Это изобретение относится к имидазохинолиновым соединениям, которые имеют тиоэфирную группу в положении 1, а также к фармацевтическим составам, содержащим такие соединения. Дополнительный аспект этого изобретения относится к использованию этих соединений в качестве иммуномодуляторов, для стимулирования биосинтеза цитокина в организме животного, а также для лечения различных болезней, включая вирусные и опухолевые заболевания.
Первые надежные данные по 1H-имидазол[4,5-с]хинолиновой циклической системе приведены Бакманом с сотр., J. Org. Chem. 15, 1278-1284 (1950), которые сообщили о синтезе 1-(6-метокси-8-хинолинил)-2-метил-1Н-имидазо[4,5-с]хинолина для его возможного применения в качестве противомалярийного средства. В последующем появились сообщения о синтезе различных замещенных 1Н-имидазо[4,5-с]хинолина. Например, Джайн с сотр., J. Med. Chem. 11, 87-92 (1968), синтезировали 1-[2-(4-пиперидил)этил]-1Н-имидазо[4,5-с]хинолин для его использования в качестве возможного противосудорожного и сердечно-сосудистого средства. Кроме того, Баранов с сотр., Chem. Abs. 85, 94362 (1976), сообщили о синтезе нескольких 2-оксоимидазо[4,5-с]хинолинов, а Берени с сотр., J. Heterocvclic Chem. 18, 1537-1540 (1981), также сообщили о синтезе нескольких 2-оксоимидазо[4,5-с]хинолинов.
Позже было установлено, что некоторые 1Н-имидазо[4,5-с]хинолин-4-амины и их 1- и 2-замещенные производные могут быть использованы в качестве противовирусных средств, бронхолитических средств и иммуномодуляторов. Эти соединения описаны среди прочих соединений в патентах США №№4689338, 4698348, 4929624, 5037986, 5268376, 5346905 и 5389640; все эти патенты приведены в списке цитируемой литературы.
Большой интерес представляют и имидазохинолиновые циклические системы. Известны некоторые 1Н-имидазо[4,5-с]нафтиридин-4-амины, 1Н-имидазо[4,5-с]пиридин-4-амины и 1Н-имидазо[4,5-с]хинолин-4-амины, имеющие эфирную группу, содержащую заместитель в положении 1. Эти соединения описаны в патентах США №№5268376, 5389640, 5494916 и WO 99/29693.
Несмотря на все эти попытки получить соединения, являющиеся модификаторами иммунной реакции, все еще сохраняется потребность в подобного рода соединениях, обладающих способностью модулировать иммунную реакцию за счет стимулирования биосинтеза цитокина или с использованием других механизмов.
Краткое изложение сущности изобретения
Найден новый класс соединений, способных стимулировать биосинтез цитокина в организме животных. В соответствии с этим данное изобретение предлагает соединения имидазохинолин-4-амина и тетрагидроимидазохинолин-4-амина, которые содержат тиоэфирную группу в 1-положении. Согласно данным ИК-спектроскопии эти соединения имеют формулы (I) и (II). Ниже приведена общая структурная формула этих соединений
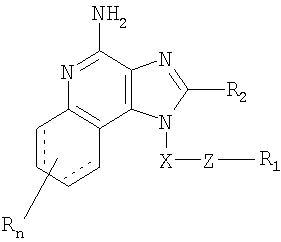
в которой заместители X, Z, R1, R2 и R определены для каждого класса соединений имеющих формулы (I) и (II).
Соединения, имеющие формулы (I) и (II), являются полезными модификаторами иммунной реакции благодаря их способности стимулировать биосинтез цитокина или иным образом модулировать иммунную реакцию при введении в организм животных. Это делает такие соединения полезными средствами для лечения различных заболеваний, таких как вирусные инфекции и опухолевые заболевания, которые вызывают такое изменение в иммунной реакции.
В изобретении также приводятся фармацевтические составы, содержащие соединения, изменяющие иммунную реакцию, и сообщается о способах стимулирования биосинтеза цитокина в организме животного, лечении вирусной инфекции и/или опухолевых заболеваний у животного путем введения в его организм соединений формулы I или формулы II.
Кроме того, приводятся способы синтеза соединений, являющихся предметом изобретения.
Подробное описание изобретения
Как указывалось выше, мы обнаружили некоторые соединения, которые стимулируют биосинтез цитокина и модифицируют иммунную реакцию в организме животных. Такие соединения имеют формулы (I) и (II), показанные ниже.
Предлагаются имидазохинолиновые соединения, содержащие тиоэфирную группу, находящуюся в положении 1, имеют формулу (I)

где X представляет собой -CHR3-, -CHR3-алкильную или -CHR3-алкенильную группу;
Z представляет собой -S-, -SO- или -SO2- группу;
R1 выбран из группы, включающей:
- алкил;
- арил;
- гетероарил;
- гетероциклил;
- алкенил;
- R4-арил;
- R4-гетероарил;
- R4-гетероциклил;
R2 выбран из группы, включающей:
- атом водорода;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил и
- алкил или алкенил, замещенные одним или большим количеством заместителей, выбранных из группы, включающей:
- ОН;
- атом галогена;
- N(R3)2;
- CO-N(R3)2;
- CO-C1-10алкил;
- СО-O-С1-10алкил;
- N3;
- арил;
- гетероарил;
- гетероциклил;
- СО-арил и
- СО-гетероарил;
каждый R3 представляет собой независимо атом водорода или C1-10-алкильную группу;
каждый R4 независимо друг от друга представляет собой алкильную или алкенильную группу;
каждый Y независимо друг от друга представляет собой -О- или S(O)0-2-;
n может иметь значение от 0 до 4 и
каждый заместитель R независимо выбран из группы, состоящей из C1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы,
или соль фармацевтического качества на основе этих групп.
Изобретение также включает тетрагидроимидазохинолиновые соединения с тиоэфирной группой, содержащей заместитель в положении 1. Такие тетрагидроимидазохинолиновые соединения могут быть представлены формулой (II):
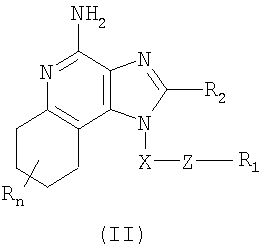
где Х представляет собой -CHR3-, -CHR3-алкильную или -CHR3-алкенильную группу;
Z представляет собой -S-, -SO- или -SO2- группу;
R1 выбран из группы, которая содержит:
- алкил;
- арил;
- гетероарил;
- гетероциклил;
- алкенил;
- R4-арил;
- R4-гетероарил;
- R4-гетероциклил;
R2 выбран из группы, включающей:
- атом водорода;
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- алкил-Y-алкил;
- алкил-Y-алкенил;
- алкил-Y-арил и
- алкильная или алкенильная группы, содержащие один или большее количество заместителей, выбранных из группы, включающей:
- ОН;
- атом галогена;
- N(R3)2;
- CO-N(R3)2;
- CO-C1-10алкил;
- СО-O-С1-10алкил;
- N3;
- арил;
- гетероарил;
- гетероциклил;
- СО-арил и
- СО-гетероарил;
каждый R3 представляет собой независимо атом водорода или C1-10алкильную группу;
каждый R4 независимо друг от друга представляет собой алкильную или алкенильную группу;
каждый Y независимо друг от друга представляет собой -О- или S(O)0-2-;
n может иметь значение от 0 до 4 и
каждый R независимо выбран из группы, состоящей из С1-10 алкильной, C1-10 алкоксильной, гидроксильной групп, атома галогена и трифторметильной группы,
или соль фармацевтического качества на основе этих групп.
Получение соединений
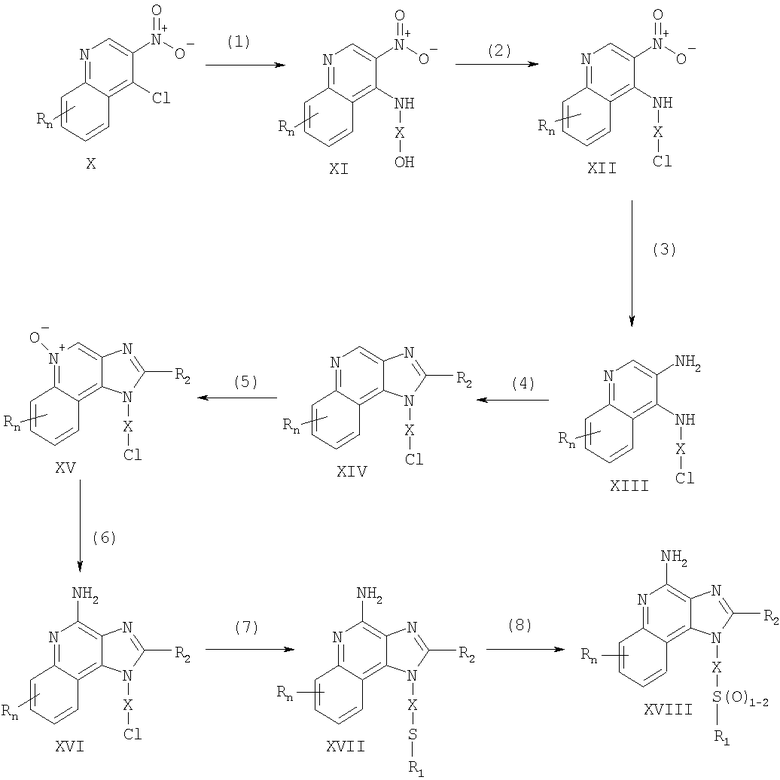
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой I реакции, в которой значения R, R1, R2, X и n определены выше.
На стадии (1) процесса (схема I реакции) 4-хлор-3-нитрохинолин, имеющий формулу X, реагирует с амином формулы HO-X-NH2, в результате чего получают 3-нитрохинолин-4-амин (соединение формулы XI). Реакцию можно проводить, добавляя амин к раствору соединения Х в подходящем растворителе, таком как хлороформ или дихлорметан, в присутствии триэтиламина и в ряде случаев при нагревании. Известны многие хинолины формулы Х (см., например, Патент США №4689338 и другие материалы, приведенные в этом патенте). Многие амины формулы HO-X-NH2 являются коммерческими продуктами, другие можно получить в соответствии с известными синтетическими способами.
На стадии (2) процесса (схема 1 реакции) 3-нитрохинолин-4-амин формулы XI хлорируют для получения 3-нитрохинолин-4-амина, имеющего формулу XII. Для этой реакции могут быть использованы обычные хлорирующие агенты. Предпочтительно проводить эту реакцию при нагревании, добавляя к соединению формулы XI раствор тионилхлорида в подходящем растворителе, таком как дихлорметан. В другом варианте эту реакцию можно проводить и в отсутствие растворителя.
На стадии (3) процесса (схема I реакции) 3-нитрохинолин-4-амин формулы XII восстанавливают до хинолин-3,4-диамина (соединение формулы XIII). Предпочтительно эту реакцию проводят, используя обычный гетерогенный катализатор процесса гидрирования, такой, например, как нанесенная на активированный уголь платина. Реакцию обычно проводят в аппарате Парра в подходящем растворителе, таком как толуол.
На стадии (4) процесса (схема I реакции) хинолин-3,4-диамин (соединение формулы XIII) обрабатывают карбоновой кислотой или эквивалентным соединением и в результате получают 1H-имидазо[4,5-с]хинолин формулы XIV. Подходящими эквивалентами карбоновой кислоты являются сложные ортоэфиры и 1,1-диалкоксиалкилалканоаты. Карбоновую кислоту или ее эквивалент выбирают таким образом, чтобы они обеспечивали введение в соединение формулы XIV желаемого заместителя R2. Например, при использовании для этой цели триэтилортоформиата будет получаться соединение, в котором R2 представляет собой атом водорода, а при использовании триметилортовалериата R2=С4Н9. Реакцию можно проводить в отсутствие растворителя или в инертном растворителе, таком как толуол. Реакцию проводят при достаточном нагревании, чтобы обеспечить удаление любого спирта или воды, образующихся в ходе этой реакции в качестве побочных продуктов. По желанию реакцию можно проводить в присутствии катализатора, такого как солянокислый пиридин.
Альтернативным образом стадию (4) можно проводить при (i) взаимодействии диамина формулы XIII с ацилгалогенидом формулы R2C(O)Cl или R2C(O)Br и (ii) последующей циклизации полученного продукта. В части (i) ацилгалогенид добавляют к раствору диамина в подходящем растворителе, таком как пиридин. Реакцию можно проводить при температуре окружающей среды. В части (ii) продукт, полученный в части (i), нагревают в пиридине в присутствии солянокислого пиридина.
На стадии (5) процесса (схема I реакции) 1Н-имидазо[4,5-с]хинолин формулы XIV подвергают окислению, в результате чего получают 1H-имидазо[4,5-с]хинолин-5N-оксид (соединение формулы XV). Для этой реакции используют обычный окисляющий агент, способный образовывать N-оксиды. Предпочтительно раствор соединения формулы XIV в подходящем растворителе, таком как дихлорметан, обрабатывают 3-хлорпероксибензойной кислотой при температуре окружающей среды.
На стадии (6) процесса (схема I реакции) 1H-имидазо[4,5-с]хинолин-5N-оксид формулы XV аминируют до 1Н-имидазо[4,5-с]хинолин-4-амина формулы XVI. Стадия (6) включает (i) взаимодействие соединения формулы XV с ацилирующим агентом и (ii) последующую обработку полученного продукта аминирующим агентом. В части (i) стадии (6) N-оксид формулы XV обрабатывают ацилирующим агентом. Подходящими ацилирующими агентами являются алкил- или арилсульфонилхлориды (например, бензолсульфонилхлорид, метансульфонилхлорид, п-толуиленсульфонилхлорид). Предпочтительными ацилирующими агентами являются арилсульфонилхлориды, причем наиболее предпочтительным ацилирующим агентом является п-толуиленсульфонилхлорид. В части (ii) стадии (6) продукт, полученный в части (i), обрабатывают избыточным количеством аминирующего агента. Подходящими аминирующими агентами являются аммиак (например, в виде гидроксида аммония) и соли аммония (например, карбонат аммония, бикарбонат аммония, фосфат аммония). Гидроксид аммония является предпочтительным аминирующим агентом. Реакцию преимущественно проводят, растворяя N-оксид формулы XV в инертном растворителе, таком как дихлорметан или хлороформ, и затем добавляя к раствору аминирующий агент, а после этого медленно добавляя ацилирующий агент.
