Область техники, к которой относится изобретение
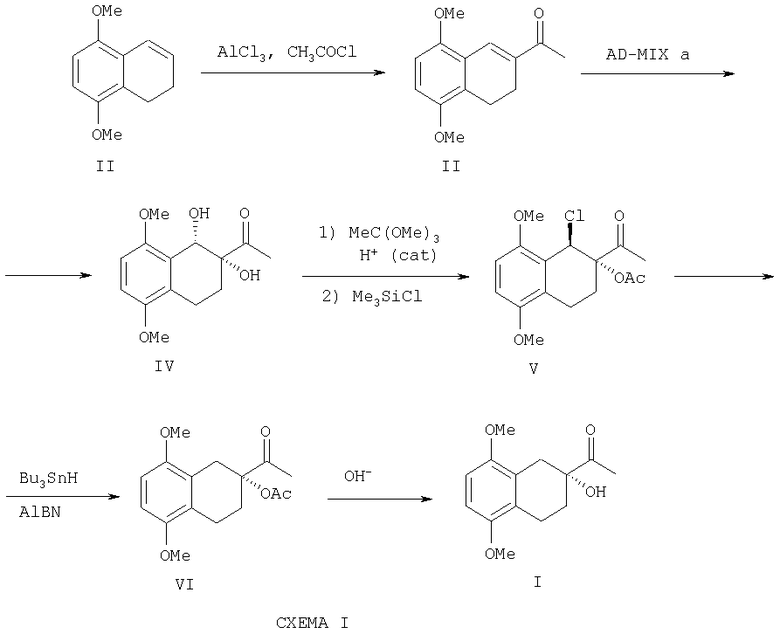
Настоящее изобретение относится к способу получения оптически активного тетралина - соединения, представляющего собой (R) 2-ацетил-2-гидрокси-5,8-диалкокси -1,2,3,4-тетрагидронафталин формулы I:
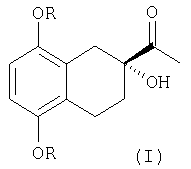
где: R=С1-3 алкил, предпочтительно метил,
получают, исходя из 5,8-диалкокси-3,4-дигидронафталина ацилированием, асимметричным дигидроксилированием, превращением в хлорацетат дегидрохлорированием и, наконец, гидролизом.
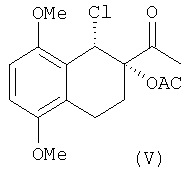
Изобретение также относится к промежуточным соединениям формулы V и VI:
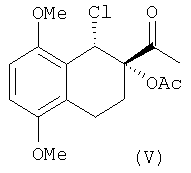
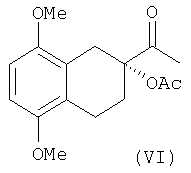
имеющим энантиомерный избыток более чем 95%.
Уровень техники
Как известно, антрациклины формулы VIII:
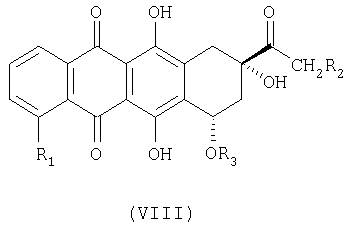
где: R1=H, ОН, ОСН3; R2=Н, ОН; R3=X, Y или Z, где
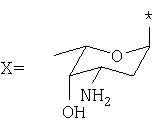
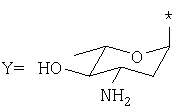
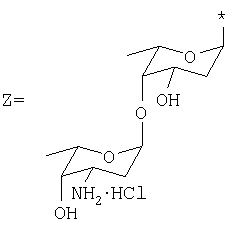
представляют собой соединения, имеющие широкое терапевтическое применение в качестве противоопухолевых лекарственных средств. Известны соединения формулы VIII, имеющие указанные выше свойства, например, дауномицин (VIII, где: R1=ОСН3, R2=Н, R3=X), доксорубицин (VIII, где: R1=ОСН3, R2=ОН, R3=X), гидрарубицин (VIII, где: R1=Н, R2=Н, R3=X) и эпирубицин (VIII, где: R1=ОСН3, R2=ОН, R3=Y), или соединения, описанные в ЕР 721456, в частности, соединение формулы VIII, в котором R1=Н, R2=ОН, R3=Z, дисахаридный антрациклин, который сейчас находится на стадии клинических испытаний.
