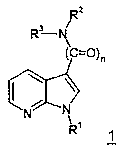
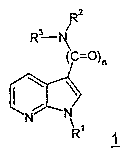
Изобретение касается замещенных 7-азоиндолов общей формулы 1,
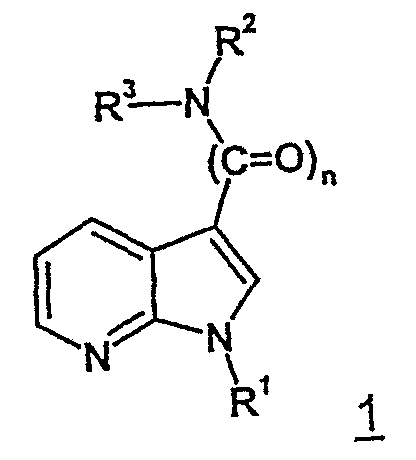
способа их получения, фармацевтических композиций, содержащих эти соединения, а также фармацевтического применения этих соединений, являющихся ингибиторами фосфодиэстеразы 4, в качестве биологически активных веществ для лечения заболеваний, которые зависят от подавления активности фосфодиэстеразы 4 в ответственных за иммунитет клетках (например, макрофагах и лимфоцитах) посредством соединений согласно изобретению.
Активация рецепторов оболочки клетки посредством передатчика приводит к активации системы "повторного сигнала" ("second messenger"-Systems). Аденилатциклаза синтезирует из AMP (аденозин-3,5-монофосфата) и GMP (гуанозин-3,5-монофосфата) активный циклический AMP (cAMP) или циклический GMP (cGMP). Они приводят, например, в гладких мышечных клетках к расслаблению или в воспаленных клетках к подавлению высвобождения или синтеза медиатора. Разрушение "повторного сигнала" cAMP и cGMP происходит посредством фосфодиэстераз (PDE). До сих пор известно 11 семейств ферментов PDE (PDE 1-11), которые различаются своей специфичностью к субстрату (cAMP, cGMP или обоим) и зависимостью от других субстратов (например, кальмодулина). Эти изоферменты обладают различными функциями в организме и по-разному проявляются в отдельных видах клеток (Beavo JA, Conti M and Heaslip RJ. Multiple cyclic nucleotide phosphodiesterases. Mol. Pharmacol. 1994, 46: 399-405; Hall IP. Isoenzyme selective phosphodiesterase inhibitors: potential clinical uses, Br. J. clin. Pharmacol. 1993, 35: 1-7). Посредством подавления различных типов изоферментов PDE это приводит к накоплению cAMP или cGMP в клетках, что может быть использовано в лечебных целях (Torphy TJ, Livi GP, Christensen SB. Novel Phosphodiesterase Inhibitors for the Therapy of Asthma, Drug News and Perspectives 1993, 6: 203-214).
В клетках, ответственных за аллергические воспаления (лимфоцитах, тучных клетках, эозинофильных гранулоцитах, макрофагах), преобладающим является фермент PDE типа 4 (Torphy, J T. and Undem, B. J. Phosphodiesterase inhibitors: new opportunities for the treatment of asthma. Thorax 1991, 46: 512-523). Подавление PDE 4 посредством пригодных ингибиторов до сих пор рассматривается как важный подход к лечению большого количества заболеваний, обусловленных аллергией (Schudt Ch, Dent G, Rabe K Phosphodiesterase Inhibitors, Academic Press, London, 1996).
Важным свойством ингибиторов фосфодиэстеразы 4 является подавление высвобождения фактора некроза опухоли а (TNFa) из воспаленнных клеток. TNFa является известным провоспалительным цитокином, который влияет на большое число биологических процессов. TNFa высвобождается, например, из активированных макрофагов, активированных Т-лимфоцитов, тучных клеток, базофилов, фибробластов, клеток эндотелия и астроцитов в головном мозге. Это само по себе способствует активированию нейтрофилов, эозинофилов, фибробластов и клеток эндотелия, вследствие чего высвобождаются различные медиаторы, разрушающие ткани. В моноцитах, макрофагах и Т-лимфоцитах TNFa вызывает повышенное продуцирование следующих провоспалительных цитокинов, как GM-CSF (фактор, стимулирующий колонию гранулоцитных макрофагов) или интерлейкин-8. Вследствие своего вызывающего воспаление и катаболического действия TNFa играет основную роль в случае многих заболеваний, как воспаления дыхательных путей, воспаления суставов, эндотоксический шок, отторжение тканей, СПИД и другие многочисленные иммунологические заболевания. Таким образом, для лечения таких заболеваний, связанных с TNFa, также пригодно ингибирование фосфодиэстеразы 4.
Хронические обструктивные заболевания легких (COPD) широко распространены среди населения и имеют также большое экономическое значение. Так, заболевания, вызванные COPD, составляют около 10-15% всех расходов, связанных с заболеваниями в развитых странах, и являются причиной около 25% смертельных случаев в США (Norman P.: COPD: New developments and therapeutic opportunities, Drug News Perspect. 11(7), 431-437, 1998), правда, большинству умерших пациентов было более 55 лет (Nolte D.: Chronische Bronchitis - eine Volkskrankheit multifaktorieller Genese. Atemw.-Lungenkrkh. 20 (5), 260-267, 1994). По оценке ВОЗ, COPD в течение ближайших 20 лет будут на третьем месте среди причин смертности.
Под клинической картиной хронически обструктивных заболеваний легких (COPD) понимают различные картины болезни хронических бронхитов с симптомами кашля и мокроты, а также прогрессирующее и необратимое ухудшение функции легких (особенно это касается выдоха). Течение заболевания сопровождается приступами и часто осложняется бактериальными инфекциями (Rennard S. I.: COPD: Overview of definitions, Epidemiology, and factors influencing its development. Chest, 113 (4) Suppl., 235S-241S, 1998). В ходе заболевания функция легких неуклонно сужается, легкое становится все более эмфизематозным, и одышка пациентов становится очевидной. Это заболевание явно причиняет вред качеству жизни пациентов (одышка, малая выносливость) и значительно сокращает продолжительность жизни. Главным фактором риска помимо факторов окружающей среды является курение (Kummer F.: Asthma und COPD. Atemw.-Lungenkrkh. 20 (5), 299-302, 1994; Rennard S. I.: COPD: Overview of definitions, Epidemiology, and factors influencing its development. Chest, 113 (4) Suppl., 235S-241S, 1998), и поэтому мужчины заболевают намного чаще, чем женщины. Однако из-за изменения жизненных привычек и роста числа курильщиц эта картина в будущем изменится.
Современная терапия направлена только на смягчение симптомов, не вмешиваясь в причины развития заболевания. Использование длительно действующих бета2-агонистов (например, сальметерола), возможно, в комбинации с мускариновыми антагонистами (например, ипратропиумом), улучшает функцию легких посредством расширения бронхов и широко применяется (Norman P: COPD: New developments and therapeutic opportunities, Drug News Perspect. 11 (7), 431-437, 1998). Большую роль при приступах COPD играют бактериальные инфекции, которые должны лечиться антибиотиками (Wilson R.: The role of infection in COPD, Chest, 113 (4) Suppl., 242S-248S, 1998; Grossman R. F.: The value of antibiotics and the outcomes of antibiotic therapy in exacerbations of COPD. Chest, 113 (4) Suppl., 249S-255S, 1998). Лечение этого заболевания до сих пор еще неудовлетворительно, особенно с точки зрения постоянного ухудшения функции легких. Новые принципы лечения, атакующие медиаторы воспаления, протеазы или молекулы сцепления, могут быть очень перспективными (Barnes P.J.: Chronic obstructive disease: new opportunities for drug development, TiPS 10 (19), 415-423, 1998).
Независимо от бактериальных инфекций, усложняющих заболевание, в бронхах находят хроническое воспаление, которое доминирует посредством нейтрофильных гранулоцитов. Для наблюдаемых структурных изменений в дыхательных путях (эмфизема) среди прочих ответственными являются медиаторы и ферменты, высвобождаемые посредством нейтрофильных гранулоцитов. Подавление активности нейтрофильных гранулоцитов, таким образом, является рациональной основой, чтобы воспрепятствовать развитию COPD (ухудшению параметров функции легких) или замедлить его. Важным стимулом для активации гранулоцитов является провоспалительный цитокин TNFa (фактор некроза опухоли). Так, известно, что TNFa стимулирует образование кислородных радикалов посредством нейтрофильных гранулоцитов (Jersmann, H.P.A.; Rathjen, D.A. and Ferrante A.: Enhancement of LPS-induced neutrophil oxygen radical production by TNFa, Infection and Immunity, 4, 1744-1747, 1998). Ингибиторы PDE 4 могут очень эффективно подавлять высвобождение TNFa из большого числа клеток и тем самым подавлять активность нейтрофильных гранулоцитов. Неспецифический ингибитор PDE пентоксифиллин в состоянии подавлять как образование кислородных радикалов, так и фагоцитозную способность нейтрофильных гранулоцитов (Wenisch, C.; Zedtwitz-Liebenstein, K.; Parschalk, B. and Graninger W.: Effect of pentoxifylline in vitro on neutrophil reactive oxygen production and phagocytic ability assessed by flow cytometry, Clin. Drug Invest., 13(2): 99-104, 1997).