На стадии (7) процесса (схема I реакции) 1Н-имидазо[4,5-с]хинолин-4-амин формулы XVI обрабатывают соединением формулы R1-SNa, в результате чего получают 1H-имидазо[4,5-с]хинолин-4-амин формулы XVII, который является производным соединением продукта формулы I. Реакцию можно проводить, добавляя к соединению формулы XVI продукт формулы R1SNa в подходящем растворителе, таком как N,N-диметилформамид или диметилсульфоксид, и нагревая полученную смесь при температуре 60-80°С. Продукт или соль фармацевтического качества на его основе можно выделить из реакционной смеси с помощью обычных способов.
На стадии (8) процесса (схема I реакции) 1Н-имидазо[4,5-с]хинолин-4-амин формулы XVII окисляют с помощью обычных окислителей до 1Н-имидазо[4,5-с]хинолин-4-амина формулы XVIII, который представляет собой производное продукта формулы I. Предпочтительно раствор соединения формулы XVII в подходящем растворителе, таком как хлороформ или дихлорметан, обрабатывают при температуре окружающей среды 3-хлорпероксибензойной кислотой. Степень окисления продукта регулируют, изменяя количество 3-хлорпероксибензойной кислоты в реакционной смеси. Так, использование приблизительно одного эквивалента окислителя будет приводить к образованию сульфоксида, в то время как в присутствии двух эквивалентов окислителя будет образовываться сульфон. Продукт или соль фармацевтического качества на его основе можно выделить из реакционной смеси с помощью обычных способов.
Схема I реакции
Указанные в изобретении соединения могут быть получены в соответствии со схемой II реакции, в которой R, R1, R2, X и n определены выше.
На стадии (1) процесса (схема II реакции) проводят реакцию между 3-нитрохинолин-4-амином формулы XII и соединением формулы R1-SNa, используя методику, применяемую на стадии (7) процесса (схема I реакции). В результате получают 3-нитрохинолин-4-амин формулы XIX.
На стадии (2) процесса (схема II реакции) 3-нитрохинолин-4-амин формулы XIX восстанавливают до хинолин-3,4-диамина (соединение формулы XX) по способу, используемому на стадии (3) процесса (схема I реакции).
Стадия (3) процесса (схема II реакции) заключается в циклизации хинолин-3,4-диамина (формулы XX) по способу, используемому на стадии (4) процесса (схема I реакции). На этой стадии образуется 1Н-имидазо[4,5-с]хинолин формулы XXI.
На стадии (4) процесса (схема II реакции) 1Н-имидазо[4,5-с]хинолин формулы XXI окисляют под действием обычного окислителя до 1Н-имидазо[4,5-с]хинолин-5N-оксида формулы XXII. Предпочтительно раствор соединения XXI в подходящем растворителе, таком как хлороформ или дихлорметан, обрабатывают при температуре окружающей среды избыточным количеством (по крайней мере, 3 эквивалента) 3-хлорпероксибензойной кислоты.
На стадии (5) процесса (схема II реакции) 1H-имидазо[4,5-с]хинолин-5N-оксид формулы XXII подвергают аминированию по способу, применяемому на стадии (6) процесса (схема I реакции). В результате получают 1H-имидазо[4,5-с]хинолин-4-амин формулы XXIII, который является производным соединением продукта формулы I. Продукт или соль фармацевтического качества на его основе можно выделить из реакционной смеси с помощью обычных способов.
Схема II реакции

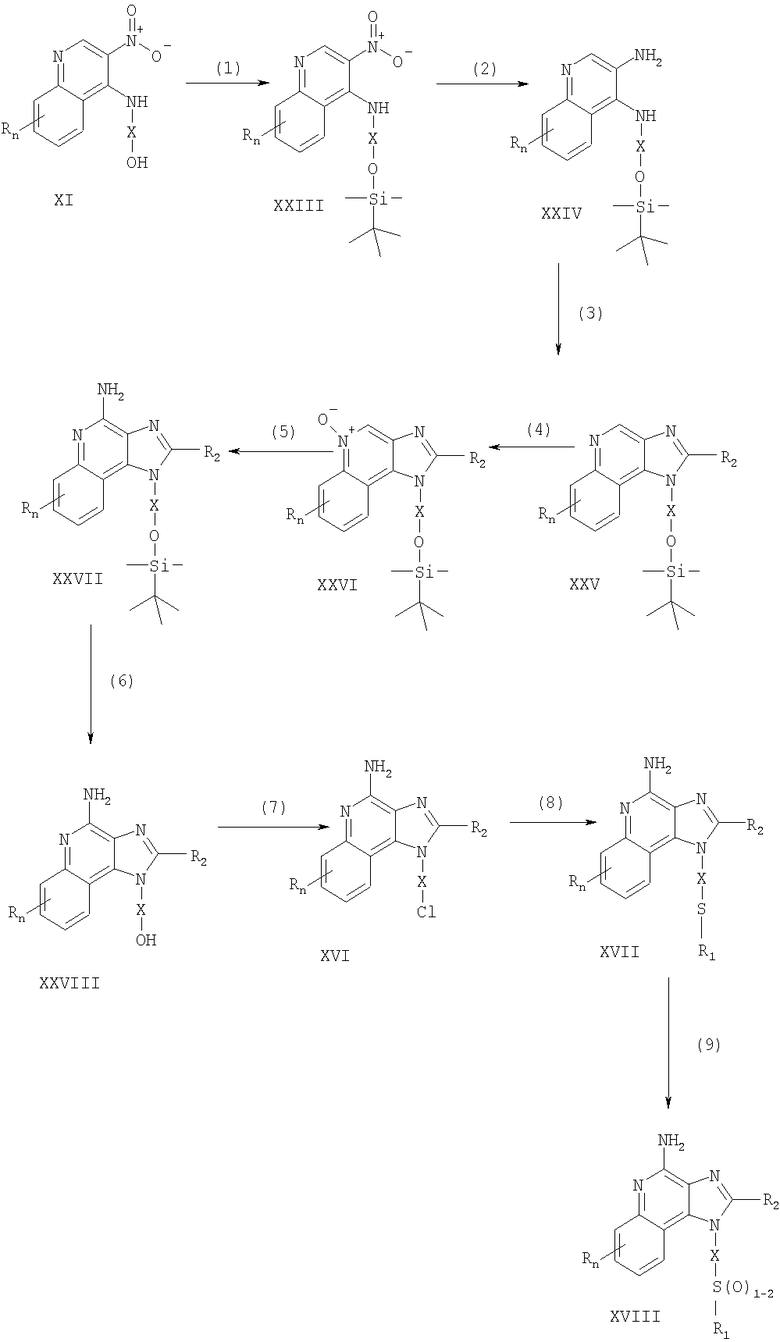
Указанные в изобретении соединения могут быть также получены в соответствии со схемой III реакции, в которой R, R1, R2, Х и n определены выше.
На стадии (1) процесса (схема III реакции) 3-нитро-4-аминохинолин-1-иловый спирт (соединение формулы XI), используя обычные способы, обрабатывают трет-бутилдиметилсилильной группой для защиты содержащихся в этом соединении функциональных групп. Предпочтительно соединение формулы XI смешивают с трет-бутилдиметилсилилхлоридом в подходящем растворителе, таком как хлороформ, в присутствии триэтиламина и каталитических количеств 4-диметиламинопиридина.
На стадии (2) процесса (схема III реакции) защищенный 3-нитро-4-аминохинолин-1-иловый спирт формулы XXIII восстанавливают до защищенного 3,4-диаминохинолин-1-илового спирта формулы XXIV по методике, используемой на стадии (3) процесса (схема I реакции).
На стадии (3) процесса (схема III реакции) проводят циклизацию защищенного 3,4-диаминохинолин-1-илового спирта формулы XXIV по методике, используемой на стадии (4) процесса (схема I реакции). В результате получают 1Н-имидазо[4,5-с]хинолин формулы XXV.
Это соединение формулы XXV на стадии (4) процесса (схема III реакции) окисляют по методике, используемой на стадии (5) процесса (схема I реакции), до 1Н-имидазо[4,5-с]хинолин-5N-оксида формулы XXVI.
На стадии (5) процесса (схема III реакции) 1Н-имидазо[4,5-с]хинолин-5N-оксид формулы XXVI аминируют по методике, используемой на стадии (6) процесса (схема I реакции), до 1H-имидазо[4,5-с]хинолин-4-амина формулы XXVII.
На стадии (6) процесса (схема III реакции) удаляют защитную группу из 1H-имидазо[4,5-с]хинолин-4-амина формулы XXVII, в результате чего получают 1H-имидазо[4,5-с]хинолин-4-амин формулы XXVIII. Предпочтительно раствор соединения формулы XXVII в подходящем растворителе, таком как тетрагидрофуран, обрабатывают тетрабутиламмонийфторидом. Известны некоторые соединения формулы XXVIII, см., например, Герстер, Патент США 4689338, и Герстер с сотр., Патент США 5605899.
На стадии (7) процесса (схема III реакции) с помощью обычных способов проводят хлорирование 1H-имидазо[4,5-с]хинолин-4-амина формулы XXVIII и в результате получают 1H-имидазо[4,5-с]хинолин-4-амин формулы XVI. С этой целью соединение формулы XXVIII можно нагревать в присутствии чистого тионилхлорида. По другому способу к раствору соединения формулы XXVIII в подходящем растворителе, таком как N,N-диметилформамид, в присутствии триэтиламина можно с определенной скоростью добавлять оксихлорид фосфора.
Стадии (8) и (9) процесса (схема III реакции) можно проводить таким же образом, как и стадии (7) и (8) процесса (схема I реакции).
Схема III реакции
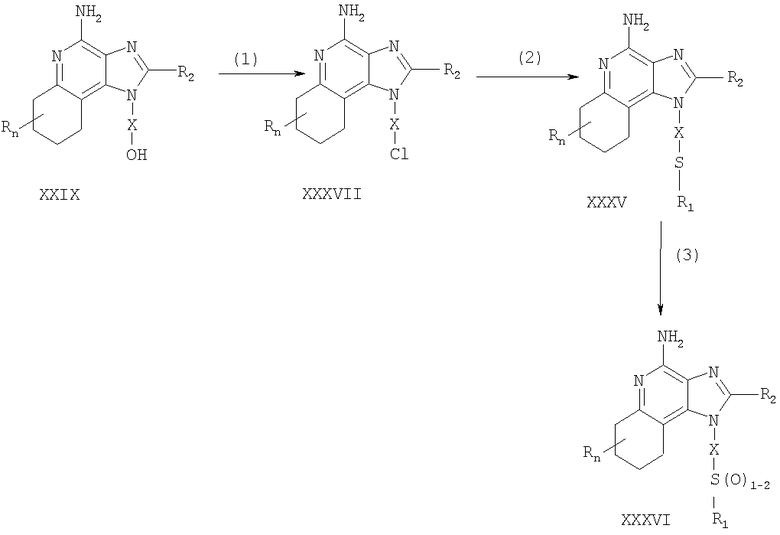
Указанные в изобретении соединения могут быть получены в соответствии со схемой IV реакции, в которой R, R1, R2, X и n определены выше, ВОС - трет-бутилкарбонильная группа.
На стадии (1) процесса (схема IV реакции) гидроксильную группу 6, 7, 8, 9-тетрагидро-1H-имидазо[4,5-с]хинолин-1-илового спирта формулы XXIX защищают трет-бутилдиметилсилильной группой, используя методику, применяемую на стадии (1) процесса (схема III реакции). Некоторые соединения формулы XXIX известны, другие такие соединения могут быть получены с помощью известных синтетических способов, см., например, Николайдес с сотр., Патент США 5352764, и Линдстром, Патент США 5693811, и цитируемую в этих патентах литературу.
На стадии (2) процесса (схема IV реакции) аминогруппу 1H-имидазо[4,5-с]хинолин-4-амина формулы XXX защищают с помощью обычных способов и получают защищенный 1Н-имидазо[4,5-с]хинолин формулы XXXI. Преимущественно соединение формулы XXX обрабатывают трет-бутилдикарбонатом в соответствующем растворителе, таком как тетрагидрофуран, в присутствии триэтиламина и 4-диметиламинопиридина. Реакцию можно проводить при повышенной температуре (60°С).
На стадии (3) процесса (схема IV реакции) проводят удаление защитной трет-бутилдиметилсилильной группы из группы формулы XXXI. Реакцию проводят в соответствии с методикой, применяемой на стадии (6) процесса (схема III реакции), и в результате получают 1H-имидазо[4,5-с]хинолин-1-иловый спирт формулы XXXII.
На стадии (4) процесса (схема IV реакции) 1H-имидазо[4,5-с]хинолин-1-иловый спирт формулы XXXII превращают в соответствующий метансульфонат формулы XXXIII. Предпочтительно раствор соединения формулы XXXII в подходящем растворителе, таком как дихлорметан, обрабатывают в присутствии триэтиламина метансульфонилхлоридом. Реакцию можно проводить при пониженной температуре (-10°С).
