Настоящее изобретение касается имидазохинолиновых соединений, имеющих простую эфирную и гетероциклильную или гетероарильную группы в положении 1, и фармацевтических составов, содержащих такие соединения. Другой аспект настоящего изобретения затрагивает применение данных соединений в качестве иммуномодуляторов для стимулирования биосинтеза цитокинов в организме животных и для лечения заболеваний, включая вирусные болезни и опухолевые заболевания.
Первый надежный отчет о циклической системе 1Н-имидазо[4,5-с]хинолина (Бакман и др., J.Org.Chem. 15, 1278-1284 (1950)) описывает синтез 1-(6-метокси-8-хинолинил)-2-метил-1H-имидазо[4,5-с]хинолина для возможного применения в качестве противомалярийного средства. Далее поступило сообщение о синтезе различных замещенных 1H-имидазо[4,5-с]хинолинов. Так, например, было синтезировано соединение 1-[2-(4-пиперидил)этил]-1Н-имидазо[4,5-с]хинолин (Джейн и др., J.Med.Chem. 11, стр. 87-92 (1968)) в качестве предполагаемого противосудорожного и сердечно-сосудистого средства. Имелись также сообщения о ряде 2-оксоимидазо[4,5-с]хинолинов (Баранов и др., Chem.Abs. 85, 94362 (1976), Берени и др., J.Heterocyclic Chem. 18,1537-1540 (1981)).
Позже было обнаружено, что некоторые 1H-имидазо[4,5-с]хинолин-4-амины и их 1- и 2-замещенные производные могут найти применение как противовирусные средства, бронхолитические средства и иммуномодуляторы. В числе прочих публикаций можно сослаться на патенты США №№4689338, 4698348, 4929624, 5037986, 5268376, 5346905 и 5389640; все эти патенты приведены здесь в качестве ссылок.
Продолжают вызывать интерес имидазохилиновые циклические системы. Известны некоторые 1Н-имидазо[4,5-с]нафтиридин-4-амины, 1Н-имидазо[4,5-с]пиридин-4-амины и 1Н-имидазо[4,5-с]хинолин-4-амины, имеющие заместитель с простой эфирной группой в положении 1. Они описаны в патентах США №№5268376, 5389640 и 5494916 и в международной заявке WO 99/29693.
По-прежнему имеется потребность в соединениях, которые обладают способностью модулировать иммунную реакцию путем стимулирования биосинтеза цитокинов или под воздействием иных механизмов.
Краткое изложение сущности изобретения
Авторами был обнаружен новый класс соединений, способных стимулировать биосинтез цитокинов в организме животных. В соответствии с этим настоящее изобретение предлагает соединения имидазо[4,5-с]хинолин-4-амина и тетрагидроимидазо[4,5-с]хинолин-4-амина, имеющие заместитель с простой эфирной группой в положении 1. Согласно данным ИК-спектроскопии, эти соединения могут быть описаны формулами (I), (II), (III) и (IV). Ниже приведена общая структурная формула этих соединений
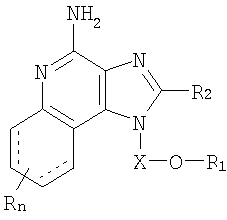
При этом X, R1, R2 и R определены для каждого класса соединений, имеющих формулы (I), (II), (III) и (IV).
Соединения, представленные формулами (I), (II), (III) и (IV), могут применяться в качестве модификаторов иммунной реакции вследствие их способности стимулировать биосинтез цитокинов и иными способами модулировать иммунную реакцию при введении в организм животных. Эти свойства делают указанные соединения полезными для лечения ряда заболеваний, таких как вирусные болезни и опухоли, реагирующие на изменения в иммунной реакции.
Настоящее изобретение касается также фармацевтических составов, содержащих соединения, модифицирующие иммунную реакцию, и способов стимулирования биосинтеза цитокинов в организме животных, лечения вирусной инфекции у животных и (или) лечения опухолевых заболеваний у животных путем введения им соединений формулы (I), (II), (III) или (IV).
Кроме того, настоящее изобретение затрагивает способы синтеза соединений, представленных в нем, и промежуточных продуктов, используемых при синтезе данных соединений.
Подробное описание изобретения
Как указывалось выше, обнаружены некоторые соединения, стимулирующие биосинтез цитокинов и модифицирующие иммунную реакцию в организме животных. Такие соединения представлены формулами (I), (II), (III) и (IV), приведенными ниже.
Имидазохинолиновые соединения, составляющие предмет настоящего изобретения и имеющие простую эфирную группу, а также гетероциклильный или гетероарильный заместитель в положении 1, представлены формулой (I):
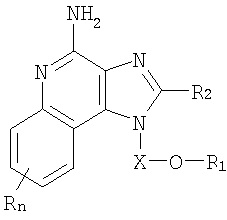
где Х представляет собой -CHR3-, -CHR3-алкильную- или -CHR3-алкенильную группу;
R1 выбран из группы, содержащей радикалы:
- гетероарил;
- гетероциклил;
- R4-гетероарил, а также
- R4-гетероциклил;
- R2 выбран из группы, содержащей радикалы:
- водород;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил, а также
- алкил или алкенил, с одним или несколькими заместителями, выбранными из группы, содержащей радикалы:
- ОН;
- галоген;
- N(R3)2;
- CO-N(R3)2;
- СО-С1-10алкил;
- CO-O-C1-10алкил;
- N3;
- арил;
- гетероарил;
- гетероциклил;
- СО-арил, а также
- СО-гетероарил;
R4 представляет собой алкил или алкенил, в который может войти одна или несколько -О-групп;
каждый R3 представляет собой независимо Н или C1-10алкил;
каждый Y представляет собой независимо -О- или -S(O)0-2-;
значение n может изменяться от 0 до 4, а также каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений. Настоящее изобретение также включает в себя имидазохинолиновые соединения, несущие простую эфирную группу в положении 1, причем заместитель с простой эфирной группой также содержит алкинильную группу и гетероциклильную или гетероарильную группу. Такие соединения представлены формулой (II):
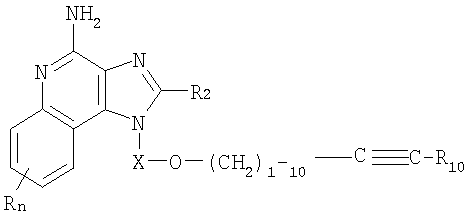
где Х представляет собой -CHR3-, -CHR3-алкильную- или -CHR3-алкенильную группу;
R10 выбран из группы, содержащей гетероарильный или гетероциклильный радикалы;
R2 выбран из группы, содержащей радикалы:
- водород;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил, а также
- алкил или алкенил с одним или несколькими заместителями, выбранными из группы, содержащей радикалы:
- ОН;
- галоген;
- N(R3)2;
- СО-N(R3)2;
- CO-C1-10алкил;
- СО-О-C1-10алкил;
- N3;
- арил;
- гетероарил;
- гетероциклил;
- СО-арил, а также
- СО-гетероарил;
n может иметь значение от 0 до 4;
каждый Rз представляет собой независимо Н или C1-10алкил;
каждый Y представляет собой независимо -О- или -S(O)0-2-, а также каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
Настоящее изобретение также включает в себя тетрагидроимидазохинолиновые соединения, несущие простую эфирную группу и гетероциклильный или гетероарильный заместитель в положении 1. Такие тетрагидроимидазохинолиновые соединения представлены формулой (III):
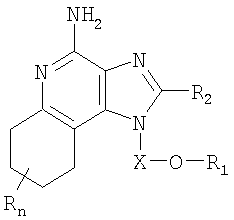
где Х представляет собой -CHR3-, -CHR3-алкильную- или -CHR3-алкенильную группу;
R1 выбран из группы, содержащей радикалы:
- гетероарил;
- гетероциклил;
- R4-гетероарил, а также
- R4-гетероциклил;
R2 выбран из группы, содержащей радикалы:
- водород;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкен ил;
- алкил-Y-арил, а также
- алкил или алкенил с одним или несколькими заместителями, выбранными из группы, содержащей радикалы:
- ОН;
- галоген;
- N(R3)2;
- СО-N(R3)2;
- CO-C1-10алкил;
- CO-O-C1-10алкил;
- N3;
- арил;
- гетероарил;
- гетероциклил;
- СО-арил, а также
- СО-гетероарил;
R4 представляет собой алкил или алкенил, в который может войти одна или несколько -О-групп;
каждый R3 представляет собой независимо Н или C1-10алкил;
каждый Y представляет собой независимо -О- или -S(O)0-2-;
n может иметь значение от 0 до 4;
каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, C1-10 алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
Дополнительным классом соединений-модификаторов иммунной реакцией в настоящем изобретении являются тетрагидроимидазохинолиновые соединения, несущие простую эфирную группу в положении 1, причем заместитель с простой эфирной группой также содержит алкинильную группу и гетероциклильную или гетероарильную группу. Такие тетрагидроимидазохинолиновые соединения представлены формулой (IV):
где
Х является -CHR3-, -CHR3-алкильной- или -CHR3-алкенильной группой;
R10 выбран из группы, содержащей гетероарильный и гетероциклильный радикалы;
R2 выбран из группы, содержащей радикалы:
- водород;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил, а также
- алкил или алкенил с одним или несколькими заместителями, выбранными из группы, содержащей радикалы:
- ОН;
- галоген;
- N(R3)2;
- CO-N(R3)2;
- CO-C1-10алкил;
- CO-O-C1-10алкил;
- N3;
- арил;
- гетероарил;
- гетероциклил;
- СО-арил, а также
- СО-гетероарил;
каждый R3 представляет собой независимо Н или C1-10алкил;
каждый Y представляет собой независимо -О- или -S(O)0-2-;
n может иметь значение от 0 до 4, а также
каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
Получение соединений
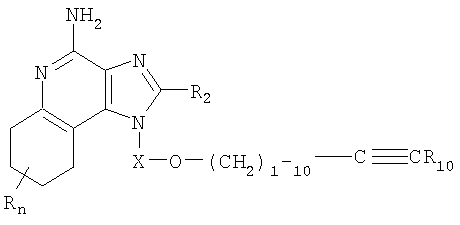
Соединения, представляющие собой предмет настоящего изобретения, можно получить по реакционной схеме I, в которой R, R2, Х и n определены выше, a R11 представляет собой алкил, замещенный гетероарильной группой, причем эта гетероарильная группа может быть незамещенной или иметь заместители, как определено ИК-спектроскопией; или же R11 может быть замещенным гетероарилом, как определено ИК-спектроскопией, с условием, что если R11 является замещенным гетероарилом, то по меньшей мере один заместитель должен быть сильной электроноакцепторной группой, расположенной в положении орто- или пара- к простой эфирной связи.
В схеме 1 реакции 4-амино-1H-имидазо[4,5-с]хинолин-1-иловый спирт (формула X) алкилируют галогенидом (формула XI), получая 1Н-имидазо[4,5-с]хинолин-4-амин (формула XII), являющийся подвидом соединения формулы I. Из спирта (формула X) в результате реакции с гидридом натрия в соответствующем растворителе, например N,N-диметилформамиде, получают алкоксид. Далее галогенид добавляют к реакционной смеси. Реакцию можно проводить при температуре окружающей среды или при осторожном нагревании (прибл. 50°С) в случае необходимости. Продукт или его соль фармацевтического качества могут быть выделен обычными способами.
Известны многие соединения формулы X, например соединения, полученные Герстером (патент США №4689338) и Герстером и др. (патент США №5605899). Сущность этих патентов включена в настоящий документ в качестве ссылок. Другие соединения могут быть получены без затруднений с применением известных приемов синтеза: см., например, патенты США №5578727 (Андре и др.), 5175296 (Герстер), 5395937 (Николаидес и др.), 5741908 (Герстер и др.), сущность которых раскрыта в настоящем документе в качестве ссылок. Многие галогениды формулы XI коммерчески доступны; прочие можно получить без затруднений с применением известных способов синтеза.
Схема I реакции
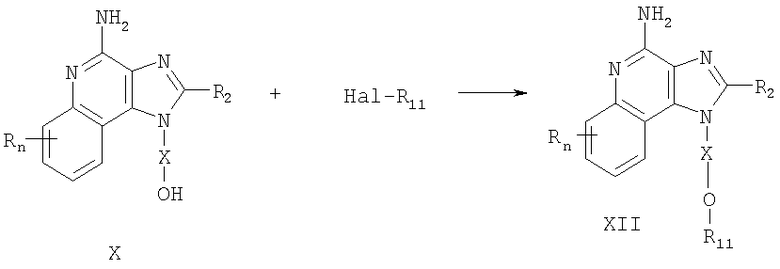
Соединения, представляющие собой предмет настоящего изобретения, можно получать в соответствии со схемой II реакции, где R, R2, R11, X и n определены выше.
На стадии (1) процесса (схема II реакции) гидроксильная группа 1H-имидазо[4,5-с]хинолин-1-илового спирта формулы XIII защищена бензильной группой. Спирт формулы XIII реагирует с гидридом натрия в соответствующем растворителе, например в N,N-диметилформамиде, образуя алкоксид. Далее алкоксид алкилируют бензилбромидом и получают соединение формулы XIV.
Реакцию можно проводить при температуре окружающей среды. Известны многие соединения формулы XIII, например соединение, полученное Герстером (патент США 4689338); другие можно получить, используя известные способы синтеза; см., например, данные Герстера и др. (патент США №5605899) и данные Герстера (патент США №5175296).
На стадии (2) процесса (схема II реакции) соединение формулы XIV окисляют до 1H-имидазо[4,5-с]хинолин-5N-оксида (формула XV) с применением обычного окислителя, способного образовывать N-оксиды. Предпочтительно проводить окисление раствора соединения формулы XIV в хлороформе или дихлорметане с применением 3-хлорпероксибензойной кислоты при температуре окружающей среды.
На стадии (3) процесса (схема II реакции) 1H-имидазо[4,5-с]хинолин-5N-оксид (формула XV) подвергают хлорированию и получают 4-хлор-1Н-имидазо[4,5-с]хинолин (формула XVI). Предпочтительно раствор соединения формулы XV в соответствующем растворителе, например в толуоле, обработать оксихлоридом фосфора при температуре окружающей среды.
На стадии (4) процесса (схема II реакции) 4-хлор-1Н-имидазо[4,5-с]хинолин формулы XVI взаимодействует с фенолом, образуя 4-фенокси-1Н-имидазо[4,5-хинолин формулы XVII. Данный фенол реагирует с гидридом натрия в соответствующем растворителе, например диглиме, образуя феноксид. Далее феноксид реагирует при повышенной температуре с соединением формулы XVI.
На стадии (5) процесса (схема II реакции) защитную бензильную группу удаляют из состава соединения формулы XVII и получают 4-фенокси-1Н-имидазо[4,5-с]хинолин-1 -иловый спирт формулы XVIII. Реакцию предпочтительно проводить путем осторожного добавления трифторуксусной кислоты к раствору соединения формулы XVII в соответствующем растворителе, таком как дихлорметан, при температуре окружающей среды.
На стадии (6) процесса (схема II реакции) 4-фенокси-1Н-имидазо[4,5-с]хинолин-1 -иловый спирт формулы XVIII алкилируют галогенидом Hal-R11, получая 4-фенокси-1Н-имидазо[4,5-с]хинолин-1-иловый простой эфир формулы XIX. Алкоксид соединения формулы XVIII получают путем добавления указанного спирта к двухфазной смеси 50 %-ного водного раствора гидроксида натрия и инертного растворителя, такого как дихлорметан, в присутствии катализатора фазового переноса, например бензилтриметиламмонийхлорида. Далее алкоксид алкилируют. Реакцию можно проводить при температуре окружающей среды.
На стадии (7) процесса (схема II реакции) 4-фенокси-1Н-имидазо[4,5-хинолин-1-иловый простой эфир формулы XIX аминируют и получают 1Н-имидазо[4,5-с]хинолин-4-амин формулы XII, являющийся подвидом соединения формулы I. Реакцию можно проводить путем добавления соединения формулы XIX к ацетату аммония и нагревания полученной смеси при температуре около 150°С. Продукт или его соль фармацевтического качества могут быть выделены с применением обычных способов.
Схема II реакции
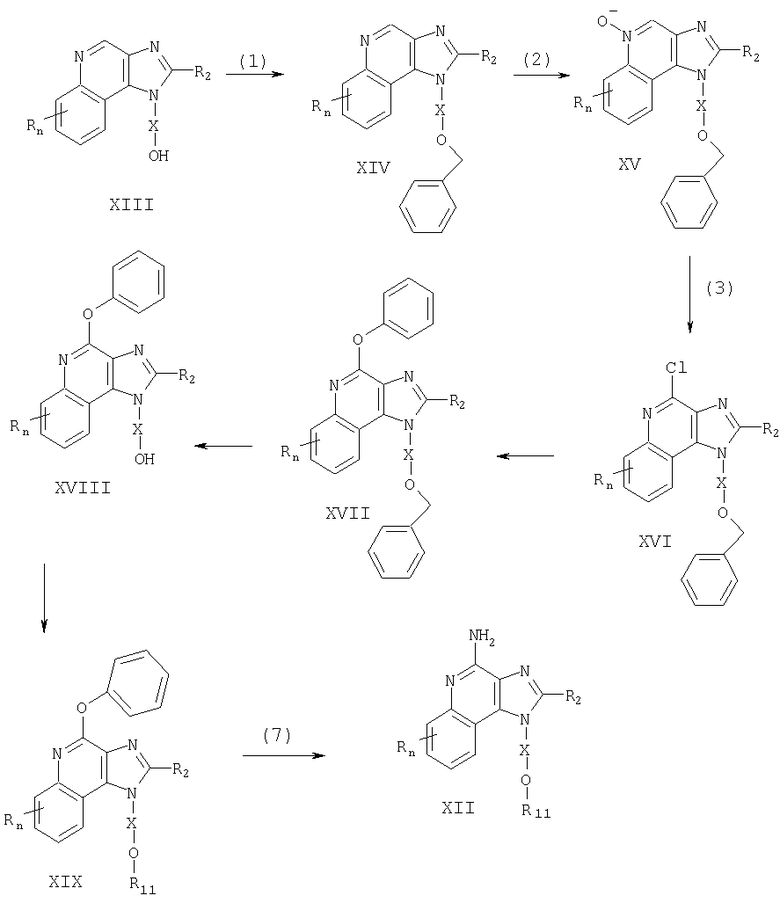
Тетрагидроимидазохинолины, представляющие собой предмет настоящего изобретения, можно получать в соответствии со схемой III реакции, где R, R2, R11, X и n определены выше.
В схеме III реакции 4-амино-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-иловый спирт формулы XX алкилируют галогенидом формулы XI и получают 6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амин формулы XXI, являющийся подвидом соединения формулы III. Спирт формулы XX реагирует с гидридом натрия в соответствующем растворителе, таком как N,N-диметилформамид, образуя алкоксид. Далее алкоксид реагирует с упомянутым галогенидом. Реакцию можно проводить при температуре окружающей среды. Продукт или его соль фармацевтического качества можно выделить, пользуясь известными способами.
Известны многие тетрагидро-1H-имидазо[4,5-с]хинолины формулы XX; см., например, патент США №5352784 (Николаидес и др.). Другие соединения можно получить, используя известные способы синтеза; см., например, патент США №5693811 (Линдстром). Сущность этих патентов раскрыта в настоящем документе в качестве ссылок.
Схема III реакции
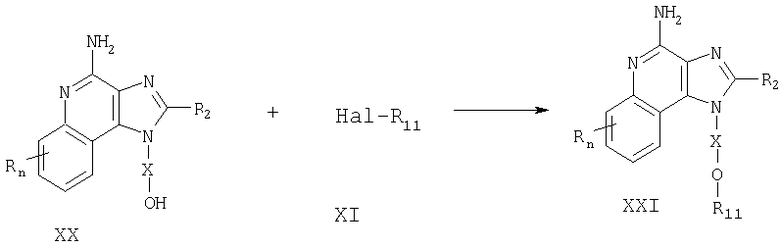
Соединения, представляющие собой предмет настоящего изобретения, могут быть получены в соответствии со схемой IV реакции, где R, R2, Х и n определены выше, a R12 является гетероарильной группой, которая может быть незамещенной или замещенной, как определено ИК-спектроскопией.
На стадии (1) процесса (схема IV реакции) 1Н-имидазо[4,5-с]хинолин-1-иловый спирт (формула XIII) алкилируют галогенидом (формула XXII), получая 1Н-имидазо[4,5-с]хинолин-1-иловый простой эфир (формула XXIII). Соединение формулы XIII и галогенид (формула XXII) взаимодействуют в двухфазной смеси 50 %-ного водного раствора гидроксида натрия и подходящего растворителя, например дихлорметана, в присутствии катализатора фазового переноса, такого как бензилтриметиламмонийхлорид. Реакцию можно проводить при температуре окружающей среды.
На стадии (2) процесса (схема IV реакции) 1Н-имидазо[4,5-с]хинолин (формула XXIII) окисляют до 1H-имидазо[4,5-с]хинолин-5N-оксида (формула XXIV) по способу, описанному на стадии (2) схемы II реакции.
На стадии (3) процесса (схема IV реакции) 1Н-имидазо[4,5-с]хинолин-5N-оксид (формула XXIV) взаимодействует с трихлорацетилизоцианатом, образуя 1Н-имидазо[4,5-с]хинолин-4-ил-ацетамид (формула XXV). Предпочтительно вводить изоцианат с осторожностью в раствор 5N-оксида в соответствующем растворителе, например дихлорметане, при температуре окружающей среды.
На стадии (4) процесса (схема IV реакции) 1Н-имидазо[4,5-с]хинолин-4-ил-ацетамид (формула XXV) гидролизуют до 1Н-имидазо[4,5-с]хинолин-4-амина (формула XXVI). Гидролиз можно выполнять обычными способами, предпочтительно путем обработки раствора соединения формулы XXV в метаноле метоксидом натрия.
На стадии (5) процесса (схема IV реакции) 1Н-имидазо[4,5-с]хинолин-4-амин (формула XXVI) реагирует с галогенидом формулы Hal-R12 в присутствии катализатора - соединения переходного металла - с образованием 1Н-имидазо[4,5-с]хинолин-4-амина (формула XXVII), являющегося подвидом соединения формулы II. Реакцию между соединением формулы XXVI и галогенидом предпочтительно осуществлять в присутствии йодида меди (I), дихлорбис(трифенилфосфин)палладия (II) и избытка триэтиламина в соответствующем растворителе, например в N,N-диметилформамиде или ацетонитриле. Предпочтительно проводить эту реакцию при повышенной температуре (60-80°С). Продукт или его соль фармацевтического качества можно выделить, пользуясь известными способами.
Схема IV реакции
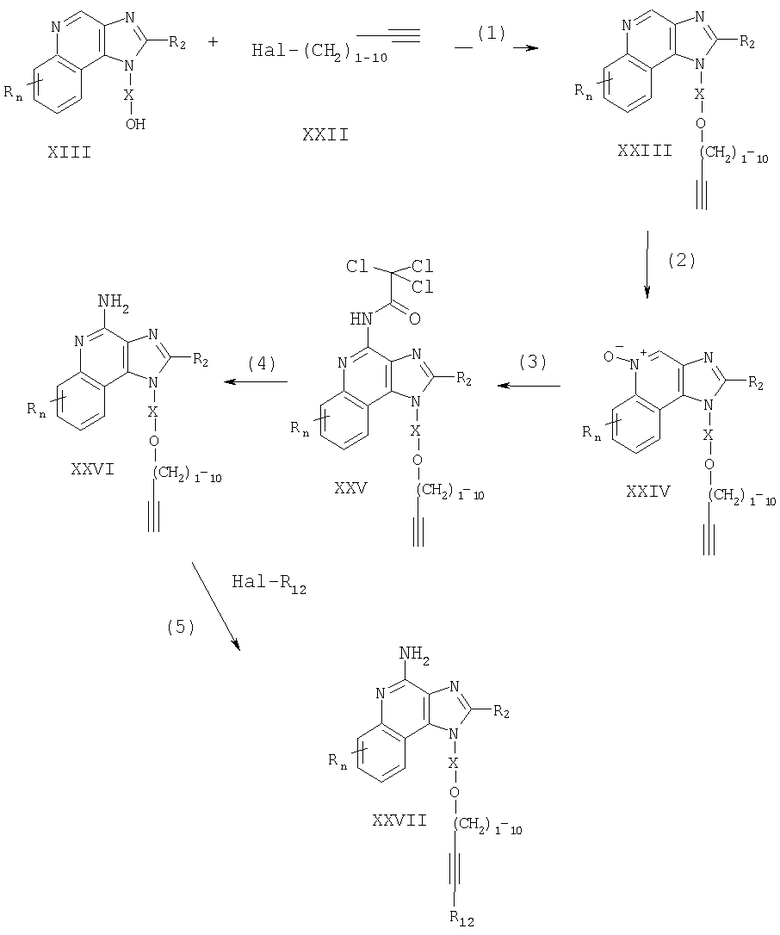
Соединения, представляющие собой предмет настоящего изобретения, можно получать в соответствии со схемой V реакции, где R, R2, R12, Х и n определены выше, а ВОС является трет-бутоксикарбонилом.
На стадии (1) процесса (схема V реакции) аминогруппа 1Н-имидазо[4,5-с]хинолин-4-амина формулы XXVI защищена трет-бутоксикарбонильными группами. Соединение формулы XXVI взаимодействует с ди-трет-бутилдикарбонатом в соответствующем растворителе, например в N,N-диметилформамиде, в присутствии 4-(диметиламино)пиридина и триэтиламина. Реакцию выполняют при повышенной температуре (80-85°С).
На стадии (2) процесса (схема V реакции) защищенный 1Н-имидазо[4,5-с]хинолин-4-амин формулы XXVIII реагирует с галогенидом формулы Hal-R12 в присутствии катализатора - соединения переходного металла - с образованием защищенного 1H-имидазо[4,5-с]хинолин-4-амина формулы XXIX. Реакцию между соединением формулы XXVIII и галогенидом предпочтительно осуществлять в присутствии йодида меди (I), дихлорбис(трифенилфосфин)палладия (II) и избытка триэтиламина в соответствующем растворителе, например в N,N-диметилформамиде или ацетонитриле. Реакция может быть проведена при температуре окружающей среды или при повышенной температуре (40-80°С).
На стадии (3) процесса (схема V реакции) защитные группы убирают путем гидролиза в кислотной среде и получают 1H-имидазо[4,5-с]хинолин-4-амин формулы XXVII, являющийся подвидом соединения формулы II. Предпочтительно соединение формулы XXIX обработать трифторуксусной кислотой в подходящем растворителе, например в дихлорметане. Реакцию можно проводить при температуре окружающей среды или пониженной (0°С) температуре. Продукт или его соль фармацевтического качества можно выделить, пользуясь известными способами.
На стадии (4) процесса (схема V реакции) алкинную связь защищенного 1Н-имидазо[4,5-с]хинолин-4-амина формулы XXIX восстанавливают до защищенного 1Н-имидазо[4,5-с]хинолин-4-амина формулы XXX. Предпочтительно проводить восстановление с применением обычно используемого гетерогенного катализатора гидрогенизации, такого как оксид платины, платина на углероде или палладий на углероде. Реакцию удобно осуществлять в аппарате Парра в соответствующем растворителе, например метаноле.
На стадии (5) процесса (схема V реакции) удаляют защитные группы соединения формулы XXX таким же способом, как на стадии (3), получая 1Н-имидазо[4,5-с]хинолин-4-амин формулы XXXI, являющийся подвидом соединения формулы I. Продукт или его соль фармацевтического качества можно выделить, пользуясь известными способами.
Схема V реакции
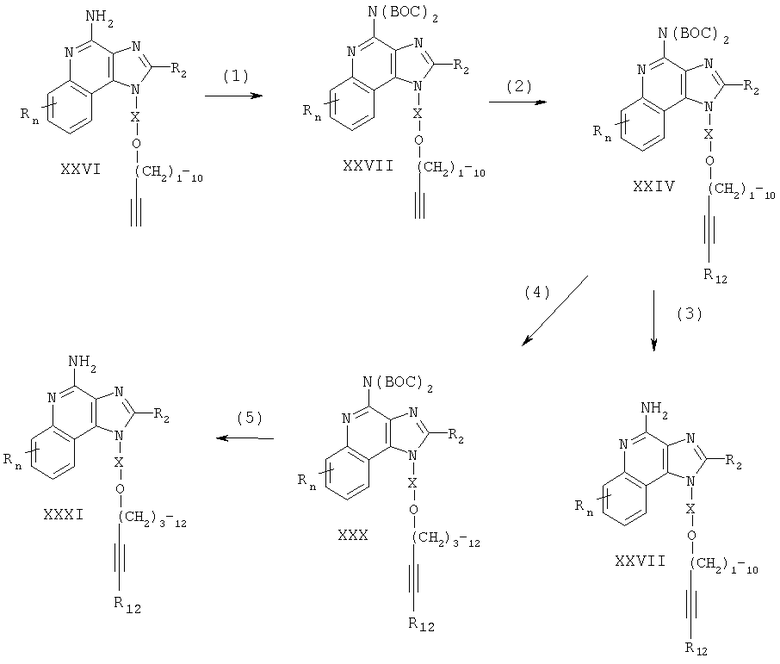
Соединения, представляющие собой предмет настоящего изобретения, можно получать в соответствии со схемой VI реакции, где R, R2, R12, X и n определены выше, a CBZ является бензилоксикарбонилом.