Синтез антрациклина формулы VIII проходит в несколько стадий и обычно осуществляется, исходя из оптически активного тетралина формулы I, который реагирует по реакции Фриделя-Крафтца со фталевым ангидридом или его производными, такими как фталоилдихлорид или метиловый эфир фталоилхлорида, с последующей циклизацией. В полученный таким образом тетрацикл вводят защиту по 13-оксо-положению с помощью этиленгликоля, бромируют по положению 7 и превращают в 7-ОН производное известными способами (Arcamone и др., Experientia, 1978, 34, 1255; Wong и др. Can. J. Chem., 1971, 49, 2712; Swenton и др., Tetrahedron, 1984, 40, 4625). После снятия защиты антрациклинон формулы VII (где R2=Н) используют как таковой или превращают в 14-ацилоксипроизводное (соединение формулы VII, в котором R2=O-ацил) известными способами. Затем соединения формулы VII гликозилируют защищенными моно- или дисахаридами, как описано в уровне техники (Arcamone и др., Experientia, 1978, 34, 1255; Terashima и др., Bull. Chem. Soc. Jpn, 1986, 59, 423) и в ЕР 721456, с получением антрациклина формулы VIII после снятия защитных групп.
В указанном выше способе или в других аналогичных способах, которые включают в качестве промежуточного соединения тетралин, ключевым промежуточным соединением является сам тетралин формулы I, как определено выше.
Этот АВ синтон (Wong и др. Can. J. Chem, 1971, 49, 2712) позволяет получить соответствующий оптически активный антрациклинон формулы VII,
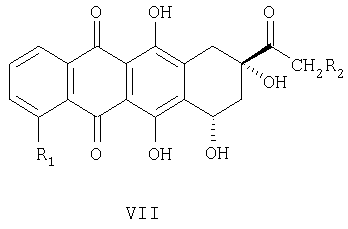
в котором R1=H, ОН, ОСН3 и R2=H, ОН, O-ацил, где ацильную группу выбирают из формила, ацетила, моно-, ди- или трихлорацетила, предпочтительно ацетила.
Как указано выше, соединение в конце превращают в необходимый антрациклин.
Стереохимия положения С-9 антрациклинона очень важна для биологической активности этих соединений, так как только соединения, имеющие (S)-конфигурацию в С-9, проявляют противоопухолевую активность.
Следовательно, тетралиновое промежуточное соединение формулы I очевидно должно обладать той же стереохимией (т.е. абсолютной конфигурацией R).
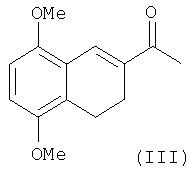
Тетралин I получают обычным способом. В соответствии со сведениями, известными из уровня техники, в виде рацемической смеси, исходя из 2-ацетил-5,8-диметокситетралина III, окислением по положению С-2 трет-бутоксидом/трет-бутанолом калия в присутствии кислорода с последующим восстановлением "in situ" (Wong и др., Can. J. Chem., 1971, 49, 2712; Gardner и др., J.Org. Chem. 1968, 33, 3294).
Соединение III получают с очень низкими выходами реакцией 5,8-диметокси-3,4-дигидронафталина II с N-N-дифенилацетамидом - POCl3 в условиях реакции Вильсмайера-Хаака, с последующим восстановлением двойной связи.
Сообщалось о некоторых попытках ацилирования соединения II, но все они были неуспешными (Rama Rao и др. hid.J.Chem. 1985, 24В, 697).
Альтернативно, соединение III получают с выходом около 50% в 4 стадии по реакции 5,8-диацетокси-3,4-дигидронафталина с ацетилхлоридом/AlCl3 и образованием хлорацетильного производного, с последующим дегидрохлорированием с помощью LiCl, гидролизом и метилированием "in situ" (Russell и др. J. Chem. Soc. Chem. Comm. 1983, 994).
