Область техники
Данное изобретение относится к способу получения промежуточного продукта, пригодного для получения (+/-)-5-[[4-[2-(5-этил-2-пиридил)этокси]фенил]метил]-2,4-тиазолидиндиона (далее называемого пиоглитазоном) формулы I

Пиоглитазон относится к тиазолидиндионовой группе противодиабетических препаратов. Данное соединение и его противодиабетические свойства были описаны в ЕР 193256. Позже было установлено, что противодиабетическое действие этого соединения заключается в снижении инсулинрезистентности, улучшении посредством этого усвояемости глюкозы без повышения секреции инсулина, в отличие от большинства других противодиабетических средств. При таких необычных свойствах данный продукт имеет очень важное значение в лечении инсулинонезависимых форм сахарного диабета. Комбинирование с инсулином или другими противодиабетическими средствами может еще более усилить данное действие.
Предпосылки изобретения
Синтез тиазолидиндионов описан в ЕР 177353, в частности получение соединений формулы VII

где В представляет собой атом кислорода или серы, R2 является алкилом, ацилом или гидроксиалкилом, представлено взаимодействием соединения формулы VIII
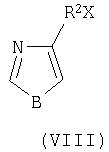
где Х означает галоген,
и фенолтиазолидиндиона формулы VI

Реакцию проводят в щелочной среде в органических растворителях (например, в диметилформамиде).
Способ, описанный в ЕР 177353, не может быть буквально использован для синтеза пиоглитазона, поскольку при взаимодействии 5-этил-2-пиридилэтилгалогенида с подходящим фенолом в щелочной среде будет преобладать реакция элиминирования, приводящая к получению винилпиридина.
В ЕР 193256 изложена другая реакция, которая может быть описана следующей схемой
Схема 1

Основным недостатком данного способа является нестандартное протекание восстановления нитрогруппы на палладии (время реакции составляет от 3 часов до нескольких дней), которое, по-видимому, зависит от содержания примесей в исходном соединении и в растворителе. Примеси, очевидно, приводят к отравлению катализатора; поэтому необходимо постепенно добавлять дополнительное количество катализатора. Увеличение времени реакции приводит к образованию большего числа примесей и снижению выходов.
Другие способы решения этой проблемы разработаны в ЕР 257781, в Chem. Pharm. Bull. 39(6) 1440-1445 (1991) и в ЕР 506273 и ЕР 816340. Общей характерной особенностью всех этих способов является реакция соединения формулы III

где Z представляет собой отщепляемую группу формулы 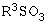 , где R3 является алкилом или арилом,
, где R3 является алкилом или арилом,
с солью щелочного металла и n-гидроксибензальдегида или n-формилфенолятом, то есть с соединением общей формулы IX
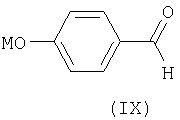
где М может быть щелочным металлом или водородом.
Продукт формулы Х

далее взаимодействует с тиазолидиндионом с образованием бензилиденового соединения формулы XI

которое превращают в пиоглитазон (формула I) восстановлением на палладии.
Способы, описанные в упомянутых выше патентах, различаются, в основном, средой, в которой проводят реакцию соединения IX с соединением III. В ЕР 257781 и в Chem. Pharm. Bull. 39(6) 1440-1445 (1991) описаны реакции в гетерогенной среде хлористый метилен-вода в присутствии катализатора межфазного переноса. В этих способах основная проблема заключается в межфазном переносе. В способе согласно ЕР 506273 впервые был выделен 4-формилфенолят щелочного металла и использован в качестве исходного соединения в последующих стадиях получения конечного продукта. Реакцию проводили в безводной среде, предпочтительно в этаноле. В ЕР 816340 описана реакция в смеси безводных растворителей, содержащей низкомолекулярный спирт и другой органический растворитель (например, толуол).
Недостаток описанных выше способов заключается в необходимости восстановления под давлением двойной связи соединения XI, то есть 5-(4-(2-(5-этил-2-пиридил)этокси)бензилиден)-2,4-тиазолидиндиона. Известны следующие способы:
- Согласно ЕР 257781 восстановление проводят водородом при катализе достаточно дорогостоящим палладием при давлении 50 кг/см2 и температуре 50°С с выходом 64%.
- Согласно ЕР 506273 при давлении 100 кг/см2 и температуре 110°С выход составляет 72%. Более высокие выходы можно получить за счет повышения давления, что повлечет за собой повышенные требования к безопасности реакции. При более высоких температурах значительно возрастает риск образования нежелательных продуктов.
- Попытка решения этих проблем предпринята в заявке WO 93/13095. Она заключается в восстановлении тетрагидроборатом натрия при катализе хлоридом кобальта.
Раскрытие изобретения
Согласно настоящему изобретению предложен способ получения противодиабетического средства формулы I