На стадии (1) процесса (схема VI реакции) аминогруппа 1Н-имидазо[4,5-с]хинолин-4-амина формулы XXVI защищена бензилоксикарбонильными группами. Соединение формулы XXVI взаимодействует с дибензилдикарбонатом в соответствующем растворителе, например в N,N-диметилформамиде. Реакцию можно проводить при температуре окружающей среды или слегка повышенной (40°С) температуре.
На стадии (2) процесса (схема VI реакции) защищенный 1H-имидазо[4,5-с]хинолин-4-амин формулы XXXII реагирует с галогенидом формулы Hal-R12 в присутствии катализатора - соединения переходного металла - с образованием защищенного 1H-имидазо[4,5-с]хинолин-4-амина формулы XXXIII. Реакцию между соединением формулы XXXII и галогенидом предпочтительно осуществлять в присутствии йодида меди (I), дихлорбис(трифенилфосфин)палладия (II) и избытка триэтиламина в соответствующем растворителе, например в N,N-диметилформамиде или ацетонитриле. Реакция может быть проведена при температуре окружающей среды или при повышенной температуре (40-80°С).
На стадии (3) процесса (схема VI реакции) защитные группы убирают путем гидролиза и в результате получают 1H-имидазо[4,5-с]хинолин-4-амин формулы XXVII, являющийся подвидом соединения формулы II. Предпочтительно соединение формулы XXXIII обработать метоксидом натрия в подходящем растворителе, например в метаноле. Реакцию можно проводить при температуре окружающей среды. Продукт или его соль фармацевтического качества можно выделить, пользуясь известными способами.
На стадии (4) процесса (схема VI реакции) защитные группы соединения формулы XXXIII удаляют гидрогенолизом; при этом алкинная связь восстанавливается, что в результате приводит к образованию 1Н-имидазо[4,5-с]хинолин-4-амина формулы XXXI, являющегося подвидом соединения формулы I. Предпочтительно проводить гидрогенолиз/восстановление с применением гидроксида палладия на углероде. Реакцию удобно осуществлять в аппарате Парра в соответствующем растворителе, например в метаноле. Продукт или его соль фармацевтического качества можно выделить, пользуясь известными способами.
Схема VI реакции
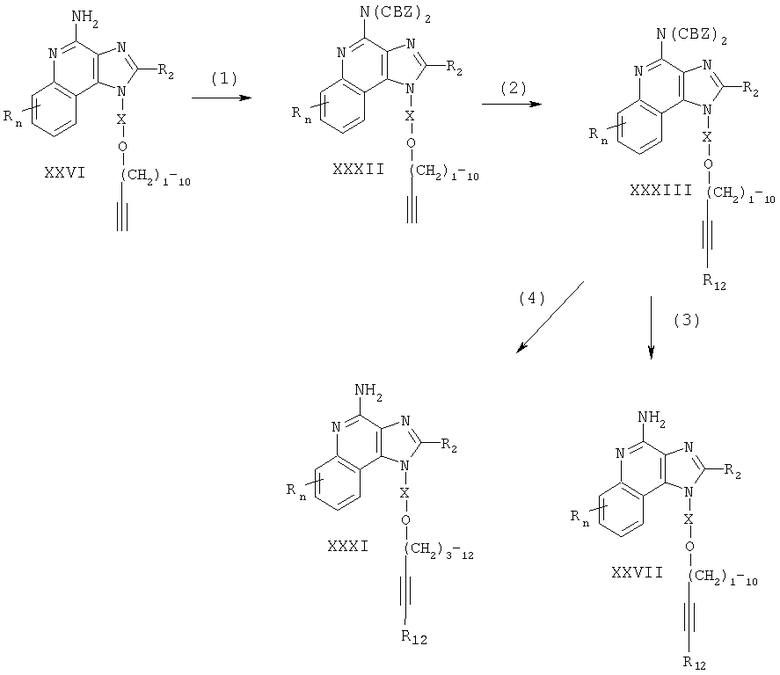
Соединения, представляющие собой предмет настоящего изобретения, можно получать в соответствии со схемой VII реакции, где R, R1, R2, X и n определены выше.
На стадии (1) процесса (схема VII реакции) 2,4-дихлор-3-нитрохинолин (формула XXXIV) реагирует с амином формулы R1-O-X-NH2, образуя 2-хлор-3-нитрохинолин-4-амин формулы XXXV. Реакцию можно проводить путем добавления амина к раствору соединения формулы XXXIV в соответствующем растворителе, например в хлороформе или дихлорметане, при возможном нагревании. Известны различные хинолины формулы XXXIV; некоторые можно получать, пользуясь известными способами (см., например, патент США №4988815, выданный на имя Андре и др., и ссылки, содержащиеся в нем).
На стадии (2) процесса (схема VII реакции) 2-хлор-3-нитрохинолин-4-амин формулы XXXV восстанавливают до 2-хлорхинолин-3,4-диамина (формула XXXVI). Реакцию предпочтительно проводить с применением обычного гетерогенного катализатора гидрогенизации, например платины на углероде или палладия на углероде. Реакцию удобно осуществлять в аппарате Парра в подходящем растворителе, например в изопропаноле или толуоле.
На стадии (3) процесса (схема VII реакции) 2-хлорхинолин-3,4-диамин (формула XXXVI) реагирует с карбоновой кислотой или ее эквивалентом, образуя 4-хлор-1Н-имидазо[4,5-с]хинолин формулы XXXVII. В число подходящих эквивалентов карбоновой кислоты входят сложные ортоэфиры и 1,1-диалкоксиалкилалканоаты. Карбоновую кислоту или ее эквивалент выбирают так, чтобы в состав соединения формулы XXXVII входил требуемый заместитель R2. Так, например, при введении триэтилортоформиата образуется соединение, в котором R2 является водородом; в присутствии триэтилортоацетата R2 представляет собой метил. Реакцию можно проводить в отсутствие растворителя или в инертном растворителе, таком как толуол. Реакция протекает с разогревом, достаточным для того, чтобы любой спирт или вода, образующиеся как побочные продукты, улетучились. В качестве варианта можно воспользоваться катализатором, таким как пиридингидрохлорид.
В качестве альтернативы можно выполнить стадию (3) путем (I) проведения реакции между диамином формулы XXXVI и ацилгалогенидом формулы R2C(O)CI и (ii) циклизации. В части (i) ацилгалогенид добавляют к раствору диамина в инертном растворителе, например в ацетонитриле, пиридине или дихлорметане. Эту реакцию можно проводить при температуре окружающей среды. В части (ii) продукт части (i) нагревают в спиртовом растворе в присутствии основания. Предпочтительно нагревать этанольный раствор продукта части (i) в присутствии избытка триэтиламина при нагревании с обратным холодильником или же нагревать в метанольном растворе аммиака. С другой стороны, если часть (i) была проведена в пиридине, то часть (ii) можно осуществить путем нагревания реакционной смеси после того, как анализ укажет на завершение части (i).
На стадии (4) процесса (схема VII реакции) 4-хлор-1H-имидазо[4,5-с]хинолин формулы XXXVII аминируют, получая 1H-имидазо[4,5-с]хинолин-4-амин формулы I. Реакцию осуществляют при нагревании (например, при температуре 125-175°С) соединения формулы XXXVII под давлением в герметизированном реакторе в присутствии раствора аммиака в алканоле. Продукт или его соль фармацевтического качества можно выделить, пользуясь известными способами.
Схема VII реакции
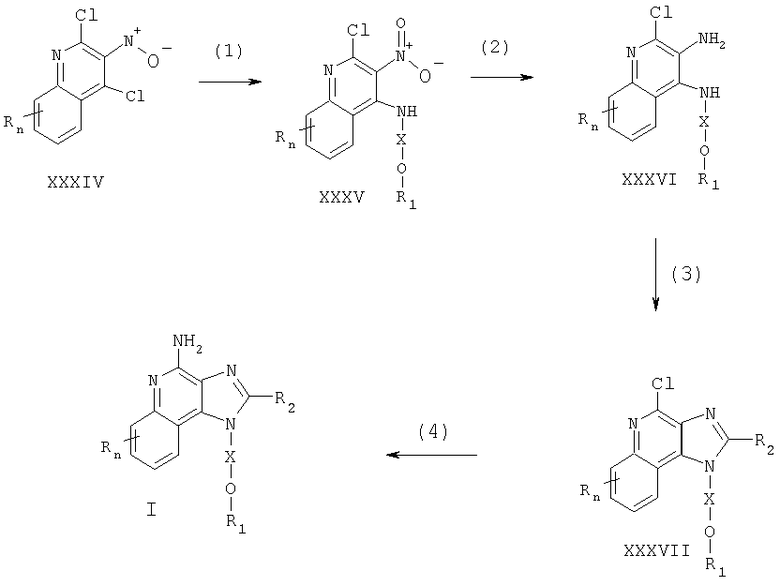
Соединения, представляющие собой предмет настоящего изобретения, можно получать в соответствии со схемой VIII реакции, где R, R1, R2, Х и n определены выше.
По схеме VIII реакции 1Н-имидазо[4,5-с]хинолин-4-амин формулы XXXVIII алкилируют галогенидом формулы XXXIX, получая 1H-имидазо[4,5-с]хинолин-4-амин формулы I. Соединение формулы XXXVIII обрабатывают гидридом натрия в соответствующем растворителе, например в N,N-диметилформамиде. Далее галогенид вводят в реакционную смесь. Реакцию можно проводить при повышенной температуре (прибл. 100°С). Алкилирование происходит по атомам азота N1 и N3; требуемый N1-изомер можно легко отделить от N3-изомера, применяя известные способы, такие как хроматографию на колонке и рекристаллизацию.
Известны многие 1H-имидазо[4,5-с]хинолин-4-амины формулы XXXVIII; другие могут быть приготовлены с использованием известных способов синтеза: см., например, патент США №5756747 (Герстер) и приведенные в нем ссылки.
Схема VIII реакции
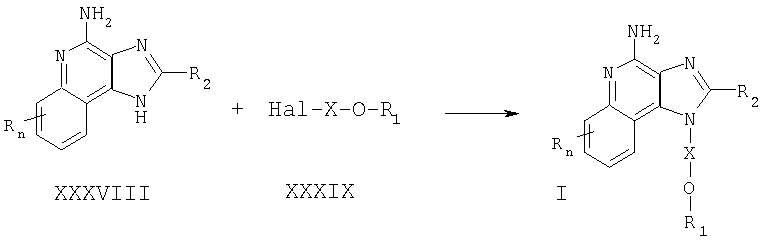
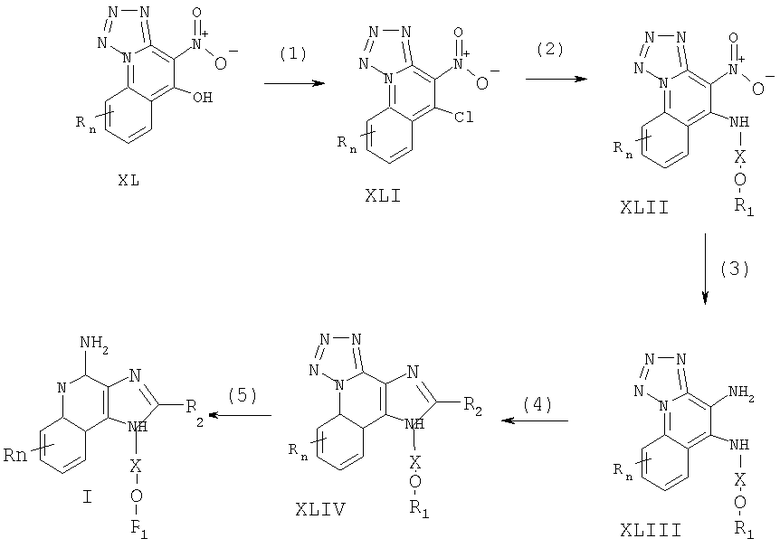
Соединения, представляющие собой предмет настоящего изобретения, можно получать в соответствии со схемой IX реакции, где R, R1, R2, Х и n определены выше.
На стадии (1) процесса (схема IX реакции) 4-нитротетразоло[1,5-а]хинолин-5-ол формулы XL хлорируют, получая 5-хлор-4-нитротетразоло[1,5-а]хинолин (формула XLI). Можно применять обычные хлорирующие вещества. Предпочтительно проводить эту реакцию с применением оксихлорида фосфора в соответствующем растворителе, например в N,N-диметилформамиде. 4-Нитротетразоло[1,5-а]хинолин-5-олы формулы XL хорошо известны или могут быть получены, используя известные способы синтеза (см., например, патент США №5741908 (Герстер) и приведенные в нем ссылки).
На стадии (2) процесса (схема IX реакции) 5-хлор-4-нитротетразоло[1,5-а]хинолин (формула XLI) реагирует с амином формулы R1-O-X-NH2, образуя 4-нитротетразоло[1,5-а]хинолин-5-амин (формула XLII). Реакцию можно выполнять путем добавления амина к раствору соединения формулы XLI в соответствующем растворителе, например в дихлорметане, в присутствии триэтиламина.
На стадии (3) процесса (схема IX реакции) 4-нитротетразоло[1,5-а]хинолин-5-амин (формула XLII) восстанавливают по способу, описанному для стадии (2) схемы VII реакции, получая тетразоло[1,5-а]хинолин-4,5-диамин (формула XLIII).
На стадии (4) процесса (схема IX реакции) тетразоло[1,5-а]хинолин-4,5-диамин (формула XLIII) циклизуют по способу, описанному для стадии (3) схемы VII реакции, получая 6Н-имидазо[4,5-с]тетразоло[1,5-а]хинолин (формула XLIV).
На стадии (5) процесса (схема IX реакции) 6Н-имидазо[4,5-с]тетразоло[1,5-а]хинолин (формула XLIV) восстанавливают, получая 1Н-имидазо[4,5-с]хинолин-4-амин формулы I. Стадия (5) включает в себя (i) взаимодействие соединения формулы XLIV с трифенилфосфином и (ii) гидролиз. Часть (i) можно выполнить путем проведения реакции между соединением формулы XLIV и трифенилфосфином в подходящем растворителе, таком как 1,2-дихлорбензол, при нагревании. Часть (ii) представляет собой гидролиз продукта части (i). Гидролиз можно выполнить, используя известные способы, такие как нагревание в присутствии воды или низшего алканола; в качестве варианта можно ввести катализатор, например гидроксид щелочного металла или низший алкоксид. Продукт или его соль фармацевтического качества можно выделить, пользуясь известными способами.
Схема IX реакции
Тетрагидроимидазохинолины, представляющие собой предмет настоящего изобретения, можно получать в соответствии со схемой Х реакции, где R, R2, R12, Х и n определены выше.
На стадии (1) процесса (схема Х реакции) 4-амино-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-иловый спирт формулы XX алкилируют по способу, описанному для схемы III реакции, применяя галогенид формулы Hal-(CH2)1-10-CH=CH, получая 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин формулы XLV.
На стадии (2) процесса (схема Х реакции) 6,7,8,9-тетрагидро-1Н-имидазо[4,5-хинолин-4-амин формулы XLV взаимодействует с галогенидом формулы Hal-R12 по способу, описанному для стадии (5) схемы IV реакции, образуя 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин формулы XLIV, являющийся подвидом соединения формулы IV. Продукт или его соль фармацевтического качества можно выделить, пользуясь известными способами.
Схема Х реакции

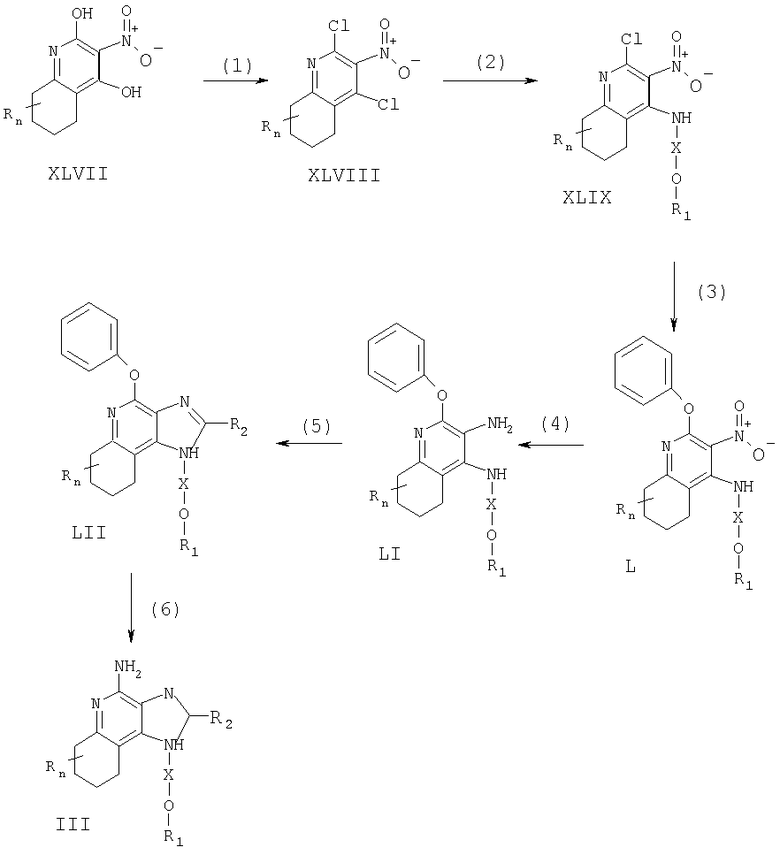
Соединения, представляющие собой предмет настоящего изобретения, можно получать в соответствии со схемой XI реакции, где R, R1, R2, Х и n определены выше.
На стадии (1) процесса (схема XI реакции) 2,4-дигидрокси-3-нитро-6,7,8,9-тетрагидрохинолин (формула XLVII) хлорируют, получая 2,4-дихлор-3-нитро-6,7,8,9-тетрагидрохинолин (формула XLVIII). Здесь можно использовать обычные хлорирующие вещества. Предпочтительно проводить реакцию путем смешивания соединения формулы XLVII с оксихлоридом фосфора с последующим нагреванием (55-85°С). Соединения формулы XLVII хорошо известны или могут быть получены, используя стандартные способы синтеза (см., например, патент США №5352784 (Николаидес и др.) и содержащиеся в нем ссылки).
На стадии (2) процесса (схема XI реакции) 2,4-дихлор-3-нитро-6,7,8,9-тетрагидрохинолин (формула XLVIII) взаимодействует с амином формулы R1-O-X-NH2, образуя 2-хлор-3-нитро-6,7,8,9-тетрагидрохинолин-4-амин (формула XLIX). Реакцию можно проводить путем добавления амина к раствору соединения формулы XLVIII в подходящем растворителе, например в N,N-диметилформамиде, при нагревании (55-65°С).
На стадии (3) процесса (схема XI реакции) 2-хлор-3-нитро-6, 7,8,9-тетрагидрохинолин-4-амин (формула XLIX) реагирует с фенолом по способу, описанному для стадии (4) схемы II реакции, с образованием 2-фенокси-3-нитро-6,7,8,9-тетрагидрохинолин-4-амина (формула L).
На стадии (4) процесса (схема XI реакции) 2-фенокси-3-нитро-6,7,8,9-тетрагидрохинолин-4-амин (формула L) восстанавливают по способу, описанному для стадии (2) схемы VII реакции, получая 2-фенокси-6,7,8,9-тетрагидрохинолин-3,4-диамин (формула LI).
На стадии (5) процесса (схема XI реакции) 2-фенокси-6,7,8,9-тетрагидрохинолин-3,4-диамин (формула LI) циклируют по способу, описанному для стадии (3) схемы VII реакции, получая 4-фенокси-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин (формула LII).
На стадии (6) процесса (схемы XI реакции) 4-фенокси-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин (формула LII) аминируют по способу, описанному для стадии (7) схемы II реакции, получая 6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амин (формула III).
Схема XI реакции
Настоящее изобретение также включает в себя новые соединения, которые можно использовать в качестве промежуточных продуктов при синтезе соединений формул (I), (II), (III) и (IV). Эти промежуточные соединения характеризуются структурными формулами (V)-(IX) и (XLIV) и описаны более подробно ниже.
Один класс промежуточных соединений имеет формулу (V):
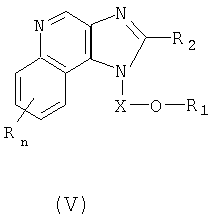
где Х является -CHR3-, -CHR3-алкильной- или -CHR3-алкенильной группой;
R1 выбран из группы, содержащей радикалы:
- гетероарил;
- гетероциклил;
- R4-гетероарил;
- R4-гетероциклил; а также
- (CH2)1-10-C=C-R10; R2 выбран из группы, содержащей радикалы:
- водород;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил, а также
- алкил или алкенил с одним или несколькими заместителями, выбранными из группы, содержащей радикалы:
- ОН;
- галоген;
- N(R3)2;
- СО-N(R3)2;
- CO-C1-10алкил;
- СО-О-C1-10алкил;
- N3;
- арил;
- гетероарил;
- гетероциклил;
- СО-арил, а также
- СО-гетероарил;
R4 представляет собой алкил или алкенил, в который может войти одна или несколько -О-групп;
каждый r3 представляет собой независимо Н или C1-10алкил;
R10 представляет собой гетероарил или гетероциклил;
каждый Y представляет собой независимо -О- или -S(O)0-2-;
n может иметь значение от 0 до 4;
каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
Еще одним классом промежуточных продуктов являются имидазохинолин-4-фенокси-соединения формулы (VI):
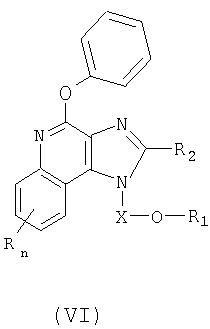
где Х является -CHR3-, -CHR3-алкильной- или -CHR3-алкенильной группой;
R1 выбран из группы, содержащей радикалы:
- гетероарил;
- гетероциклил;
- R4-гетероарил;
- R4-гетероциклил, а также
- (CH2)1-10-C=C-R10;
R2 выбран из группы, содержащей радикалы:
- водород;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил; а также
- алкил или алкенил с одним или несколькими заместителями, выбранными из группы, содержащей радикалы:
- ОН;
- галоген;
- N(R3)2;
- CO-N(R3)2;
- CO-C1-10алкил;
- CO-O-C1-10алкил;
- N3;
- арил;
- гетероарил;
- гетероциклил;
- СО-арил; а также
- СО-гетероарил;
R4 представляет собой алкил или алкенил, в который может войти одна или несколько -О-групп;
каждый R3 представляет собой независимо Н или C1-10алкил;
R10 представляет собой гетероарил или гетероциклил;
каждый Y представляет собой независимо -О- или -S(O)0-2-;
n может иметь значение от 0 до 4; а также
каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
Еще один класс промежуточных продуктов имеет формулу (VII):
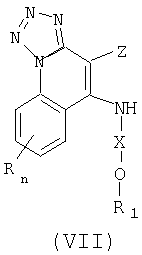
где Z представляет собой NH2 или NO2;
Х представляет собой -CHR3-, -CHR3-алкильную- или -CHR3-алкенильную группу;
R1 выбран из группы, содержащей радикалы:
- гетероарил;
- гетероциклил;
- R4-гетероарил; а также
- R4-гетероциклил;
R4 представляет собой алкил или алкенил, в который может войти одна или несколько -О-групп;
каждый R3 представляет собой независимо Н или C1-10алкил;
n может иметь значение от 0 до 4; а также
каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
Существует класс промежуточных продуктов, представленных формулой (XLIV):
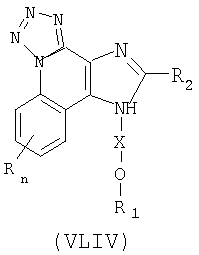
где Х представляет собой -CHR3-, -CHR3-алкильную или -CHR3-алкенильную группу;
R1 выбран из группы, содержащей радикалы:
- гетероарил;
- гетероциклил;
- R4-гетероарил; а также
- R4-гетероциклил;
R2 выбран из группы, содержащей радикалы:
- водород;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил; а также
- алкил или алкенил с одним или несколькими заместителями, выбранными из группы, содержащей радикалы:
- ОН;
- галоген;
- N(R3)2;
- CO-N(R3)2;
- CO-C1-10алкил;
- СО-О-C1-10алкил;
- N3;
- арил;
- гетероарил;
- гетероциклил;
- СО-арил; а также
- СО-гетероарил;
R4 представляет собой алкил или алкенил, в который может войти одна или несколько -О-групп;
каждый R3 представляет собой независимо Н или C1-10алкил;
каждый Y представляет собой независимо -О- или -S(O)0-2-;
n может иметь значение от 0 до 4; а также
каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
Дополнительный класс промежуточных продуктов имеет формулу (VIII):
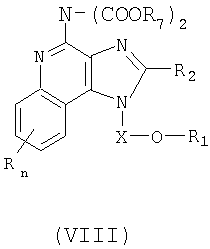
где X представляет собой CHR3-, -CHRз-алкильную или -CHR3-
алкенильную группу;
R1 выбран из группы, содержащей радикалы:
- гетероарил;
- гетероциклил;
- R4-гетероарил; а также
- R4-гетероциклил;
R2 выбран из группы, содержащей радикалы:
- водород;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил, а также
- алкил или алкенил с одним или двумя заместителями, выбранными из группы, содержащей радикалы:
- ОН;
- галоген;
- N(R3)2;
- СО-N(R3)2;
- CO-C1-10алкил;
- СО-O-C1-10алкил;
- N3;
- арил;
- гетероарил;
- гетероциклил;
- СО-арил, а также
- СО-гетероарил;
R4 представляет собой алкил или алкенил, в который может войти одна или несколько -О-групп;
каждый R3 представляет собой независимо Н или C1-10алкил;
каждый Y представляет собой независимо -О- или -S(O)0-2-;
n может иметь значение от 0 до 4; а также каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
R7 представляет собой трет-бутил или бензил;
или же соль фармацевтического качества указанных соединений.
Еще один класс промежуточных продуктов составляют имидазохинолин-4-хлор-соединения формулы (IX):
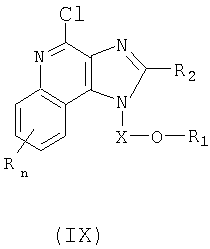
где Х представляет собой CHR3-, -CHR3-алкильную или -CHR3-алкенильную группу;
R1 выбран из группы, содержащей радикалы:
- гетероарил;
- гетероциклил;
- R4-гетероарил; а также
- R4-гетероциклил;
R2 выбран из группы, содержащей радикалы:
- водород;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил; а также
- алкил или алкенил с одним или несколькими заместителями, выбранными из группы, содержащей радикалы:
- ОН;
- галоген;
- N(R3)2;
- СО-N(R3)2;
- CO-C1-10алкил;
- CO-O-C1-10алкил;
- N3;
- арил;
- гетероарил;
- гетероциклил;
- СО-арил; а также
- СО-гетероарил;
R4 представляет собой алкил или алкенил, в который может войти одна или несколько -О-групп;
каждый R3 представляет собой независимо Н или C1-10алкил;
каждый Y представляет собой независимо -О- или -S(O)0-2-;
n может иметь значение от 0 до 4; а также
каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, С1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
Используемые в тексте термины «алкил», «алкенил» и префикс «алк-» включают в себя как группы с прямой и разветвленной цепью, так и циклические группы, т.е. циклоалкильные и циклоалкенильные остатки. Если иное не оговорено специально, эти группы содержат от 1 до 20 атомов углерода, причем алкенильные группы содержат от 2 до 20 атомов углерода. Суммарное число атомов углерода в группах, которым отдается предпочтение, может доходить до 10. Циклические группы могут содержать один цикл или несколько циклов; предпочтение отдается числу атомов углерода в цикле от 3 до 10. В качестве примера можно назвать такие остатки, как циклопропил, циклопентил, циклогексил, циклопропилметил и адамантил.
Следует также добавить, что алкильная и алкенильная части -Х-групп могут не иметь заместителей или содержат заместители (один или большее число); эти заместители выбраны из групп, включающих в себя алкил, алкенил, арил, гетероарил, гетероциклил, арилалкил, гетероарилалкил и гетероциклилалкил.
Термин «галогеналкил» включает в себя группы, замещенные одним или несколькими атомами галогена, в том числе и перфорированные группы. Это утверждение верно и в отношении групп, в наименовании которых имеется префикс «гало-». Примерами приемлемых галогеналкильных групп являются хлорметильный, трифторметильный и аналогичные остатки.