Уже известны различные ингибиторы PDE 4. Преимущественно при этом речь идет о производных ксантина, аналогах ролипрама или производных нитракуазона (обзор в: Karisson J-A, Aldos D. Phosphodiesterase 4 inhibitors for the treatment of asthma, Exp. Opin. Ther. Patents 1997, 7: 989-1003). Ни одно из этих соединений до сих пор не было доведено до клинического использования. Было определенно установлено, что известные ингибиторы PDE 4 также характеризуются различными побочными действиями, как тошнота и рвота, которые до сих пор не могли быть устранены в достаточной мере. Поэтому существует потребность в открытии новых ингибиторов PDE 4 с лучшим терапевтическим спектром действия.
Применение 7-азаиндолов для получения новых биологически активных веществ для различного назначения определений до сих пор описано только в относительно малых случаях.
В японском патенте JP 10120681 (Fujisawa Pharmaceutical Co., LTD.) заявлены 5- и 7-азаиндолы общей формулы,
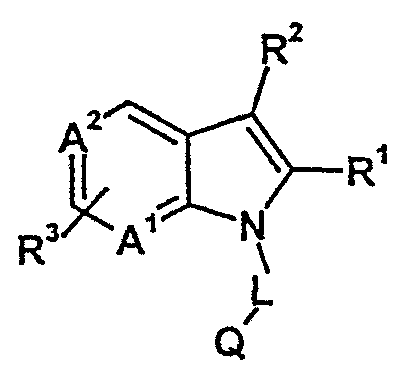
причем R1 обозначает водород или низшие алкильные группы, R2 может обозначать водород, галоген, короткие алкильные группы, циклоалкильные группы, алкилкарбонильные группы или алканоильные группы, R3 обозначает алканоильные группы, защищенные карбоксильные группы, цианогруппу или замещенные карбамоильные группы. L обозначает низший алкиленовый мостик. Q обозначает замещенные ароматические или гетероциклы. А1 и А2 обозначают: один - N, а другой - СН. Эти соединения отличаются от соединений согласно изобретению особенно в отношении заместителей R2 и R3, частично - R1 и А2. Описанные соединения заявлены в качестве ингибиторов фосфодиэстеразы (PDE 5), специфичной cGMP. В качестве областей применения называются различные сердечно-сосудистые заболевания, бронхит, астма, ринит, импотенция, диабетические осложнения и глаукома.
L.N. Yakhontov, S.S. Liberman, D.M. Krasnokutskaya et al., Khim.-Farm. Zh. 8 (11), 1974, 5-9, описали синтезы различных 3-аминоалкил-4-азаиндолов и 3-аминоалкил-7-азаиндолов. Для 3-(2-аминоэтил)-7-азаиндолов описано депрессивное или антидепрессивное действие. Для 3-аминометил-7-азаиндолов установлено действие, понижающее кровяное давление.
A.J. Verbiscar, J. Med. Chem. 15 (2), 1972, 149-52, описывает соединение формулы:
с антималярийным действием.
В патенте GB 1141949 (Sterling Drug Inc.) описан синтез различных 2-(имидазолин-2-ил)-алкил-7-азаиндолов или 3-(имидазолин-2-ил)-алкил-7-азаиндолов из соответствующих 2- или 3-цианоалкил-7-азаиндолов и для этих соединений указано применение в качестве сосудосуживающих средств.
До сих пор действие 7-азаиндолов в качестве ингибиторов PDE 4 полностью неизвестно.
Изобретение касается замещенных 7-азаиндолов общей формулы 1:
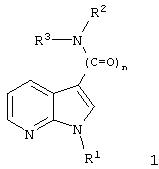
где
n может быть равно 1 или 2, и
R1 обозначает
-С1-С10-алкил, неразветвленный или разветвленный,
в случае необходимости, одно- или многократно замещенный группами -ОН, -SH, -NH2, -NH-C1-С6-алкил, -N(C1-С6-алкил)2, -NH-C6-С14-арил, -N(C6-С14-арил)2, -N(C1-С6-алкил)(C6-С14-арил),
-NO2, -CN, -F, -Cl, -Br, -I, -O-C1-С6-алкил, -O-C6-С14-арил, -S-C1-С6-алкил, -S-C6-С14-арил, -SO3H, -SO2-C1-С6-алкил, -SO2-C6-С14-арил, -OSO2-C1-С6-алкил, -OSO2-C6-С14-арил, -COOH, -(CO)-C1-С5-алкил, с моно-, би- или трициклическими насыщенными или одно- или многократно ненасыщенными карбоциклами с 3-14 членами в кольце, с моно-, би- или трициклическими насыщенными или одно- или многократно ненасыщенными гетероциклами с 5-15 членами в кольце и 1-6 гетероатомами, предпочтительно N, O и S,
причем C1-С6-арильные группы и карбоциклические и гетероциклические заместители, со своей стороны, в случае необходимости, одно- или многократно могут быть замещены R4,
-С2-С10-алкенил, одно- или многократно ненасыщенный, неразветвленный или разветвленный, в случае необходимости, одно- или многократно замещенный группами -ОН, -SH, -NH2, -NH-C1-С6-алкил, -N(C1-С6-алкил)2, -NH-C6-С14-арил, -N(C6-С14-арил)2, -N(C1-С6-алкил)(C6-С14-арил), -NO2, -CN, -F, -Cl, -Br, -I, -O-C1-С6-алкил, -O-C6-С14-арил, -S-C1-С6-алкил, -S-C6-С14-арил, -SO3H, -SO2-C1-С6-алкил, -SO2-C6-С14-арил, -OSO2-C1-С6-алкил, -OSO2-C6-С14-арил, -COOH, -(CO) -C1-С5-алкил, с моно-, би- или трициклическими насыщенными или одно- или многократно ненасыщенными карбоциклами с 3-14 членами в кольце, с моно-, би- или трициклическими насыщенными или одно- или многократно ненасыщенными гетероциклами с 5-15 членами в кольце и 1-6 гетероатомами, предпочтительно N, O и S,
причем C6-С14-арильные группы и карбоциклические и гетероциклические заместители, со своей стороны, в случае необходимости, одно- или многократно могут быть замещены R4.
R2 и R3 могут быть одинаковыми или разными, причем только один из них может быть водородом. Далее, R2 и R3 могут обозначать
-C1-С5-алкил, в случае необходимости, одно- или многократно замещенный группами: -ОН, -SH, -NH2, -NH-C1-С6-алкил, -N(C1-С6-алкил)2, -NO2, -CN, -F, -Cl, -Br, -I, -O-C1-С6-алкил, -S-C1-С6-алкил, -фенил, -пиридил, -фенил, в случае необходимости, одно- или многократно замещенный группами -ОН, -SH, -NH2, -NH-C1-С3-алкил, -N(C1-С3-алкил)2, -NO2, -CN, -COOH, -COO-C1-С3-алкил, -F, -Cl, -Br, -O-C1-С3-алкил, -S-C1-С3-алкил, -пиридил, в случае необходимости, одно- или многократно замещенный группами -NO2, -CN, -COOH, -COO-C1-С3-алкил, -Cl, -Br, -O-C1-С3-алкил, -S-C1-С3-алкил, а также
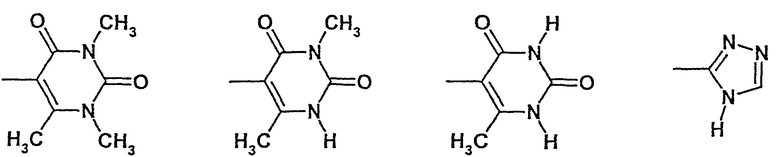
Вместе группа -NR2R3 может обозначать:
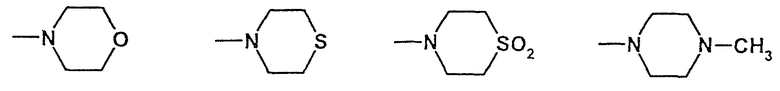
R4 обозначает
-H, -ОН, -SH, -NH2, -NH-C1-С6-алкил, -N(C1-С6-алкил)2, -NH-C6-С14-арил, -N(C6-С14-арил)2, -N(C1-С6-алкил)(C6-С14-арил), -NHCO-C1-С6-алкил, -NO2, -CN, -COOH, -COO-C1-С6-алкил, -(CO)-C1-С6-алкил, -(CS)-C1-С6-алкил, -F, -Cl, -Br, -I, -O-C1-С6-алкил, -O-C6-С14-арил, -S-C1-С6-алкил, -S-C6-С14-арил, -SO-C1-С6-алкил, -SO2-C1-С6-алкил.
