Предлагаемое изобретение относится к области разработки способов получения термостойких взрывчатых веществ (ТВВ), которые находят применение в средствах инициирования и передачи детонационного импульса для создания устройств, используемых при проведении торпедировочно-прострелочных работ в нефтегазодобывающей промышленности в скважинах с эксплуатационной температурой выше 200°С.
По комплексу эксплуатационных и физико-химических характеристик одним из наиболее перспективных ТВВ является 2,4,6,4',6',2",4",6"-октанитро-мета-терфенил (сокращенное название - октанит). Октанит сочетает требуемую мощность с высокой максимальной эксплуатационной температурой 290-300°С, обладает удовлетворительной восприимчивостью к инициирующим импульсам, не теряет своих свойств при эксплуатации длительное время (50-100 часов) в замкнутом объеме при температуре 280-290°С, сохраняет работоспособность при контакте со скважинной средой при температуре 250°С и давлении 150 МПа.
Известен способ получения октанитро-мета-терфенила [патент США 3592860 от 13.07.1971 г.], взятый за прототип, взаимодействием 2,4,6-тринитрогалоидбензолов (пикрилгалоидов) с 1,5-дигалоид-2,4-динитробензолами по реакции Ульмана в присутствии порошкообразной меди в ароматических растворителях. Полученные результаты при проведении реакции взаимодействия в среде нитробензола представлены в таблице 1.
Приведенные в патенте в качестве оптимальных по выходу целевого продукта исходных компонентов синтеза октанита бром- и йодполинитропроизводных бензола являются дефицитными и дорогостоящими реактивами, которые не выпускаются в промышленных масштабах. Наиболее приемлемым является использование доступных хлорполинитропроизводных бензола-1,5-дихлор-24-динитробензола и пикрилхлорида, стоимость которых более чем на порядок ниже стоимости бром- и йодполинитропроизводных. Однако данные таблицы 1 свидетельствуют, что при использовании в предлагаемых условиях синтеза в качестве исходных компонентов хлорполинитропроизводных бензола необходимы жесткие температурные условия реакции (188÷200°С), и выход целевого продукта составляет в этом случае только 6% от теоретически возможного.
В процессе синтеза октанита наряду с основным веществом в конденсированной фазе выделяются непрореагировавший медный порошок, галогениды, побочные продукты реакции (гексанитродифенил, полинитрополифенилены и др.). Очистка октанита от сопутствующих веществ осуществлялась в следующей последовательности:
- обработкой смеси конденсированных продуктов 20%-ной соляной кислотой;
- кристаллизацией путем растворения в диметилсульфоксиде и высаживанием из раствора метиловым спиртом;
- повторной кристаллизацией путем растворения продукта в нитробензоле при 200°С;
- отмывкой нитробензола от кристаллизованного октанита метанолом;
- сушкой октанита при 140-150°С под вакуумом для полного удаления нитробензола.
Данный способ получения октанита имеет ряд недостатков:
- высокую температуру, необходимую для осуществления взаимодействия галоидполинитробензолов по Ульману с образованием октанита;
- практическую невозможность использования для получения октанита наиболее доступных реагентов - 1,5-дихлор-2,4-динитробензола с пикрилхлоридом (температура реакции 188÷200°С, выход 6%);
- сложную многостадийную очистку октанита с использованием большого спектра реактивов (соляная кислота, диметилсульфоксид, нитробензол, метанол).
Совокупность недостатков разработанного способа практически исключает возможность использования его в опытно-промышленном производстве.
Задачей настоящего изобретения является разработка способа получения октанита, позволяющего:
- проводить процесс при оптимальных для производства взрывчатых веществ температурах;
- использовать недефицитные и дешевые исходные компоненты;
- применять технологичные, менее материалоемкие методы очистки;
- повысить выход октанита.