Используемый здесь термин «арил» относится к карбоциклическим ароматическим циклам или к системам циклов. В числе примеров арильных групп можно назвать фенил, нафтил, дифенил, фторфенил и инденил. Термин «гетероарил» относится к ароматическим циклам или системам циклов, содержащим хотя бы один гетероатом (например, О, S, N) в цикле. В число используемых гетероарильных радикалов входят фурил, тиенил, пиридил, хинолинил, изохинолинил, индолил, изоиндолил, триазолил, пирролил, тетразолил, имидазолил, пиразолил, оксазолил, тиазолил, бензофуранил, бензотиофенил, карбазолил, бензоксазолил, пиримидинил, хиноксалинил, бензимидазолил, бензотиазолил, нафтиридинил, изоксазолил, изотиазолил, хиназолинил, пуринил и т.д.
Термин «гетероциклил» включает в себя неароматические циклы или системы циклов, содержащие хотя бы один гетероатом (например, О, S, N) в цикле. Сюда относятся все полностью насыщенные и частично ненасыщенные производные вышеупомянутых гетероарильных групп. В числе примеров гетероциклических групп можно назвать пирролидинил, тетрагидрофуранил, морфолинил, тиоморфолинил, пиперидинил, пиперазинил, тиазолидинил, имидазолидинил, изотиазолидинил и т.д.
Арильные, гетероарильные и гетероциклильные группы могут быть незамещенными или замещенными; в последнем случае они могут содержать один или несколько заместителей, независимо выбранных из группы, состоящей из таких радикалов, как алкил, алкокси, алкилтио, галогеналкил, галогеналкокси, галогеналкилтио, галоген, нитро, гидрокси, меркапто, циано, карбокси, формил, арил, арилокси, арилтио, арилалкокси, арилалкилтио, гетероарил, гетероарилокси, гетероарилтио, гетероарилалкокси, гетероарилалкилтио, амино, алкиламино, диалкиламино, гетероциклил, гетероциклоалкил, алкилкарбонил, алкенилкарбонил, алкоксикарбонил, галогеналкилкарбонил, галогеналкоксикарбонил, алкилтиокарбонил, арилкарбонил, гетероарилкарбонил, арилоксикарбонил, гетероарилоксикарбонил, арилтиокарбонил, гетероарилтиокарбонил, алканоилокси, алканоилтио, алканоиламино, ароилокси, ароилтио, ароиламино, алкиламиносульфонил, алкилсульфонил, арилсульфонил, гетероарилсульфонил, арилдиазинил, алкилсульфониламино, алкиленсульфониламино, арилсульфониламино, арилалкилсульфониламино, гетероарилсульфониламино, гетероалкилсульфониламино, алкилкарбониламино, алкенилкарбониламино, арилкарбониламино, арилалкилкарбониламино, гетероарилкарбониламино, гетероарилалкилкарбониламино, алкиламинокарбониламино, алкениламинокарбониламино, ариламинокарбониламино, арилалкиламинокарбонил, гетероариламинокарбониламино, гетероарилалкиламинокарбониламино; в то же время в случае гетероциклильных групп не допускается наличия таких радикалов, как гетероциклил, алкилкарбонил, алкенилкарбонил, галогеналкилкарбонил, арилкарбонил, гетероарилкарбонил, алкилтиокарбонил, арилтиокарбонил, гетероарилкарбонил, алкиламиносульфонил, алкилсульфонил, арилсульфонил, гетероарилсульфонил. Если о каких-либо иных группах говорят, что они «замещены» или «возможно замещены», то эти группы также могут содержать один или несколько вышеупомянутых заместителей.
В целом некоторым заместителям отдано большее предпочтение. Так, например, в число предпочтительных гетероарильных групп входят радикалы: 2-пиридин, 3-пиридин, 4-пиридин, 2-пиримидин и 5-пиримидин. Предпочтительно, чтобы заместители R отсутствовали (т.е. n=0). В число предпочтительных групп R2 входят водород, алкильные группы с 1-4 атомами углерода (это метил, этил, пропил, изопропил, н-бутил, втор-бутил, изобутил и циклопропилметил), метоксиэтил и этоксиметил. Один или несколько таких предпочтительных заместителей при их наличии могут находиться в составе соединений, являющихся предметом настоящего изобретения, в любой комбинации.
Настоящее изобретение включает в себя описанные здесь соединения в любой фармацевтически доступной форме, включая изомеры (например, диастереоизомеры и энантиомеры), соли, сольваты, полиморфные варианты и т.д. В частности, если соединение является оптически активным, настоящее изобретение включает в себя каждый из энантиомеров соединения, а также рацемические смеси энантиомеров.
Фармацевтические составы и биологическая активность
Фармацевтические составы, составляющие предмет настоящего изобретения, содержат терапевтически эффективные количества описанного выше соединения в сочетании с фармацевтически доступным носителем.
Термин «терапевтически эффективное количество» означает то количество соединения, которое достаточно для достижения терапевтического эффекта, такого как стимулирование синтеза цитокинов, проявление противоопухолевой активности и (или) проявление противовирусной активности. Хотя точное количество активного соединения, примененного в фармацевтической композиции, составляющей предмет настоящего изобретения, может меняться в зависимости от факторов, известных тем, кто является специалистом в этой области (например, физическая и химическая природа соединения, природа носителя и предполагаемый режим дозирования), предполагается, что составы, являющиеся предметом настоящего изобретения, будут содержать достаточное количество активного ингредиента для создания дозы соединения от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно от приблизительно 10 мкг/кг до приблизительно 5 мг/кг при расчете на массу тела пациента. Могут быть использованы любые известные лекарственные формы, такие как таблетки, пастилки, парентеральные препараты, сиропы, кремы, мази, аэрозольные препараты, чрезкожные пластыри, пластыри на слизистой оболочке и т.д.
Соединения, являющиеся предметом настоящего изобретения, можно применять как единственное терапевтическое средство в схеме лечения или же данные соединения можно применять в виде сочетания с одним или несколькими активными веществами, включая дополнительные модификаторы иммунной реакции, противовирусные вещества, антибиотики и т.д.
Было показано, что соединения, являющиеся предметом настоящего изобретения, индуцируют синтез определенных цитокинов в экспериментах, выполненных в соответствии с условиями испытаний, описанными ниже. Результаты испытаний показывают, что указанные соединения могут использоваться в качестве модификаторов иммунной реакции; они могут изменять иммунную реакцию разными путями, что делает эти соединения весьма полезными при лечении различных заболеваний.
В число цитокинов, синтез которых может быть стимулирован применением соединений, являющихся предметом настоящего изобретения, обычно включают интерферон-α (ИФ-α) и (или) фактор некроза опухолей-α (ФНО-α), а также некоторые интерлейкины (ИЛ). В группу цитокинов, биосинтез которых может индуцироваться соединениями, являющимися предметом настоящего изобретения, входят ИФ-α, ФНО-α, ИЛ-1, ИЛ-6, ИЛ-10 и ИЛ-12, а также некоторые другие цитокины. Среди прочих эффектов эти и иные цитокины могут ингибировать размножение вирусов и рост опухолевых клеток, что делает данные соединения полезными при лечении вирусных заболеваний и опухолей. В связи со сказанным настоящее изобретение предлагает способ стимулирования биосинтеза цитокинов в организме животного путем введения эффективного количества соединения или состава, являющихся предметом настоящего изобретения, в организм животного.
Было обнаружено, что некоторые соединения, являющиеся предметом настоящего изобретения, предпочтительно индуцируют экспрессию ИФ-α в популяции кроветворных клеток, таких как ОКПК (одноядерные клетки периферической крови) и клетки pDC2 (дендритные клетки-предшественники - тип 2), без сопутствующего синтеза значительных количеств цитокинов, сопровождающих воспаление.
В дополнение к способности индуцировать синтез цитокинов соединения, являющиеся предметом настоящего изобретения, воздействуют и на другие аспекты врожденной иммунной реакции. Так, например, возможна стимуляция активности натуральных клеток-киллеров, вероятно, вследствие индукции цитокинов. Данные соединения могут также активировать макрофаги, что, в свою очередь, стимулирует секрецию оксида азота и синтез дополнительных количеств цитокинов. Кроме того, эти соединения могут вызвать пролиферацию и дифференциацию В-лимфоцитов.
Соединения, являющиеся предметом настоящего изобретения, также воздействуют на приобретенную иммунную реакцию. Например, хотя как полагают, они не оказывают непосредственного влияния на Т-клетки или напрямую на образование цитокинов Т-клеток, синтез цитокина ИФ-γ Т-хелперов типа 1 (Тх1) индуцируется косвенным образом, а синтез цитокинов ИЛ-4, ИЛ-5 и ИЛ-13 Т-хелперов типа 2 (Тх2) ингибируется после введения этих соединений. Данная активность означает, что эти соединения применимы для лечения заболеваний, при которых необходима повышающая регуляция Тх1-реакции и (или) понижающая регуляция Тх2-реакции. Ввиду способности предлагаемых в настоящем изобретении соединений ингибировать Тх2-иммунную реакцию, можно ожидать, что они применимы при лечении атопических заболеваний, например атопического дерматита, астмы, аллергии, аллергического ринита, системной красной волчанки; они могут также применяться как адъюванты вакцин для развития клеточно-опосредованного иммунитета. Возможно их применение для лечения рецидивирующих заболеваний, вызванных микроскопическими грибами, и хламидиоза.
Эффекты, связанные с модифицированием иммунной реакции данными соединениями, делают возможным их применение при лечении широкого спектра заболеваний. Вследствие своей способности индуцировать синтез цитокинов, таких как ИФ-α и (или) ФНО-α, эти соединения особенно полезны при лечении вирусных заболеваний и опухолей. Иммуномодулирующая активность данных соединений дает основания полагать, что соединения, являющиеся предметом настоящего изобретения, могут найти применение при лечении таких заболеваний, как вирусные, включая, помимо прочего, остроконечные кондиломы, простые бородавки, подошвенные бородавки, гепатит В, гепатит С, заболеваний, вызванных вирусом простого герпеса типа I и типа II, контагиозный моллюск, натуральная оспа, в особенности черная натуральная оспа, заболеваний, вызванных риновирусом, аденовирусом, вирусом гриппа и вирусом парагриппа, ВИЧ, ЦМВ, вирусом ветряной оспы. Возможно применение этих соединений в случае интраэпителиальных неоплазий, таких как цервикальная интраэпителиальная неоплазия. Возможно воздействие на болезни, вызванные вирусом папилломы человека, и сопутствующие неоплазий; на заболевания, вызванные микроскопическими грибами (Candida, Aspergillus), в том числе криптококковый менингит. Данные соединения могут найти применение при лечении таких опухолевых заболеваний, как базально-клеточный рак, лейкозный ретикулоэндотелиоз, саркома Капоши, почечно-клеточный рак, плоскоклеточный рак, миелобластный лейкоз, множественная миелома, меланома, не-ходжкиновская лимфома, кожная Т-клеточная лимфома и другие виды рака. Возможно применение при лечении паразитарных заболеваний: пневмоцистоза, вызванного микроорганизмом Pneumocystis caiinii, криптоспоридиоза, гистоплазмоза, токсоплазмоза, трипаносомоза, лейшманиоза, бактериальных инфекций, например туберкулеза и заболевания, вызванного микроорганизмом Mycobacterium avium. В число прочих заболеваний, которые можно лечить соединениями, являющимися предметом настоящего изобретения, входят лучевой кератоз, экзема, эозинофилия, эссенциальная тромбоцитемия, проказа, множественный склероз, синдром Омена, дискоидная красная волчанка, болезнь Боуэна, папулез Боуэна, гнездная алопеция, торможение образования келоидов и прочих послеоперационных рубцов. Следует добавить, что данные соединения могут усиливать или стимулировать заживление ран, включая хронические повреждения. Данные соединения могут быть применены для лечения заболеваний, вызванных условно-патогенными микроорганизмами, и опухолей, возникающих после подавления клеточно-опосредованного иммунитета, например, у пациентов после трансплантации органов, у онкологических больных и ВИЧ-зараженных.
Количество соединения, эффективное при стимулировании биосинтеза цитокинов, - это масса, достаточная для того, чтобы стимулировать один или несколько типов клеток, например моноциты, макрофаги, дендритные клетки и В-клетки, к продуцированию определенного количества одного или нескольких цитокинов, таких как ИФ-α, ФНО-α, ИЛ-1, ИЛ-6, ИЛ-10 и ИЛ-12, превышающего фоновый уровень этих цитокинов. Точное количество зависит от факторов, известных специалистам в данной области, но предполагается, что доза будет составлять от приблизительно 100 нг/кг до приблизительно 50 нг/кг, предпочтительно от приблизительно 10 мкг/кг до приблизительно 5 мг/кг. Настоящее изобретение также предлагает способ лечения вирусных инфекций у животных и способ лечения опухолевых заболеваний животных путем применения эффективного количества соединения или состава, являющихся предметом настоящего изобретения. Масса вещества, эффективная при лечении или торможении вирусной инфекции, является количеством, вызывающим уменьшение интенсивности одного или нескольких проявлений вирусной инфекции, таких как вирусные поражения, вирусная нагрузка, скорость размножения вирусов и смертность, по сравнению с показателями контрольных животных, не прошедших лечения. Точное количество зависит от факторов, известных специалистам в этой области, но предполагается, что доза будет составлять от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно от приблизительно 10 мкг/кг до приблизительно 5 мг/кг. Масса соединения, эффективная при лечении опухолевых заболеваний, является количеством, вызывающим уменьшение размеров опухоли или числа очагов заболевания. Опять-таки точное количество зависит от факторов, известных специалистам в этой области, но предполагается, что доза будет составлять от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно от приблизительно 10 мкг/кг до приблизительно 5 мг/кг.
Далее описание настоящего изобретения иллюстрируется примерами, приведенными ниже; примеры ни в коем случае не ограничивают общие рамки изобретения.
В приведенных ниже примерах некоторые соединения были подвергнуты очистке способом полупрепаративной ВЭЖХ. Применяли два способа, описанных ниже. В данных способах был использован прибор А-100 Gilson-6, снабженный интерфейсом 900 Series Intelligent Interface. Фракции, полученные способом полупрепаративной ВЭЖХ, анализировали на приборе LC-APCI/MS. Отдельные фракции объединяли и подвергали лиофильной сушке с целью получения трифторацетатной соли требуемого соединения.
Способ А
Колонка: Microsorb C18, 21,4×250 мм, размер частиц 8 мкм, размер пор 60 Å;
расход 10 мл/мин. Градиентное элюирование от 2 до 95 % компонента В в течение 25 мин, выдержка на уровне 95 % компонента В в течение 5 мин, где А=0,1 %-ный раствор трифторуксусной кислоты в воде, В=0,1 %-ный раствор трифторуксусной кислоты в ацетонитриле. Пик детектировали на длине волны 254 нм с целью включения коллектора фракций.
Способ В
Колонка: Phenomenex Capcell PakC18, 35×20 мм, размер частиц 5 мкм, расход 20 мл/мин. Градиентное элюирование от 5 до 95 % компонента В в течение 10 мин, выдержка на уровне 95 % компонента В в течение 2 мин, где А=0,1 %-ный раствор трифторуксусной кислоты в воде, В=0,1 %-ный раствор трифторуксусной кислоты в ацетонитриле. Пик детектировали на длине волны 254 нм с целью включения коллектора фракций.
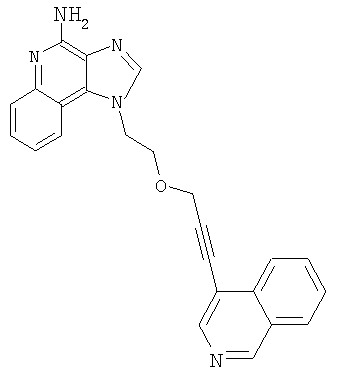
Пример 1
1-(2-{[3-Изохинолин-4-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
2-(1Н-Имидазо[4,5-с]хинолин-1-ил)-1-этанол (28,5 г, 0,133 моля) добавляли отдельными порциями в течение 1 часа к смеси гидроксида натрия (240 мл, 50 %-ный раствор), дихлорметана (240 мл), пропаргилбромида (39,6 г 80 %-ного препарата, 0,266 моля) и бензилтриметиламмонийхлорида (2,46 г, 0,013 ммоля). Полученную реакционную смесь перемешивали при температуре окружающей среды в течение 16 часов; в конце этого периода смесь была гомогенной. После разделения слоев водную фракцию экстрагировали дополнительным количеством дихлорметана. Органические фракции соединяли, промывали водой, высушивали над сульфатом магния и концентрировали при пониженном давлении. Полученный остаток обрабатывали диэтиловым эфиром при перемешивании. При фильтровании получили твердое вещество оранжевого цвета. Это вещество рекристаллизовывали из этилацетата и получили 19,8 г 2-(1H-имидазо[4,5-с]хинолин-1-ил)этил(2-пропинил)ового простого эфира в виде твердого кристаллического вещества желтого цвета (температура плавления 124-126°С).
Данные элементного анализа: Рассчитано для С15Н13N3О: С 71,70 %, Н 5,21 %, N 16,72 %. Найдено: С 71,85 %, Н 5,25 %, N 16,90 %.
Данные 1Н ЯМР (300 МГц, ДМСО): δ 9,21 (с, 1 Н); 8,44 (м, 1 Н); 8,36 (с, 1 Н); 8,18 (м, 1 Н); 7,71 (м, 2 Н); 4,93 (т, J=5,1 Гц, 2 Н); 4,14 (д, J=2,4 Гц, 2 Н); 3,98 (т, J=5,1 Гц, 2 Н); 3,35 (т, J=2,2 Гц, 1 Н).
Данные масс-спектрометрии высокого разрешения HRMS(ESI): Рассчитано для C15H14N3O (MH+): 252,1137. Найдено: 252,1141.
Часть В
2-(1Н-Имидазо[4,5-с]хинолин-1-ил)этил(2-пропинил)овый простой эфир (19,7 г, 78,4 ммолей) смешивали с хлороформом и охлаждали до 0°С. После добавления 3-хлорпероксибензойной кислоты (15,7 г 57-86 %-ного препарата) смесь перемешивали в течение 0,5 часа. Смесь оставляли до достижения ею температуры окружающей среды; к концу этого периода весь материал был в растворе. Анализ, выполненный способом тонкослойной хроматографии (ТСХ), показал, что в системе все еще присутствовал исходный материал; поэтому добавили дополнительное количество 3-хлорпероксибензойной кислоты (две отдельные порции по 4 г). Спустя 0,5 часа после добавления второй порции ТСХ не обнаружила наличия исходного материала. Реакционную смесь экстрагировали 10 %-ным гидроксидом натрия. Водную фракцию экстрагировали несколько раз дихлорметаном. Органические фракции соединяли, высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении, получив 18,5 г 1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-5N-оксида в виде маслянистой жидкости желтого цвета.
Данные масс-спектрометрии высокого разрешения HRMS(ESI): Рассчитано для
C15H14N3O2 (MH+): 268,1086. Найдено: 268,1098.
Часть С
Трихлорацетилизоцианат (15,5 г, 82,2 ммоля) добавляли по каплям в смесь 1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-5N-оксида (18,3 г, 68,5 ммоля) и дихлорметана (300 мл) в атмосфере азота. При этом наблюдали энергичное выделение углекислоты. Спустя приблизительно 0,5 часа весь материал был в растворе. Реакционный раствор перемешивали в течение почти 1 часа; после этого ТСХ обнаружила наличие небольшого количества исходного вещества. Было добавлено дополнительное количество трихлорацетилизоцианата (4,5 г); спустя 1 час ТСХ показала, что реакция завершена. Летучие вещества были удалены путем понижения давления; при этом получили N-{1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-ил}-2,2,2-трихлорацетамид в виде твердого вещества бледно-желтого цвета.
Часть D
Дихлорметан (150 мл) добавляли к смеси твердого вещества из Части С и метанола (200 мл); все твердое вещество растворилось. Далее ввели метоксид натрия (50 г 25 %-ного раствора в метаноле) и перемешивали реакционный раствор в течение ночи при температуре окружающей среды. Полученный осадок отделяли фильтрованием. Фильтрат концентрировали до объема, равного приблизительно 100 мл, и фильтрованием отделили дополнительное количество осадка. Оба осадка соединили и высушивали в вакуумной печи при температуре 60°С в течение 16 часов. Было получено 16,4 г 1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества почти белого цвета, (температура плавления 225-227°С.)
Данные элементного анализа: Рассчитано для C15H14N4O(H2O)1/4: С 66,53 %, Н 5,40 %, N 20,69 %. Найдено: С 66,33 %, Н 5,18 %, N 21,12 %.
Данные 1H ЯМР (300 МГц, ДМСО): δ 8,13 (с, 1 Н); 8,08 (шд, J=7,8 Гц, 1 Н); 7,62 (шд, J=8,3 Гц, 1 Н); 7,44 (шт, J=7,6 Гц, 1 Н); 7,24 (шт, J=7,5 Гц, 1 Н); 6,54 (с, 2 Н); 4,81 (т, J=5,4 Гц, 2 Н); 4,14 (д, J=2,4 Гц, 2 Н); 3,93 (т, J=5,1 Гц, 2 Н); 3,38 (т, J=2,4 Гц, 1 H).
Данные масс-спектрометрии высокого разрешения HRMS(ESI): Рассчитано для C15H15N4O (MH+): 267,1246. Найдено: 267,1253.
Часть Е
В атмосфере азота смешали 1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-хинолин-4-амин (16 г, 60,1 ммоля), ди-трет-бутилдикарбонат (32,7 г, 150 ммолей), триэтиламин (21 мл, 150 молей), N,N-диметилформамид (150 мл) и 4-(диметиламино)пиридин (0,1 г) и смесь нагрели до 80-85°С. Спустя приблизительно 1 час смесь стала гомогенной; ТСХ показала, что непрореагировавшее исходное вещество осталось в очень малом количестве. Раствор нагревали еще в течение 1 часа. Раствор разбавили этилацетатом и водой. После разделения слоев водную фракцию экстрагировали этилацетатом. Органические фракции соединили, промыли водой и рассолом, высушили над сульфатом магния, отфильтровали и концентрировали при пониженном давлении. Было получено твердое вещество бледного оранжево-желтого цвета. Это вещество растерли с диэтиловым эфиром, получив 22,6 г N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества почти белого цвета (температура плавления 139-142°С).
Данные элементного анализа: Рассчитано для
C25H30N4O5: С 64,36 %, Н 6,48 %, N 12,01 %. Найдено: С 64,40 %, Н 6,43 %, N 12,06 %.
Данные 1H ЯМР (300 МГц, ДМСО): δ 8,44 (м, 1 Н); 8,35 (с, 1 Н); 8,08 (м, 1 Н); 7,73 (м, 2 Н); 4,94 (т, J=4,9 Гц, 2 Н); 4,12 (д, J=2,4 Гц, 2 Н); 3,98 (т, J=5,1 Гц, 2 Н); 3,31 (T,J=2,4 Гц, 1 Н); 1,34 (с, 18 Н).
Данные масс-спектрометрии высокого разрешения HRMS(ESI): Рассчитано для C25H31N4O5 (MH+): 467,2294. Найдено: 467,2307.
Часть F
В атмосфере азота смешали N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин (1,0 г, 2,14 ммоля), триэтиламин (0,8 мл, 5,56 ммоля) и N,N-диметилформамид (25 мл) и раствор нагрели до 80-85°С. Далее добавили дихлорбис(трифенилфосфин)палладий (II) (0,08 г, 0,11 моля), йодид меди (I) (0,04 г, 0,21 ммоля) и 4-бромизохинолин (0,49 г, 2,35 ммоля). Спустя 3 часа анализ способом ВЭЖХ в обращенной фазе с градиентом ацетонитрил/вода показал, что реакция прошла полностью. Реакционный раствор медленно вылили в воду при энергичном перемешивании. Полученный осадок кремового цвета отделили фильтрованием, промыли водой и высушивали в вакуумной печи (< 40°С) в течение 16 часов. Был получен N,N-(бис-трет-бутоксикарбонил)-1-[2-{[3-изохинолин-4-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амин в количестве 1,21 г.
Данные масс-спектрометрии высокого разрешения HRMS(EI): Рассчитано для
С34Н35N5O5 (М+): 594,2716. Найдено: 594,2732.
Часть G
Продукт Части F добавляли порциями к смеси дихлорметана (5 мл) и трифторуксусной кислоты (5 мл) в атмосфере азота. Полученный раствор перемешивали при температуре окружающей среды в течение 2 часа; к концу этого периода ТСХ показала, что реакция завершена. Растворители удалили при пониженном давлении. Остаток обработали смесью дихлорметан-метанол (прибл. 4:1) и 20 %-ным раствором гидроксида натрия, после чего разделили слои. Водную фракцию экстрагировали смесью дихлорметан-метанол (прибл. 4:1). Органические фракции соединили, высушили над сульфатом магния, отфильтровали и концентрировали при пониженном давлении. Продукт очистили способом высокоскоростной хроматографии, получив 0,15 г 1-(2-{[3-изохинолин-4-ил)-2-пропинил]окси}этил)-1H-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества почти белого цвета (температура плавления (с разложением) свыше 205°С).
Данные 1H ЯМР (300 МГц, ДМСО): δ 9,30 (с, 1 Н); 8,43 (с, 1 Н); 8,35 (с, 1 Н); 8,19 (м, 2 Н); 7,88 (шд, J=8,0 Гц, 1 Н); 7,65-7,80 (м, 4 Н); 7,60 (д, J=8,3 Гц, 1 Н); 7,49 (т, J=7,8 Гц, 1 Н); 7,34 (т, J=7,8 Гц, 1 Н); 4,93 (т, J=4,9 Гц, 2 Н); 4,57 (с, 2 Н); 4,14 (т, J=5,1 Гц, 2Н).
Данные масс-спектрометрии высокого разрешения HRMS(ESI): Рассчитано для
C24H19N5O (MH+): 394,1668. Найдено: 394,1669.
Пример 2
1-(2-{[3-(1,3-Тиазол-2-ил)-2-пропинил]окси}этил)-1H-имидазо[4,5-с]хинолин-4-амин
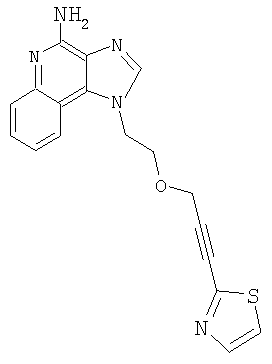
Часть А
Используя общий способ, описанный в Примере 1, Часть F, провели реакцию между N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амином (1,0 г, 2,14 ммоля) и 2-бромтиазолом и получили 0,97 г N,N-(бис-трет-бутоксикарбонил)-1-(2-{[3-(1,3-тиазол-2-ил)-2-пропинил]окси}этил)-1H-имидазо[4,5-с]хинолин-4-амина в виде стеклообразного твердого вещества желтого цвета.
Данные масс-спектрометрии MS(CI): 550, 450, 350.
Часть В
Используя общий способ, описанный в Примере 1, Часть G, провели гидролиз продукта Части В, получив 0,11 г 1-(2-{[3-(1,3-тиазол-2-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амина в виде белого твердого вещества (температура плавления 157-159°С).
Данные элементного анализа: Рассчитано для C18H15N5OS·(H2O)1/4: С 61,09 %, Н 4,42 %, N 19,79 %. Найдено: С 61,06 %, Н 4,37 %, N 19,53 %.
Данные 1H ЯМР (500 МГц, ДМСО): δ 8,18 (с, 1 Н); 8,11 (д, J=7,9 Гц, 1 Н); 7,89 (дд, J=17,7; 2,9 Гц, 1 Н); 7,62 (д, J=7,9 Гц, 1 Н); 7,43 (т, J=7,5 Гц, 1 Н); 7,23 (т, J=7,5 Гц, 1 Н); 6,64 (с, 2 Н); 4,83 (м, 2 Н); 4,50 (с, 2 Н); 4,01 (м, 2 Н).
Данные масс-спектрометрии высокого разрешения HRMS(EI): Рассчитано для
C18H15N5OS (M+): 349,0997. Найдено: 349,0988.
Пример 3
1-{2-[3-(1Н-Пиразол-4-ил)пропокси]этил}-1H-имидазо[4,5-с]хинолин-4-амин
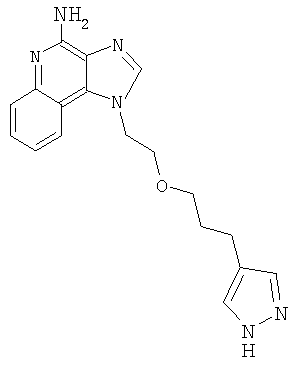
Часть А
N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-4-амин (2,25 г, 4,82 ммоля) смешали с триэтиламином (1,34 мл, 9,64 ммоля), 4-йодпиразолом (1,02 г, 5,30 ммоля) и безводным ацетонитрилом (40 мл). Через эту смесь пропускали азот в течение 10 мин. Добавили дихлорбис(трифенилфофсин)палладий (II) (68 мг, 0,096 моля) и йодид меди (I) (37 мг, 0,192 ммоля); раствор нагрели до 40°С. Спустя 1 час анализ по способу ВЭЖХ в обращенной фазе показал, что реакция не произошла. Реакционный раствор нагрели приблизительно до 90°С. Спустя 4 часа анализ по способу ВЭЖХ показал, что реакция была полностью завершена. Летучие вещества удалили при пониженном давлении. Остаток очистили способом высокоскоростной хроматографии (элюент: дихлорметан-метанол 9:1) и получили 1,2 г N,N-(бис-трет-бутоксикарбонил)-1-(2-{[3-(1 Н-пиразол-4-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества белого цвета.
