Настоящее изобретение относится к нитроксипроизводным гидроксибензойной кислоты в модифицированной физической форме и к фармацевтическим композициям на их основе, причем вышеупомянутые производные имеют общую формулу
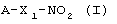
где А означает производное гидроксибензойной кислоты, как определено здесь ниже, X1 означает связывающий бивалентный, радикал как определено здесь ниже, причем вышеупомянутые композиции способны вызывать за очень короткое время, порядка 2-2,5 часов, пик плазматической концентрации производного гидроксибензойной кислоты, обозначенного как А.
Композиции настоящего изобретения могут быть использованы для получения пероральной лекарственной формы, пригодной для быстрого вызывания фармакологического эффекта.
Хорошо известно, что фармакологически активные вещества, когда вводятся перорально, вызывают общий эффект только после прохождения процесса абсорбции через стенки желудочно-кишечного тракта. Процесс абсорбции лекарства представляет собой сложное явление, которое зависит от различных факторов, среди которых жирорастворимссть и водорастворимость. Трудно теоретически предсказать, практически это невозможно знать, какой является оптимальная комбинация этих факторов для получения максимального пика абсорбции активного начала за короткий срок, в основном за 2-2,5 часа.
Как правило, терапевтический эффект лекарства, которое
проявляет свою активность посредством системного пути, когда вводится перорально, зависит, в частности, от следующих факторов:
- абсорбция лекарства через желудочно-кишечную стенку;
- концентрация в крови;
- возможное взаимодействие между целевыми тканями.
В частности, для лекарств, имеющих противовоспалительную и аналгезирующую активность, необходимым фактором является быстрота действия, то есть начало действия должно проявляться в относительно короткий промежуток времени после приема.
Нитропроизводные формулы (I) в немодифицированной физической форме согласно настоящему изобретению известны из патентных заявок WO 95/30641 и WO 97/16405 на имя Заявителя. Эти соединения по отношению к противовоспалительным лекарствам-предшественникам имеют общую сопоставимую или более высокую эффективность, но они имеют преимущество в том, что проявляют сниженные побочные эффекты. Недостаток этих продуктов состоит в том, что они не показывают таких физико-химических свойств, которые обеспечили бы пик максимальной абсорбции в крови за период времени по большей части 2,5 часа. Фармакокинетические исследования, проведенные Заявителем с использованием обычных фармацевтических форм для перорального использования нитропроизводных формулы (I), необработанных, как представлено в настоящем изобретении, показали, что пик концентрации в крови отсутствует в упомянутый короткий промежуток времени, следовательно, этот продукт не проявляет своевременно свои терапевтические свойства. Смотри примеры, показывающие, что кровяной пик имеет место после слишком долгого времени после приема примерно 6 часов.
WO 92/01668 описывает производные бензойной кислоты, имеющие сердечно-сосудистую активность, причем вышеупомянутые соединения содержат группу - ONO2, связанную с остатком бензойной кислоты посредством бивалентного связывающего мостика, который не содержит какого-либо фенильного кольца.
WO 98/57967 описывает итраконазол, проявляющий улучшенную растворимость в связи с уменьшением размера частиц и изменением его кристалличности из кристаллической в аморфную. Для получения упомянутого улучшенного итраконазола соединение растворяют в органическом растворителе и растворенное высушивают для получения порошкообразного итраконазала.
Патент США 5,597,847 описывает нитроксибутильные производные диклофенака.
Патент США 5,621,000 описывает нитроксибутильные производные кетопрофена.
Ощущалась необходимость иметь доступные фармацевтические композиции для перорального использования, содержащие нитропроизводные формулы (I), такие, чтобы давать максимальный пик концентрации в плазме (Сmах) в короткий период времени, такой, чтобы tmax (tmax является тем временем, при котором появляется Сmах) составляло 2,5 часа по большей мере предпочтительно ниже или равное 2 часам.
Заявитель обнаружил, что эту техническую проблему можно решить посредством соединений и их композиций для перорального использования, как показано здесь ниже.