В 7-азаиндолах формулы 1 согласно изобретению остаток R1 предпочтительно является С1-С10-алкильным остатком. Такой алкильный остаток может быть линейным, разветвленным или циклическим и предпочтительно является линейным. Особенно предпочтительны алкильные остатки с 1 до 6, еще более предпочтительны алкильные остатки с 1 до 4 атомов углерода. В следующей предпочтительной форме выполнения R1 обозначает С2-С10, в особенности С2-С6, и наиболее предпочтительно -С2-С4-алкильный остаток. Алкенильный остаток может быть одно- или многократно, например, дву- или трехкратно ненасыщенным. В случае алкенильного остатка речь может идти о неразветвленном, разветвленном или циклическом углеводородном остатке. Особенно предпочтительными являются остатки R1, в которых алкильный или алкенильный остаток одно- или многократно, например, двукратно, трехкратно, четырехкратно или пятикратно замещены. Особенно предпочтителен остаток R1, замещенный С1-алкильным (например, метильным) остатком. Из вышеназванных заместителей для алкильной или алкенильной группы остатка R1 особенно предпочтительными заместителями являются -OH, -F, -Cl, -Br, -I, -C1-C4-алкоксигруппа. Далее, предпочтительны заместители, в которых, в случае необходимости, имеющийся алкильный остаток содержит 1 до 4 атомов углерода и, в случае необходимости, имеющийся арильный остаток содержит 6 до 10 атомов углерода. Из карбоциклов предпочтительным является фенильный остаток, в особенности замещенный фенильный остаток, предпочтительно замещенный -F, -Cl, -Br, -I, -C1-C6-алкокси- или гидроксигруппой. Из гетероциклов предпочтительны такие, которые характеризуются по крайней мере одним гетероатомом, выбранным из группы N, O или S. Особенно предпочтительным у гетероциклов является пиридильный остаток, а также изоксазольный остаток, особенно 3,5-диметилизоксазольный остаток. Примером конденсированных карбоциклических заместителей является нафтильный остаток.
Особенно предпочтительно R1 обозначает группу, включающую циклический углеводородный остаток, как, например, циклопропилметил-, линейный углеводород, как, например, н-гексил-, линейный углеводород, замещенный алкоксильным остатком, как, например, 2-метоксиэтил-, разветвленный углеводородный остаток, как, например, изобутил-, ненасыщенный углеводородный остаток, как, например, 2-метилпропен-3-ил-, или углеводородный остаток, содержащий ароматическую группу, которая, в случае необходимости, может быть замещена, как, например, 4-фторбензил-, 3-метоксибензил-, 4-метоксибензил-, 4-хлорбензил-, 4-метилбензил-, 3-гидроксибензил- или 4-гидроксибензил, группу, содержащую гетероароматический углеводородный остаток, как, например, 4-пиридилметил- или 3,5-диметилизоксазол-4-метил-, или группу, содержащую конденсированный ароматический углеводород, как, например, 1-нафтилметил. Заместители у атома азота, R2 иR3, в предпочтительной форме выполнения могут представлять собой, в случае необходимости, замещенный С1-С5, в особенности С1-С3, и особенно предпочтительно С1 (соответственно метильный) алкильный остаток.
Один из остатков R2 или/и R3 предпочтительно обозначает остаток, включающий гетероароматический углеводород, как, например, 4-пиридилметил-, причем гетероароматический углеводород может быть замещен предпочтительно галогеном, как, например, 3,5-дихлор-4-пиридил-. В следующей предпочтительной форме выполнения для остатков R2 или/и R3 речь идет о морфолиноостатках. Далее, предпочтительными являются остатки R2 и R3, включающие ароматический углеводород, предпочтительно замещенный, в особенности галогеном или карбоксигруппой, как, например, 2,6-дихлорфенил-, 4-карбоксифенил-, 4-этоксикарбонилфенил-, 3,4-диметоксифенил-. Далее, как R2, так и R3, предпочтительно обозначают метоксиэтил-. В следующей предпочтительной форме выполнения R2 или R3 обозначает остаток:
или вместе группа -NR2R3:
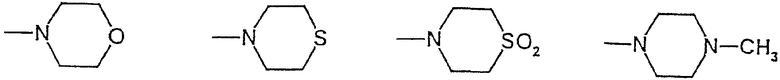
Далее изобретение касается физиологически совместимых солей соединений согласно формуле 1.
Физиологически совместимые соли получают обычным способом посредством нейтрализации оснований неорганическими или органическими кислотами или посредством нейтрализации кислот неорганическими или органическими основаниями. В качестве неорганических кислот имеют в виду, например, соляную кислоту, серную кислоту, фосфорную кислоту или бромистоводородную кислоту, в качестве органических кислот, например, карбоновые, сульфо- или сульфоновые кислоты, как уксусная кислота, винная кислота, молочная кислота, пропионовая кислота, гликолевая кислота, малоновая кислота, малеиновая кислота, фумаровая кислота, дубильная кислота, сукциновая кислота, альгининовая кислота, бензойная кислота, 2-феноксибензойная кислота, 2-ацетоксибензойная кислота, коричная кислота, миндальная кислота, лимонная кислота, яблочная кислота, салициловая кислота, 3-аминосалициловая кислота, аскорбиновая кислота, эмбоновая кислота, никотиновая кислота, изоникотиновая кислота, щавелевая кислота, аминокислоты, метансульфокислота, этансульфокислота, 2-гидроксиэтансульфокислота, этан-1,2-дисульфокислота, бензолсульфокислота, 4-метилбензолсульфокислота или нафталин-2-сульфокислота. В качестве неорганических оснований имеют в виду, например, гидроксид натрия, гидроксид калия, аммиак, а также в качестве органических оснований - амины, однако предпочтительно третичные амины, как триметиламин, триэтиламин, пиридин, N,N-диметиланилин, хинолин, изохинолин, а-пиколин, b-пиколин, g-пиколин, хинальдин или пиримидин.
Далее, физиологически совместимые соли соединений согласно формуле 1 могли быть получены благодаря тому, что производные, имеющие третичные аминогруппы, переводятся в соответствующие четвертичные аммониевые соли известным образом с помощью кватернизирующего средства. Под кватернизирующим средством имеют в виду, например, алкилгалогениды, как метилиодид, этилбромид и н-пропилхлорид, а также арилалкилгалогениды как бензилхлорид или 2-фенилэтилбромид.
Далее, изобретение касается соединений формулы 1, содержащих асимметрический атом углерода, D-формы, L-формы и D, L-смесей, а также, в случае нескольких асимметрических атомов углерода - диастереомерных форм. Соединения формулы 1, которые содержат асимметрические атомы углерода и, как правило, образуются в виде рацематов, могут быть разделены на оптически активные изомеры известным образом, например, оптически активной кислотой. Возможно также с самого начала использовать оптически активное исходное вещество, причем в таком случае в качестве конечного продукта получается соответствующее оптически активное или диастереомерное соединение.
Для соединений согласно изобретению были обнаружены фармакологически значимые свойства, которые могут быть терапевтически использованы.
Соединения согласно изобретению являются ингибиторами высвобождения TNFa.
Поэтому соединения могут быть использованы для подавления высвобождения TNFa.
Итак, объектом данного изобретения являются соединения согласно формуле 1 и их соли, а также фармацевтические композиции, содержащие эти соединения или их соли, которые могут быть использованы для лечения заболеваний, при которых необходимо ингибирование TNFa. К этим заболеваниям относятся, например, воспаления суставов, включая артрит и ревматоидный артрит, а также другие артритные заболевания, как ревматоидный спондилез и остеоартрит. Следующей возможностью использования является лечение пациентов, которые больны остеопорозом, сепсисом, септическим шоком, грамотрицательным сепсисом, токсическим синдромом шока, синдромом удушья, астмой или другими хроническими легочными заболеваниями, болезнями резорбции костей или реакциями отторжения трансплантата, или другими аутоиммунными заболеваниями, как красная волчанка, рассеянный склероз, гломерулонефрит и увеит, инсулинозависимый сахарный диабет, а также хроническая демиелинизация.
Кроме того, соединения согласно изобретению также могут быть использованы для лечения инфекций, как вирусные инфекции и паразитарные инфекции, например, для лечения малярии, лейшманиоза, лихорадки, вызванной инфекцией, инфекционной миалгии, СПИДа и кахексии.