включающий конденсацию 4-замещенного фенола или фенолята общей формулы II
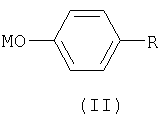
где R представляет собой органический радикал, содержащий аминогруппу, выбранный из группы, включающей группу общей формулы
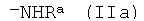
где Ra является водородом или защитной группой, которую удаляют перед дальнейшей обработкой, и группу общей формулы
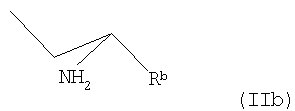
где Rb представляет собой карбоксильную группу в виде свободной кислоты либо в виде соли или эфира, и М является водородом или щелочным металлом,
с пиридиновым основанием общей формулы III
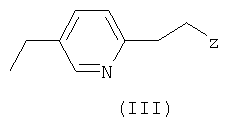
где Z - отщепляемая группа, отличная от галогена,
где проводят следующие операции:
(a) диазотирование аминогруппы, имеющейся в органическом радикале R,
(b) превращение диазотированного радикала R в производное 2-галогенпропионата или 2-галогенпропионитрила формулы
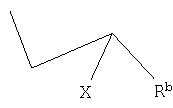
где Rb определен выше, а Х является галогеном,
(c) циклизацию производного 2-галогенпропионата или 2-галогенпропионитрила с тиомочевиной,
(d) гидролиз полученного имина;
причем в том случае, когда R представляет собой группу (IIa) сначала проводят конденсацию, а потом операции (а)-(d) с получением средства формулы (I), или в том случае, когда R представляет собой группу (IIb), то сначала проводят операции (а)-(d), а затем конденсацию с пиридиновым основанием общей формулы III с получением средства формулы I.
Один из вариантов способа заключается в использовании исходного соединения формулы II, где R представляет собой группу формулы , где Ra - как определено выше, и исходного соединения формулы III, в котором Z - как определено выше, для получения соединения формулы V
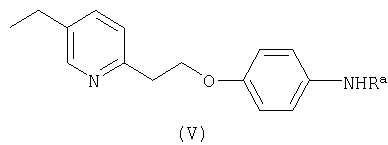
где Ra - как определено выше, которое затем гидролизуют с получением соединения формулы IV

где m означает 0 для свободного амина или 1 для аммониевой соли, R0 - остаток неорганической или органической кислоты, такой как галоген, HSO4 -, NO3 -, НСО3 -, R1COO- или R1SO3 -, где R1 представляет собой водород или алкил.
В другом варианте осуществления заявленного способа используется исходное соединение формулы II, где R представляет собой группу формулы
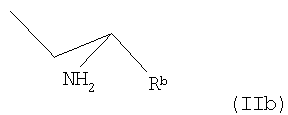
где Rb - как определено в п.1,
причем соединение формулы II перед конденсацией подвергают операциям от (а) до (d), получая соединение формулы (VI)

где Q представляет собой кислород или иминогруппу NH,
после чего проводят его конденсацию с пиридиновым основанием общей формулы III.
Согласно заявленному способу гидролиз соединения формулы V проводят в кислой среде.
А также возможно провести гидролиз соединения формулы V в щелочной среде.
Когда гидролиз проводят в кислой среде, то используют смесь этанола и концентрированной соляной или бромистоводородной кислоты при объемном отношении от 1:3 до 1:1 при кипячении в течение от 2 до 5 часов.
Согласно заявленному способу реакцию соединений II и III можно проводить в гетерогенной смеси органического растворителя и воды в присутствии катализатора межфазного переноса.
Предпочтительно проводить реакцию в смеси хлористого метилена и воды в присутствии хлорида бензилтрибутиламмония.
Согласно заявленному способу конденсацию можно проводить в полярном органическом растворителе при температуре от 50 до 130°С в присутствии щелочи.
Согласно изобретению также предложено соединение формулы V
где Ra является защитной группой, выбранной из группы, включающей ацил, n-алкоксикарбонил, трет-бутоксикарбонил, бензилоксикарбонил, 9-флуоренилметоксикарбонил, аллилоксикарбонил, 2-цианоэтоксикарбонил в качестве промежуточного продукта для получения соединения формулы I.
Согласно еще одному аспекту изобретения предложено соединение формулы V
где Ra - группа СОСН3.
Диазотирование
Диазотирование проводят исходя из гидробромида соответствующего производного анилина или аминокислоты, добавляя по каплям раствор нитрита натрия в воде, при температуре около 5°С.
Превращение в галогенпроизводное общей формулы

