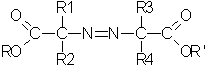
в которой:
R1, R2, R3, R4 и R и R' определены выше применительно к солям простых азоиминоэфиров.
Предметом изобретения также являются эфиры азокарбоновых кислот формулы III':
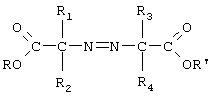
в которой:
R1, R2, R3, R4 и R и R' определены выше применительно к солям смешанных простых азоиминоэфиров (а именно: R и R' отличны один от другого).
При получении сложных эфиров в конце превращения азонитрила (который в конце реакции может быть определен известным методом с помощью ИК-анализа полосы CN) проводят гидролиз соли простого азоиминоэфира без ее выделения или после ее выделения фильтрацией. В конце реакции получают две прозрачные фазы. Их оставляют для расслаивания до полного разделения фаз. Водную фазу удаляют, а оставшуюся органическую фазу, содержащую эфир азокарбоновой кислоты, концентрируют при пониженном давлении, удаляя растворитель. В другом варианте выделение конечного продукта состоит в удалении после гидролиза реакционного растворителя в азеотропе с водой при пониженном давлении с последующим отделением верхней органической фазы.
Для получения конечного продукта удовлетворительной чистоты важно свести к минимуму количество азонитрила, остающегося в конечном продукте, поскольку продукты разложения азонитрилов (в частности в случае 2,2'-азобисизобутиронитрила с образованием тетраметилсукцинонитрила) являются токсичными. В случае выделения гидрохлорида азоиминоэфира фильтрацией с промывкой органическим растворителем (таким, как циклогексан, толуол) происходит очистка, поскольку азонитрил растворим в этих растворителях. После этого может быть проведен гидролиз путем постепенного прибавления фильтрационного осадка к воде при температуре от 15 до 50°С, предпочтительно от 25 до 35°С.
С другой стороны, для того, чтобы избежать стадии фильтрации малоустойчивого продукта, заявитель разработал гидролиз, осуществляемый постепенным прибавлением воды к суспензии гидрохлорида простого азоиминоэфира в реакционном растворителе или прибавлением этой суспензии к воде при регулируемой температуре, например при температуре от 15 до 50°С, предпочтительно от 25 до 35°С.
Настоящее изобретение относится также к способу получения жидкой композиции эфиров (смешанных и/или симметричных) азокарбоновых кислот. Этот способ включает первую стадию синтеза солей простых азоиминоэфиров либо реакцией азонитрила, либо синтезом азонитрила in situ из соответствующего гидразонитрила в присутствии смеси спиртов, как описано выше, затем гидролиз полученных солей и отделение органической фазы, содержащей сложные эфиры, любым подходящим известным способом. Полученные таким образом жидкие композиции обладают тем преимуществом, что являются жидкими при температурах, близких к комнатной, и в некоторых случаях (до -20°С) с ними легко работать, они не пылят, не являются токсичными или цианосодержащими. Для синтеза этих композиций подходят описанные в отношении способа получения эфиров азокарбоновых кислот различные пути осуществления (добавление воды к суспензии гидрохлорида азоиминоэфира, прибавление этой суспензии к воде или фильтрация гидрохлорида азоиминоэфира и добавление отфильтрованного осадка к воде). В качестве исходного вещества возможно использование гидразонитрила вместо азонитрила; в этом случае в качестве реакционного растворителя используют предпочтительно хлорбензол. Способ улучшить образование смешанного сложного эфира состоит во введении в реакцию сначала более тяжелого спирта, а потом - наименее тяжелого. Заявитель также показал, что этот синтез смешанных сложных эфиров также зависит от добавленного избытка НСl при той же исходной смеси спиртов.
Также предметом настоящего изобретения является способ получения инициаторов полимеризации, включающий синтез эфира азокарбоновой кислоты способом, описанным выше, и, в некоторых случаях, к превращению этих эфиров в инициаторы с применением известных способов. Изобретение распространяется также на получение всех соединений, которые могут быть образованы из эфиров азокарбоновых кислот, таких как соответствующие спирты и ацетаты, алканы, кислоты и соответствующие амиды.
Более предпочтительными солями смешанных азоиминоэфиров являются такие, которые получены из смеси спиртов, выбранных их метанола, этанола, н-пропанола и н-бутанола, преимущественно из смеси, содержащей по крайней мере метанол.
Эфиры азокарбоновых кислот, получаемые из этих смешанных простых эфиров, также составляют часть настоящего изобретения.
