Область техники, к которой относится изобретение
Изобретение относится к катализаторам превращений углеводородов и, в частности, касается катализаторов превращения углеводородов на основе синтетических мезопористых кристаллических материалов и способа их получения. Предложенные катализаторы могут быть использованы в процессах изомеризации легких бензиновых фракций, изодепарафинизации дизельных и масляных фракций, алкилировании изоалканов алкенами, гидрокрекинге и других процессах переработки нефтяного сырья.
Уровень техники
В настоящее время внимание исследователей сосредоточено на разработке каталитических систем на основе оксида циркония, модифицированных анионными добавками и содержащих металлы VIII группы элементов, которые превосходят по своим характеристикам известные катализаторы, используемые в различных областях нефтепереработки и нефтехимии.
Известно, что активность катализаторов превращения углеводородов увеличивается при повышении концентрации активных центров. Это может быть достигнуто повышением удельной поверхности катализатора, а также повышением доступности активных центров молекулам реагента при увеличении радиуса пор.
Известен сульфатированный цирконий-оксидный катализатор, полученный традиционным методом, включающим осаждение геля оксида циркония из солей цирконила, его пропитку сульфатом аммония, прокаливание при 500°С и нанесение металлов VIII группы методом влажной пропитки (V.C.F.Holm, G.C.Bailey, Патент США №3032599). Полученный катализатор имеет относительно низкую активность, которая связана с низкой удельной поверхностью (˜100 м2/г).
Известен способ синтеза сульфатированных цирконий-оксидных катализаторов, содержащих металлы VIII группы, являющийся модификацией традиционного метода осаждения-пропитки в виде замены сульфата аммония раствором серной кислоты при сохранении остальных стадий, что приводит к формированию катализатора с удельной поверхностью до 150 м2/г (К.Matsuzawa, EP 0925830). Недостатком катализатора, полученного данным способом, является сравнительно невысокая активность.
Известен способ получения катализатора, включающий формирование геля оксида циркония промыванием и выдерживанием реакционной смеси в среде ацетона или метанола или совместным осаждением гидратированного оксида циркония и алюминия, последующую пропитку сульфатом аммония и нанесение платины на оксидный носитель методом пропитки по влагоемкости (G.Szabo, P.Nascimento, A.Milan, S.Decker, J.Denayer, J.-P. Dath, Патент США №6448198). Удельная поверхность катализаторов, полученных по этому способу, составляет 150-160 м2/г, общий объем пор до 0,2-0,25 см3/г при диаметре пор до 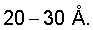
Известен способ синтеза сульфатированного цирконий-оксидного катализатора, содержащего элемент VIII группы, основанный на золь-гель методе, включающий синтез золя циркония в водном растворе аммиака в присутствии гидроксипропил-метилцеллюлозы и крахмала, формирование микросферической или сферической структуры геля, его желирование, промывание и высушивание с последующей пропиткой водным раствором сульфата аммония и элементами VIII группы и прокаливанием при 450-700°С (М.Marella, M.Tomaselli, L.Maergalli, F.Pinna, Патент США №6180556). Удельная поверхность катализаторов, полученных по этому способу, достигает 235 м2/г, общий объем пор 0,17 см3/г при диаметре пор 1,4-2,3 нм. К недостаткам катализаторов, полученных вышеупомянутыми способами, можно отнести низкую термическую стабильность из-за невозможности формирования регулярной широкопористой структуры с контролируемым радиусом пор.