осуществляют, в зависимости от типа исходного соединения II, либо напрямую в процессе диазотирования в присутствии избытка галогенид-ионов, либо добавляя акрилат или акрилонитрил и оксид меди к раствору полной соли диазония.
Циклизацию с тиомочевиной проводят в этаноле при его температуре кипения с последующим выделением продукта.
Последующий гидролиз с образованием тиазолидиндионового цикла осуществляют с помощью соляной кислоты при ее температуре кипения.
Конденсацию с 2-(5-этил-2-пиридил)этильным производным проводят в присутствии основания в виде карбоната, гидроксида или гидрида щелочного металла в органическом растворителе или гетерогенной смеси органического растворителя с водой. При проведении реакции в органическом растворителе температуру варьируют от 50 до 130°С. После завершения реакции растворитель отгоняют или, если используют высококипящий растворитель, к смеси добавляют воду. Продукт экстрагируют этилацетатом.
Предпочтительно Ra в формуле (IIa) может быть ацильной группой, производным низших алифатических кислот (от C1 до С4), как, например, ацетил, или производным низших ароматических кислот, как, например, бензоил.
Соединение (IV) может быть с успехом использовано для получения пиоглитазона
Гидролиз соединения (V) проводят неорганической или органической кислотой общей формулы  или основанием.
или основанием.
При гидролизе основанием, например, смесью гидроксида калия и этанола при температуре кипения соединение IV представляет собой 4-(2-(5-этил-2-пиридил)этокси)анилин, то есть свободное основание (m равно 0).
При кислотном гидролизе могут быть использованы, например, галогеновые кислоты, такие как соляная кислота; соединение IV, образовавшееся в ходе реакции, представляет собой гидрохлорид 4-(2-(5-этил-2-пиридил)этокси)анилина, который в процессе выделения превращают в свободное основание (m=0). С точки зрения последующей стадии синтеза пиоглитазона (диазотирования) бромистоводородная кислота является наиболее предпочтительной для гидролиза соединений формулы V, где R0 представляет собой Br, и продукт формулы IV, гидробромид 4-(2-(5-этил-2-пиридил)этокси)анилина, не требует выделения. Кислородсодержащие неорганические кислоты, такие как серная кислота, также могут использоваться для гидролиза. Предпочтительные органические кислоты включают алкил- или арилсульфоновые кислоты (такие как метансульфокислота). Для гидролиза могут быть использованы многочисленные карбоновые кислоты, такие как муравьиная или уксусная.
Соединение формулы IV далее обрабатывают известным способом, то есть диазотируют, проводят реакцию Меервейна, циклизуют с тиомочевиной и проводят гидролиз имина. Этот способ описан в ЕР 193256 и представлен здесь Схемой 1.
В случае наиболее распространенной защитной группы, когда Ra является ацетилом, ее разложение проводят смесью концентрированной бромистоводородной кислоты и этанола в объемном отношении 1:3.
В том случае, когда исходным соединением является аминокислота тирозин, недорогое природное сырье формулы
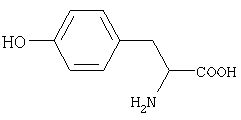
то конденсацию промежуточного продукта VI с основанием III можно проводить в органическом полярном растворителе, таком как диметилформамид или диметилсульфоксид. Температуру реакции выбирают в зависимости от использованного основания в интервале от 70 до 130°С. Время реакции, также в зависимости от выбранного основания, варьируют от 0,5 до 3 часов. Согласно обычному осуществлению после завершения реакции реакционную массу разбавляют водой и продукт экстрагируют, как правило, этилацетатом.
Полный синтез пиоглитазона для описанного выше конкретного случая может быть представлен схемой 2:
Схема 2