Другой реакционный путь для получения предшественника III, описанный в уровне техники, включает пять стадий, исходя из 5,8-дигидрокси-1,4-дигидронафталина с общим выходом около 50% (Giles и др. S.Afr.J.Chem, 1990, 43, 87).
Рацемический тетралин I далее превращают в чистое энантиомерное соединение обычными методами, применяемыми для расщепления рацематов, через диастереоизомерные основания Шиффа по ацетильной боковой цепи с помощью (-)-1-фенилэтиламина (Arcamone и др. ВР 02691/75, 1975). Альтернативно энантиомерное чистое соединение получают кинетическим расщеплением через асимметричное эпоксидирование по Шарплессу, с последующим окислением образовавшегося аллилового спирта, полученного при восстановлении 2-ацетил-5,8-диметокси-3,4-дигидронафталина (Sharpless и др. J. Am. Chem. Soc. 1981, 103, 6237). Другой способ получения оптически чистого тетралина заключается в стереоселективном восстановлении рацемической смеси пекарскими дрожжами до смесей диастереоизомерных диолов, с последующим хроматографическим разделением и повторным окислением (Terashima и др., Chem. Pharm. Bull. 1984, 32, 4328).
Асимметричный синтез тетралина I, исходя из предшественника III, энантиоселективным дигидроксилированием, описан в М.Nakajima и др. Tetrahedron, 1993, 49, 10807, но некоторые стадии и конечное содержание тетралина I и в особенности использование тетраоксида осмия в стехиометрических количествах, вместо каталитических, а также использование определенного количества дорогих хиральных аминов (всегда в стехиометрических количествах) при температуре -110°С, сильно затрудняет промышленное применение этого способа.
Другие асимметричные синтезы АВ синтона с помощью хиральных соединений или соединений, включающих хиральные производные природных соединений, описаны в уровне техники, но все эти синтезы являются очень сложными и непригодными для промышленного использования (Krohn, Angew. Chem.Int. Ed. Engl., 1986, 25, 790).
Сущность изобретения
Настоящее изобретение относится к способу получения антрациклинов формулы VIII, как она определена выше, в котором оптически активный тетралин формулы I, как он определен выше, получают стереоселективным синтезом, исходя из 5,8-диалкокси-3,4-дигидронафталина II, который в противоположность способам, использующим трудно осуществимое расщепление рацемической смеси и приводящим к выходам менее 30%, имеет преимущество, заключающееся в получении ключевого промежуточного соединения I с намного большим выходом по сравнению с выходом, известным из уровня техники, и легко применимо в промышленности.
В частности, хотя в уровне техники попытки ацилирования соединения II описывались как бесполезные или неинтересные из-за низкого выхода (Rama Rao и др. Ind. J.Chem. 1985, 24В, 697, Russell и др. J. Chem. Soc. Chem. Comm. 1983, 994, Giles и др. S.Afr.J.Chem, 1990, 43, 87), 5,8-диалкокси-3,4-дигидронафталин (соединение формулы II, в котором R представляет собой С1-3 алкильную группу, предпочтительно метильную) может быть неожиданно ацилирован всего лишь в одну стадию в присутствии ацилхлорида и треххлористого алюминия, с образованием соответствующего ацильного производного III. Более того, это новое применение методики энантиоселективного каталитического дигидроксилирования олефинов (Sharpless и др., Chem. Rev. 1994, 94, 2483) с получением ненасыщенного ацильного производного, позволяет получить оптически активный диол IV с хорошим выходом. Соединение далее превращают в соответствующее 1-хлор-2-ацетилпроизводное методом Шарплесса (Sharpless и др. Tetrahedron, 1992, 48, 10515) и дегалогенируют известными методами, например каталитическим гидрированием или в присутствии олова, при этом радикальный предшественник может быть непосредственно дегидроксилирован каталитическим восстановлением. Конечный гидролиз эфирной группы позволяет получить соединение I с хорошим выходом и с высокой оптической чистотой.