Наиболее близким к предложенному изобретению является катализатор на основе сульфатированного ZrO2 с мезопористой структурой, приготовленный двухэтапным методом (W.M.H.Sachtler, Y.Y.Huang, Патент США №5786294). На первом этапе проводят гидротермальную кристаллизацию смеси Zr(OPri)4 и С16Н33NH2 в растворе вода-этанол-ацетилацетон, отделение твердого материала центрифугированием и удаление органической матрицы экстракцией этанолом. На втором этапе проводят стабилизацию водным раствором серной кислоты и прокаливание с последующим нанесением металлов VIII группы (Pt, Pd, Rh, Ru, Ni). На рентгенограмме катализатора наблюдается рефлекс, соответствующий межплоскостному расстоянию в 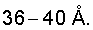 Удельная поверхность катализатора составляет 91-347 м2/г, а объем пор равен 0,16-0,31 см3/г. К недостаткам данного способа следует отнести то, что полученный катализатор мезопористой структуры не обладает достаточной стабильностью в процессе термообработки, а также использование дорогих и нетехнологичных исходных веществ (Zr(OPri)4,) и процессов (а именно экстракция этанолом, центрифугирование).
Удельная поверхность катализатора составляет 91-347 м2/г, а объем пор равен 0,16-0,31 см3/г. К недостаткам данного способа следует отнести то, что полученный катализатор мезопористой структуры не обладает достаточной стабильностью в процессе термообработки, а также использование дорогих и нетехнологичных исходных веществ (Zr(OPri)4,) и процессов (а именно экстракция этанолом, центрифугирование).
Раскрытие изобретения
Предложенное изобретение направлено на создание катализатора превращения углеводородов на основе мезопористого оксида циркония, содержащего металл VIII группы элементов, обладающего высокой удельной поверхностью (более 200-300 м2/г) и объемом пор (не менее 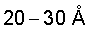 ) и повышенной термической стабильностью, позволяющей выдерживать многократные циклы регенерации (например, удаление кокса прокаливанием на воздухе) при высоких температурах, а также способа получения катализатора исходя из выпускаемых в промышленности доступных крупнотоннажных продуктов.
) и повышенной термической стабильностью, позволяющей выдерживать многократные циклы регенерации (например, удаление кокса прокаливанием на воздухе) при высоких температурах, а также способа получения катализатора исходя из выпускаемых в промышленности доступных крупнотоннажных продуктов.
В соответствии с этим объектом предложенного изобретения является катализатор превращения углеводородов, имеющий состав металл VIII группы/SO4 2-/ZrO2-ЭОх, где Э=элемент III или IV группы периодической таблицы Д.И.Менделеева, х=1,5 или 2, содержание SO4 2- составляет 0,1-10 мас.%, мольное соотношение ZrO2:ЭОх=1:(0,1-1,0), и имеющий мезопористую кристаллическую структуру с удельной поверхностью 300-800 м2/г и суммарным объемом пор 0,3-0,8 см3/г.
Другим объектом изобретения является способ получения катализатора превращения углеводородов, включающий осаждение соединений циркония в гидротермальных условиях в присутствии сурфактанта с получением мезопористой фазы, ее стабилизацию стабилизирующим агентом, прокаливание и введение металла VIII группы периодической таблицы Д.И.Менделеева, причем в качестве соединений циркония используют гидроксид циркония или цирконила, в качестве стабилизирующего агента используют элементы III или IV группы, а после стабилизации, при необходимости, осуществляют регулирование кислотности.
Предпочтительно гидроксид циркония или цирконила получают осаждением из них соответствующих солей.
В частном варианте осуществления изобретения гидроксид циркония или цирконила предпочтительно осаждают водным раствором аммиака.
В другом частном случае осуществления изобретения осаждение гидроксида циркония или цирконила в гидротермальных условиях осуществляют из композиции ZrOa(OH)b-H2SO4-H2O, где а=0-2, b=0-4.
В частном случае осуществления изобретения металл VIII группы вводят ионным обменом или пропиткой.
В еще одном частном случае осуществления изобретения регулирование кислотности осуществляют обработкой водными растворами кислот и/или их солей, при этом в качестве солей используют, например, аммониевые соли.
Предпочтительным режимом прокаливания катализатора является температурный диапазон 450-900°С, при этом сохраняются его пористые характеристики.