Если R является защищенным амином формулы  , наиболее предпочтительным будет случай, когда Ra - ацетил, а соединение формулы II представляет собой широко используемый парацетомол.
, наиболее предпочтительным будет случай, когда Ra - ацетил, а соединение формулы II представляет собой широко используемый парацетомол.
Конденсацию в этом случае предпочтительно проводить в этаноле при 50°С. После завершения реакции этанол отгоняют и продукт экстрагируют этилацетатом.
Полная последовательность этой предпочтительной методики со ссылкой на соответствующие Примеры представлена Схемой 3:
Схема 3
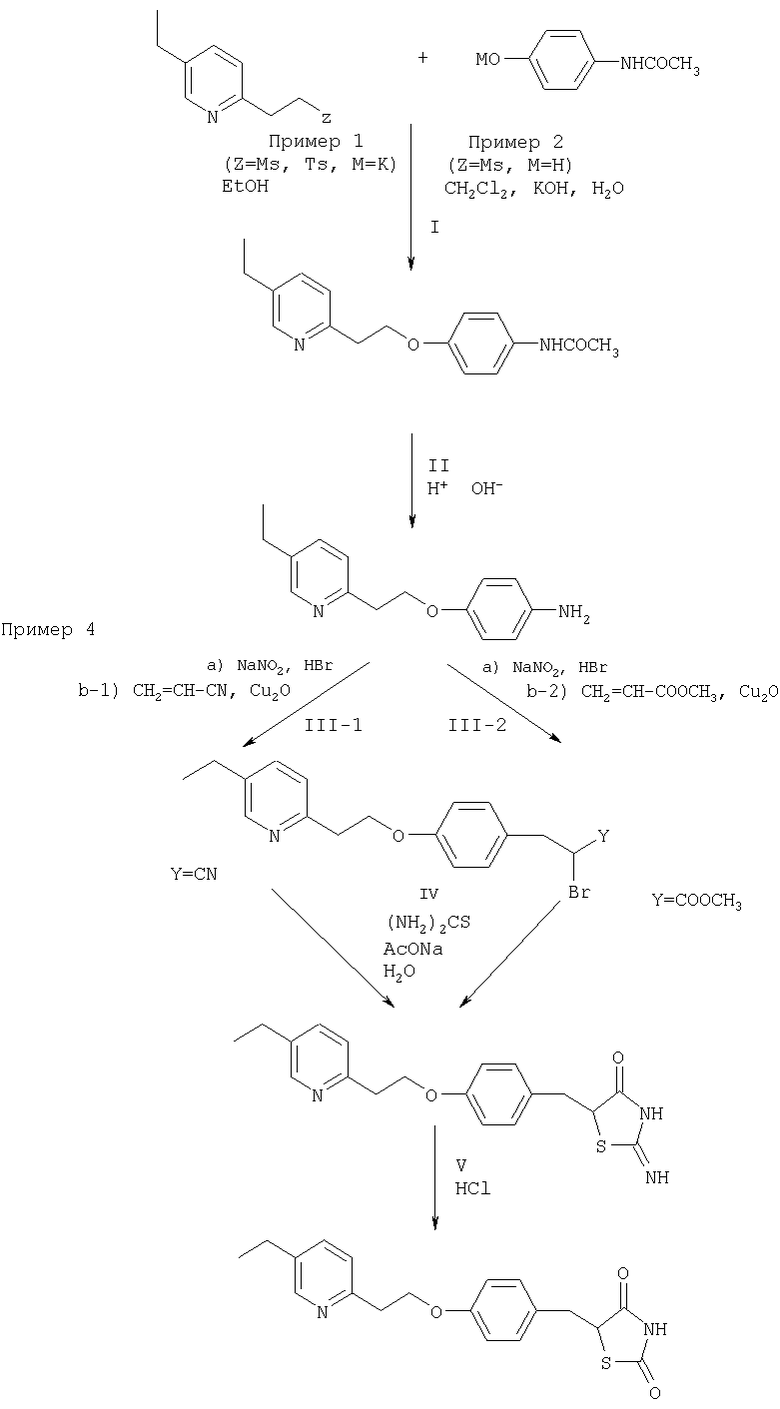
Примеры
Следующие примеры дополнительно иллюстрируют данное изобретение.
Пример сравнения - Получение пиоглитазона согласно ЕР 193256
a) Получение 4-(2-(5-этил-2-пиридил)этокси)нитробензола
К раствору 2-(5-этил-2-пиридил)этанола (12 г) и 4-фторнитробензола (11,2 г) в диметилформамиде (80 мл) порциями при охлаждении на ледяной бане прибавляют гидрид натрия (3,9 г). Эту смесь перемешивают 1 час при охлаждении на ледяной бане, 30 минут при комнатной температуре, выливают в ледяную воду и экстрагируют этилацетатом. Сушка органического слоя и отгонка растворителя дают 4-(2-(5-этил-2-пиридил)этокси)нитробензол в виде кристаллов желтого цвета (21,4 г, 98,2%). После перекристаллизации из смеси эфира и гексана получают кристаллы бледно-желтого цвета (т.пл. 45-47°С). Спектр ЯМР1Н (CDCl3), δ, м.д.: 1,25 (3Н, т, J=7,6), 2,65 (2Н, кв, J=7,6), 3,28 (2Н, т, J=6,7), 4,47 (2Н, т, J=6,7), 6,95 (2Н, м), 7,19 (1Н, д, J=7,9), 7,49 (1Н, дд, J=2,4; 8,0), 8,17 (2Н, м), 8,41 (1Н, д, J=2,2).
b) Получение 4-(2-(5-этил-2-пиридил)этокси)анилина
Раствор 4-(2-(5-этил-2-пиридил)этокси)нитробензола (23 г) гидрируют при атмосферном давлении в присутствии 10% Pd на угле (50% влажности, 2,5 г). Катализатор отделяют фильтрацией. После отгонки растворителя при пониженном давлении получают 4-(2-(5-этил-2-пиридил)этокси)анилин в виде технического масла (20 г, 98%).
c) Получение метил-2-бром-3-(4-(2-(5-этил-2-пиридил)этокси)фенил)пропионата
К раствору 4-(2-(5-этил-2-пиридил)этокси)анилина (20,4 г) в смеси метанола (70 мл) и ацетона (180 мл) прибавляют 47% бромистоводородную кислоту (36 мл). Смесь охлаждают. Раствор нитрита натрия (6,12 г) в воде (18 мл) прибавляют по каплям при температуре от 0 до 5°С. Смесь перемешивают при этой температуре в течение еще 20 минут, после чего прибавляют метилакрилат (43,2 мл) и повышают температуру до 38°С. К этой смеси небольшими порциями при перемешивании прибавляют оксид меди (0,72 г). Реакционную массу перемешивают до окончания выделения азота. Затем ее концентрируют при пониженном давлении, концентрат подщелачивают добавлением концентрированного раствора аммиака (36 мл) и экстрагируют этилацетатом. Органический слой промывают водой и сушат над сульфатом натрия. После концентрирования получают метил-2-бром-3-(4-(2-(5-этил-2-пиридил)этокси)фенил)пропионат в виде технического масла (28,4 г; 86,2%). Спектр ЯМР1Н (CDCl3), δ, м.д.: 1,21 (3Н, т, J=7), 2,60 (2Н, кв, J=7), 3,0-3,6 (4Н, м), 3,66 (3Н, м), 4,30 (2Н, т, J=7), 4,3 (1Н, м), 6,7-7,5 (6Н, м), 8,35 (1Н, д, J=2).
d) Получение 5-(4-(2-(5-этил-2-пиридил)этокси)бензил)-2-имино-4-тиазолидинона