Часть В
Продукт Части А смешали с метанолом (прибл. 20 мл) и катализатором (0,25 г палладия (10 %) на углероде). Смесь подвергали гидрогенизации в течение 4 часов; к концу этого периода анализ способом жидкостной хроматографии с масс-спектрометрическим детектором показал снижение количеств алкена и алкана. Добавили еще порцию катализатора (0,25 г) и продолжали гидрогенизацию в течение 2 дней. В итоге анализ указанным способом показал, что в системе нет исходного вещества или алкена. Смесь отфильтровали и фильтрат промыли метанолом. После концентрирования получили твердое вещество, которое далее подвергли очистке способом высокоскоростной хроматографии (элюент: дихлорметан-метанол 9:1) и получили 0,9 г N,N-(бис-трет-бутоксикарбонил)-1-{2-[3-(1 Н-пиразол-4-ил)пропокси] этил}-1H-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества белого цвета.
Данные 1H ЯМР (300 МГц, ДМСО): δ 12,43 (шс, 1 Н); 8,48 (шд, J=7,1 Гц, 1 Н); 8,37 (с, 1 Н); 8,08 (шд, J=7,3 Гц, 1 Н); 7,72 (м, 2 Н); 7,30 (шс, 1 Н); 7,14 (шс, 1 Н); 4,92 (т, J=4,9 Гц, 2 Н); 3,88 (шд, J=4,9 Гц, 2 Н); 2,22 (т, J=7,8 Гц, 2 Н); 1,56 (м, 2 Н);1,31 (с, 18 Н).
Данные масс-спектрометрии MS(CI): 537, 437, 337.
Часть С
В атмосфере азота добавили трифторуксусную кислоту к смеси N,N-(бис-трет-бутоксикарбонил)-1-{2-[3-(1 Н-пиразол-4-ил)пропокси] этил}-1Н-имидазо[4,5-с]хинолин-4-амина (0,5 г, 0,93 ммоля) и дихлорметана (5 мл). Полученный раствор перемешивали в течение 16 часов; в конце этого периода анализ способом жидкостной хроматографии с масс-спектрометрическим детектором показал, что реакция завершена. Растворители удалили при пониженном давлении. Остаток растворили в этилацетате (прибл. 10 мл), после чего добавили триэтиламин (2 мл). Образовался осадок; реакционную смесь перемешивали еще в течение 2 часов. Твердый продукт отделили фильтрованием и очистили способом высокоскоростной хроматографии (элюент: дихлорметан-метанол от 9:1 до 8:2) и получили 0,18 г 1-{2-[3-(1H-пиразо-4-лил)пропокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества белого цвета (температура плавления 163-169°С).
Данные элементного анализа: Рассчитано для C18H20N6O·(CF3CO2H)0.15: С 62,18 %, Н 5,75 %, F 2,42 %, N 23,77 %. Найдено: С 61,86 %, Н 5,70 %, F 2,52 %, N 23,44 %.
Данные 1H ЯМР (300 МГц, ДМСО): δ 12,50 (шс, 1 Н); 8,20 (с, 1 Н); 8,15 (д, J=8,3 Гц, 1 Н); 7,66 (д, J=8,3 Гц, 1 Н); 7,49 (т, J=7,6 Гц, 1 Н); 7,29 (т, J=7,6 Гц, 1 Н); 7,15-7,40 (шс, 2 Н); 7,00 (шс, 2 Н); 4,81 (т, J=4,6 Гц, 2 Н); 3,84 (т, J=4,6 Гц, 2 Н); 3,34 (т, J=6,1 Гц, 2 Н); 2,27 (т, J=7,6 Гц, 2 Н); 1,60 (м, 2 Н).
Пример 4
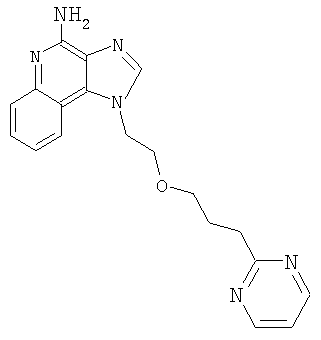
1-[2-(3-Пиримидин-2-илпропокси)этил]-1H-имидазо[4,5-с]хинолин-4-амин
Часть А
Дибензилдикарбонат (50 г, 174 ммоля) добавили в атмосфере азота к смеси 1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина (16,4 г, 61,6 ммоля) и безводного N,N-диметилформамида (200 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 часов; при этом смесь стала гомогенной. Реакционную смесь разделили между этилацетатом и водой. После разделения слоев водный слой экстрагировали этилацетатом. Органические фракции соединили, промыли водой и рассолом, высушили над сульфатом магния, отфильтровали и концентрировали при пониженном давлении, получив полутвердую массу. Эту массу растерли в диэтиловом эфире, получив 27,4 г N,N-(бис-бензилоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества белого цвета.
Часть В
N,N-(Бис-бензилоксикарбонил)-1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-c]хинолин-4-амин (1,00 г, 1,87 ммоля) смешали с безводным ацетонитрилом (10 мл), триэтиламином (0,68 мл, 4,86 ммоля) и 2-бромпиримидином (0,327 г, 2,06 ммоля). В смесь добавили йодид меди (I) (0,014 г) и дихлорбис(трифенилфосфин)палладий (II) (0,026 г) в атмосфере азота. Реакционную смесь выдержали при температуре окружающей среды в течение 15 мин, после чего нагревали при температуре 80°С в течение 1,5 часов. Далее реакционную смесь разбавили этилацетатом и водой. Водный слой отделили и провели экстракцию этилацетатом до тех пор, пока в водном слое не осталось никаких веществ, обладающих флуоресценцией в УФ-свете. Органические фракции соединили, промыли водным раствором бикарбоната натрия и рассолом, высушили над сульфатом магния, отфильтровали и концентрировали при пониженном давлении. Остаток очистили способом хроматографии на колонке при элюировании смесью этилацетат-метанол (98:2), получив 0,68 г смеси защищенного одним бензилоксикарбонильным остатком и двумя такими остатками 1-{2-[(3-пиримидин-2-илпроп-2-инил)окси]этил}-1 Н-имидазо[4,5-с]хинолин-4-амина.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,78 (д, J=4,9 Гц, 2 Н); 8,49 (м, 1 Н); 8,45 (с, 1 Н); 8,12 (м, 1 Н); 7,73-7,78 (м, 2 Н); 7,50 (т, J=4,9 Гц, 1 Н); 7,23-7,28 (м, 6 Н); 7,14-7,17 (м, 4 Н); 5,20 (с, 4 Н); 5,02 (т, J=5,0 Гц, 2 Н); 4,51 (с, 2 Н); 4,10 (т, J=5,0 Гц, 2 Н).
Данные масс-спектрометрии MS(CI): Рассчитано для С35Н8N5О5: m/z 613 (MH+), 569, 461, 345.
Часть С
Продукт Части В смешали с гидроксидом палладия (0,25 г, 20 %, на углероде) и метанолом (25 мл); гидрогенизацию проводили при давлении 3,3 бар в течение 3 часов при температуре окружающей среды. Реакционную смесь оставили постоять на выходные дни; к концу этого периода анализ указал на наличие какого-то продукта с защищенными аминными группами. Реакционную смесь отфильтровали с целью удаления катализатора; фильтрат обрабатывали метоксидом натрия (1 мл 25 %-ного раствора в метаноле) в течение приблизительно 16 часов для удаления защитных групп. Далее реакционную смесь концентрировали при пониженном давлении. Остаток очищали способом колоночной хроматографии при элюировании смесью этилацетат-метанол-гексан (1:1:1), получив 0,235 г твердого вещества. Этот продукт перемешали с горячим толуолом и отфильтровали с целью удаления нерастворимых примесей. Фильтрат концентрировали при пониженном давлении. Остаток растирали с изопропанолом и этилацетатом, получив 61 мг 1-[2-(3-пиримидин-2-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества с температурой плавления 126-127°С.
Данные элементного анализа: Рассчитано для C19H20N6O: С 65,5 %, Н 5,79 %, N 24,12 %. Найдено: С 65,65 %, Н 5,78 %, N 24,15 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,66 (д, J=4,7 Гц, 2 Н); 8,14 (с, 1 Н); 8,08 (д, J=8,0 Гц, 1 Н); 7,62 (д, J=8,2 Гц, 1 Н); 7,45 (т, J=7,0 Гц, 1 Н); 7,24-7,31 (м, 2 Н); 6,58 (с, 2 Н); 4,77 (т, J=4,7 Гц, 2 Н); 3,84 (т, J=4,5 Гц, 2 Н); 3,42 (т, J=6,2 Гц, 2 Н); 2,82 (т, J=7,5 Гц, 2 Н); 1,89 (м, 2 Н).
Данные ИК-спектроскопии (KBr): 3302, 3187, 2868, 1637, 1561, 1418, 1139 см-1.
Данные масс-спектрометрии высокого разрешения HRMS (EI): Рассчитано для C19H20N6O (M+): 348,1699. Найдено: 348,1700.
Пример 5
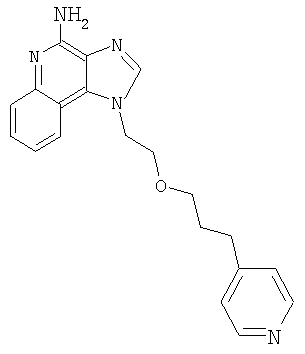
1-[2-(3-Пиридин-4-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
Используя общий способ, описанный в Примере 4, Часть В, N,N-(бис-бензилоксикарбонил)-1-[2-(2-пропинилокси)этил]-1N-имидазо[4,5-с]хинолин-4-амин (2,00 г, 3,74 ммоля) обработали 4-бромпиридином (0,8 г, 4,12 ммоля), получив 1,47 г смеси защищенного одним бензилоксикарбонильным остатком и двумя такими остатками 1-{2-[(3-пиридин-4-илпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,46 (м, 3 Н); 8,43 (с, 1 Н); 8,12 (м, 1 Н); 7,72-7,76 (м, 2 Н); 7,22-7,28 (м, 5 Н); 7,14-7,17 (м, 6Н); 5,18 (с, 4 Н); 5,00 (т, J=5,0 Гц, 2 Н); 4,45 (с, 2 Н); 4,12 (т, J = 4,0 Гц, 2 Н).
Данные масс-спектрометрии MS(CI): Рассчитано для С36Н29N5О5: m/z 612 (MH+), 568, 344.
Часть В
К метанольному раствору продукта Части А (прибл. 10 мл) добавили гидроксид палладия (0,57 г, 20 % на углероде). Продукт гидрогенизировали в течение 5 часов при давлении 3,5 бар. Далее добавили дополнительное количество катализатора (0,07 г) и продолжали гидрогенизацию еще в течение 1 часа. Реакционную смесь отфильтровали с целью удаления катализатора; осадок на фильтре тщательно промыли метанолом. Фильтрат концентрировали при пониженном давлении. Остаток очистили способом хроматографии на колонке; элюат: смесь этилацетата, метанола и гексана (6:3:1). Далее остаток растерли с диэтиловым эфиром, получив твердое вещество. Это вещество очистили способом хроматографии на колонке; элюат: смесь дихлорметана и метанола с гидроксидом аммония (9:1). Получили 0,20 г 1-[2-(3-пиридин-4-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества с температурой плавления 160-162°С.
Данные элементного анализа: Рассчитано для C20H21N5O: С 69,14 %, Н 6,09 %, N 20,16 %. Найдено: С 69,17 %, Н 6,09 %, N 19,79 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6):δ 8,29 (дд, J=2,6; 1,8 Гц, 2 Н); 8,18 (с, 1 H); 8,11 (A,J=8,2 Гц,1 Н); 7,62(дд J=7,1; 1,4 Гц, 1 H); 7,45 (дт, J=6,9; 1,7 Гц, 1 Н); 7,23 (дт, J=6,7; 1,3 Гц, 1 H); 6,91 (дд, J=4,4; 1,3 Гц, 2 H); 6,62 (с, 2 H); 4,81 (т, J=5,0 Гц, 2 H); 3,82 (т, J=5,0 Гц, 2 H); 2,38 (т, J=7,6 Гц, 2 H); 3,28 (т, J=6,1 Гц, 2 Н); 1,64 (м, 2 Н).
Данные ИК-спектроскопии (KBr): 3418, 3100, 1698, 1595, 1531, 1094, 767 см-1.
Данные масс-спектрометрии высокого разрешения HRMS (EI): Рассчитано для C20H21N5O (M+): 347,1746. Найдено: 347,1747.
Пример 6
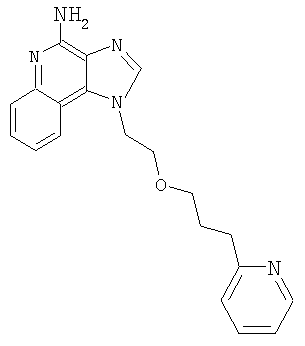
1-[2-(3-Пиридин-2-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
N,N-(бисбензилоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин (2,5 г, 4,68 ммоля) смешали в атмосфере азота с безводным ацетонитрилом (20 мл), триэтиламином (1,7 мл, 12,2 ммоля) и 2-бромпиридином (0,5 мл, 5,14 ммоля). Полученную гомогенную смесь нагрели до 40°С. Далее добавили йодид меди (I) (0,036 г) и дихлорбис(трифенилфосфин)палладий (II) (0,066 г). Через 18,5 часов реакционную смесь разделили между этилацетатом и водным раствором бикарбоната натрия. Органическую фракцию промыли рассолом, высушили над сульфатом магния и концентрировали при пониженном давлении. Остаток очистили способом хроматографии на колонке; элюент: смесь гексана и этилацетата (1:9). Получили 0,9 г смеси защищенного одним бензилоксикарбонильным остатком и двумя такими остатками 1-{2-[(3-пиридин-2-илпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,50-8,54 (м, 2 Н); 8,44 (с, 1 Н); 8,12 (м, 1 Н); 7,71-7,77 (м, 3 Н); 7,34-7,39 (м, 1 Н); 7,23-7,29 (м, 7 Н); 7,14-7,17 (м, 4 Н); 5,19 (с, 4 Н); 5,01 (т, J=4,6 Гц, 2 Н); 4,46 (с, 2 Н); 4,10 (т, J=4,8 Гц, 2 Н).
Данные масс-спектрометрии MS(CI): Рассчитано для C36H29N5O5: m/z 612 (MH+), 568, 460.
Часть В
К метанольному раствору продукта Части А добавили гидроксид палладия (0,776 г, 20 % на углероде). Продукт гидрогенизировали в течение 2,5 часов при давлении 3,2 бар. Реакционную смесь отфильтровали с целью удаления катализатора; осадок на фильтре тщательно промыли метанолом. Фильтрат концентрировали при пониженном давлении, получив стеклообразное твердое вещество. Это вещество растерли с диэтиловым эфиром и гексаном, содержащим небольшое количество толуола. Полученный порошок отделили фильтрованием и высушили в течение ночи в вакуумной печи при температуре 78°С. Данное вещество очистили способом хроматографии на колонке; элюат: смесь дихлорметана и метанола (9:1), куда было добавлено несколько капель гидроксида аммония. Получили 25 мг 1-[2-(3-пиридин-2-илпропокси)этил]-1H-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества с температурой плавления 138-140°С.
Данные элементного анализа: Рассчитано для C20H21N5O·(H2O)1/5: С 68,43 %,
Н 6,15 %, N 19,95 %. Найдено: С 68,47 %, Н 5,95 %, N 19,63 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,41 (д, J = 4,4 Гц, 1 Н); 8,16 (с, 1 Н); 8,10 (д, J=7,7 Гц, 1 Н); 7,63 (д, J=8,4 Гц, 1 Н); 7,54 (дт, J=9,7; 1,7 Гц, 1 Н); 7,43 (т, J=7,3 Гц, 1 Н); 7,24 (т, J=7,5 Гц, 1 Н); 7,13 (т, J=5,5 Гц, 1 Н); 6,93 (д, J=7,6 Гц, 1 Н); 6,59 (с, 2 Н); 4,77 (т, J=5,1 Гц, 2 Н); 3,82 (т, J=5,5 Гц, 2 Н); 3,34 (т, J=6,3 Гц, 2 Н); 2,57 (т, J=7,3 Гц, 2 Н); 1,75 (м, 2 Н).
Данные ИК-спектроскопии (KBr): 3361, 3302, 3188, 1638, 1526, 1119, 751 см-1.
Данные масс-спектрометрии высокого разрешения HRMS (EI): Рассчитано для C20H21N5O (M+): 347,1746. Найдено: 347,1747.
Пример 7
1-{2-[3-(1,3-Тиазол-2-ил)пропокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин
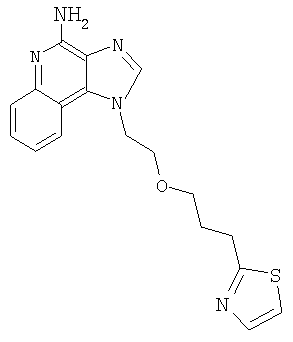
Часть А
N,N-(Бисбензилоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин (3,25 г, 6,08 ммоля) смешали в атмосфере азота с безводным N,N-диметилформамидом (15 мл), триэтиламином (2,2 мл, 15,8 ммолей) и 2-бромтиазолом (0,6 мл, 6,69 ммоля). Полученную смесь нагрели до 80°С. Далее добавили йодид меди (I) (0,046 г) и дихлорбис(трифенилфосфин)палладий (II) (0,085 г). Через 2 часа реакцию остановили и удалили растворитель. Продукт очистили способом хроматографии на колонке; элюент: смесь этилацетата и гексана (8:2). Получили приблизительно 2 г смеси защищенного одним бензилоксикарбонильным остатком и двумя такими остатками 1-(2-{[3-(1,3-тиазол-2-ил)проп-2-инил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амина.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,47-8,50 (м, 1 Н); 8,44 (с, 1 Н); 8,11 (м, 1 Н); 7,89 (д, J=3,2 Гц, 1 Н); 7,85 (д, J=3,3 Гц, 1 Н); 7,31-7,77 (м, 2 Н); 7,23-7,28 (м, 6 Н); 7,14-7,17 (м, 4 Н); 5,20 (с, 4 Н); 5,00 (т, J=5,0 Гц, 2 Н); 4,52 (с, 2 Н), 4,09 (T, J=5,5 Гц, 2 H).
Данные масс-спектрометрии MS(CI): Рассчитано для C24H27N5O5S: m/z 618 (MH+), 475, 466.
Часть В
К метанольному раствору продукта Части А добавили гидроксид палладия (прибл. 2 г, 20 % на углероде). Продукт гидрогенизировали в течение 3 часов при давлении 3,2 бар. Дважды добавляли дополнительные количества катализатора (0,3 г) и гидрогенизацию продолжали до достижения суммарного времени прибл. 25 часов. Реакционную смесь отфильтровали с целью удаления катализатора. Фильтрат концентрировали при пониженном давлении, получив 1,3 г защищенного одним бензилоксикарбонильным остатком 1-{2-[3-(1,3-тиазол-2-ил)пропокси]этил}-1H-имидазо[4,5-с]хинолин-4-амина. Это вещество смешали с метанолом (5 мл) и метоксидом натрия (25 мл 25 %-ного раствора в метаноле). Полученную смесь перемешивали в течение 3 дней. Далее реакционную смесь концентрировали при пониженном давлении и очищали способом хроматографии на колонке. Очищенный продукт растирали с диэтиловым эфиром и высушивали. Получили 0,28 г 1-{2-[3-(1,3-тиазол-2-ил)пропокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества с температурой плавления 134-135°С.
Данные элементного анализа: Рассчитано для C18H19N5OS: С 61,17 %, Н 5,42 %, N 19,81 %. Найдено: С 61,20 %, Н 5,23 %, N 19,51 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,16 (с, 1 Н); 8,10 (д, J=8,4 Гц, 1 Н); 7,63 (м, 2 Н); 7,51 (д, J=3,3Гц, 1 Н); 7,43 (т, J=7,1 Гц, 1 Н); 7,23 (т, J=8,0 Гц, 1 Н); 6,58 (с, 2 Н); 4,79 (т, J=4,7 Гц, 2 Н); 3,84 (т, J=4,8 Гц, 2 Н); 3,4 (т, J=6,0 Гц, 2 Н); 2,86 (т, J= 7,8 Гц, 2 Н); 1,83 (м, 2 Н).
Данные ИК-спектроскопии (KBr): 3458, 3358, 3295, 3191, 1640, 1538, 1121, 752 см-1.
Данные масс-спектрометрии высокого разрешения HRMS (EI): Рассчитано для C18H19N5OS (M+): 353,1310. Найдено: 353,1308.
Пример 8
Бис(трифторацетат) 1-[2-(3-пиридин-3-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина
Часть А
В атмосфере азота N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин (1,75 г, 3,75 ммоля) смешали с 3-йодпиридином (0,85 г, 4,13 ммоля), триэтиламином (1,4 мл) и ацетонитрилом (15 мл) и нагрели до 60°С. Далее добавили йодид меди (I) (0,03 г, 0,15 ммоля) и дихлорбис(трифенилфосфин)палладий (II) (0,05 г, 0,075 ммоля). Реакция завершилась за 30 мин. Растворители удалили при пониженном давлении. Продукт очистили способом хроматографии на колонке (элюирование с силикагеля сначала дихлорметаном, а затем смесью дихлорметан-метанол (98:2)). Получили 1,26 г смеси защищенного двумя бензилоксикарбонильными остатками и незащищенного 1-{2-[(3-пиридин-3-илпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолин-4-амина.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,54 (шс, 1 Н); 8,44-8,48 (м, 2 Н); 8,4 (с, 1 Н); 8,06 (м, 1 Н); 7,69-7,73 (м, 2 Н); 7,54 (д, J=7,6 Гц, 1 Н); 7,35 (м, 1 Н); 4,99 (т, J=4,8 Гц, 2 Н); 4,40 (с, 2 Н); 4,09 (т, J=5,0 Гц, 2 Н); 1,31 (с, 18 Н).
Данные масс-спектрометрии MS(CI): Рассчитано для С30Н33N5О5: m/z 544 (MH+), 444, 344.
Часть В
К метанольному раствору продукта Части А добавили катализатор (0,7 г палладия, 10 % на углероде). Продукт гидрогенизировали в течение 2 часов при давлении 3,2 бар. Катализатор удалили фильтрованием; фильтрат концентрировали при пониженном давлении, получив 0,67 г смеси защищенного двумя бензилоксикарбонильными остатками и незащищенного 1-[2-(3-пиридин-3-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,50 (д, J=7,3 Гц, 1 Н); 8,39-8,486 (м, 2 Н); 8,29 (с, 1 Н); 8,07 (д, J=8,4 Гц, 1 Н); 7,71-7,75 (м, 2 Н); 7,46 (д, J=8,5 Гц, 1 Н); 7,31 (м, 1 Н); 4,94 (т, J=4,6 Гц, 2 Н); 3,88 (т, J=5,0 Гц, 2 Н); 3,32 (т, J=5,9 Гц, 2 Н); 2,38 (т, J=7,5 Гц, 2 Н); 1,63 (м, 2 Н); 1,30 (с, 18 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для С30Н37N5О5: m/z 548 (MH+), 448, 348.
Часть С
Продукт Части В смешали с безводным дихлорметаном (5 мл) и трифторуксусной кислотой (5 мл) в атмосфере азота. Реакционную смесь перемешивали при температуре окружающей среды в течение 1 часа. Растворители удалили при пониженном давлении. Остаток растерли с диэтиловым эфиром, отфильтровали и высушили в вакуумной печи, получив твердое вещество желтовато-коричневого цвета. Продукт подвергли рекристаллизации сначала из изопропанола, а затем из смеси дихлорметан-метанол, получив 0,40 г бис(трифторацетат) 1-[2-(3-пиридин-3-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина с температурой плавления 134-136°С.
Данные элементного анализа: Рассчитано для C20H21N5O·(C2HF3O2)2·(H2O)1/2:С 50,08 %, Н 4,23 %, N 12,03 %. Найдено: С 49,87 %, Н 3,82 %, N 12,16 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 9,00-9,25 (шс, 2 Н); 8,50 (с, 2 Н); 8,37 (д, J=7,7 Гц, 1 Н); 7,82 (д, J=7,4 Гц, 1 Н); 7,73-7,75 (м, 2 Н); 7,55-7,58 (м, 2 Н); 4,90 (т, J=4,9 Гц, 2 Н); 3,86 (т, J=4,8 Гц, 2 Н); 3,35 (т, J=6,1 Гц, 2 Н); 2,49 (т, J=7,0 Гц, 2 Н); 1,67 (м, 2 Н).
Данные ИК-спектроскопии (KBr): 3421, 3212, 2885, 1699,1199, 1120, 720 см-1.
Данные масс-спектрометрии высокого разрешения HRMS (EI): Рассчитано для C20H21N5O (M+): 347,1746. Найдено: 347,1743.
Пример 9
1-[2-(3-Пиримидин-5-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
Пользуясь общим способом, описанным в Примере 8, Часть А, за исключением того, что температура при реакции была повышена до 80°С, провели реакцию между N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-4-амином (2,5 г, 5,36 ммоля) и 5-бромпиримидином (0,94 г, 5,89 ммоля), получив 1,59 г N,N-(бис-трет-бутоксикарбонил)-1-{2-[(3-пиримидин-5-илпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 9,19 (с, 1 Н); 8,64 (с, 2 Н); 8,44-8,47 (м, 1 Н); 8,40 (с, 1 Н); 8,02-8,06 (м, 1 Н); 7,68-7,72 (м, 2 Н); 4,99 (т, J=5,0 Гц, 2 Н); 4,43 (с, 2 Н); 4,10 (т, J=5,2 Гц, 2 Н); 1,32 (с, 18 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для С29H32N6O5: m/z 545 (MH+), 445, 345.
Часть В
К метанольному раствору продукта Части А добавили катализатор (последовательно 5 % платины на углероде, гидроксид палладия и 10 % палладия на углероде). В результате гидрогенизации получили 0,60 г N,N-(бис-трет-бутоксикарбонил)-1-[2-(3-пиримидин-5-илпропокси)этил]-1H-имидазо[4,5-с]хинолин-4-амина.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,98 (с, 1 Н); 8,48-8,52 (м, 1 Н); 8,45 (с, 2 Н); 8,40 (с, 1 Н); 8,06-8,09 (м, 1 Н); 7,70-7,74 (м, 2 Н); 4,94 (т, J=5,1 Гц, 2 Н); 3,89 (т, J=5,0 Гц, 2 Н); 3,34 (м, 2 Н); 2,34 (т, J=7,3 Гц, 2 Н); 1,64 (м, 2 Н); 1,29 (с, 18 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для С29Н36N6О6: m/z 549 (МН+), 449, 349.
Часть С
Пользуясь общим способом, описанным в Примере 8, Часть С, провели гидролиз продукта Части В, получив 0,14 г 1-[2-(3-пиримидин-5-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина с температурой плавления 159-161°С.
Данные элементного анализа: Рассчитано для C19H20N6O·(C2HF3O2)1/10·(H2O)1/4: С 63,27 %, Н 5,70 %, N 23,06 %. Найдено: С 63,47 %, Н 5,35 %, N 22,88 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,98 (с, 1 Н); 8,48 (с, 2 Н); 8,19 (с, 1 Н); 8,15 (д, J=8,0 Гц, 1 Н); 7,63 (д, J=8,6 Гц, 1 Н); 7,46 (т, J=6,0 Гц, 1 Н); 7,28 (т, J=8,4 Гц, 1 Н); 6,79 (с, 2 Н); 4,81 (т, J=4,8 Гц, 2 Н); 3,84 (т, J=5,1 Гц, 2 Н); 3,35 (т, J=6,0 Гц, 2 Н); 2,43 (т, J=7,4 Гц, 2 Н); 1,69 (м, 2 Н).
Данные ИК-спектроскопии (KBr): 3310, 3132, 1647, 1582, 1531, 1403, 1117 см-1.
Данные масс-спеюрометрии высокого разрешения HRMS (EI): Рассчитано для
C19H20N6O (М+): 348,1699. Найдено: 348,1695.
Пример 10
1-{2-[(1-Бензил-1H-1,2,3-триазол-4-ил)метокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина гидрохлорид
1-{2-[(1-Бензил-1H-1,2,3-триазол-5-ил)метокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина гидрохлорид
Часть А
N,N-(Бисбензилоксикарбонил)-1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-c]хинолин-4-амин (1,5 г, 2,81 ммоля) смешали с безводным толуолом (30 мл) и бензилазидом (1,3 мл, 9,12 ммоля) и нагревали в масляной бане при температуре 100°С в течение 28 часов. Реакционную смесь концентрировали при пониженном давлении, получив неочищенный продукт в виде маслянистой жидкости коричневого цвета.
Часть В
Метоксид натрия (2,19 мл 25 %-ного раствора в метаноле, 9,52 ммоля) добавили к смеси продукта Части А и метанола (20 мл). Смесь перемешивали при температуре окружающей среды в течение ночи, после чего ее концентрировали при пониженном давлении и получили маслянистую жидкость темного цвета.