На стадии (5) процесса (схема IV реакции) метансульфонат формулы XXXIII обрабатывают тиолом формулы R1SH, в результате чего получают тиоэфир формулы XXXIV. Предпочтительно раствор соединения формулы XXXIII в подходящем растворителе, таком как N,N-диметилформамид, обрабатывают тиолом в присутствии триэтиламина. Реакцию можно проводить при повышенной температуре (80°С).
На стадии (6) процесса (схема IV реакции) в результате гидролиза, протекающего в кислотных условиях, осуществляют удаление защитных трет-бутилкарбонильных групп. В результате получают 1Н-имидазо[4,5-с]хинолин-4-амин формулы XXXV, представляющий собой производное соединение продукта формулы II. Предпочтительно соединение формулы XXXIV в соответствующем растворителе, таком как дихлорметан, обрабатывают при температуре окружающей среды раствором соляной кислоты в диоксане. Продукт или соль фармацевтического качества на его основе можно выделить из реакционной смеси с помощью обычных способов.
На стадии (7) процесса (схема IV реакции) тиоэфир формулы XXXV окисляют по способу, используемому на стадии (8) процесса (схема I реакции), до сульфона или сульфоксида формулы XXXVI. Эти вещества представляют собой производное соединение продукта формулы II. Продукт или соль фармацевтического качества на его основе можно выделить из реакционной смеси с помощью обычных способов.
Схема IV реакции

Указанные в изобретении соединения могут быть получены в соответствии со схемой V реакции, в которой R, R1, R2, Х и n определены выше.
На стадии (1) процесса (схема V реакции) 6, 7, 8, 9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1 -иловый спирт формулы XXIX хлорируют по способу, применяемому на стадии (7) процесса (схема III реакции), и в результате получают соединение формулы XXXVII.
На стадии (2) процесса (схема V реакции) продукт формулы XXXVII обрабатывают соединением формулы R1-SNa, используя методику, применяемую на стадии (7) процесса (схема I реакции), и в результате получают тиоэфир формулы XXXV, представляющий собой производное соединение продукта формулы II. Продукт или соль фармацевтического качества на его основе можно выделить из реакционной смеси с помощью обычных способов.
На стадии (3) процесса (схема V реакции) тиоэфир формулы XXXV окисляют по способу, применяемому на стадии (8) процесса (схема I реакции), до сульфона или сульфоксида формулы XXXVI, которые представляют собой производное соединение продукта формулы II. Продукт или соль фармацевтического качества на его основе можно выделить из реакционной смеси с помощью обычных способов.
Схема V реакции
Используемые здесь термины «алкил», «алкиленил» и приставка «алк-» относятся как к линейным, так и разветвленным цепным группам, а также к циклическим группам, например циклоалкильным и циклоалкенильным группам. Если не указано иначе, эти группы содержат от 1 до 20 атомов углерода, а алкенильные группы от 2 до 20 атомов углерода. Предпочтительно эти группы содержат до 10 атомов углерода. Циклические группы могут быть либо моноциклическими, либо полициклическими и преимущественно содержать в цикле от 3 до 10 атомов углерода. Примерами циклических групп являются циклопропильная, циклопропилметильная, циклопентильная, циклогексильная и адамантильная группы.
Кроме того, алкильная и алкенильная часть групп -Х- могут быть либо незамещенными, либо содержать один или большее количество заместителей, причем эти заместители выбраны из группы, состоящей из алкильной, алкенильной, арильной, гетероарильной, гетероциклильной, арилалкильной, гетероарилалкильной и гетероциклоалкильной группировок.
Термин «галоидоалкил» включает группы, которые в качестве заместителя имеют один или большее количество галоидных атомов, включая перфторированные группы. Такое определение относится также к группам, которые включают приставку «гало». Примерами подходящих галоидоалкильных групп являются хлорметильная, трифторметильная и подобные группы.
Термин «арил», используемый в данном описании, включает в себя карбоциклические ароматические группы или циклические системы. Примеры арильных групп включают фенильную, нафтильную, бифенильную, флуоренильную и инденильную группы. Термин «гетероарил» относится к ароматическим циклам или циклическим системам, содержащим в кольце, по крайней мере, один гетероатом (например, О, S, N). Подходящие гетероарильные группы включают фурильную, тиенильную, пиридильную, хинолинильную, изохинолильную, индолильную, изоиндолильную, триазольную, пирролильную, тетразолильную имидазолильную, пиразолильную, оксазолильную, тиазолильную, бензофуранильную, бензотиофенильную, карбазолильную, бензоксазолильную, пиримидинильную, хиноксалинильную, бензимидазолильную, нафтиридинильную, изоксазолильную, изотиазолильную, хиназолинильную, пуринильную и подобные группы.
В состав «гетероциклильных» соединений входят неароматические циклы или циклические системы, содержащие в кольце, по крайней мере, один гетероатом (например, О, S, N). К этим соединениям относятся все полностью насыщенные и частично ненасыщенные производные указанных выше гетероарильных групп. Примерами гетероциклических групп являются пирролидинильная, тетрагидрофуранильная, морфолинильная, тиоморфолинильная, пиперидинильная, пиперазинильная, тиазолидинильная, имидазолидинильная, изотиазолидинильная и подобные группы.
Арильная, гетероарильная и гетероциклильная группы могут быть как незамещенными, так и содержащими один или более заместителей, независимо выбранных из группы, содержащей алкильную, алкоксильную, алкилтионильную, галоидоалкильную, галоидоалкоксильную, галоидоалкилтионильную группы, атом галогена, нитрильную, гидроксильную, меркапто- и цианогруппы, карбоксильную, формильную, арильную, арилоксильную, арилтионильную, арилалкоксильную, арилалкилтионильную, гетероарильную, гетероарилоксильную, гетероарилтионильную, гетероарилалкоксильную, гетероарилалкилтионильную, амино-, алкиламино-, диалкиламино-, гетероциклильную, гетероциклоалкильную, алкилкарбонильную, алкенилкарбонильную, алкоксикарбонильную, галоалкилкарбонильную, галоалкоксикарбонильную, алкилтиокарбонильную, арилкарбонильную, гетероарилкарбонильную, арилоксикарбонильную, гетероарилоксикарбонильную, арилтиокарбонильную, гетероарилтиокарбонильную, алканоилоксильную, алканоилтионильную, алканоиламиновую, арилкарбонилоксильную, арилкарбонилтионильную, алкиламиносульфонильную, алкилсульфонильную, арилсульфонильную, гетероарилсульфонильную, арилдиазинильную, алкилсульфониламино-, арилсульфониламино-, арилалкилсульфониламино-, алкилкарбониламино-, алкенилкарбониламино-, арилкарбониламино, арилалкилкарбониламино-, гетероарилкарбониламино-, гетероарилалкикарбониламино-, алкилсульфониламино-, алкенилсульфониламино-, арилсульфониламино-, арилалкилсульфониламино-, гетероарилсульфониламино-, гетероарилалкилсульфониламино-, алкиламинокарбониламино-, алкениламинокарбониламино-, ариламинокарбониламино-, арилалкиламинокарбониламино-, гетероариламинокарбониламино-, гетероарилалкилкарбониламиногруппы; кроме того, в случае гетероциклильных групп - оксогруппы. В том случае, когда любые другие группы идентифицированы как «замещенные» или «иногда замещенные», эти группы также могут иметь в качестве заместителя одну или большее количество перечисленных выше группировок.
Обычно лишь некоторые заместители являются предпочтительными. Например, предпочтительными Х группами являются этиленовая и н-бутиленовая группы, а предпочтительными заместителями R1 являются алкильная и арильная группы, причем предпочтительными арильными группами являются фенильная или замещенная фенильная группы. Предпочтительно, чтобы заместитель R отсутствовал (т.е. n=0). Предпочтительные R2 группы включают атом водорода, алкильные группы, содержащие от 1 до 4 углеродных атомов (т.е. метильная, этильная, пропильная, изопропильная, н-бутильная, втор-бутильная, изобутильная и циклопропилметильная группы), метоксиметильную и этоксиметильную группы. Один или большее количество этих предпочтительных заместителей, если они вообще присутствуют, могут содержаться в предлагаемых в изобретении соединениях в любых комбинациях.
Данное изобретение включает описанные выше соединения в любой их фармацевтически доступной форме, включая изомеры, такие как диастереомеры и энантиомеры, соли, сольваты, полиморфные формы и т.п. В частности, если соединение является оптически активным, в изобретении описаны каждый энантиомер этого соединения, а также рацемическая смесь энантиомеров.
Фармацевтические составы и биологическая активность
Предлагаемые в изобретении фармацевтические составы содержат терапевтически эффективные количества описанных выше соединений в сочетании с фармацевтически приемлемым носителем.
Используемый здесь термин «терапевтически эффективное количество» означает количество соединения, достаточное для оказания терапевтического эффекта, такого как стимулирование образования цитокина, противоопухолевая активность и/или противовирусная активность. Хотя точное количество активного вещества, используемого в фармацевтическом составе, предлагаемом в изобретении, широко варьируется в зависимости от различных факторов, таких как физическая и химическая природа вещества, а также природа носителя и предполагаемая доза, полагают, что предлагаемые в изобретении композиции будут содержать достаточное количество активного ингредиента, чтобы обеспечить дозу в пределах от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно от приблизительно 10 мкг/кг до приблизительно 5 мг соединения на 1 кг веса пациента. Составы могут использоваться в любых обычных лекарственных формах, таких как таблетки, лепешки, парентеральные композиции, сиропы, кремы, мази, аэрозольные составы, различного рода пластыри и т.п.
Предлагаемые в изобретении соединения могут вводиться в организм в виде одного прописанного в рецепте терапевтического средства или в виде комбинации с одним или большим количеством других активных агентов, таких как дополнительные модификаторы иммунной реакции, противовирусные препараты, антибиотики и т.п.
Установлено, что предлагаемые в изобретении соединения стимулируют образование некоторых цитокинов в опытах, выполненных в соответствии с приведенными ниже способами испытания. Эти результаты показывают, что предлагаемые соединения представляют собой полезные модификаторы иммунной реакции, т.е. они способны модулировать иммунную реакцию целым рядом различных способов, оказывая тем самым помощь в излечении различных расстройств.
Цитокины, образующиеся при введении предлагаемых в данном изобретении соединений, обычно включают интерферон-α (IFN-α) и/или фактор-α некроза опухоли (TNF-α), а также некоторые интерлейкины (IL). В частности, эти соединения вызывают образование IFN-α, TNF-α, IL-1, IL-6, IL-10 и IL-12, а также различных других цитокинов. Помимо других эффектов цитокины замедляют образование вирусов и рост опухолевых клеток, что делает предлагаемые в изобретении соединения полезными при лечении опухолей и вирусных заболеваний. Таким образом, настоящее изобретение предоставляет способ стимулирования биосинтеза цитокина при введении в организм животного эффективных количеств соединений или композиций, предлагаемых в изобретении.
Установлено, что некоторые предлагаемые в изобретении соединения предпочтительно стимулируют участие IFN-α в изменении численности кроветворных клеток, таких как РВМС (одноядерные клетки периферийной крови), содержащих pDC2 клетки (предшественников дендритных клеток типа 2), не вызывая значительного сопутствующего воспалительного цитокинеза.
Помимо присущей им способности стимулировать образование цитокинов эти соединения оказывают влияние и на другие аспекты врожденной иммунной реакции. Например, может быть стимулирована естественная активность клетки-убийцы, причем этот эффект может быть обусловлен образованием цитокина. Соединения могут также активировать макрофаги, которые, в свою очередь, стимулируют секрецию оксида азота и образование дополнительных цитокинов. Кроме того, эти соединения могут вызывать разрастание и дифференциацию В-лимфоцитов.
Предлагаемые в изобретении соединения оказывают также влияние на приобретенную иммунную реакцию. Например, хотя, как полагают, предлагаемые соединения не оказывают непосредственного влияния на Т-лимфоциты или на образование Т-лимфоцитных цитокинов, эти соединения оказывают косвенное влияние на образование цитокина IFN-γ из фаг-помощника типа 1 Т-лимфоцита (Th1), а образование Тh2 цитокинов IL-4, IL-5 и IL-13 замедляется при введении соединений, предлагаемых в настоящем изобретении. Эти результаты показывают, что предлагаемые в данном изобретении соединения оказывают помощь при лечении заболеваний, при которых требуется увеличение количества Th1 и/или уменьшение количества Тh2. Учитывая способность соединений, предлагаемых в данном изобретении, замедлять иммунную реакцию Т-фаг-помощника типа 2, можно ожидать, что эти соединения окажутся полезными при лечении аллергических заболеваний, например атопических дерматитов, астмы, аллергии, аллергических ринитов, системной красной волчанки, а также в качестве вспомогательной вакцины для увеличения иммунитета, а возможно, и для лечения повторяющихся грибковых заболеваний и хламидий.