Объектом настоящего изобретения является способ получения фармацевтической композиции, обладающей противовоспалительной и анальгезирующей активностью, для перорального введения, включающей в качестве активного начала вышеупомянутые соединения
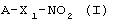
имеющие полностью состояние аморфности или частично состояние аморфности, включающий в себя следующие этапы:
- перемешивание соединения формулы (I), по меньшей мере, с одним наполнителем, способным придать состояние аморфности полученной смеси, выбранном из группы, состоящей из C5-C6 полиспиртов, моно- и дисахаридов и их производных, олигосахаридов, содержащих от 3 до 10 моносахаридных единиц и их производных, полисахаридов, их производных, включая их соли, циклодекстринов и их производных, нециклических производных β-циклодекстрина, полимеров и сополимеров на основе винильных мономерных звеньев, и/или содержащих карбоксильную функцию, или метакрильных мономеров, причем массовое соотношение между количеством соединения формулы (I) и количеством, по меньшей мере, одного наполнителя находится в пределах 1:20 и 1:0,5,
- достижение состояния аморфности полученной смеси совместным измельчением, перемешиванием, распылительной сушкой, лиофилизацией, где в формуле (I)
А=R(COX),
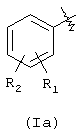
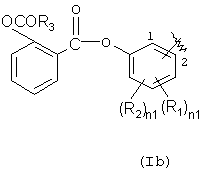
Х=О, NH, NR1c, где R1c является линейным или разветвленным C1-С10 алкилом, R выбран из следующих радикалов:
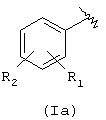
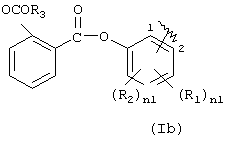
где R1 означает ОСОR3 группу; где R3 означает метил, этил или линейный или разветвленный С3-С5 алкил,
R2 означает водород, гидрокси, галоген, линейный или разветвленный C1-C4 алкил, линейный или разветвленный C1-C4 алкоксил; линейный или разветвленный C1-C4 перфторалкил, моно- или ди-(C1-C4) алкиламино;
R1 и R2 вместе означают диоксиметиленовую группу с тем условием, что когда Х=NH, тогда Y означает этилен и R2=H, как определено здесь ниже;
R1 не может быть ОСОR3 в положении 2, когда R3 означает метил;
n1 означает целое число и представляет собой 0 или 1.
X1 означает бивалентный связывающий мостик, выбранный из следующего:
YO:
Y=линейный или разветвленный, когда возможно C1-C20, алкилен; или замещенный или незамещенный
C5-C7 циклоалкилен, или X1 выбран из следующего:
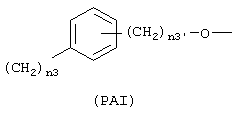
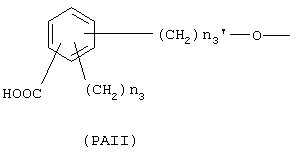
где n3 означает целое число от 0 до 3, n3' означает целое число от 1 до 3;
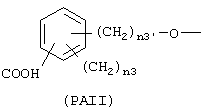
где n3 и n3' имеют следующие вышеупомянутые значения;
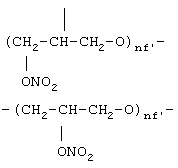
где nf’ означает целое число от 1 до 6,
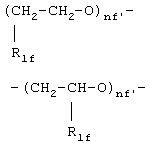
где R1f=H, СН3 и nf' является таким, как определено выше;
с тем условием, что когда R=(Ia), R1 означает ОСОR3 группу, где R3 означает метил или этил и у выбран из группы, состоящей из линейного или разветвленного C1-C20 алкилена, Х не может быть NH или NR1c, с тем условием, что когда R=(Iа), R1 означает ацетокси в орто положении по отношению к -СО-группе и R2 означает водород; Х=0; X1 является ароматическим радикалом формулы (PAI), где n3'=1 и n3=0 и
когда соединение формулы (I) представляет эфир 3-(нитрооксиметил)фенил ацетилсалициловой кислоты.
Степень аморфности может быть измерена хорошо известными методами, такими как, например, дифференциальная сканирующая калориметрия (ДСК), RX, инфракрасная спектроскопия и так далее. Частичная аморфность означает, что в фармацевтических композициях настоящего изобретения соединения формулы (I), как правило, аморфны, по крайней мере, на 5%, предпочтительно на 10% и наиболее предпочтительно, по крайней мере, на 80%, как измерено ДСК.
Степень аморфности определяют ДСК как изменение (уменьшение) площади эндотермического пика плавления активного начала. Когда аморфность является полной, характеристика в виде пика плавления активного начала формулы (I) по существу исчезает. Это означает, что происходит изменение энтальпии, связанной с пиком плавления.
Тест по измерению степени аморфности согласно настоящему изобретению представляет собой следующее: в количество нитропроизводного формулы (I) добавляют гидроксипропил-β-циклодекстрин в молярном соотношении 1:2; 43 г соединения формулы (I) растворяют в 5 л этилового спирта; полученный таким образом раствор смешивают при комнатной температуре с 5 л деионизированной воды, содержащей 7% масс/об (350 г) гидроксипропил-β-циклодекстрина. Этот водно-спиртовой раствор обрабатывают в устройстве для распылительной сушки LabPlant SD-05 Spray-Drying горячим потоком воздуха с температурой 60°С на входе, поддерживая поток воздуха таким образом, чтобы температура на выходе составляла 45°С; потерю кристалличности оценивают на порошке (5-10 мг) путем ДСК метода и изменение площади пика определяют сравнением с площадью пика предшественника, обработанного в тех же условиях без добавления циклодекстрина.
Показательный тест уменьшения кристалличности соединений формулы (I) основан на определении их скорости растворения в воде.