Краткое описание чертежей
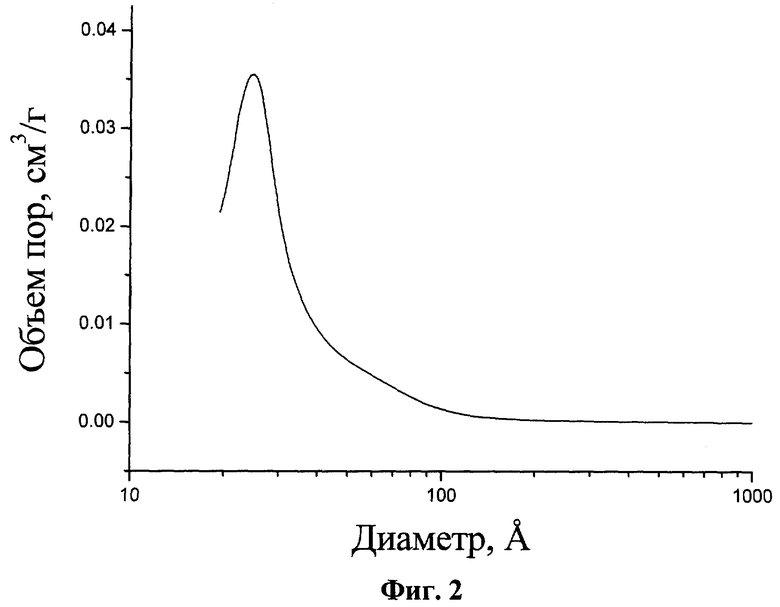
На Фиг.1 представлена рентгенограмма соответствующего предлагаемому изобретению катализатора превращения углеводородов на основе оксида циркония, прокаленного на воздухе при 550°.
На Фиг.2 представлено распределение пор по размерам для соответствующего предлагаемому изобретению катализатора на основе оксида циркония, прокаленного на воздухе при 550°, полученное по данным адсорбционных измерений.
Осуществление изобретения
Способ получения катализатора включает на первом этапе приготовление композиции, состоящей из гидратированного оксида циркония, сульфат анионов и воды, посредством осаждения гидратированной оксидной фазы из растворимых солей циркония или цирконила с их последующим растворением в концентрированной серной кислоте и разбавлением водой. Полученную композицию подвергают переосаждению в гидротермальных условиях в присутствии катионных сурфактантов, используемых в качестве матрицы, с формированием мезопористой структуры. На втором этапе проводят стабилизацию мезопористой структуры обработкой соединениями элементов III-IV группы, взятых в определенных отношениях к мезопористой кристаллической фазе. На третьем этапе, при необходимости, производят регулирование кислотно-основных свойств мезопористого материала дополнительной обработкой кислотами или их солями, а также осуществляют прокаливание. На четвертом этапе проводят нанесение металла VIII группы пропиткой или ионным обменом.
Приготовление катализатора, соответствующего предложенному изобретению, осуществляют следующим способом.
На первом этапе раствор соли циркония или цирконила (ZrOCl2, ZrO(NO3)2) обрабатывают водным раствором аммиака в мольном соотношении 1 Zr : 4NH3. Полученный осадок отделяют, промывают водой, а затем обрабатывают серной кислотой в мольном соотношении 1 Zr:(1,5-2,5)SO4 2- до полного растворения осадка. К полученному раствору добавляют водный раствор катионного сурфактанта галогенида алкилтриметиламмония ((Сn(СН3)3NHal, где n=10-20, Hal=Cl, Br)) в мольных соотношениях 1 Zr:(0,2-0,5)Сn(СН3)3NHal:(400-600)Н2O и выдерживают при температуре 40-120°С в течение 20-100 часов. По окончании кристаллизации полученный мезопористый кристаллический материал на основе оксида циркония, содержащий органическую матрицу, фильтруют, промывают и сушат.