Модифицирующее воздействие соединений на иммунную реакцию делает их полезными при лечении большого числа различных заболеваний. Вследствие своей способности стимулировать образование цитокинов, таких как IFN-α и/или TNF-α, предлагаемые в данном изобретении соединения особенно полезны при лечении вирусных заболеваний и опухолей. Эта иммуномодулирующая активность дает основания полагать, что предлагаемые в изобретении соединения могут быть полезными при лечении таких болезней (но не только их), как вирусные заболевания, например остроконечная кондилома, обычные бородавки, подошвенные бородавки, гепатит В, гепатит С, герпетическая лихорадка типа I и типа II, контагиозный моллюск, оспа, особенно натуральная оспа, ВИЧ, цитомегаловирус, вирус варицелисзостер, риновирус, аденовирус, грипп и парагрипп, цервикальная интраэпителиальная неоплазия, человеческая вирусная папиллома и ассоциированные опухоли; грибковые заболевания, например кандидоз, аспергиллез, криптококковые менингиты; заболевания, связанные с появлением новообразований, например базалиома, лейкемия «волосистых» клеток, саркома Капоши, рак клеток почечного эпителия, рак клеток простого сквамозного эпителия, лейкемия миелопоэза, множественная миелома, меланома, неходжкинская лимфома, кожная лимфома и другие виды онкологических заболеваний; паразитарные заболевания, например пневмоцистоз, криптоспоридиоз, гистоплазмоз, токсоплазмоз, трипаносомная инфекция, лейшманиоз; бактериальные инфекции, например туберкулез и mycobacterium avium. С помощью предлагаемых соединений можно, кроме того, лечить старческий кератоз, экзему, эозинофилию, эссенциальную тромбоцитаэмию, проказу, множественный склероз, синдром Оммена, дискоидную волчанку, болезнь Боуена и боуеноидный папулез, облысение, ингибирование образования келоидных швов после хирургических операций и других типов постхирургических шрамов. Кроме того, эти соединения могут стимулировать заживление ран, включая хронические раны. Они могут быть полезны также для лечения оппортунистических инфекций и опухолей, которые появляются после подавления иммунитета, например, у онкологических и ВИЧ больных, а также после операций по пересадке органов.
Эффективным количеством введенного соединения, которое предназначено для стимулирования биосинтеза цитокинов, является такое количество, которое оказывается достаточным, чтобы побудить один или большее число типов клеток, таких как моноциты, макрофаги, дендритные клетки и В-клетки, к образованию одного или большего числа цитокинов, таких как, например, IFN-α, TNF-α, IL-1, IL-6, IL-10 и IL-12, в количестве, превышающем фоновый уровень таких цитокинов. Точное количество соединения будет изменяться в зависимости от различных факторов, но ожидается, что эффективная доза этого соединения будет находиться в интервале от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно в интервале между приблизительно 10 мкг/кг до приблизительно 5 мг/кг. Изобретение предлагает также способ лечения вирусной инфекции и онкологических заболеваний у животного путем введения в организм животного эффективного количества предлагаемого в изобретении соединения или композиции на его основе. Эффективным количеством соединения для лечения или подавления вирусной инфекции является такое его количество, которое будет вызывать уменьшение одного или большего числа симптомов вирусной инфекции, таких как вирусные поражения, вирусная нагрузка, скорость образования вирусов, и увеличивать их смертность по сравнению с ситуацией, наблюдаемой для контрольных животных, не принимающих эти соединения. Точное количество соединения будет изменяться в широких пределах в зависимости от различных факторов, но ожидается, что доза этого соединения должна находиться в интервале от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно в интервале между приблизительно 10 мкг/кг до приблизительно 5 мг/кг. Эффективным количеством соединения для лечения онкологического заболевания является такое его количество, которое будет вызывать уменьшение размера опухоли или числа опухолей. И в этом случае точное количество соединения будет изменяться в широких пределах в зависимости от различных факторов, но ожидается, что доза этого соединения должна находиться в интервале от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно в интервале между приблизительно 10 мкг/кг до приблизительно 5 мг/кг.
Предлагаемое изобретение иллюстрируется ниже различными примерами. Эти примеры приведены только в качестве иллюстрации и ни в коей мере не ограничивают общие рамки изобретения.
Пример 1
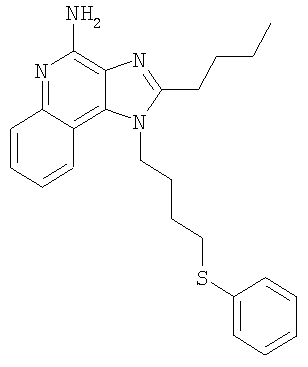
2-Бутил-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
В круглодонную колбу, снабженную магнитной мешалкой, загружают 109,70 г (525,87 ммоля) 4-хлор-3-нитрохинолина и 500 мл дихлорметана. К раствору добавляют 79,82 г (788,81 ммоля) триэтиламина и 46,87 г (525,87 ммоля) 4-амино-1-бутанола и в результате получают гомогенный темно-желтый раствор. Для завершения реакции реакционную смесь кипятят с обратным холодильником в течение 30 минут. После этого раствор охлаждают и затем осуществляют его перераспределение между хлороформом и насыщенным водным раствором хлористого аммония. Водный и органический слои разделяют и водный слой экстрагируют хлороформом. Органические слои объединяют и концентрируют при пониженном давлении, в результате чего получают 104,67 г (400,60 ммоля) 4-[(3-нитрохинолин-4-ил)амино]бутан-1-ола в виде темно-желтого твердого продукта. Этот материал используют без дальнейшей очистки.
Часть В
В круглодонную колбу, снабженную магнитной мешалкой, загружают 5,0 г (19,14 ммоля) 4-[(3-нитрохинолин-4-ил)амино]бутан-1-ола, 2,91 г (28,71 ммоля) триэтиламина, 3,75 г (24,9 ммоль) трет-бутилдиметилсилилхлорида, 0,10 г 4-диметиламинопиридина и 40 мл хлороформа. В результате получают темно-желтый раствор. Этот раствор перемешивают при температуре окружающей среды в течение 2 часов. Раствор распределяют между этилацетатом и насыщенным водным раствором хлористого аммония. Водный и органический слои разделяют, органический слой промывают насыщенным водным раствором бикарбоната натрия, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения 6,05 г (16,11 ммоля) желто-зеленого твердого N-(4-{[трет-бутил(диметил)силил]окси}бутил)-3-нитрохинолин-4-амина. Этот продукт используют без дальнейшей очистки. MS (масс-спектр)(Cl) для С19H29N3O3Si m/z 376 (MH+), 342,210.
Часть С
В аппарат Парра загружают 6,05 г (16,11 ммоля) N-(4-{[трет-бутил(диметил)силил]окси}бутил)-3-нитрохинолин-4-амина, 3,0 г активированного угля, содержащего 5% нанесенной на него платины, и 32 мл толуола. Реакционную смесь встряхивают в атмосфере водорода (при его давлении 3,5 кг/см2 (50 фунт/дюйм2)) в течение 1 часа, после чего добавляют еще 3,0 г катализатора, 15 мл толуола и встряхивание продолжают при давлении 3,5 кг/см3 (50 фунт/дюйм2) еще в течение 1 часа. Катализатор отфильтровывают на бумажном фильтре и фильтр промывают толуолом (50 мл). Фильтраты объединяют вместе и концентрируют при пониженном давлении, в результате чего получают 5,57 г (16,11 ммоля) темного маслообразного N-(4-{[трет-бутил(диметил)силил]окси}бутил)хинолин-3,4-диамина. Этот продукт используют без дальнейшей очистки.
Часть D
В круглодонную колбу, снабженную магнитной мешалкой, загружают 5,57 г (16,11 ммоля) N-(4-{[трет-бутил(диметил)силил]окси}бутил)хинолин-3,4-диамина, 5,23 г (32, 22 ммоля) триметилортовалериата и 47 мл толуола. Реакционную смесь кипятят с обратным холодильником и при этом медленно отгоняют метанол, образующийся в качестве побочного продукта. Реакция завершается после 15 часов кипячения. После этого смесь охлаждают и летучие продукты удаляют при пониженном давлении, в результате чего получают густой коричневый маслообразный 2-бутил-1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1H-имидазо[4,5-с]хинолин (4,65 г, 11,30 ммоля). Этот продукт используют без дальнейшей очистки. MS (Cl) для C24H37N3OSi m/z 412 (MH+), 298.
Часть Е
В круглодонную колбу, снабженную магнитной мешалкой, загружают 4,65 г (11,30 ммоля) 2-бутил-1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1Н-имидазо[4,5-с]хинолина и 57 мл хлороформа. К раствору в течение 15 минут добавляют 2,78 г (2,43 ммоля) твердой 3-хлорпербензойной кислоты и смесь перемешивают при температуре окружающей среды еще в течение 1 часа. Затем к реакционной смеси добавляют дополнительно 0,5 г (2,9 ммоля) 3-хлорпербензойной кислоты, и через 30 минут после этого происходит полное израсходование исходного материала. Раствор распределяют между хлороформом и насыщенным водным раствором бикарбоната натрия. Разделяют водный и органический слои. Органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении, в результате чего получают 4,83 г (11,30 ммоля) 2-бутил-1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1H-имидазо[4,5-с]хинолин-5N-оксида в виде темного масла. Этот продукт используют без дальнейшей очистки.
Часть F
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают (11,30 ммоля) 2-бутил-1-(4-[трет-бутил(диметил)силил]окси}бутил)-1Н-имидазо[4,5-с]хинолин-5N-оксида и 57 мл безводного диметилформамида. К раствору медленно по каплям добавляют 1,91 г (12,43 ммоля) оксихлорида фосфора и после окончания добавления получают гомогенный раствор. Реакция заканчивается после 1,5 часов перемешивания при температуре окружающей среды. Полученную реакционную смесь распределяют между дихлорметаном и насыщенным водным раствором бикарбоната натрия. Разделяют водный и органический слои. Органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении, в результате чего получают 3,65 г (10,42 ммоля) твердого коричневого 2-бутил-4-хлор-1-(4-хлорбутил)-1Н-имидазо[4,5-с]хинолина. Этот продукт используют без дальнейшей очистки. MS (Cl) для С18H21Cl2N3 m/z 376 (MH+), 314.
Часть G
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 1,18 г (3,37 ммоля) 2-бутил-4-хлор-1-(4-хлорбутил)-1H-имидазо[4,5-с]хинолина, 0,56 г (5,05 ммоля) бензолтиола, 0,68 г (6,74 ммоля) триэтиламина и 15 мл диметилформамида. Смесь нагревают до 80°С для получения гомогенного раствора и этот раствор выдерживают при 80°С в течение 2,5 часов. Согласно данным высокоэффективной жидкостной хроматографии полученный продукт не содержит исходного материала и представляет собой 3:1 смесь 2-бутил-4-хлор-1-[4-(фенилтио)бутил]-1H-имидазо[4,5-с]хинолина и 2-бутил-(4-фенилтио)-1-[4-(фенилтио)бутил]-1H-имидазо[4,5-с]хинолина. Раствор охлаждают и распределяют между этилацетатом и насыщенным водным раствором бикарбоната натрия. Разделяют водный и органический слои. Органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении, в результате чего получают 1,43 г (3:1) смеси указанных выше продуктов. Материал используют без дальнейшей очистки.
Часть Н
Смесь(3:1) 2-бутил-4-хлор-1-[4-(фенилтио)бутил]-1H-имидазо[4,5-с]хинолина и 2-бутил-(4-фенилтио)-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолина (общее количество смеси 1,38 г) и 30 мл 7%-ного раствора аммиака в метаноле нагревают в автоклаве при 150°С. Реакция завершается в течение 5 часов. Летучие продукты удаляют при пониженном давлении. Остаток перемешивают с водой и подщелачивают раствор до рН 10, добавляя к нему твердый карбонат натрия. Водный раствор трижды экстрагируют хлороформом. Объединенные органические фракции промывают насыщенным водным раствором бикарбоната натрия и рассолом, после чего сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения желтого твердого кристаллического продукта. 0,8 г этого продукта растворяют в 50 мл этилацетата и раствор нагревают до кипения. Добавляют 0,4 г активированного угля и полученную смесь кипятят с обратным холодильником в течение 5 минут, после чего активированный уголь отфильтровывают на бумажном фильтре. В результате получают бесцветный раствор. Раствор концентрируют при пониженном давлении до получения твердого продукта, который перекристаллизовывают из этилацетата и гексанов. В результате получают 0,51 г (1,25 ммоля) 2-бутил-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде белых игольчатых кристаллов. Температура плавления этого продукта 118-120°С.
Анализ. Рассчитано для C24H28N4S: %С 71,25; %Н 6,98; %N 13,85. Найдено: %С 71,12;%Н 6,81; %N 13,62.
1H ЯМР (300 МГц, ДМСО) δ 8,02 (дублет, J=8,3 Гц, 1Н); δ 7,61 (дублет, J=8,3 Гц, 1Н); δ 7,41 (триплет, J=8,3 Гц, 1Н); δ 7,16-7,30 (мультиплет, 6Н); δ 6,46 (широкий синглет, 2Н); δ 4,52 (триплет, J=7,6 Гц, 2Н); δ 3,02 (триплет, J= 7,3 Гц, 2Н); δ 2,89 (триплет, J=7,8 Гц, 2Н); δ 1,95 (мультиплет, 2Н); δ 1,75 (мультиплет, 4Н); δ 1,43 (секстет, J=7,3 Гц, 2Н); δ 0,94 (триплет, J=7,3 Гц, ЗН).
MS (Cl) для C24H28N4S m/z 405 (MH+), 282, 241.
Пример 2
Солянокислый 2-бутил-1-[2-(фенилтио)этил]-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амин

Часть А
В круглодонную колбу, снабженную магнитной мешалкой, загружают 1,0 г (3,47 ммоля) 2-(4-амино-2-бутил-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил)этанола, 1,62 г (10,75 ммоля) трет-бутилдиметилсилилхлорида, 1,58 г (15,62 ммоля) триэтиламина, 0,1 г 4-диметиламинопиридина и 30 мл хлороформа. Полученную гетерогенную смесь перемешивают при 60°С в течение 2 часов. По окончании реакции полученный раствор распределяют между этилацетатом и насыщенным водным раствором хлористого аммония. Разделяют водный и органический слои. Органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении, в результате чего получают 1,79 г (3:1) смеси 2-бутил-1-(2-{[трет-бутил(диметил)силил]окси}этил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина и 2-бутил-N-[трет-бутил(диметил)силил]-1-(2-{[трет-бутил(диметил)силил]окси}этил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина в виде темно-коричневого масла. Материал используют без дальнейшей очистки.