Следующие далее примеры приведены в качестве иллюстрации и ни в коей мере не ограничивают изобретение.
ПРИМЕРЫ
Пример 1
К смеси, содержащей 600 г толуола и 76,8 г метанола или 110,4 г этанола (2,4 моль), добавляют 164 г (1 моль) 2,2'-азобисизобутиронитрила. Охлаждая смесь до 15-20°C, добавляют в течение 4-5 ч газообразную хлористоводородную кислоту. Производят перемешивание в течение 16 ч при приблизительно 20°С.
Смесь фильтруют и дважды промывают осадок 100 г циклогексана. Осадок гидрохлорида азоиминоэфира медленно прибавляют к 600 г воды при температуре приблизительно 30°С. После перемешивания в течение 1 часа при приблизительно 30°С реакционную смесь охлаждают до приблизительно 10°С. Водную и органическую фазы разделяют декантацией органической фазы и экстрагированием водной фазы 100 г циклогексана. Полученные таким образом органические фазы объединяют и досуха упаривают при пониженном давлении и температуре приблизительно 35°С.
Этот способ осуществлен для разных значений молярного отношения HCl/азонитрил, указанных в приведенной ниже таблице 1. В этой таблице приведены полученные выходы сложных азоэфиров и природа спирта.
Полученные результаты ясно показывают, что отношение HCl/азонитрил сильно влияет на выход реакции и зависит от природы спирта.
Этот результат был подтвержден при воспроизведении примеров синтеза азоиминоэфира, описанных в ЕР 230586, с варьированием природы спирта в приведенном ниже примере 2.
Пример 2
К смеси 640 г толуола и 76,8 г метанола или 110,4 г этанола (2,4 моль) добавляют 166 г (1 моль) 2,2'-гидразобисизобутиронитрила. Охлаждая смесь до 15-20°С, добавляют 74,6 г хлора (1,05 моль, что приводит в результате взаимодействия с 1 моль гидразонитрила к образованию in situ 2 моль НСl). Производят перемешивание в течение 5 час при 25°С и затем 15 час при приблизительно 20°С. Последующие стадии проводят в условиях, идентичных описанным в предыдущем примере. Отношение HCl/азонитрил составляет здесь 2,6, что соответствует сумме 2,0 моль, образованных in situ, и 0,6 моль добавленных.
С метанолом выход сложного бисметилового эфира 2,2'-азобисизомасляной кислоты равен 88%, в то время как с этанолом выход сложного бисэтилового эфира 2,2'-азобисизомасляной кислоты составляет только 25%.
Те же опыты, проведенные исходя из азотистоводородной кислоты, в которых после хлорирования добавляют хлористоводородную кислоту (0,6 моль в случае метанола и 2,4 моль в случае этанола, что приводит к отношениям R соответственно 2,6 и 4,4), позволили достичь выходов сложного азоэфира соответственно 91 и 90%). В этом случае после добавления хлора добавляют при температуре 15-20°С 21,9 г (0,6 моль) хлористоводородной кислоты в течение приблизительно 1 часа в опыте с метанолом и 87,6 г (2,4 моль) хлористоводородной кислоты в течение приблизительно 2 час в опыте с этанолом. После этого производят перемешивание в течение 15 час при 20°С. Последующие стадии идентичны описанным выше.
Пример 3
К 600 г толуола добавляют 200 г (1 моль) влажного 2,2'-гидразобисизобутиронитрила, нагревая смесь приблизительно до 35°С, чтобы обеспечить растворение гидразобисизобутиронитрила. Отделяют нижнюю водную фазу в количестве 31 г. Удаляют растворенную в толуоле воду азеотропной отгонкой в вакууме. Добавляют 110,4 г (2,4 моль) этанола и 0,2 г бромида натрия, используемого в качестве катализатора хлорирования. Охлаждая смесь до приблизительно 20°С, прибавляют в течение 2 час 74,6 г хлора (1,05 моль, что приводит к образованию in situ 2 моль НСl) и затем в течение 3 час 87,6 г (2,4 моль) хлористоводородной кислоты. Отношение R равно приблизительно 4,4. Производят перемешивание реакционной смеси в течение ночи при 20°С. После этого вливают полученную таким образом смесь в 600 г подогретой до 25°С воды, не превышая температуру 30°С. Перемешивают смесь 1 час при 30°С, удаляют толуол азеотропной отгонкой в вакууме при 35°С и отделяют верхнюю органическую фазу. Выход сложного бисэтилового эфира 2,2'-азобисизомасляной кислоты составляет 91%, а содержание азонитрила в конечном сложном бисметиловом эфире 2,2'-азобисизомасляной кислоты составляет 0,3%.