Подробное описание изобретения
На схеме I представлен способ получения тетралина формулы I, в котором R=СН3. В этом случае исходным продуктом является 5,8-диметокси-3,4-дигидронафталин II, полученный известными способами, исходя из бутадиена и п-хинона (Fieser и др., J.Am.Chem.Soc., 1948, 70, 3151).
Несмотря на то, что в уровне техники попытки ацилирования соединения II описаны как бесполезные или неинтересные из-за низких выходов, 5,8-диметокси-3,4-дигидронафталин II обрабатывают ацетилхлоридом в присутствии избытка треххлористого алюминия, предпочтительно 5-9 молей треххлористого алюминия на один моль ацилхлорида, при температуре -35°÷25°С, предпочтительно при 0°С. После обычной обработки и кристаллизации из этилацетата, 2-ацетил-5,8-диметокси-3,4-дигидронафталин получают с выходами более чем 70%.
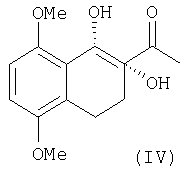
Соединение III стереоселективно превращают в диол IV асимметричным дигидроксилированием по Шарплессу, которое описано в уровне техники для других, олефиновых субстратов (Sharpless и др., Chem. Rev. 1994, 94, 2483). Реактивом, используемым на этой стадии, является AD-смесь α, содержащая каталитическое количество осмиата калия (0,52 г на кг AD-смеси α (каталог Aldrich, реактив 39275-8, а также J. Org. Chem. 1992, 57, 2768) с последующим прибавлением соли осмия (K2OsO2(OH)4) и метансульфон-амида.
Соль осмия всегда присутствует в каталитическом количестве по отношению к субстрату. Реакцию проводят при низкой температуре, от -4 до +20°С, предпочтительно при 0°С, с выходом 70%, с энантиомерным избытком более чем 95%.
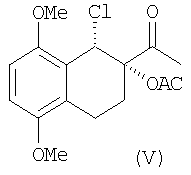
Оптически активный диол IV превращают в хлорацетат V через образование "in situ" циклического промежуточного соединения с помощью триметилортоацетата в присутствии кислотного катализатора с последующей обработкой триметилсилилхлоридом в соответствии со способом, уже описанным в уровне техники для различных диолов (Sharpless и др. Tetrahedron, 1992, 48, 10515).
Эта стадия проходит с выходами более 80%.
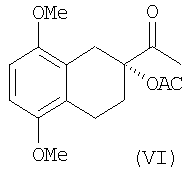
Восстановление хлорацетата в ацетат VI может быть осуществлена фотохимической или термической обработкой в присутствии трибутилгидрида олова и радикальных предшественников, таких как AIBN или ВРО, или каталитическим гидрированием.
Выходы составляют более 80%.
Ацетат может быть получен непосредственно каталитическим восстановлением диола IV.
Гидролиз ацетата может осуществляться с помощью ионообменных смол с количественным выходом. Альтернативно известными способами проведения гидролиза ацетатов являются такие, как обработка метоксидом натрия или гидроксидом натрия.
То, что показано на Схеме I, может легко применяться для синтеза всех соединений формулы I при использовании соответствующих исходных продуктов.
Следовательно, тетралин I, который является объектом настоящего изобретения, получают только в 4-5 стадий с намного большим общим выходом, чем выход, который получают в известных способах.
Более того, описанные условия реакции делают возможным промышленное применение процесса. Последующие стадии способа через антрациклинон до конечного антрациклина осуществляют так, как описано в уровне техники.
Процесс, в соответствии с которым осуществляют настоящее изобретение, будет лучше понятен из проведенных в дальнейшем примеров, которые относятся к Схеме 1, т.е. получению тетралина формулы I, в которой R=СН3.