Соединения согласно изобретению являются ингибиторами фосфодиэстеразы 4.
Поэтому соединения согласно изобретению могут быть использованы для подавления фосфодиэстеразы 4.
Итак, объектом этого изобретения являются соединения согласно формуле 1 и их соли, а также фармацевтические препараты, содержащие эти соединения или их соли, которые могут быть использованы для лечения заболеваний, при которых необходимо ингибирование фосфодиэстеразы 4.
Так, соединения согласно изобретению могут быть использованы в качестве бронходилататоров и для профилактики астмы. Далее, соединения формулы 1 являются ингибиторами накопления эозинофилов и их активности. Вследствие этого соединения согласно изобретению могут быть использованы также при заболеваниях, при которых играют роль эозинофилы. К этим заболеваниям относятся, например, воспалительные заболевания дыхательных путей, как бронхиальная астма, аллергический ринит, аллергический конъюнктивит, атопический дерматит, экзема, аллергический ангиит, воспаления, которым способствуют эозинофилы, как эозинофильный фасцит, эозинофильная пневмония и PIE -синдром (легочная эозинофильная инфильтрация), крапивница, язвенный колит, болезнь Крона и пролиферативные заболевания кожи, как псориаз или кератоз.
Объектом данного изобретения является также способность соединений согласно формуле 1 и их солей ингибировать как высвобождение TNFa in vitro, так и индуцированную LPS (липополисахаридами) легочную нейтрофильную инфильтрацию у крыс in vivo. Совокупность этих обнаруженных фармакологически важных свойств обосновывает то, что соединения согласно формуле 1 и их соли, а также фармацевтические препараты, содержащие эти соединения или их соли, могут быть терапевтически использованы для лечения хронически обструктивных легочных заболеваний.
Далее, соединения согласно изобретению обладают нейрозащитными свойствами и могут использоваться для лечения заболеваний, при которых требуется нейрозащитное действие. Такими заболеваниями являются, например, старческое слабоумие (болезнь Альцгеймера), потеря памяти, болезнь Паркинсона, депрессии, инсульт и интермиттирующая хромота.
Следующей возможностью использования соединений согласно изобретению является профилактика и лечение болезней простаты, как, например, доброкачественная гиперплазия простаты, поллакиурия, ноктурия (Nocturie), а также лечение недержания, колик, вызванных мочевыми камнями и мужских и женских сексуальных дисфункций.
В заключение, соединения согласно изобретению также могут быть использованы для ингибирования возникновения лекарственной зависимости при неоднократном приеме анальгетиков, как, например, морфий, а также для уменьшения развития толерантности при неоднократном приеме этих анальгетиков.
Для получения лекарственных средств наряду с обычными вспомогательными веществами, носителями и добавками используют эффективную дозу соединений согласно изобретению или их солей.
Дозировка биологически активных веществ может варьироваться в зависимости от способа приема, возраста, веса пациента, вида и тяжести заболевания, подвергаемого лечению, и подобных факторов.
Дневная доза может вводиться в виде разовой дозы, принимаемой однократно, или разделенной на 2 или несколько дневных доз и составляет, как правило, 0,001-100 мг.
Под формами введения имеют в виду оральные, парентеральные, внутривенные, подкожные, локальные, ингаляционные и интраназальные композиции.
Для применения используют обычные галеновые препаративные готовые формы, как таблетки, драже, капсулы, диспергируемые порошки, грануляты, водные растворы, водные или масляные суспензии, сиропы, соки или капли.
Твердые лекарственные формы могут содержать инертные ингредиенты и носители, как, например, карбонат кальция, фосфат кальция, фосфат натрия, лактоза, крахмал, маннит, альгинат, желатина, гуаран, стеарат магния или стеарат алюминия, метилцеллюлоза, тальк, высокодисперсные кремневые кислоты, силиконовое масло, высокомолекулярные жирные кислоты (как стеариновая кислота), желатина, агар-агар или растительные или животные жиры и масла, твердые высокомолекулярные полимеры (как полиэтиленгликоль); оральные препаративные готовые формы могут содержать, при желании, дополнительные придающие вкус вещества и/или подсластители.
Жидкие лекарственные формы могут быть стерилизованы и/или, в случае необходимости, содержать вспомогательные вещества, как консерванты, стабилизаторы, смачивающие средства, пенетрационные средства, эмульгаторы, разбрызгивающие средства, агенты растворения, соли, сахара или спирты сахаров для регулирования осмотического давления или для буферности и/или регуляторы вязкости.
Такого рода добавками являются, например, тартратный или цитратный буфер, этанол, комплексообразователь (как этилендиамин-тетрауксусная кислота и ее нетоксичные соли). Для регулирования вязкости используют высокомолекулярные полимеры, как, например, жидкий полиэтиленоксид, микрокристаллическая целлюлоза, карбоксиметилцеллюлоза, поливинилпирролидон, декстран или желатина. Твердыми носителями являются, например, крахмал, лактоза, маннит, метилцеллюлоза, тальк, высокодисперсная кремневая кислота, высокомолекулярные жирные кислоты (как стеариновая кислота), желатина, агар-агар, фосфат кальция, стеарат магния, животные и растительные жиры, твердые высокомолекулярные полимеры, как полиэтиленгликоль.
Масляные суспензии для парентерального или локального применения могут быть растительными, синтетическими или полусинтетическими маслами, как, например, жидкие сложные эфиры жирных кислот, при необходимости, с 8 до 22 С-атомами в цепи жирных кислот, например, пальмитиновой, лауриновой, тридециловой, маргариновой, стеариновой, арахиновой, миристиновой, бегеновой, пентадециловой, линолевой, элаидиновой, брассидиновой, эруковой или олеиновой кислот, которые этерифицицированы одно- до трехатомными спиртами с 1 до 6 С-атомами, как, например, метанол, этанол, пропанол, бутанол, пентанол или их изомеры, гликоль или глицерин. Подобными эфирами жирных кислот являются, например, коммерчески доступные миглиоль, изопропилмиристат, изопропилпальмитат, изопропилстеарат, эфир полиэтиленгликоля (ПЭГ) и 6-каприновой кислоты, сложные эфиры каприл/каприновой кислоты и насыщенных жирных спиртов, полиоксиэтиленглицеринтриолеаты, этилолеат, воскоподобные сложные эфиры жирных кислот, как искусственный жир уточной крестцовой железы, изопропиловый эфир кислот кокосового масла, олеиловый эфир олеиновой кислоты, дециловый эфир олеиновой кислоты, этиловый эфир молочной кислоты, дибутилфталат, диизопропиловый эфир адипиновой кислоты, полиоловые эфиры жирных кислот и другие. Также пригодны силиконовые масла различной вязкости или жирные спирты, как изотридециловый спирт, 2-октилдодеканол, цетилстеариловый спирт или олеиловый спирт, жирные кислоты, как, например, олеиновая кислота. Далее, могут применяться растительные масла, как касторовое масло, миндальное масло, оливковое масло, кунжутное масло, хлопковое масло, арахисовое масло или соевое масло.
Под растворителем, гелеобразователем и агентом растворения имеют в виду воду или смешивающиеся с водой растворители. Пригодными являются, например, спирты, как, например, этанол или изопропиловый спирт, бензиловый спирт, 2-октилдодеканол, полиэтиленгликоли, фталаты, адипаты, пропиленгликоль, глицерин, ди- или трипропиленгликоль, воски, мелилцеллозольв, целлозольв, сложные эфиры, морфолин, диоксан, диметилсульфоксид, диметилформамид, тетрагидрофуран, циклогексанон и т. д.
В качестве пленкообразователя могут применяться простые эфиры целлюлозы, которые могут растворяться или набухать как в воде, так и в органических растворителях, как, например, гидроксипропилметилцеллюлоза, метилцеллюлоза, этилцеллюлоза или растворимые крахмалы.Также вполне возможны смешанные формы между геле- и пленкообразователями. Здесь, прежде всего, подходят для использования ионные макромолекулы, как, например, натрийкарбоксиметилцеллюлоза, полиакриловая кислота, полиметакриловая кислота и их соли, амилопектинсемигликолят натрия, альгиновая кислота или пропиленгликольальгинат в виде натриевой соли, гуммиарабик, ксантановая резина, гуаран или карраген.