Сущность предлагаемого изобретения заключается:
- в изыскании условий активации реакции взаимодействия 1,5-дигалоид-2,4-динитробензолов с пикрилгалоидами в среде 1,2-дихлорэтана за счет введения в реакционную массу одновременно с порошком меди эквимолекулярных количеств активирующих добавок апротонных растворителей, например диметилформамида, или диметилсульфоксида, или диметилацетамида, или N-метилпирролидона, или ацетонитрила в соотношении 1-3 моля активирующей добавки на моль исходного 1,5-дигалоид-2,4-динитробензола;
- в использовании азотной кислоты для выделения и очистки октанита из смеси продуктов реакции.
Технический результат проведения взаимодействия 1,5-дигалоидбензолов с пикрилгалоидами в среде дихлорэтана в присутствии эквимолекулярных количеств активирующих добавок апротонных растворителей достигается существенным снижением уровня необходимой для начала образования октанита температуры. Реакция начинается при 75°С, и ее результат практически не зависит от реакционной способности галоида. В таблице 2 приведены данные по зависимости выхода октанита от условий проведения реакции Ульмана в среде 1,2-дихлорэтана.
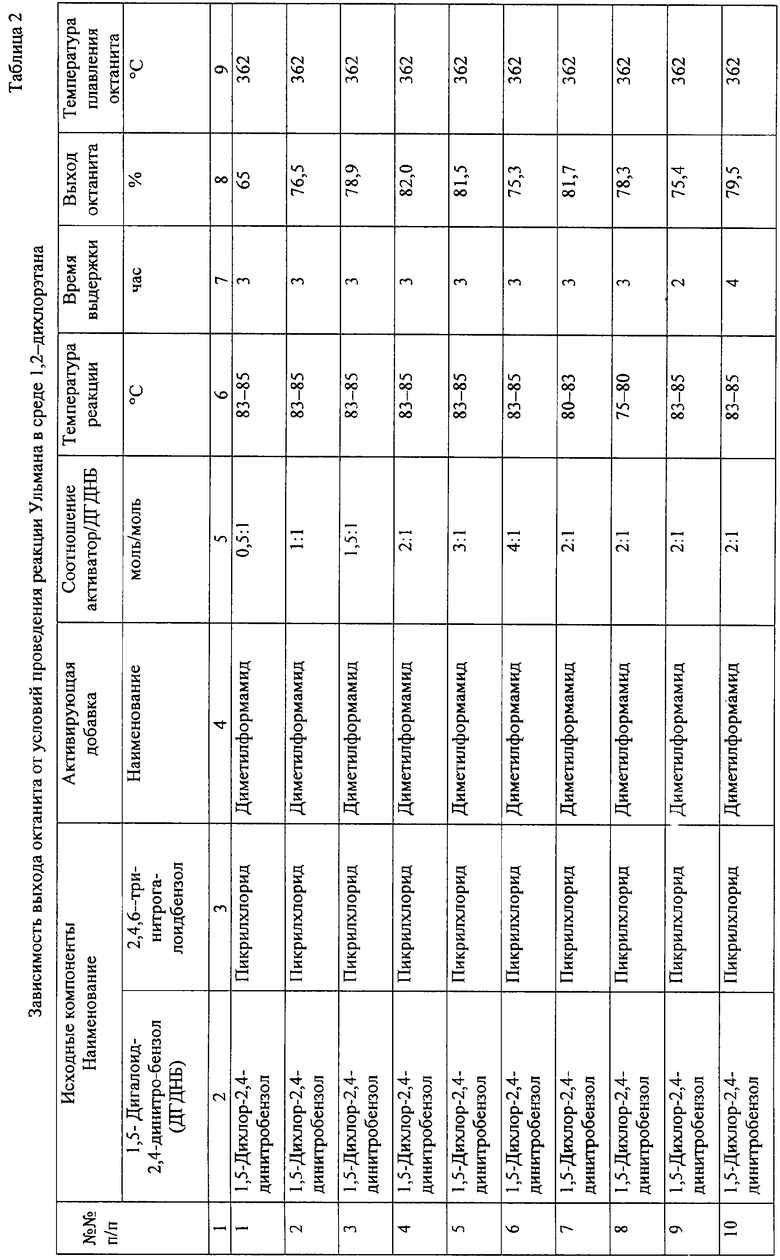
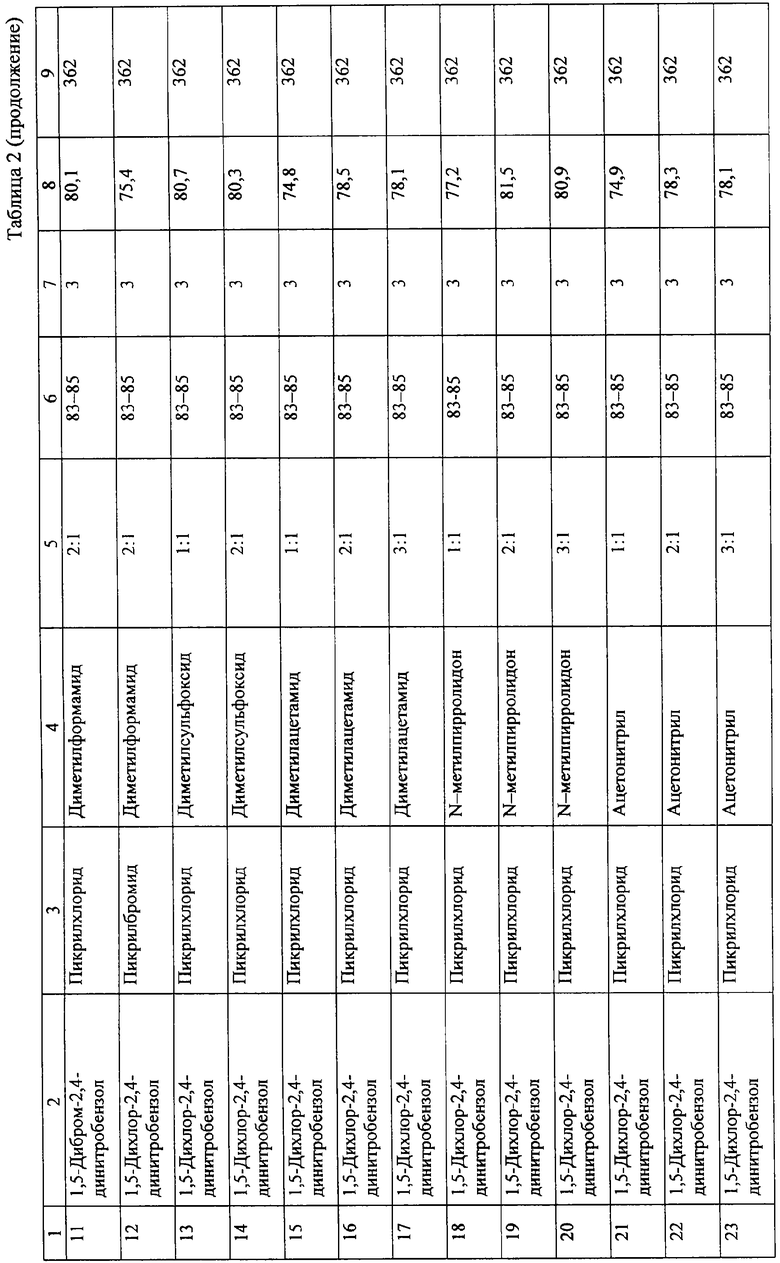
Результаты взаимодействия 1,5-дигалоид-2,4-динитробензолов с пикрилгалоидами в среде дихлорэтана определяются количеством вводимой активирующей добавки, температурой реакции и временем выдержки. Уменьшение содержания активирующей добавки в реакционной среде ниже соотношения 1 моль активатора: 1 моль 1,5-дигалоид-2,4-динитробензола, а также увеличение содержания свыше соотношения 3 моля активатора: 1 моль 1,5-дигалоид-2,4-динитробензола ведут к снижению выхода октанита. Необходимая температура взаимодействия и выход октанита аналогичны в случае использования бромполинитропроизводных и хлорполинитропроизводных бензола.
Максимальный выход октанита достигается при 3-часовой выдержке реакционной массы при температуре 83÷85°С. Уменьшение или увеличение времени выдержки сопровождается снижением выхода октанита.