ПРИМЕР 1
Синтез соединения III
К суспензии треххлористого алюминия (449 г) в дихлорметане (2 л) в токе азота добавляют по каплям ацетилхлорид (380 мл) при температуре 0°С. После 30 минут перемешивания при температуре 0°С к полученному раствору медленно по каплям добавляют раствор 5,8-диметокси-3,4-дигидронафталина II (80 г) в дихлорметане (2,5 л). Через 30 минут перемешивания при температуре 0°С смесь гидролизуют льдом. После отделения органической фазы и промывания с помощью HCl 1N (3×6 л), Н2О (3×4 л) и рассола (2×4 л) растворитель упаривают под вакуумом при температуре 40°С, что дает твердый осадок желтого цвета (98 г). Кристаллизация из кипящего этилацетата позволяет получить 71 г желаемого соединения III. Выход 73%.
1Н ЯМР (CDCI3): 2.44 (с, 3Н, Н10); 2.53 (м, 2Н, Н6); 2.80 (м, 2Н, Н5); 3.30, 3.84 (2 с, 6Н, ОСН3); 6.75 (дд, 2Н, H2+Н3); 7.81 (м, 1H1 H8);
13С ЯМР (CDCI3): 19.9, 20.5 (C5, С6); 25.3 (C10); 55.9. 56.1 (ОСН3); 108.5, 113.2 (C2, С3); 122.6, 127.2 (C4a, C8a); 131.5 (C8); 137.2 (С7); 150.4, 151.0 (C1, C4); 198.8 (C9).
ТСЖХ: Rf 0.80 (петролейный эфир/этилацетат = 80/20).
ВЭЖХ: время удержания=8.9 минут (условия: колонка Lichrospher 100 RP 18 (5 мкм, 250×4 мм).
СН3CN/Н2O+0.1% TFA(трифторуксусная кислота)=60/40; 1 мл/минута; λ=214 нм; 20 мкл раствора 1 мг/10 мл).
ПРИМЕР 2
Синтез соединения IV
К раствору AD смеси α (600 г) и K2OsO2(OH)4 (1 г) в воде (2 л) добавляют трет-бутанол (2.15 л), метансульфонамид (40.7 г), гидрокарбонат натрия (109 г). Смесь перемешивают до полного растворения твердых исходных реагентов, охлаждают до температуры 0°С, добавляют 2-ацетил-5,8-диметокси-3,4-дигидронафталин (100 г) и энергично перемешивают в течение 96 часов, общее количество K2OsO2(OH)4 составляет 1,312 г.
После завершения вступления в реакцию исходного, как показывает ТСЖХ (петролейный эфир/этилацетат = 80/20), добавляют порциями 630 г гидросульфита натрия и после часа перемешивания добавляют 4 л AcOEt, после чего фазы отделяют. Органическую фазу промывают NaOH 1N (1×2 л), Н2О (1×2 л) и упаривают в вакууме. Полученное твердое вещество растворяют в 750 мл СН2Cl2 и раствор экстрагируют H2SO4, 3% насыщенным раствором K2SO4 (4×200 мл), NaHCO3 (твердый) (1×300 мл) и Н2О (1×300 мл). Органическую фазу, высушенную над безводным MgSO4, упаривают в вакууме, получая твердый осадок.
Продукт кристаллизуют из смеси AcOEt/циклогексан = 1/1, фильтруют и высушивают в вакууме. Получают 78.7 г кристаллического твердого вещества. Выход: 70.5%
1H ЯМР (CDCI3): 1.87 (м, 2Н, Н6); 2.38 (с, 3Н, Н10); 2.79 (м, 2Н, Н5); 3.78, 3.84 (2 с, 6Н, ОСН3); 3.81 (м, 1Н, H8); 4.87 (д, 1Н, OH8); 5.29 (с, 1Н, ОН7); 6.71 (с, 2Н, Н2+Н3).
13С ЯМР (CDCI3): 19.1 (С10); 25.8 (C5); 28.7 (С6); 55.7, 55.7 (ОСН3); 68.5 (C8); 78.7 (С7); 108.0, 108.8 (С2, С3); 125.8, 127.1 (C4a, C8a); 151.1, 152.3 (C1, C4); 214.2 (C9).