В качестве дальнейших вспомогательных средств для препаративных готовых форм могут быть введены: глицерин, парафины различной вязкости, триэтаноламин, коллаген, аллантоин, новантисоловая кислота. Также для препаративных готовых форм может быть необходимо применение тензидов, эмульгаторов или смачивающих агентов, как, например, лаурилсульфат натрия, сульфаты простых эфиров жирных спиртов, ди-Na-N-лаурил-β-иминодипропионат, полиоксиэтилированное касторовое масло или сорбитан-моноолеат, сорбитан-моностеарат, полисорбаты (например, Tween), цетиловый спирт, лецитин, глицеринмоностеарат, полиоксиэтиленстеарат, алкилфеноловые эфиры полигликолей, цетилтриметиламмоний хлорид или соли моноэтаноламина и эфира моно-/диалкилполигликоля и ортофосфорной кислоты. Также, в случае необходимости, для композиций желаемых препаративных форм могут требоваться стабилизаторы, как монтмориллонит или коллоидные кремневые кислоты для стабилизации эмульсий, или активные вещества для предотвращения разложения, как антиоксиданты, например, токоферол или бутилгидроксианизол, или консерванты, как сложный эфир п-гидроксибензойной кислоты.
Препараты для парентерального введения могут находиться в отдельных дозированных единичных формах, как, например, ампулы или флаконы. Преимущественно используют растворы биологически активных веществ, предпочтительно водные растворы и, прежде всего, изотонические растворы, а также суспензии. Эти формы инъекций могут использоваться в виде готового препарата или приготовлены только непосредственно перед использованием посредством смешивания активного соединения, например лиофилизата, в случае необходимости, с другим твердым носителем, с желаемым растворителем или суспендирующим агентом.
Интраназальные препараты могут находиться в виде водных или масляных растворов или в виде водных или масляных суспензий. Они также могут находиться в виде лиофилизатов, которые перед использованием смешиваются с пригодным растворителем или суспендирующим агентом.
Получение, фасовка и закупоривание препаратов происходит в общепринятых антимикробных и асептических условиях.
Далее, изобретение касается способа получения соединений согласно изобретению.
Согласно изобретению получают соединения общей формулы 1 с приведенными ранее значениями R1, R2, R3 и n=1,
тем, что 7-азаиндол-3-карбоновые кислоты формулы 2 с идентичным значением R1
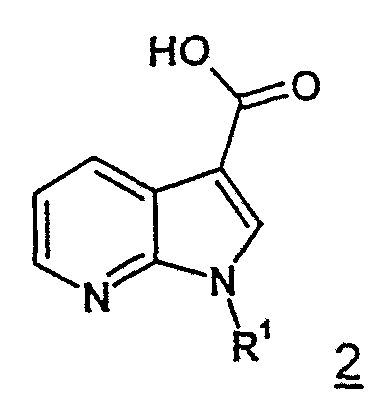
самим по себе известным способом посредством хлорангидридов кислот, предпочтительно тионилхлоридом или хлорангидридом щавелевой кислоты, прежде всего переводят в аналогичные хлорангидриды 7-азаиндол-3-карбоновых кислот формулы 3.
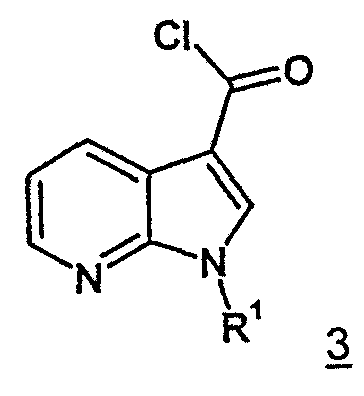
Из выделенных хлорангидридов 7-азаиндол-3-карбоновых кислот формулы 3 затем посредством взаимодействия с первичным или вторичным амином получаются соединения согласно изобретению общей формулы 1 с приведенными ранее значениями R1, R2, R3 и n=1. Реакция предпочтительно проходит в присутствии вспомогательного основания. В качестве вспомогательных оснований могут быть использованы избыток амина, использованного при реакции в качестве реагента, третичный амин, предпочтительно пиридин или триэтиламин, а также неорганические основания, предпочтительно гидроксиды щелочных металлов или гидриды щелочных металлов.
Согласно изобретению соединения общей формулы 1 с приведенными ранее значениями R1, R2, R3 и n=2 получают
тем, что 7-азаиндолы формулы 4 с идентичным значением R1
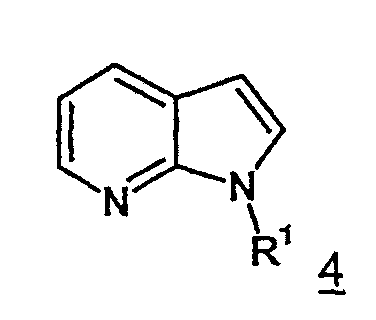
самим по себе известным способом посредством ацилирования хлорангидридом щавелевой кислоты, прежде всего, переводят в аналогичные хлорангидриды 7-азаиндол-3-ил- глиоксиловой кислоты формулы 5.
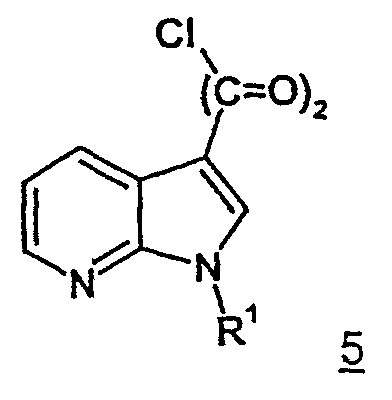
Из выделенных хлорангидридов 7-азаиндол-3-ил- глиоксиловой кислоты формулы 5 затем посредством взаимодействия с первичным или вторичным амином получают соединения согласно изобретению общей формулы 1 с приведенными ранее значениями R1, R2, R3 и n=2. Реакция предпочтительно проходит в присутствии вспомогательного основания. В качестве вспомогательных оснований могут быть использованы избыток амина, использованного при взаимодействии в качестве реагента, третичный амин, предпочтительно пиридин или триэтиламин, а также неорганические основания, предпочтительно гидроксиды щелочных металлов или гидриды щелочных металлов.
Примеры выполнения
Вариант способа получения соединений согласно изобретению формулы 1, где n=1:
Пример 1. Амид N-(4-пиридилметил)-1-циклопропилметил-7-азаиндол-3-карбоновой кислоты
1,87 г 1-циклопропилметил-7-азаиндол-3-карбоновой кислоты (8,6 ммоль) суспендируют в 15 мл дихлорметана. При охлаждении водой добавляют 1,8 мл хлорангидрида щавелевой кислоты (17,4 ммоль). Реакционную смесь перемешивают в течение 8 часов. При этом выкристаллизовывается хлорангидрид 1-циклопропилметил-7-азаиндол-3-карбоновой кислоты. Его выделяют и растворяют в 18 мл тетрагидрофурана (ТГФ).
1,14 г гидрида натрия (60%-ного) суспендируют в 21 мл ТГФ. При перемешивании при температуре около 10°С прикапывают раствор 0,93 г 4-аминометилпиридина (8,6 ммоль) в 21 мл ТГФ. Через приблизительно 15 минут к реакционной смеси прикапывают ранее полученный раствор хлорангидрида 1-циклопропилметил-7-азаиндол-3-карбоновой кислоты. После этого всю смесь кипятят с обратным холодильником 3 часа. После охлаждения к реакционной смеси прибавляют 36 мл этилового эфира уксусной кислоты и 36 мл воды. Фазы разделяют и органическую фазу промывают водой. Растворитель отгоняют, и осадок перекристаллизовывают из этанола.
Выход: 1,3 г (50% от теории).
Температура плавления: 187-189°С.
При использовании данного способа получения может быть получено большое число других соединений формулы 1, где n=1, примеры которых приведены далее:
Вариант способа получения соединений согласно изобретению формулы 1, где n=2:
Пример 12. Амид N-(3,5-дихлорпиридин-4-ил)-[1-(3-метоксибензил)-7-азаиндол-3-ил]глиоксиловой кислоты.
3,57 г 1-(3-метоксибензил)-7-азаиндола (15 ммоль) растворяют в 50 мл трет. бутилметилового эфира. При 0°С при перемешивании прикапывают раствор 1,54 мл хлорангидрида щавелевой кислоты (18 ммоль) в 10 мл трет. бутилметилового эфира. После этого смесь кипятят 2 часа с обратным холодильником. Затем растворитель отгоняют под вакуумом. Образующийся хлорангидрид 1-(3-метоксибензил)-7-азаиндол-3-илглиоксиловой кислоты получают в виде твердого осадка, который суспендируют в 50 мл тетрагидрофурана (ТГФ).