Жидкость подвергли очистке способом хроматографии на колонке (элюент: 5 % метанола в дихлорметане) и получили маслянистую жидкость светло-желтого цвета. Эту жидкость обработали хлористым водородом (1,0 М) с получением твердого вещества розового цвета. Это вещество подвергли двойной рекристаллизации из ацетонитрила и высушивали в вакуумной печи при температуре 80°С в течение 2 часов. Получили 0,12 г смеси региоизомеров требуемого продукта, содержащей как 1-{2-[(1-бензил-1Н-1,2,3-триазол-4-ил)метокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина гидрохлорид, так и
1-{2-[(1-бензил-1Н-1,2,3-триазол-5-ил)метокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина гидрохлорид в виде кристаллического вещества светло-розового цвета с температурой плавления 209-211°С.
Данные элементного анализа: Рассчитано для С22Н21N7О·0,951HCl·0,615Н2O: С 59,35 %, Н 5,25 %, N 22,02 %. Найдено: С 59,46 %, Н 5,16 %, N 22,05 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 9,00 (шс, 2 Н); 8,46 (с, 1 Н); 8,28 (д, J=7,8 Гц, 1 Н); 7,98 (с, 1 Н); 7,83 (шд, J=7,8 Гц, 1 Н); 7,71 (шт, J=7,8 Гц, 1 Н); 7,50 (шт, J=7,7 Гц, 1 Н); 7,20-7,40 (м, 5 Н); 5,52 (с, 1,88 Н); 5,39 (с, 0,12 Н); 4,88 (т, J=4,9 Гц, 2 Н); 4,52 (с, 2 Н); 3,95 (т, J=4,9 Гц, 1,88 Н); 3,87 (т, J=5,1 Гц, 0,12 Н).
Данные ИК-спектроскопии (KBr): 3152, 2638, 1672, 1605, 1126 см-1.
Данные масс-спектрометрии высокого разрешения HRMS (EI): Рассчитано для
С22Н21N7О (М+): 399,1808. Найдено: 399,1802.
Пример 11
1-[2-({1-[Фенилсульфанил)метил]-1Н-1,2,3-триазол-4-ил}метокси)этил]-1Н-имидазо[4,5-с]
хинолин-4-амин 1-[2-({1-[Фенилсульфанил)метил]-1Н-1,2,3-триазол-5-ил}метокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
Безводный толуол (20 мл) смешали в атмосфере азота с N,N-(бис-трет-бутоксикарбонил)-1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-c]хинолин-4-амином (1,0 г, 2,1 ммоля) и азидометилфенилсульфидом (0,61 мл, 4,3 ммоля) и нагревали с обратным холодильником в течение 72 часов. Реакционную смесь оставили для охлаждения до температуры окружающей среды, после чего концентрировали при пониженном давлении, получив маслянистую жидкость коричневого цвета. Данный продукт очистили способом хроматографии на колонке (элюент: смесь этилацетата и гексана 80:20), получив 0,95 г продукта в виде прозрачной маслянистой жидкости.
Данные масс-спектрометрии MS (CI): Рассчитано для С32Н37N7O5S: m/z 632 (МН+), 532, 458, 432.
Часть В
Раствор продукта Части А в безводном дихлорметане (15 мл) добавили к смеси трифторуксусной кислоты (7,4 мл) и безводного дихлорметана (6 мл), заранее охлажденной до температуры 0°С. Реакционную смесь выдерживали в ледяной бане в течение 2 часов, после чего оставили для нагрева до температуры окружающей среды. Через 6 часов реакционную смесь промыли 20 %-ным гидроксидом натрия. Водную фракцию экстрагировали дихлорметаном. Органические фракции соединили, промыли водой, высушили над сульфатом магния, отфильтровали и концентрировали при пониженном давлении с получением маслянистой жидкости зеленого цвета. Эту жидкость очистили способом хроматографии на колонке при элюировании смесью 5 % метанола и дихлорметана, получив твердое кристаллическое вещество зеленого цвета. Это вещество рекристаллизировали из изопропанола, получив 0,12 г смеси региоизомеров требуемого продукта, то есть смеси, содержащей как 1-[2-({1-[(фенилсульфанил)метил]-1Н-1,2,3-триазол-4-ил}метокси)этил]-1Н-имидазо[4,5-c]хинолин-4-амин, так и 1-[2-({1-[(фенилсульфанил)метил]-1H-1,2,3-триазол-5-ил}метокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин. Продукт представлял собой твердое вещество почти белого цвета с температурой плавления 182-184°С.
Данные элементного анализа: Рассчитано для C22H21N7OS: С 61,24 %, Н 4,91 %, N 22,72 %. Найдено: С 60,94 %, Н 4,94 %, N 22,38 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,00-8,20 (м, 2 Н); 7,87 (с, 0,8 Н); 7,60-7,65 (м, 1 Н); 7,52 (с, 0,2 Н); 7,40-7,50 (м, 1 Н); 7,20-7,40 (м, 6 Н); 6,65 (с, 2 Н); 5,87 (с, 1,6 Н); 5,65 (с, 0,4 Н); 4,83 (шт, J=4,6 Гц, 0,4 Н); 4,78 (шт, J=4,9 Гц, 1,6 Н); 4,49 (с, 1,6 Н); 4,42 (с, 0,4 Н); 3,80-3,90 (м, 2 Н).
Данные ИК-спектроскопии (KBr): 3322, 3205, 1643, 1527, 1095 см-1.
Данные масс-спектрометрии высокого разрешения HRMS (EI): Рассчитано для C22H21N7OS (M+): 431,1528. Найдено: 431,1522.
Пример 12
1-[2-(Бензо[b]фуран-2-илметокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
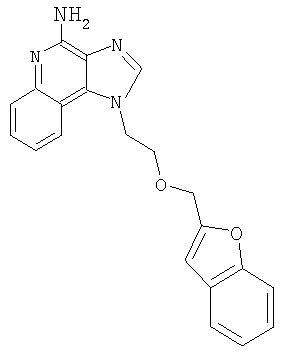
Часть А
Бензилтриметиламмонийхлорид (0,436 г) и пропаргилбромид (6,07 мл, 80 %-ный продукт) добавили при перемешивании к смеси дихлорметана (185 мл) и водного раствора гидроксида натрия (60 мл, 50 %-ный продукт). К полученному раствору добавили 2-(1Н-имидазо[4,5-с]хинолин-1-ил)этанол (10,0 г, 46,9 ммоля). Реакционную смесь перемешивали при температуре окружающей среды в течение 21 часов; к концу этого периода анализ способом ВЭЖХ показал, что в системе остались непрореагировавшие вещества. Было добавлено дополнительное эквивалентное количество пропаргилбромида, и реакционную смесь перемешивали еще в течение 46 часов. К реакционной смеси добавили воду, после чего разделили слои. Водную фракцию экстрагировали дихлорметаном. Органические фракции соединили, промыли водой и рассолом, высушили над сульфатом магния и концентрировали при пониженном давлении, получив твердое вещество темно-коричневого цвета. Продукт был очищен способом скоростной хроматографии (продукт на силикагеле элюировали смесью метанола (5 %) и дихлорметана), получив 7,0 г 1-[2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолина в виде твердого вещества коричневатого цвета.
Часть В
1-[2-(пропинилокси)этил]-1H-имидазо[4,5-с]хинолин (1,0 г, 4,21 ммоля) смешали с триэтиламином (1,53 мл, 10,96 ммоля) и безводным ацетонитрилом (20 мл) в атмосфере азота, после чего смесь нагрели до 60°С. К смеси добавили 2-йодфенол (1,02 г, 4,63 ммоля), йодид меди (I) (0,08 г) и дихлорбис(трифенилфосфин)палладий (II) (0,148 г). Через 4 часа анализ способом ТСХ (5 % метанола в дихлорметане) показал, что реакция прошла полностью. Реакционную смесь отфильтровали через слой целита (Celite®) для удаления катализатора. Фильтрат концентрировали при пониженном давлении и получили маслянистую жидкость. Эту жидкость очистили способом высокоскоростной хроматографии (вещество на силикагеле элюировали 3 %-ной смесью метанола и дихлорметана), получив 0,91 г 1-[2-(бензо[b]фуран-2-илметокси)этил]-1H-имидазо[4,5-с]хинолина в виде маслянистой жидкости желтого цвета.
Часть С
3-Хлорпероксибензойную кислоту (0,65 г) добавляли по частям в течение 5 мин к раствору продукта Части В в хлороформе (15 мл). За ходом реакции следили при помощи ТСХ. Добавили дополнительное количество 3-хлорпероксибензойной кислоты (2×0,2 г). Спустя 1,5 часа реакционную смесь дважды промыли водным раствором бикарбоната натрия, проэкстрагировали хлороформом, промыли рассолом и концентрировали при пониженном давлении, получив 1-[2-(бензо[b]фуран-2-илметокси)этил]-1H-имидазо[4,5-с]хинолин-5N-оксид. Продукт хранили в атмосфере азота при пониженной температуре в течение выходных дней.
Часть D
К раствору N-оксида (часть С) в дихлорметане (15 мл) в атмосфере азота медленно добавили при помощи шприца трихлорацетилизоцианат (0,60 г, 3,18 ммоля). Летучие вещества удалили при пониженном давлении; получили 2,2,2-трихлор-{1-[2-(бензо[b]фуран-2-илметокси)этил]-1Н-имидазо[4,5-с]хинолин-4-ил}ацетамид в виде твердого вещества желтовато-коричневого цвета. Этот продукт растворили в метаноле (15 мл). Далее добавили метоксид натрия (2,04 мл, 9,01 ммоля); полученный раствор перемешивали в течение 48 часов. Белый осадок отделили фильтрованием, после чего его рекристаллизовали из ацетонитрила, получив 0,22 г 1-[2-(бензо[b]фуран-2-илметокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде игл почти белого цвета с температурой плавления 201-203°С.
Данные элементного анализа: Рассчитано для C21H18N4O2: С 70,38 %, Н 5,06 %, N 15,63 %. Найдено: С 70,36 %, Н 4,80 %, N 15,51 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,16 (с, 1 Н); 8,06 (д, J=7,8 Гц, 1 Н); 7,49-7,62 (м, 3 Н); 7,42 (м, 1 Н); 7,16-7.31 (м, 3 Н); 6,76 (с, 1 Н); 6,58 (шс, 2 Н); 4,83 (т, J=5,4 Гц, 2 Н); 4,61 (с, 2 Н); 3,97 (т, J=5,1 Гц, 2 Н).
Данные ИК-спектроскопии (KBr): 3455, 3069, 1583, 1530, 1397, 1254, 1088 см-1.
Данные масс-спектрометрии высокого разрешения HRMS (EI): Рассчитано для C21H18N4O2 (M+): 358,1430. Найдено: 358,1428.
Пример 13
1-[2-(Пиридин-3-илметокси)этил]-1H-имидазо[4,5-с]хинолин-4-амина гидрохлорид
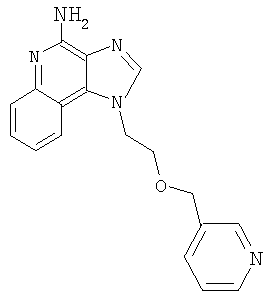
Часть А
В атмосфере азота гидрид натрия (16,88 г, 60 %-ный раствор в минеральном масле, 422 ммолей) добавляли порциями к раствору 2-(1Н-имидазо[4,5-с]хинолин-1-ил)этанола (60,0 г, 281 ммоль) в безводном N,N-диметилформамиде (600 мл). Алкоксид перемешивали в течение приблизительно 1,5 часов. Далее медленно добавляли бензилбромид (50,2 мл, 422 ммолей) в течение почти 30 мин. Реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Растворитель удалили при пониженном давлении. Остаток был обработан этилацетатом, промыт водой несколько раз, промыт рассолом, высушен над сульфатом магния и концентрирован при пониженном давлении. В результате получили 1-[2-(бензилокси)этил]-1Н-имидазо[4,5-с]хинолин в виде маслянистой жидкости темного цвета.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 9,22 (с, 1 Н); 8,42 (с, 1 Н); 8,40 (с, 1 Н); 8,17 (м, 1 Н); 7,69 (м, 2 Н); 7,10-7,22 (м, 5 Н); 4,95 (т, J=5,1 Гц, 2 Н); 4,45 (с, 2 Н); 3,93 (т, J=5,1 Гц, 2 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для С19Н17N3О: m/z 304 (МН+), 214.
Часть В
3-Хлорпероксибензойную кислоту (69,36 г с максимальным содержанием 77 %) добавляли порциями к раствору 1-[2-(бензилокси)этил]-1Н-имидазо[4,5-c]хинолина (85,36 г, 281 ммолей) в хлороформе (800 мл) в течение 15 мин. Через 1 час анализ способом ТСХ (10 % метанола в дихлорметане) показал, что реакция прошла полностью. Реакционную смесь дважды промыли насыщенным раствором бикарбоната натрия, промыли рассолом, высушили над сульфатом магния и концентрировали при пониженном давлении, получив твердое вещество. Это вещество суспендировали в диэтиловом эфире и отделили фильтрованием, получив 1-[2-(бензилокси)этил]-1Н-имидазо[4,5-с]хинолин-5N-оксид в виде твердого продукта темно-желтого цвета.
Часть С
К смеси 1-[2-(бензилокси)этил]-1H-имидазо[4,5-с]хинолин-5N-оксида (40,0 г, 125 ммолей) и безводного толуола (600 мл) медленно добавили оксихлорид фосфора (12,84 мл, 138 ммолей). Реакционную смесь перемешивали в течение приблизительно 30 мин, после чего летучие вещества были удалены при пониженном давлении. Полученную маслянистую жидкость красного цвета растворили в дихлорметане, дважды промыли насыщенным раствором бикарбоната натрия и концентрировали при пониженном давлении. Попытка рекристаллизации продукта из этилацетата закончилась образованием смолообразной массы. Продукт смешали с этилацетатом (500 мл), после чего добавили триэтиламин (25,34 г, 250 молей). Раствор охладили в ледяной бане и осадок отделили фильтрованием. Вскоре после фильтрования этот продукт вновь превратился в маслянистую жидкость. Данную жидкость смешали с дихлорметаном, соединили с фильтратом и концентрировали при пониженном давлении, получив маслянистую жидкость. Полученную жидкость распределили между дихлорметаном и 15 %-ным гидроксидом натрия. Органическую фракцию промыли рассолом, высушили над сульфатом магния и концентрировали при пониженном давлении, получив маслянистый продукт. Этот продукт подвергли очистке способом высокоскоростной хроматографии (элюирование с силикагеля сначала дихлорметаном, далее 2 %-ным метанолом в дихлорметане и в конце 5 %-ным метанолом в дихлорметане) и получили приблизительно 21 г 1-[2-(бензилокси)этил]-4-хлор-1Н-имидазо[4,5-с]хинолина.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,47 (с, 1 Н); 8,42 (дд, J=8,3; 1,5 Гц, 1 Н); 8,08 (дд, J=8,3; 1,5 Гц, 1 Н); 7,72 (м, 2 Н); 7,04-7,17 (м, 5 Н); 4,96 (т, J=5,1 Гц, 2 Н); 4,44 (с, 2 Н); 3,92 (т, J=5,1 Гц, 2 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для C19H16CIN3O: m/z 338 (MH+), 309, 248, 214.
Часть D
Фенол (6,21 г, 66 ммолей) добавляли порциями к охлажденной суспензии гидрида натрия (2,79 г 60%-ного раствора в минеральном масле, 69,7 ммоля) в диглиме (25 мл). По окончании выделения пузырьков добавили (одной порцией) раствор продукта Части С в диглиме (10 мл). Полученный раствор нагрели до 110°С и перемешивали в течении ночи. Анализ способом ТСХ (3 %-ный метанол в дихлорметане) показал, что реакция прошла полностью. Раствор охладили до 0°С, получив коричневый осадок. Диглим удалили декантацией. Твердую фазу суспендировали в гексане и отделили фильтрованием. Далее твердую фазу суспендировали в воде, отделили фильтрованием и высушили в печи в течение ночи. Твердый продукт рекристаллизовали из изопропанола, получив 19,3 г 1-[2-(бензилокси)этил]-4-фенокси-1H-имидазо[4,5-с]хинолина в виде твердого вещества.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,39 (с, 1 Н); 8,32 (дд, J=8,3; 1,5 Гц, 1 Н); 7,69 (дд, J=8,3; 1,5 Гц, 1 Н); 7,46-7,59 (м, 4 Н); 7,12-7,33 (м, 8 Н); 4,93 (т, J=5,1 Гц, 2 Н); 4,47 (с, 2 Н); 3,94 (т, J=5,1 Гц, 2 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для С25Н21N3О2: m/z 396 (MH+), 306, 288.
Часть Е
В атмосфере азота трифторуксусную кислоту (29,0 г) добавляли по каплям к раствору 1-[2-(бензилокси)этил]-4-фенокси-1Н-имидазо[4,5-с]хинолина (7,65 г) в безводном дихлорметане (200 мл). После того как анализ способом ТСХ (5 % метанола в дихлорметане) показал, что реакция прошла полностью, данную смесь концентрировали при пониженном давлении, получив маслянистую жидкость. Жидкость растворили в этилацетате; далее добавили триэтиламин (10 экв.). Раствор разбавили дополнительным количеством этилацетата, промыли водой и рассолом, высушили над сульфатом магния и концентрировали при пониженном давлении. Продукт рекристаллизовали из этилацетата, получив приблизительно 4,8 г 2-(4-фенокси-1H-имидазо[4,5-с]хинолин-1-ил)этанола в виде рыхлого твердого вещества белого цвета.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,34 (м, 1 Н); 8,32 (м, 1 Н); 7,70 (м, 1 Н); 7,46-7,60 (м, 4 Н); 7,25-7,31 (м, 3 Н); 5,06 (т, J=5,4 Гц, 1 Н); 4,76 (т, J=5,4 Гц, 2 Н); 3,90 (к, J=5,4 Гц, 2 Н).
Часть F
К раствору, содержащему 2-(4-фенокси-1H-имидазо[4,5-с]хинолин-1-ил)этанол (0,7 г, 2,29 ммоля), бензилтриметиламмонийхлорид (приблизительно 0,03 г), гидроксид натрия (28 мл 50 %-ного продукта) и дихлорметан (28 мл), добавили при перемешивании 3-(бромметил)пиридингидробромид (0,638 г, 2,52 ммоля) в виде одной порции. Через 3 часа анализ способом ТСХ (5 % метанола в дихлорметане) показал, что реакция завершено полностью. Реакционную смесь разбавили водой (100 мл) и дихлорметаном (100 мл). После разделения слоев водную фракцию экстрагировали дихлорметаном (100 мл). Органические фракции соединили, промыли рассолом, высушили над сульфатом магния и концентрировали при пониженном давлении, получив твердое вещество темно-желтого цвета. Этот продукт очистили способом высокоскоростной хроматографии (вещество на силикагеле элюировали 5 %-ным раствором метанола в дихлорметане), получив 0,74 г 4-фенокси-1-[2-(пиридин-3-илметокси)этил]-1H-имидазо[4,5-с]хинолина в виде твердого вещества ярко-желтого цвета.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,42 (дд, J=4,9; 2,0 Гц, 1 Н); 8,38 (с, 2 Н); 8,33 (дд, J=8,3; 1,5 Гц, 1 Н); 7,70 (дд, J=8,3; 2,0 Гц, 1 Н); 7,46-7,59 (м, 5 Н); 7,22-7,33 (м, 4 Н); 4,96 (т, J=4,9 Гц, 2 Н); 4,52 (с, 2 Н); 3,98 (т, J=4,9 Гц, 2 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для C24H20N4O2: m/z 397 (MH+), 306, 288, 212, 110.
Часть G
4-Фенокси-1-[2-(пиридин-3-илметокси)этил]-1Н-имидазо[4,5-с]хинолин (0,67 г, 1,69 ммоля) смешали с ацетатом аммония (1,30 г, 16,9 ммоля) и нагрели до 150°С. Через 5 часов анализ способом ТСХ (10 % метанола в дихлорметане) указал на наличие в системе исходного вещества. Было добавлено дополнительное количество ацетата аммония (5 г). Через 1 час анализ способом ТСХ показал, что реакция прошла полностью. Реакционную смесь оставили на ночь для охлаждения до температуры окружающей среды. Полученный продукт в виде маслянистой жидкости коричневого цвета смешали с водой (100 мл) и сдвинули рН в щелочную сторону (рН 9) путем добавления бикарбоната натрия. Продукт экстрагировали дихлорметаном (2×100 мл). Экстракты соединили, промыли рассолом, высушили над сульфатом магния и концентрировали при пониженном давлении, получив смолистое вещество почти белого цвета. Этот продукт очистили способом высокоскоростной хроматографии (вещество на силикагеле элюировали 10 %-ным раствором метанола в дихлорметане), получив 0,40 г смолистого вещества белого цвета. Это вещество растворили в метаноле (10 мл). К раствору добавили по каплям смесь хлористого водорода и диэтилового эфира (5 экв.) и перемешивали данную массу в течение 1 часа. Образовавшийся осадок отделили фильтрованием, промыли диэтиловым эфиром и высушили в вакуумной печи, получив 0,358 г гидрохлорида 1-[2-(пиридин-3-илметокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества светло-желтого цвета с температурой плавления 229-231°С.
Данные элементного анализа: Рассчитано для C18H17N5O·2,75 HCl·0,4 H2O: С 50,62 %, Н 4,85 %, N 16,40 %. Найдено: С 50,44 %, Н 4,96 %, N 16,19 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,78 (д, J=5,4 Гц, 1 Н); 8,71 (с, 1 Н); 8,56 (с, 1 Н); 8,34 (д, J=8,3 Гц, 1 Н); 8,26 (д, J=7,8 Гц, 1 Н); 7,90 (дд J=7,8; 5,9 Гц, 1 Н); 7,84 (д, J=8,3 Гц, 1 Н); 7,72 (т, J=7,8 Гц, 1 Н); 7,54 (т, J=7,8 Гц, 1 Н); 4,98 (т, J=4,9 Гц, 2 Н); 4,69 (с, 2 Н); 4,04 (т, J=4,9 Гц, 2 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для C18H17N5O·HCl(11/4)·H2O(2/5): m/z 321 (MH+), 229.
Пример 14
1-[2-(Пиридин-2-илметокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
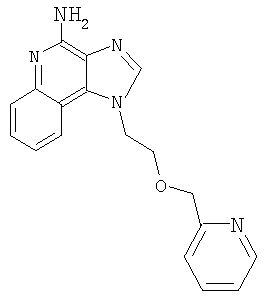
Часть А
Используя общий способ, описанный в Примере 13, Часть F, провели реакцию между 2-(4-фенокси-1Н-имидазо[4,5-с]хинолин-1-ил)этанолом (0,9 г, 2,95 ммоля) и солянокислым 2-пиколилхлоридом (0,53 г, 3,24 ммоля); продукт очистили, получив 0,65 г 4-фенокси-1-[2-(пиридин-2-илметокси)этил]-1H-имидазо[4,5-с]хинолина.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,41 (м, 2 Н); 8,34 (дд, J=8,3; 1,5 Гц, 1 Н); 7,70 (дд J=8,3; 1,5 Гц, 2 Н); 7,46-7,66 (м, 5 Н); 7,18-7,33 (м, 4 Н); 7,30 (д, J=7,8 Гц, 1 Н); 4,98 (т, J=4,9 Гц, 2 Н); 4,55 (с, 2 Н); 4,04 (т, J=4,9 Гц, 2 Н).
Часть В
Продукт Части А и ацетат аммония смешали и нагрели до 150°С. Через 5 часов анализ способом ТСХ (10 % метанола в дихлорметане) показал, что реакция прошла полностью. Реакционную смесь оставили для охлаждения до температуры окружающей среды, добавили воды (100 мл) и сдвинули рН в щелочную сторону (рН 9) путем добавления бикарбоната натрия. Полученный белый осадок отделили фильтрованием и суспендировали в диэтиловом эфире. Затем продукт белого цвета отделили фильтрованием и рекристаллизовали из ацетонитрила, получив 0,18 г 1-[2-(пиридин-2-илметокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества белого цвета с температурой плавления 196-198°С.
Данные элементного анализа: Рассчитано для C18H17N5O: С 67,70 %, Н 5,37 %, N 21,93 %. Найдено: С 67,86 %, Н 5,31 %, N 22,13 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,43 (д, J=4,9 Гц, 2 Н); 8,20 (с, 1 Н); 8,09 (д, J=6,8 Гц, 1 Н); 7,63 (дт, J=8,3; 1,5 Гц, 2 Н); 7,43 (дт, J=8,3; 1,5 Гц, 1 Н); 7,19-7,24 (м, 2 Н); 7,12 (д, J=7,8 Гц, 1 Н); 6,53 (шс, 2 Н); 4,87 (т, J=5,1 Гц, 2 Н); 4,54 (с, 2 Н); 3,99 (т, J=5,1 Гц, 2 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для C18H17N5O: m/z 320 (MH+), 229, 211.
Пример 15
1-[2-(Пиридин-4-илметокси)этил]-1H-имидазо[4,5-с]хинолин-4-амин
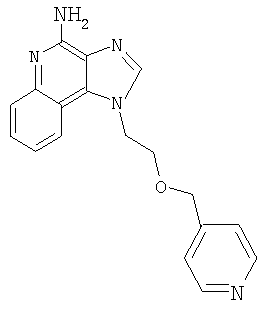
Часть А
Используя общий способ, описанный в Примере 13, Часть F, провели реакцию между 2-(4-фенокси-1Н-имидазо[4,5-с]хинолин-1-ил)этанолом (1,1 г, 3,61 ммоля) и солянокислым 4-пиколилхлоридом (0,649 г, 3,96 ммоля); продукт очистили, получив приблизительно 0,3 г 4-фенокси-1-[2-(пиридин-4-илметокси)этил]-1Н-имидазо[4,5-с]хинолина.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,41 (с, 1 Н); 8,39 (с, 1 Н); 8,34 (дд, J=7,8; 1,5 Гц, 2 Н); 7,70 (дд, J=7,8; 1,5 Гц, 1 Н); 7,46-7,60 (м, 4 Н); 7,25 7,33 (м, 3 Н); 7,10 (д, J=5,9 Гц, 2 Н); 5,00 (т, J=4,9 Гц, 2 Н); 4,53 (с, 2 Н); 4,00 (т, J=4,9 Гц, 2 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для C24H20N4O2: m/z 397 (MH+), 306, 288, 212, 110.
Часть В
Используя общий способ, описанный в Примере 14, Часть В, провели аминирование 4-фенокси-1 -[2-(пиридин-4-илметокси)этил]-1H-имидазо[4,5-с]хинолина (0,25 г), получив 0,14 г 1-[2-(пиридин-4-илметокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества с температурой плавления 159-161°С.
Данные элементного анализа: Рассчитано для C18H17N5O: С 67,70 %, Н 5,37 %, N 21,93 %. Найдено: С 67,37 %, Н 5,31 %, N 22,49 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,40 (дд, J=4,4; 1,5 Гц, 2 Н); 8,20 (с, 1 Н); 8,09 (д, J=8,3 Гц, 1 Н); 7,62 (д, J=8,3 Гц, 1 Н); 7,43 (м, 1 Н); 7,21 (м, 1 Н); 7,10 (д, J=5,4 Гц, 1 Н); 6,54 (шс, 2 Н); 4,87 (т, J=5,1 Гц, 2 Н); 4,51 (с, 2 Н); 3,94 (т, J=5,1 Гц, 2 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для C18H17N5O: m/z 320 (MH+), 229, 136.
Пример 16
1-{2-[(3,5-Диметилизоксазол-4-ил)метокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин
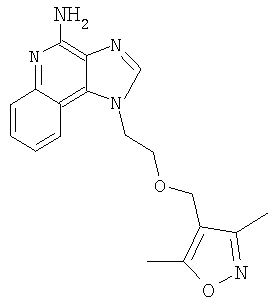
Часть А
Используя общий способ, описанный в Примере 13, Часть F, провели реакцию между 2-(4-фенокси-1Н-имидазо[4,5-с]хинолин-1-ил)этанолом (0,82 г, 2,69 ммоля) и 4-(хлорметил)-3,5-диметилизоксазолом (0,43 г, 2,95 ммоля); продукт очистили, получив 0,59 г 1-{2-[(3,5-диметилизоксазол-4-ил)метокси]этил}-4-фенокси-1H-имидазо[4,5-с]хинолин-4-амина в виде твердого пенистого вещества белого цвета.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,29-8,32 (м, 2 Н); 7,70 (дд J=7,8; 1,5 Гц, 1 Н); 7,46-7,60 (м, 4 Н); 7,25-7,32 (м, 3 Н); 4,89 (т, J=5,1 Гц, 2 Н); 4,24 (с, 2 Н); 3,89 (т, J=4,9 Гц, 2 Н); 2,16 (с, 3Н); 1,93 (с, 3Н).
Данные масс-спектрометр и и MS (CI): Рассчитано для C24H22N4O3: m/z 415 (MH+), 306, 212, 112.