ТСЖХ: Rf 0.25 (петролейный эфир/этилацетат = 80/20)
ВЭЖХ: время удержания = 4.1 минут (условия: колонка Lichrospher 100 RP 18 (5 мкм) 250×4 мм СН3CN/Н20+0.1% TFA=50/50; 1 мл/минута; λ=214 нм; 20 мкл раствора 2.8 мг/10 мл).
Энантиомерный выход = 98%, определен с помощью хиральной ВЭЖХ (условия: колонка Chiralcel OD 250 X 4.6 мм;
н-гексан/EtOH=90/10; 1 мл/минута; λ=214 нм; 20 мкл раствора 1.3 мг/10 мл)
температура плавления = 141-143°С.
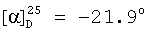 (с=1.0, CHCl3)
(с=1.0, CHCl3)
ПРИМЕР 3
Синтез соединения V
К раствору диола (77 г) в СН2Cl2 (600 мл), в атмосфере азота добавляют триметилортоацетат (59.3 мл) и 4-толуолсульфонат пиридиния (2 г). Раствор перемешивают при комнатной температуре в течение 24 часов. Затем растворитель упаривают в вакууме с получением твердого осадка. Твердое вещество, растворенное в СН2Cl2 (600 мл) добавляют в атмосфере азота к триметилсилилхлориду (65 мл). Реакционную смесь перемешивают при комнатной температуре в течение часа и после упаривания растворителя в вакууме, обрабатывают циклогексаном (400 мл) при энергичном перемешивании в течение 3 часов. Твердое вещество отфильтровывают и высушивают в вакууме. Получают 98.1 г желаемого продукта (количественный выход).
1H ЯМР (CDCI3): 1.96 (с, 3Н, Н10); 1.97-3.15 (м, 4Н, H5+Н6); 2.43 (с, 3Н, Н12); 3.81, 3.87 (2 с, 6Н, ОСН3); 5.35 (д, 1Н, H8); 6.76 (дд, 2Н, Н2+Н3);
13С ЯМР (CDCI3): 19.6 (С11); 20.2, 20.5 (C5, С6); 26.3 (С12); 52.9 (C8); 55.6, 56.0 (ОСН3); 82.9 (C7); 108.3, 110.1 (С2, С3); 123.2, 125.4 (C4a, C8a); 150.7, 151.6 (C1, С4); 169.6 (С11); 204.2 (С9).
ТСЖХ:Rf=0.55 (петролейный эфир/AcOEt=75/25)
температура плавления = 128-138°С.
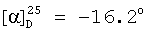 (с=1.0, СН2Cl2).
(с=1.0, СН2Cl2).
ПРИМЕР 4
Синтез соединения VI
К раствору хлорацетата (97.3 г) в толуоле (2 л) добавляют в токе азота AIBN (1.5 г) и трибутилгидрида олова (225 мл). Смесь перемешивают при облучении вольфрамовой лампой мощностью 200 ватт в течение 24 часов и после этого экстрагируют водой (500 мл). Органическую фазу отделяют, высушивают и упаривают в вакууме. Осадок обрабатывают циклогексаном (500 мл) при перемешивании, фильтруют и высушивают в вакууме при температуре 40°С. Получают 67.8 г желаемого продукта в виде твердого вещества белого цвета. Выход: 80.3%.
1H ЯМР (CDCI3): 1.95,2.50 (2 м, 2Н, Н6); 2.05 (1.3Н, Н10); 2.22 (1.3Н, Н12); 2.40, 2.90 (2 м, 2Н, Н5); 3.00 (дд, 2Н, H8); 3.77, 3.80 (2 с, 6Н, ОСН3); 6.66 (м, 2Н, H2+Н3).
13С ЯМР (CDCI3): 19.5, 21.0 (C5, С6); 24.0 (C10); 26.7 (С12); 30.2 (C8); 55.6, 55.5 (ОСН3); 83.6 (С7); 107.0, 107.2 (C2, С3); 122.7, 125.1 (C4a, C8a); 150.9, 151.4 (C1, C4); 170.5 (С11); 206.5 (С9).