Смесь метил-2-бром-3-(4-(2-(5-этил-2-пиридил)этокси)фенил)пропионата (28,4 г), тиомочевины (5,5 г), ацетата натрия (6 г) и этанола (200 мл) кипятят с обратным холодильником в течение 3 часов. Смесь концентрируют и концентрат нейтрализуют насыщенным раствором гидрокарбоната натрия. После добавления воды (80 мл) и эфира (40 мл) выпадает осадок. После фильтрации получают кристаллы 5-(4-(2-(5-этил-2-пиридил)этокси)бензил)-2-имино-4-тиазолидинона бежевого цвета (21,4 г; 85,9%; т.пл. 178-182°С). Спектр ЯМР1Н (CDCl3), δ, м.д.: 1,2 (3Н, т, J=7,6), 2,61 (2Н, кв, J=7,6), 2,85 (1H, дд, J=9,4; 14,3), 3,14 (2Н, т, J=6,7), 3,31 (1H, дд, J=4,1; 14,2), 4,31 (2Н, т, J=6,7), 4,52 (1H, дд, J=4,1; 9,4), 6,84 (2Н, м), 7,14 (2Н, м), 7,27 (1H, 6д, J=7,9), 7,57 (1H, 6дд, J=2,3; 7,9), 8,38 (1Н, 6д, J=2,2), 8,7 (2Н, 6с).
e) Получение 5-(4-(2-(5-этил-2-пиридил)этокси)бензил)-2,4-тиазолидиндиона
Раствор 5-(4-(2-(5-этил-2-пиридил)этокси)бензил)-2-имино-4-тиазолидинона (21,4 г) в 2М соляной кислоте (200 мл) кипятят с обратным холодильником в течение 6 часов, после чего концентрируют при пониженном давлении. Остаток после упаривания нейтрализуют насыщенным раствором гидрокарбоната натрия, из смеси отфильтровывают технический продукт. После перекристаллизации из смеси диметилформамида и воды получают кристаллический 5-(4-(2-(5-этил-2-пиридил)этокси)бензил)-2,4-тиазолидиндион (10,6 г; 56,1%; т.пл. 170-173°С). Спектр ЯМР1Н (CDCl3), δ, м.д.: 1,24 (3Н, т, J=7,6), 2,64 (2Н, кв, J=7,6), 3,04 (1H, дд, J=9,4; 14,3), 3,21 (2Н, т, J=6,6), 3,39 (1H, дд, J=4,0; 14,2), 4,33 (2Н, т, J=6,6), 4,48 (1H, дд, J=4,0; 9,3), 6,83 (2Н, м), 7,12 (2Н, м), 7,27 (1H, д, J=8,0), 7,50 (1H, дд, J=2,3; 8,0), 8,37 (1H, д, J=1,9).
Пример 1: Получение 4-(2-(5-этил-2-пиридил)этокси)анилина
Получение 4-(2-(5-этил-2-пиридил)этокси)ацетанилида (конденсация - две разные исходные соли)
a) Смесь 2-(5-этил-2-пиридил)этилмезилата (3,64 г) и 4-ацетамидофенолята калия (3,32 г) в этаноле (18 мл) перемешивают в течение 6 часов при 60°С. Смесь концентрируют и экстрагируют этилацетатом и водой. Органический слой трижды промывают 0,2М раствором гидроксида натрия и дважды водой, после чего сушат над сульфатом натрия. После отгонки растворителя и промывания остатка эфиром получают кристаллический 4-(2-(5-этил-2-пиридил)этокси)ацетанилид (2,32 г; 51,5%; т.пл. 85-87°С).
b) К раствору 2-(5-этил-2-пиридил)этилтозилата (15,27 г) в этаноле (50 мл) прибавляют в течение 1,5 часов при 50-60°С раствор 4-ацетамидофенолята калия (9,47 г) в этаноле (40 мл). Реакционную массу перемешивают при этой температуре в течение еще 6 часов, концентрируют и экстрагируют этилацетатом (150 мл) и водой. Органический слой промывают 0,2М раствором гидроксида натрия (4×100 мл) и водой (2×100 мл) и сушат над сульфатом натрия. После отгонки растворителя и промывания остатка эфиром получают кристаллический 4-(2-(5-этил-2-пиридил)этокси)ацетанилид (6 г; 42,3%; т.пл. 85-87°С).
Получение 4-(2-(5-этил-2-пиридил)этокси)анилина (три различные модификации)
a) Раствор 4-(2-(5-этил-2-пиридил)этокси)ацетанилида (13 г) в смеси этанола (80 мл) и концентрированной соляной кислоты (80 мл) кипятят с обратным холодильником в течение 2,5 часов. Реакционную массу нейтрализуют водным раствором гидроксида натрия и экстрагируют этилацетатом. Органический слой промывают водой и сушат над сульфатом магния. После отгонки этилацетата получают 4-(2-(5-этил-2-пиридил)этокси)анилин (9,6 г; 88,1%) в виде масла.