Часть В
Используя общий способ, описанный в Примере 14, Часть В, провели аминирование продукта Части А, получив 0,39 г 1-{2-[(3,5-диметилизоксазол-4-ил)метокси]этил}-1H-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества белого цвета с температурой плавления 213-215°С.
Данные элементного анализа: Рассчитано для С18Н19N5О2: С 64,08 %, Н 5,68 %, N 20,76 %. Найдено: С 64,02 %, Н 5,53 %, N 21,01 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,12 (с, 1 Н); 8,05 (дд, J=8,3; 1,0 Гц, 1 Н); 7,61 (дд, J=8,3; 1,0 Гц, 1 Н); 7,43 (м, 1 Н); 7,21 (м, 1 Н); 6,52 (шс, 2 Н); 4,79 (т, J=5,1 Гц, 2 Н); 4,23 (с, 2 Н); 3,85 (т, J=5,1 Гц, 2 Н); 2,20 (с, 3 Н); 1,97 (с, 3 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для C18H19N5O2: m/z 338 (MH+), 229, 112.
Пример 17
1-(2-{[3-(Пиримидин-2-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амина трифторацетат
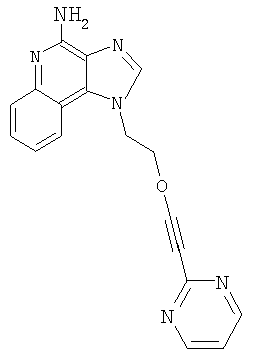
В атмосфере азота смешали 1-[2-(-2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин (1,0 г, 3,7 ммоля), триэтиламин (1,0 г, 9,7 ммоля) и безводный N,N-диметилформамид (20 мл). Полученный раствор нагрели до 65°С. В смесь добавили йодид меди (I) (0,07 г, 0,4 ммоля), дихлорбис(трифенилфосфин)палладий (II) (0,13 г, 0,2 ммоля) и 2-бромпиримидин (0,65 г, 4,1 ммоля). Реакционную смесь перемешивали в течение ночи при температуре 65°С. N,N-диметилформамид удалили при пониженном давлении, получив неочищенный продукт в виде смолы. Этот продукт очистили сначала способом хроматографии на колонке (вещество на силикагеле элюировали дихлорметаном), а затем способом полупрепаративной хроматографии (способ А), получив 0,05 г трифторацетата 1-(2-{[3-(пиримидин-2-ил)-2-пропинил]окси}этил)-1H-имидазо[4,5-с]хинолин-4-амина в виде губчатого твердого вещества белого цвета с температурой плавления 214-215°С.
Данные элементного анализа: Рассчитано для C19H16N6O2·1,5С2HF3О2·0,3Н2O: С 50,67 %, Н 3,51 %, N 16,12 %. Найдено: С 50,67 %, Н 3,11 %, N 16,14 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ (10-6) 9,169 (с, 1 Н); 8,646 (с, 2 Н); 8,497 (с, 1 Н); 8,328 (д, J=8,3 Гц, 1 Н); 7,773 (д, J=6,9 Гц, 1 Н); 7,703 (т, J=6,7 Гц, 1 Н); 7,558 (т, J=7,2 Гц, 1 Н); 4,942 (т, J=4,8 Гц, 2 Н); 4,447 (с, 2 Н); 4,073 (т, J=4,9 Гц, 2 Н).
Пример 18
1-(2-{[3-(Пирид-4-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амина бис(трифторацетат)
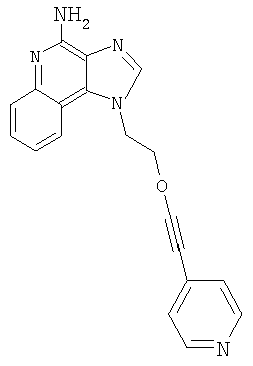
В атмосфере азота при температуре окружающей среды перемешивали 1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-4-амин (0,5 г, 1,9 ммоля), триэтиламин (0,5 г, 4,9 ммоля), йодид меди (I) (0,036 г, 0,2 ммоля), 4-бромпиридин (0,51 г, 2,6 ммоля) и ацетонитрил (20 мл). Далее добавили дихлорбис(трифенилфосфин)палладий (II) (0,066 г, 0,1 ммоля). Реакционную смесь оставили на ночь при нагреве с обратным холодильником. Ацетонитрил удалили при пониженном давлении. Остаток смешали с дихлорметаном и метанолом, а затем раствор пропустили через колонку с оксидом алюминия, обладающим основными свойствами. Фракции соединили и концентрировали при пониженном давлении. Продукт растерли с ацетонитрилом. Полученное твердое вещество отделили фильтрованием и очистили способом полупрепаративной ВЭЖХ (Способ А), получив 0,1 г бис(трифторацетата) 1-(2-{[3-(пирид-4-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амина в виде рыхлого твердого вещества серого цвета с температурой плавления 135°С (с разложением).
Данные элементного анализа: Рассчитано для C20H17N5O·2,0С2HF3О2·0,5Н2О: С 49,66 %, Н 3,47 %, N 12,06 %. Найдено: С 49,59 %, Н 3,51 %, N 12,22 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ (10-6) 9,063 (шс, 1 Н); 8,551 (д, J=5,2 Гц, 2 Н); 8,498 (с, 1 Н); 8,335 (д, J=7,7 Гц, 1 Н); 7,795 (д, J=6,9 Гц, 1 Н); 7,727 (т, J=8,3 Гц, 1 Н); 7,564 (т, J=8,3 Гц, 1 Н); 7,139 (д, J=5,7 Гц, 2 Н); 4,942 (т, J=4,8 Гц, 2 Н); 4,427 (с, 2 Н); 4,056 (т, J=4,8 Гц, 2 Н).
Пример 19
1-(2-{[3-(Фур-3-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амина трифторацетат
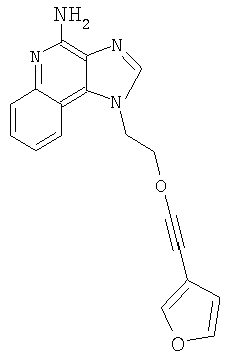
В атмосфере азота при температуре окружающей среды соединили и перемешивали 1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-4-амин (0,5 г, 1,9 ммоля), триэтиламин (0,5 г, 4,9 ммоля), йодид меди (I) (0,036 г, 0,2 ммоля), 3-бромфуран (0,38 г, 2,6 ммоля) и безводный N,N-диметилформамид (20 мл). Далее добавили дихлорбис(трифенилфосфин)палладий (II) (0,066 г, 0,1 ммоля). Реакционную смесь нагревали в течение ночи при температуре 80°С. После охлаждения до температуры окружающей среды реакционную смесь разбавили дихлорметаном. Полученный мелкодисперсный осадок коричневого цвета отделили фильтрованием. Фильтрат концентрировали при пониженном давлении. Остаток растворили в минимальном количестве N,N-диметилформамида, а затем раствор пропустили через колонку с силикагелем. Соответствующие фракции соединили и концентрировали при пониженном давлении. Продукт очистили способом полупрепаративной ВЭЖХ (Способ А), получив 0,1 г трифторацетата 1-(2-{[3-(фур-3-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амина в виде рыхлого твердого вещества цвета слоновой кости с температурой плавления 160-162°С.
Данные элементного анализа: Рассчитано для C19H16N4O2·С2HF3О2·0,25Н2О: С 55,94 %, Н 3,91 %, N 12,42 %. Найдено: С 55,57 %, Н 3,43 %, N 12,45 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ (10-6) 8,859 (шс, 2 Н); 8,473 (с, 1 Н); 8,32 (д, J=8,3 Гц, 1 Н); 7,849 (с, 1 Н); 7,813 (д, J=7,3 Гц, 1 Н); 7,714 (т, J=8,5 Гц, 1 Н); 7,697 (д, J=2 Гц, 1 Н); 7,551 (т, J=6,8 Гц, 1 Н); 6,409 (д, J=1,9 Гц, 1 Н); 4,919 (т, J=5,5 Гц, 2 Н); 4,337 (с, 2 Н); 4,002 (т, J=4,8 Гц, 2 Н).
Пример 20
4-{3-[2-(4-Амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]-пропин-1-ил}-
тиофен-2-илкарбоксальдегида трифторацетат
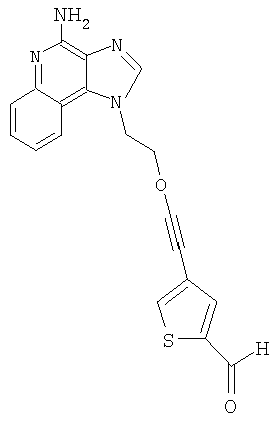
Пользуясь общим способом примера 20, провели реакцию между 1-[2-(2-пропинилокси)этил]-1H-имидазо[4,5-с]хинолин-4-амином (0,5 г, 1,9 ммоля) и 3-бром-2-тиофенкарбоксальдегидом (0,5 г, 2,6 ммоля); продукт подвергли очистке способом полупрепаративной ВЭЖХ (Способ А), получив 0,13 г трифторацетата 4-{3-[2-(4-амино-1H-имидазо[4,5-c]хинолин-1-ил)этокси]-пропин-1-ил}-тиофен-2-илкарбоксальдегида в виде рыхлого твердого вещества цвета слоновой кости с температурой плавления 195°С.
Данные элементного анализа: Рассчитано для C20H16N4O2S·С2HF3O2: С 53,88 %, Н 3,49 %, N 11,42 %. Найдено: С 54,16 %, Н 3,21 %, N 11,36 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6):δ (10-6) 9,874 (с, 1 Н); 8,972 (шс, 2 Н); 8,483 (с, 1 Н); 8,322 (д, J=7,9 Гц, 1 Н); 8,076 (с, 1 Н); 7,771 (д, J=8,3 Гц, 1 Н); 7,736 (с, 1 Н); 7,71 (т, J=8,4 Гц, 1 Н); 7,555 (т, J=6,9 Гц, 1 Н); 4,928 (т, J=5,3 Гц, 2 Н); 4,371 (с, 2 Н); 4,043 (т, J=4,8 Гц, 2 Н).
Пример 21
1-(2-{[3-(Пирид-2-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амина трифторацетат
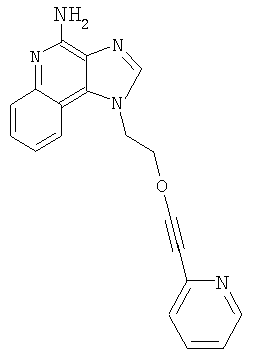
Пользуясь общим способом, описанным в Примере 19, провели реакцию между 1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амином (0,5 г, 1,9 ммоля) и 2-бромпиридином (0,51 г, 2,6 ммоля); продукт подвергли очистке способом полупрепаративной ВЭЖХ (Способ А), получив 0,1 г трифторацетата 1-(2-{[3-(пирид-2-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амина в виде рыхлого твердого вещества серого цвета с температурой плавления 129-131°С.
Данные элементного анализа: Рассчитано для C20H17N5O·1,752HFзO2·0,25Н2O: С 51,56 %, Н 3,55 %, N 12,80 %. Найдено: С 51,80 %, Н 3,20 %, N 13,11 %.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ (10-6) 9,013 (шс, 2 Н); 8,516 (с, 1 Н); 8,495 (с, 1 Н); 8,331 (д, J=8,2 Гц, 1 Н); 7,75 (м, 3 Н); 7,553 (т, J=8,2 Гц, 1 Н); 7,375 (дд, J=7,8; 4,9 Гц, 1 Н); 7,23 (д, J=7,8 Гц, 1 Н); 4,944 (т, J=5,4 Гц, 2 Н); 4,418 (с, 2 Н); 4,059 (т, J=4,8 Гц, 2 Н).
Примеры 22-26
Соединения, представленные в нижеследующей таблице, были получены по общему способу синтеза для схемы 1 реакции:
4-Амино-1H-имидазо[4,5-с]хинолин-1-иловый спирт (25 мг) помещали во флакон вместимостью 7,4 мл. Далее добавили гидрид натрия (1,2 экв. 60 %-ного продукта в минеральном масле) и N,N-диметилформамид (1 мл). Флакон поместили в ультразвуковой излучатель приблизительно на 15-30 мин при температуре окружающей среды; при этом образовывался алкоксид. Далее вводили галогенид (1,2 экв.) и флакон снова поместили в ультразвуковой излучатель приблизительно на 15-120 мин при температуре окружающей среды. Реакционную смесь анализировали способом жидкостной хроматографии с масс-спектрометрическим детектированием для подтверждения получения требуемого продукта. Реакционную смесь очищали способом полупрепаративной ВЭЖХ. Полученные при этом фракции анализировали способом жидкостной хроматографии (LC-APCI/MS). Соответствующие фракции соединяли и подвергали лиофильной сушке, при этом получили соль трифторацетата требуемого соединения; для подтверждения проводили точные масс-спектроскопические и 1H ЯМР-спектроскопические измерения. В нижеприведенной таблице показаны структура свободного основания, теоретическая масса (ТМ), измеренная масса (ИМ) или номинальная масса (НМ).
ИМ=326.1739
ИМ=392.1584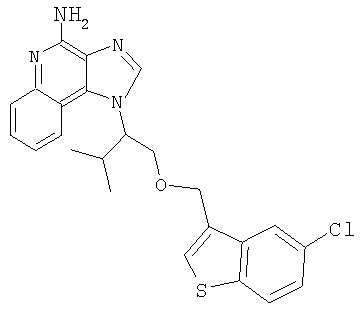
ИМ=450.1285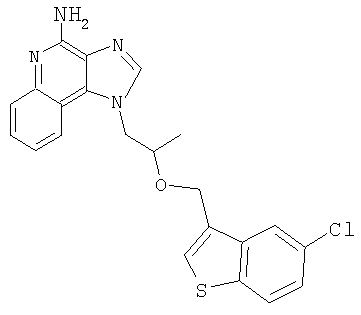
ИМ=422.0966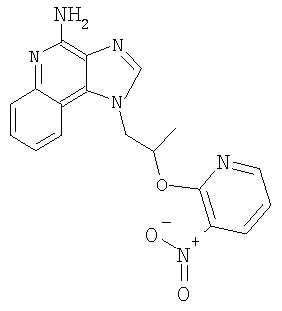
НМ[M+H]+l=365
Пример 27
1-{2-Метил-1-[(пирид-2-илокси)метил]пропил}-1H-имидазо[4,5-с]хинолин-4-амина трифторацетат
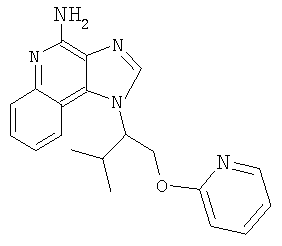
Пользуясь общим способом, описанным выше в Примерах 22-26, провели реакцию между 2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)-3-метилбутан-1-олом и 2-(трифторметилсульфонилокси)пиридином; продукт подвергли очистке способом полупрепаративной ВЭЖХ (Способ А) и получили трифторацетат 1-{2-метил-1-[(пирид-2-илокси)метил]пропил}-1 Н-имидазо[4,5-с]хинолин-4-амина. ТМ=347,1746, ИМ=347,1740.
Пример 28
1-{1-[(Пирид-2-илокси)метил]пропил}-1Н-имидазо[4,5-с]хинолин-4-амина трифторацетат
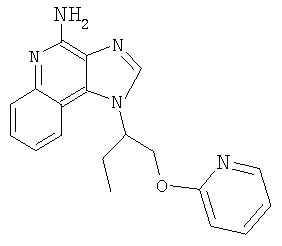
Пользуясь общим способом, описанным выше в Примерах 22-26, провели реакцию между 2-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)-3-метилбутан-1-олом и 2-(трифторметилсульфонилокси)пиридином; продукт подвергли очистке способом полупрепаративной ВЭЖХ (Способ В) и получили трифторацетат 1-{1-[(пирид-2-илокси)метил]пропил}-1Н-имидазо[4,5-с]хинолин-4-амина. ТМ=333,1590, ИМ=333,1598.
Пример 29
1-[2-(9Н-Карбазол-3-илокси)пропил]-1H-имидазо[4,5-с]хинолин-4-амина трифторацетат
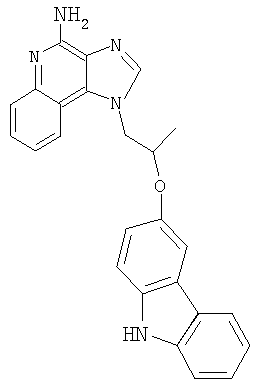
В стеклянный флакон вместимостью 7,4 мл, содержащий 2-гидроксикарбазол (38 мг, 2 экв.), ввели 1 мл раствора, приготовленного растворением 0,5 г 1-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)пропан-2-ола в N,N-диметилформамиде (20 мл). Далее во флакон добавили трифенилфосфин (54 мг, 2 экв.), растворенный в N,N-диметилформамиде (1 мл). Полученную взвесь подвергли ультразвуковому облучению с тем, чтобы растворить фенол. После этого добавили диэтилазидокарбоксилат (36 мг, 2 экв.). Реакционную смесь вновь подвергли ультразвуковому облучению в течение 30 мин, после чего система находилась всю ночь при температуре окружающей среды в аппарате для встряхивания. Растворитель удалили, а остаток подвергли очистке способом полупрепаративной ВЭЖХ (Способ А). Требуемое соединение получили в виде соли трифторацетата. ТМ=407, НМ(М+Н]+1=408.
Пример 30
1-{2-[(3-Тиен-2-илпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолин-4-амин
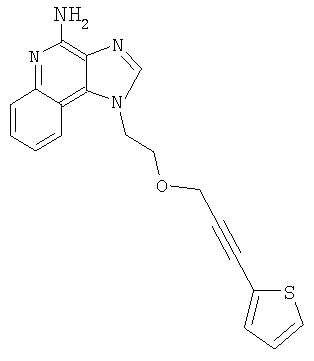
Часть А
Пользуясь общим способом, описанным в Примере 1, Часть В, 2(1 Н-имидазо[4,5-с]хинолин-1-ил)этил-(2-пропинил)овый эфир окислили, получив 67,5 г
1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-5N-оксида в виде твердого вещества желтовато-коричневого цвета.
Данные масс-спектрометрии MS (CI): Рассчитано для С15Н14N3O2: m/z 268 (MH+) 0,252, 214.
Часть В
В сухую круглодонную колбу загрузили магнитную мешалку, 1-[2-(2-пропинилокси)этил]-1Н-имидазо[4,5-с]хинолин-5N-оксид (57,5 г, 215,1 ммоля), безводный толуол (200 мл) и безводный диметилформамид (400 мл) в атмосфере азота. К смеси по каплям добавляли оксихлорид фосфора (23 мл, 247,4 ммолей) в течение 20 мин, что привело к некоторому тепловыделению (прибл. 40°С). Реакцию считали завершенной спустя 1,25 часа при температуре окружающей среды. Летучие вещества удалили при пониженном давлении, а оставшуюся твердую фазу распределили между хлороформом и 10 %-ным водным раствором карбоната натрия. Водный слой экстрагировали хлороформом; органические фракции соединяли, высушивали над безводным сульфатом магния, отфильтровывали и концентрировали при пониженном давлении. Полученное твердое вещество коричневого цвета высушивали при температуре 60°С в вакууме в течение 4 часов, получив 36,6 г 4-хлор-1-[2-(проп-2-инилокси)этил]-1Н-имидазо[4,5-с]хинолина в виде порошка.
Данные масс-спектрометрии MS (CI): Рассчитано для С15Н12ClN3О: m/z 286 (МН+), 246, 204.
Часть С
В сухую круглодонную колбу загрузили магнитную мешалку, гидрид натрия (8,15 г, 203,9 ммоль) и безводный 2-метоксилэтиловый эфир (диглим, 100 мл) в атмосфере азота при температуре окружающей среды. К смеси добавляли твердый компонент - фенол (20,7 г, 220,2 ммолей) - несколькими порциями; полученный раствор перемешивали в атмосфере азота до прекращения газовыделения. Далее добавили 4-хлор-1-[2-(проп-2-инилокси)этил]-1Н-имидазо[4,5-с]хинолин (46,6 г, 163,1 ммоля) и раствор нагрели до 110°С. Через 15,5 часов к реакционному раствору добавили заранее изготовленный раствор феноксида натрия (фенол - 5 г, 53,1 ммоля; гидрид натрия - 1,91 г, 47,8 ммоля) в диглиме (20 мл) и температуру повысили до 165°С. Реакция считается полностью завершенной через 1 час при температуре 165°С. После охлаждения до температуры ниже 70°С летучие вещества удалили при пониженном давлении, а полученное твердое вещество коричневого цвета распределили между хлороформом и насыщенным водным раствором карбоната натрия. Водную фракцию экстрагировали хлороформом, а соединенные органические фракции высушили над безводным сульфатом магния, отфильтровали и концентрировали при пониженном давлении, получив твердое вещество коричневого цвета. Это вещество рекристаллизовали из ацетонитрила, содержавшего небольшое количество диметилформамида, и получили 25 г 4-фенокси-1-[2-(проп-2-инилокси)этил]-1H-имидазо[4,5-с]хинолина в виде кристаллического вещества.
Данные масс-спектрометрии MS (CI): Рассчитано для C21H17N3O2: m/z 344 (МН+), 306, 288.
Часть D
Пользуясь общим способом, описанным в Примере 3, Часть А, провели реакцию между 4-фенокси-1-[2-(проп-2-инилокси)этил]-1Н-имидазо[4,5-с]хинолином (10 г, 29,4 ммоля) и 2-йодтиофеном (3,6 мл, 32,3 ммоля). Стеклообразный продукт, полученный при хроматографической очистке на силикагеле (дихлорметан-метанол 98:2), растерли с эфиром. Получили 5,3 г 4-фенокси-1-{2-(3-тиен-3-илпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолина в виде серого порошка.
Данные масс-спектрометрии MS (CI): Рассчитано для C25H19N3O2S: m/z426 (МН+), 306, 288.
Часть Е
4-Фенокси-1-{2-(3-тиен-3-илпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин (3,2 г, 7,52 ммоля) сплавили в атмосфере азота с ацетатом аммония (32 г, 415 ммолей) в сухой круглодонной колбе, нагретой до 150°С. Через 2 часа добавили дополнительное количество ацетата аммония (10 г, 129 ммолей). Реакцию считали завершенной в течение суммарного времени, равного 4 часам. Расплав охладили до температуры окружающей среды и довели рН до щелочного значения (прибл. 13), добавив 1 N водный раствор гидроксида калия. Водную смесь экстрагировали дихлорметаном (3 х); соединенные органические фракции промыли рассолом, высушили над безводным сульфатом магния, отфильтровали и сконцентрировали при пониженном давлении. Полученное твердое вещество очистили способом хроматографии на силикагеле (дихлорметан-метанол 98:2) и растерли с эфиром. Синтезировали 0,812 г 1-{2-[(3-тиен-2-илпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина в виде белого порошка с температурой плавления 148-150°С.
Данные элементного анализа: Рассчитано для C19H16N4OS: С 65,50 %, Н 4,63 %, N 16,08 %. Найдено: С 65,42 %, Н 4,65 %, N 16,11 %.
Данные 1H ЯМР (300 МГц, ДМСО): δ 8,16 (c, 1 Н); 8,11 (д, J=6,8 Гц, 1 Н); 7,60 (м, 2 Н); 7,43 (т, J=6,8 Гц, 1 Н); 7,20-7,25 (м, 2 Н); 7,04 (дд, J=4,9; 3,9 Гц, 1 Н); 6,58 (с, 2 Н); 4,84 (т, J=5,4 Гц, 2 Н); 4,41 (с, 2 Н); 3,99 (т, J=5,4 Гц, 2 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для C19H16N4OS: m/z 349 (МН+), 229, 185.
Пример 31
1-{2-[(1-метил-1Н-индол-2-ил)метокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин
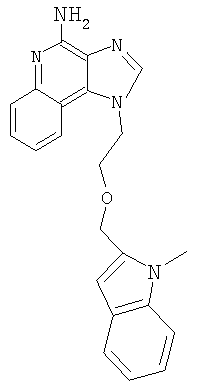
Часть А
Пользуясь общим способом, описанным в Примере 30, Часть D, провели реакцию между 4-фенокси-1-[2-(проп-2-инилокси)этил]-1H-имидазо[4,5-с]хинолином (3,16 г, 9,20 ммоля) и 2-йод-N,N-диметиланилином (2,5 г, 10,1 ммоля), получив 1,0 г 1-{2-[(1-метил-1Н-индол-2-ил)метокси]этил}-4-фенокси-1H-имидазо[4,5-с]хинолина в виде твердого кристаллического вещества бледно-желтого цвета.
Данные масс-спектрометрии MS (CI): Рассчитано для C28H24N4O2: m/z 449 (MH+), 306, 186.
Часть В
1-{2-[(1-Метил-1Н-индол-2-ил)метокси]этил}-4-фенокси-1Н-имидазо[4,5-с]хинолин (0,78 г, 1,74 ммоля) частично превратили в требуемый продукт путем воздействия аммиаком (7 % в метаноле, 20 мл) в течение 52 часов при температуре 160°С в бомбе. Летучие вещества удалили при пониженном давлении; в систему добавили дополнительное количество аммиака (7 % в метаноле, 20 мл) для полного израсходования исходного вещества; реакция продолжалась еще 80 часов при температуре 160°С. Твердое вещество удалили фильтрованием, а фильтрат концентрировали при пониженном вакууме. Полученный продукт очищали способом хроматографии на силикагеле (дихлорметан-метанол 9:1). После хроматографии провели рекристаллизацию этого твердого вещества из диметилформамида, получив 0,121 г 1-{2-[(1-метил-1Н-индол-2-ил)метокси]этил}-1H-имидазо[4,5-с]хинолин-4-амина в виде плоских белых кристаллов с температурой плавления 243-245°С.
Данные элементного анализа: Рассчитано для С22Н21N5O(С3Н7ON)0,20: С 70,50 %, Н 5,81 %, N 18,75 %. Найдено: С 70,72 %, Н 5,70 %, N 18,36 %.
Данные 1H ЯМР (300 МГц, ДМСО): δ 8,13 (с, 1 Н); 8,05 (д, J=8,3 Гц, 1 Н); 7,60 (д, J=9,3 Гц, 1 Н); 7,36-7,47 (м, 3 Н); 7,10-7,20 (м, 2 Н); 6,98 (т, J=7,3 Гц, 1 Н); 6,58 (шс, 2 Н); 6,36 (с, 1 Н); 4,82 (т, J=4,9 Гц, 2 Н); 4,64 (с, 2 Н); 3,92 (т, J=4,9 Гц, 2 Н); 3,52 (с, 3 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для C22H21N5O: m/z 372 (МН+), 229, 144.
Пример 32
1-{2-[(3-тиен-2-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
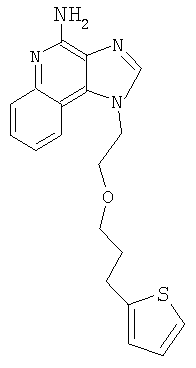
Часть А
В сухую круглодонную колбу загрузили магнитную мешалку, 2-(1Н-имидазо[4,5-с]хинолин-1-ил)этил-(2-пропинил)овый эфир (11,78 г, 46,88 ммолей), безводный триэтиламин (14 мл, 121,9 ммоля), 2-йодтиофен (5,7 мл, 51,57 ммоля) и безводный диметилформамид (130 мл) в атмосфере азота и нагрели смесь до 80°С. Через 5 мин сразу добавили дихлорбис(трифенилфосфин)палладий (II) (0,658 г, 0,937 моля) и йодид меди (I) (0,357 г, 1,875 ммоля). Считали, что реакция завершилась через 50 мин. Летучие вещества удалили при пониженном давлении, а остающуюся твердую фазу распределили между дихлорметаном и 0,5 N водным раствором гидроксида калия. Водную фракцию экстрагировали дихлорметаном (3 х); соединенные органические фракции высушивали над безводным сульфатом магния, отфильтровывали и концентрировали при пониженном давлении, получив твердое вещество коричневого цвета. В результате очистки способом хроматографии на силикагеле (элюент: смесь дихлорметан-метанол 98:2) образовалось стеклообразное твердое вещество. После растирания с эфиром получили 9,5 г 1-{2-[(3-тиен-2-илпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолина в виде твердого вещества желтовато-коричневого цвета.
Данные масс-спектрометрии MS (CI): Рассчитано для C19H15N3OS: m/z 334 (MH+), 290, 214.
Часть В
Пользуясь общим способом, описанным в Примере 3, Часть В, восстановили 1-{2-[(3-тиен-2-илпроп-2-инил)окси]этил}-1H-имидазо[4,5-с]хинолин (9,5 г, 28,49 ммоля) палладием на углероде (10 %, 1 г) в среде метанола (25 мл), получив 9,1 г 1-[2-(3-тиен-2-илпропокси)этил]-1Н-имидазо[4,5-с]хинолина в виде маслянистой жидкости коричневого цвета.
Данные масс-спектрометр и и MS (CI): Рассчитано для C19H19N3OS: m/z 338 (MH+), 214.