ТСЖХ:Rf=0.28 (толуол/этилацетат=95/5)
ВЭЖХ: время удержания = 7.4 минут (условия: колонка Lichrospher 100 RP 18 (5 мкм) 250×4 мм, CH3CN/H2O+0.1% TFA=60/40; 1 мл/минута; λ=214 нм; 20 мкл раствора 1.2 мг/10 мл).
Т.пл.: 110-118°С.
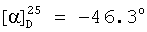 (с=1.0, CHCl3).
(с=1.0, CHCl3).
ПРИМЕР 5
Синтез соединения I
К раствору ацетата (66 г) в метаноле (5 л) добавляют смолу Амберлит IRA-400 (ОН) (183 мл), предварительно активированную обработкой 30% NaOH (8×400 мл) и промытую водой (5×400 мл) и метанолом (4×400 мл). Реакционную смесь перемешивают в течение ночи при комнатной температуре. После удаления смолы с помощью фильрования и упаривания растворителя в вакууме получают твердый осадок, который после кристаллизации из циклогексана/этилацетата, фильтрования и высушивания дает 51.85 г сухого желаемого продукта. Выход: 92%.
1Н ЯМР (CDCI3): 1.89 (м, 2Н, Н6); 2.33 (с, 3Н, Н10); 2.91 (м, 4Н, Н5+H6); 3.65 (с, 1H, ОН); 3.77, 3.80 (2 с, 6Н, ОСН3); 6.66 (с, 2Н, Н2+Н3).
13С ЯМР (CDCI3): 19.2 (C5); 23.9 (C10); 29.7, 32.4 (С6, C8); 55.5, 55.6 (ОСН3); 76.4 (C7); 107.0, 107.4 (C2, С3); 122.7, 125.5 (C4a, C8a); 151.1, 151.6 (C1, C4); 212.3 (C9).
ТСЖХ:Rf=0.27 (петролейный эфир/этилацетат = 80/20)
ВЭЖХ: время удержания = 5.9 минут (условия: колонка Lichrospher 100 RP 18 (5 мкм) 250×4 mm, CH3CN/H2O+0.1% TFA=50/50; 1 мл/минута; λ=214 нм; 20 мкл раствора 2.5 мг/мл).
Энантиомерный выход = >99%, определенный с помощью хиральной ВЭЖХ (условия: колонка Chiralcel OD 250 X 4.6 мм;
н-гексан/EtOH=90/10; 1 мл/минута; λ=214 нм; 20 мкл раствора 1.35 мг/10 мл) температура плавления: 126-129°С.
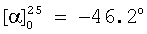 (с=1.0, CHCl3)
(с=1.0, CHCl3)
Пример 6 (как продолжение примера IV)
Раствор диола (IV (20 г) в ледяной уксусной кислоте (200 мл) обрабатывают 5% Pd/C (4 г) и полученную суспензию гидрогенизируют перемешивая в течение 7 час при 50°С. К полученной суспензии добавляют СН2Cl2 (100 мл). Смесь фильтруют через кремниевую подушку и фильтрат концентрируют. Затем добавляют воду (600 мл) и смесь перемешивают в течение часа при комнатной температуре. Твердое вещество отфильтровывают и высушивают под вакуумом. Получают 16,47 г продукта (VI) (выход: 75%).