b) Смесь 4-(2-(5-этил-2-пиридил)этокси)ацетанилида (15,5 г), 4М гидроксида калия (150 мл) и этанола (150 мл) кипятят с обратным холодильником в течение 20 часов, после чего этанол отгоняют. Смесь экстрагируют этилацетатом; органический слой промывают водой и сушат над сульфатом натрия. После отгонки растворителя получают продукт в виде масла, 4-(2-(5-этил-2-пиридил)этокси)анилин (11,03 г; 80%).
c) К раствору 4-(2-(5-этил-2-пиридил)этокси)ацетанилида (6 г) в этаноле (100 мл) прибавляют концентрированную бромистоводородную кислоту (36 мл). Смесь кипятят с обратным холодильником в течение 4 часов. После отгонки этанола остаточную смесь используют на следующей стадии синтеза без выделения.
Пример 2: 4-(2-(5-этил-2-пиридил)этокси)анилин
Получение 4-(2-(5-этил-2-пиридил)этокси)ацетанилида
К раствору 2-(5-этил-2-пиридил)этилмезилата (1,7 г) и хлорида бензилтрибутиламмония (0,5 г) в хлористом метилене (10 мл) прибавляют раствор 4-гидроксиацетанилида (1,2 г) и карбоната калия (1,1 г) в воде (10 мл). После перемешивания в течение 9 часов и нагревания до 75°С отделяют органический слой, промывают его водой и сушат над сульфатом натрия. После отгонки растворителя и промывания остатка эфиром получают кристаллический 4-(2-(5-этил-2-пиридил)этокси)ацетанилид (1,15 г; 54,8%; 82-85°С).
Получение 4-(2-(5-этил-2-пиридил)этокси)анилина
4-(2-(5-этил-2-пиридил)этокси)ацетанилин получают гидролизом 4-(2-(5-этил-2-пиридил)этокси)ацетанилида, как описано в Примере 1.
Пример 3: Получение 5-[[4-[2-(5-этил-2-пиридил)этокси]фенил]метил]-2,4-тиазолидиндиона (пиоглитазона)
Получение 2-бром-3-(4-гидроксифенил)пропионовой кислоты
Тирозин (20 г) растворяют в HBr (100 мл, 48%, разбавленной 200 мл воды). К охлажденному раствору по каплям прибавляют NaNO2 в 30 мл воды. Продукт экстрагируют этилацетатом и после обычной обработки получают 2-бром-3-(4-гидроксифенил)пропионовую кислоту, которую используют на следующей стадии без очистки.
Получение 5-(4-гидроксибензил)-1,3-тиазолидин-2,4-диона
2-Бром-3-(4-гидроксифенил)пропионовую кислоту кипятят с обратным холодильником в этаноле (150 мл) с тиомочевиной (14,7 г) и ацетатом натрия (16,32 г). Через 3,5 часа растворитель отгоняют. Остаток размешивают в воде, отфильтровывают и промывают эфиром. Получают продукт (17,15 г) с температурой плавления 180,5-183,6°С. Полученный 5-(4-гидроксибензил)-2-имино-1,3-тиазолидин-4-он (16 г) растворяют в метоксиэтаноле (150 мл) и прибавляют HCl (27 мл). Реакционную массу кипятят с обратным холодильником в течение 4 часов. После отгонки растворителя остаток смешивают с водой. Продукт экстрагируют из воды этилацетатом. Продукт может быть перекристаллизован из смеси этилацетат-гептан. Получают 9,5 г 5-(4-гидроксибензил)-1,3-тиазолидин-2,4-диона с температурой плавления 150,9-152,5°С.
Получение 5-[[4-[2-(5-этил-2-пиридил)этокси]фенил]метил]-2,4-тиазолидиндиона
0,5 г 5-(4-гидроксибензил)-1,3-тиазолидин-2,4-диона растворяют в этаноле (7 мл), содержащем КОН (0,25 г), и перемешивают при комнатной температуре в течение 2 часов. К полученной соли прибавляют 2-(5-этил-2-пиридил)этилтозилат, после чего реакционную смесь кипятят с обратным холодильником в течение 5 часов. После охлаждения выпавшую соль отфильтровывают, регулируют рН и отгоняют растворитель. Полутвердый остаток смешивают с водой, отфильтровывают кристаллическую часть 5-[[4-[2-(5-этил-2-пиридил)этокси]фенил]метил]-2,4-тиазолидиндиона (0,18 г) и перекристаллизовывают из смеси ДМФА-вода.