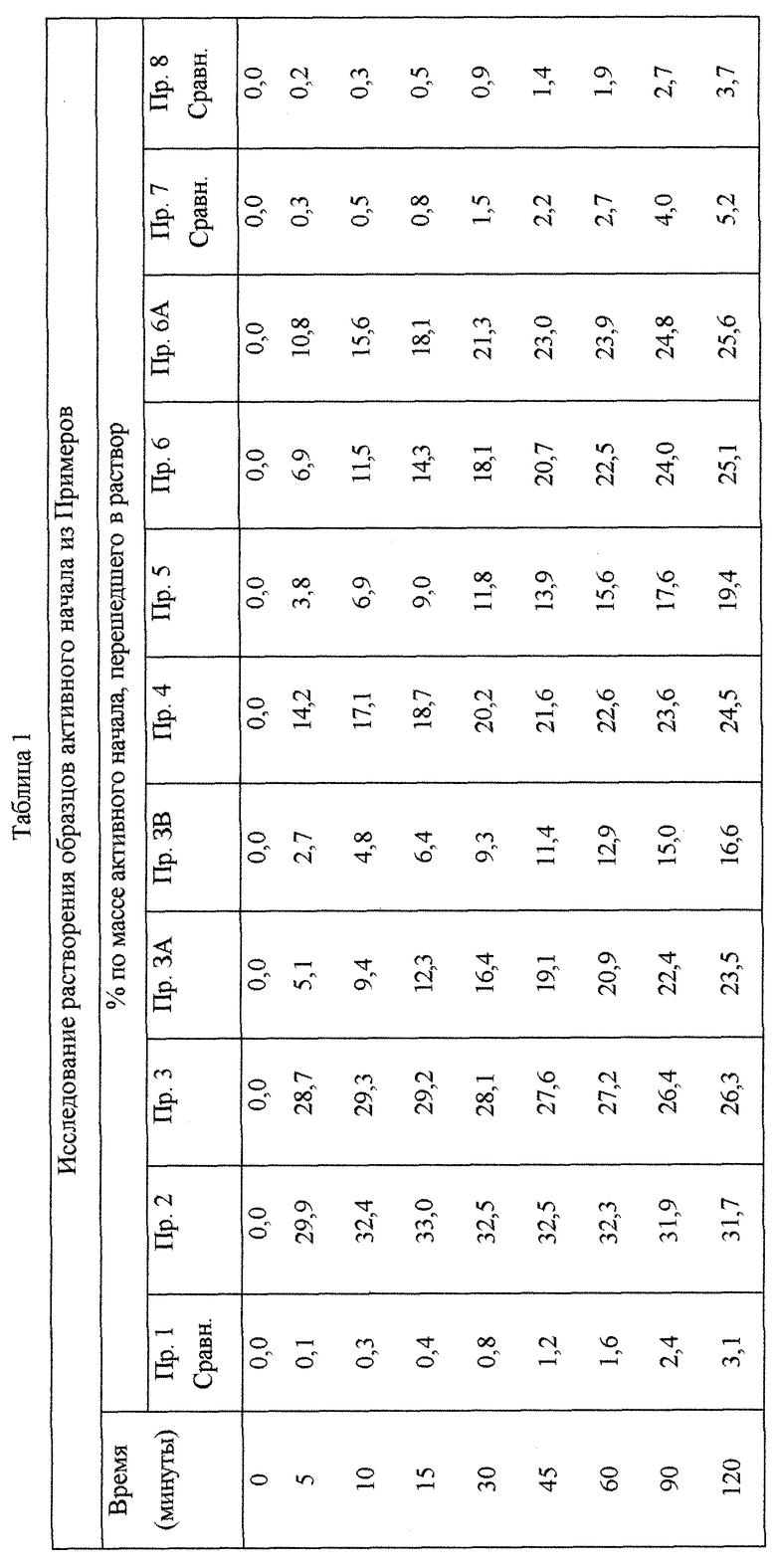
Испытание на скорость растворение проводят в устройстве для растворения согласно United States Pharmakopeia 23 при использовании объема деионизированной воды, равного 1000 мл. Скорость лопастной мешалки составляет 100 об/мин и температура равна 37±0,5°С.
В маленьком стеклянном сосуде взвешивают точное количество каждого образца таким образом, чтобы он содержал количество активного начала, равное 30 мг, которое непосредственно вводят в сосуд, содержащий деионизированную воду. В заранее определенное время, соответственно 5, 10, 15, 30, 45, 50, 60, 90 и 120 минут от начала теста, определяют количество нитропроизводного, перешедшего в раствор, измеряя его концентрацию масс/об посредством ультрафиолетовой спектроскопии при длине волны 235 нм, используя калибровочную кривую. Данные выражены как процент нитропроизводного, перешедшего в раствор в зависимости от времени. Количество соединения формулы (I), перешедшего в раствор за время 10 минут, по крайней мере, в 10 раз выше по отношению к количеству перешедшего в раствор нитропроизводного не в аморфной форме.
Как говорилось, композиции изобретения к удивлению и неожиданно способны вызывать за короткое время, порядка 2-2,5 часов, пик концентрации в плазме производного А гидроксибензойной кислоты.
Заявитель с удивлением и неожиданно обнаружил, что когда нитропроизводные формулы (I) представлены в фармацевтических композициях изобретения в полностью или частично аморфной форме, вследствие желудочно-кишечной абсорбции высокую плазматическую концентрацию получают за очень короткое время, максимальный пик концентрации в плазме получают по большей части за период времени 2,5 часа.