| название | год | авторы | номер документа |
|---|---|---|---|
| 3-ОКСО-3,9-ДИГИДРО-1Н-ХРОМЕНО[2,3-c]ПИРРОЛЫ В КАЧЕСТВЕ АКТИВАТОРОВ ГЛЮКОКИНАЗЫ | 2011 |
|
RU2603191C2 |
| СПОСОБ ПОЛУЧЕНИЯ (S)-(-)-6-БЕНЗИЛОКСИ-3,4-ДИГИДРО-2,5,7,8-ТЕТРАМЕТИЛ-2Н-1-БЕНЗОПИРАН-2-ИЛМЕТАНОЛА | 2010 |
|
RU2443695C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-АМИНО-5,8-ДИМЕТОКСИ[1,2,4]ТРИАЗОЛО[1,5-c]ПИРИМИДИНА ИЗ 4-ХЛОР-2,5-ДИМЕТОКСИПИРИМИДИНА | 2013 |
|
RU2635352C2 |
| СПОСОБ ПОЛУЧЕНИЯ АНТРАЦИКЛИНОНОВ | 1990 |
|
RU2077526C1 |
| (E)-2-(4-{[3-(2,4-ДИМЕТОКСИФЕНИЛ)АКРИЛАМИДО]МЕТИЛ}-1H-1,2,3-ТРИАЗОЛ-1-ИЛ)-2-ИЗОПРОПИЛ-9-(4-МЕТИЛПИПЕРАЗИН-1-ИЛ)-3,7-ДИОКСО-3,7-ДИГИДРО-2H-ФУРО[3,2-g]ХРОМЕН, ОБЛАДАЮЩИЙ АНАЛЬГЕТИЧЕСКОЙ АКТИВНОСТЬЮ | 2014 |
|
RU2549574C1 |
| Способ получения 2-амино-4-арил-6-гексил-7-гидрокси-4H-хромен-3-карбонитрилов | 2022 |
|
RU2802631C1 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ 3,4-ДИАЛКОКСИ-2,5-ТИОФЕНДИКАРБОНОВОЙ КИСЛОТЫ | 2021 |
|
RU2778232C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-ЗАМЕЩЕННЫХ 3,3-ДИМЕТИЛ-3,4-ДИГИДРОИЗОХИНОЛИНОВ | 2001 |
|
RU2213735C2 |
| 4-ЗАМЕЩЕННЫЕ АНТРАЦИКЛИНОНЫ И ГЛИКОЗИД АНТРАЦИКЛИНА | 1991 |
|
RU2024483C1 |
| Способ получения 4-арилзамещенных 7-гидрокси-6'-фенил-5',6'-дигидро-1'Н-спиро[хроман-2,4'-пиримидин]-2'(3'H)-онов | 2023 |
|
RU2818006C1 |
Оптически активный тетралин формулы I
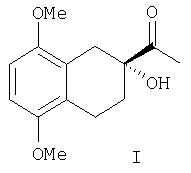
получают, исходя из 5,8-диметокси-3,4-дигидронафталина согласно следующим стадиям:
- ацилирование в присутствии избытка AlCl3 при температуре -35÷25°С;
- энантиоселективное дигидроксилирование по Шарплессу в присутствии каталитического количества соли осмия;
- получение 1-хлор-2-ацетилпроизводного;
- дигидрохлорирование и последующий гидролиз.
Технический результат - значительное увеличение выхода целевого вещества. 2 н. и 3 з.п. ф-лы.
заключающийся в том, что
а) 5,8-диалкокси-3,4-дигидронафталин формулы (II)
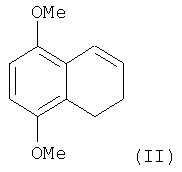
подвергают взаимодействию с ацетилхлоридом в присутствии молярного избытка AlCl3 по отношению к ацетилхлориду при температуре в интервале -35÷25°С, с образованием соответствующего ацильного производного формулы (III)
b) ацильное производное (III) стереоселективно дигидроксилируют по Шарплессу, где реактивом является AD смесь а, с последующим прибавлением соли осмия К2O2Os(ОН)4 и метансульфонамида, с образованием диола (IV)
с) диол формулы (IV) или непосредственно восстанавливают в ацетат (VI)
в присутствии ледяной уксусной кислоты, или предварительно с помощью реакции с триэтилортоацетатом и триметилсилилхлоридом переводят в 1-хлор-2-ацетилпроизводное формулы (V)
которое восстанавливают известными методами;
d) ацетат (VI) гидролизуют до тетралина формулы (I).
| Yong S.Rho | |||
| et al | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Chein | |||
| Bull | |||
| Korean | |||
| Chem | |||
| Soc. | |||
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| A.V.Rama Rao et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Indian Journal of Chemistry | |||
| Приспособление для установки двигателя в топках с получающими возвратно-поступательное перемещение колосниками | 1917 |
|
SU1985A1 |
| A.V.Rama Rao et al | |||
| A | |||

