Настоящее изобретение касается имидазохинолиновых соединений, имеющих простую эфирную и мочевинную группы в положении 1, и фармацевтических составов, содержащих такие соединения. Другой аспект настоящего изобретения затрагивает применение данных соединений в качестве иммуномодуляторов для стимулирования биосинтеза цитокинов в организме животных и для лечения заболеваний, включая вирусные болезни и опухолевые заболевания.
Первый надежный отчет о циклической системе 1Н-имидазо[4,5-с]хинолина (Бакман и др., J. Orq. Chem. 15, 1278-1284 (1950)) описывает синтез 1-(6-метокси-8-хинолинил)-2-метил-1Н-имидазо[4,5-с]хинолина для возможного применения в качестве противомалярийного средства. Далее поступило сообщение о синтезе различных замещенных 1H-имидазо[4,5-с]хинолинов. Так, например, было синтезировано соединение 1-[2-(4-пиперидил)этил]-1Н-имидазо[4,5-с]хинолин (Джейн и др., J. Med. Chem. 11, p.87-92 (1968)) в качестве предполагаемого противосудорожного и сердечно-сосудистого средства. Имелись также сообщения о ряде 2-оксоимидазо[4,5-с]хинолинов (Баранов и др., Chem. Abs. 85, 94362 (1976), Берени и др., J. Heterocyclic Chem. 18, 1537-1540 (1981)).
Позднее было обнаружено, что некоторые 1H-имидазо[4,5-с]хинолин-4-амины и их 1- и 2-замещенные производные могут найти применение как противовирусные средства, бронхолитические средства и иммуномодуляторы. В числе прочих публикаций можно сослаться на патенты США №4689338, 4698348, 4929624, 5037986, 5268376, 5346905 и 5389640; все эти патенты приведены здесь в качестве ссылок.
Продолжает вызывать интерес циклическая система имидазохинолина.
Известны некоторые 1H-имидазо[4,5-с]нафтиридин-4-амины, 1H-имидазо[4,5-с]пиридин-4-амины и 1Н-имидазо[4,5-с]хинолин-4-амины, имеющие заместитель с простой эфирной группой в положении 1. Они описаны в патентах США №5268376, 5389640 и 5494916 и в международной заявке WO 99/29693.
Несмотря на указанные попытки выявить соединения, полезные в качестве модификаторов иммунной реакции, по-прежнему имеется потребность в соединениях, которые обладают способностью модулировать иммунную реакцию путем стимулирования биосинтеза цитокинов или под действием иных механизмов.
Краткое описание сущности изобретения
Авторами был обнаружен новый класс соединений, способных стимулировать биосинтез цитокинов в организме животных. Настоящее изобретение поэтому касается соединений имидазохинолин-4-амина и тетрагидроимидазохинолин-4-амина, имеющих заместитель с простой эфирной и мочевинной группами в положении 1. Данные соединения могут быть описаны формулами (I) и (II); подробности их строения приведены ниже. Общая структурная формула этих соединений такова:
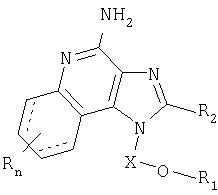
При этом X, R1, R2 и R определены для каждого класса соединений, имеющих формулы (I) и (II).
Соединения, представленные формулами (I) и (II), могут применяться в качестве модификаторов иммунной реакции вследствие их способности стимулировать биосинтез цитокинов и иными способами модулировать иммунную реакцию при введении в организм животных. Эти свойства делают указанные соединения полезными для лечения ряда заболеваний, таких как вирусные болезни и опухоли, реагирующие на изменения в иммунной реакции.
Настоящее изобретение касается фармацевтических составов, содержащих соединения, модифицирующие иммунную реакцию, и способов стимулирования биосинтеза цитокинов в организме животных, лечения вирусной инфекции у животных и(или) лечения опухолевых заболеваний у животных путем введения животным соединений формулы (I) или (II).
Кроме того, настоящее изобретение затрагивает способы синтеза соединений, представленных в нем, и промежуточных продуктов, используемых при синтезе данных соединений.
Подробное описание изобретения
Как было упомянуто выше, обнаружены некоторые соединения, стимулирующие биосинтез цитокинов и модифицирующие иммунную реакцию в организме животных. Такие соединения представлены формулами (I) и (II), приведенными ниже.
Имидазохинолиновые соединения, составляющие предмет изобретения и имеющие простую эфирную и мочевинную группы в положении 1, представлены формулой (I):
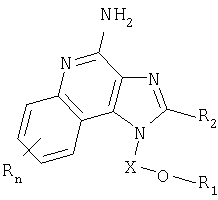
Где X представляет собой -CHR5-, -CHR5-алкильную или -CHR5-алкенильную группу;
R1 выбран из группы, содержащей радикалы:
-R4-NR8-CR3-NR5-Z-R6-алкил;
-R4-NR8-CR3-NR5-Z-R6-алкенил;
-R4-NR8-CR3-NR5-Z-R6-арил;
-R4-NR8-CR3-NR5-Z-R6-гетероарил;
-R4-NR8-CR3-NR5-Z-R6-гетероциклил;
-R4-NR8-CR3-NR5R7;
-R4-NR8-CR3-NR9-Z-R6-алкил;
-R4-NR8-CR3-NR9-Z-R6-алкенил;
-R4-NR8-CR3-NR9-Z-R6-арил;
-R4-NR8-CR3-NR9-Z-R6-гетероарил;
-R4-NR8-CR3-NR9-Z-R6-гетероциклил;
R2 выбран из группы, содержащей радикалы:
-водород;
-алкил;
-алкенил;
-арил;
-гетероарил;
-гетероциклил;
-алкил-Y-алкил;
-алкил-Y-алкенил;
-алкил-Y-арил; а также
-алкил или алкенил, с одним или несколькими заместителями, выбранными из группы, содержащей радикалы:
-ОН;
-галоген;
-N(R5)2;
-CO-N(R5)2;
-СО-С1-10алкил;
-СО-O-С1-10алкил;
-N3;
-арил;
-гетероарил;
-гетероциклил;
-СО-арил; а также
-СО-гетероарил;
каждый R3 представляет собой =O или =S;
каждый R4 представляет собой независимо алкил или алкенил, в который может войти одна или несколько -O-групп;
каждый R5 представляет собой независимо Н или C1-10алкил;
R6 представляет собой связь или же алкил или алкенил, который может прерваться одной или несколькими -O-группами;
R7 представляет собой Н или C1-10алкил, в который может войти гетероатом, или же R7 и R5 могут соединиться, образуя цикл;
R8 представляет собой Н, C1-10алкил или арилалкил или же R4 и R8 могут соединиться, образуя цикл;
R9 представляет собой C1-10алкил, который может соединиться с R8 с образованием цикла;
каждый Y представляет собой независимо -О- или -S(O)0-2-;
Z представляет собой связь, -СО- или -SO2-;
n может иметь значение от 0 до 4;
каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
Настоящее изобретение также включает в себя тетрагидроимидазохинолиновые соединения, несущие простую эфирную группу и заместитель, содержащий мочевину, в положении 1. Такие тетрагидроимидазохинолиновые соединения представлены формулой (II):
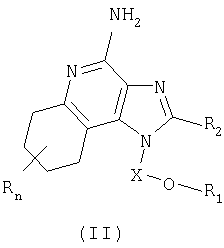
Где Х представляет собой -CHR5-, -CHR5-алкильную или -CHR5-алкенильную группу;
R1 выбран из группы, содержащей радикалы:
-R4-NR8-CR3-NR5-Z-R6-алкил;
-R4-NR8-CR3-NR5-Z-R6-алкенил;
-R4-NR8-CR3-NR5-Z-R6-арил;
-R4-NR8-CR3-NR5-Z-R6-гетероарил
-R4-NR8-CR3-NR5-Z-R6-гетероциклил
-R4-NR8-CR3-NR5R7
-R4-NR8-CR3-NR9-Z-R6-арил
-R4-NR8-CR3-NR9-Z-R6-алкенил;
-R4-NR8-CR3-NR9-Z-R6-арил;
-R4-NR8-CR3-NR9-Z-R6-гетероарил; а также
-R4-NR8-CR3-NR9-Z-R6-гетероциклил;
R2 выбран из группы, содержащей радикалы:
-водород;
-алкил;
-алкенил;
-арил;
-гетероарил;
-гетероциклил;
-алкил-Y-алкил;
-алкил-Y-алкенил;
-алкил-Y-арил; а также
-алкил или алкенил, с одним или несколькими заместителями, выбранными из группы, содержащей радикалы:
-ОН;
-галоген;
-N(R5)2;
-CO-N(R5)2;
-CO-C1-10алкил;
-CO-O-C1-10алкил;
-N3;
-арил;
-гетероарил;
-гетероциклил;
-СО-арил; а также
-СО-гетероарил;
каждый R3 представляет собой =O или =S;
каждый R4 представляет собой независимо алкил или алкенил, в который может войти одна или несколько -O-групп;
каждый R5 представляет собой независимо Н или C1-10алкил;
R6 представляет собой связь или же алкил или алкенил, который может прерваться одной или несколькими -O-группами;
R7 представляет собой Н или С1-10алкил, в который может войти гетероатом, или же R5 и R7 могут соединиться, образуя цикл;
R8 представляет собой Н, С1-10алкил или арилалкил или же R4 и R8 могут соединиться, образуя цикл;
R9 представляет собой C1-10алкил, который может соединиться с R8, образуя цикл;
каждый Y представляет собой независимо -О- или -S(O)0-2-;
Z представляет собой связь, -СО- или -SO2-;
n может иметь значение от 0 до 4;
каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
Получение соединений
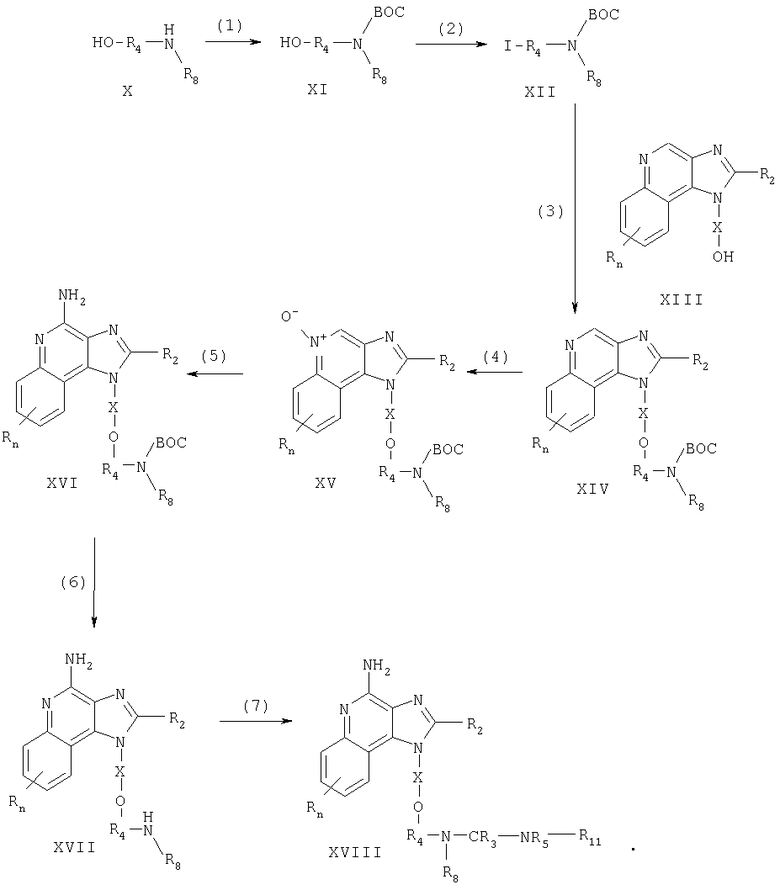
Соединения, представляющие собой предмет настоящего изобретения, можно получать по схеме реакции I, где R, R2, R3, R4, R5, R8, Х и n определены выше, ВОС представляет собой трет-бутоксикарбонил, a R11 это -Z-R6-алкил, -Z-R6-алкенил, -Z-R6-арил, -Z-R6-гетероарил, -Z-R6-гетероциклил или же R11 представляет собой R7, причем значения R6, R7 и Z определены выше.
На стадии (1) процесса (схема I реакции) аминогруппа аминоспирта формулы X защищена трет-бутоксикарбонильной группой. Раствор аминоспирта в тетрагидрофуране обрабатывают ди-трет-бутилдикарбонатом в присутствии основания, например, гидроксида натрия. Многие аминоспирты формулы Х коммерчески доступны; другие могут быть получены с применением известных способов синтеза.
На стадии (2) процесса (схема I реакции) защищенный аминоспирт формулы XI превращают в йодид (формула XII). Йод вводят в раствор трифенилфосфина и имидазола в дихлорметане; далее добавляют раствор защищенного аминоспирта формулы XI в дихлорметане. Реакцию проводят при температуре окружающей среды.
На стадии (3) процесса (схема I реакции) 1H-имидазо[4,5-с]хинолин-1-иловый спирт формулы XIII алкилируют йодидом формулы XII с получением 1Н-имидазо[4,5-с]хинолин-1-илового простого эфира формулы XIV. Спирт формулы XIII обрабатывают гидридом натрия в соответствующем растворителе, таком как N,N-диметилформамиде образуя алкоксид. При температуре окружающей среды к раствору алкоксида добавляют указанный йодид. По завершении добавления реакционную смесь перемешивают при повышенной температуре (прибл. 100°С). Известны многие соединения формулы XIII; см., например, патент США 4689338 (Герстер). Другие соединения могут быть получены без затруднений по известным способам синтеза, см., например, патенты США №5605899 (Герстер и др.) и 5175296 (Герстер).
На стадии (4) процесса (схема I реакции) 1H-имидазо[4,5-с]хинолин-1-иловый эфир формулы XIV окисляют до 1Н-имидазо[4,5-с]хинолин-5N-оксида формулы XV с применением обычно используемого окислителя, способного образовывать N-оксиды. Предпочтительно окислять раствор соединения формулы XIV в хлороформе 3-хлорпероксибензойной кислотой при температуре окружающей среды.
На стадии (5) процесса (схема I реакции) 1Н-имидазо[4,5-с]хинолин-5N-оксид формулы XV аминируют с получением 1Н-имидазо[4,5-с]хинолин-4-амина формулы XVI. В стадию (5) входят следующие части: (i) реакция соединения формулы XV с ацилирующим веществом и (ii) реакция полученного продукта с аминирующим веществом. Часть (I) стадии (5) вступает в реакцию N-оксида формулы XV с ацилирующим веществом. В число подходящих ацилирующих веществ входят алкил- или арилсульфонилхлориды (например, бензолсульфонилхлорид, метансульфонилхлорид, п-толуолсульфонилхлорид). Предпочтение отдается арилсульфонилхлоридам. Наиболее применим n-толуолсульфонилхлорид. Часть (ii) стадии (5) включает в себя взаимодействие продукта части (i) с избытком аминирующего вещества. В число подходящих аминирующих веществ входят аммиак (например, в виде гидроксида аммония) и соли аммония (например, карбонат аммония, бикарбонат аммония, фосфат аммония). Предпочтение отдается гидроксиду аммония. Реакцию предпочтительно проводить путем растворения N-оксида формулы XV в инертном растворителе (таком, как дихлорметан или 1,2-дихлорэтан) при нагревании, если это необходимо, добавления аминирующего вещества к раствору и медленного введения ацилирующего вещества. В качестве варианта можно проводить реакцию в автоклаве при повышенной температуре (85-100°С).
На стадии (6) процесса (схема I реакции) защитную группу удаляют путем гидролиза в кислотных условиях, получая 1H-имидазо[4,5-с]хинолин-4-амин формулы XVII. Предпочтительно обрабатывать соединение формулы XVI соляной кислотой с этанолом при температуре окружающей среды или при осторожном нагревании.
На стадии (7) процесса (схема I реакции) 1H-имидазо[4,5-с]хинолин-4-амин формулы XVII превращают в производное мочевины или тиомочевины формулы XVIII с применением известных способов синтеза. Так, например, соединение формулы XVII можно обработать изоцианатом формулы R12-N=C=O, где R12 это -R6-алкил, -R6-алкенил, -R6-арил, R6-гетероарил или -R6-гетероциклил. Реакцию можно проводить путем добавления раствора изоцианата в соответствующем растворителе, таком как дихлорметан или 1-метил-2-пирролидинон, к раствору соединения формулы XVII при температуре окружающей среды. В качестве варианта можно обработать соединение формулы XVII тиоизоцианатом формулы R12-N=C=S, ацилизоцианатом формулы R12-C(O)-N=C=O, сульфонилизоцианатом формулы R12-S(O2)-N=C=O или карбамоилхлоридом формулы R13-N-C(O)Cl, где R13 это R12 или R7. Продукт или его соль фармацевтического качества можно выделить по стандартным способам.