Опыты, проведенные в тех же условиях, но с заменой этанола на метанол с добавлением 0,6 моль (вместо 2,4) хлористоводородной кислоты, дали 0,8% азонитрила в конечном продукте. Без добавления НСl в конечном продукте остается 5% азонитрила.
Пример 4
К 750 г хлорбензола добавляют 200 г (1 моль) влажного 2,2'-гидразобисизобутиронитрила, нагревая смесь приблизительно до 30°C, чтобы обеспечить растворение гидразобисизобутиронитрила. Отделяют верхнюю водную фазу в количестве 30 г. Удаляют растворенную в хлорбензоле воду азеотропной отгонкой в вакууме. Добавляют 76,8 г (2,4 моль) метанола и 0,2 г бромида натрия. Охлаждая смесь до приблизительно 15°С, добавляют 74,6 г хлора (1,05 моль, что приводит к образованию in situ 2,0 моль НСl) и затем 21,9 г (0,6 моль) хлористоводородной кислоты. Реакционную смесь перемешивают 18 час при приблизительно 20°С, вливают в течение 1 часа в 600 г воды и дают температуре достичь приблизительно 30°С, после чего производят охлаждение, чтобы температура не превышала это значение. Перемешивают смесь 1 час при приблизительно 30°С, получая прозрачные водную и органическую фазы. Удаляют хлорбензол азеотропной отгонкой в вакууме при 35°С и отделяют верхнюю органическую фазу. Выход полученного таким образом сложного бисметилового эфира 2,2'-азобисизомасляной кислоты составляет 92%.
Пример 5
К смеси 600 г толуола, 51,2 г (1,6 моль) метанола и 36,8 г (0,8 моль) этанола добавляют 164 г (1 моль) 2,2'-азобисизобутиронитрила. Охлаждая смесь до 15-20°C, добавляют в течение 4-5 час газообразный хлористый водород. Реакционную смесь перемешивают 16 час при 20°С и вливают в течение 1 часа в 600 г воды, давая температуре подняться до приблизительно 30°С. Образовавшуюся смесь перемешивают 1 час при 30°С, получая прозрачные водную и органическую фазы. Удаляют толуол азеотропной отгонкой в вакууме при температуре приблизительно 35°С, после чего отделяют верхнюю органическую фазу.
Получают близкие к 85% выходы продуктов, являющихся прозрачными при комнатной температуре, состав которых различается в зависимости от количества использованной хлористоводородной кислоты и которые застывают при охлаждении до различной температуры.
В - метилэтиловый эфир 2,2'-азобисизомасляной кислоты
С - этиловый эфир 2,2'-азобисизомасляной кислоты
Молярное отношение рассчитывают, деля общее число групп -СООСН3 на общее число групп -COOC2H5, которые присутствуют в конечной смеси.
В порядке сравнения: полученные отдельно смеси продуктов А (твердое вещество с температурой плавления 26-29°С) и С (жидкость до -20°С) не являются жидкостями при комнатной температуре при тех же молярных отношениях -СООСН3/-СООС2Н5, а представляют собой смеси твердое вещество/жидкость и быстро застывают при охлаждении до 15°С.
Варьированием соотношения спиртов варьируют состав смесей. Так, работая как описано выше, но с 4,3 моль хлористоводородной кислоты и со спиртом:
a) метанол 1,2 моль=38,4 г+этанол 1,2 моль=55,2 г;
b) метанол 1,6 моль=51,2 г+этанол 0,8 моль=36,8 г;
c) метанол 2,0 моль=64 г+этанол 0,4 моль=18,4 г,
получают следующие результаты, где А, В и С имеют указанные выше значения.
В порядке сравнения: продукт, полученный смешением А и С при молярном отношении 1,5, дает смесь твердое вещество/жидкость, начиная с приблизительно 15°C, и при молярном отношении 3,6 застывает при приблизительно 15°С.