Пример 4: Получение 5-[[4-[2-(5-этил-2-пиридил)этокси]фенил]метил]-2,4-тиазолидиндиона (пиоглитазона) - второй способ
Получение нитрила 2-бром-3-[4-(2-(5-этил-2-пиридил)этокси)фенил]пропионовой кислоты
К раствору бромида 4-(2-(5-этил-2-пиридил)этокси)фениламмония (52,8 ммоль) в бромистоводородной кислоте (полученному гидролизом 4-(2-(5-этил-2-пиридил)этокси)ацетанилида в соответствии с Примером 1с) прибавляют 25 мл 48% бромистоводородной кислоты, 50 мл метанола и 100 мл ацетона, после чего реакционную массу охлаждают до 0°С. К смеси по каплям прибавляют раствор нитрита натрия (4,5 г) в воде (10 мл) при температуре от 0 до 5°С. Массу перемешивают при 5°С в течение еще 20 минут, затем прибавляют акрилонитрил и повышают температуру до 38°С. Небольшими порциями прибавляют оксид меди (0,5 г). Смесь перемешивают до прекращения выделения азота, затем концентрируют при пониженном давлении, концентрат подщелачивают водным раствором аммиака и экстрагируют этилацетатом. Этилацетатный слой промывают водой и сушат над сульфатом натрия. После отгонки растворителя получают нитрил 2-бром-3-[4-(2-(5-этил-2-пиридил)этокси)фенил]пропионовой кислоты в виде технического масла (18,7 г, 99% от теоретического).
Получение 5-[4-(2-(5-этил-2-пиридил)этокси)бензил]-2-имино-4-тиазолидинона
Смесь нитрила 2-бром-3-[4-(2-(5-этил-2-пиридил)этокси)фенил]пропионовой кислоты (18,7 г), тиомочевины (3,9 г), ацетата натрия (4,2 г) и этанола (110 мл) кипятят с обратным холодильником в течение 6 часов, затем концентрируют при пониженном давлении, прибавляют этилацетат (30 мл) и насыщенный раствор гидрокарбоната натрия (100 мл). Смесь оставляют на ночь, затем фильтруют и выпавшие кристаллы бежевого цвета промывают эфиром и получают 5-[4-(2-(5-этил-2-пиридил)этокси)бензил]-2-имино-4-тиазолидинона (11,2 г, 59% от теоретического).
Далее 5-[4-(2-(5-этил-2-пиридил)этокси)бензил]-2-имино-4-тиазолидинон обрабатывают таким же образом, как описано в Примере 3.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТИАЗОЛИДИНДИОНА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2129553C1 |
| СПОСОБ ПОЛУЧЕНИЯ {2-[4-(АЛЬФА-ФЕНИЛ-П-ХЛОРБЕНЗИЛ)ПИПЕРАЗИН-1-ИЛ]-ЭТОКСИ}-УКСУСНОЙ КИСЛОТЫ И НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2248974C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ, И СПОСОБ ПОЛУЧЕНИЯ АНТИАНГИОГЕННОГО ЭФФЕКТА И/ИЛИ ЭФФЕКТА СНИЖЕНИЯ СОСУДИСТОЙ ПРОНИЦАЕМОСТИ С ИХ ПРИМЕНЕНИЕМ | 1996 |
|
RU2194701C2 |
| 1,3-ДИИОКСАНОНОВЫЕ ПРОИЗВОДНЫЕ АЛКЕНОВОЙ КИСЛОТЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 1989 |
|
RU2040525C1 |
| Способ получения производных тиазолидиндиона или их натриевых солей | 1985 |
|
SU1496634A3 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИЛИ ИХ СОЛИ С КИСЛОТАМИ ИЛИ ОСНОВАНИЯМИ | 1990 |
|
RU2049784C1 |
| СПОСОБ ПОЛУЧЕНИЯ РЕТИНАЛЯ ИЛИ АДДУКТА (ПОЛНОСТЬЮ-Е)-РЕТИНАЛЯ С ГИДРОХИНОНОМ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 2002 |
|
RU2318805C2 |
| ПРОИЗВОДНЫЕ 1,3-ДИОКСАНАЛКЕНОВОЙ КИСЛОТЫ | 1989 |
|
RU2045526C1 |
| ПРОИЗВОДНЫЕ ИНДАНУКСУСНОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2314298C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, ИХ ТАУТОМЕРЫ ИЛИ ИХ СОЛИ И ЛЕКАРСТВЕННОЕ СРЕДСТВО С АНТАГОНИСТИЧЕСКИМ В ОТНОШЕНИИ АНГИОТЕНЗИНА II ДЕЙСТВИЕМ | 1993 |
|
RU2126401C1 |
Изобретение относится к способу получения противодиабетического средства пиоглитазона формулы I