Частично или полностью аморфные нитропроизводные формулы (I) получают обработкой нитропроизводных одним или более наполнителей, способных делать аморфными вышеупомянутые нитропроизводные.
Методики, используемые для перевода в аморфное состояние, представляют собой, например, совместное измельчение, перемешивание, распылительную сушку, лиофилизацию, предпочтительно распылительную сушку и совместное измельчение.
В частности, в методике распылительной сушки активное начало растворено в растворителе, например в спиртах, и таким образом полученный органический раствор смешивают при комнатной температуре с раствором или суспензией наполнителя, способного вызвать аморфное состояние соединений формулы (I). Раствор или суспензию, полученные после смешивания, обрабатывают в устройстве для распылительной сушки. Для этой и других методик смотри специальные Примеры.
Наполнители предпочтительно относятся к одному или более классов, упомянутых здесь позже: C5-C6 полиспирты, моно- и дисахариды и их производные, олигосахариды, содержащие от 3 до 10 моносахаридных остатков и их производные, полисахариды, их производные, включая их соли, циклодекстирины и их производные, нециклические аналоги циклодекстрина, например нециклические производные β-циклодекстрина, полимеры и сополимеры на основе винильных мономерных звеньев, и/или содержащих карбоксильную функцию, или метакрильных мономеров.
Примерами С5-С6 полиспиртов являются сорбит, маннит; примерами моносахаридов и из производных являются глюкоза, фруктоза, манноза, галактоза, галактоза глюкозамин; примером дисахаридов является лактоза, сахароза, мальтоза и т.д.; примерами полисахаридов и их производных являются микрокристаллическая целлюлоза, гидроксипропилцеллюлоза, гидроксиэтилцеллюлоза, гидроксиметилцеллюлоза, метилцеллюлоза, этилцеллюлоза, карбоксиметилцеллюлоза и их соли, предпочтительно соли натрия и калия, и их сшитые формы, ацетат целлюлозы, ацетофталат целлюлозы и их эфиры, крахмал и производные, такие как, например, карбоксиметилкрахмал натрия, растворимый крахмал, гелеобразный крахмал; примерами циклодекстринов и их производных являются диметил-β-циклодекстрин, 2-гидрокси-этил-β-циклодекстрин, 2-гидроксипропил-β-циклодекстрин, 3-гидроксипропил-β-циклодекстрин, триметил-β-циклодекстрин.
Объектом настоящего изобретения являются фармацевтические композиции для перорального применения, содержащие в качестве активного начала частично или полностью аморфные соединения формулы (I) и содержащие, по крайней мере, один из вышеуказанных наполнителей.
Заявителем было обнаружено, что композиции изобретения показывают улучшенную скорость растворения в воде, определенную вышеуказанным тестом на растворение.
В композициях согласно настоящему изобретению соотношение между количеством по массе нитропроизводных формулы (I) и наполнителей, способных переводить в аморфное состояние нитропроизводные, как правило, находится в пределах 1:20 и 1:0,5, предпочтительно 1:0,7 и 1:10.
Как уже говорилось, композиции для перорального применения настоящего изобретения способны вызывать за очень короткое время, порядка 2-2,5 часов, пик концентрации в плазме производного гидроксибензойной кислоты, как определено в А, и также было обнаружено, что они способны вызывать следующие фармакокинетические эффекты:
увеличивать плазматическую Смакc (максимальную концентрацию) производного гидроксибензойной кислоты, как определено в А, после однократного введения по отношению к необработанному (в неаморфном состоянии) продукту согласно настоящему изобретению;
увеличивать по крайней мере на 20%, предпочтительно на 50% площадь, описанную кривой плазматических концентраций в интервале 0-3,5 часов после введения.
Когда в частично или полностью аморфном продукте формулы (I) R=(Ia), производное гидроксибензойной кислоты, как определено в А, является салициловой кислотой (смотри Примеры).
Нитропроизводные формулы (I), где R является радикалом формулы Iа) или Ib), получают согласно известным способам предшествующего уровня техники. Смотри, например, способы, описанные в патентных заявках на имя Заявителя WO 95/30641, WO 97/16405 или в международной заявке на патент РСТ/ЕРОО/00353.
Следующие примеры иллюстрируют изобретение, не ограничивая его область.
ПРИМЕРЫ
А) Исследование соединения формулы (I) посредством ДСК.
Исследование проведено на примере 3-(нитроксиметил)фенилэфира ацетилсалициловой кислоты, вещества, использованного в примерах, описанных далее, которое получали согласно Примеру 1 международной заявки на патент РСТ/ЕР00/00353.
Специфическую энтальпию плавления ΔН (Дж/г) данного продукта как такового и продукта, обработанного способами, способными уменьшить его кристалличность (состав), определяют ДСК анализом.
Процент потери кристалличности оценивают следующим уравнением:

Параметры сканирования, выбранные для анализа ДСК, являются следующими:
Диапазон сканирования: от 30°С мин до 330°С макс.