Схема I реакции
Соединения, представляющие собой предмет настоящего изобретения, можно получать в соответствии со схемой II реакции, где R, R2, R3, R4, R5, R8, X и n определены выше, а ВОС представляет собой трет-бутоксикарбонил.
На стадии (1) процесса (схема II реакции) аминогруппа аминоспирта формулы XIX защищена трет-бутоксикарбонильной группой. Раствор аминоспирта в тетрагидрофуране обрабатывают ди-трет-бутилдикарбонатом в присутствии основания, например гидроксида натрия. Многие аминоспирты формулы XIX коммерчески доступны; другие могут быть получены с применением известных способов синтеза.
На стадии (2) процесса (схема II реакции) защищенный аминоспирт формулы XX превращают в метансульфонат (формула XXI). Раствор соединения формулы XX в соответствующем растворителе, например в дихлорметане, обрабатывают метансульфонилхлоридом в присутствии основания, например триэтиламина. Реакцию можно проводить при пониженной температуре (0°С).
На стадии (3а) процесса (схема II реакции) метансульфонат формулы XXI превращают в азид формулы XXII. К раствору соединения формулы XXI в подходящем растворителе, таком как N,N-диметилформамид, добавляют азид натрия. Реакцию можно проводить при повышенной температуре (80-100°С).
На стадии (3b) процесса (схема II реакции) соединение формулы XXII алкилируют галогенидом формулы Hal-R8, получая соединения формулы XXIII. Для соединений, у которых R8 представляет собой водород, данный этап пропускают. Проводят реакцию между соединением формулы XXII и гидридом натрия в подходящем растворителе, таком как N,N-диметилформамид или тетрагидрофуран, с образованием аниона, после чего осуществляют взаимодействие с галогенидом. Реакцию можно проводить при температуре окружающей среды.
На стадии (4) процесса (схема II реакции) азид формулы XXII или XXIII восстанавливают до амина формулы XXIV. Предпочтительно проводить восстановление с применением обычного гетерогенного катализатора гидрогенизации, например палладия на углероде. Реакцию удобно выполнять в аппарате Парра в соответствующем растворителе, например в метаноле или изопропаноле.
На стадии (5) процесса (схема II реакции) 4-хлор-3-нитрохинолин формулы XXV реагирует с амином формулы XXIV, образуя 3-нитрохинолин формулы XXVI. Реакцию можно выполнять путем добавления амина формулы XXIV к раствору соединения формулы XXV в соответствующем растворителе, например в дихлорметане, в присутствии основания, например триэтиламина. Известно много хинолинов формулы XXV; такие хинолины можно также получать, используя известные способы: см., например, патент США 4689338 (Герстер) и данные в нем ссылки.
На стадии (6) процесса (схема II реакции) 3-нитрохинолин формулы XXVI восстанавливают до 3-аминохинолина формулы XXVII. Предпочтительно проводить восстановление с применением обычного гетерогенного катализатора гидрогенизации, например палладия на углероде. Реакцию удобно выполнять в аппарате Парра в соответствующем растворителе, например в толуоле.
На стадии (7) процесса (схема II реакции) соединение формулы XXVII реагирует с карбоновой кислотой или ее эквивалентом, образуя 1Н-имидазо[4,5-финолин формулы XIV. В число подходящих эквивалентов карбоновой кислоты входят сложные ортоэфиры и 1,1-диалкоксиалкилалканоаты. Карбоновую кислоту или ее эквивалент выбирают так, чтобы в состав соединения формулы XIV входил желаемый заместитель R2. Так, например, при введении триэтилортоформиата образуется соединение, в котором R2 представляет собой водород; в присутствии триэтилортовалерата R2 представляет собой бутил. Реакцию можно проводить в отсутствие растворителя или в инертном растворителе, таком как толуол. Реакция протекает с разогревом, достаточным для того, чтобы любой спирт или вода, образующиеся как побочные продукты, улетучились. В качестве варианта можно ввести каталитическое количество пиридингидрохлорида.
В качестве альтернативы можно выполнить стадию (7) путем (i) проведения реакции между соединением формулы XXVII и ацилгалогенидом формулы R2C(O)Cl и (ii) циклизации. В части (i) ацилгалогенид добавляют к раствору соединения формулы XXVII в инертном растворителе, например в ацетонитриле или дихлорметане. Эту реакцию можно проводить при температуре окружающей среды или пониженной температуре. В части (ii) продукт части (i) нагревают в спиртовом растворе в присутствии основания. Предпочтительно нагревать этанольный раствор продукта части (i) в присутствии избытка триэтиламина с обратным холодильником или же нагревать в метанольном растворе аммиака.
Стадии (8), (9), (10) и (11) проводят так же, как стадии (4), (5), (6) и (7) процесса (схема I реакции).
Схема II реакции
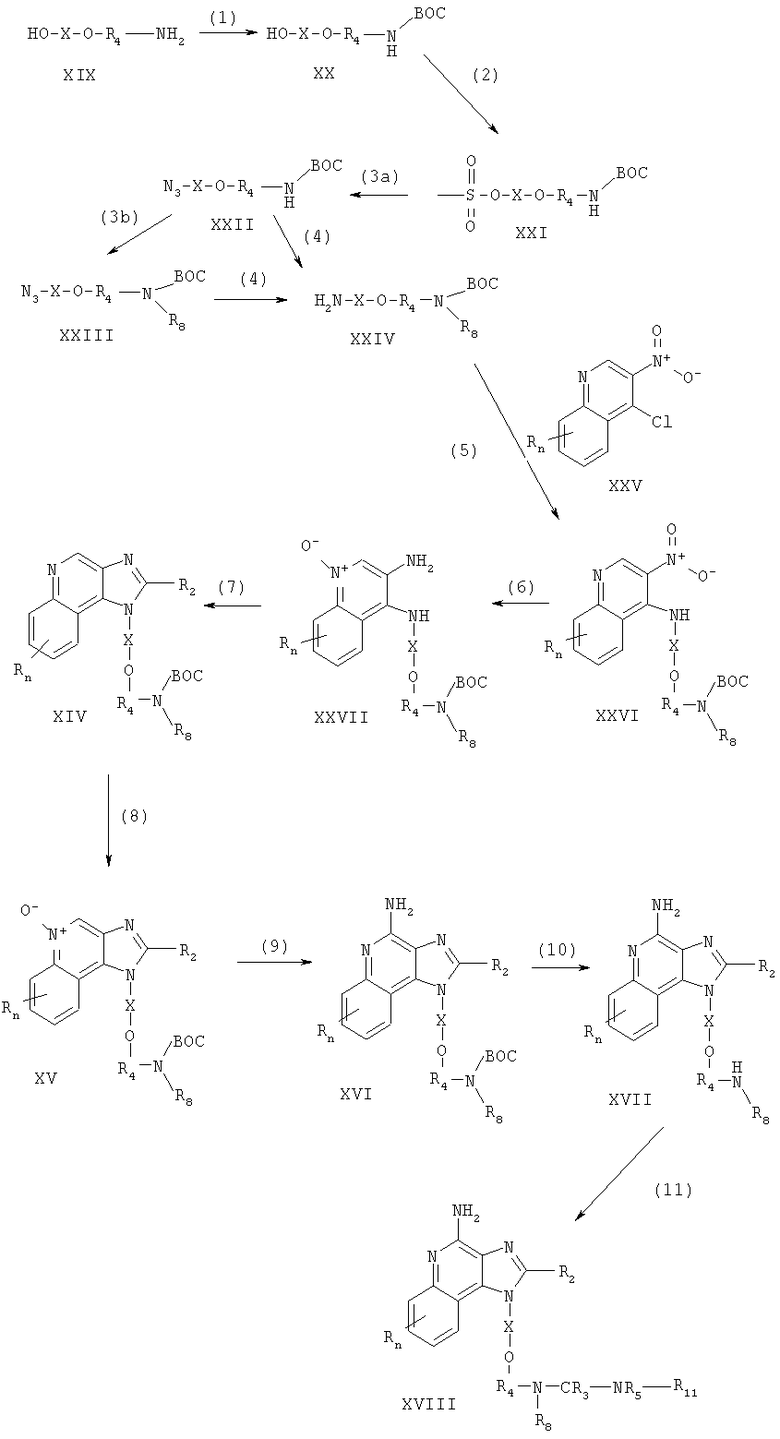
Соединения, представляющие собой предмет настоящего изобретения, можно получать по схеме III реакции, где R, R2, R3, R4, R5, R8, R11, Х и n определены выше.
На стадии (1) процесса (схема III реакции) 1H-имидазо[4,5-с]хинолин-4-амин формулы XVII восстанавливают до 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина формулы XXVIII. Предпочтительно проводить восстановление путем суспендирования или растворения соединения формулы XVII в трифторуксусной кислоте, добавления каталитического количества оксида платины (IV) и последующей гидрогенизации. Реакцию удобно выполнять в аппарате Парра.
Стадию (2) проводят так же, как стадию (7) процесса (схема I реакции) получая 6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амин формулы XXIX. Продукт или его соль фармацевтического качества можно выделить по стандартным способам.
Реакционная схема III
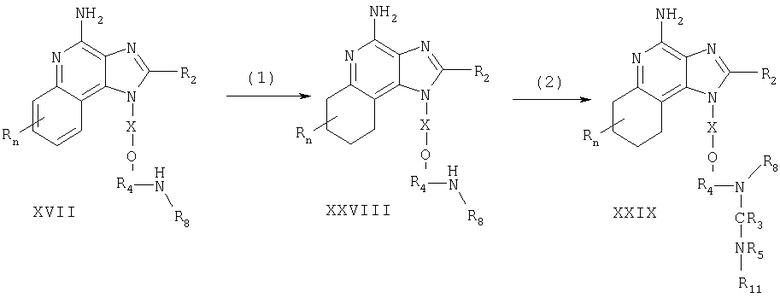
Соединения, представляющие собой предмет настоящего изобретения, можно также получать в соответствии со схемой IV реакции, где R, R2, Х и n определены выше.
На стадии (1) процесса (схема IV реакции) 4-хлор-3-нитрохинолин формулы XXV реагирует с амином формулы R1-O-X-NH2, образуя 3-нитрохинолин-4-амин формулы XXX. Реакцию можно проводить путем добавления амина к раствору соединения формулы XXV в подходящем растворителе, таком как хлороформ или дихлорметан, с возможным нагревом. Многие хинолины формулы XXV хорошо известны: см., например патент США 4689338 (Герстер) и приведенные там ссылки.
На стадии (2) процесса (схема IV реакции) 3-нитрохинолин-4-амин формулы XXX восстанавливают с применением способа, описанного для стадии (6) процесса (схема II реакции), получая хинолин-3,4-диамин формулы XXXI.
На стадии (3) процесса (схема IV реакции) хинолин-3,4-диамин формулы XXXI подвергают циклизации по способу, описанному для стадии (7) процесса (схема II реакции), получая 1H-имидазо[4,5-с]хинолин формулы XXXII.
На стадии (4) процесса (схема IV реакции) 1H-имидазо[4,5-с]хинолин формулы XXXII окисляют по способу, описанному для стадии (4) процесса (схема I реакции), получая 1H-имидазо[4,5-с]хинолин-5N-оксид формулы XXXIII.
На стадии (5) процесса (схема IV реакции) 1Н-имидазо[4,5-с]хинолин-5N-оксид формулы XXXIII аминируют по способу, описанному для стадии (5) процесса (схема I реакции) с получением 1Н-имидазо[4,5-с]хинолин-4-амина формулы I. Продукт или его соль фармацевтического качества можно выделить по стандартным способам.
Схема IV реакции
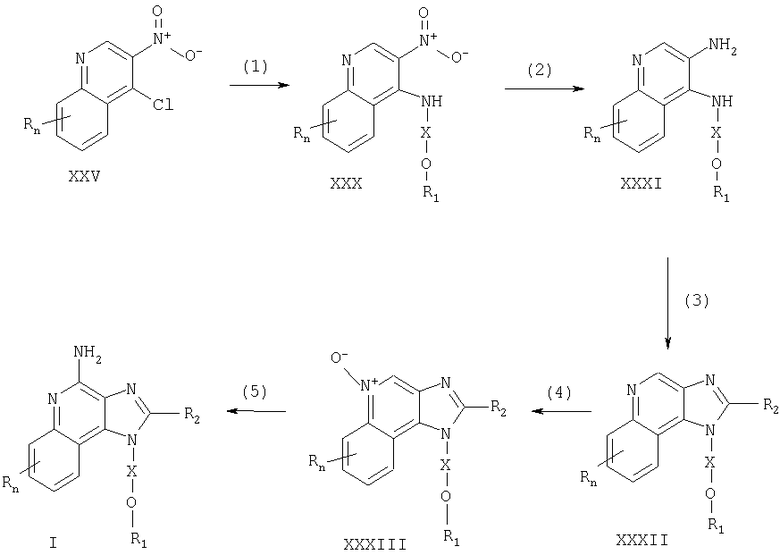
Соединения, представляющие собой предмет настоящего изобретения, можно получать в соответствии со схемой V реакции, где R, R2, R3, R4, R5, R8, R11, Х и n определены выше.
На стадии (1) процесса (схема V реакции) группу ВОС удаляют из соединения формулы XIV, применяя способ, описанный для стадии (6) процесса (схема I реакции), с получением 1H-имидазо[4,5-с]хинолина формулы XXXIV.
На стадии (2) процесса (схема V реакции) 1Н-имидазо[4,5-с]хинолин формулы XXXIV превращают в производное мочевины или тиомочевины формулы XXXV, применяя способ, описанный для стадии (7) процесса (схема I реакции).
На стадии (3) процесса (схема V реакции) 1H-имидазо[4,5-с]хинолин формулы XXXV окисляют согласно способу, описанному для стадии (4) процесса (схема I реакции), с получением 1Н-имидазо[4,5-с]хинолин-5N-оксида формулы XXXVI.
На стадии (4) процесса (схема V реакции) 1H-имидазо[4,5-с]хинолин-5N-оксид формулы XXXVI окисляют по способу, описанному для стадии (5) процесса (схема I реакции), с получением 1H-имидазо[4,5-с]хинолин-4-амина формулы XVIII. Продукт или его соль фармацевтического качества можно выделить по стандартным способам.
Схема V реакции

Настоящее изобретение также включает в себя новые соединения, используемые в качестве промежуточных продуктов синтеза соединений с формулами (I) и (II). Эти промежуточные соединения характеризуются структурными формулами (III) и (IV) и подробно описаны ниже.