К суспензии 2 г гидрида натрия в 20 мл ТГФ при -5°С прикапывают раствор 2,4 г 4-амино-3,5-дихлорпиридина (15 ммоль) в 30 мл ТГФ. При перемешивании смесь в течение 1 часа термостатируют при 20°С. Затем при температуре около 0°С прикапывают ранее полученную суспензию хлорангидрида 1-(3-метоксибензил)-7-азаиндол-3-илглиоксиловой кислоты. В заключение реакционную смесь кипятят 4 часа с обратным холодильником. Растворитель отгоняют под вакуумом. Осадок смешивают с 50 мл этилового эфира уксусной кислоты и 50 мл воды. Фазы разделяют. Органическую фазу промывают водой. Растворитель отгоняют под вакуумом. Осадок перекристаллизовывают из изопропанола.
Выход: 3,5 г (51,5% от теории).
Температура плавления: 165-167°С.
При использовании данного способа получения может быть получено большое число других соединений формулы 1, где n=2, примеры которых приведены далее:
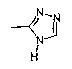
Соединения согласно изобретению являются сильными ингибиторами фосфодиэстеразы 4 и высвобождения TNFa. Их терапевтический потенциал подтверждается in vivo, например, посредством подавления астматической реакции поздней фазы (эозинофилии), а также посредством влияния сосудистой проницаемости, индуцированной аллергеном, у активно сенсибилизированных крыс Brown-Norway.
Ингибирование фосфодиэстеразы
Активность PDE 4 определяют при препарировании ферментов из человеческих полиморфноядерных лимфоцитов (PMNL), активность PDE 2, 3 и 5 с PDE из человеческих тромбоцитов. Человеческую кровь стабилизировали против коагуляции цитратом. Посредством центрифугирования при 700g в течение 20 минут при комнатной температуре обогащенную тромбоцитами плазму в надосадочной жидкости отделяют от эритроцитов и лейкоцитов. Тромбоциты посредством ультразвука подвергают лизису и используют в анализе PDE 3 и PDE 5. Для определения активности PDE 2 очищают цитозольную фракцию тромбоцитов с помощью анионообменной колонки посредством градиента NaCl и получают пик PDE 2 для анализа. PMNL для определения PDE 4 выделяют посредством последующей седиментации декстрана и последующего градиентного центрифугирования с конволютом с фиколом (Ficoll-Paque). После двукратной промывки клеток еще содержащиеся эритроциты подвергают лизису посредством добавления 10 мл гипотонического буфера (155 мМ NH4Cl, 10мМ NaHCO3, 0,1мМ EDTA, pH=7,4) в течение 6 минут при 4°С. Пока еще неповрежденные PMNL еще 2 раза промывают PBS и подвергают лизису посредством ультразвука. Надосадочная жидкость после часового центрифугирования при 4°С при 48000g содержит цитозольную фракцию PDE 4 и используется для определения PDE 4.
Активность фосфодиэстеразы определяют с некоторыми модификациями по методу Томпсона и др. (Thompson, W.J.; Appleman, M.M., Assay of cyclic nucleotide phosphodiesterase and resolution of multiple molecular forms of the enzyme. Adv. Cycl. Nucl. Res. 1979, 10, 69-92).
Реакционные смеси содержат 50 мМ Tris-HCl (pH 7,4), 5 мМ MgCl2, ингибиторы в варьирующихся концентрациях, соответствующие ферменты, подвергнутые препарированию, а также другие компоненты, необходимые для учета отдельных изоферментов (смотри ниже). Посредством добавления субстрата 0,5 мкМ [3H]-cAMP или [3H]-cGMP (около 6000 CMP/тест) начинают реакцию. Конечный объем составляет 100 мл. Тестируемые вещества используют в качестве маточных растворов в ДМСО. Концентрация ДМСО в реакционной смеси составляет 1% об./об. При этой концентрации ДМСО не оказывает влияния на активность PDE. После начала реакции посредством добавления субстрата образцы инкубируют в течение 30 минут при 37°С. Посредством нагревания тестируемых пробирок в течение 2 минут при 110оС реакцию останавливают. Образцы на следующие 10 минут оставляют на льду. После добавления 30 мкл 5'-нуклеотидазы (1 мг/мл, из суспензии змеиного яда Crotalus adamanteus) следует инкубация в течение 10 минут при 37°С. Процесс в образцах прекращают на льду, по мере надобности добавляют 400 мкл смеси Dowex-вода-этанол (1+1+1), хорошо перемешивают и вновь 15 минут инкубируют на льду. Реакционные сосуды центрифугируют 20 минут при 3000g. 200 мкл аликвоты надосадочной жидкости переводят непосредственно в сцинтилляционный сосуд. После добавления 3 мл сцинтиллятора измеряют образцы в бета-сцинтилляционном счетчике.
Для определения активности PDE 4, 3 и 2 в качестве субстрата используют [3H]-cAMP, для определения активности PDE 5 используют [3H]-cGMP. Неспецифические активности ферментов, по мере надобности, определяют в присутствии 100 мкМ Rolipram для PDE 4 и в присутствии 100 мкМ IBMX для определения PDE 3 и 5 и вычитают из тестируемых величин. Исходные смеси для инкубации анализируемой PDE 3 содержат 10 мкМ Rolipram, чтобы подавить возможные загрязнения посредством PDE 4. PDE 2 тестируют посредством анализа SPA фирмы Amersham. Анализ проводят в присутствии активатора PDE 2 (5 мкМ cGMP).
Для соединений согласно изобретению относительно ингибирования фосфодиэстеразы 4 было определено значение IC50 в области от 10-9 до 10-5 М. Селективность по отношению к PDE типов 2, 3 и 5 имела фактор 100-10000.
Результаты подавления PDE 4 для выбранных примеров выполнения обобщены в следующей таблице:
Подавление высвобождения TNFa из клеток носовых полипов
Последовательность опыта в основном соответствует методу, описанному Campbell A.M. и Bousquet J. (Anti-allergic activity of H1-blockers. Int. Arch. Allergy Immunol., 1993, 101, 308-310). Исходный материал представлял собой носовые полипы (OP-материал) пациентов, подвергавшихся хирургическому лечению.
Ткань промывали RPMI 1640 и затем обрабатывали протеазой (2,0 мг/мл), коллагеназой (1,5 мг/мл), гиалуронидазой (0,75 мг/мл) и DNA-зой (0,05 мг/мл) через 2 ч при 37°С (1 г ткани в 4 мл RPMI 1640 с ферментом). Полученные клетки, смесь из клеток эпителия, моноцитов, макрофагов, лимфоцитов, фибробластов и гранулоцитов фильтруют и посредством повторяющегося центрифугирования промывают питательным раствором, пассивно сенсибилизируют добавлением человеческого IgE и суспензию клеток используют в концентрации 2 млн клеток/мл в RPMI 1640 (добавляя антибиотики, 10% плодной телячьей сыворотки, 2 мМ глутамина и 25 мМ Hepes). Эту суспензию распределяют в 6-луночном планшете для клеточных культур (1 мл на лунку). Клетки предварительно инкубируют с испытуемыми веществами в различных конечных концентрациях 30 минут и затем стимулируют добавлением анти-IgE (7,2 мкг/мл) для высвобождения TNFa. Максимальное высвобождение происходит в питательной среде приблизительно через 18 часов. В этот период клетки инкубируют при 37°С и 5% СО2. Питательную среду (надосадочную жидкость) получают посредством центрифугирования (5 мин 4000 об/мин) и хранят при -70°С вплоть до определения цитокинов. Определение TNF происходит в надосадочной жидкости с так называемыми сэндвичевыми структурами ELISAs (Grundmaterial Pharmingen), с которыми могут быть обнаружены концентрации цитокина в области от 30-1000 пг/мл.
Клетки, не стимулированные анти-IgE, едва продуцируют TNFa, стимулированные клетки, напротив, продуцируют большие количества TNFa, что, например, может быть уменьшено посредством ингибиторов PDE 4 в зависимости от дозы. Из процентного подавления (высвобождение TNFa клеток, стимулированных анти-IgE, =100%) испытуемыми веществами при различных концентрациях рассчитывают IC50 (концентрацию 50%-ного ингибирования).
Для соединений согласно изобретению было определено значение IC50 в области от 10-7 до 10-5 М.
Результаты подавления высвобождения TNFa для выбранных примеров выполнения обобщены в следующей таблице:
Подавление поздней фазы эозинофилии через 48 ч после ингаляционного овальбуминового вызова у активно сенсибилизированных крыс Brown-Norway.