На втором этапе проводят стабилизацию мезопористой кристаллической фазы оксида циркония. Для этого смесь, содержащую исходную форму мезопористой кристаллической фазы, композицию R4NOH-SiO2(Al2О3) (где R=Me, Et) и воду в мольном соотношении 1 Zr:(0,1-1,0):SiO2(Al2О3):(0,1-1,0)R4NOH:(100-300)H2O, выдерживают при температуре 40-100°С в течение 4-60 ч. По окончании кристаллизации исходную форму мезопористой кристаллической фазы оксида циркония фильтруют, промывают и сушат.
На третьем этапе при необходимости полученный материал обрабатывают водным раствором кислот НnА (например, серной, вольфрамфосфорной и др.) или их аммониевых солей при мольном отношении 1 ZrO2:(0,01-0,1)Аn-. Материал сушат и прокаливают при температуре 500-700°С в течение 4-8 часов.
На четвертом этапе осуществляют нанесение металлов VIII группы из водных растворов хлоридных или аммиачных комплексов металлов VIII группы ионным обменом или пропиткой. Полученный катализатор сушат и прокаливают при 400°С в течение 2 ч.
Полученный катализатор превращения углеводородов, соответствующий предложенному изобретению, существенно превосходит по своим характеристикам известные катализаторы, а именно имеет удельную поверхность 600-650 м /г и суммарный объем пор 0,5-0,6 см3/г. Химический состав в мольном отношении равен 1 ZrO2:(0,1-1,0)SiO2(Ab2О3):(0,01-0,1)SO4 2-:(0,0005-0,05) металл VIII группы. Рентгенограмму катализатора характеризует наличие рефлекса, соответствующего межплоскостному расстоянию 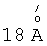 (Фиг.1), что указывает на формирование структурированной мезопористой фазы. Данный рефлекс сохраняется после прокаливания при 550-600°С, что свидетельствует о сохранности мезопористой структуры. Средний диаметр пор заявляемого материала составляет
(Фиг.1), что указывает на формирование структурированной мезопористой фазы. Данный рефлекс сохраняется после прокаливания при 550-600°С, что свидетельствует о сохранности мезопористой структуры. Средний диаметр пор заявляемого материала составляет 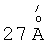 (Фиг.2).
(Фиг.2).
Осуществление предложенного способа получения катализатора, соответствующего предложенному изобретению, иллюстрируется Примерами 1-6. Условия приготовления катализатора сведены в Таблице 1. Физико-химические характеристики катализаторов, полученных после прокаливания на воздухе при 550°С, приготовленных в соответствии с предложенным способом и катализатора, известного из US Патента 5786294, сведены в Таблице 2.
Катализаторы, соответствующие изобретению, были испытаны в реакции изомеризации линейных алканов С4-С7. Перед испытаниями катализатор восстанавливают в токе водорода при температуре 200°С. Все катализаторы проявляют высокую активность. В частности, выход изомеров при превращении н-бутана и н-гексана при 1 атм превышает 30% и 70% соответственно.
Условия приготовления мезопористых катализаторов
Физико-химические характеристики мезопористых катализаторов после прокаливания при 550°С
Измерение физико-химических характеристик целевых продуктов проводили следующим образом.
Удельная поверхность и объем пор были измерены на адсорбционном порозиметре Micromeritics ASAP 2020 методом адсорбции азота. Удельная поверхность рассчитана по модели BET (Брунауэр-Эммет-Тэллер) при относительном парциальном давлении Р/Р0=0,2. Общий объем пор и распределение пор по радиусам рассчитан по адсорбционной кривой с использованием модели BJH (Баррет-Джойнер-Халенда) при относительном парциальном давлении Р/Р0=0,99.
Рентгенограммы образцов катализаторов, соответствующих предложенному изобретению, сделаны на приборе X'Pert PRO PANAlytical в монохроматизированном CuKα излучении. Расчет значений межплоскостных расстояний d проведен по формуле d=λ/2sinθ, где θ - угол максимума рефлекса.