Один класс промежуточных соединений имеет формулу (III):
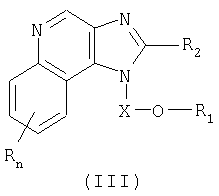
Где Х представляет собой -CHR5-, -CHR5-алкильную или -CHR5-алкенильную группу;
R1 выбран из группы, содержащей радикалы:
-R4-NR8-CR3-NR5-Z-R6-алкил;
-R4-NR8-CR3-NR5-Z-R6-алкенил;
-R4-NR8-CR3-NR5-Z-R6-арил;
-R4-NR8-CR3-NR5-Z-R6-гетероарил;
-R4-NR8-CR3-NR5-Z-R6-гетероциклил; а также
-R4-NR8-CR3-NR5R7;
-R4-NR8-CR3-NR9-Z-R6-алкил;
-R4-NR8-CR3-NR9-Z-R6-алкенил;
-R4-NR8-CR3-NR9-Z-R6-арил;
-R4-NR8-CR3-NR9-Z-R6-гетероциклил; а также
-R4-NR8-CR3-NR9-Z-R6-гетероциклил;
R2 выбран из группы, содержащей радикалы:
-водород;
-алкил;
-алкенил;
-арил;
-гетероарил;
-гетероциклил;
-алкил-Y-алкил;
-алкил-Y-алкенил;
-алкил-Y-арил; а также
-алкил или алкенил, с одним или несколькими заместителями, выбранными из группы, содержащей радикалы:
-ОН;
-галоген;
-N(R5)2;
-CO-N(R5)2;
-СО-С1-10алкил;
-СО-O-С1-10алкил;
-N3;
-арил;
-гетероарил;
-гетероциклил;
-СО-арил; а также
-СО-гетероарил;
каждый R3 представляет собой =O или =S;
каждый R4 представляет собой независимо алкил или алкенил, в который может войти одна или несколько -O-групп;
каждый R5 представляет собой независимо Н или C1-10алкил;
R6 представляет собой связь, или же алкил, или алкенил, который может прерваться одной или несколькими -O-группами;
R7 представляет собой Н или C1-10алкил, в который может войти гетероатом, или же R7 и R5 могут соединиться, образуя цикл;
R8 представляет собой Н, C1-10алкил, или арилалкил; или же R4 и R8 могут соединиться, образуя цикл;
R9 представляет собой C1-10алкил, который может соединиться с R8 с образованием цикла;
каждый Y представляет собой независимо -О- или -S(O)0-2-;
Z представляет собой связь, -СО- или -SO2-;
n может иметь значение от 0 до 4;
каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
Настоящее изобретение также включает в себя соединения имидазохинолин-N-оксида формулы (IV):
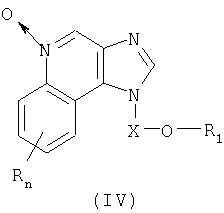
Где X представляет собой -CHR5-, -CHR5-алкиленовую или -CHR5-алкениленовую группу;
R1 выбран из группы, содержащей радикалы:
-R4-NR8-CR3-NR5-Z-R6-алкил;
-R4-NR8-CR3-NR5-Z-R6-алкенил;
-R4-NR8-CR3-NR5-Z-R6-арил;
-R4-NR8-CR3-NR5-Z-R6-гетероарил;
-R4-NR8-CR3-NR5-Z-R6-гетероциклил;
-R4-NR8-CR3-NR5R7;
-R4-NR8-CR3-NR9-Z-R6-алкил;
-R4-NR8-CR3-NR9-Z-R6-алкенил;
-R4-NR8-CR3-NR9-Z-R6-арил;
-R4-NR8-CR3-NR9-Z-R6-гетероарил; а также
-R4-NR8-CR3-NR9-Z-R6-гетероциклил;
каждый Y представляет собой независимо -О- или -S(О)0-2-;
Z представляет собой связь, -СО- или -SO2-;
каждый R4 представляет собой независимо алкил или алкенил, в который может войти одна или несколько -О-групп;
каждый R5 представляет собой независимо Н или С1-10алкил;
R6 представляет собой связь, или же алкил, или алкенил, который может прерваться одной или несколькими -О-группами;
R7 представляет собой Н или С1-10алкил, в который может войти гетероатом, или же R7 и R5 могут соединиться, образуя цикл;
R8 представляет собой Н, C1-10алкил, или арилалкил; или же R4 и R6 могут соединиться, образуя цикл;
R9 представляет собой C1-10алкил, который может соединиться с R8 с образованием цикла;
n может иметь значение от 0 до 4;
каждый R выбран независимо из группы, состоящей из радикалов C1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
Применяемые в тексте термины «алкил», «алкенил» и префикс «алк-» включают в себя как группы с прямой и разветвленной цепью, так и циклические группы, т.е. циклоалкильные и циклоалкенильные остатки. Если иное не оговорено специально, эти группы содержат от 1 до 20 атомов углерода, причем алкенильные группы содержат от 2 до 20 атомов углерода. Суммарное число атомов углерода в группах, которым отдается предпочтение, может доходить до 10. Циклические группы могут содержать один цикл или несколько циклов; предпочтение отдается числу атомов углерода в цикле от 3 до 10. В числе примеров можно назвать такие остатки, как циклопропил, циклопропилметил, циклопентил, циклогексил, и адамантил.
Следует также добавить, что алкильная и алкенильная части -Х-групп могут не иметь заместителей или содержат заместители (один или большее число); эти заместители выбраны из групп, включающих в себя алкил, алкенил, арил, гетероарил, гетероциклил, арилалкил, гетероарилалкил и гетероциклилалкил.
Термин «галогеналкил» включает в себя группы, замещенные одним или несколькими атомами галогена, в том числе и перфторированные группы. Это утверждение верно и в отношении групп, в наименование которых входит префикс «гало-». Примерами приемлемых галогеналкильных групп являются хлорметильный, трифторметильный и аналогичные остатки.
Применяемый здесь термин «арил» относится к карбоциклическим ароматическим циклам или к системам циклов. В числе примеров арильных групп можно назвать фенил, нафтил, дифенил, фторфенил и инденил. Термин «гетероарил» касается ароматических циклов или систем циклов, содержащих хотя бы один гетероатом (например, О, S, N) в цикле. В число используемых гетероарильных групп входят фурил, тиенил, пиридил, хинолинил, изохинолинил, индолил, изоиндолил, триазолил, пирролил, тетразолил, имидазолил, пиразолил, оксазолил, тиазолил, бензофуранил, бензотиофенил, карбазолил, бензоксазолил, пиримидинил, бензимидазолил, хиноксалинил, бензотиазолил, нафтиридинил, изоксазолил, изотиазолил, пуринил, хиназолинил и т.д.
Термин «гетероциклил» включает в себя неароматические циклы или системы циклов, содержащие хотя бы один гетероатом (например, О, S, N) в цикле. Сюда относятся все полностью насыщенные и частично ненасыщенные производные вышеупомянутых гетероарильных групп. В числе примеров гетероциклических групп можно назвать пирролидинил, тетрагидрофуранил, морфолинил, тиоморфолинил, пиперидинил, пиперазинил, тиазолидинил, имидазолидинил, изотиазолидинил, и т.д.
Арильные, гетероарильные и гетероциклильные группы могут быть незамещенными или замещенными; в последнем случае они могут содержать один заместитель или несколько заместителей, независимо выбранных из группы, состоящей из таких радикалов, как алкил, алкокси, алкилтио, галогеналкил, галогеналкокси, галогеналкилтио, галоген, нитро, гидрокси, меркапто, циано, карбокси, формил, арил, арилокси, арилтио, арилалкокси, арилалкилтио, гетероарил, гетероарилокси, гетероарилтио, гетероарилалкокси, гетероарилалкилтио, амино, алкиламино, диалкиламино, гетероциклил, гетероциклоалкил, алкилкарбонил, алкенилкарбонил, алкоксикарбонил, галогеналкилкарбонил, галогеналкоксикарбонил, алкилтиокарбонил, арилкарбонил, гетероарилкарбонил, арилоксикарбонил, гетероарилоксикарбонил, арилтиокарбонил, гетероарилтиокарбонил, алканоилокси, алканоилтио, алканоиламино, арилкарбонилокси, арилкарбонилтио, алкиламиносульфонил, алкилсульфонил, арилсульфонил, гетероарилсульфонил, арилдиазинил, алкилсульфониламино, арилсульфониламино, арилалкилсульфониламино, алкилкарбониламино, алкенилкарбониламино, арилкарбониламино, арилалкилкарбониламино, гетероарилкарбониламино, гетероарилалкилкарбониламино, алкилсульфониламино, алкенилсульфониламино, арилсульфониламино, арилалкилсульфониламино, гетероарилсульфониламино, гетероарилалкилсульфониламино, алкиламинокарбониламино, алкениламинокарбониламино, ариламинокарбониламино, арилалкиламинокарбониламино, гетероариламинокарбониламино, гетероарилалкиламинокарбониламино и, в случае гетероциклильного остатка, оксо. Если о каких-либо иных группах говорят, что они «замещены» или «возможно замещены», то эти группы также могут содержать один или несколько из вышеупомянутых заместителей.
В целом некоторым заместителям отдано большее предпочтение. Так, например, в число предпочитаемых R1-групп входят R4-NR8-CR3-NR5-Z-R6-алкил и R4-NR8-CR3-NR5-Z-R6-арил, где алкильная и арильная группы могут не содержать заместителей или содержать их; предпочтительно, чтобы R4 являлся этиленом или н-бутиленом или же R4 и R8 могут соединяться, образуя цикл. Предпочтительно отсутствие R-заместителей, т.е. n=0. В число предпочитаемых групп R2 входят водород, алкильные группы с 1-4 атомами углерода (это метил, этил, пропил, изопропил, н-бутил, втор-бутил, изобутил и циклопропилметил), метоксиэтил и этоксиметил. Для замещенных групп, таких как замещенные алкильные или замещенные арильные остатки, в число предпочитаемых заместителей входят галоген, нитрил, метокси, метилтио, трифторметил и трифторметокси. Один или несколько из этих предпочитаемых заместителей при их наличии могут находиться в составе соединений, являющихся предметом настоящего изобретения, в любой комбинации.
Настоящее изобретение включает в себя описанные здесь соединения в любой фармацевтически доступной форме, включая изомеры (например, диастереомеры и энантиомеры), соли, сольваты, полиморфные варианты и т.д. В особенности, если соединение является оптически активным, настоящее изобретение включает в себя каждый из энантиомеров соединения, а также рацемические смеси энантиомеров.
Фармацевтические составы и биологическая активность
Фармацевтические составы, являющиеся предметом настоящего изобретения, содержат терапевтически эффективные количества описанного выше соединения в сочетании с фармацевтически доступным носителем.
Термин «терапевтически эффективное количество» означает то количество соединения, которое достаточно для достижения терапевтического эффекта, такого как стимулирование синтеза цитокинов, проявление противоопухолевой активности и(или) проявление противовирусной активности. Хотя точное количество активного соединения, примененного в фармацевтическом составе, являющемся предметом настоящего изобретения, может меняться в зависимости от факторов, известных тем, кто является специалистом в этой области (например, физическая и химическая природа соединения, природа носителя и предполагаемый режим дозирования), предполагается, что составы, являющиеся предметом настоящего изобретения, будут содержать достаточное количество активного ингредиента для создания дозы соединения от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно от приблизительно 10 мкг/кг до приблизительно 5 мг/кг при расчете на массу тела пациента. Могут быть использованы любые известные лекарственные формы, такие как таблетки, пастилки, парентеральные препараты, сиропы, кремы, мази, аэрозольные препараты, чрезкожные пластыри, пластыри на слизистой оболочке и т.д.
Соединения, являющиеся предметом настоящего изобретения, можно применять как единственное терапевтическое средство в схеме лечения или же данные соединения можно применять в виде сочетания одного с другим или с иными активными веществами, включая дополнительные модификаторы иммунной реакции, противовирусные вещества, антибиотики и т.д.
Было показано, что соединения, являющиеся предметом настоящего изобретения, стимулируют синтез определенных цитокинов в экспериментах, выполненных в соответствии с условиями испытаний, описанными ниже. Результаты испытаний показывают, что указанные соединения используются в качестве модификаторов иммунной реакции; они могут изменять иммунную реакцию разными путями, что делает эти соединения весьма полезными при лечении различных заболеваний.
В число цитокинов, синтез которых может быть стимулирован применением соединений, являющихся предметом настоящего изобретения, обычно включают интерферон-α (ИФ-α) и(или) фактор некроза опухолей-α (ФНО-α), а также некоторые интерлейкины (ИЛ). В группу цитокинов, биосинтез которых может стимулироваться соединениями, являющимися предметом настоящего изобретения, входят ИФ-α, ФНО-α, ИЛ-1, ИЛ-6, ИЛ-10 и ИЛ-12, а также некоторые другие цитокины. Среди прочих эффектов эти и иные цитокины могут ингибировать размножение вирусов и рост опухолевых клеток, что делает данные соединения полезными при лечении вирусных заболеваний и опухолей. В связи со сказанным настоящее изобретение касается способа стимулирования биосинтеза цитокинов в организме животного путем введения эффективного количества соединения, являющегося предметом настоящего изобретения, или состава в организм животного.
Было обнаружено, что некоторые соединения, являющиеся предметом настоящего изобретения, в большей степени стимулируют экспрессию ИФ-α в популяции кроветворных клеток, таких как ОКПК (одноядерные клетки периферической крови) и клетки pDC2 (дендритные клетки-предшественники тип 2) без сопутствующего синтеза значительных количеств цитокинов, сопровождающих воспаление.
Вдобавок к способности стимулировать синтез цитокинов соединения, являющиеся предметом настоящего изобретения, воздействуют и на другие аспекты врожденной иммунной реакции. Так, например, возможна стимуляция активности натуральных клеток-киллеров, вероятно вследствие стимуляции цитокинов. Данные соединения могут также активировать макрофаги, что в свою очередь стимулирует секрецию оксида азота и синтез дополнительных количеств цитокинов. Кроме того, эти соединения могут вызвать пролиферацию и дифференциацию В-лимфоцитов.
Соединения, являющиеся предметом настоящего изобретения, также воздействуют на приобретенную иммунную реакцию. Например, хотя и не полагают, что они оказывают непосредственно влияние на Т-клетки или напрямую индуцируют цитокины Т-клеток, синтез цитокина ИФ-γ Т-хелперов типа 1 (Т×1) стимулируется косвенным образом, а синтез цитокинов ИЛ-4, ИЛ-5 и ИЛ-13 Т-хелперов типа 2 (Т×2) ингибируется после введения этих соединений. Данная активность означает, что эти соединения применимы для лечения заболеваний, при которых необходима повышающая регуляция Т×1-ответа и (или) понижающая регуляция Т×2-ответа. Ввиду способности данных соединений ингибировать Т×2 иммунную реакцию, можно ожидать, что они применимы при лечении атопических заболеваний, например атопического дерматита, астмы, аллергии, аллергического ринита, системной красной волчанки; они могут также применяться как адъюванты вакцин для развития клеточно-опосредованного иммунитета. Возможно применение для лечения рецидивирующих заболеваний, вызванных микроскопическими грибами, и хламидиоза.
Эффекты, связанные с модифицированием иммунной реакции данными соединениями, делают возможным их применение при лечении широкого спектра заболеваний. Вследствие своей способности стимулировать синтез цитокинов, таких как ИФ-α и(или) ФНО-α, эти соединения особенно применимы при лечении вирусных заболеваний и опухолей. Иммуномодулирующая активность данных соединений дает основания полагать, что соединения, являющиеся предметом настоящего изобретения, могут найти применение при лечении таких заболеваний, как вирусные, включая остроконечные кондиломы, простые бородавки, подошвенные бородавки, гепатит В, гепатит С, заболеваний, вызванных вирусом простого герпеса типа I и типа II, контагиозный моллюск, натуральная оспа, в особенности черная натуральная оспа, заболеваний, вызванных ВИЧ, ЦМВ, вирусом ветряной оспы, заболеваний, вызванных риновирусом, аденовирусом, вирусом гриппа и вирусом парагриппа (приведенные здесь перечни не являются исчерпывающими). Возможно применение этих соединений в случае интраэпителиальных неоплазий, таких как цервикальная интраэпителиальная неоплазия. Возможно воздействие на болезни, вызванные вирусом папилломы человека, и сопутствующие неоплазий; на заболевания, вызванные микроскопическими грибами (Candida, Aspergillus), в том числе криптококковый менингит. Данные соединения могут найти применение при лечении таких опухолевых заболеваний, как базально-клеточный рак, лейкозный ретикулоэндотелиоз, саркома Капоши, почечно-клеточный рак, плоскоклеточный рак, миелобластный лейкоз, множественная миелома, меланома, неходжкиновская лимфома, кожная Т-клеточная лимфома и другие виды рака. Возможно применение при лечении паразитарных заболеваний: пневмоцистоза, вызванного микроорганизмом Pneumocystis canriii, криптоспоридиоза, гистоплазмоза, токсоплазмоза, трипаносомоза, лейшманиоза, бактериальных инфекций, например туберкулеза и заболевания, вызванного микроорганизмом Mycobacterium avium. В число прочих заболеваний, которые можно лечить соединениями, являющимися предметом настоящего изобретения, входят лучевой кератоз, экзема, эозинофилия, эссенциальная тромбоцитемия, проказа, множественный склероз, синдром Омена, дискоидная красная волчанка, болезнь Боуэна, папулез Боуэна, гнездная алопеция, торможение образования келоидов и прочих послеоперационных рубцов. Следует добавить, что данные соединения могут усиливать или стимулировать заживление ран, включая хронические повреждения. Данные соединения могут быть применены для лечения заболеваний, вызванных условно-патогенными микроорганизмами, и опухолей, возникающих после подавления клеточно-опосредованного иммунитета, например у пациентов после трансплантации органов, у раковых больных и ВИЧ-инфицированных.