Использование азотной кислоты для очистки октанита приводит к упрощению процесса и исключению необходимости использования для этой цели большого количества агрессивных и высокотоксичных компонентов.
Очистку октанита предлагается проводить последовательно в 55÷60%-ной и 90%-ной азотной кислоте.
Непрореагировавшая медь и галогениды меди в 55÷60% азотной кислоте превращаются в водорастворимую азотнокислую медь, отделяющуюся от смеси нерастворимых в воде полинитрополифенилов в виде водного раствора.
Сопутствующие октаниту полинитрополифенильные соединения отделяются в результате отмывки их из смеси с октанитом 90%-ной азотной кислотой. Октанит в 90%-ной азотной кислоте практически нерастворим и выделяется из нее в чистом виде. Очищенный октанит подвергается водной промывке при 70÷80°С для удаления остаточной кислоты.
Сушка октанита осуществляется при температуре 80÷90°С в вакуум-сушильных шкафах до постоянной массы.
В качестве медного компонента реакции взаимодействия 1,5-дигалоид-2,4-динитробензолов с пикрилгалоидами по реакции Ульмана использовался порошок меди марки ПМС-1 или ПМС-2 (ГОСТ 4960-75).
Пример получения 2,4,6,4',6',2",4",6"-октанитро-мета-терфенила
В реактор с 300 мл сухого 1,2-дихлорэтана загружают 23,7 г (0,1 моль) 1,5-дихлор-2,4-динитробензола и 62 г (0,25 моль) пикрилхлорида. Раствор в реакторе нагревают до кипения и отгоняют азеотроп дихлорэтана с водой до достижения в парах температуры кипения дихлорэтана (82÷83°С). Раствор охлаждают до температуры 60±5°С, в реактор заливают 15,2 мл (2 моль) диметилформамида и загружают порциями 63 г (1 моль) порошка меди марки ПМС-1.
Суспензию в реакторе при интенсивном перемешивании нагревают до кипения (83÷85°С) и дают выдержку 3 часа. Далее реакционную массу в реакторе охлаждают до 15÷20°С, смесь конденсированных продуктов отфильтровывают от маточного раствора на вакуум-фильтре и промывают 50 мл дихлорэтана от смолистых примесей.
Промытую смесь продуктов конденсации порциями дозируют при перемешивании и температуре не выше 50°С к 350 мл 55÷60%-ной азотной кислоты. Реакционную массу нагревают до 70°С и дают часовую выдержку. К реакционной массе дозируют 350 мл воды при температуре не выше 70°С и дают получасовую выдержку.
Реакционную массу охлаждают до 15÷20°С, осадок отфильтровывают от маточного раствора, промывают на вакуум-фильтре водой до исчезновения голубого окрашивания промывной воды, подсушивают на вакуум-фильтре до влажности не более 20%. Выход октанита составил 67 г.
Влажный продукт порциями дозируют к 670 мл 98%-ной азотной кислоты (плотность 1,5 г/см3) при перемешивании и фактической температуре. Суспензию в реакторе нагревают до 75°С и дают часовую выдержку при 70÷80°С. Далее к реакционной массе при температуре 70÷80°С порциями в течение часа дозируют воду до достижения концентрации кислоты 90% (˜90 мл). Суспензия в реакторе самопроизвольно охлаждается до 15-20°С, и октанит офильтровывают от маточной кислоты на вакуум-фильтре. Октанит на вакуум-фильтре промывают 30 мл 70%-ной азотной кислоты и водой до нейтральной реакции промывных вод, затем на вакуум-фильтре подсушивают до влажности не более 20%. Выход влажного октанита составил 60,5 г.
Влажный октанит загружают в 600 мл воды, суспензию нагревают до 80÷90°С и дают 2-часовую выдержку при данной температуре и перемешивании. После охлаждения суспензии до 40÷50°С октанит отфильтровывают от промывной воды, промывают на вакуум-фильтре 100 мл воды и подсушивают до влажности не более 20%.