Каталитическая активность образцов изучена в реакциях превращения линейных алканов C4-C7 в проточной установке с неподвижньм слоем катализатора при давлениях 0,1-3,5 МПа и температурах 160-380°С. Скорость подачи сырья составляет 0,5-2 ч-1, объемное соотношение водород: сырье = 400-1000, каталитическую активность тестируют через 30 мин после установления требуемого режима.
Пример 1.
Синтез катализатора проводят в четыре этапа.
На первом этапе 10,12 г (0,03 моль) хлорида цирконила (ZrOCl2·8H2O) растворяют в 60 г воды. Полученный раствор обрабатывают 4,4 мл 25%-ного водного раствора NH4ОН до полного осаждения гидратированной оксидной фазы. Осадок отделяют фильтрованием, промывают на фильтре водой и затем обрабатывают 6,16 г (0,06 моль) концентрированной серной кислоты до полного растворения гидратированной оксидной фазы. В полученную смесь добавляют 37,1 г воды. Полученный раствор по каплям при интенсивном перемешивании в течение 30 мин приливают к раствору сурфактанта, содержащему 6,13 г (0,016 моль) бромида гексадецилтриметиламмония в 175 г воды. Смесь выдерживают при температуре 95°С в течение 48 часов. Продукт остужают до комнатной температуры, отделяют фильтрованием, промывают на фильтре водой и сушат при комнатной температуре в течение 48 ч.
На втором этапе проводят стабилизацию мезопористой фазы. 10 г сухого продукта, полученного на первом этапе, суспендируют в 50 г воды. К полученной суспензии при перемешивании добавляют раствор, содержащий 2,93 г (0,045 моль) оксида кремния, 32,96 мл 20%-ного раствора (0,045 моль) гидроксида тетраэтиламмония в воде и 15 г воды. Мольное отношение Zr:Si составляет 1,7. Смесь выдерживают при температуре 90°С в течение 30 часов. Продукт остужают до комнатной температуры, отделяют фильтрованием, промывают на фильтре водой и сушат при комнатной температуре в течение 48 ч.
На третьем этапе катализатор прокаливают при 550°С.
На четвертом этапе осуществляют нанесение платины методом циркуляционной пропитки из раствора платинохлористоводородной кислоты. 10 г сухого продукта, полученного на предыдущем этапе, пропитывают 50 мл раствора, содержащего 0,05 г платины в виде H2PtCl6. Пропитку проводят в течение 1 ч при 50°С, затем раствор отделяют и катализатор сушат при комнатной температуре в течение 24 ч и при 100°С в течение 6 ч. Катализатор прокаливают в токе воздуха при 400°С в течение 3 ч.
Пример 2.
Синтез проводят по Примеру 1. На первом этапе в качестве сурфактанта используют смесь 21,55 мл 25%-ного раствора хлорида гексадецилтриметиламмония в 159 мл воды.
Пример 3.
Синтез катализатора проводят в четыре этапа. Первый, второй и четвертый этапы проводят по Примеру 1.
На третьем этапе перед прокаливанием дополнительно проводят регулирование кислотных свойств. 10 г сухого продукта, полученного на втором этапе, обрабатывают раствором 0,5 г (0,005 моль) серной кислоты в 50 г воды. Смесь выдерживают при комнатной температуре в течение 1 ч, затем упаривают и сушат при 100°С в течение 2 ч. Продукт прокаливают в токе воздуха при температуре 550°С в течение 6 ч.
Пример 4.
Синтез проводят по Примеру 3. На втором этапе мольное отношение Zr:Si составляет 1,7. Время стабилизации 30 часов. Регулирование кислотности на третьем этапе проводят раствором 0,66 г (0,005 моль) сульфата аммония в 50 г воды.
Пример 5.
Синтез проводят по Примеру 3 за исключением того, что на первом этапе в качестве исходного соединения берут сульфат циркония.