Количество соединения, эффективное при стимулировании биосинтеза цитокинов, это масса, достаточная для того, чтобы стимулировать один тип клетки или несколько типов клеток, например моноциты, макрофаги, дендритные клетки и В-клетки, к продуцированию определенного количества одного или нескольких цитокинов, таких как ИФ-α, ФНО-α, ИЛ-1, ИЛ-6, ИЛ-10 и ИЛ-12, превышающего фоновый уровень. Точное количество зависит от факторов, известных специалистам в данной области, но предполагается, что доза будет составлять от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно от приблизительно 10 мкг/кг до приблизительно 5 мг/кг. Настоящее изобретение также затрагивает способ лечения вирусных инфекций у животных и способ лечения опухолевых заболеваний у животных путем применения эффективного количества соединения или состава, являющихся предметом настоящего изобретения. Масса вещества, эффективная при лечении или торможении вирусной инфекции, является количеством, вызывающим уменьшение интенсивности одного или нескольких проявлений вирусной инфекции, таких как вирусные поражения, вирусная нагрузка, скорость размножения вирусов и смертность, по сравнению с показателями контрольных животных, не прошедших лечения. Точное количество зависит от факторов, известных специалистам в этой области, но предполагается, что доза будет составлять от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно от приблизительно 10 мкг/кг до приблизительно 5 мг/кг. Масса соединения, эффективная при лечении опухолевых заболеваний, является количеством, вызывающим уменьшение размеров опухоли или числа очагов заболевания. Точное количество зависит от факторов, известных специалистам в этой области, но предполагается, что доза будет составлять от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно от приблизительно 10 мкг/кг до приблизительно 5 мг/кг.
Далее описание настоящего изобретения иллюстрируется примерами, приведенными ниже; примеры ни в коем случае не ограничивают общие рамки применения изобретения.
В приведенных далее примерах некоторые соединения были подвергнуты очистке способом полупрепаративной ВЭЖХ. Применяли автоматическую систему очистки Waters Fraction Lynx. Фракции, полученные способом полупрепаративной ВЭЖХ, анализировали на газохроматографическом приборе Micromass LC-TOFMS; соответствующие фракции соединяли и высушивали в центрифуге, получая трифторацетатные соли необходимых соединений. Структуры соединений подтверждали способом 1H ЯМР.
Колонка: Phenomenex Luna C18(2), 10×50 мм, размер частиц 5 мкм, размер пор 100 Å. Расход 25 мл/мин. Градиентное элюирование: 5-65% компонента В в течение 4 мин, 65-95% компонента В в течение 0,1 мин, выдержка на уровне 95% компонента В в течение 0,4 мин. А представляет собой 0,05%-ную смесь трифторуксусной кислоты и воды, В представляет собой 0,05%-ную смесь трифторуксусной кислоты и ацетонитрила. Сбор фракций осуществляется по масс-селективному сигналу.
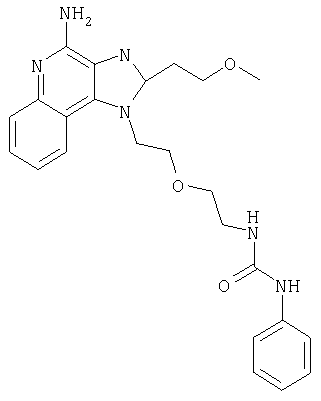
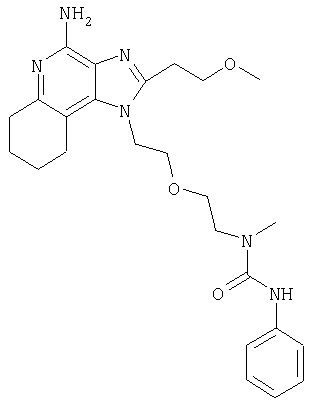
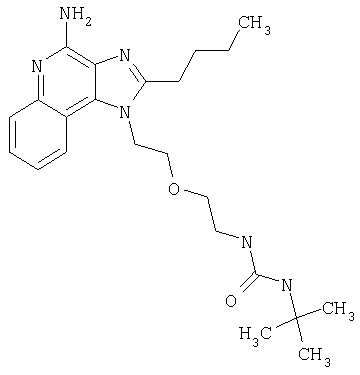
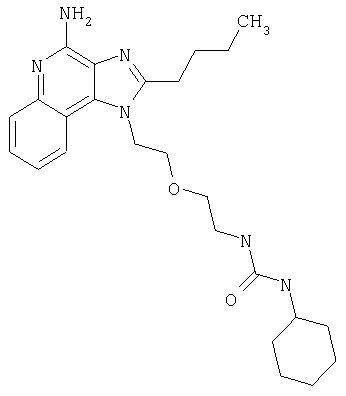
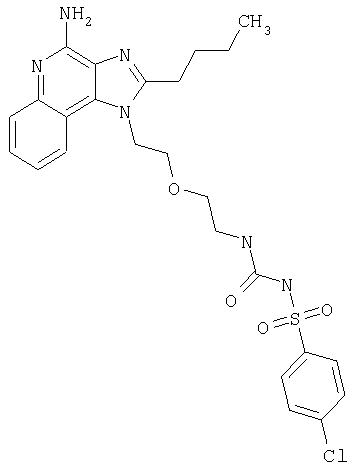
Пример 1
N-(2-{2-[4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил)-N'-фенилмочевина
Часть А
В атмосфере азота охлаждали раствор 2-2(2-аминоэтокси)этанола (29,0 г, 0,276 моля) в 180 мл тетрагидрофурана (ТГФ) до температуры 0°С и обрабатывали 140 мл 2N раствора NaOH. Далее добавляли по каплям в течение 1 часа раствор ди-трет-бутилдикарбоната (60,2 г, 0,276 моля) в 180 мл ТГФ при быстром перемешивании. Реакционную смесь оставили для нагрева до комнатной температуры и перемешивали еще в течение 18 часов. После этого ТГФ удаляли при пониженном давлении, а рН остающегося водного шлама доводили до 3, добавляя 150 мл 1М раствора H2SO4. Продукт экстрагировали этилацетатом (300 мл, 100 мл), и соединенные органические слои промывали водой (2×) и рассолом. Органическую часть высушивали над Na2SO4 и концентрировали, получая трет-бутил-2-(2-гидроксиэтокси)этилкарбамат в виде бесцветной маслянистой жидкости (47,1 г).
Часть В
В атмосфере азота охладили быстро перемешиваемый раствор трет-бутил-2-(2-гидроксиэтокси)этилкарбамата (47,1 г, 0,230 моля) в 1 л безводного СН2Cl2 до температуры 0°С и обработали триэтиламином (48,0 мл, 0,345 моля). Далее по каплям добавляли метансульфонилхлорид (19,6 мл, 0,253 моля) в течение 30 мин. Реакционную смесь оставили для нагрева до комнатной температуры и перемешивали еще в течение 22 часов. Реакцию останавливали, добавляя 500 мл насыщенного раствора NaHCO3; органический слой отделяли. Органическую фазу промывали водой (3×500 мл) и рассолом. Органическую часть высушивали над Na2SO4 и концентрировали, получая 2-{2-[(трет-бутоксикапбонил)амино]этокси}этилметансульфонат в виде маслянистой жидкости коричневого цвета (63,5 г).
Часть С
Перемешиваемый раствор 2-{2-[(трет-бутоксикарбонил)амино]этокси}этилметансульфоната (63,5 г, 0,224 моля) в 400 мл N,N-диметилформамида (ДМФ) обрабатывали NaN3 (16,1 г, 0,247 моля), и реакционную смесь нагрели до 90°С в атмосфере азота. Через 5 часов раствор охладили до комнатной температуры и обработали 500 мл холодной воды. Далее реакционную смесь экстрагировали Et2O (3×300 мл). Совмещенные органические экстракты промывали водой (4×100 мл) и рассолом (2×100 мл). Органическую часть высушивали над MgSO4 и концентрировали, получая 52,0 г трет-бутил-2-(2-азидоэтокси)этилкарбамат в виде маслянистой жидкости светло-коричневого цвета.
Часть D
Раствор трет-бутил-2-(2-азидоэтокси)этилкарбамата (47,0 г, 0,204 моля) в МеОН обрабатывали 4 г Pd (10%) на углероде и встряхивали в атмосфере водорода (3 бар) в течение 24 часов. Раствор фильтровали через слой целита (Celite) и концентрировали, получая 35,3 г неочищенный трет-бутил-2-(2-аминоэтокси)этилкарбамат в виде бесцветной жидкости, которую использовали без дальнейшей очистки.
Часть Е
В атмосфере азота перемешиваемый раствор 4-хлор-3-нитрохинолина (31,4 г, 0,151 моля) в 500 мл безводного СН2Cl2 обрабатывали триэтиламином (43 мл, 0,308 моля) и трет-бутил-2-(2-аминоэтокси)этилкарбаматом (0,151 моля). После перемешивания в течение ночи реакционную смесь промывали водой (2×300 мл) и рассолом (300 мл). Органическую часть высушивали над Na2SO4 и концентрировали, получив твердое вещество ярко-желтого цвета. При рекристаллизации из смеси этилацетат-гексан получили 43,6 г трет-бутил-2-{2-[(3-нитрохинолин-4-ил)амино]этокси}этилкарбамата в виде кристаллов ярко-желтого цвета.
Часть F
Раствор трет-бутил-2-{2-[(3-нитрохинолин-4-ил)амино]этокси}этилкарбамата (7,52 г, 20,0 ммоля) в толуоле обрабатывали 1,5 г Pt (5%) на углероде и встряхивали в атмосфере водорода (3 бар) в течение 24 часов. Раствор фильтровали через слой целита и концентрировали, получив 6,92 г неочищенный трет-бутил-2-{2-[(3-аминохинолин-4-ил)амино]этокси}этилкарбамат в виде сиропообразной жидкости желтого цвета.
Часть G
Раствор трет-бутил-2-{2-[(3-нитрохинолин-4-ил)амино]этокси}этилкарбамата (10,2 г, 29,5 ммоля) в 250 мл безводного CH2Cl2 охлаждали до температуры 0°С и обрабатывали триэтиламином (4,18 мл, 30,0 ммоля). Далее по каплям добавляли метоксипропионилхлорид (3,30 мл, 30,3 ммоля) в течение 5 мин. Реакционную смесь нагревали до комнатной температуры и продолжали перемешивание еще в течение 1 часа. После этого реакционную смесь концентрировали при пониженном давлении с получением твердого вещества оранжевого цвета. Этот продукт растворили в 250 мл EtOH и добавили 12,5 мл триэтиламина. Смесь нагревали с обратным холодильником при перемешивании в атмосфере азота в течение ночи, после чего смесь концентрировали досуха при пониженном давлении и обработали 300 мл Et2O. Смесь отфильтровывали и фильтрат концентрировали при пониженном давлении с получением твердого вещества коричневого цвета. Вещество растворили в 200 мл горячего МеОН и обработали активированным углем. Горячий раствор отфильтровали и концентрировали, получив 11,1 г трет-бутил-2-{2-[2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этилкарбамат в виде сиропообразной жидкости желтого цвета.
Часть Н
Раствор трет-бутил-2-{2-[2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил]этокси}этилкарбамата (10,22 г, 24,7 ммоля) в 250 мл CHCl3 обрабатывали 3-хлорпероксибензойной кислотой (77%, 9,12 г, 40,8 ммоля). После перемешивания в течение 30 мин реакционную смесь промывали 1%-ным раствором Na2CO3 (2×75 мл) и рассолом. Органический слой высушивали над Na2SO4 и концентрировали, получив 10,6 г трет-бутил-2-{2-[2-(2-метоксиэтил)-5-оксидо-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этилкарбамат в виде оранжевой пены. Этот продукт использовали без дальнейшей очистки.
Часть I
Раствор трет-бутил-2-{2-[2-(2-метоксиэтил)-5-оксидо-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этилкарбамата (10,6 г, 24,6 ммоля) в 100 мл 1,2-дихлорэтана нагрели до 60°С и обработали 10 мл концентрированного раствора NH4ОН. При быстром перемешивании добавляли твердый п-толуолсульфонилхлорид (7,05 г, 37,0 ммолей) в течение 10 мин. Реакционную смесь обработали дополнительным количеством концентрированного раствора NH4OH (1 мл) и поместили в автоклав; нагревание продолжалось еще в течение 2 часов. Реакционную смесь охладили и обработали 100 мл CHCl2. Далее реакционную смесь промывали водой, 1%-ным раствором Na2CO3 (2×) и рассолом. Органическую часть высушивали над Na2SO4 и концентрировали, получив 10,6 г трет-бутил-2-{2-[4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этилкарбамата в виде коричневой пены.
Часть J
Трет-бутил-2-{2-[4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этилкарбамат (10,6 г, 24,6 ммоля) обрабатывали 75 мл 2М раствора HCl в EtOH; смесь нагревали с обратным холодильником при перемешивании. Через 1,5 часа реакционную смесь охладили и отфильтровали с получением твердого смолообразного вещества. Это твердое вещество промыли EtOH и Et2O, высушили в вакууме и получили солянокислую соль в виде твердого вещества светло-коричневого цвета. Свободное основание получили путем растворения соли в 50 мл воды и обработки 10%-ным раствором NaOH. Далее водную суспензию концентрировали досуха, а остаток обработали CHCl3. Полученные соли удалили фильтрованием; фильтрат концентрировали и получили 3,82 г 1-[2-(2-аминоэтокси)этил]-2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-4-амина в виде порошка желтовато-коричневого цвета.
Данные масс-спектрометрии MS: 330 (М+Н)+.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,10 (д, J=8,1 Гц, 1Н); 7,66 (д, J=8,2 Гц, 1Н); 7,40 (м, 1Н); 7,25 (м, 1Н); 6,88 (шс, 2Н); 4,78 (т, J=5,4 Гц, 2Н); 3,89 (т, J=4,8 Гц, 2Н); 3,84 (т, J=6,9 Гц, 2Н); 3,54 (т, J=5,4 Гц, 2Н); 3,31 (с, 3Н); 3,23 (т, J=6,6 Гц, 2Н); 2,88 (т, J=5,3 Гц, 2Н).
Часть К
1-[2-(2-аминоэтокси)этил]-2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-4-амин (750 мг, 2,28 ммоля) растворили в 30 мл безводного СН2Cl2 и охладили до 0°С в атмосфере азота. К реакционной массе добавили фенилизоцианат (247 мкл, 2,28 ммоль) и Et3N (0,64 мл, 4,56 ммоль) и дали системе медленно нагреться до комнатной температуры. После 2-х часового перемешивания реакционную смесь концентрировали при пониженном давлении и получили твердое вещество желтого цвета. Это вещество растворили в минимальном количестве СН2Cl2, после чего добавляли EtOAc до помутнения раствора. Эту смесь поставили в морозильную камеру на ночь; в результате образовались белые кристаллы. Кристаллы отделили фильтрованием и высушили под вакуумом; получили 126 мг N-(2-{2-[4-амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил]этокси}этил)-N'-фенилмочевины. Температура плавления 171,0-174,0°С.
Данные масс-спектрометрии MS: 449 (М+Н)+.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,50 (с, 1Н); 8,05 (д, J=7,7 Гц, 1Н); 7,62 (д, J=8,8 Гц, 1Н); 7,44-7,18 (м, 3Н); 7,27-7,18 (м, 3Н); 6,88 (т, J=7,3 Гц, 1Н); 6,54 (с, 2Н); 6,12 (т, J=5,5 Гц, 2Н); 4,76 (т, J=4,8 Гц, 2Н); 3,88 (т, J=5,3 Гц, 2Н); 3,81 (т, J=6,7 Гц, 2Н); 3,40 (т, J=6,0 Гц, 2Н); 3,28 (с, 3Н); 3,25-3,14 (м, 4Н).
Данные 13С ЯМР (75 МГц, ДМСО-d6): δ 155,5; 152,0; 144,9; 140,8; 132,7; 129,0; 126,8; 126,5; 121,5; 121,4; 120,5; 117,9; 115,1; 70,5; 69,4; 58,4; 45,5; 27,6.
Данные элементного анализа: Рассчитано для С24Н28N6О3.0,21 Н2О: С 63,73%, Н 6,33%, N 18,58%. Найдено: С 63,33%, Н 6,28%, N 18,67%.