Скорость сканирования: 10 К/мин
ДСК след от сложного эфира как такового показывает эндотермический пик плавления Т=64,7°С с соответствующей ΔН=99,81 Джоуль/г.
В) Изучение скорости растворения соединения формулы (I) 3-(нитроксиметил)фенилэфира ацетилсалициловой кислоты
Изучение скорости растворения 3-(нитроксиметил)фенилэфира ацетилсалициловой кислоты как таковую и в препаратах, содержащих его согласно примерам, описанным далее, проводят на оборудовании для растворения согласно USP (Фармакопея США) XXIII.
Опыт проводят, используя объем деионизированной воды, равный 1000 мл. Угловая скорость лопастной мешалки составляет 100 об/мин и температура равна 37±0,5°С.
В небольшом стеклянном сосуде взвешивают точное количество каждого препарата так, чтобы оно содержало количество активного начала, равное 30 мг, которое вводят непосредственно в сосуд. Через заранее определенное время, соответственно через 5, 10,15, 30, 45, 50, 60, 90 и 120 мин от начала теста определяют количество соединения, перешедшего в раствор, измеряя его концентрацию масс/об посредством УФ-спектрометрии при длине волны 235 нм, используя калибровочную кривую. Данные выражены как процент активного начала, перешедшего в раствор в зависимости от времени.
Пример 1 (сравнительный)
Скорость растворения 3-(нитроксиметил)фенилэфира полностью кристаллической ацетилсалициловой кислоты
Перекристаллизованный из изопропанола продукт является полностью кристаллическим, и анализ ДСК демонстрирует эндотермический пик при Т=64,7°С с ΔН 879,47 Дж/г.
30 мг вышеупомянутого соединения переносят в устройство для определения скорости растворения. В таблице 1 представлены проценты соединений, перешедших в раствор, измеренные в значения времени, указанные в вышеописанном тесте. Через 10 мин количество активного начала, перешедшего в раствор, составляет 0,3%.
Пример 2
Получение 3-(нитроксиметил)фенилэфира полностью аморфной ацетилсалициловой кислоты путем обработки распылительной сушкой соединения с примесью гидроксипропил-β-циклодекстрина в массовом соотношении соединение: циклодекстрин 1:8,33, соответствующем молярному соотношению 1:2.
43 г соединения растворяют в 5 л этилового спирта.
Полученный таким образом органический раствор смешивают при комнатной температуре с 5 л деионизированной воды, содержащей 7% масс/об (350 г) гидроксипропил-β-циклодекстрина. Этот водно-спиртовой раствор обрабатывают в установке для распылительной сушки LabPlant SD-05 Spray-Drying потоком горячего воздуха на входе при температуре 60°С, поддерживая поток воздуха таковым, чтобы позволить температуре на выходе составлять 45°С.
Потеря соединением кристалличности, измеренная на полученном порошке согласно ДСК методу, составляет 100%. Фактически на ДСК отпечатке эндотермический пик плавления отсутствует.
Количество порошка, соответствующее 30 мг активного начала, взвешивают и переносят в устройство для определения скорости растворения. В таблице 1 приведены проценты соединения, перешедшего в раствор, измеренные для значений времени, указанных в вышеуказанном опыте на растворение в соответствии со взвешенным количеством. Спустя 10 минут % активного начала, перешедшего в раствор, составляет 32%.
Пример 3
Получение 3-(нитроксиметил)фенилэфира частично аморфной ацетилсалициловой кислоты путем обработки распылительной сушкой соединения с примесью лактозы в соотношении 1 (соединение): 9 (лактоза) по массе.
Растворяют 10 г соединения в 1,5 л этилового спирта. Полученный органический раствор смешивают с 1,5 л деионизированной воды, содержащей 6% масс/об (90 г) лактозы.
Водно-спиртовой раствор обрабатывают в устройстве для распылительной сушки, устанавливая на входе температуру горячего потока воздуха, равную 70°С, и поддерживая поток воздуха таким образом, чтобы обеспечить температуру на выходе, равную примерно 50°С.
Потеря кристалличности, измеренная согласно методу ДСК в таким образом полученном порошке, составляет 87% (ΔН 12,58 Дж/г).
Количество порошка, соответствующее 30 мг активного начала, взвешивают и переносят в устройство для определения скорости растворения. В таблице 1 приведены проценты соединения, перешедшего в раствор, измеренные для значений времени, указанных в вышеуказанном опыте на растворение. Спустя 10 минут % активного начала, перешедшего в раствор, составляет 30%.
Пример 3А
Получение 3-(нитроксиметил)фенилэфира частично аморфной ацетилсалициловой кислоты путем обработки распылительной сушкой соединения с примесью микрокристаллической целлюлозы в соотношении 1:0,7.
Растворяют 10 г соединения в 1,5 л этилового спирта. К этому раствору добавляют 0,670 л 1% масс/об водной суспензии микрокристаллической целлюлозы (Avicel PH 101). Суспензию подвергают распылительной сушке, используя температуру воздуха на входе 70°С и поддерживая поток воздуха таким образом, чтобы обеспечить температуру на выходе примерно 50°С.