Часть В
В круглодонную колбу, снабженную магнитной мешалкой, загружают 1,6 г 3:1 смеси 2-бутил-1-(2-{[трет-бутил(диметил)силил]окси}этил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амина и 2-бутил-N-[трет-бутил(диметил)силил]-1-(2-{[трет-бутил(диметил)силил]окси}этил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амина и 85 мл 1 М раствора уксусной кислоты в дихлорметане. Полученный гомогенный продукт перемешивают при температуре окружающей среды в течение 30 минут. По окончании реакции раствор распределяют между хлороформом и рассолом. Разделяют водный и органический слои. Органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения темно-коричневого масла. Этот продукт подвергают очистке на хроматографической колонке, заполненной силикагелем, используя в качестве элюента тройную смесь (95/4/1) дихлорметана/метанола/водного раствора гидроксида аммония (14,8 М). В результате получают 1,24 г (3,10 ммоля) 2-бутил-1-(2-{[трет-бутил(диметил)силил]окси}этил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина в виде бесцветного масла.
Часть С
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 0,83 г (2,06 ммоля) 2-бутил-1-(2-{[трет-бутил(диметил)силил]окси}этил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина, 1,79 г (8,24 ммоля) ди-трет-бутилдикарбоната, 0,52 г (5,15 ммоль) триэтиламина, 0,1 г 4-диметиламинопиридина и 21 мл тетрагидрофурана. При нагревании до 60°С смесь превращается в гомогенный раствор, который выдерживают при этой температуре в течение 2,5 часов. За это время реакция полностью завершается. Раствор охлаждают до температуры окружающей среды и добавляют к нему 2,27 мл (2,27 ммоль) 1 М раствора тетрабутиламмонийфторида в тетрагидрофуране. Реакция завершается при перемешивании реакционной смеси при температуре окружающей среды в течение 30 минут. Раствор распределяют между этилацетатом и насыщенным водным раствором хлористого аммония. Разделяют водный и органический слои. Органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения светло-желтого твердого продукта. Этот продукт подвергают очистке на хроматографической колонке, заполненной силикагелем, используя в качестве элюента смесь (95/5) дихлорметана/метанола. В результате получают 0,55 г (1,13 ммоля) ди(трет-бутил)-2-бутил-1-(2-гидроксиэтил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-илимидодикарбоната в виде прозрачного клейкого вещества.
Часть D
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 0,55 г (1,13 ммоля) ди(трет-бутил)-2-бутил-1-(2-гидроксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-илимидодикарбоната и 11 мл безводного дихлорметана. Полученный гомогенный раствор охлаждают до -10°С, используя для этой цели смесь метанола со льдом. К охлажденному раствору добавляют 0,23 г (2,26 ммоля) триэтиламина и 0,19 г (1,70 ммоля) метансульфонилхлорида. Реакция полностью протекает при перемешивании реакционной смеси при этой температуре в течение 15 мин. Полученную смесь распределяют между этилацетатом и насыщенным водным раствором хлористого аммония. Разделяют водный и органический слои. Органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения 0,61 г (1,08 ммоля) 2-{4-[бис(трет-бутоксикарбонил)амино]-2-бутил-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-1-ил}этилметансульфоната в виде желтого твердого клейкого вещества. Этот материал используют без дальнейшей очистки. MS (Cl) для C27H42N4O7S m/z 567 (MH+), 467, 367, 271.
Часть Е
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 0,61 г (1,08 ммоля) 2-{4-[бис(трет-бутоксикарбонил)амино]-2-бутил-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-1-ил}этилметансульфоната, 0,21 г (1,88 ммоля) бензолтиола, 0,25 г (2,43 ммоля) триэтиламина и 11 мл безводного диметилформамида. При нагревании до 80°С смесь превращается в гомогенный темно-желтый раствор, который выдерживают при такой температуре в течение 2,5 часов. За это время реакция полностью завершается. Раствор охлаждают и распределяют между этилацетатом и насыщенным водным раствором хлористого аммония. Разделяют водный и органический слои. Органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения желтого масла. Этот продукт подвергают очистке на хроматографической колонке, заполненной силикагелем, используя в качестве элюента смесь 95/5 дихлорметана/метанола. В результате получают 0,54 г (0,93 ммоля) ди(трет-бутил)-2-бутил-1-[2-(фенилтио)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-илимидодикарбоната в виде светло-желтого масла. MS (Cl) для C32H44N4O4S m/z 581 (MH+), 481, 381, 245.
Часть F
В круглодонную колбу, снабженную магнитной мешалкой, загружают 0,50 г (0,86 ммоля) ди(трет-бутил)-2-бутил-1-[2-(фенилтио)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-илимидодикарбоната, 5 мл 4 М раствора соляной кислоты в диоксане и 5 мл дихлорметана. Реакция завершается при перемешивании реакционной смеси при температуре окружающей среды в течение 2 часов. Летучие продукты удаляют при пониженном давлении и в результате получают твердое вещество беловатого цвета. После перекристаллизации этого материала из ацетонитрила получают 0,17 г (1,30 ммоля) солянокислого 2-бутил-1-[2-(фенилтио)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина в виде пушистых белых игольчатых кристаллов. Температура плавления 237-238°С.
Анализ. Рассчитано для C22H28N4S·(H2O)1/4·(HCl)2: %C 57,70; %Н 6,71; %N 12,23.
Найдено: %C 57,62; %Н 6,57; %N 12,41.
1H ЯМР (300 МГц, ДМСО) δ 7,81 (широкий синглет, 2Н); δ 7,61 (дублет, J=8,3 Гц, 1Н); δ 7,22-7,39 (мультиплет, 5Н); δ 4,64 (триплет, J=6,8 Гц, 2Н); δ 3,40 (триплет, J=6,8 Гц, 2Н); δ 2,75 (мультиплет, 6Н); δ 1,71 (мультиплет, 6Н); δ 1,34 (секстет, J=7,3 Гц, 2Н); δ 0,89 (триплет, J=7,3 Гц, 3Н).
MS (Cl) для C22H28N4S·(H2O)1/4·(HCI)2 m/z 381 (MH+), 245, 137.
Пример 3
2-Бутил-1-[4-(фенилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин

Часть А
Используя основной способ, применяемый в примере 1, часть Е, проводят окисление 16,0 г (38,87 ммоля) 2-бутил-1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1H-имидазо[4,5-с]хинолина до 2-бутил-1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1H-имидазо[4,5-с]хинолин-5N-оксида. В результате получают 16,61 г (38,87 ммоля) этого коричневатого твердого продукта, который используют без дополнительной очистки.
Часть В
В круглодонную колбу, снабженную магнитной мешалкой, загружают 16,61 г (38,87 ммоля) 2-бутил-1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1Н-имидазо[4,5-с]хинолин-5N-оксида, 75 мл 14,8 М раствора гидроксида аммония в воде и 200 мл хлороформа. К быстро перемешиваемому раствору в несколько порций добавляют 8,15 г (42,76 ммоль) п-толуиленсульфонилхлорида. При этом наблюдается небольшое повышение температуры. Реакция завершается после перемешивания реакционной смеси при температуре окружающей среды в течение 10 минут. Полученный раствор распределяют между хлороформом и насыщенным водным раствором бикарбоната натрия. Разделяют водный и органический слои. Органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения беловатого твердого продукта. Этот продукт обрабатывают этиловым эфиром и после фильтрования получают 9,3 г (21,80 ммоля) 2-бутил-1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1Н-имидазо[4,5-с]хинолин-4-амина в виде мелкого белого порошка. Этот материал используют без дальнейшей очистки.
Часть С
В круглодонную колбу, снабженную магнитной мешалкой, загружают 9,2 г (21,56 ммоля) 2-бутил-1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1Н-имидазо[4,5-с]хинолин-4-амина, 23,72 мл (23,72 ммоля) 1 М раствора тетрабутиламмонийфторида в тетрагидрофуране и 100 мл безводного тетрагидрофурана. В результате получают гомогенный светло-оранжевый раствор. Для завершения реакции реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. При добавлении 100 мл воды к полученной смеси наблюдается ее слабый разогрев. Летучие продукты удаляют при пониженном давлении до выпадения из раствора твердого осадка. Этот твердый осадок отфильтровывают и промывают 20 мл воды и 20 мл ацетона. После обработки полученного материала 50 мл этилового эфира получают 6,12 г (19,59 ммоля) 4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутан-1-ола в виде мелкого белого порошка. Температура плавления этого продукта 184-186°С.
Анализ. Рассчитано для C18H24N4O: %С 69,20; %Н 7,74; %N 17,93. Найдено: %С 69,05; %Н 8,02; %N 18,03.
MS (Cl) для C18H24N4O m/z 313 (MH+).
Часть D
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 7,3 г (23,37 ммоля) 4-(4-амино-2-бутил-1H-имидазо[4,5-с]хинолин-1-ил)бутан-1-ола, 3,55 г (35,06 ммоля) триэтиламина и 93 мл безводного диметилформамида. К перемешиваемому раствору по каплям добавляют 3,94 г (25,70 ммоля) оксихлорида фосфора. При этом наблюдается разогрев смеси и образуется темно-желтая гетерогенная смесь, которая при нагревании до 60°С становится гомогенной. Смесь перемешивают при 60°С в течение 5 часов; этого времени достаточно для полного исчерпания исходного продукта. После этого летучие продукты удаляют при пониженном давлении и в результате получают темно-коричневый маслообразный продукт. Этот продукт распределяют между хлороформом и насыщенным водным раствором бикарбоната натрия. Органический и водный слои разделяют и водный слой экстрагируют хлороформом. Объединенные органические фракции концентрируют при пониженном давлении до получения беловатого твердого продукта, представляющего собой 7,70 г (2:1) смеси N'-[2-бутил-1-(4-хлорбутил)-1Н-имидазо[4,5-с]хинолин-4-ил]-N,N-диметилимидоформамида и 2-бутил-1-(4-хлорбутил)-1Н-имидазо[4,5-с]хинолин-4-амина. Этот продукт используют без дополнительной очистки.
Часть Е
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 1,3 г (2:1) смеси N'-[2-бутил-1-(4-хлорбутил)-1Н-имидазо[4,5-с]-хинолин-4-ил]-N,N-диметилимидоформамида и 2-бутил-1-(4-хлорбутил)-1H-имидазо[4,5-с]хинолин-4-амина, 1,67 г (10,11 ммоля) натриевой соли бензолсульфоновой кислоты и 15 мл безводного диметилформамида. Полученную смесь нагревают до 100°С, в результате чего получают гомогенный раствор, который для завершения реакции нагревают при этой температуре в течение 90 часов. Раствор охлаждают и распределяют его между хлороформом и водой. Органический и водный слои разделяют. Органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения смолы темно-желтого цвета. Этот материал растворяют в смеси 20 мл метанола и 3,02 мл (12,1 ммоля) 4 М раствора соляной кислоты в диоксане. Для завершения реакции светло-оранжевый раствор перемешивают при температуре окружающей среды в течение 12 часов. После этого отгоняют летучие продукты и получают продукт в виде смолы светло-желтого цвета. Этот продукт распределяют между хлороформом и насыщенным водным раствором бикарбоната натрия. Слои отделяют друг от друга и водный слой экстрагируют хлороформом. Органические слои соединяют вместе, промывают рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения светло-желтого твердого продукта. Очистка этого продукта хроматографическим способом на силикагеле (95/5 дихлорметан/метанол) дает беловатый твердый продукт. 0,63 г этого продукта растворяют в 50 мл этилацетата и раствор доводят до кипения. К полученной смеси добавляют 0,6 г активированного угля и кипятят ее с обратным холодильником в течение 5 минут. После удаления активированного угля на бумажном фильтре получают бесцветный раствор, который концентрируют при пониженном давлении до получения твердого продукта. Перекристаллизация этого продукта из этилацетата и гексанов дает 0,37 г (0,85 ммоля) 2-бутил-1-[4-(фенилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде пушистого белого продукта. Температура плавления 179-180°С.
Анализ. Рассчитано для C24H28N4O2S: %С 66,03; %Н 6,46; %N 12,83. Найдено: %С 65,88; %Н 6,49; %N 12,76.
1H ЯМР (300 МГц, ДМСО) δ 7,98 (дублет, J=8,3 Гц, 1Н); δ 7,82 (мультиплет, 2Н); δ 7,73 (дублет, J=7,3 Гц, 1Н); δ 7,62 (мультиплет, ЗН); δ 7,41 (триплет, J=7,6 Гц, 1Н); δ 7,22 (триплет, J=7,6 Гц, 1Н); δ 6,45 (широкий синглет, 2Н); δ 4,51 (триплет, J=7,3 Гц, 2Н); δ 3,90 (триплет, J=7,8 Гц, 2Н); δ 2,86 (триплет, J=7,6 Гц, 3Н); δ 1,69-1,90 (мультиплет, 6Н); δ 1,43 (секстет, J=7,3 Гц, 2Н); δ 0,95 (триплет, J=7,3 Гц, 3Н). MS (Cl) для C24H28N4O2S m/z 437 (МН+), 295.
Пример 4
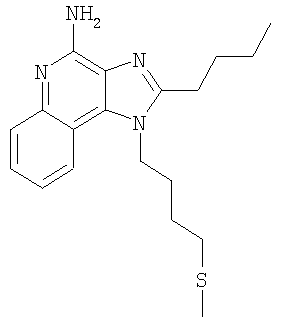
2-Бутил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
В круглодонную колбу, снабженную магнитной мешалкой, загружают 6,17 г (2:1) смеси N'-[2-бутил-1-(4-хлорбутил)-1H-имидазо[4,5-с]-хинолин-4-ил]-N,N-диметилимидоформамида и 2-бутил-1-(4-хлорбутил)-1H-имидазо[4,5-с]хинолин-4-амина, 21,15 мл (84,56 ммоля) 4 М раствора соляной кислоты в диоксане и 200 мл метанола. Для завершения реакции полученный светло-оранжевый раствор перемешивают при температуре окружающей среды в течение 43 часов. Летучие продукты отгоняют при пониженном давлении, и полученный светло-желтый твердый продукт распределяют между хлороформом и насыщенным водным раствором бикарбоната натрия. Слои отделяют друг от друга и водный слой экстрагируют хлороформом. Органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения 4,65 г (14,05 ммоля) 2-бутил-1-(4-хлорбутил)-1H-имидазо[4,5-с]хинолин-4-амина в виде беловатого твердого вещества. Этот продукт используют в дальнейшем без дополнительной очистки. MS (Cl) для C18H23CIN4 m/z 331 (MH+), 295.