Пример 6
а) К смеси 600 г толуола, 164 г 2,2'-азобисизобутиронитрила и 36,8 г (0,8 моль) этанола добавляют в течение 2 час, охлаждая смесь до 15-20°С, 64,2 г (1,76 моль) газообразного хлористого водорода. Перемешивают смесь 4 часа при 20°С. Добавляют после этого 51,2 г (1,6 моль) метанола и затем, охлаждая смесь до 15-20°С, добавляют в течение 2 час 30 мин 92,7 г (2,54 моль) газообразного хлористого водорода. Перемешивают реакционную смесь в течение 15 час при приблизительно 15°С. Полученную суспензию вливают в 600 г подогретой до приблизительно 25°С воды, не превышая температуру 30°С, перемешивают приблизительно 1 час при 30°С и удаляют толуол азеотропной отгонкой в вакууме при температуре приблизительно 35°С. После этого отделяют верхнюю органическую фазу.
b) В тех же условиях проводят следующую реакцию: в первой половине реакции добавляют 55,2 г (1,2 моль) этанола и 96,4 г (2,64 моль) газообразного хлористого водорода и затем, во второй половине реакции, 38,4 г (1,2 моль) метанола и 60,6 г (1,66 моль) газообразного хлористого водорода.
Физические характеристики полученных таким образом композиций даны в приведенной ниже таблице 4.
При добавлении в первую очередь более тяжелого спирта отмечается понижение температуры застывания и температуры, при которой образуется гомогенная жидкость (см. для сравнения пример 5 (а) и (b)).
Изобретение относится к получению сложного эфира азокарбоновой кислоты. Способ включает синтез гидрохлорида простого азоиминоэфира путем реакции азонитрила со спиртом и хлористоводородной кислотой в ароматическом растворителе при молярном отношении R=HCl/азонитрил от 2 до 6, если спиртом является метанол, и от 3 до 6, если спиртом является этанол или высший спирт. Затем осуществляют гидролиз полученного гидрохлорида простого азоиминоэфира в присутствии воды. Гидролиз осуществляют путем постепенного добавления воды к реакционной смеси или приливанием реакционной смеси к воде при температуре от 15 до 50°С. Предложен способ получения жидкой смеси сложных эфиров азокарбоновых кислот, включающий синтез гидрохлоридов простых азоиминоэфиров, гидролиз полученных солей в присутствии воды и выделение органической фазы, содержащей эфиры кислот. Предложена жидкая смесь сложных эфиров азокарбоновых кислот. Предложены соли смешанных простых азоиминоэфиров, соответствующие формуле II':
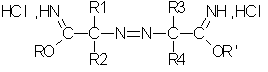
в которой R1, R2, R3 и R4 обозначают С1-С4-алкильные группы; R и R' отличны один от другого и независимо выбираются из группы, в которую входят нормальные или разветвленные алифатические С1-С4-радикалы. Также предложены смешанные эфиры азокарбоновых кислот, отвечающие формуле III':
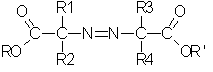
в которой R1, R2, R3, R4 обозначают С1-С4-алкильные группы, а R и R' отличаются друг от друга и независимо выбраны из группы, в которую входят нормальные или разветвленные алифатические С1-С4-радикалы. Технический результат - увеличение выхода и чистоты продукта. 5 н. и 18 з.п. ф-лы, 4 табл.
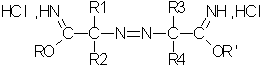
в которой R1, R2, R3 и R4 обозначают С1-С4-алкильные группы;
R и R', одинаковые или разные, независимо выбирают из группы, в которую входят нормальные или разветвленные алифатические C1-C4-радикалы.
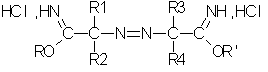
в которой R1, R2, R3 и R4 обозначают С1-С4-алкильные группы;
R и R' отличны один от другого и независимо выбираются из группы, в которую входят нормальные или разветвленные алифатические С1-С4-радикалы.
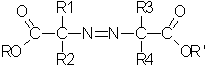
в которой R1, R2, R3, R4 обозначают С1-С4-алкильные группы, а R и R' отличаются друг от друга и независимо выбраны из группы, в которую входят нормальные или разветвленные алифатические С1-С4-радикалы, или имеют значения, определенные в п.21, при условии, что смешанный эфир азокарбоновой кислоты не является метилбутилизобутиратом.
| KELLY, DAVID P | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Устройство для регистрации максимальных значений малых перемещений в применении к измерительным приборам (сейсмометрам, тензометрам, клинометрам и т.п.) | 1948 |
|
SU80275A1 |
| EP 230586 A1, 05.08.1987 | |||
| -Аминоэтиламид азо-бис-изомасляной кислоты в качестве инициатора радикалной полимеризации и способ его получения | 1975 |
|
SU569561A1 |