Количество порошка, соответствующее 30 мг активного начала, взвешивают и переносят в устройство для определения скорости растворения. В таблице 1 приведены проценты соединения, перешедшего в раствор, измеренные для значений времени, указанных в вышеуказанном опыте на растворение. Спустя 10 минут % активного начала, перешедшего в раствор, составляет 9,4%. Потеря кристалличности составляет 10%.
Пример 3В
Получение 3-(нитроксиметил)фенилэфира частично аморфной ацетилсалициловой кислоты обработкой путем совместного измельчения с примесью микрокристаллической целлюлозы и лаурилсульфата натрия в массовом соотношении активное начало:
микрокристаллическая целлюлоза: лаурилсульфат натрия 1:0,5:0,1.
10 г активного начала смешивают в ступке с 1 г лаурилсульфата натрия в течение 5 минут и затем с 5 г микрокристаллической целлюлозы. Порошкообразную смесь принудительно измельчают пестиком в течение 30 минут.
Опыт на растворение проводят, используя 48 мг полученной смеси, равной 30 мг активного начала.
Проценты соединения, перешедшего в раствор, за промежутки времени, указанные в описанном выше тесте на растворение, приведены в таблице 1. % активного начала, перешедшего в раствор через 10 минут, равен 4,8%.
Анализ ДСК порошка показывает потерю кристалличности активного начала на 6%.
Пример 4
Получение 3-(нитроксиметил)фенилового эфира частично кристаллической ацетилсалициловой кислоты путем обработки распылительной сушкой соединения с примесью лактозы в соотношении 1 (соединение): 4 (лактоза) по массе.
Растворяют 10 г соединения в 1,5 л этилового спирта. Полученный органический раствор смешивают с 0,75 л деионизированной воды, содержащей 6% масс/об (45 г) лактозы. Водно-спиртовой раствор обрабатывают в устройстве для распылительной сушки, используя температуру воздуха на входе 70°С и поддерживая поток воздуха таким образом, чтобы обеспечить температуру на выходе примерно 50°С.
Потеря кристалличности, измеренная для таким образом полученного порошка согласно способу ДСК, составляет 83% (ΔН 16,44 Дж/г).
Количество полученного порошка, соответствующее 30 мг активного начала, взвешивают и переносят в устройство для определения скорости растворения. В таблице 1 приведены проценты соединения, перешедшего в раствор, измеренные для значений времени, указанных в описанном выше опыте на растворение. Спустя 10 минут % активного начала, перешедшего в раствор, составляет 17,1%.
Пример 5
Получение 3-(нитроксиметил)фенилэфира частично аморфной ацетилсалициловой кислоты обработкой путем совместного измельчения с примесью гидроксипропил-β-циклодекстрина в соотношении соединение: циклодекстрин 1:4,16 по массе, соответствующем молярному соотношению 1:1.
10 г соединения смешивают с 41,6 г циклодекстрина. Смесь совместно измельчают в ступке в течение 30 минут.
Потеря кристалличности, измеренная для таким образом полученного порошка согласно способу ДСК, составляет 43% (ΔН 56,5 Дж/г).
Количество полученного порошка, соответствующее 30 мг активного начала, взвешивают и переносят в устройство для определения скорости растворения. В таблице 1 приведены проценты соединения, перешедшего в раствор, измеренные для значений времени, упомянутых в опыте на растворение, указанном в В). Спустя 10 минут % активного начала, перешедшего в раствор, составляет 6,9%.
Пример 6
Получение 3-(нитроксиметил)фенилового эфира частично аморфной ацетилсалициловой кислоты обработкой соединения перемешиванием с примесью лактозы в соотношении 1 (соединение): 9 (лактоза) по массе.
5 г соединения непосредственно смешивают с 45 г лактозы. Смесь смешивают с 10 мл 50% этанола в воде и затем оставляют высушиваться под вакуумом, создаваемым водоструйным насосом, при комнатной температуре в течение 24 часов. Высушенный продукт просеивают через сито с 600 мкм ячейками перед анализом.
Потеря кристалличности, измеренная для таким образом полученного порошка согласно способу ДСК, составляет 7% (ΔН 92,34 Дж/г).
Количество полученного порошка, соответствующее 30 мг активного начала, взвешивают и переносят в устройство для определения скорости растворения. В Таблице 1 приведены проценты соединения, перешедшего в раствор, измеренные для значений времени, упомянутых в опыте на растворение, указанном в В). Спустя 10 минут % активного начала, перешедшего в раствор, составляет 11,5%.
Пример 6А
Пример 6 повторяют, но используют смесь, содержащую также гидроксипропил-β-циклодекстрин, в соотношении активное начало: лактоза: гидроксипропил-β-циклодекстрин 1: 0,5: 0,2 по массе.
1000 г активного начала смешивают с 500 г лактозы и 200 г гидроксипропил-β-циклодекстрина. Смесь перемешивают механической мешалкой с 100 мл 3% масс/об раствора поливинилпирролидона в воде/изопропиловом спирте 1:1, используя последовательное добавление.