На первом этапе 15 г (0,042 моль) Zr(SO4)2·4H2O растворяют в 50 г воды. Полученный раствор по каплям при интенсивном перемешивании в течение 30 мин приливают к раствору 8,24 г (0,021 моль) бромида гексадецилтриметиламмония в 235 г воды. Смесь выдерживают при температуре 95°С в течение 48 часов. Продукт остужают до комнатной температуры, отделяют фильтрованием, промывают на фильтре водой и сушат при комнатной температуре в течение 48 ч.
Второй, третий и четвертый этапы проводят по Примеру 3.
Пример 6.
Первый, второй и третий этапы проводят по Примеру 3. На четвертом этапе проводят нанесение палладия из раствора палладийхлористоводородной кислоты. 10 г сухого продукта, полученного на третьем этапе, пропитывают 50 мл раствора, содержащего 0,05 г Pd в виде H2PtCl4. Пропитку проводят в течение 1 ч при 50°С. Раствор отделяют и катализатор сушат при комнатной температуре 24 ч и при 100°С в течение 6 ч. Катализатор прокаливают в токе воздуха при 400°С в течение 3 ч.
Промышленная применимость
Катализаторы, соответствующие предложенному изобретению, представляют большой интерес для использования в процессах изомеризации легких бензиновых фракций, изодепарафинизации дизельных и масляных фракций, алкилировании изоалканов алкенами, гидрокрекинге и др. Интерес определяется высокой термической стабильностью и дополнительными характеристиками, обусловленными особенностями кристаллической структуры, сочетающей широкие поры (свыше 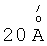 ), унифицированные по размерам, большие величины удельной поверхности и объема пор, а также возможность формирования специфических активных центров на внутренней поверхности пор.
), унифицированные по размерам, большие величины удельной поверхности и объема пор, а также возможность формирования специфических активных центров на внутренней поверхности пор.
| название | год | авторы | номер документа |
|---|---|---|---|
| МЕЗОПОРИСТЫЙ МАТЕРИАЛ НА ОСНОВЕ ОКСИДА ЦИРКОНИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2005 |
|
RU2280504C1 |
| СПОСОБ ПОЛУЧЕНИЯ НОСИТЕЛЯ КАТАЛИЗАТОРА ПРЕВРАЩЕНИЙ УГЛЕВОДОРОДНОГО СЫРЬЯ НА ОСНОВЕ МЕЗОПОРИСТОГО МАТЕРИАЛА | 2015 |
|
RU2584951C1 |
| МАТЕРИАЛ НА ОСНОВЕ ДИОКСИДА ЦИРКОНИЯ И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 1997 |
|
RU2129989C1 |
| КАТАЛИЗАТОР ОКИСЛЕНИЯ УГЛЕВОДОРОДОВ В КИСЛОРОДСОДЕРЖАЩЕМ ГАЗЕ (ВАРИАНТЫ) И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2009 |