Часть В
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 1,5 г (4,53 ммоля) 2-бутил-1-(4-хлорбутил)-1H-имидазо[4,5-с]хинолин-4-амина, 0,48 г (6,80 ммоля) тиометилата натрия и 18 мл безводного диметилформамида. Для получения гомогенного раствора смесь нагревают до 60°С и затем выдерживают при этой температуре в течение 16 часов, чтобы обеспечить полное израсходование исходного материала. Раствор охлаждают и распределяют между хлороформом и водой. Водный и органический слои отделяют друг от друга и органический слой промывают насыщенным водным раствором бикарбоната натрия. Объединенные водные фракции экстрагируют хлороформом. Объединенные органические фракции промывают рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения темно-коричневого масла. После хроматографической очистки на силикагеле (90/10 дихлорметан/метанол) продукт получают в виде светло-желтого твердого вещества. Перекристаллизация этого продукта из диметилформамида и воды дает 0,83 г (2,42 ммоля) 2-бутил-1-[4-(метилтио)бутил]-1H-имидазо[4,5-с]хинолин-4-амина в виде светло-желтых игольчатых кристаллов. Температура плавления 127-130°С.
Анализ. Рассчитано для C19H26N4S: %С 66,63; %Н 7,65; %N 16,36. Найдено: %С 66,68; %Н 7,53; %N 16,35.
1H ЯМР (500 МГц, ДМСО) δ 8,04 (дублет, J=8,3 Гц, 1H); δ 7,61 (дублет, J=8,3 Гц, 1Н); δ 7,41 (триплет, J=8,3 Гц, 1Н); δ 7,25 (триплет, J=8,3 Гц, 1Н); δ 6,46 (широкий синглет, 2Н); δ 4,52 (триплет, J=7,6 Гц, 2Н); δ 2,92 (триплет, J=7,8 Гц, 2Н); δ 2,53 (триплет, J=7,3 Гц, 2Н); δ 2,01 (синглет, 3Н); δ 1,90 (мультиплет, 2Н); δ 1,80 (квинтет, J=7,8 Гц, 2Н); δ 1,71 (квинтет, J=7,3 Гц, 2Н); δ 1,46 (секстет, J=7,3 Гц, 2Н); δ 0,96 (триплет, J=7,3 Гц, 3Н).
MS (Cl) для C19H26N4S m/z 343 (MH+), 295, 241.
Пример 5
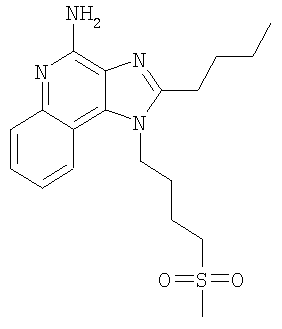
2-Бутил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
В круглодонную колбу, снабженную магнитной мешалкой, загружают 1,2 г (3,50 ммоля) 2-бутил-1-[4-(метилтио)бутил]-1H-имидазо[4,5-с]хинолин-4-амина и 18 мл хлороформа. К полученному раствору в течение 15 минут добавляют 1,72 г (7,71 ммоля) твердой 3-хлорпербензойной кислоты. Реакция завершается при перемешивании при температуре окружающей среды в течение 5 минут. Раствор распределяют между хлороформом и 1%-ным водным раствором карбоната натрия. Водный и органический слои отделяют друг от друга и органический слой промывают рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения светло-коричневого твердого продукта. После хроматографической очистки на силикагеле (90/10 дихлорметан/метанол) продукт получают в виде беловатого твердого вещества. Перекристаллизация этого продукта из ацетонитрила и воды дает 0,61 г (1,63 ммоля) 2-бутил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде беловатых игольчатых кристаллов. Температура плавления 164-165°С.
Анализ. Рассчитано для C19H26N4O2S: %С 60,94; %Н 7,00; %N 14,96. Найдено: %С 60,71; %Н 6,94; %N 14,94.
1H ЯМР (300 МГц, ДМСО) δ 8,03 (дублет, J=8,3 Гц, 1H); δ 7,61 (дублет, J=8,3 Гц, 1Н); δ 7,42 (триплет, J=8,3 Гц, 1Н); δ 7,26 (триплет, J=8,3 Гц, 1Н); δ 6,46 (широкий синглет, 2Н); δ 4,56 (триплет, J=7,6 Гц, 2Н); δ 3,21 (триплет, J=7,3 Гц, 2Н); δ 2,96 (синглет, 3Н); δ 2,93 (триплет, J=7,8 Гц, 2Н); δ 1,91 (мультиплет, 4Н); δ 1,81 (квинтет, J=7,8 Гц, 2Н); δ 1,71 (квинтет, J=7,3 Гц, 2Н); δ 1,45 (секстет, J=7,3 Гц, 2Н); δ 0,96 (триплет, J=7,3 Гц, 3Н).
MS (Cl) для C19H26N4O2S m/z 375 (MH+), 295.
Пример 6
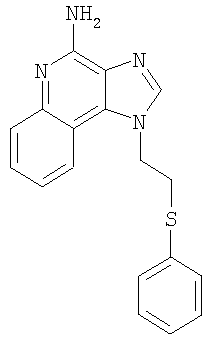
1-[2-(Фенилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 8,46 г (37,06 ммоля) 2-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)этанола и 68,99 г (57,99 ммоля) тионилхлорида. Реакционную смесь нагревают до 80°С и полученный гомогенный раствор выдерживают при этой температуре в течение 2 часов. За это время происходит полное исчерпание исходного материала. Раствор охлаждают и добавляют к нему 400 мл воды. При перемешивании к раствору добавляют твердый карбонат натрия до достижения рН=10, при этом происходит осаждение твердого осадка из раствора. Этот твердый продукт отфильтровывают и получают 7,86 г (31,86 ммоля) 1-(2-хлорэтил)-1H-имидазо[4,5-с]хинолин-4-амина в виде беловатого твердого продукта. Полученный продукт используют в дальнейшем без дополнительной очистки.
Часть В
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 2,0 г (8,11 ммоля) 1-(2-хлорэтил)-1Н-имидазо[4,5-с]хинолин-4-амина, 1,79 г (12,16 ммоля) бензолтиолата натрия и 40 мл безводного диметилсульфоксида. Реакционную смесь нагревают до 100°С и полученный гомогенный раствор выдерживают при этой температуре в течение 30 минут. За это время происходит полное израсходование исходного материала. Горячий раствор выливают при быстром перемешивании в 300 мл воды, в результате чего выпадает твердый осадок. Этот твердый продукт отфильтровывают и получают беловатое твердое вещество, после обработки которого ацетонитрилом получают 2,08 г (6,49 ммоля) беловатого порошка 1-[2-(фенилтио)этил]-1H-имидазо[4,5-с]хинолин-4-амина. Температура плавления этого продукта 233-235°С.
Анализ. Рассчитано для C18H16N4S: %С 67,47; %Н 5,03; %N 17,49. Найдено: %С 67,20; %Н 4,95; %N 17,52.
1H ЯМР (300 МГц, ДМСО) δ 8,14 (синглет, 1Н); 4 7,76 (дублет, J=8,3 Гц, 1H); δ 7,60 (триплет, J=8,3 Гц, 1Н); δ 7,28 - 7,44 (мультиплет, 6Н); δ 7,12 (триплет, J=8,3 Гц, 1Н); δ 6,58 (широкий синглет, 2Н); δ 4,79 (триплет, J=6,8 Гц, 2Н); δ 3,48 (триплет, J=6,8 Гц, 2Н).
MS (Cl) для C18H16N4S m/z 321 (MH+), 185, 137.
Пример 7
1-[4-(Фенилсульфонил)бутил]-1H-имидазо[4,5-c]хинолин-4-амин

Часть А
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 20,0 г (55,04 ммоля) N,N-дибензил-1Н-имидазо[4,5-с]хинолин-4-амина, 3,3 г (82,56 ммоля) 60%-ной дисперсии гидрида натрия и 275 мл безводного диметилформамида. Смесь перемешивают при температуре окружающей среды в течение 2 часов, после чего добавляют к ней 19,23 г (88,06 ммоля) 4-хлор-1-иодбутана. Для полного завершения реакции полученный гомогенный раствор перемешивают при температуре окружающей среды в течение 48 часов, чтобы обеспечить полное израсходывание исходного материала. Раствор распределяют между этилацетатом и водой. Водный и органический слои отделяют друг от друга и органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения светло-желтого твердого продукта. После перекристаллизации из этилацетата и гексанов получают 20,7 г (45,49 ммоля) N,N-дибензил-1-(4-хлорбутил)-1Н-имидазо[4,5-с]хинолин-4-амина в виде белых игольчатых кристаллов. MS(Cl) для C28H27CIN4 m/z 455 (MH+), 365, 329, 239.
Часть В
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 7,0 г (15,38 ммоля) N,N-дибензил-1-(4-хлорбутил)-1H-имидазо[4,5-с]хинолин-4-амина, 3,46 г (26,15 ммоля) бензолтиолата натрия и 77 мл безводного диметилформамида. Реакционную смесь нагревают до 60°С и полученный гомогенный раствор выдерживают при этой температуре в течение 4 часов; за это время происходит полное израсходование исходного материала. Охлажденный раствор распределяют между этилацетатом и водой. Водный и органический слои отделяют друг от друга и органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения бесцветного масла. Этот продукт подвергают хроматографической очистке на силикагеле (80/20 гексаны/этилацетат), в результате чего получают 7,5 г (14,19 ммоля) N,N-дибензил-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина в виде бесцветного масла. MS (Cl) для C34H32N4S m/z 529 (МН+), 439, 349.
Часть С
В круглодонную колбу, снабженную магнитной мешалкой, загружают 3,64 г (6,88 ммоля) N,N-дибензил-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина и 34 мл хлороформа. К полученному раствору в течение 5 минут добавляют 3,39 г (15,14 ммоль) твердой 3-хлорпербензойной кислоты. Для завершения реакции смесь перемешивают при температуре окружающей среды в течение 5 минут. Раствор распределяют между хлороформом и 1%-ным водным раствором карбоната натрия. Водный и органический слои отделяют друг от друга и органический слой промывают рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения красной смолы. Этот продукт подвергают хроматографической очистке на силикагеле (в качестве элюента используют дихлорметан) и в результате получают 2,85 г (5,08 ммоля) N,N-дибензил-1-[4-(фенилсульфонил)бутил]-1H-имидазо[4,5-с]хинолин-4-амина в виде бледно-розовой смолы. MS (Cl) для С34Н32N4O2S m/z 561 (МН+), 471, 381.
Часть D
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 1,0 г (1,78 ммоля) N,N-дибензил-1-[4-(фенилсульфонил)бутил]-1H-имидазо[4,5-с]хинолин-4-амина, 2,68 г (17,83 ммоля) трифторметануксусной кислоты и 14 мл безводного дихлорметана. Для завершения реакции смесь перемешивают при температуре окружающей среды в течение 24 часов. Смесь распределяют между хлороформом и избытком 20%-ного водного раствора гидроксида натрия. Водный и органический слои отделяют друг от друга. Водный слой трижды экстрагируют хлороформом. Органические фракции объединяют вместе и концентрируют при пониженном давлении до получения светло-коричневого твердого продукта. Этот продукт подвергают хроматографической очистке на силикагеле (90/10 дихлорметан/метанол) и в результате получают мелкий белый порошок, после перекристаллизации которого из ацетонитрила получают 0,32 г (0,84 ммоля) 1-[4-(фенилсульфонил)бутил]-1H-имидазо[4,5-с]хинолин-4-амина в виде белых игольчатых кристаллов. Температура плавления этого продукта 175-177°С.
Анализ. Рассчитано для C20H20N4O2S: %С 63,14; %Н 5,30; %N 14,73. Найдено: %С 63,14; %Н 5,24; %N 14,77.
1H ЯМР (300 МГц, ДМСО) δ 8,15 (синглет, 1Н); δ 8,01 (дублет, J=8,3 Гц, 1Н); δ 7,80 (мультиплет, 2Н); δ 7,71 (мультиплет, 1Н); δ 7,60 (мультиплет, 3Н); δ 7,44 (триплет, J=8,3 Гц, 1Н); δ 7,24 (триплет, J=8,3 Гц, 1Н); δ 6,59 (широкий синглет, 2Н); δ 4,59 (триплет, J=6,8 Гц, 2Н); δ 3,38 (триплет, J=7,8 Гц, 2Н); δ 1,93 (мультиплет, 2Н); δ 1,58 (мультиплет, 2Н).
MS (Cl) для C20H20N4O2S m/z 381 (МН+), 239.