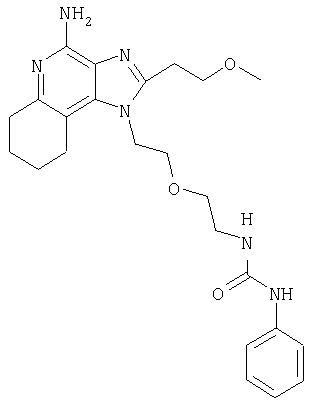
Пример 2
N-(2-{2-[4-амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил)-N'-фенилмочевина
Часть А
1-[2-(2-Аминоэтокси)этил]-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амин (10,0 г, 27,3 ммоль) растворили в 50 мл трифторуксусной кислоты и обработали PtO2 (1,0 г). Реакционную смесь встряхивали в атмосфере водорода (3 бар). Спустя 4 дня добавили дополнительное количество PtO2 (0,5 г) и продолжали гидрогенизацию еще в течение 3 дней. Реакционную смесь отфильтровали через целит и концентрировали при пониженном давлении, получив маслянистую жидкость коричневого цвета. Жидкость растворили в 200 мл воды и придали раствору основную реакцию (рН˜11) путем добавления 10%-ного раствора NaOH. После экстракции CHCl3 (5×75 мл) соединенные органические слои высушили над Na2SO4 и концентрировали, получив 5,17 г 1-[2-(2-аминоэтокси)этил]-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина в виде твердого вещества желтовато-коричневого цвета.
Данные масс-спектрометрии MS: 334 (М+Н)+.
Данные 1H ЯМР (300 МГц, CDCl3): δ 5,19 (с, 2Н); 4,49 (т, J=5,4 Гц, 2Н); 3,84 (т, J=6,6 Гц, 2Н); 3,71 (т, J=5,4 Гц, 2Н); 3,36 (т, J=5,2 Гц, 2Н); 3,51 (с, 3Н); 3,15 (т, J=6,6 Гц, 2Н); 2,95 (м, 2Н); 2,82 (м, 2Н); 2,76 (т, J=5,1 Гц, 2Н); 1,84 (м, 4Н); 1,47 (шс, 2Н).
Часть В
1-[2-(2-аминоэтокси)этил]-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин (919 мг, 2,76 ммоля) растворили в 30 мл безводного CH2Cl2 и охладили до 0°С в атмосфере азота. Далее в реакционную смесь добавили фенилизоцианат (300 мкл, 2,76 ммоля) и Et3N (0,77 мл, 5,51 ммоля); смеси дали медленно нагреться до комнатной температуры. После перемешивания, длившегося всю ночь, реакцию остановили путем добавления насыщенного раствора NaHCO3 (30 мл). Органический слой отделили и промыли водой и рассолом, высушили над Na2SO4 и концентрировали при пониженном давлении с получением твердого вещества желтого цвета. Это вещество растерли с Et2O (30 мл), куда было добавлено несколько капель МеОН. Твердый продукт отделили фильтрованием и высушили под вакуумом, получив 460 мг N-(2-{2-[4-амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил)-N'-фенилмочевины в виде белого порошка. Температура плавления 180-182°С.
Данные масс-спектрометрии MS: 453 (М+Н)+.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,51 (с, 1Н); 7,37 (д, J=7,7 Гц, 2Н); 7,19 (т, J=8,2 Гц, 2Н); 6,86 (т, J=7,7 Гц, 1Н); 6,11 (т, J=5,5 Гц, 2Н); 5,70 (с, 2Н); 4,43 (т, J=5,1 Гц, 2Н); 3,78-3,69 (м, 4Н); 3,39 (т, J=5,6 Гц, 2Н); 3,25 (с, 3Н); 3,19 (м, 2Н); 3,10 (т, J=6,8 Гц, 2Н); 2,91 (м, 2Н); 2,64 (м, 2Н); 1,72 (м, 4Н).
Данные 13С ЯМР (75 МГц, ДМСО-d6): δ 155,5; 151,3; 149,3; 146,3; 140,8; 138,5; 129,0; 125,0; 121,4; 118,0; 105,6; 70,6; 70,5; 70,4; 58,4; 44,6; 39,2; 32,7; 27,6; 23,8; 23,1; 23,0.
Данные элементного анализа: Рассчитано для С24Н32N6О3: С 63,70%, Н 7,13%, N 18,57%. Найдено: С 63,33%, Н 7,16%, N 18,66%.
Пример 3
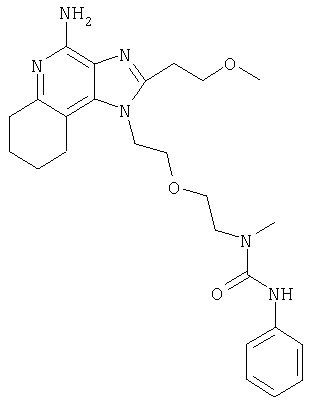
N-(2-{2-[4-амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил]этокси}этил)-N-метил-N'-фенилмочевина
Часть А
Гидрид натрия (60%-ная дисперсия в масле, 9,1 г, 228 ммолей) поместили в круглодонную колбу и промыли гексаном (3×) в атмосфере азота. Высушенный гидрид натрия растворили в 800 мл безводного ТГФ. К перемешиваемому раствору гидрида натрия добавляли в течение 40 мин раствор трет-бутил-2-(2-азидоэтокси)этилкарбамата (41,9 г, 182 ммолей). По завершении добавления реакционную смесь перемешивали еще в течение 20 мин, после чего добавили метилйодид (13,6 мл, 218 ммолей). После перемешивания, длившегося всю ночь, реакцию остановили путем добавления 300 мл насыщенного раствора NaHCO3. Далее в реакционную смесь внесли 200 мл воды и 1 л Et2O. Органическую фазу отделили и промыли водой и рассолом. Органическую часть высушили над MgSO4 и концентрировали при пониженном давлении, получив 41,9 г трет-бутил-2-(2-азидоэтокси)этил(метил)карбамата в виде жидкости желтого цвета.
Часть В
Раствор трет-бутил-2-(2-азидоэтокси)этил(метил)карбамата (41,9 г, 170 ммолей) в 600 мл МеОН обрабатывали 2,5 г Pd (10%) на углероде, встряхивая в атмосфере водорода (3 бар) в течение 24 часов. Далее раствор отфильтровали через целит и концентрировали с получением 37,2 неочищенного трет-бутил-2-(2-аминоэтокси)этил(метил)карбамата в виде жидкости светло-желтого цвета.
Часть С
В атмосфере азота к раствору 4-хлор-3-нитрохинолина (32,3 г, 155 ммоль) в 400 мл безводного CH2Cl2 добавили при перемешивании триэтиламин (43,1 мл, 310 ммоль) и трет-бутил-2-(2-аминоэтокси)этил(метил)карбамат (37,2 г, 171 ммолей). После перемешивания в течение ночи реакционную смесь промыли водой (2×300 мл) и рассолом (300 мл). Органическую часть высушили над Na2SO4 и концентрировали, получив маслянистую жидкость коричневого цвета. После очистки способом хроматографии на колонке (SiO2, элюирование смесью этилацетат-гексан от 33 до 67%) получили 46,7 г трет-бутилметил(2-{2-[(3-нитрохинолин-4-ил)амино]этокси}этил)карбамата в виде твердого вещества желтого цвета.
Часть D
Раствор трет-бутилметил(2-{2-[(3-нитрохинолин-4-ил)амино]этокси}этил)карбамата (6,56 г, 16,8 ммоля) в 75 мл толуола обрабатывали 0,5 г Pt (5%) на углероде, встряхивая в атмосфере водорода (3 бар) в течение 24 часов. Раствор отфильтровали через целит и концентрировали, получив неочищенный трет-бутил-2-{2-[(3-аминохинолин-4-ил)амино]этокси}этил(метил)карбамат в виде сиропообразной жидкости оранжевого цвета. Продукт использовали без дальнейшей очистки.
Часть Е
Раствор трет-бутил-2-{2-[(3-аминохинолин-4-ил)амино]этокси}этил(метил)карбамата (6,05 г, 16,8 ммоля) в 200 мл безводного CH2Cl2 охладили до 0°С и обработали триэтиламином (2,40 мл, 17,2 ммоля). Далее по каплям добавляли метоксипропионилхлорид (1,72 мл, 17,2 ммоля) в течение 5 мин. Реакционную смесь нагрели до комнатной температуры и перемешивание продолжали еще в течение 3 часов. Далее реакционную смесь концентрировали при пониженном давлении с получением твердого вещества оранжевого цвета. Данное вещество растворили в 200 мл EtoH и к нему добавили 7,2 мл триэтиламина. Смесь нагревали с обратным холодильником при перемешивании в течение ночи в атмосфере азота. После этого реакционную смесь концентрировали досуха при пониженном давлении и обработали 300 мл Et2O. Далее смесь фильтровали; фильтрат концентрировали при пониженном давлении с получением твердого вещества коричневого цвета. Это вещество растворили в 300 мл СН2Cl2 и промыли водой и рассолом. Органическую часть высушивали над Na2SO4 и концентрировали, получив маслянистую жидкость коричневого цвета. Эту жидкость растворили в 100 мл горячего МеОН и обработали активированным углем. Горячий раствор отфильтровали и концентрировали, получив 7,20 г трет-бутил-2-{2-[2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил(метил)карбамата в виде сиропообразной жидкости желтого цвета.
Часть F
Раствор трет-бутил-2-{2-[2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил]этокси}этил(метил)карбамата (7,20 г, 16,8 ммоля) в 200 мл CH2Cl2 обрабатывали 3-хлорпероксибензойной кислотой (77%, 4,32 г, 19,3 ммоля). После перемешивания в течение 6 часов реакционную смесь обработали насыщенным раствором NaHCO3 и разделили образовавшиеся слои. Органическую часть промыли водой и рассолом, высушили над Na2SO4 и концентрировали, получив 7,05 г трет-бутил-2-{2-[2-(2-метоксиэтил)-5-оксидо-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил(метил)карбамата в виде твердого вещества светло-коричневого цвета.
Часть G
Раствор трет-бутил-2-{2-[2-(2-метоксиэтил)-5-оксидо-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил(метил)карбамата (7,05 г, 15,9 ммоля) в 100 мл 1,2-дихлорэтана нагрели до 80°С и обработали 5 мл концентрированного раствора NH4OH. При быстром перемешивании к раствору добавляли твердый п-толуолсульфонилхлорид (3,33 г, 17,5 ммол) в течение 10 мин. Далее к реакционной смеси добавили дополнительное количество концентрированного раствора NH4OH (5 мл) и поместили смесь в автоклав; нагревание продолжалось еще в течение 4 часов. Далее реакционную смесь охладили и обработали 100 мл CH2Cl2. После этого реакционную смесь промыли водой, 1%-ным раствором Na2СО3 (3×) и рассолом. Органическую часть высушили над Na2SO4 и концентрировали, получив 6,50 г трет-бутил-2-{2-[4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил(метил)карбамата в виде маслянистой жидкости коричневого цвета.
Часть Н
Трет-бутил-2-{2-[4-амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил]этокси}этил(метил)карбамат (6,50 г, 14,7 ммоля) растворили в 100 мл EtOH и обработали 20 мл 2М раствора HCl в EtOH; смесь нагревали с обратным холодильником при перемешивании. Через 6 часов реакционную смесь охладили и отфильтровали, получив смолообразное вещество. Это вещество промыли EtOH и Et2O и высушили под вакуумом, получив солянокислую соль в виде порошка светло-коричневого цвета. Свободное основание получили путем растворения соли в 50 мл воды и обработки 5 мл концентрированного NH4OH. Водную суспензию экстрагировали CH2Cl2 (5×50 мл). Соединенные органические слои высушили над Na2SO4 и концентрировали, получив 3,93 г 2-(2-метоксиэтил)-1-{2-[2-(метиламино)этокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амина в виде порошка светло-коричневого цвета.
Данные масс-спектрометрии MS: 344 (М+Н)+.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,07 (д, J=7,7 Гц, 1Н); 7,62 (дд, J=1,0; 8,3 Гц, 1Н);7,42(ддд, J=1,0; 7,1;8,2 Гц, 1 Н); 7,22 (ддд, J=1,1; 7,1; 8,2 Гц, 1Н); 6,49 (с, 2Н); 4,75 (т, J=5,1 Гц, 2Н); 3,83 (т, J=6,8 Гц, 4Н); 3,35 (т, J=5,6 Гц, 2Н); 3,30 (с, 3Н); 3,21 (т, J=6,9 Гц, 2Н); 2,45 (т, J=5,6 Гц, 2Н); 2,12 (с, 3Н).
Часть I
2-(2-Метоксиэтил)-1-{2-[2-(метиламино)этокси]этил}-1Н-имидазо[4,5-финолин-4-амин (929 мг, 2,71 ммоля) растворили в 30 мл безводного CH2Cl2 и обработали фенилизоцианатом (300 мкл, 2,76 ммоля). После перемешивания в течение ночи в атмосфере азота реакционную смесь концентрировали при пониженном давлении. При очистке способом хроматографии на колонке (SiO2, элюент 3%-ный раствор МеОН в CHCl3, насыщенный водным раствором NH4OH) получили продукт в виде твердого вещества белого цвета. При кристаллизации из смеси Н2O-МеОН получили 610 мг N-(2-{2-[4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил-N-метил-N'-фенилмочевины в виде белых чешуйчатых кристаллов. Температура плавления 184,8-185,8°С.
Данные масс-спектрометрии MS: 463 (М+Н)+.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,16 (с, 1Н); 8,06 (д, J=7,7 Гц, 1Н); 7,61 (дд, J=1,0; 8,3 Гц, 1Н); 7,43-7,38 (м, 3Н); 7,25-7,17 (м, 3Н); 6,91 (т, J=7,3 Гц, 1Н); 6,47 (с, 2Н); 4,76 (т, J=5,0 Гц, 2Н); 3,88 (т, J=5,1 Гц, 2Н); 3,78 (т, J=6,8 Гц, 2Н); 3,48(т, J=5,2 Гц, 2Н); 3,39 (т, J=5,4 Гц, 2Н); 3,27 (с, 3Н); 3,20 (т, J=6,8 Гц, 2Н); 2,82 (с, 3Н).
Данные 13С ЯМР (75 МГц, ДМСО-d6): δ 155,6; 152,0; 151,9; 145,1; 140,9; 132,7; 128,5; 126,7; 126,6; 122,0; 121,4; 120,5; 120,1; 115,1; 70,5; 69,6; 69,4; 58,4; 47,7; 45,5; 35,4; 27,6.
Данные элементного анализа: Рассчитано для C25H30N6O3.0,12 H2O С 64,62%, Н 6,56%, N 18,08%. Найдено: С 64,69%, Н 6,65%, N 18,09%.
Пример 4
N-(2-{2-[4-амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил]этокси}этил)-N-метил-N'-фенилмочевина
Часть А
2-(2-Метоксиэтил)-1-{2-[2-(метиламино)этокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин (4,22 г, 12,3 ммоля) растворили в 25 мл трифторуксусной кислоты и обработали PtO2 (0,5 г). Реакционную смесь встряхивали в атмосфере водорода (3 бар). Через 4 дня добавили дополнительное количество PtO3 (0,5 г) и продолжали гидрогенизацию еще в течение 3 дней. Реакционную смесь отфильтровали через целит и концентрировали при пониженном давлении с получением маслянистой жидкости желтого цвета. Эту жидкость растворили в 50 мл воды и проэкстрагировали 50 мл CHCl3. Органическую часть отделили и отбросили. Водной части придали основную реакцию (рН прибл. 12), добавляя 10%-ный раствор NaOH. Далее провели экстракцию CHCl3 (6×50 мл) и соединенные органические слои высушили над Na2SO4 и концентрировали, получив маслянистую жидкость коричневого цвета. Эту жидкость растворили в 100 мл горячего МеОН и обработали 1 г активированного угля. Горячий раствор отфильтровали через целит и концентрировали досуха. Полученное смолистое вещество высаживали несколько раз из Et2O; продукт 2-(2-метоксиэтил)-1-{2-[2- (метиламино)этокси]этил}-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин(3,19 г) в виде порошка почти белого цвета.
Данные масс-спектрометрии MS: 348 (М+Н)+.
Данные 1H ЯМР (300 МГц, CDCl3): δ 4,84 (с, 2 Н); 4,48 (т, J=5,7 Гц, 2 Н); 3,84 (т, J=6,7 Гц, 2Н); 3,70 (т, J=5,7 Гц, 2Н); 3,46 (т, J=5,1 Гц, 2Н); 3,36 (с, 3Н); 3,14 (т, J=6,7 Гц, 2Н); 2,96 (м, 2Н); 2,83 (м, 2Н); 2,65 (т, J=5,1 Гц, 2Н); 2,36 (с, 3Н); 1,85 (м, 4Н).