Перемешанную смесь экструдируют через гранулирующую головку и высушивают в сушильном аппарате при температуре 40°С. Высушенный гранулят пропускают через сито вибрационного гранулятора для того, чтобы привести гранулометрию к единообразию.
Количество полученного порошка, соответствующего 30 мг активного начала, взвешивают и переносят в устройство для определения скорости растворения. В таблице 1 приведены проценты соединения, перешедшего в раствор, измеренные для значений времени, упомянутых в опыте на растворение, указанном в В). Спустя 10 минут % активного начала, перешедшего в раствор, составляет 15,6%. Потеря кристалличности составляет 39,4%.
Пример 7 (сравнительный)
Распылительная сушка активного начала как такового
16 г 3-(нитроксиметил)фенилового эфира ацетилсалициловой кислоты растворяют в 3 л смеси этиловый спирт/вода 80/20. Водно-спиртовой раствор обрабатывают в устройстве для распылительной сушки горячим потоком воздуха на входе, поддерживая поток воздуха таким образом, чтобы обеспечить температуру на выходе примерно 45°С.
ДСК анализ полученного порошка показал, что соединение получают в полностью кристаллической форме (ΔН 100,5 Дж/г).
30 мг порошка активного начала взвешивают и переносят в устройство для определения скорости растворения. В таблице 1 приведены проценты соединения, перешедшего в раствор, измеренные для значений времени, упомянутых в опыте на растворение, описанном в А). Спустя 10 минут % активного начала, перешедшего в раствор, составляет 0,5% и ,следовательно, по существу не отличается от полученного в опыте на растворение активного начала как такового (Пример 1).
Пример 8 (сравнительный)
Получение таблеток согласно предшествующему уровню техники, содержащих: 300 мг 3-(нитроксиметил)фенилового эфира ацетилсалициловой кислоты, 143,7 мг микрокристаллической целлюлозы, 3 мг талька, 3 мг стеарата магния и 0,3 мг диоксида кремния.
300 мг активного начала смешивают в ступке с 0,3 г коллоидного диоксида кремния, 143,7 г микрокристаллической целлюлозы, добавленной аликвотами, согласно последующему способу разбавления. Затем добавляют 3 г талька. Смесь переносят в порошковый смеситель (Turbula) и перемешивают в течение 10 минут. Добавляют 3 г стеарата магния и продолжают перемешивание в течение дополнительных 5 минут. Порошковую смесь непосредственно прессуют ротационным компрессором (Officine Ronchi), оборудованным дыропробойными штампами (с диаметром 9,5 мм; радиусом закругления 9 мм), получая таблетки, имеющие средний вес 450 мг и среднее сопротивление разрушению 10 кг. Полученные таким образом таблетки толкут в ступке до получения порошка, способного проходить через сито с ячейками 200 мкм. Количество полученного порошка, соответствующего 30 мг активного начала, взвешивают и переносят в устройство для определения скорости растворения. В таблице 1 приведены проценты соединения, перешедшего в раствор, измеренные для значений времени, упомянутых в опыте на растворение, описанном в В). Спустя 10 минут % активного начала, перешедшего в раствор, составляет 0,34%.
ДСК анализ не показывает какого-либо случая аморфности активного начала в результате прессования.
Исследования in vivo
Пример 9
Фармакокинетика у животных при использовании фармацевтической композиции согласно настоящему изобретению, описанной в примере 3В.
Однократную дозу 80 мг/кг, равную 50 мг/кг активного начала, фармацевтической композиции (порошка) примера 3В в водной суспензии (5 мл/кг), вводили перорально группе из 10 крыс, весящих 180-200 г.
Образцы 0,5 мл крови брали из хвостовой вены животных через 0,5, 1, 1,5, 2, 4, 8, 12 и 24 часа после введения.
Образцы переносили в гепаринизированные пробирки и центрифугировали в течение 15 минут при комнатной температуре. В 100 мкл аликвоту сыворотки добавляли 25 мкл раствора внутреннего стандарта (полученного растворением 10 мг напроксена в 100 мл ацетонитрила). Образец впрыскивают в установку для ВЭЖХ Hewlett Packard series 1050, имеющую детектор изменяющихся длин волн, насос, самопробоотборник с 5 мкм ODS (10×0,46 см) колонкой, соединенной последовательно с 5 мкм ODS (С 18-250×4 мм) колонкой. Подвижная фаза представляет собой ацетонитрил/ уксусную кислоту 3% в соотношении 60/40 по объему. Расход составляет 0,8 мл/мин. Все анализы проводят при комнатной температуре и измерения эффективны при длине волны 234 нм. Значение Сmах составляет 61,7 мкг/мл в момент времени 2 часа.
Пример 10 (сравнительный)
Фармакокинетика у животных при использовании фармакокинетической композиции, описанной в примере 8
Однократную дозу 75 мг/кг, равную 50 мг/кг активного начала, порошка, полученного из истолченных таблеток, описанных в примере 8, в водной суспензии (5 мл/кг) вводят перорально в группу из 10 крыс, весящих 180-200 г.