включающему конденсацию 4-замещенного фенола или фенолята общей формулы II
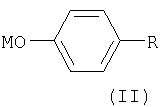
где R представляет собой органический радикал, содержащий аминогруппу, выбранный из группы, включающей группу общей формулы  , где Ra является водородом или защитной группой, которую удаляют перед дальнейшей обработкой, и группу общей формулы
, где Ra является водородом или защитной группой, которую удаляют перед дальнейшей обработкой, и группу общей формулы

где Rb представляет собой карбоксильную группу в виде свободной кислоты либо в виде соли или эфира, и М является водородом или щелочным металлом, с пиридиновым основанием общей формулы III

где Z - отщепляемая группа, отличная от галогена, где проводят следующие операции: (а) диазотирование аминогруппы, имеющейся в органическом радикале R, (b) превращение диазотированного радикала R в производное 2-галогенпропионата или 2-галогенпропионитрила формулы

где Rb определен выше, а Х является галогеном, (с) циклизацию производного 2-галогенпропионата или 2-галогенпропионитрила с тиомочевиной, и (d) гидролиз полученного имина; причем в том случае, когда R представляет собой группу (IIa) сначала проводят конденсацию, а потом операции (a)-(d) с получением средства формулы I, или в том случае, когда R представляет собой группу (IIb), то сначала проводят операции (а)-(d), а затем конденсацию с пиридиновым основанием общей формулы III с получением средства формулы I. Также описаны соединения формулы V
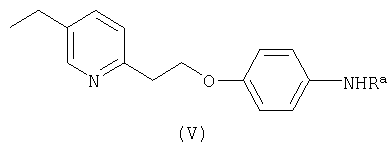
где Ra является защитной группой, выбранной из группы, включающей ацил, n-алкоксикарбонил, трет-бутоксикарбонил, бензилоксикарбонил, 9-флуоренилметоксикарбонил, аллилоксикарбонил, 2-цианоэтоксикарбонил в качестве промежуточного продукта для получения соединения формулы I. 3 н. и 9 з.п. ф-лы.

включающий конденсацию 4-замещенного фенола или фенолята общей формулы II

где R представляет собой органический радикал, содержащий аминогруппу, выбранный из группы, включающей
группу общей формулы
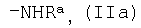
где Ra является водородом или защитной группой, которую удаляют перед дальнейшей обработкой, и
группу общей формулы
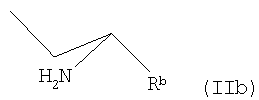
где Rb представляет собой карбоксильную группу в виде свободной кислоты либо в виде соли или эфира, и М является водородом или щелочным металлом,
с пиридиновым основанием общей формулы III
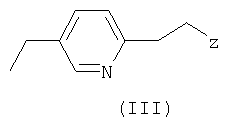
где Z - отщепляемая группа, отличная от галогена, где проводят следующие операции:
(a) диазотирование аминогруппы, имеющейся в органическом радикале R,
(b) превращение диазотированного радикала R в производное 2-галогенпропионата или 2-галогенпропионитрила формулы

где Rb определен выше, а Х является галогеном,
(c) циклизацию производного 2-галогенпропионата или 2-галогенпропионитрила с тиомочевиной,
(d) гидролиз полученного имина,
причем в том случае, когда R представляет собой группу (IIa), сначала проводят конденсацию, а потом операции (a)-(d) с получением средства формулы I, или
в том случае, когда R представляет собой группу (IIb), то сначала проводят операции (a)-(d), а затем конденсацию с пиридиновым основанием общей формулы III с получением средства формулы I.
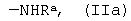
где Ra - как определено в п.1,
и исходное соединение формулы III, в котором Z - как определено в п.1,
для получения соединения формулы V

где Ra - как определено выше,
которое затем гидролизуют с получением соединения формулы IV

где m означает 0 для свободного амина или 1 для аммониевой соли, R0 - остаток неорганической или органической кислоты, такой как галоген, HSO4 -, NO3 -, НСО3 -, R1COO- или R1SO3 -, где R1 представляет собой водород или алкил.

где Rb - как определено в п.1,
причем соединение формулы II перед конденсацией подвергают операциям от (а) до (d), получая соединение формулы (VI)

где Q представляет собой кислород или иминогруппу NH,
после чего проводят его конденсацию с пиридиновым основанием общей формулы III.
где Ra является защитной группой, выбранной из группы, включающей ацил, n-алкоксикарбонил, трет-бутоксикарбонил, бензилоксикарбонил, 9-флуоренилметоксикарбонил, аллилоксикарбонил, 2-цианоэтоксикарбонил в качестве промежуточного продукта для получения соединения формулы I по п.1.
где Ra - группа СОСН3.
| ЕР 0816340 A1, 07.01.1998 | |||
| СПОСОБ ОБЕЗЖИРИВАНИЯ КИСЛОРОДОПРОВОДОБ | 0 |
|
SU193256A1 |
| Способ получения производных тиазолидиндиона | 1986 |
|
SU1436876A3 |
| Способ крашения тканей | 1922 |
|
SU62A1 |