Влажный октанит сушат в сушильных шкафах при температуре 80÷90°С при атмосферном давлении или под вакуумом до постоянной массы в течение 15-18 часов. Выход 2,4,6,4',6',2",4",6"-октанитро-мета-терфенила составил 48,5 г (82% от теоретического).
Полученный данным способом октанит имеет температуру плавления (разложения) 362°С. Элементный состав - найдено, % С 36,60; Н 1,00; N 19, 70
C18H6O16N8
Вычислено, %: С-36,62; Н 1,07, N 18,92
По данным тонкослойной жидкостной хроматографии (ТСХ) на "Silufol" (элюент-бензол:ацетон 10:1) полученное вещество по Rf не отличается от образца, изготовленного по патенту, и не содержит примесей.
Молекулярная масса, определенная методом масс-спектрального анализа, для полученного соединения (C18H6O16N8 590,31) составляет 589.
По аналогии с приведенным примером осуществляется взаимодействие пикрилхлорида с 1,5-дигалоид-2,4-динитробензолами в присутствии других перечисленных активирующих добавок апротонных растворителей. Результаты опытов представлены в таблице 2.
Разработанный способ получения апробирован в условиях производства для наработки опытных и промышленных партий октанита.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2,4,6,4',6',2'',4'',6''-ОКТАНИТРО-МЕТА-ТЕРФЕНИЛА | 2014 |
|
RU2562271C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,4,6-ТРИ(2,4,6-ТРИНИТРОФЕНИЛ)-1,3,5-ТРИАЗИНА | 2012 |
|
RU2484087C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-БРОМ-2-АМИНО-4-НИТРОФЕНОЛА | 1991 |
|
RU2053223C1 |
| СПОСОБ НИТРОВАНИЯ БЕНЗОЛА | 1994 |
|
RU2087463C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-БРОМ-2,4-ДИНИТРОФЕНОЛА | 1991 |
|
RU2053221C1 |
| СПОСОБ НИТРОВАНИЯ БЕНЗОЛА НА ЦЕОЛИТНЫХ КАТАЛИЗАТОРАХ | 1995 |
|
RU2095342C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ 3-(4-(2,4-ДИФТОРБЕНЗИЛОКСИ)-3-БРОМ-6-МЕТИЛ-2-ОКСОПИРИДИН-1(2Н)-ИЛ)-N,4-ДИМЕТИЛБЕНЗАМИДА | 2007 |
|
RU2411236C1 |
| Способ получения динитропроизводных дифениловых и трифениловых эфиров | 2017 |
|
RU2671581C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-БРОМ-4,6-ДИНИТРОАНИЛИНА | 1991 |
|
RU2030391C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-БРОМ-2-АМИНО-4-НИТРОФЕНОЛА | 1992 |
|
RU2053224C1 |
Изобретение относится к области разработки способов получения термостойких взрывчатых веществ, нашедших применение в термостойких средствах инициирования и передачи детонации, используемых в нефте- и газодобывающей промышленности. Сущность изобретения заключается в изыскании условий активации взаимодействия 1,5-дигалоид-2,4-динитробензолов с 2,4,6-тринитрогалоидбензолами в среде дихлорэтана по реакции Ульмана за счет введения в реакционную массу эквимолекулярных количеств активирующих добавок апротонных растворителей, выбранных из диметилформамида, диметилсульфоксида, диметилацетамида, N-метилпирролидона, ацетонитрила, при соотношении 1÷3 моль активатора на моль 1,5-дигалоид-2,4-динитробензола, и использовании азотной кислоты для выделения октанита из смеси продуктов реакции, обеспечивается снижение уровня температуры, необходимой для образования целевого продукта, до 83÷85°С, возможность использования для его производства наиболее доступных хлорполинитробензолов и повышение выхода до 82% от теоретического. 2 з.п. ф-лы, 2 табл.
| US 3592860 А, 13.07.1971 | |||
| Краткий энциклопедический словарь под ред | |||
| ак | |||
| Жукова Б.П | |||
| Энергетические конденсированные системы | |||
| М., "Янус-К", 2000, с.333 | |||
| US 3403185 А, 24.09.1968 | |||
| US 3320320 А, 16.05.1967. |