Пример 8
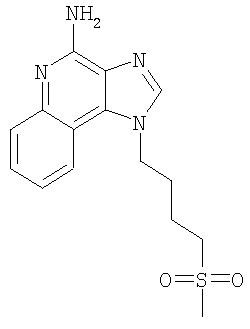
1-[4-(Метилсульфонил)бутил]-1H-имидазо[4,5-c]хинолин-4-амин
Часть А
Используя основной способ, применяемый в примере 7, часть В, N,N-дибензил-1-(4-хлорбутил)-1Н-имидазо[4,5-с]хинолин-4-амин (5,0 г, 10,99 ммоля) под действием тиометилата натрия превращают в N,N-дибензил-1-[4-(метилтио)бутил]-1H-имидазо[4,5-с]хинолин-4-амин (1,16 г, 16,48 ммоля). Этот продукт подвергают хроматографической очистке на силикагеле (80/20 гексаны/этилацетат), и в результате получают 4,91 г (10,52 ммоля) целевого продукта в виде бесцветного масла. MS (Cl) для C29H30N4S m/z 467 (MH+), 377, 287, 185.
Часть В
Используя основной способ, применяемый в примере 7, часть С, проводят окисление N,N-дибензил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина (4,91 г, 15,52 ммоля) в N,N-дибензил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин, после хроматографической очистки которого на силикагеле (80/20 гексаны/этилацетат) получают 4,53 г (9,08 ммоля) целевого твердого продукта светло-оранжевого цвета. MS(Cl) для C29H30N4O2S m/z 499 (MH+), 409, 319.
Часть С
Используя основной способ, применяемый в примере 7, часть D, N,N-дибензил-1-[4-(метилсульфонил)бутил]-1H-имидазо[4,5-с]хинолин-4-амин (4,53 г, 9,08 ммоля) превращают в 1-[4-(метилсульфонил)бутил]-1H-имидазо[4,5-с]хинолин-4-амин. Этот продукт перекристализовывают из метанола и воды и получают 1,33 г (4,18 ммоля) целевого продукта в виде белых игольчатых кристаллов. Температура плавления этого продукта составляет 203-204°С.
Анализ. Рассчитано для C15H18N4O2S: %С 56,58; %Н 5,70; %N 17,60. Найдено: %С 56,33; %Н 5,63; %N 17,41.
1H ЯМР (300 МГц, ДМСО) δ 8,22 (синглет, 1Н); δ 8,06 (дублет, J=8,3 Гц, 1Н); δ 7,62 (дублет, J=8,3 Гц, 1Н); δ 7,45 (триплет, J=8,3 Гц, 1Н); δ 7,27 (триплет, J=8,3 Гц, 1Н); δ 6,59 (широкий синглет, 2Н); δ 4,65 (триплет, J=6,8 Гц, 2Н); δ 3,19 (триплет, J=7,8 Гц, 2Н); 62,93 (синглет, 3Н); δ 1,99 (мультиплет, 2Н); δ 1,74 (мультиплет, 2Н). MS (Cl) для C15H18N4O2S m/z 319 (MH+), 239.
Пример 9
1-[4-(Фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин

Часть А
Используя основной способ, применяемый в примере 1, часть D, под действием триэтилортоформиата (65,11 г, 439,35 ммоля) проводят циклизацию N-(4-{[трет-бугил(диметил)силил]окси}бутил)хинолин-3,4-диамина (101,21 г, 292,90 ммоль) до 1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1H-имидазо[4,5-с]хинолина. 75,0 г (210,93 ммоля) этого продукта было выделено из реакционной смеси в виде коричневого масла. В дальнейшем этот продукт используют без дополнительной очистки.
Часть В
Используя основной способ, применяемый в примере 1, часть Е, 42,2 г (118,69 ммоля) 1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1H-имидазо[4,5-с]хинолина окисляют до 1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1H-имидазо[4,5-с]хинолин-5N-оксида (44,10 г, 118,69 ммоля), который выделяют из реакционной смеси в виде желтовато-коричневого твердого продукта и используют без дополнительной очистки.
Часть С
Используя основной способ, применяемый в примере 3, часть В, проводят аминирование 44,10 г (118,69 ммоля) 1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1H-имидазо[4,5-с]хинолин-5N-оксида до 1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1Н-имидазо[4,5-с]хинолин-4-амина. После обработки реакционной смеси этиловым эфиром из нее отфильтровывают 21,54 г (58,12 ммоля) целевого амина в виде светло-коричневого твердого вещества. Этот продукт в дальнейшем используют без дополнительной очистки.
Часть D
Используя основной способ, применяемый в примере 3, часть С, 21,5 г (58,02 ммоля) 1-(4-{[трет-бутил(диметил)силил]окси}бутил)-1H-имидазо[4,5-с]хинолин-4-амина превращают в 4-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)бутан-1-ол. Полученную реакционную смесь обрабатывают холодным метанолом (0°С) и после фильтрации получают 13,92 г (54,30 ммоля) целевого спирта, который используют без дополнительной очистки. MS (Cl) для C14H16N4O m/z 257 (MH+), 185.
Часть Е
Используя основной способ, применяемый в примере 6, часть А, 4-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)бутан-1-ол (5,0 г, 19,51 ммоля) хлорируют для получения 1-(4-хлорбутил)-1Н-имидазо[4,5-с]хинолин-4-амина (4,92 г, 17,91 ммоля), который выделяют из реакционной смеси в виде беловатого твердого вещества и используют без дополнительной очистки.
Часть F
Используя основной способ, применяемый в примере 6, часть В, только при несколько более низкой температуре (80°С) 1-(4-хлорбутил)-1Н-имидазо[4,5-с]хинолин-4-амин (1,5 г, 5,46 ммоля) превращают в 1-[4-(фенилтио)бутил]-1H-имидазо[4,5-с]хинолин-4-амин. Полученный в результате реакции твердый продукт (1,53 г) растворяют в 90 мл ацетонитрила и кипятят с обратным холодильником. К реакционной смеси добавляют 0,9 г активированного угля и смесь кипятят в течение 5 минут, активированный уголь удаляют на бумажном фильтре и в результате получают бесцветный раствор, из которого выделяют 0,86 г (2,47 ммоля) целевого продукта - 1-[4-(фенилтио)бутил]-1H-имидазо-[4,5-с]хинолин-4-амина в виде белых игольчатых кристаллов. Температура плавления продукта 158-160°С.
Анализ. Рассчитано для C20H20N4S: %С 68,94; %Н 5,79; %N 16,08. Найдено: %С 68,70;%Н 5,74; %N 16,08.
1H ЯМР (300 МГц, ДМСО) δ 8,18 (синглет, 1Н); δ 8,05 (дублет, J=8,3 Гц, 1Н); δ 7,63 (дублет, J=8,3 Гц, 1Н); δ 7,45 (триплет, J=8,3 Гц, 1Н); δ 7,26 (мультиплет, 5Н); δ 7,14-7,19 (мультиплет, 1H); δ 6,60 (широкий синглет, 2Н); δ 4,62 (триплет, J=6,8 Гц, 2Н); δ 3,00 (триплет, J=7,3 Гц, 2Н); δ 2,00 (мультиплет, 2Н); δ 1,61 (мультиплет, 2Н).
MS (Cl) для C20H20N4S m/z 349 (M+), 185.
Пример 10
1-[4-(Метилтио)бутил-1Н-имидазо[4,5-c]хинолин-4-амин

Часть А
Используя основной способ, применяемый в примере 6, часть В, только при несколько более низкой температуре (80°С) и под действием тиометилата натрия (0,88 г, 12,56 ммоля) вместо бензолтиолата натрия, 1-(4-хлорбутил)-1H-имидазо[4,5-с]хинолин-4-амин (1,5 г, 5,46 ммоля) превращают в 1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин. Полученный в результате реакции твердый продукт (1,26 г) растворяют в 40 мл ацетонитрила и раствор нагревают до кипения. К раствору добавляют 0,7 г активированного угля и смесь кипятят с обратным холодильником в течение 5 минут, после чего активированный уголь удаляют на бумажном фильтре и в результате получают бесцветный раствор. Этот раствор концентрируют при пониженном давлении до получения твердого продукта, после перекристаллизации которого из ацетонитрила образуется 0,66 г (2,30 ммоля) целевого продукта в виде белых игольчатых кристаллов. Температура плавления 1-[4-(метилтио)бутил]-1H-имидазо[4,5-с]хинолин-4-амина составляет 163-164°С.
Анализ. Рассчитано для C15H18N4S: %С 62,91; %Н 6,34; %N 19,56. Найдено: %С 62,70; %Н 6,19; %N 19,45.
1H ЯМР (300 МГц, ДМСО) δ 8,21 (синглет, 1Н); δ 8,06 (дублет, J=8,3 Гц, 1Н); δ 7,62 (дублет, J=8,3 Гц, 1Н); δ 7,44 (триплет, J=8,3 Гц, 1Н); δ 7,26 (триплет, J=8,3 Гц, 1Н); δ 6,59 (широкий синглет, 2Н); δ 4,62 (триплет, J=7,6 Гц, 2Н); δ 2,50 (триплет, J=6,8 Гц, 2Н); δ 1,99 (синглет, 3Н); δ 1,95 (квинтет, J=7,3 Гц, 2Н); δ 1,59 (квинтет, J=7,3 Гц, 2Н).
MS (Cl) для C15H18N4S m/z 287 (MH+), 185.
Пример 11
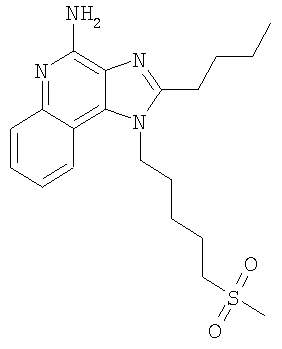
2-Бутил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
Используя основной способ, применяемый в примере 1, часть А, 4-хлор-3-нитрохинолин (107,7 г, 525,87 ммоля) под действием 5-амино-1-пентанола (79,82 г, 788,81 ммоля) (а не под действием 4-аминобутанола, как в примере 1) превращают в 5-[(3-нитрохинолин-4-ил)амино]пентан-1-ол. Полученный темно-желтый твердый продукт (117,22 г, 425,77 ммоля) используют в дальнейшем без дополнительной очистки. MS (Cl) для C14H17N3O3 m/z 412 (MH+), 224.
Часть В
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 5,0 г (18,16 ммоля) 5-[(3-нитрохинолин-4-ил)амино]пентан-1-ола и 40,78 г (0,34 ммоля) тионилхлорида. Реакционную смесь нагревают до 80°С для получения гомогенного раствора и затем выдерживают при этой температуре в течение 1 часа до полного израсходования исходного материала. Летучие продукты отгоняют при пониженном давлении, образующееся масло перемешивают с водой и раствор подщелачивают до рН 10, добавляя к нему твердый карбонат натрия. Полученный твердый продукт отфильтровывают и получают 4,80 г (16,35 ммоля) N-(5-хлорпентил)-3-нитрохинолин-4 амина, который используют в дальнейшем без дополнительной очистки.
Часть С
По способу, приведенному в примере 6, часть В, но при температуре 80°С и при использовании тиометилата натрия (1,43 г, 19,40 ммоля) вместо бензолтиолата натрия, N-(5-хлорпентил)-3-нитрохинолин-4-амин (4,75 г, 16,17 ммоля) превращают в N-[5-(метилтио)пентил]-3-нитрохинолин-4 амин. В результате реакции получают 3,28 г (10,74 ммоля) целевого светло-желтого твердого продукта, который используют без дополнительной очистки. MS (Cl) для C15H19N3O2S m/z 306 (MH+), 272, 117.
Часть D
Используя основной способ, применяемый в примере 1, часть С, восстановливают 3,20 г (10,48 ммоля) N-[5-метилтио)пентил]-3-нитрохинолин-4 амина до 2,89 г (10,48 ммоля) N4-[5-(метилтио)пентил]хинолин-3,4-диамина, который выделяют в виде коричневого масла и используют без последующей очистки.
Часть Е
Используя основной способ, применяемый в примере 1, часть D, проводят циклизацию N4-[5-(метиолтио)пентил]хинолин-3,4-диамина (2,89 г, 10,48 ммоля) до 2-бутил-1-[5-(метилтио)пентил]-1H-имидазо[4,5-с]хинолина. После хроматографической очистки на силикагеле (в качестве элюента используют этилацетат) получают 2,10 г (6,15 ммоля) целевого продукта в виде светло-коричневого масла.
Часть F
В круглодонную колбу, снабженную магнитной мешалкой, загружают 2,1 г (6,15 ммоля) 2-бутил-1-[5-(метилтио)пентил]-1H-имидазо[4,5-с]хинолина и 31 мл хлороформа. К полученному раствору в течение 10 минут добавляют 4,41 г (19,68 ммоля) твердой 3-хлорпербензойной кислоты. Реакции смесь перемешивают при температуре окружающей среды в течение 30 минут до полного израсходования исходного материала. Раствор распределяют между хлороформом и насыщенным водным раствором бикарбоната натрия. Водный и органический слои отделяют друг от друга и органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения 2,40 г (6,15 ммоля) желтовато-коричневого твердого вещества, представляющего собой 2-бутил-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-5N-оксид. Этот продукт использовали в дальнейшем без дополнительной очистки.
Часть G
Используя основной способ, применяемый в примере 3, часть В, проводят аминирование 2,40 г (6,15 ммоля) 2-бутил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-5N-оксида до 2-бутил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амина. Полученный твердый продукт (2,24 г) растворяют в 40 мл ацетонитрила и раствор доводят до кипения. После этого добавляют 1 г активированного угля и полученную смесь кипятят с обратным холодильником в течение 5 минут, после чего активированный уголь удаляют на бумажном фильтре и в результате получают светло-коричневый раствор, при охлаждении которого выпадают 0,90 г (2,32 ммоля) белых игольчатых кристаллов 2-бутил-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-4-амина. Температура плавления этого продукта 173-175°С.
Анализ. Рассчитано для C20H28N4O2S: %С 61,83; %Н 7,26; %N 14,42. Найдено: %С 61,58; %Н 7,27; %N 14,36.