Часть В
2-(2-Метоксиэтил)-1-{2-[2-(метиламино)этокси]этил}-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин (750 мг, 2,16 ммоля) растворили в 30 мл безводного СН2Cl2 и обработали фенилизоцианатом (239 мкл, 2,20 ммоля). После перемешивания в течение ночи в атмосфере азота реакционную смесь концентрировали при пониженном давлении. При кристаллизации из смеси EtOAc-СН2Cl2 получили 170 мг N-(2-{2-[4-амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил]этокси}этил-N-метил-N'-фенилмочевины в виде белых пушистых кристаллов. Температура плавления 167,7-170,0°С.
Данные масс-спектрометрии MS: 467 (М+Н)+.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,17 (с, 1Н); 7,43 (д, J=7,6 Гц, 2Н); 7,21 (т, J=7,9 Гц, 2Н); 6,91 (т, J=7,3 Гц, 1Н); 5,65 (с, 2Н); 4,43 (т, J=5,0 Гц, 2Н); 3,72 (т, J=7,0 Гц, 2Н); 3,70 (т, J=5,2 Гц, 2Н); 3,46-3,41 (м, 4Н); 3,24 (с, 3Н); 3,07(т, J=6,9 Гц, 2Н); 2,92 (м, 2Н); 2,85 (с, 3Н); 2,64 (м, 2Н); 1,72 (м, 4Н).
Данные 13С ЯМР (75 МГц, ДМСО-d6): δ 155,6; 151,2; 149,3; 146,3; 140,9; 138,4; 128,5; 124,9; 122,0; 120,1; 105,5; 70,7; 70,5; 69,5; 58,4; 48,0; 44,6; 35,5; 32,8; 27,6; 23,8; 23,1; 23,0.
Данные элементного анализа: Рассчитано для С25Н34N6О3: С 64,36%, Н 7,35%, N 18,01%. Найдено: С 64,04%, Н 7,38%, N 18,02%.
Пример 5
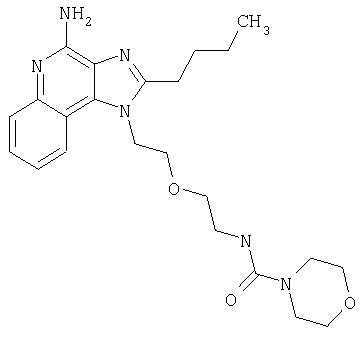
N-(2-{2-[4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил)морфолин-4-карбоксамид
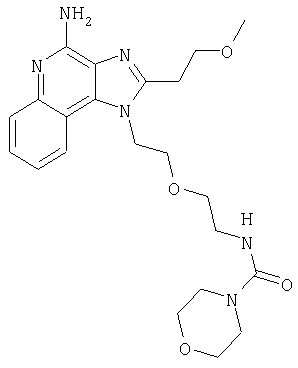
В атмосфере азота 1-[2-(2-аминоэтокси)этил]-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амин (0,75 г, 2,3 ммоля) растворили в дихлорметане (30 мл) и триэтиламине (0,64 мл, 4,6 ммоля) при осторожном нагревании и энергичном перемешивании. Раствор охладили в бане с ледяной водой, после чего в раствор по каплям ввели 4-морфолинкарбонилхлорид (0,27 мл, 2,3 ммоля). Охлаждающую баню удалили и продолжали перемешивание еще в течение 4 часов. Реакцию остановили путем добавления насыщенного раствора бикарбоната натрия (25 мл). После разделения фаз органический слой промыли водой (3×25 мл) и рассолом (25 мл), высушили над Na2SO4, отфильтровали и концентрировали, получив желтую пену. Продукт рекристаллизовали из смеси дихлорметана и этилацетата. Кристаллы продукта растирали с эфиром (2×5 мл) с целью удаления остаточного количества растворителя. После сушки в вакуумной печи получили 200 мг N-(2-{2-[4-амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил]этокси}этил)морфолин-4-карбоксамида в виде кристаллического вещества желтовато-коричневого цвета.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,06 (д, J=8,1 Гц, 1Н); 7,61 (д, J=7,3 Гц, 1Н); 7,42 (т, J=7,2 Гц, 1Н); 7,23 (т, J=7,8 Гц, 1Н); 6,51 (с, 2Н); 6,33 (т, J=5,0 Гц, 1Н); 4,74 (т, J=4,3 Гц, 2Н); 3,85-3,81 (м, 4Н); 3,49 (т, J=4,3 Гц, 4Н); 3,33 (т, J=5,9 Гц, 2Н); 3,30 (с, 3Н); 3,21 (т, J=6,8 Гц, 2Н); 3,14 (т, J=4,5 Гц, 4Н); 3,08 (т, J=6,0 Гц, 2Н).
Данные 13С ЯМР (75 МГц, ДМСО-d6): δ 157,8; 151,9; 145,0; 132,7; 126,7; 126,6; 121,4; 120,5; 115,1; 70,4; 70,2; 69,2; 58,4; 45,5; 44,0; 27,6.
Данные элементного анализа: Рассчитано для С22Н30N6O4: С 59,71%, Н 6,83%, N 18.99%. Найдено: С 59,71%, Н 6,80%, N 18,78%.
Данные масс-спектрометрии MS: 443 (М+Н).
Пример 6
N-(2-{2-[4-амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил]этокси}этил)-М-метилморфолин-4-карбоксамид
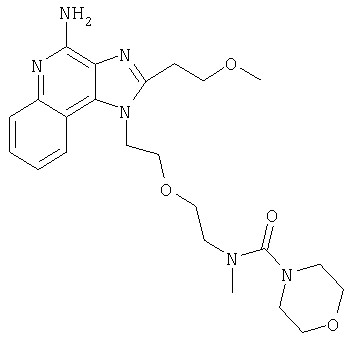
В атмосфере азота 2-(2-метоксиэтил)-1-{2-[2-(метиламино)этокси]этил}-1Н-имидазо[4,5-с]хинолин-4-амин (802 мг, 2,34 ммоля) растворили в 30 мл безводного СН2Cl2 и охладили до 0°С. При перемешивании добавили Et3N (0,65 мл, 4,68 ммоль) и морфолинкарбонилхлорид (273 мкл, 2,34 ммоля); реакционную смесь оставили на ночь для разогрева до комнатной температуры. Реакцию остановили, добавляя насыщенный раствор NaHCO3 (30 мл) и СН2Cl2 (30 мл). Органический слой после отделения промыли водой и рассолом, высушили над Na2SO4 и концентрировали при пониженном давлении. При очистке способом хроматографии на колонке (SiO2, элюент 2-5%-ный раствор МеОН в CHCl3, насыщенный водным раствором NH4OH) получили продукт в виде бесцветной пены. При кристаллизации из EtOAc получили 640 мг N-(2-{2-[4-амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил]этокси}этил-N-метилморфолин-4-карбоксамида в виде белых кристаллов. Температура плавления 121,8-122,3°С.
Данные масс-спектрометрии MS: 457 (М+Н)+.
Данные 1H ЯМР (500 МГц, ДМСО-d6): δ 8,06 (дд, J=0,9; 8,3 Гц, 1Н); 7,61 (дд, J=1,1; 8,3 Гц, 1Н); 7,41 (ддд, J=1,2; 7,0; 8,3 Гц, 1Н); 7,22 (ддд, J=1,3; 7,0; 8,1 Гц, 1Н); 6,44 (с, 2Н); 4,74 (т, J=5,2 Гц, 2Н); 3,84 (т, J=5,2 Гц, 2Н); 3,82 (т, J=6,9 Гц, 2Н); 3,50-3,43 (м, 6Н); 3,30 (с, 3Н); 3,20 (т, J=6,9 Гц, 2Н); 3,16 (т, J=5,5 Гц, 2Н); 2,88 (т, J=4,7 Гц, 4Н); 2,59 (с, 3Н).
Данные 13С ЯМР (75 МГц, ДМСО-d6): δ 163,8; 152,0; 151,8; 145,2; 132,7; 126,7; 121,3; 120,6; 115,1; 70,4; 69,4; 68,9; 66,1; 58,5; 49,1; 47,3; 45,5; 36,9; 27,7.
Данные элементного анализа: Рассчитано для C23H32N6O4: С 60,51%, Н 7,07%, N 18,41%. Найдено: С 60,56%, Н 6,85%, N 18,19%.
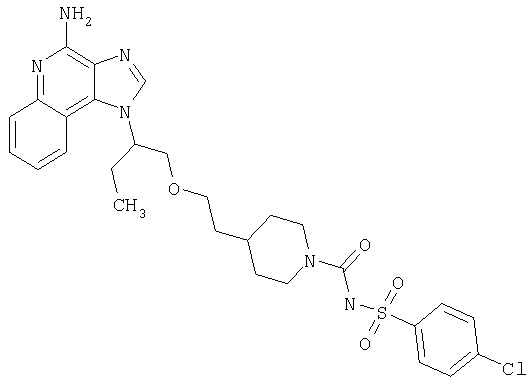
Примеры 7-21
Часть А
Раствор трет-бутил-2-{2-[(3-аминохинолин-4-ил)амино]этокси}этилкарбамата (3,46 г, 10,0 ммоля) в 50 мл толуола обработали триэтилортовалератом (2,5 мл, 14,5 ммоля) и реакционную смесь стали нагревать с обратным холодильником. Далее добавили 25 мг пиридинийгидрохлорида и нагревание с обратным холодильником продолжили еще 4 ч. Реакционную смесь концентрировали досуха при пониженном давлении. Продукт растворили в 50 мл CH2Cl2 и промыли насыщенным раствором NaHCO3, водой и рассолом. Органическую часть высушили над Na2SO4 и концентрировали, получив маслянистую жидкость зеленого цвета. Эту жидкость растворили в 50 мл горячего МеОН и обработали активированным углем. Горячий раствор отфильтровали и концентрировали, получив 4,12 г трет-бутил-2-[2-(2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]этилкарбамата в виде маслянистой жидкости желтого цвета.
Часть В
Раствор трет-бутил-2-[2-(2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]этилкарбамата (4,12 г, 10,0 ммолей) в 50 мл CH2Cl2 обработали 3-хлорпероксибензойной кислотой (77%, 2,5 г, 11,2 ммоля). После перемешивания в течение 5 часов реакционную смесь обработали насыщенным раствором NaHCO3 и разделили слои. Органическую часть промыли водой и рассолом, высушили над NaSO4 и концентрировали, получив 3,68 г трет-бутил 2-[2-(2-бутил-5-оксидо-1H-имидазо[4,5-с]хинолин-1-ил)этокси]этилкарбамата в виде пены светло-коричневого цвета.
Часть С
Раствор трет-бутил 2-[2-(2-бутил-5-оксидо-1H-имидазо[4,5-с]хинолин-1-ил)этокси]этилкарбамата (3,68 г, 8,60 ммоль) в 100 мл 1,2-дихлорэтана нагрели до 80°С и обработали 10 мл концентрированного раствора NH4OH. При быстром перемешивании в раствор добавляли твердый п-толуолсульфонилхлорид (1,87 г, 9,81 ммоля) в течение 10 мин. Реакционную смесь поместили в автоклав и нагревание продолжали еще в течение 2 часов. После этого реакционную смесь охладили и обработали 100 мл CH2Cl2. Далее реакционную смесь промыли водой, 1%-ным раствором Na2CO2 (3×) и рассолом. Органическую часть высушили над Na2SO4 и концентрировали, получив 3,68 г трет-бутил 2-[2-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]этилкарбамата в виде пены светло-коричневого цвета.
Часть D
Трет-бутил 2-[2-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]этилкарбамат (3,68 г, 8,60 ммоль) суспендировали в 20 мл 2М раствора HCl в EtOH и суспензию нагревали с обратным холодильником при перемешивании. Через 3 часа реакционную смесь концентрировали, получив твердый остаток. Остаток растирали с горячим EtOH (50 мл) и отфильтровали, получив 2,90 г продукта в виде солянокислой соли. Свободное основание получили путем растворения соли в 50 мл воды и обработки 5 мл концентрированного NH2ОН. Эту водную суспензию экстрагировали CH2Cl2 (3×50 мл). Соединенные органические слои высушили над Na2SO4 и концентрировали, получив 1-[2-(2-аминоэтокси)этил]-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амина в виде порошка желтовато-коричневого цвета.
Данные масс-спектрометрии MS: 328 (М+Н)+.
Данные 1H ЯМР (300 МГц, CDCl3): δ 7,95 (д, J=8,3 Гц, 1Н); 7,83 (д, J=8,4 Гц, 1Н); 7,50 (м, 1Н); 7,30 (м, 1Н); 5,41 (с, 2Н); 4,69 (т, J=5,6 Гц, 2Н); 3,93 (т, J=5,6 Гц, 2Н); 3,39 (т, J=5,1 Гц, 2Н); 2,97 (т, J=7,9 Гц, 2Н); 2,76 (т, J=5,1 Гц, 2Н); 1,89 (м, 2Н); 1,52 (м, 2Н); 1,26 (шс, 2Н); 1,01 (т, J=7,3 Гц, 3Н).
Часть Е
Соединения, приведенные в нижеследующей таблице 1, были приготовлены по стадии (7) процесса (схема 1 реакции) (см. выше) с учетом следующих общих принципов.
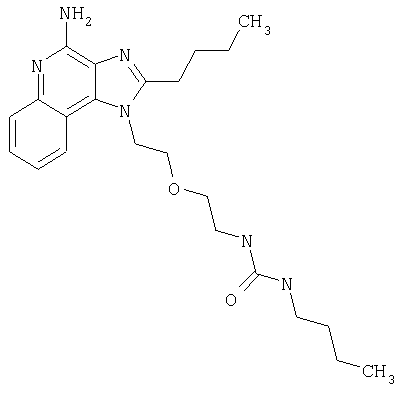
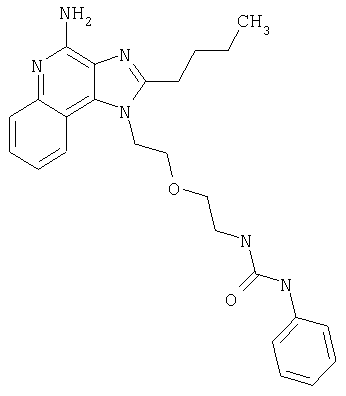
Изоцианат (84 мкмоль) ввели в пробирку с раствором 1-[2-(2-аминоэтокси)этил]-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амина (25 мг, 76 мкмолей) в дихлорметане (5 мл). Пробирку закрыли и поместили в аппарат для встряхивания при температуре окружающей среды; встряхивание продолжалось в течение 20 часов. Растворитель удалили вакуумным центрифугированием. Остаток очистили способом полупрепаративной ВЭЖХ; подробности процедуры описаны выше. В таблице приведены структура свободного основания и данные о наблюдаемой точной массе (М+Н).