Часть С
Пользуясь общим способом, описанным в Примере 1, Часть В, окислили 1-[2-(3-тиен-2-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин, получив 4,4 г неочищенного 1-[2-(3-тиен-2-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-5N-оксида в виде неочищенного твердого вещества желтовато-коричневого цвета.
Данные масс-спектрометрии MS (CI): Рассчитано для C19H19N3O2S: m/z 354 (MH+), 338, 214.
Часть D
Пользуясь общим способом, описанным в Примере 1, Часть С, провели реакцию между 1-[2-(3-тиен-2-илпропокси)этил]-1H-имидазо[4,5-с]хинолин-5N-оксидом (4,4 г, 12,45 ммоля) и трихлорацетилизоцианатом (1,8 мл, 14,9 ммоля), получив неочищенный 2,2,2-трихлор-N-{1-[2-(3-тиен-2-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-ил}ацетамид в виде неочищенного стеклообразного вещества.
Часть Е
В сухую круглодонную колбу загрузили магнитную мешалку, 2,2,2-трихлор-N-{1-[2-(3-тиен-2-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-ил}ацетамид и метоксид натрия (25 % в метаноле, 11 мл, 49,8 ммоля) при температуре окружающей среды. Считали, что реакция завершилась через 30 часов. Летучие вещества удалили при пониженном давлении, а образовавшуюся маслянистую жидкость очистили способами хроматографии на силикагеле (элюент: смесь дихлорметан-метанол 95:5), полупрепаративной ВЭЖХ (Способ А) и рекристаллизации из смеси этилацетат-гексан, получив 43 мг 1-[2-(3-тиен-2-илпропокси)этил]-1H-имидазо[4,5-с]хинолин-4-амина в виде белого кристаллического вещества с температурой плавления 130,1-131,6°С.
Данные элементного анализа: Рассчитано для C19H20N4OS·(Н2O)0,30: С 63,77 %, Н 5,80 %, N 15,66 %. Найдено: С 63,84 %, Н 5,79 %, N 15,57 %.
Данные 1H ЯМР (300 МГц, ДМСО): δ 8,16 (с, 1 Н); 8,12 (д, J=8,3 Гц, 1 Н); 7,62 (д, J=8,3 Гц, 1 Н); 7,43 (т, J=7,3 Гц, 1 Н); 7,20-7,26 (м, 2 Н); 6,84 (дд, J=4,9; 3,4 Гц, 1 Н); 6,62 (д, J=2,4 Гц, 1 Н); 6,58 (с, 2 Н); 4,79 (т, J=5,4 Гц, 2 Н); 3,83 (т, J=5,4 Гц, 2 Н); 3,35 (т, J=6,4 Гц, 2 Н); 2,64 (т, J=7,8 Гц, 2 Н); 1,69 (п, J=6,8; 6,3 Гц, 2 Н).
Данные масс-спектрометрии MS (CI): Рассчитано для C19H20N4OS: m/z 353 (МН+), 211,185.
Пример 33
1-[2-(3-Пиридин-3-илпропокси)этил]-1H-имидазо[4,5-с]хинолин-4-амин
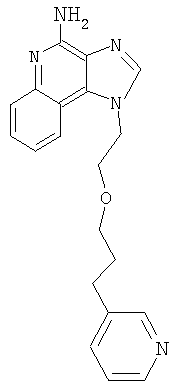
Часть А
В атмосфере азота раствор 2-аминобензойной кислоты (100,0 г, 0,73 моля) в уксусном ангидриде (400 мл, 4,2 моля) нагревали с обратным холодильником в течение 2 часов. Реакционную смесь охладили до комнатной температуры и удалили растворитель вакуумной обработкой. Остаток растворили в ледяной уксусной кислоте (500 мл), после чего добавили NaN3 (49,77 г, 0,77 моля). Смесь оставили на ночь при перемешивании (комнатная температура), после чего уксусную кислоту удалили вакуумной обработкой. Остаток растворили в 10 %-ном растворе NaOH (500 мл) и нагревали с обратным холодильником в течение 3,5 часов. Реакционную смесь охладили до комнатной температуры и затем вылили в смесь ледяной воды (2 л) и HCl (150 мл). Образовавшийся твердый осадок белого цвета собрали вакуумной фильтрацией. Осадок высушили в вакууме, получив 130,5 г 2-(5-метил-1H-тетразол-1-ил)бензойной кислоты.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,12 (д, J=7,2 Гц, 1 Н); 7,91-7,78 (м, 2 Н); 7,72 (д, J=7,4 Гц, 1 Н); 2,37 (с, 3 Н).
Данные масс-спектрометрии MS (Cl): m/e 205 (МН+), 162 (М-N3).
Часть В
В атмосфере азота растворили 2-(5-метил-1Н-тетразол-1-ил)бензойную кислоту (89,7 г, 0,44 моля) в ацетоне (1 л), после чего добавили карбонат цезия (214,7 г, 0,66 моля) при энергичном перемешивании. Далее по каплям ввели этилйодид (70,3 мл, 0,88 моля) и реакционную смесь нагревали с обратным холодильником в течение 4 часов. Реакционную смесь отфильтровали после ее охлаждения до комнатной температуры. Ацетон удалили при пониженном давлении и получили твердое вещество желтого цвета, которое далее растворили в дихлорметане (800 мл) и промыли насыщенным раствором бикарбоната натрия (200 мл). Органическую фракцию высушили над сульфатом натрия (Na2SO4), отфильтровали и концентрировали, получив 92,7 г этил-2-(5-метил-1Н-тетразол-1-ил)бензоата в виде твердого вещества светло-желтого цвета.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,14 (д, J=7,8 Гц, 1 Н); 7,89 (м, 2Н); 7,79 (д, J=7,9 Гц, 1 Н); 4,08 (к, J=7,4 Гц, 2 Н); 2,40 (с, 3 Н); 1,04 (т, J=6,9 Гц, 3 Н).
Данные масс-спектрометрии MS (Cl): m/e 233 (МН+), 159.
Часть С
В атмосфере азота растворили этил-2-(5-метил-1Н-тетразол-1-ил)бензоат (92,7 г, 0,34-моля) в N,N-диметилформамиде (600 мл); раствор охладили в бане с ледяной водой. К раствору медленно добавили этоксид калия (67,2 г, 0,80 моля). Спустя несколько минут баню с ледяной водой удалили и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Далее в реакционную смесь добавили 100 мл воды, после этого приблизительно 300-400 мл растворителя удалили в вакууме. Остаток реакционной смеси вылили в раствор ледяной уксусной кислоты (125 мл) в ледяной воде (2 л). Образовавшийся осадок и смесь разбавили дополнительным количеством воды (3 л). Твердую фазу отделили вакуумным фильтрованием, получив 63,25 г тетразоло[1,5-а]хинолин-5-ола в виде твердого вещества желтого цвета.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,54 (д, J=8,4 Гц, 1 Н); 8,27 (д, J=8,1 Гц, 1 Н); 7,99 (т, J=7,4 Гц, 1 Н); 7,80 (т, J=7,2 Гц, 1 Н); 7,04 (с, 1 Н). Данные масс-спектрометрии MS (Cl): m/e 187 (МН+), 159.
Часть D
Тетразоло[1,5-а]хинолин-5-ол (63,25 г, 0,34 моля) добавили к ледяной уксусной кислоте (630 мл); образовалась густая суспензия почти белого цвета. Смесь энергично перемешивали при медленном добавлении азотной кислоты (23,6 мл, 0,37 моля, 70 %-ный раствор). Далее реакционную смесь нагрели с 25 до 80°С в течение 15 мин. Образовался желтый осадок; реакцию продолжали при температуре 80°С в течение 5 мин. Смесь медленно охладили до 0°С. Твердую фазу отделили фильтрованием и высушили под вакуумом, получив 60,0 г 4-нитро-тетразоло[1,5-а]хинолин-5-ола в виде твердого вещества желтого цвета.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,35 (д, J=3,9 Гц, 1 Н); 8,32 (д, J=3,2 Гц, 1 Н); 7,90 (т, J=7,3 Гц, 1 Н); 7,68 (т, J=8,2 Гц, 1 Н).
Данные масс-спектрометрии MS (CI): m/e 232 (МН+), 204.
Часть Е
В атмосфере азота медленно ввели POCl3 (16,42 мл, 0,17 моля) в охлажденную до 0°С колбу, содержащую N,N-диметилформамид (100 мл). Полученный раствор медленно нагрели до комнатной температуры, после чего его стали по каплям добавлять к суспензии 4-нитро-тетразоло[1,5-а]хинолин-5-ола в N,N-диметилформамиде (300 мл). Реакционную смесь нагревали до температуры 100°С в течение 30 мин. Полученный оранжево-красный раствор быстро охладили, влив в него 1 л ледяной воды. Образовавшийся желтый осадок отделили фильтрованием, растворили в хлороформе (прибл. 750 мл), высушили над Na2SO4, отфильтровали и концентрировали в вакууме, получив 33,74 г 5-хлор-4-нитротетразоло[1,5-а]хинолина в виде твердого вещества желтого цвета.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,78 (д, J = 8,2 Гц, 1 Н); 8,57 (д, J = 8,3 Гц, 1 Н); 8,29 - 8,22 (м, 1 Н); 8,09 - 8,03 (м, 1 Н).
Данные масс-спектрометрии MS (CI): m/e 250 (МН+).
Часть F
Смешали 5-хлор-4-нитротетразоло[1,5-а]хинолин (28,86 г, 0,11 моля), дихлорметан (600 мл) и триэтиламин (21,14 мл, 0,11 моля) и полученный раствор охладили до 0°С. К раствору добавили по каплям 2-(3-пиридин-3-илпропокси)этиламин (22,9 г, 0,13 моля). Смесь оставили медленно нагреваться до комнатной температуры, после чего ее перемешивали при комнатной температуре в течение 1 часа и далее нагревали с обратным холодильником в течение 2 часов. Реакционную смесь охладили до комнатной температуры и добавили холодной воды (200 мл). После разделения фаз водный слой экстрагировали дихлорметаном (3 х 50 мл). Соединенные органические фракции промыли рассолом (100 мл), высушили над Na2SO4, отфильтровали и концентрировали, получив твердое вещество желтого цвета. Это вещество суспендировали в этаноле (150 мл) и отфильтровали. Получили 34,3 г 4-нитро-N-[2-(3-пиридин-3-илпропокси)этил]тетразоло[1,5-а]хинолин-5-амина.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 10,27 (шс, 1 Н); 8,69 (д, J=8,3 Гц, 1 Н); 8,54 (д, J=8,3 Гц, 1 Н); 8,37 (шс, 2 Н); 8,08 (т J=7,7 Гц, 1 Н); 7,81 (т, J=7,2 Гц, 1 Н); 7,57 (д, J=7,3 Гц, 1 Н); 7,27 (дд, J=7,9; 5,0 Гц, 1 Н); 3,92 (м, 2 Н); 3,71 (т, J=5,4 Гц, 2 Н); 3,47 (т, J=6,0 Гц, 2 Н); 2,62 (т, J=7,4 Гц, 2 Н); 1,82 (м, 2 Н).
Данные масс-спектрометрии MS (CI): m/e 394 (MH+), 366.
Часть G
4-Нитро-N-[2-(3-пиридин-3-илпропокси)этил]тетразоло[1,5-а]хинолин-5-амин (34,3 г, 87,2 ммоля) ввели в автоклав вместимостью 3 л, изготовленный из нержавеющей стали; в автоклаве содержался этанол (1,25 л абсолютного спирта). Добавили платину на углероде (3,00 г, 5 % масс.) и автоклав поместили в аппарат Парра для гидрогенизации. Реакционную смесь встряхивали в течение 24 часов в атмосфере водорода (3,15 бар). Далее катализатор отделили фильтрованием через слой целита (Celite™); слой целита несколько раз промыли этанолом. Фильтрат концентрировали в вакууме, получив 30,8 г N5-[2-(3-пиридин-3-илпропокси)этил]тетразоло[1,5-а]хинолин-4,5-диамина в виде маслянистой жидкости оранжево-красного цвета.
Данные масс-спектрометрии MS (CI): m/e 364 (MH+), 336.
Часть Н
Триэтилортоформиат (21,1 мл, 127 ммолей) добавили к раствору N5-[2-(3-пиридин-3-илпропокси)этил]тетразоло[1,5-а]хинолин-4,5-диамина (30,8 г, 84,7 ммоля) в 1,2-дихлорэтане (750 мл); реакционную смесь нагревали с обратным холодильником в течение 3 часов. Реакционную смесь охладили до комнатной температуры и разбавили насыщенным раствором бикарбоната натрия (200 мл). После разделения фаз водный слой экстрагировали дихлорметаном (3×75 мл). Соединенные органические фракции промыли рассолом (200 мл), высушили над Na2SO4, концентрировали, получив твердое вещество оранжевого цвета. Это вещество растерли в диэтиловом эфире и отфильтровали, получив 28,7 г 6-[2-(3-пиридин-3-илпропокси)этил]-6Н-имидазо[4,5-с]тетразоло[1,5-а]хинолина в виде твердого вещества желтовато-коричневого/оранжевого цвета.
Данные 1H ЯМР (300 МГц, CDCl3): δ 8,71 (дд, J=8,1; 1,3Гц, 1 Н); 8,38 (дд, J=4,8; 1,5 Гц, 1 Н); 8,30 (д, J=2,1 Гц, 1 Н); 8,20 (д, J=7,7 Гц, 1 Н); 8,07 (с, 1 Н); 7,73 (м, 2 Н); 7,32 (дт, J=7,8; 1,9 Гц, 1 Н); 7,13 (дд, J=7,7; 4,8 Гц, 1 Н); 4,81 (т, J=5,1 Гц, 2 Н); 3,96 (т, J=5,1 Гц, 2 Н); 3,42 (т, J=6,2 Гц, 2 Н); 2,52 (т, J=7,5 Гц, 2 Н); 1,82-1,74 (м, 2 Н).
Данные масс-спектрометрии MS (CI): m/e 374 (MH+).
Часть I
Трифенилфосфин (27,0 г, 115 ммолей) добавили к раствору 6-[2-(3-пиридин-3-илпропокси)этил]-6Н-имидазо[4,5-с]тетразоло[1,5-а]хинолина (28,7 г, 76,9 ммоля) в 1,2-дихлорбензоле (1 л). Смесь нагревали с обратным холодильником в течение ночи. Полученный темно-красный раствор охладили до комнатной температуры и обработали 1N HCl (225 мл). Образовался осадок желтовато-коричневого цвета. Смесь концентрировали под вакуумом, получив темно-красное/коричневое твердое вещество. К данному веществу прибавили 500 мл воды и смесь энергично перемешивали. В результате избытка трифенилфосфина и оксида трифенилфосфина образовался осадок, который удалили фильтрованием под вакуумом. Твердую фазу промыли несколькими порциями воды, и в заключение - разбавленной HCl (смесь 1 N HCl - вода 1:5). Коричнево-красный фильтрат собрали, промыли эфиром (3 х 150 мл) и обрабатывали 10 %-ным раствором NaOH до достижения рН 12. Продукт, выделившийся в виде осадка желтовато-коричневого цвета, отделили фильтрованием. Очистку продукта выполнили путем обработки (2 х) активированным углем (Darco-G60) в метаноле с обратным холодильником. Активированный уголь отделили фильтрованием. Требуемый продукт образовался в виде осадка при концентрировании фильтрата. Твердый продукт отделили путем вакуумного фильтрования, промыли диэтиловым эфиром и высушили под вакуумом, получив 17 г 1-[2-(3-пиридин-3-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества желтовато-коричневого цвета с температурой плавления 125,0-128,0°С.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,33 (д, J=4,8 Гц, 1 Н); 8,24 (с, 1 Н); 8,18 (с, 1 Н); 8,13 (д, J=7,7 Гц, 1 Н); 7,63 (д, J=8,4 Гц, 1 Н); 7,45 (т, J=6,8 Гц, 1 Н); 7,33-7,21 (м, 2 Н); 7,16 (дд, J=7,7; 4,8 Гц, 1 Н); 6,62 (с, 2 Н); 4,80 (т, J=4,8 Гц, 2 Н); 3,82 (т, J=4,9 Гц, 2 Н); 3,30 (т, J=6,3 Гц, 2 Н); 2,39 (т, J=7,3 Гц, 2 Н); 1,64 (м, 2Н).
Данные масс-спектрометр и и MS (Cl): m/e 348 (МН+).
Данные элементного анализа: Рассчитано для С20Н21N5O·0,08H2O: С 68,89 %, Н 6,11 %, N 20,09 %. Найдено: С 68,49 %, Н 5,95 %, N 20,08 %.
Пример 34
2-Метил-1-[2-(3-пиридин-3-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
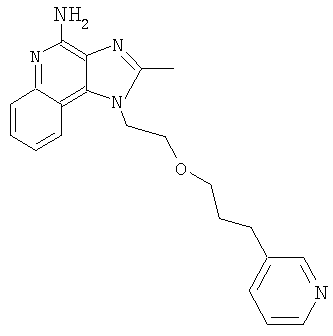
Часть А
В атмосфере азота растворили N5-[2-(3-пиридин-3-илпропокси)этил]тетразоло[1,5-а]хинолин-4,5-диамин (0,70 г, 1,92 ммоля) в 1,2-дихлорэтане (15 мл). Через шприц добавили триэтилортоацетат (0,53 мл, 2,88 ммоля и реакционную смесь подвергли нагреву с обратным холодильником в течение 3 часов. Анализ способом тонкослойной хроматографии (смесь хлороформ-метанол 95:5) показал, что диамин прореагировал полностью.
Реакцию остановили путем добавления воды (15 мл). После разделения фаз водную фракцию экстрагировали дихлорметаном (3х10 мл). Соединенные органические фракции промыли рассолом (15 мл), высушили над Na2SO4, отфильтровали и концентрировали под вакуумом, получив 0,73 г 5-метил-6-[2-(3-пиридин-3-илпропокси)этил]-6Н-имидазо[4,5-с]тетразоло[1,5-а]хинолина в виде маслянистой жидкости красного цвета. Этот продукт использовали без дальнейшей очистки.
Данные масс-спектрометрии MS (CI): m/e 388 (М+Н).
Часть В
В атмосфере азота растворили 5-метил-6-[2-(3-пиридин-3-илпропокси)этил]-6Н-имидазо[4,5-с]тетразоло[1,5-а]хинолин (0,73 г, 1,89 ммоля) и трифенилфосфин (0,64 г, 2,84 ммоля) в 1,2-дихлорбензоле (15 мл). Реакционную смесь нагревали с обратным холодильником в течение 18 часов. После охлаждения до температуры окружающей среды растворитель удалили под вакуумом. Остаток обработали смесью 1 N HCl-вода (30 мл); при энергичном перемешивании получили суспензию почти белого цвета. Твердую фазу отобрали путем фильтрования. Оставшийся желтый фильтрат обрабатывали 10 %-ным раствором гидроксида натрия в воде до достижения рН 11. Далее фильтрат экстрагировали дихлорметаном (3 х 25 мл). Соединенные органические фракции промыли рассолом (25 мл), высушили над Na2SO4, отфильтровали и концентрировали под вакуумом, получив маслянистую жидкость оранжевого цвета. Жидкость растворили в минимальном количестве дихлорметана и разбавили эфиром, что привело к образованию осадка. Твердый продукт рекристаллизовали из н-пропилацетата, получив 0,16 г 2-метил-1-[2-(3-пиридин-3-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества желтовато-коричневого цвета с температурой плавления 145,0-146,0°С.
Данные 1H ЯМР (300 МГц, CDCl3): δ 8,40 (д, J=4,9 Гц, 1 Н); 8,34 (д, J=2,0 Гц, 1 Н); 7,93 (д, J=7,4 Гц, 1 Н); 7,84 (д, J=8,4 Гц, 1 Н); 7,52 (т, J=7,2 Гц, 1 Н); 7,31 (т, J=6,6 Гц, 1 Н); 7,22 (д, J=7,8 Гц, 1 Н); 7,09 (дд, J=4,8; 7,8 Гц, 1 Н); 5,46 (шс, 2 Н); 4,67 (т, J=5,3 Гц, 2 Н); 3,90 (т, J=5,3 Гц, 2 Н); 3,34 (т, J=6,2 Гц, 2 Н); 2,70 (с, 3 Н); 2,52 (т, J=8,0 Гц, 2 Н); 1,76 (м, 2 Н).
Данные масс-спектрометрии MS (Cl): m/e 362 (М+Н).
Данные элементного анализа: Рассчитано для C21H23N5O: С 69,78 %, Н 6,41 %, N 19,38%. Найдено: С 69,40 %, Н 6,38 %, N 19,00 %.
Пример 35
2-Бутил-1-[2-(3-пиридин-3-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
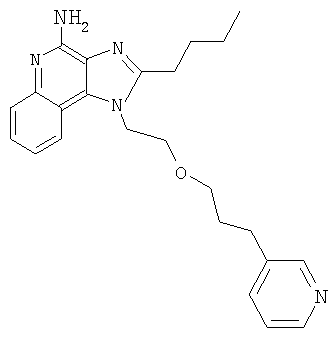
Часть А
В атмосфере азота растворили N5-[2-(3-пиридин-3-илпропокси)этил]тетразоло[1,5-а]хинолин-4,5-диамин (2,48 г, 6,82 ммоля) в толуоле (40 мл). Через шприц добавили триметилортовалерат (1,29 мл, 7,51 ммоля). В реакционную смесь ввели каталитическое количество гидрохлорида пиридина; колбу снабдили ловушкой Дина-Старка. Смесь нагревали с обратным холодильником; летучие вещества собирали в ловушке. Через 4 часа реакционную смесь охладили до комнатной температуры; реакцию остановили путем добавления воды (30 мл). После разделения фаз водную фазу экстрагировали этилацетатом (3×15 мл). Соединенные органические фракции промыли рассолом (25 мл), высушили над Na2SO4, отфильтровали и концентрировали под вакуумом, получив маслянистую жидкость красно-коричневого цвета. Жидкость очистили способом высокоскоростной хроматографии на колонке (силикагель; градиент смеси этилацетат-гексан от 2:1 до 95:5), получив 1,98 г 2-бутил-6-[2-(3-пиридин-3-илпропокси)этил]-6H-имидазо[4,5-с]тетразоло[1,5-а]хинолина в виде маслянистого вещества оранжевого цвета.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,53 (д, J=8,2 Гц, 1 Н); 8,41 (д, J=7,9 Гц, 1 Н); 8,37 (д, J=5,0 Гц, 1 Н); 8,31 (д, J=1,9 Гц, 1 Н); 7,84 (д, J= 7,2 Гц, 1 Н); 7,75 (т, J=7,5 Гц, 1 Н); 7,48 (д, J=7,9 Гц, 1 Н); 7,23 (дд, J=7,8; 4,9 Гц, 1 Н); 3,63 (т, J=5,0 Гц, 2 Н); 3,56 (т, J=4,9 Гц, 2 Н); 3,36 (т, J=6,3 Гц, 2 Н); 2,51 (м, 2 Н); 2,12 (т, J=7,7 Гц, 2 Н); 1,73 (пентет, J=7,4 Гц, 2 Н); 1,45 (пентет, J=7,7 Гц, 2 Н); 1,12 (м, 2 Н); 0,68 (т, J=7,4 Гц, 3 Н).
Данные масс-спектрометрии MS (CI): m/e 430 (М+Н).
Часть В
2-Бутил-6-[2-(3-пиридин-3-илпропокси)этил]-6H-имидазо[4,5-с]тетразоло[1,5-а]хинолин (1,98 г, 4,61 ммоля) подвергли общей процедуре обработки, описанной в Примере 35. При рекристаллизации из изопропанола получили 1,09 г 2-бутил-1-[2-(3-пиридин-3-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества бежевого цвета.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,33 (д, J=4,3 Гц, 1 Н); 8,24 (с, 1 Н); 8,08 (д, J=8,7 Гц, 1 Н); 7,61 (д, J=8,0 Гц, 1 Н); 7,41 (т, J=7,7 Гц, 1 Н); 7,30-7,20 (м, 2 Н); 7,15 (дд, J=7,4; 4,8 Гц, 1 Н); 6,44 (шс, 2 Н); 4,74 (т, J=5,4 Гц, 2 Н); 3,82 (т, J=5,3 Гц, 2 Н); 3,27 (т, J=5,9 Гц, 2 Н); 2,97 (т, J=7,4 Гц, 2 Н); 2,41 (т, J=7,5 Гц, 2 Н); 1,84 (пентет, J=7,4 Гц, 2 Н); 1,64 (пентет, J=7,2 Гц, 2 Н); 1,46 (м, 2 Н); 0,95 (т, J=7,3 Гц, 3 Н).
Данные масс-спектрометр и и MS (Cl): m/e 404 (М + Н).
Данные элементного анализа: Рассчитано для C24H29N5O: С 71,44 %, Н 7,24 %, N 17,36%. Найдено: С 71,23 %, Н 6,98 %, N 17,05 %.
Пример 36
2-(2-Метоксиэтил)-1-[2-(3-пиридин-3-илпропокси)этил]-1H-имидазо[4,5-с]хинолин-4-амин
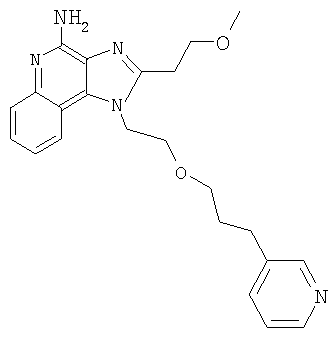
Часть А
В атмосфере азота смешали N5-(3-пиридин-3-илпропокси)этил]тетразоло[1,5-а]хинолин-4,5-диамин (2,48 г, 6,82 ммоля), 1,2-дихлорэтан (30 мл) и триэтиламин (1,14 мл, 8,2 ммоля). Смесь охладили в бане с ледяной водой. Далее в раствор по каплям добавили 3-метоксипропионилхлорид (0,92 г, 7,5 ммолей). Баню убрали и реакционную смесь перемешивали в течение 18 часов. Реакцию затормозили путем добавления воды (30 мл). После разделения фаз водную фазу экстрагировали дихлорметаном (2×15 мл). Соединенные органические фракции промыли рассолом (20 мл), высушили над Na2SO4, отфильтровали и концентрировали, получив 3,16 г сиропообразной жидкости оранжевого цвета. Анализ продукта способом жидкостной хроматографии с масс-спектрометрическим детектором указал на наличие смеси моно- и биацилированного вещества. Это вещество использовали без последующей очистки.
Часть В
В атмосфере азота смешали продукт Части А (3,16 г), толуол (40 мл) и пиридингидрохлорид (50 мг, 0,4 ммоля). Смесь нагревали с обратным холодильником в течение 4 часов. Летучие вещества собирали в ловушке Дина-Старка. Реакционную смесь охладили до температуры окружающей среды и далее разбавили водой (30 мл). После разделения фаз водную фазу экстрагировали дихлорметаном (3×20 мл). Соединенные органические фракции промыли рассолом (20 мл), высушили над Na2SO4, отфильтровали и концентрировали, получив пену оранжевого цвета. Продукт очистили способом хроматографии на колонке (силикагель; градиент CHCl3-МеОН до 9:1) и рекристаллизовали из 2-пропанола, получив 0,35 г 5-(2-метоксиэтил)-6-[2-(3-пиридин-3-илпропокси)этил]-6Н-имидазо[4,5-с]тетразоло[1,5-а]хинолина в виде твердого вещества бежевого цвета.
Часть С
5-(2-Метоксиэтил)-6-[2-(3-пиридин-3-илпропокси)этил]-6Н-имидазо[4,5-с]тетразоло[1,5-а]хинолин (0,35 г, 0,80 ммоля) обработали трифенилфосфином (0,28 г, 1,20 ммоля), пользуясь общей процедурой, описанной в Части В Примера 35. Неочищенный продукт кристаллизовали из эфира, получив 90 мг 2-(2-метоксиэтил)-1-[2-(3-пиридин-3-илпропокси)этил]-1H-имидазо[4,5-с]хинолин-4-амина в виде кристаллов почти белого цвета.
Данные 1H ЯМР (300 МГц, CDCl3): δ 8,39 (дд, J=4,8; 1,5 Гц, 1 Н); 8,33 (д, J=1,8 Гц, 1 Н); 7,94 (д, J=7,4 Гц, 1 Н); 7,83 (д, J=8,3 Гц, 1 Н); 7,52-7,47 (м, 1 Н); 7,32-7,21 (м, 2 Н); 7,09 (дд, J=7,0; 4,8 Гц, 1 Н); 5,59 (шс, 2 Н); 4,74 (т, J=5,4 Гц, 2 Н); 3,90 (т, J=6,6 Гц, 2 Н); 3,87 (т, J=5,4 Гц, 2 Н); 3,38 (с, 3 Н); 3,33 (т, J=6,1 Гц, 2 Н); 3,28 (т, J=6,6 Гц, 2 Н); 2,51 (т, J=7,4 Гц, 2 Н); 1,81-1,71 (м, 2 Н).