Образцы 0,5 мл крови брали из хвостовой вены животных через 1,5, 3, 6, 12 и 24 часа после введения. Затем действовали, как описано в предшествующем примере 8. Значение Сmах составляет 53,2 мкг/мл в момент времени 6 часов.
Изобретение относится к способу получения фармацевтической композиции, обладающей противовоспалительной и анальгезирующей активностью, для перорального введения, содержащей соединение формулы A-X1-NO2 (I), имеющее полное состояние аморфности или частично состояние аморфности, включающий в себя следующие этапы: перемешивание соединения формулы (I), по меньшей мере, с одним наполнителем, способным придать состояние аморфности полученной смеси, выбранном из группы, состоящей из С5-С6 полиспиртов, моно- и дисахаридов и их производных, олигосахаридов, содержащих от 3 до 10 моносахаридных единиц и их производных, полисахаридов, их производных, включая их соли, циклодекстринов и их производных, нециклических производных β - циклодекстрина, полимеров и сополимеров на основе винильных мономерных звеньев, и/или содержащих карбоксильную функцию, или метакрильных мономеров, причем массовое соотношение между количеством соединения формулы (I) и количеством, по меньшей мере, одного наполнителя находится в пределах 1:20 и 1:0,5, и достижение состояния аморфности полученной смеси совместным измельчением, перемешиванием, распылительной сушкой, лиофилизацией, где в формуле (I) А, Х1 такие, как определено в п.1 формулы изобретения. Изобретение также относится к фармацевтической композиции, обладающей противовоспалительной и анальгезирующей активностью. Технический результат – получение фармацевтической композиции для перорального введения и лекарственных средств на ее основе в целях лечения воспалений. 3 н. и 7 з.п. ф-лы, 1 табл.
A-X1-NO2(I)
имеющее полное состояние аморфности или частично состояние аморфности, включающий в себя следующие этапы:
- перемешивание соединения формулы (I), по меньшей мере, с одним наполнителем, способным придать состояние аморфности полученной смеси, выбранном из группы, состоящей из С5-С6 полиспиртов, моно- и дисахаридов и их производных, олигосахаридов, содержащих от 3 до 10 моносахаридных единиц и их производных, полисахаридов, их производных, включая их соли, циклодекстринов и их производных, не циклических производных β-циклодекстрина, полимеров и сополимеров на основе винильных мономерных звеньев, и/или содержащих карбоксильную функцию, или метакрильных мономеров, причем массовое соотношение между количеством соединения формулы (I) и количеством, по меньшей мере, одного наполнителя находится в пределах 1:20 и 1:0,5
и
- достижение состояния аморфности полученной смеси совместным измельчением, перемешиванием, распылительной сушкой, лиофилизацией, где в формуле (I)
A=R(COX),
Х=O, NH, NR1c, где R1c является линейным или разветвленным C1 - С10 алкилом, R выбран из следующих радикалов:
где
R1 означает OCOR3 группу; где R3 означает метил, этил или разветвленный С3-С5 алкил;
R2 означает водород, гидрокси, галоген, линейный или разветвленный C1-С4 алкил, линейный или разветвленный С1-С4 алкоксил, линейный или разветвленный С1-С4 перфторалкил, моно- или ди-(С1-С4) алкиламино;
R1 и R2 вместе означают диоксиметиленовую группу с тем условием, что когда Х=NH, тогда Y означает этилен и R2=Н как определено ниже;
R1 не может быть OCOR3 в положении 2, когда R3 означает метил;
n1 означает целое число и представляет собой 0 или 1;
X1 означает бивалентный связующий мостик, выбранный из следующего:
YO:
Y = линейный или разветвленный C1-C20 алкилен; или замещенный или незамещенный С5-С7 циклоалкилен:
или X1 выбран из следующего:
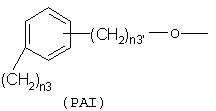
где n3 означает целое число от 0 до 3, n3' означает целое число от 1 до 3;
где n3 и n3' имеют следующие вышеупомянутые значения;
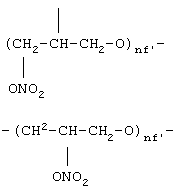
где nf' означает целое число от 1 до 6;
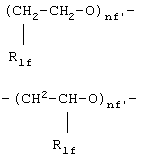
где R1f =H, СН3 и nf' является таким как определено выше;
с тем условием, что когда R= (1a), R1 означает OCOR3 группу, где R3 означает метил или этил, и Y выбран из группы, состоящей из линейного или разветвленного C1-С20 алкилена, Х не может быть NH или NR1c.
| WO 9530641 A1, 16.11.1995 | |||
| WO 9716405 A1, 09.05.1997 | |||
| WO 9201668 A1, 06.02.1992 | |||
| WO 9857967 A1, 23.12.1998.RU 2109009 C1, 20.04.1998 | |||
| НИТРОЭФИРЫ, ОБЛАДАЮЩИЕ ФАРМАКОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ, И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2127723C1 |