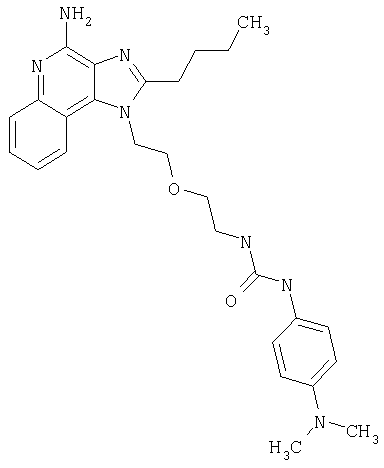
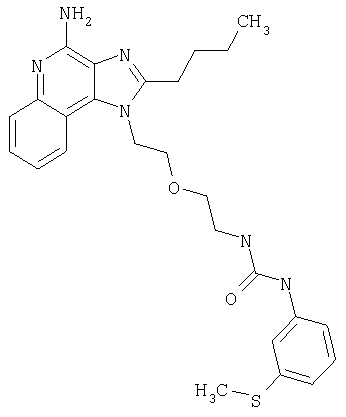

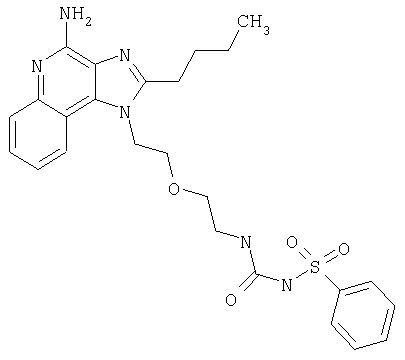
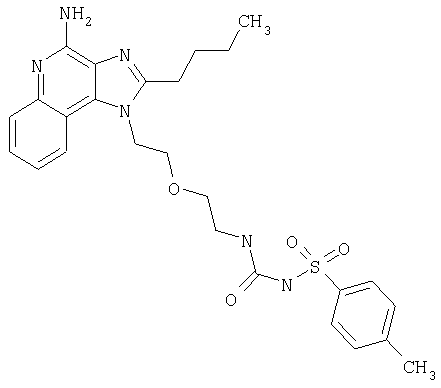
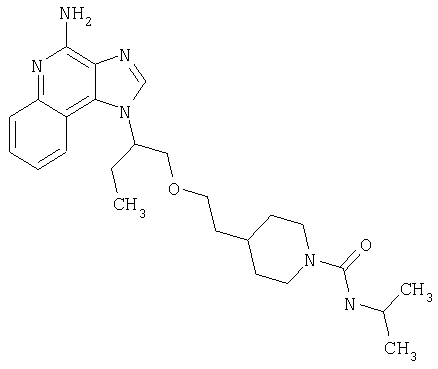
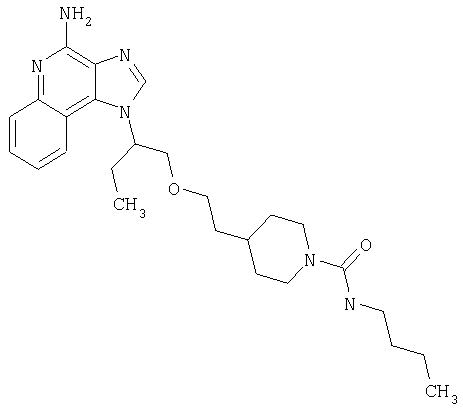
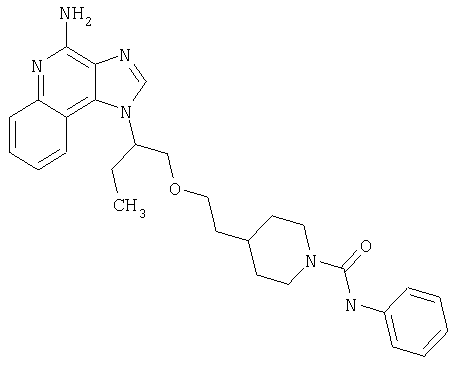
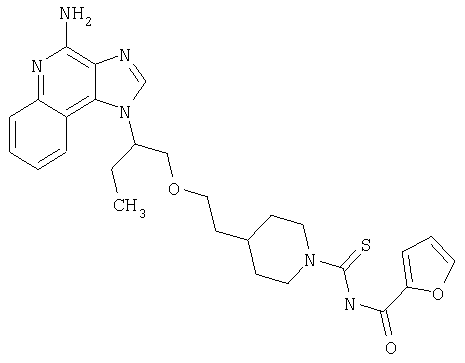
Примеры 22-36
Часть А
С учетом общеупотребительного способа, описанного в Части А для Примеров 7-21, провели реакцию между 4-пиперидинэтанолом (10 г, 77,4 ммоль) и ди-трет-бутилдикарбонатом (17,7 г, 81,3 ммоля), получив 13,1 г трет-бутил-4-(2-гидроксиэтил)пиперидин-1-карбоксилата в виде прозрачной маслянистой жидкости.
Часть В
К раствору имидазола (3,89 г, 57,1 ммоля) и трифенилфосфина (14,98 г, 57,1 ммоля) в дихлорметане (350 мл) добавили йод (7,97 г) тремя порциями. Через 5 мин ввели раствор продукта, полученного в Части А, в дихлорметане (70 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Далее добавили дополнительное количество йода (7,97 г) и перемешивание при температуре окружающей среды продолжили еще в течение 1 часа. Реакционную смесь промыли насыщенным раствором тиосульфата натрия (2×) и рассолом, высушили над сульфатом натрия, отфильтровали и концентрировали при пониженном давлении, получив маслянистый остаток. Остаток очистили способом хроматографии на колонке (силикагель, элюент 20%-ный раствор этилацетата в гексане), получив 15,52 г трет-бутил-4-(2-йодэтил)пиперидин-1-карбоксилата в виде маслянистой жидкости светло-желтого цвета.
Часть С
В атмосфере азота 2-(1Н-имидазо[4,5-с]хинолин-1-ил)бутан-1-ол (6,5 г, 26,9 ммоля) добавили тремя порциями к суспензии гидрида натрия (1,4 г, 60%, 35,0 ммолей) в безводном N,N-диметилформамиде. Реакционную массу перемешивали в течение 45 мин; к концу этого периода выделение газа закончилось. По каплям прибавляли трет-бутил-4-(2-йодэтил)пиперидин-1-карбоксилат (10,05 г, 29,6 ммоля) в течение 15 мин. Реакционную смесь перемешивали при температуре окружающей среды в течение 2,5 часов; далее массу нагрели до 100°С и оставили при перемешивании на ночь. Анализ способом ВЭЖХ показал, что реакция завершена приблизительно на 35%. В смесь ввели насыщенный раствор хлорида аммония и полученную смесь перемешивали еще в течение 20 мин, после чего провели экстракцию этилацетатом (2×). Полученные экстракты промыли водой (2×) и рассолом, соединили, высушили над сульфатом натрия, отфильтровали и концентрировали при пониженном давлении, получив маслянистую жидкость коричневого цвета. Эту жидкость очистили способом хроматографии на колонке (силикагель; элюировали последовательно 30%-ным раствором этилацетата в гексане, 50%-ным раствором этилацетата в гексане и этилацетатом), получив 2,2 г трет-бутил 4-{2-[2-(1Н-имидазо[4,5-с]хинолин-1-ил)бутокси]этил}пиперидин-1-карбоксилата.
Часть D
С учетом общеупотребительного способа, описанного в Части Н для Примеров 7-21, окислили продукт Части С, получив трет-бутил 4-{2-[2-(5-оксидо-1H-имидазо[4,5-с]хинолин-1-ил)бутокси]этил}пиперидин-1-карбоксилата в виде маслянистой жидкости.
Часть Е
Раствор гидроксида аммония (20 мл) добавили к раствору продукта Части D в дихлорметане (20 мл). Далее добавили раствор тозилхлорида (0,99 г, 5,2 ммоля) в дихлорметане (10 мл) в течение 5 мин. Полученную двухфазную реакционную смесь оставили на ночь при перемешивании. Реакционную смесь разбавили хлороформом и насыщенным раствором бикарбоната натрия. После разделения слоев органический слой высушили над сульфатом натрия, отфильтровали и концентрировали при пониженном давлении, получив стеклообразную массу коричневого цвета. Эту массу очистили способом хроматографии на колонке (силикагель; элюировали последовательно 50%-ным раствором этилацетата в гексане и этилацетатом), получив 1,0 г трет-бутил 4-{2-[2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)бутокси]этил}пиперидин-1-карбоксилата в виде стеклообразной пены желтого цвета.
Часть F
В атмосфере азота трет-бутил 4-{2-[2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)бутокси]этил}пиперидин-1-карбоксилат (1,00 г, 2,1 ммоля) смешали с 2 N раствором соляной кислоты в этаноле (10 мл, 20 ммолей); раствор перемешивали при температуре окружающей среды в течение 14 часов. Растворитель удалили под вакуумом, а полученное твердое вещество желтовато-коричневого цвета растворили в воде. Далее добавляли насыщенный водный раствор карбоната натрия до достижения рН 10. После экстракции дихлорметаном (3×) органические фракции соединили, промыли рассолом, высушили (Na2SO4), отфильтровали и убрали большую часть растворителя вакуумной обработкой. Далее добавили гексан, что привело к образованию осадка. После вакуумного фильтрования получили 0,5 г 1-{1-[(2-пиперидин-4-илэтокси)метил]пропил}-1H-имидазо[4,5-с]хинолин-4-амина в виде порошка желтовато-коричневого цвета.
Данные 1H ЯМР (300 МГц, ДМСО-d6): δ 8,34 (шс, 1Н); 8,19 (д, J=8,49 Гц, 1Н); 7,61 (дд, J=8,31; 1,13 Гц, 1Н); 7,45-7,39 (м, 1Н); 7,25-7,19 (м, 1Н); 6,55 (с, 2Н); 5,25-5,15 (м, 1Н); 4,00-3,80 (м, 2Н); 3,5-3,3 (м, 2Н); 2,8-2,64 (м, 2Н); 2,22-2,11 (м, 2Н); 2,09-1,99 (м, 2Н); 1,8-1,63 (шс, 1Н); 1,37-1,0 (м, 5Н); 0,95-0,7 (м, 5Н).
Данные 13С ЯМР (75 МГц, ДМСО-d6): (152,8; 145,8; 140,6; 133,0; 127,8; 127,0; 126,9; 121,3; 121,0; 115,5; 71,8; 68,1; 58,4; 46,1; 36,3; 33,1; 32,7; 24,5; 9,9.
Данные масс-спектрометрии MS(CI): m/e 368,2459 (368,2450 при расчете для С21Н30N50).
Часть G
Соединения, приведенные в нижеследующей таблице 2, были приготовлены в соответствии со стадией (7) процесса (схема I реакции) (см. выше) с учетом следующих общих принципов.
Изоцианат или изотиоцианат (75 мкмолей) ввели в пробирку с раствором 1-{1-[(2-пиперидин-4-илэтокси)метил]пропил}-1H-имидазо[4,5-с]хинолин-4-амина (25 мг, 68 мкмоль) в дихлорметане (5 мл). Пробирку закрыли и поместили в аппарат для встряхивания при температуре окружающей среды; встряхивание продолжалось 20 часов. Растворитель удалили вакуумным центрифугированием. Остаток очистили способом полупрепаративной ВЭЖХ; подробности процедуры описаны выше. В таблице приведены структура свободного основания и данные о наблюдаемой точной массе (М+Н).
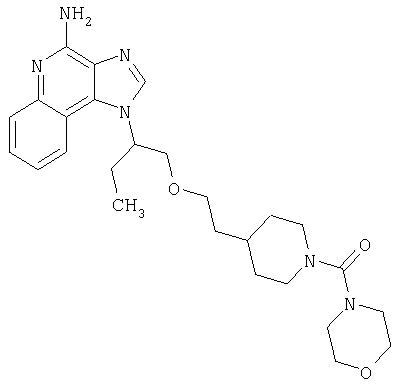
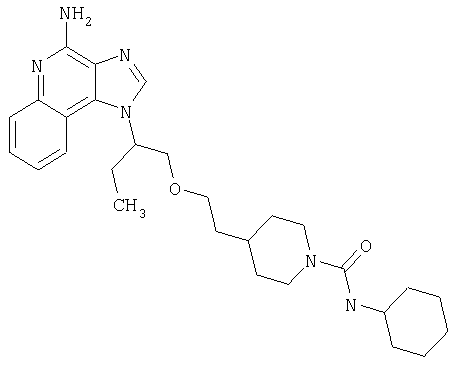
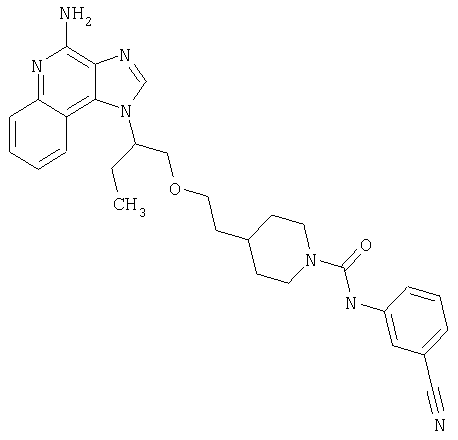
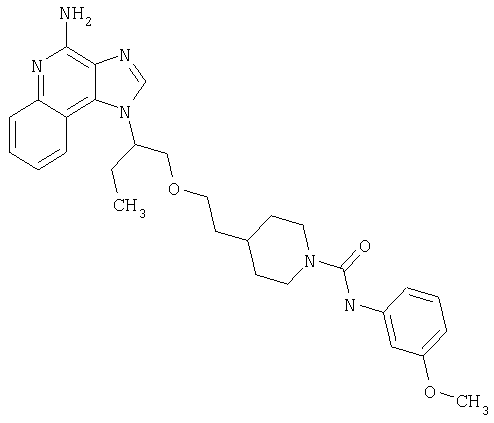
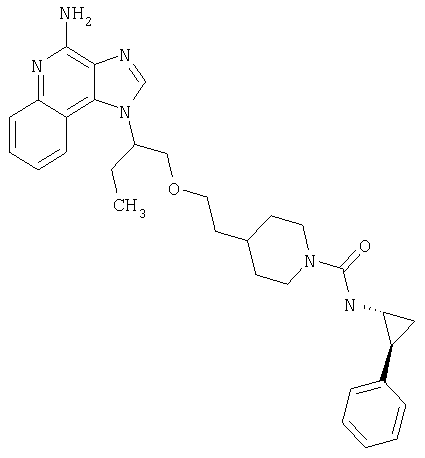

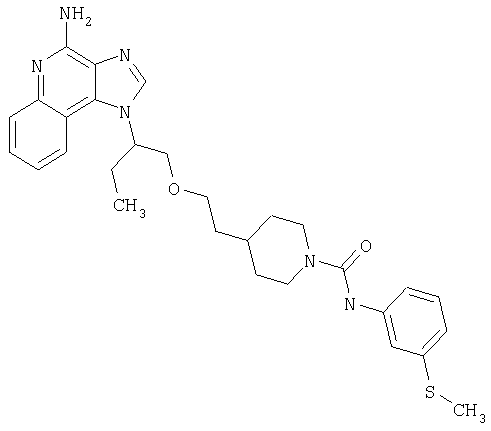
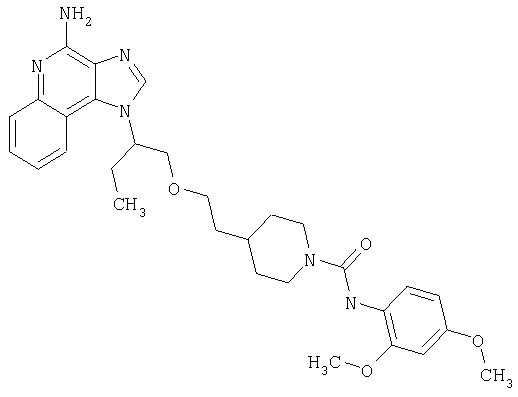
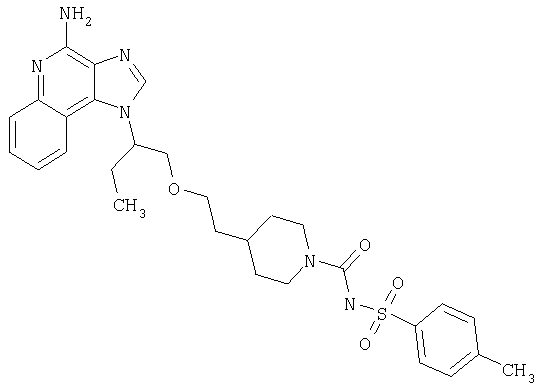

Примеры 37-44
Часть А
Раствор трет-бутил 2-{2-[(3-аминохинолин-4-ил)амино]этокси}этилкарбамата (6,92 г, 20,0 ммолей) в 100 мл толуола обработали триэтилортоформиатом (4,65 мл, 28,0 ммолей), и реакционную смесь стали нагревать с обратным холодильником. Далее добавили 100 мг пиридинийгидрохлорида и нагревание с обратным холодильником продолжили еще в течение 2 часов. Реакционную смесь концентрировали досуха при пониженном давлении. Продукт растворили в 200 мл CH2Cl2 и промыли насыщенным раствором NaHCO3, водой и рассолом. Органическую часть высушили над Na2SO4 и концентрировали, получив маслянистую жидкость зеленого цвета. Эту жидкость растворили в 200 мл горячего МеОН и обработали активированным углем (10 г). Горячий раствор отфильтровали и концентрировали, получив 5,25 г трет-бутил 2-[2-(1H-имидазо[4,5-с]хинолин-1-ил)этокси]этилкарбамата в виде маслянистой жидкости светло-желтого цвета.
Часть В
Раствор трет-бутил 2-[2-(1Н-имидазо[4,5-с]хинолин-1-ил)этокси]этилкарбамата (5,25 г, 14,7 ммолей) в 200 мл CH2Cl2 обработали 3-хлорпероксибензойной кислотой (77%, 3,63 г, 16,3 ммоля). После перемешивания в течение ночи реакционную смесь обработали насыщенным раствором NaHCO3 и разделили слои. Органическую часть промыли водой и рассолом, высушили над Na2SO4 и концентрировали, получив 4,60 г трет-бутил 2-[2-(5-оксидо-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]этилкарбамата в виде пены светло-коричневого цвета.