Данные 13С ЯМР (75 МГц, CDCl3): δ 152,4; 151,6; 150,2; 147,8; 145,2; 137,1; 136,2; 133,7; 127,7; 127,4; 123,6; 122,5; 120,0; 115,9; 71,2; 70,6; 69,6; 59,4; 46,1; 31,1:29,6; 28,7.
Данные масс-спектрометрии MS (CI): m/e 406,2242 (рассчитано для C23H28N5O2 406,2243, М+Н).
Пример 37
2-Метил-1-[2-(3-пиридин-3-илпропокси)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
В круглодонную колбу вместимостью 200 мл загрузили 3-нитро-5,6,7,8-тетрагидрохинолин-2,4-диол (10 г, 0,048 моля) и оксихлорид фосфора (100 мл, 1,07 моля, 22 экв.). Реакционную смесь нагрели до 80°С и перемешивали при этой температуре в течение 6 часов. Реакцию остановили путем медленного вливания реакционной смеси в воду (1500 мл). Далее провели экстракцию дихлорметаном (4 х 100 мл). Органические фракции соединили, высушили над сульфатом магния и концентрировали, получив 2,4-дихлор-3-нитро-5,6,7,8-тетрагидрохинолин в виде твердого вещества светло-коричневого цвета (10,6 г, 91 %) с температурой плавления 63-64°С. Данные тонкослойной хроматографии (10 % MeOH/CH2Cl2): Rf=0,84.
Часть В
В круглодонную колбу вместимостью 200 мл загрузили 2,4-дихлор-3-нитро-5,6,7,8-тетрагидрохинолин (10 г, 0,04 моля), триэтиламин (6,1 г, 0,06 моля, 1,5 экв.) и безводный N,N-диметилформамид (100 мл). К этому раствору добавили 2-(3-пиридин-3-илпропокси)этанамин (7,3 г, 0,04 моля). Реакционную смесь нагрели до 55°С и перемешивали при этой температуре в течение ночи. Реакцию остановили путем вливания реакционной смеси в воду (1000 мл). Далее провели экстракцию смесью гексан-этилацетат (1:1) (4×200 мл). Органические фракции соединили, промыли рассолом (300 мл) и концентрировали, получив 2-хлор-3-нитро-N-[2-(3-пиридин-3-илпропокси)этил]-5,6,7,8-тетрагидрохинолин-4-амин в виде сиропообразной жидкости оранжевого цвета (14,8 г, 94 %). Данные тонкослойной хроматографии (10 % MeOH/CH2Cl2): Rf=0,84.
Часть С
В круглодонную колбу вместимостью 200 мл загрузили 60 %-ный гидрид натрия (2,5 г, 0,06 моля, 1,7 экв.) и промыли содержимое колбы гексаном (50 мл). Далее медленно добавили раствор фенола (5,7 г, 0,06 моля, 1,6 экв.) в диглиме (25 мл). Реакцию проводили при перемешивании в течение 1,5 часов (комнатная температура). К анионному раствору фенола медленно добавили раствор 2-хлор-3-нитро-N-[2-(3-пиридин-3-илпропокси)этил]-5,6,7,8-тетрагидрохинолин-4-амина (14,8 г, 0,04 моля) в диглиме (25 мл). Реакционную смесь нагрели при перемешиваний до 60°С и продолжали перемешивать при этой температуре в течение ночи. Реакцию остановили путем выливания реакционной смеси на лед (1000 мл). Продукт находился в системе в виде маслянистой жидкости. Далее смесь экстрагировали дихлорметаном (4×100 мл). Соединенные экстракты концентрировали досуха. Остаток растворили в смеси гексан-этилацетат (1:1) (250 мл) и промыли водой (2×50 мл). Органический слой концентрировали досуха. Было обнаружено, что остаток содержит избыток фенола. Фенол удалили путем растворения остатка в диэтиловом эфире (500 мл) и перемешивания с 10 %-ным раствором гидроксида натрия (250 мл) в течение ночи. После разделения слоев эфирную фракцию концентрировали, получив 3-нитро-2-фенокси-N-[2-(3-пиридин-3-илпропокси)этил]-5,6,7,8-тетрагидрохинолин-4-амин в виде сиропообразной жидкости бледно-оранжевого цвета (12,0 г, 71 %). Данные тонкослойной хроматографии (10 % MeOH/CH2Cl2): Rf=0,58.
Часть D
В сосуд Парра вместимостью 500 мл загрузили раствор 3-нитро-2-фенокси-N-[2-(3-пиридин-3-илпропокси)этил]-5,6,7,8-тетрагидрохинолин-4-амина в толуоле (150 мл) и катализатор (5 % Pt на углероде, 1,1 г), поместили сосуд в аппарат Парра и подали водород (3,8 бар). Реакцию проводили при встряхивании в течение 4 часов при контроле способом ВЭЖХ. По истечении указанного времени реакция не прошла полностью. В сосуд Парра было добавлено дополнительное количество катализатора (1,0 г, 5 % Pt на углероде), вновь ввели водород и реакцию продолжали при встряхивании в течение ночи, что привело к завершению процесса. Реакционную смесь отфильтровали через целит и промыли толуолом (500 мл). Фильтрат концентрировали, получив 2-фенокси-N4-[2-(3-пиридин-3-илпропокси)этил]-5,6,7,8-тетрагидрохинолин-3,4-диамин в виде сиропообразной жидкости желтого цвета (8,2 г, 74 %). Данные тонкослойной хроматографии (10 % MeOH/CH2Cl2): Rf=0,48.
Данные масс-спектрометрии: М+1=419,2.
Часть Е
В круглодонную колбу вместимостью 200 мл загрузили 2-фенокси-N4-[2-(3-пиридин-3-илпропокси)этил]-5,6,7,8-тетрагидрохинолин-3,4-диамин (4,1 г, 0,0098 моля) и пиридин (40 мл) при комнатной температуре. Далее медленно добавили ацетилхлорид (0,8 г, 0,011 моля, 1,1 экв.). Реакцию проводили при перемешивании при комнатной температуре. Через 2 часа проверили полноту реакции и обнаружили наличие лишь амидного промежуточного продукта. Реакционную смесь нагрели с обратным холодильником и оставили в таком состоянии на ночь. При концентрировании получили сиропообразную жидкость темно-желтого цвета. Жидкость растворили в этилацетате (300 мл) и промыли водой (2×100 мл). Слой этилацетата концентрировали, получив 2-метил-4-фенокси-1-[2-(3-пиридин-3-илпропокси)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин в виде сиропообразной жидкости оранжевого цвета (3,8 г, 88 %). Данные тонкослойной хроматографии (10% МеОН/CH2Cl2): Rf=0,34.
Данные масс-спектрометрии: М+1=443,2.
Часть F
В круглодонную колбу вместимостью 200 мл загрузили 2-метил-4-фенокси-1-[2-(3-пиридин-3-илпропокси)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин (3,7 г, 0,0084 моля) и ацетат аммония (37 г, 0,48 моля, 57 экв.); реакционную смесь нагрели до 150°С. В течение 20 мин реакционная смесь была гомогенной. Реакционную смесь перемешивали при указанной температуре в течение ночи. Через 24 часа проверили полноту реакции и нашли, что реакция еще не завершена. Реакцию продолжали в течение выходных дней, после чего реакционную смесь охладили, растворили в 1 N HCl (250 мл) и промыли диэтиловым эфиром (200 мл). Водный слой после доведения до рН 11 добавлением гидроксида натрия экстрагировали дихлорметаном (3 х 100 мл). Соединенные органические фракции сконцентрировали, получив сиропообразную жидкость оранжевого цвета. Эту жидкость очистили способом хроматографии на колонке (10 % MeOH/CH2Cl2). Соответствующие фракции соединили и сконцентрировали, получив сиропообразную жидкость бледно-оранжевого цвета. Эта жидкость оказалась смесью требуемого продукта и N-ацетилированного производного. Смесь обработали 1 N HCl с обратным холодильником в течение 1 часа. Смесь охладили, рН довели до 11 и провели экстракцию дихлорметаном. Органическую фазу концентрировали досуха. Остаток очистили способом хроматографии на колонке; получили 2-метил-1-[2-(3-пиридин-3-илпропокси)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин в виде твердого вещества бледно-золотистого цвета (0,07 г, 2 %). Данные тонкослойной хроматографии (10% MeOH/CH2Cl2): Rf=0,05. Температура плавления 140-141°С.
СТИМУЛИРОВАНИЕ СИНТЕЗА ЦИТОКИНОВ В КЛЕТКАХ ЧЕЛОВЕКА
Для оценки кинетики стимулирования синтеза цитокинов применяют систему ин-витро с клетками крови человека. Активность синтеза измеряют по количеству интерферона и фактора некроза опухолей (α) (ИФ и ФНО соответственно), секретируемых в культуральную среду, как описано Тестерманом и др. (Testerman et al., Cytokine Induction by the Immunomodulators Imiquimod and S-27609, Journal of Leukocyte Biology, 58, 365-372, September, 1995).
Подготовка клеток крови для выращивания в культуральной среде
Цельную кровь, взятую у здоровых доноров из вены, помещают в вакуумированные пробирки, содержащие ЭДТУ. Одноядерные клетки периферической крови (ОКПК) отделяют от цельной крови путем центрифугирования в градиенте плотности с использованием реагента Histopaque®-1077. ОКПК дважды промывают сбалансированным раствором солей Хэнка и далее суспендируют в количестве (3-4)×106 кл/мл в полной среде RPMI. Суспензию ОКПК вносят на плоские стерильные планшеты для культуры тканей с 48 лунками (Costar, Кембридж, шт. Массачусетс или Becton Dickinson Labware, Линкольн Парк, шт. Нью-Джерси); в лунках содержатся равные объемы полной среды RPMI, содержащей исследуемое соединение.
Приготовление исследуемого соединения
Соединения солюбилизируют в диметилсульфоксиде (ДМСО). Концентрация в ДМСО не должна превышать конечного значения (1 %) при добавлении в лунки.
Инкубация
Раствор исследуемого соединения (60 мкМ) вводят в первую лунку, содержащую полную среду RPMI, а в остальные лунки вводят растворы после серийных трехкратных разведений. Далее в лунки добавляют суспензию ОКПК в равных объемах, получая требуемый диапазон концентраций исследуемого соединения (0,12-30 мкМ). Конечная концентрация суспензии ОКПК составляет (1,5-2)×106 кл/мл. Планшеты накрывают стерильными пластиковыми крышками, осторожно перемешивают и инкубируют в течение 18-24 часов при температуре 37°С в атмосфере, содержащей 5 % углекислоты.
Разделение
После инкубации планшеты центрифугируют в течение 5-10 мин при скорости 1000 мин-1 (прибл. 200g) и температуре 4°С. Надосадочную жидкость, не содержащую клеток, удаляют при помощи полипропиленовой пипетки и переносят в стерильные полипропиленовые пробирки. Образцы хранят до выполнения анализов при температуре от -30 до -70°С. Выполняют анализы на интерферон (α) и фактор некроза опухолей (α) способом ЭЛАЙЗА.
Анализы на интерферон (α) и Фактор некроза опухолей (α) способом ЭЛАЙЗА
Концентрацию интерферона (α) определяли при помощи набора ЭЛАЙЗА под названием Human Multi-Species Kit производства фирмы Biomedical Laboratories, Нью-Брансвик, шт. Нью-Джерси. Результаты выражали в пг/мл.
Концентрацию фактора некроза опухолей (α) (ФНО) определяли при помощи наборов ЭЛАЙЗА производства фирм Genzyme, Кембридж, шт. Массачусетс; R&D Systems, Миннеаполис, шт. Миннесота; Pharmingen, Сан-Диего, шт. Калифорния. Результаты выражали в пг/мл.
Приведенная ниже таблица дает самые низкие концентрации, которые, как было обнаружено, индуцируют синтез интерферона и индуцируют синтез фактора некроза опухолей для каждого соединения. Звездочка* означает, что стимулирования не было отмечено при любой концентрации исследуемых соединений. В целом самая высокая исследованная концентрация составляла 10 - 30 мкМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| АРИЛЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ВИРУСНОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ОПУХОЛЕВОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2308456C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНЫ | 2001 |
|
RU2294934C2 |
| КАРБАМИДЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНОВЫЕ ЭФИРЫ | 2001 |
|
RU2302418C2 |
| АМИДОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2000 |
|
RU2295343C2 |
| ТИОЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2001 |
|
RU2315049C2 |
| N-{2-[4-АМИНО-2-(ЭТОКСИМЕТИЛ)-1Н-ИМИДАЗО-[4,5-с]-ХИНОЛИН-1-ИЛ]-1,1-ДИМЕТИЛЭТИЛ}-МЕТАНСУЛЬФОНАМИД И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2004 |
|
RU2374246C2 |
| МОЧЕВИНОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2265020C2 |
| ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2248975C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2586974C2 |
| ТИОЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2304143C2 |
Изобретение относится к имидазохинолинам, имеющим формулу (I) и (II), а также тетрагидроимидазохинолинам, имеющим формулу (III), в которых радикалы и символы имеют значения, указанные в формуле изобретения. Данные соединения и фармацевтические составы на их основе, представляющие предмет настоящего изобретения, могут стимулировать биосинтез различных цитокинов, в частности α-интерферона, и использоваться в лечении некоторых заболеваний, включая вирусные болезни и опухолевые заболевания. Объектами изобретения также являются способы лечения вирусных и опухолевых заболеваний и промежуточные соединения. 25 н. и 16 з.п. ф-лы, 2 табл.
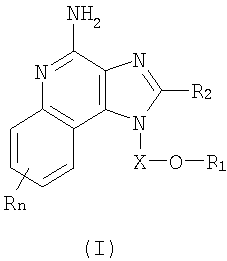
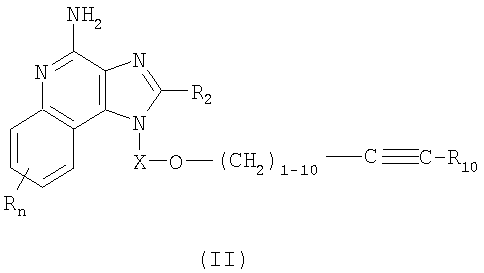
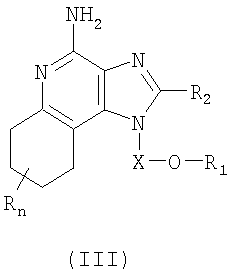
1. Имидазохинолины, имеющие формулу (I):
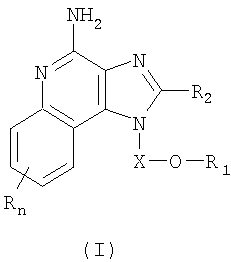
где Х представляет собой -CHR3- или -CHR3-алкильную группу;
R1 выбран из группы, содержащей радикалы:
гетероарил, при этом гетероарил представляет собой ароматическое кольцо или системы колец, содержащие, по меньшей мере, один атом азота;
R4-гетероарил, при этом гетероарил представляет собой ароматическое кольцо или системы колец, содержащие, по меньшей мере, один гетероатом, выбранный из О, S, и N; и
R4-гетероциклил, при этом гетероциклил представляет собой неароматическое кольцо, содержащее, по меньшей мере, один атом кислорода;
R2 выбран из группы, содержащей радикалы:
водород;
С1-10алкил и
С1-10алкил-Y-С1-10алкил;
R4 представляет собой С1-10алкил;
каждый R3 представляет собой независимо Н или С1-10алкил;
каждый Y представляет собой -O-;
n может иметь значение, равное от 0 до 4;
и каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, С1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
2. Соединение или соль по п.1, отличающиеся тем, что R1 представляет собой 2-пиридил, 3-карбазолил или -(CH2)1-3-гетероарил.
3. Соединение или соль по п.2, отличающиеся тем, что гетероарил выбран из группы, состоящей из следующих радикалов: 2-пиридил, 3-пиридил, 4-пиридил, 2-тиазолил, 2-пиримидинил, 4-пиримидинил, 4-триазолил, 2-бензофуранил, 2-индолил, 3-карбазолил, 2-фуранил, 4-изохинолинил, 4-изоксазолил и 4-пиразолил.
4. Соединение или соль по п.1, отличающиеся тем, что Х представляет собой -СН(С1-10алкил)(алкил)-, причем алкильные группы могут быть идентичными или разными.
5. Соединение или соль по п.1, отличающиеся тем, что Х представляет собой -СН2-СН2-.
6. Соединение или соль по п.1, отличающиеся тем, что Х представляет собой -СН(С2Н5)(СН2)-.
7. Соединение или соль по п.1, отличающиеся тем, что R2 представляет собой Н.
8. Соединение или соль по п.1, отличающиеся тем, что R2 представляет собой С1-10алкил.
9. Соединение или соль по п.1, отличающиеся тем, что R2 представляет собой - С1-10алкил-О-С1-10алкил.
10. Имидазохинолины, имеющие формулу (II):

где Х представляет собой -CHR3- или -CHR3-алкильную группу;
R10 выбран из группы, состоящей из гетероарила, при этом гетероарил представляет собой ароматическое кольцо или системы колец, содержащие, по меньшей мере, один гетероатом, выбранный из О, S и N;
R2 выбран из группы, содержащей
водород;
n может иметь значение, равное от 0 до 4;
каждый R3 представляет собой независимо Н или С1-10алкил; и каждый R выбран независимо из группы, состоящей из радикалов С1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
11. Соединение или соль по п.10, отличающиеся тем, что гетероарильный радикал выбран из группы, состоящей из следующих радикалов: 2-пиридил, 3-пиридил, 4-пиридил, 2-тиазолил, 3-фуранил, 2-тиенил и 2-пиримидинил.
12. Соединение или соль по п.10, отличающиеся тем, что Х представляет собой -СН(С1-10алкил)(алкил)-, причем алкильные группы могут быть идентичными или разными.
13. Соединение или соль по п.10, отличающиеся тем, что Х представляет собой -СН2-СН2-.
14. Соединение или соль по п.10, отличающиеся тем, что Х представляет собой -СН(С2Н5)(СН2)-.
15. Соединение, выбранное из группы, состоящей из следующих веществ:
1-(2-{[3-(изохинолин-4-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амин;
1-(2-{[3-(1,3-тиазол-2-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амин;
1-{2-[3-(1Н-4-пиразолил)пропокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(3-пиримидин-2-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(3-пиридин-4-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(3-пиридин-2-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-{2-[3-(1,3-тиазол-2-ил)пропокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(3-пиридин-3-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(3-пиримидин-5-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-{2-[(1-бензил-1Н-1,2,3-триазол-4-ил)метокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
1-{2-[(1-бензил-1Н-1,2,3-триазол-5-ил)метокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-({1-[(фенилсульфанил)метил]-1Н-1,2,3-триазол-4-ил}метокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-({1-[(фенилсульфанил)метил]-1Н-1,2,3-триазол-5-ил}метокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(бензо[b]фуран-2-илметокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(пиридин-3-илметокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(пиридин-2-илметокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(пиридин-4-илметокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-{2-[(3,5-диметилизоксазол-4-ил)метокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
1-(2-{[3-(пиримидин-2-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амин;
1-(2-{[3-(пирид-4-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амин;
1-(2-{[3-(фур-3-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амин;
4-{3-[2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]-пропин-1-ил}тиофен-2-илкарбоксальдегид;
1-(2-{[3-(пирид-2-ил)-2-пропинил]окси}этил)-1Н-имидазо[4,5-с]хинолин-4-амин;
1-{2-метил-1-[(пирид-2-илокси)метил]пропил}-1Н-имидазо[4,5-c]хинолин-4-амин;
1-{1-[(пирид-2-илокси)метил]пропил}-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(9Н-карбазол-3-илокси)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-{2-[(3-тиен-2-илпроп-2-инил)окси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
1-{2-[(1-метил-1Н-индол-2-ил)метокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(3-тиен-2-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[2-(3-пиридин-3-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[2-(3-пиридин-3-илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(тетрагидрофуран-2-илметокси)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-{2-[(5-хлор-1-бензотиен-3-ил)метокси]пропил}-1Н-имидазо[4,5-с]хинолин-4-амин;
1-{2-[(3-нитропиридин-2-ил)окси]пропил}-1Н-имидазо[4,5-с]хинолин-4-амин;
1-(2-метил-1-{[(3-нитропиридин-2-ил)окси]метил}пропил)-1Н-имидазо[4,5-с]хинолин-4-амин;
1-(1-{[(5-хлор-1-бензотиен-3-ил)метокси]метил}-2-метилпропил)-1Н-имидазо[4,5-с]хинолин-4-амин;
2-(2-метоксиэтил)-1-[2-(3-пиридин-3 -илпропокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин; а также
2-метил-1-[2-(3-пиридин-3-илпропокси)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин, или его соль фармацевтического качества.
16. Тетрагидроимидазохинолины, имеющие формулу (III)
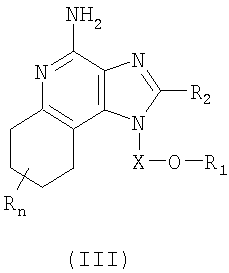
где X представляет собой -CHR3- или -CHR3-алкильную группу;
R1 выбран из группы, содержащей радикалы:
R4-гетероарил, при этом гетероарил представляет собой ароматическое кольцо или системы колец, содержащие, по меньшей мере, один атом азота;
R2 представляет собой
C1-10алкил;
R4 представляет собой
С1-10алкил;
каждый R3 представляет собой Н;
n может иметь значение, равное от 0 до 4;
и каждый R выбран независимо из группы, состоящей из радикалов С1-10алкил, С1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
17. Фармацевтический состав для индуцирования биосинтеза цитокинов, содержащий терапевтически эффективное количество соединения или соли по п.1 и фармацевтически доступный носитель.
18. Фармацевтический состав для индуцирования биосинтеза цитокинов, содержащий терапевтически эффективное количество соединения или соли по п.10 и фармацевтически доступный носитель.
19. Фармацевтический состав для индуцирования биосинтеза цитокинов, содержащий терапевтически эффективное количество соединения или соли по п.16 и фармацевтически доступный носитель.
20. Способ стимулирования биосинтеза цитокинов в организме животных, заключающийся во введении терапевтически эффективного количества соединения или соли по п.1 в организм животного.
21. Способ по п.20, отличающийся тем, что цитокин представляет собой α-интерферон.
22. Способ стимулирования биосинтеза цитокинов в организме животных, заключающийся во введении терапевтически эффективного количества соединения или соли по п.10 в организм животного.
23. Способ по п.22, отличающийся тем, что цитокин представляет собой α-интерферон.
24. Способ лечения вирусного заболевания у животного, заключающийся во введении терапевтически эффективного количества соединения или соли по п.1 в организм животного.
25. Способ лечения опухолевого заболевания у животного, заключающийся во введении терапевтически эффективного количества соединения или соли по п.1 в организм животного.
26. Способ лечения вирусного заболевания у животного, заключающийся во введении терапевтически эффективного количества соединения или соли по п.10 в организм животного.
27. Способ лечения опухолевого заболевания у животного, заключающийся во введении терапевтически эффективного количества соединения или соли по п.10 в организм животного.
28. Способ стимулирования биосинтеза цитокинов в организме животных, заключающийся во введении терапевтически эффективного количества соединения или соли по п.15 в организм животного.
29. Способ по п.28, отличающийся тем, что цитокин представляет собой α-интерферон.
30. Способ лечения вирусного заболевания у животного, заключающийся во введении терапевтически эффективного количества соединения или соли по п.15 в организм животного.
31. Способ лечения опухолевого заболевания у животного, заключающийся во введении терапевтически эффективного количества соединения или соли по п.15 в организм животного.
32. Промежуточное соединение, имеющее формулу (V):
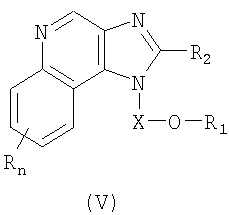
где Х представляет собой -CHR3- или -CHR3-алкильную группу;
R1 выбран из группы, содержащей радикалы:
R4-гетероарил, при этом гетероарил представляет собой ароматическое кольцо или системы колец, содержащие, по меньшей мере, один гетероатом, выбранный из О и S; и
-(CH2)1-10-C≡C-R10;
R2 выбран из группы, содержащей
водород;
R4 представляет собой -С1-10алкил;
каждый R3 представляет собой Н;
R10 представляет собой гетероарил, при этом гетероарил представляет собой ароматическое кольцо или кольцо, включающее в себя, по меньшей мере, один атом серы;
n имеет значение, равное от 0 до 4;
и каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, С1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
33. Промежуточное соединение, имеющее формулу (VI):
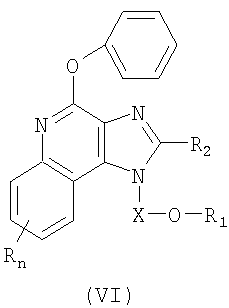
где Х представляет собой -CHR3- или -CHR3-алкильную группу;
R1 выбран из группы, содержащей радикалы:
R4-гетероарил, при этом гетероарил представляет собой ароматическое кольцо или системы колец, содержащие, по меньшей мере, один гетероатом, выбранный из О и N; и
-(CH2)1-10-C≡C-R10;
R2 выбран из группы, содержащей
водород;
R4 представляет собой -С1-10алкил;
каждый R3 представляет собой Н;
R10 представляет собой гетероарил, при этом гетероарил представляет собой ароматическое кольцо или кольцо, включающее в себя, по меньшей мере, один атом серы;
n имеет значение, равное от 0 до 4;
и каждый R выбран независимо из группы, состоящей из радикалов С1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
34. Промежуточное соединение, имеющее формулу (VIII):
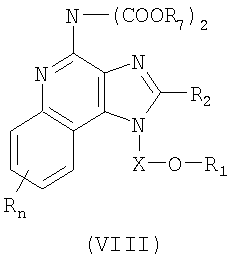
где X представляет собой -CHR3-, или -CHR3-алкильную группу;
R1 выбран из группы, содержащей радикалы:
R4-гетероарил, при этом гетероарил представляет собой ароматическое кольцо или системы колец, содержащие, по меньшей мере, один атом азота;
R2 выбран из группы, содержащей
водород;
R4 представляет собой -C1-10алкил;
каждый R3 представляет собой Н;
n может иметь значение, равное от 0 до 4;
каждый R выбран независимо из группы, состоящей из радикалов С1-10алкил, С1-10алкокси, гидрокси, галоген и трифторметил;
R7 представляет собой трет-бутил или бензил;
или же соль фармацевтического качества указанных соединений.
35. Фармацевтический состав, предназначенный для лечения вирусных, опухолевых, атопических заболеваний, а также предназначенный для использования в качестве адъювантов вакцин, содержащий терапевтически эффективное количество соединения или его соли по п.16 и фармацевтически доступный носитель.
36. Способ индуцирования биосинтеза цитокинов в организме животных, заключающийся во введении терапевтически эффективного количества соединения или соли по п.16 в организм животного.
37. Способ по п.36, отличающийся тем, что цитокин представляет собой α-интерферон.
38. Способ лечения вирусного заболевания у животного, заключающийся во введении терапевтически эффективного количества соединения или соли по п.16 в организм животного.
39. Способ лечения опухолевого заболевания у животного, заключающийся во введении терапевтически эффективного количества соединения или соли по п.16 в организм животного.
40. Промежуточное соединение, имеющее формулу (VII):
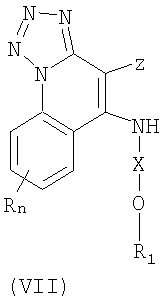
где Z представляет собой NH2 или NO2;
Х представляет собой -CHR3- или -CHR3-алкильную группу;
R1 выбран из группы, содержащей радикалы:
R4-гетероарил, при этом гетероарил представляет собой ароматическое кольцо или систему кольца, включающего в себя, по меньшей мере, один атом азота;
R4 представляет собой -C1-10алкил;
каждый rs представляет собой независимо Н;
n может иметь значение от 0 до 4; и
каждый R выбран независимо из группы, состоящей из радикалов
C1-10алкил, С1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
41. Промежуточное соединение, имеющее формулу (XLIV):
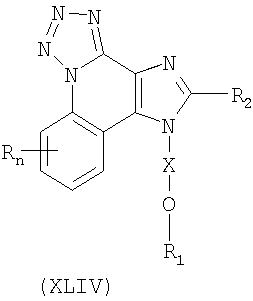
где Х представляет собой -CHR3- или -CHR3-алкильную группу;
R1 выбран из группы, содержащей радикалы:
R4-гетероарил, при этом гетероарил представляет собой ароматическое кольцо, содержащее, по меньшей мере, один атом азота;
R2 выбран из группы, содержащей радикалы:
водород;
С1-10алкил и
С1-10алкил-Y-С1-10алкил;
R4 представляет собой -С1-10алкил;
каждый R3 представляет собой Н;
каждый Y представляет собой -O-;
n может иметь значение, равное от 0 до 4;
и каждый R выбран независимо из группы, состоящей из радикалов С1-10алкил, С1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| ПРОИЗВОДНОЕ 1,3-ДИГИДРО-2H-ИМИДАЗО/4,5-B/ХИНОЛИН-2-ОНА В КАЧЕСТВЕ ИНГИБИТОРА ФОСФОДИЭСТЕРАЗЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ ДЛЯ НЕГО, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1992 |
|
RU2127273C1 |