Подавление легочной эозинофильной инфильтрации с помощью веществ согласно изобретению испытывают на активно сенсибилизированных против овальбумина (OVA) мужских особях крыс Brown-Norway (200-250 г). Сенсибилизация происходит посредством подкожных инъекций суспензии 10 мкг OVA вместе с 20 мг гидроксида алюминия в качестве вспомогательного средства в 0,5 мл физиологического раствора поваренной соли, на животное в 1, 14 и 21 день. Дополнительно к этому животным вводили через одинаковые периоды времени вакцину Bordetella pertussis с разбавлением на животное 0,25 мл внутрибрюшинно. На 28 день эксперимента в открытые плексигласовые боксы объемом 1 л по отдельности помещаются животные, у которых голова и нос закрыты специальными устройствами (Expositionsgerdt). Животных подвергают воздействию аэрозоля из 1,0%-ной овальбуминовой суспензии (аллергический вызов). Овальбуминовый аэрозоль образуется посредством распылителя (Bird micro nebulizer, Palm Springs CA, USA), действующим с помощью сжатого воздуха (0,2 МПа). Время выдержки составляет 1 час, причем контрольные животные опрыскиваются аэрозолем из 0,9%-ного раствора поваренной соли также в течение 1 часа.
Через 48 часов аллергеновый вызов приводит к массовому внедрению эозинофильных гранулоцитов в легкие животных. К этому периоду животным вводили сверхдозу наркотического средства этилуретана (1,5 г/кг веса тела внутрибрюшинно) и проводили бронхоальвеолярное промывание (BAL) с 3Ч4 мл балансового раствора Хэнкса. Общее число клеток и некоторое количество эозинофильных гранулоцитов промывной жидкости после BAL затем определяют с помощью автоматического прибора дифференцирования клеток (Bayer Diagnostics Technicon H1E). Для каждого животного рассчитывают эозинофилы (EOS) в BAL в млн/животное: EOS/мкл·регенерированное BAL (BAL-Recovery) (мл)=EOS/животное.
В каждом тесте имеется 2 контрольные группы (опрыскивание физиологическим раствором поваренной соли и опрыскивание раствором овальбумина).
Процентное подавление эозинофилов в группе, подвергнутой лечению веществами, рассчитывают по следующей формуле:
{((OVAC-SC)-(OVAD-SC))/(OVAC-SC)}·100%=% подавления(SC=контрольная группа, обработанная индифферентной основой лекарственного препарата и 0,9%-ным раствором поваренной соли; OVAC=контрольная группа, обработанная индифферентной основой лекарственного препарата и 1%-ной суспензией овальбумина; OVAD=испытуемая группа, подвергнутая лечению активным веществом и обработанная 1%-ной суспензией овальбумина).
Тестируемые вещества применяют внутрибрюшинно или орально в виде суспензии в 10% полиэтиленгликоле 300 и 0,5%-ной 5-гидроксиэтилцеллюлозе за 2 часа перед вводом аллергена. Контрольные группы лечат соответственно форме применения тестируемого вещества индифферентной основой лекарственного препарата.
Соединения согласно изобретению подавляют позднюю фазу эозинофилии после внутрибрюшинного введения от 10 мг/кг на 30-100% и после орального приема 30 мг/кг на 30-75%.
Таким образом, соединения согласно изобретению особенно пригодны для получения лекарственных средств для лечения заболеваний, связанных с действием эозинофилов.
Результаты подавления эозинофилии для выбранных примеров выполнения обобщены в следующей таблице:
Подавление легочной нейтрофилии, индуцированной липополисахаридами (LPS), у крыс Lewis.
Подавление легочной нейтрофильной инфильтрации посредством веществ согласно изобретению испытывают на мужских особях крыс Lewis (200-250 г). В день эксперимента в открытые плексигласовые боксы объемом 1 л по отдельности помещаются животные, у которых голова и нос закрыты специальными устройствами. Животных подвергают воздействию аэрозоля из липополисахаридной суспензии (100 мкг LPS/мл 0,1% раствора гидроксиламина) в PBS (провокация LPS). Аэрозоль LPS/гидроксиламин образуется посредством распылителя (Bird micro nebulizer, Palm Springs CA, USA), действующего с помощью сжатого воздуха (0,2 МПа). Время выдержки составляет 40 минут, причем контрольные животные опрыскиваются аэрозолем из 0,1%-ного раствора гидроксиламина в PBS также в течение 40 минут.
Через 6 часов провокация LPS приводит к максимальному массовому внедрению нейтрофильных гранулоцитов в легкие животных. К этому периоду животным вводят сверхдозу наркотического средства этилуретана (1,5 г/кг веса тела внутрибрюшинно) и проводят бронхоальвеолярное промывание (BAL) с 3Ч4 мл балансового раствора Хэнкса. Общее число клеток и некоторое количество нейтрофильных гранулоцитов промывной жидкости BAL затем определяют с помощью автоматического прибора дифференцирования клеток (Bayer Diagnostics Technicon H1E). Для каждого животного рассчитывают нейтрофилы (NEUTRO) в BAL в млн/животное: NEUTRO/мкл·регенерированное BAL (мл)=NEUTRO/животное.
В каждом тесте имеется 2 контрольные группы (опрыскивание 0,1%-ного раствором гидроксиламина в PBS и опрыскивание 100 мкг LPS/мл 0,1%-ного раствора гидроксиламина в PBS).
Процентное подавление нейтрофилов в экспериментальной группе, подвергнутой лечению веществами, рассчитывают по следующей формуле:
{((LPSC-SC)-(LPSD-SC))/(LPSC-SC)}·100%=% подавления
(SC=контрольная группа, обработанная индифферентной основой лекарственного препарата и 0,1%-ным раствором гидроксиламина; LPSC=контрольная группа, обработанная индифферентной основой лекарственного препарата и LPS (100 мкг/мл 0,1% раствора гидроксиламина); LPSD = испытуемая группа, подвергнутая лечению активным веществом и обработанная LPS (100 мкг/мл 0,1% раствора гидроксиламина).
Тестируемые вещества применяют орально в виде суспензии в 10% полиэтиленгликоле 300 и 0,5%-ной 5-гидроксиэтилцеллюлозе за 2 часа перед провокацией LPS. Контрольные группы лечат соответственно форме применения тестируемого вещества индифферентной основой лекарственного препарата.
Соединения согласно изобретению подавляют нейтрофилию после орального приема 1 мг/кг на 40-90% и, таким образом, особенно пригодны для получения лекарственных средств для лечения заболеваний, связанных с действием нейтрофилов.
Результаты подавления нейтрофилии для выбранных примеров выполнения обобщены в следующей таблице:
| название | год | авторы | номер документа |
|---|---|---|---|
| 7-АЗАИНДОЛЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2004 |
|
RU2349592C2 |
| ДИГИДРОБЕНЗОФУРАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2138498C1 |
| ГИДРОКСИИНДОЛЫ, ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ 4 И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1999 |
|
RU2217422C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИИНДОЛИЛГЛИОКСИЛАМИДОВ ВЫСОКОЙ ЧИСТОТЫ | 2003 |
|
RU2315046C2 |
| ПРОИЗВОДНЫЕ ФТАЛАЗИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ 4 | 2003 |
|
RU2329262C2 |
| ПРОИЗВОДНЫЕ 1-ФЕНИЛ-2-ПИРИДИНИЛ-АЛКИЛОВЫХ СПИРТОВ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2013 |
|
RU2637945C2 |
| ПРОИЗВОДНЫЕ ИНДОЛЬНОГО РЯДА В КАЧЕСТВЕ ИНГИБИТОРОВ p38 КИНАЗЫ | 2000 |
|
RU2278115C2 |
| ПРОИЗВОДНЫЕ 1-ФЕНИЛ-2-ПИРИДИНИЛАЛКИЛЬНЫХ СПИРТОВ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2012 |
|
RU2626956C2 |
| ПРОИЗВОДНЫЕ ИНДОЛИЛ-3-ГЛИОКСИЛОВОЙ КИСЛОТЫ - СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО (ВАРИАНТЫ) | 1999 |
|
RU2262339C2 |
| АМИДНОЕ ПРОИЗВОДНОЕ ПИРАЗОЛА | 2015 |
|
RU2658827C2 |
Описываются новые 7-азаиндолы общей формулы I
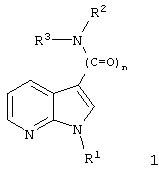
где n - равно 1 или 2, и
R1 - С2-С10-алкенил, одно- или многократно ненасыщенный, неразветвленный или разветвленный;
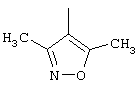
C1-C10-алкил, неразветвленный или разветвленный, незамещенный или однократно замещенный C1-С6-алкокси, нафтилом, пиридинилом, С3-С6-циклоалкилом, фенилом, который в свою очередь может быть замещен C1-С6-алкилом, галогеном, C1-С6-алкокси или гидрокси, или радикалом
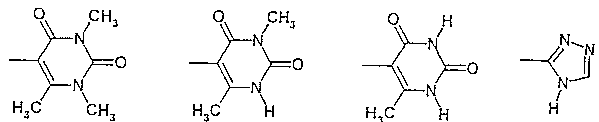
R2 и R3 - одинаковы или различны, причем только один из них может быть водородом, и обозначают C1-С5-алкил, возможно замещенный -О-C1-С6-алкилом или пиридилом, фенил, возможно дважды замещенный -F, -Cl, -Br, -O-C1-С3-алкилом или однократно замещенный -СООН или -COO-C1-С3-алкилом, пиридил, возможно замещенный дважды -Cl, -Br, или группы формулы
или R2 и R3 вместе N-атомом означают:
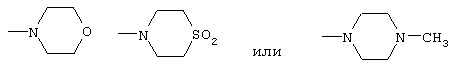
при условии, что, если n=1, одновременно не означают: R1=С1-6-алкил; R2=Н или С1-6-алкил и R3=

где R и R' независимо означают Cl или Br, обладающие ингибирующей активностью в отношении фосфодиэстеразы 4, лекарственное средство, их содержащее, способы получения и применение данных соединений для получения лекарственных средств. 9 н. и 8 з.п., 6 табл.