Часть С
Раствор трет-бутил 2-[2-(5-оксидо-1Н-имидазо[4,5-с]хинолин-1-ил)этокси]этилкарбамата (4,60 г, 12,4 ммоля) в 150 мл 1,2-дихлорэтана нагрели до 80°С и обработали 10 мл концентрированного раствора NH4OH. При быстром перемешивании добавляли твердый п-толуолсульфонилхлорид (2,71 г, 14,2 ммоля) в течение 10 мин. Реакционную смесь обработали дополнительным количеством (2 мл) концентрированного раствора NH4OH и поместили в автоклав; нагревание продолжали еще в течение 3 часов. После этого реакционную смесь охладили и обработали 100 мл CH2Cl2. Далее реакционную смесь промыли водой, 1%-ным раствором Na2СО3 (3×) и рассолом. Органическую часть высушили над Na2SO4 и концентрировали, получив 4,56 г трет-бутил 2-[2-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)этокси]этилкарбамата в виде пены светло-коричневого цвета.
Часть D
Трет-бутил 2-[2-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)этокси]этилкарбамат (4,56 г, 12,3 ммоль) растворили в 100 мл EtOH и обработали 30 мл 2 М раствора HCl в EtOH. Смесь нагревали с обратным холодильником при перемешивании. Через 3 часа реакционную смесь концентрировали, получив твердый остаток. Остаток растирали с горячим EtOH (100 мл) и отфильтровали, получив продукт в виде солянокислой соли. Свободное основание получили путем растворения соли в 50 мл воды и обработки 5 мл концентрированного NH4OH. Эту водную суспензию экстрагировали СН2Cl2 (5×50 мл). Соединенные органические слои высушили над Na2SO4 и концентрировали, получив 1,35 г 1-[2-(2-аминоэтокси)этил]-1H-имидазо[4,5-с]хинолин-4-амина в виде порошка желтовато-коричневого цвета.
Данные масс-спектрометрии MS: 272 (М+Н)+.
Данные 1H ЯМР (300 МГц, CDCl3): δ 7,98 (д, J=8,2 Гц, 1Н); 7,88 (д, J=8,4 Гц, 1Н); 7,84 (д, J=8,4 Гц, 1Н); 7,54 (м, 1 Н); 7,32 (м, 1 Н); 5,43 (с, 2 Н); 4,74 (т, J=5,2 Гц, 2 Н); 3,97 (т, J=5,2 Гц, 2 Н); 3,42 (т, J=5,1 Гц, 2 Н); 2,78 (т, J=5,1 Гц, 2Н); 1,10 (шс, 2Н).
Часть Е
Соединения, приведенные в нижеследующей таблице 3, были приготовлены в соответствии со стадией (7) процесса (схема 1 реакции) (см. выше) с учетом следующих общих принципов.
1-[2-(2-Аминоэтокси)этил]-1Н-имидазо[4,5-с]хинолин-4-амин (20 мг, 74 мкмолей) смешали в пробирке с 1-метил-2-пирролидоном (5 мл) и приготовили раствор путем ультразвукового облучения при нагреве. Далее добавили изоцианат (81 мкмолей), пробирку закрыли и поместили в аппарат для встряхивания при температуре окружающей среды; встряхивание продолжалось в течение 20 часов. Растворитель удалили вакуумным центрифугированием. Остаток очистили способом полупрепаративной ВЭЖХ; подробности процедуры описаны выше. В таблице приведены структура свободного основания и данные о наблюдаемой точной массе (М+Н).
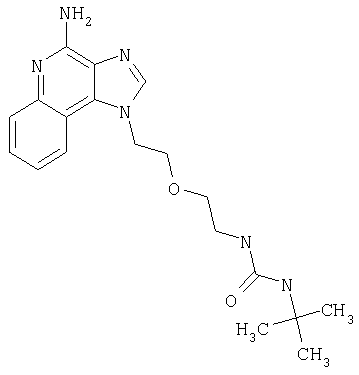
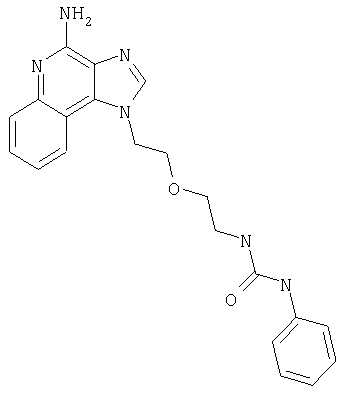
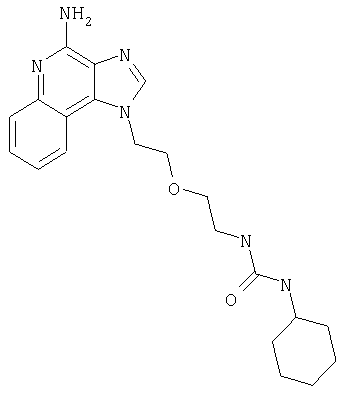
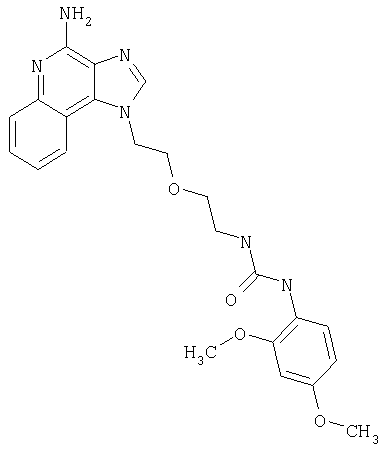
СТИМУЛИРОВАНИЕ СИНТЕЗА ЦИТОКИНОВ В КЛЕТКАХ ЧЕЛОВЕКА
Для оценки кинетики стимулирования синтеза цитокинов применяют систему ин-витро с клетками крови человека. Активность синтеза измеряют по количеству интерферона и фактора некроза опухолей (α) (ИФ и ФНО соответственно), секретируемых в культуральную среду, как описано Тестерманом и др. (Testerman et al., Cytokine Induction by the Immunomodulators Imiquimod and S-27609, Journal of Leukocyte Biology, 58, 365-372, September, 1995).
Подготовка клеток крови для выращивания в культуральной среде
Цельную кровь, взятую у здоровых доноров из вены, помещают в вакуумированные пробирки, содержащие ЭДТУ. Одноядерные клетки периферической крови (ОКПК) отделяют от цельной крови путем центрифугирования в градиенте плотности с использованием реагента Histopaque®-1077. ОКПК дважды промывают сбалансированным раствором солей Хэнка и далее суспендируют в количестве (3-4)×106 кл/мл в полной среде RPMI. Суспензию ОКПК вносят на плоские стерильные планшеты для культуры тканей с 48 лунками (Costar, Cambridge, МА или Becton Dickinson Labware, Lincoln Park, NJ); в лунках содержатся равные объемы полной среды RPMI, содержащей исследуемое соединение.
Приготовление исследуемого соединения
Соединения солюбилизируют в диметилсульфоксиде (ДМСО). Концентрация в ДМСО не должна превышать конечного значения (1%) при добавлении в лунки.
Инкубация
Раствор исследуемого соединения вводят в первую лунку, содержащую полную среду RPMI, а в остальных лунках проводят серийные разведения. Далее добавляют суспензию ОКПК в равных объемах, получая требуемый диапазон концентраций исследуемого соединения. Конечная концентрация суспензии ОКПК составляет (1,5-2)×106 кл/мл. Планшеты накрывают стерильными пластиковыми крышками, осторожно перемешивают и инкубируют в течение 18-24 часов при температуре 37°С в атмосфере, содержащей 5% углекислоты.
Разделение
После инкубации планшеты центрифугируют в течение 5-10 мин при скорости 1000 мин-1 (прибл. 200 g) и температуре 4°С. Надосадочную жидкость, не содержащую клеток, удаляют при помощи стерильной полипропиленовой пипетки и переносят в стерильные полипропиленовые пробирки. Образцы хранят до анализов при температуре от -30 до -70°С. Выполняют анализы на интерферон (α) и фактор некроза опухолей (α) способом ЭЛАЙЗА.
Анализы на интерферон (α) и фактор некроза опухолей (α) способом ЭЛАЙЗА
Концентрацию интерферона (α) определяли при помощи набора ЭЛАЙЗА под названием Human Multi-Species Kit производства фирмы PBL Biomedical Laboratories, New Brunswick, NJ. Результаты выражали в пг/мл.
Концентрацию фактора некроза опухолей (α) (ФНО) определяли при помощи наборов ЭЛАЙЗА производства фирм Genzyme, Cambridge, MA; R&D Systems, Minneapolis, MN; Pharmingen, San Diego, CA. Результаты выражали в пг/мл.
Приведенная ниже таблица 4 дает самые низкие концентрации, которые, как было обнаружено, стимулируют синтез интерферона и стимулируют синтез фактора некроза опухолей для каждого соединения. Звездочка * означает, что стимулирования не было отмечено при любой концентрации исследуемых соединений. В целом самая высокая исследованная концентрация составляла 10-30 мкМ.
Индуцирование синтеза цитокинов в человеческих клетках
Изобретение относится к карбамидзамещенным имидазохинолиновым эфирам. Описывается соединение формулы (I):
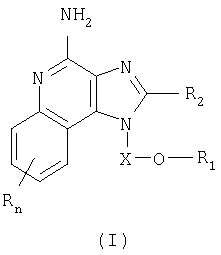
Х представляет собой -CHR5-, -CHR5-алкильную группу; R1 выбран из группы, содержащей радикалы: -R4-NR8-CR3-NR5-Z-R6-алкил; -R4-NR8-CR3-NR5-Z-R6-фенил; -R4-NR8-CR3-NR5-Z-R6-фуранил; -R4-NR8-CR3-NR5R7; при этом фенил замещается или не замещается одним или более заместителями, выбранными из группы, состоящей из метила; метоксила; метилтио; циано; водорода; диметиламина и ацетила; R2 выбран из группы, содержащий радикалы: -водород; -алкил; -алкил-Y-алкил; R3 представляет собой =O или =S; R4 представляет собой алкил, в который может войти одна или несколько -O-групп; каждый R5 представляет собой Н или C1-10алкил; R6 представляет собой связь или алкил; R7 соединяется с R5, образуя цикл; R8 представляет собой Н, С1-10алкил или R4 и R8 могут соединяться, образуя морфолиновое кольцо; Y представляет собой -O-; Z представляет собой связь, -СО- или -SO2-; n имеет значение 0; каждый R выбран независимо из группы, состоящей из радикалов С1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил; или же соль фармацевтического качества указанных соединений. Также описываются соединения общей формулы (II), промежуточные соединения общих формул (III) и (IV), фармацевтические составы на основе соединений формул (I) и (II) являющиеся иммуномодуляторами для биосинтеза цитокинов, способы стимулирования биосинтеза цитокинов на основе соединений формул (I) и (II), способы лечения вирусного заболевания на основе соединений формул (I) и (II), способы лечения опухолевого заболевания на основе соединений формул (I) и (II). Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 13 н. и 10 з.п. ф-лы, 4 табл.
X представляет собой -CHR5-, -CHR5-алкильную группу, R1 выбран из группы, содержащей радикалы:
-R4-NR8-CR3-NR5-Z-R6-алкил;
-R4-NR8-CR3-NR5-Z-R6-фенил;
-R4-NR8-CR3-NR5-Z-R6-фуранил;
-R4-NR8-CR3-NR5R7;
при этом фенил замещается или не замещается одним или более заместителями, выбранными из группы, состоящей из метила; метоксила; метилтио; циано; водорода; диметиламина и ацетила;
R2 выбран из группы, содержащий радикалы:
-водород;
-алкил; и
-алкил-Y-алкил;
R3 представляет собой =O или=S;
R4 представляет собой алкил, в который может войти одна или несколько -O-групп; каждый R5 представляет собой Н или С1-10алкил;
R6 представляет собой связь или алкил;
R7 соединяется с R5, образуя морфолиновое кольцо;
R8 представляет собой Н, С1-10алкил или же R4 и R8 могут соединяться, образуя цикл;
Y представляет собой -O-;
Z представляет собой связь, -СО- или -SO2-;
n имеет значение 0;
каждый R выбран независимо из группы, состоящей из радикалов С1-10алкил, C1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
N-(2-{2-[4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил)-N'-фенилмочевина;
N-(2-{2-[4-амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил)-N'-фенилмочевина;
N-(2-{2-[4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил)-N-метил-N'-фенилмочевина;
N-(2-{2-[4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил)-N-метилморфолин-4-карбоксамид;
N-(2-{2-[4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил)-N-метил-N'-фенилмочевина;
N-(2-{2-[4-амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-1-ил]этокси}этил)-N-метил-N'-фенилмочевина;
и его соль фармацевтического качества.
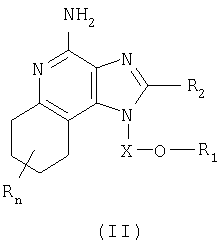
X представляет собой -CHR5-, -CHR5-алкил;
R1 выбран из группы, содержащей:
-R4-NR8-CR3-NR5-Z-R6-фенил;
R2 выбран из группы, содержащей:
-водород;
-алкил; и
-алкил-Y-алкил;
R3 представляет собой =O;
R4 представляет собой алкил;
R5 представляет собой Н;
R6 представляет собой связь;
R8 представляет собой H, С1-10алкил;
Y представляет собой -O-;
Z представляет собой связь;
n имеет значение 0;
каждый R выбран независимо из группы, состоящей из радикалов С1-10алкил, С1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
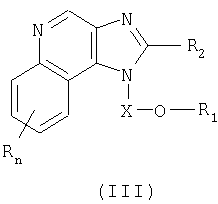
X представляет собой -CHR5-, -CHR5-алкил;
R1 выбран из группы, содержащей радикалы:
-R4-NR8-CR3-NR5-Z-R6-алкил;
-R4-NR8-CR3-NR5-Z-R6-фенил;
-R4-NR8-CR3-NR5-Z-R6-фуранил;
-R4-NR8-CR3-NR5R7;
при этом фенил замещается или не замещается одним или более заместителями, выбранными из группы, состоящей из метила; метоксила; метилтио; циано; водорода; диметиламина и ацетила;
R2 выбран из группы, содержащий радикалы:
-водород;
-алкил; и
-алкил-Y-алкил;
R3 представляет собой =O или =S;
R4 представляет собой алкил, в который может войти одна или несколько -O-групп;
каждый R5 представляет собой независимо Н или С1-10алкил;
R6 представляет собой связь или алкил;
R7 соединяется с R5, образуя морфолиновое кольцо;
R8 представляет собой Н или С1-10алкил и может соединяться с R4, образуя цикл;
Y представляет собой -O-;
Z представляет собой связь, -СО- или -SO2-;
n имеет значение 0;
каждый R выбран независимо из группы, состоящей из радикалов С1-10алкил, С1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
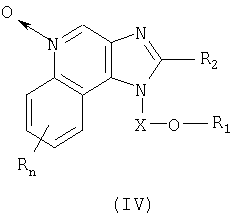
Х представляет собой -CHR5-, -CHR5-алкилен;
R1 выбран из группы, содержащей радикалы:
-R4-NR8-CR3-NR5-Z-R6-алкил;
-R4-NR8-CR3-NR5-Z-R6-фенил;
-R4-NR8-CR3-NR5-Z-R6-фуранил;
-R4-NR8-CR3-NR5R7;
при этом фенил замещается или не замещается одним или более заместителями, выбранными из группы, состоящей из метила; метоксила; метилтио; циано; водорода; диметиламина и ацетила;
Y представляет собой -O-;
Z представляет собой связь, -СО- или SO2-;
R4 представляет собой алкил, в который может войти одна или несколько -O-групп;
каждый R5 представляет собой независимо Н или С1-10алкил;
R6 представляет собой связь или алкил;
R7 соединяется с R5, образуя морфолиновое кольцо;
R8 представляет собой Н, С1-10алкил или же R4 и R8 могут соединяться, образуя цикл;
n имеет значение 0;
каждый R выбран независимо из группы, состоящей из радикалов С1-10алкил, С1-10алкокси, гидрокси, галоген и трифторметил;
или же соль фармацевтического качества указанных соединений.
| WO 9215582 A1, 17.09.1992 | |||
| WO 9502598 A1, 26.01.1995 | |||
| WO 9305042 A1, 18.03.1993 | |||
| RU 2000114496 А1, 10.07.2002. |