|
RU2402379C1 |
| Катализатор для осуществления процесса Фишера-Тропша в компактном варианте и способ его получения (варианты) | 2015 |
|
RU2610526C2 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА ИЗОМЕРИЗАЦИИ Н-БУТАНА В ИЗОБУТАН | 2002 |
|
RU2236291C1 |
| ОКСИДНЫЙ КАТАЛИЗАТОР ДЛЯ ИЗОМЕРИЗАЦИИ ЛЕГКИХ БЕНЗИНОВЫХ ФРАКЦИЙ | 2012 |
|
RU2486005C1 |
| МИКРОСФЕРИЧЕСКИЙ ЦЕОЛИТСОДЕРЖАЩИЙ КАТАЛИЗАТОР ДЛЯ ПРЕВРАЩЕНИЯ АЛИФАТИЧЕСКИХ УГЛЕВОДОРОДОВ C- C | 1992 |
|
RU2019290C1 |
| СПОСОБ КОНВЕРСИИ УГЛЕВОДОРОДОВ, КАТАЛИЗАТОР ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ С МИКРО-МЕЗОПОРИСТОЙ СТРУКТУРОЙ И СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА | 2005 |
|
RU2288034C1 |
| КАТАЛИЗАТОР АРОМАТИЗАЦИИ МЕТАНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ КОНВЕРСИИ МЕТАНА С ПОЛУЧЕНИЕМ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2015 |
|
RU2585289C1 |
Изобретение относится к катализаторам превращений углеводородов и, в частности, касается катализаторов на основе синтетических мезопористых кристаллических материалов и способа их получения. Описан катализатор превращения углеводородов, имеющий состав металл VIII группы/SO4 2-/ZrO2-ЭОx, где Э=элемент III или IV группы периодической таблицы Д.И.Менделеева, х=1,5 или 2, содержание SO4 2- составляет 0,1-10 мас.%, мольное соотношение ZrO2:ЭОx=1:(0,1-1,0), и имеющий мезопористую кристаллическую структуру с удельной поверхностью 300-800 м2/г и суммарным объемом пор 0,3-0,8 см3/г, а также способ его получения. Способ включает осаждение соединений циркония, в качестве которых используют гидроксид циркония или цирконила, в гидротермальных условиях в присутствии сурфактанта с получением мезопористой фазы, стабилизацию ее стабилизирующим агентом - элементами III и IV группы, после стабилизации, при необходимости, осуществляют регулирование кислотности и затем введение металла VIII группы. Технический эффект - повышение удельной поверхности и термической стабильности, упрощение технологии. 2 н. и 7 з.п. ф-лы, 2 ил., 2 табл.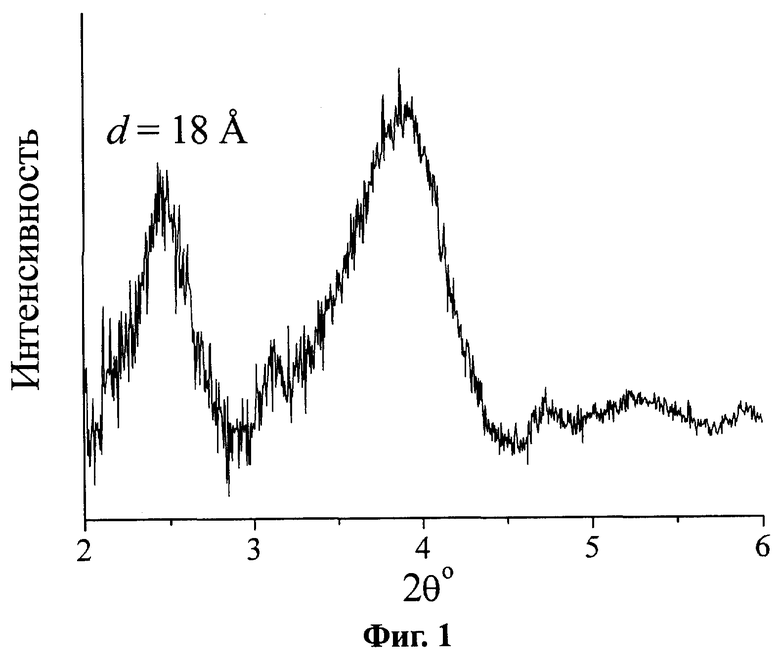
| US 5786294 А, 28.07.1998 | |||
| СПОСОБ ПОЛУЧЕНИЯ НОСИТЕЛЯ ДЛЯ КАТАЛИЗАТОРОВ | 1997 |
|
RU2157729C2 |
| НЕОРГАНИЧЕСКИЙ СФЕРОГРАНУЛИРОВАННЫЙ ПОРИСТЫЙ СОРБЕНТ НА ОСНОВЕ ГИДРОКСИДА ЦИРКОНИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1994 |
|
RU2064825C1 |
| ЕР 0908232 В1, 18.12.2002 | |||
| Фильтр для очистки газов | 2016 |
|
RU2629070C1 |