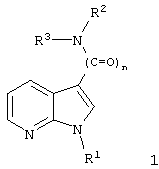
где n означает целое число 1 или 2;
R1 обозначает С2-С10-алкенил, одно- или многократно ненасыщенный, неразветвленный или разветвленный; C1-С10-алкил, неразветвленный или разветвленный, незамещенный или однократно замещенный C1-С6-алкокси, нафтилом, пиридинилом, C3-С6-циклоалкилом, фенилом, который в свою очередь может быть замещен C1-С6-алкилом, галогеном, C1-С6-алкокси или гидрокси или радикалом
R2 и R3 могут быть одинаковыми или разными, причем только один из них может быть водородом, и обозначают C1-С6-алкил, незамещенный или однократно замещенный группой -O-C1-С6-алкил или пиридил; фенил, незамещенный или дважды замещенный -F, -Cl, -Br, -O-C1-С3-алкилом или однократно замещенный -СООН или -COO-C1-С3-алкилом; пиридил, незамещенный или дважды замещенный -Cl, -Br, а также
или вместе группа -NR2R3 означает
при условии, что, если n=1, одновременно не означают
R1- C1-6-алкил,
R2 - Н или C1-6-алкил и
R3
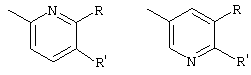
причем R, R' независимо друг от друга обозначают Cl или Br,
а также их физиологически совместимые соли.
амид N-(4-пиридилметил)-1 -циклопропилметил-7-азаиндол-3-карбоновой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-1-изобутил-7-азаиндол-3-карбоновой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-1-гексил-7-азаиндол-3-карбоновой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-1-циклопропилметил-7-азаиндол-3-карбоновой кислоты,
амид N-(4-пиридилметил)-1-(4-фторбензил)-7-азаиндол-3-карбоновой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-1-(4-фторбензил)-7-азаиндол-3-карбоновой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-1-(4-метоксибензил)-7-азаиндол-3-карбоновой кислоты,
амид N-(4-пиридилметил)-1-(4-хлорбензил)-7-азаиндол-3-карбоновой кислоты,
морфолид 1-(4-фторбензил)-7-азаиндол-3-карбоновой кислоты,
амид N-(2,6-дихлорфенил)-1-(2-метилпропен-3-ил)-7-азаиндол-3-карбоновой кислоты и
амид N-(3,5-дихлорпиридин-4-ил)-1-(4-пиридилметил)-7-азаиндол-3-карбоновой кислоты.
амид N-(3,5-дихлорпиридин-4-ил)-[1-(3-метоксибензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид гидрохлорид N-(4-пиридил)-[1-(4-фторбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-[1-(4-фторбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид гидрохлорид N-(4-пиридил)-[1-(4-хлорбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-[1-(4-хлорбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-[1-(4-метоксибензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(2,6-дихлорфенил)-[1-(4-хлорбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(4-карбоксифенил)-[1-(4-фторбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(4-этоксикарбонилфенил)-[1-(4-фторбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(3,4-диметоксифенил)-[1-(4-фторбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-[1-(4-метилбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-[1-(4-гидроксибензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-[1-(3-гидроксибензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-(1-циклопропилметил-7-азаиндол-3-ил)глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-(1-гексил-7-азаиндол-3-ил)глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-(1-изобутил-7-азаиндол-3-ил)глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-[1-(2-метилпропен-3-ил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-[1-(2-метоксиэтил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-[1-(1-нафтилметил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-[1-(4-пиридилметил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(3,5-дихлорпиридин-4-ил)-[1-(3,5-диметилизоксазол-4-ил-метил)-7-азаиндол-3-ил]глиоксиловой кислоты,
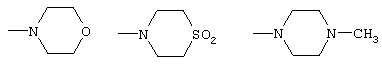
амид N,N-бис(2-метоксиэтил)-[1-(4-фторбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
морфолид [1-(4-фторбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
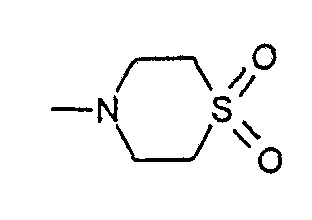
S,S-диоксотиоморфолид[1-(4-фторбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
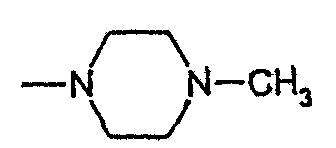
4-метилпиперазид 1-(4-фторбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
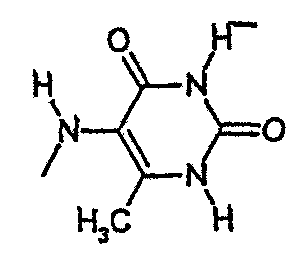
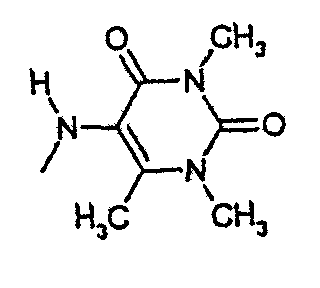
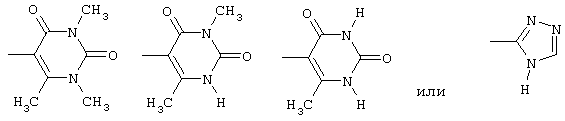
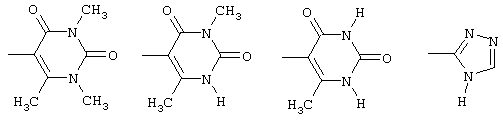
амид N-(6-метилурацил-5-ил)-[1-(4-фторбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
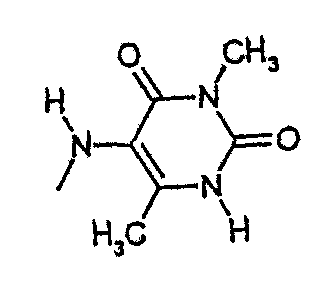
амид N-(3,6-диметилурацил-5-ил)-[1-(4-фторбензил)-7-азаиндол-3-ил]глиоксиловой кислоты,
амид N-(1,3,6-триметилурацил-5-ил)-[1-(4-фторбензил)-7-азаиндол-3-ил]глиоксиловой кислоты и
амид N-(1,2,4-4Н-триазол-3-ил)-[1-(4-фторбензил)-7-азаиндол-3-ил]глиоксиловой кислоты.
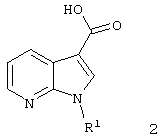
где R1 имеет указанные в п.1 значения,
посредством хлорангидридов кислот переводят в соответствующие хлорангидриды 7-азаиндол-3-карбоновых кислот и затем посредством реакции с первичными или вторичными аминами превращают в соединения согласно изобретению формулы 1, где R1 R2 и R3 имеют указанные в п.1 значения и n=1.
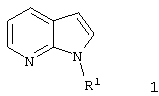
где R1 имеет указанные в п.1 значения,
посредством хлорангидрида щавелевой кислоты переводят в аналогичные хлорангидриды 7-азаиндол-3-ил-глиоксиловой кислоты и затем посредством реакции с первичными или вторичными аминами превращают в соединения формулы 1, где R1, R2 и R3 имеют указанные в п.1 значения и n=2.
| WO 9611929 A, 25.04.1996 | |||
| GB 1141949 A, 05.02.1969 | |||
| RU 94040390 C1, 27.05.1997 | |||
| ПИРАЗОЛО- И ПИРРОЛОПИРИДИНЫ | 1995 |
|
RU2135498C1 |
| ИМИДЫ КАК ИНГИБИТОРЫ TNF-АЛЬФА | 1994 |
|
RU2174516C2 |