1H ЯМР (300 МГц, ДМСО) δ 8,01 (дублет, J=8,3 Гц, 1Н); δ 7,61 (дублет, J=8,3 Гц, 1Н); δ 7,41 (триплет, J=8,3 Гц, 1Н); δ 7,26 (триплет, J=8,3 Гц, 1Н); δ 6,45 (широкий синглет, 2Н); δ 4,51 (триплет, J=7,6 Гц, 2Н); δ 3,10 (триплет, J=7,8 Гц, 2Н); δ 2,92 (синглет, 3Н), δ 2,92 (триплет, J=7,3 Гц, 2Н); δ 1,76 (мультиплет, 6Н); δ 1,54 (мультиплет, 2Н); δ 1,46 (секстет, J=7,3 Гц, 2Н); δ 0,99 (триплет, J=7,3 Гц, 3Н). MS (Cl) для C20H28N4O2S m/z 389 (МН+).
Пример 12
2-Метил-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-4-амин
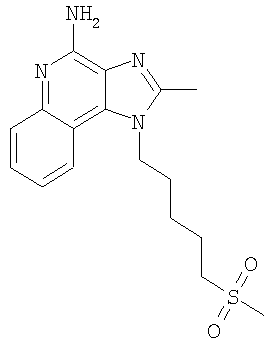
Часть А
Используя основной способ, применяемый в примере 1, часть D, в присутствии 1,1,1-триметоксиэтана (2,95 г, 24,6 ммоля) и солянокислого пиридина (0,1 г) проводят циклизацию 4,53 г (16,37 ммоля) N4-[5-(метилтио)пентил]хинолин-3,4-диамина до 2-метил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолина. Полученную смесь обрабатывают этиловым эфиром и после фильтрации получают 3,78 г (12,62 ммоля) светло-коричневого целевого продукта, который используют без дополнительной очистки.
Часть В
Используя основной способ, применяемый в примере 11, часть F, 3,78 г (12,62 ммоля) 2-метил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолина окисляют до 2-метил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-5N-оксида (4,38 г, 12,62 ммоля), который выделяют в виде твердого желтовато-коричневого продукта и используют без дополнительной очистки.
Часть С
Используя основной способ, применяемый в примере 3, часть В, проводят аминирование 2-метил-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-5N-оксида (4,38 г, 12,62 ммоля) до 2-метил-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-4-амина. Полученный твердый продукт обрабатывают ацетонитрилом и отделяют от раствора фильтрованием. В результате получают 0,8 г (2,31 ммоля) беловатого продукта, температура плавления которого составляет 235-240°С.
Анализ. Рассчитано для C17H22N4O2S: %С 58,94; %Н 6,40; %N 16,17. Найдено: %С 58,77; %Н 6,34; %N 16,39.
1H ЯМР (300 МГц, ДМСО) δ 8,02 (дублет, J=8,3 Гц, 1Н); δ 7,60 (дублет, J=8,3 Гц, 1Н); δ 7,41 (триплет, J=8,3 Гц, 1Н); δ 7,25 (триплет, J=8,3 Гц, 1Н); δ 6,49 (широкий синглет, 2Н); δ 4,50 (триплет, J=7,3 Гц, 2Н); δ 3,12 (триплет, J=7,8 Гц, 2Н); δ 2,92 (синглет, 3Н), δ 2,61 синглет, 3Н); δ 1,86 (мультиплет, 2Н); δ 1,74 (мультиплет, 2Н); δ 1,53 (мультиплет, 2Н).
MS(Cl) для C17H22N4O2S m/z 347 (MH+), 267.
Пример 13
2-Этил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин
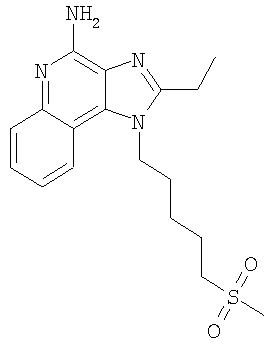
Часть А
Используя основной способ, применяемый в примере 1, часть D, в присутствии триэтилортопропионата (4,3 г, 24,56 ммоля) и солянокислого пиридина (0,1 г) проводят циклизацию 4,53 г (16,37 ммоля) N4-[5-(метилтио)пентил]хинолин-3,4-диамина до 2-этил-1-[5-(метилтио)пентил]-1H-имидазо[4,5-с]хинолина. Полученную смесь обрабатывают этиловым эфиром и после фильтрации получают 3,25 г (10,37 ммоля) беловатого порошка, который используют без дополнительной очистки.
Часть В
Используя основной способ, применяемый в примере 11, часть F, 3,25 г (10,37 ммоля) 2-этил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолина окисляют до 2-этил-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-5N-оксида (3,75 г, 10,37 ммоля), который выделяют в виде твердого желтовато-коричневого продукта и используют без дополнительной очистки.
Часть С
Используя основной способ, применяемый в примере 3, часть В, проводят аминирование 2-этил-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-5N-оксида (3,75 г, 10,38 ммоля) до 2-этил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амина. Полученный твердый продукт перекристаллизовывают последовательно из этанола и ацетонитрила и в результате получают 1,4 г (3,88 ммоля) продукта в виде беловатых игольчатых кристаллов. Температура плавления этого продукта составляет 189-191°С.
Анализ. Рассчитано для C18H24N4O2S: %С 59,98; %Н, 6,71; %N, 15,54. Найдено: %С 59,71; %Н, 6,68; %N, 15,64.
1H ЯМР (300 МГц, ДМСО) δ 8,01 (дублет, J=8,3 Гц, 1Н); δ 7,61 (дублет, J=8,3 Гц, 1Н); δ 7,42 (триплет, J=8,3 Гц, 1Н); δ 7,26 (триплет, J=8,3 Гц, 1Н); δ 6,45 (широкий синглет, 2Н); δ 4,50 (триплет, J=7,6 Гц, 2Н); δ 3,10 (триплет, J=7,8 Гц, 2Н); δ 2,95 (квинтет, J=7,3 Гц, 2Н); δ 2,92 (синглет, 3Н), δ 1,85 (мультиплет, 2Н); δ 1,74 (мультиплет, 2Н); δ 1,55 (мультиплет, 2Н); δ 1,38 (триплет, J=7,3 Гц, 3Н).
MS (Cl) для C18H24N4O2S m/z 361 (MI-H+), 281.
Пример 14
1-[5-(Метилсульфонил)пентил]-1H-имидазо[4,5-c]хинолин-4-амин

Часть А
Используя основной способ, применяемый в примере 1, часть D, в присутствии триэтилформиата (3,64 г, 24,56 ммоля) и солянокислого пиридина (0,1 г) проводят циклизацию 4,53 г (16,37 ммоля) N4-[5-(метилтио)пентил]хинолин-3,4-диамина до 1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолина. 4,05 г (14,19 ммоля) продукта в виде коричневого масла выделяют из реакционной смеси и используют без дополнительной очистки.
Часть В
Используя основной способ, применяемый в примере 11, часть F, 4,05 г (14,19 ммоля) 1-[5-(метилтио)пентил]-1H-имидазо[4,5-с]хинолина окисляют до 1-[5-(метилсульфонил)-пентил]-1H-имидазо[4,5-с]хинолин-5N-оксида (4,73 г, 14,19 ммоля), который выделяют в виде твердого желтовато-коричневого продукта и используют без дополнительной очистки.
Часть С
Используя основной способ, применяемый в примере 3, часть В, проводят аминирование 1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-5N-оксида (4,73 г, 14,19 ммоля) до 1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-4-амина. Полученный продукт подвергают хроматографической очистке на силикагеле (95/5 дихлорметан/метанол) и в результате получают светло-желтое твердое вещество. Это вещество перекристаллизовывают из диметилформамида и получают 0,43 г (1,29 ммоля) целевого продукта в виде светло-желтых гранул. Температура плавления этого продукта составляет 199-201°С.
Анализ. Рассчитано для C16H20N4O2S: %С 57,81; %Н 6,06; %N 16,85. Найдено: %С 57,01; %Н 6,06; %N 16,70.
1H ЯМР (300 МГц, ДМСО) δ 8,20 (синглет, 1Н); δ 8,04 (дублет, J=8,3 Гц, 1Н); δ 7,62 (дублет, J=8,3 Гц, 1Н); δ 7,44 (триплет, J=8,3 Гц, 1Н); δ 7,27 (триплет, J=8,3 Гц, 1Н); δ 6,57 (широкий синглет, 2Н); δ 4,61 (триплет, J=6,8 Гц, 2Н); δ 3,09 (триплет, J=7,8 Гц, 2Н); δ 2,92 (синглет, 3Н), δ 1,91 (квинтет, J=7,6 Гц, 2Н); δ 1,73 (мультиплет, 2Н); δ 1,45 (мультиплет, 2Н).
MS (Cl) для C16H20N4O2S m/z 333 (МН+).
Пример 15
2-Гексил-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-c]хинолин-4-амин
Часть А
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 3,17 г (11,46 ммоля) N4-[5-(метилтио)пентил]хинолин-3,4-диамина и 46 мл безводного пиридина. Полученный гомогенный раствор охлаждают на ледяной бане до 0°С и к охлажденному раствору добавляют 1,87 г (12,61 ммоля) гептаноилхлорида. Для завершения реакции смесь перемешивают при температуре окружающей среды в течение 1 часа. Летучие продукты удаляют при пониженном давлении, а оставшееся масло распределяют между хлороформом и водой. Водный и органический слои отделяют друг от друга и органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения N-(4-{[5-(метилтио)пентил]амино}хинолин-3-ил)гептанамида (4,44 г, 11,46 ммоля), который выделяют в виде коричневого масла и используют без дополнительной очистки.
Часть В
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают N-(4-{[5-(метилтио)пентил]амино}хинолин-3-ил)гептанамид (4,44 г, 11,46 ммоля), 0,13 г (1,15 ммоля) солянокислого пиридина и 50 мл безводного пиридина, для завершения реакции смесь при кипении перемешивают с обратным холодильником в течение 1,5 часов. Раствор охлаждают и распределяют между этилацетатом и водой. Водный и органический слои отделяют друг от друга и органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения 4,0 г (10,82 ммоля) 2-гексил-1-[5-(метилтио)пентил]-1H-имидазо[4,5-с]хинолина в виде коричневого масла, которое используют без дополнительной очистки.
Часть С
Используя основной способ, применяемый в примере 11, часть F, 4,0 г (10,82 ммоля) 2-гексил-1-[5-(метилтио)пентил]-1H-имидазо[4,5-c]хинолина окисляют до 2-гексил-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-5N-оксида (4,52 г, 10,82 ммоля), который выделяют в виде твердого желтовато-коричневого продукта и используют без дополнительной очистки.
Часть D
Используя основной способ, применяемый в примере 3, часть В, проводят аминирование 2-гексил-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-5N-оксида (4,0 г, 10,82 ммоля) до 2-гексил-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-4-амина. Полученный продукт перекристаллизовывают из ацетонитрила и получают 2,25 г (5,40 ммоля) целевого продукта в виде беловатых игольчатых кристаллов. Температура плавления этого продукта составляет 168-171°С.
Анализ. Рассчитано для C22H32N4O2S: %С 63,43; %Н 7,74; %N 13,45. Найдено: %С 63,06; %Н 7,66; %N 13,81.
1H ЯМР (300 МГц, ДМСО) δ 8,01 (дублет, J=8,3 Гц, 1Н); δ 7,62 (дублет, J=8,3 Гц, 1Н); δ 7,42 (триплет, J=8,3 Гц, 1Н); δ 7,26 (триплет, J=8,3 Гц, 1Н); δ 6,51 (широкий синглет, 2Н); δ 4,51 (триплет, J=7,3 Гц, 2Н); δ 3,10 (триплет, J=7,8 Гц, 2Н); δ 2,93 (синглет, 3Н), δ 1,71-1,87 (мультиплет, 6Н); δ 1,54 (мультиплет, 2Н); δ 1,44 (мультиплет, 2Н); δ 1,33 (мультиплет, 4Н); δ 0,89 (триплет, J=7,3 Гц, 3Н).
MS (Cl) для C22H32N4O2S m/z 417 (МН+), 337.
Пример 16
2-(2-Метоксиэтил)-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-4-амин
Часть А
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 3,56 г (12,93 ммоля) N4-[5-(метилтио)пентил]хинолин-3,4-диамина и 52 мл безводного пиридина. Полученный гомогенный раствор охлаждают на ледяной бане до 0°С и к охлажденному раствору добавляют 2,74 г (22,36 ммоля) хлорангидрида 3-метоксипропионовой кислоты, по окончании добавления хлорангидрида реакционную смесь нагревают до кипения и кипятят ее с обратным холодильником в течение 14 часов. В течение этого времени происходит полное израсходование ацилированного промежуточного продукта. Смесь охлаждают и распределяют между хлороформом и насыщенным водным раствором хлористого аммония. Водный и органический слои отделяют друг от друга и органический слой промывают насыщенным водным раствором бикарбоната натрия, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения 3,0 г (8,73 ммоля) 2-(2-метоксиэтил)-1-[5-(метилтио)пентил]-1H-имидазо[4,5-с]хинолина, который выделяют в виде коричневого масла и используют без дополнительной очистки.
Часть В
Используя основной способ, применяемый в примере 11, часть F, 3,0 г (8,73 ммоля) 2-(2-метоксиэтил)-1-[5-(метилтио)пентил]-1H-имидазо[4,5-с]хинолина окисляют до 2-(2-метоксиэтил)-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-5N-оксида (3,41 г, 8,73 ммоля), который выделяют в виде твердого желтовато-коричневого продукта и используют без дополнительной очистки.
Часть С
Используя основной способ, применяемый в примере 3, часть В, проводят аминирование 2-(2-метоксиэтил)-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-5N-оксида (3,41 г, 8,73 ммоля) до 2-(2-метоксиэтил)-1-[5-(метилсульфонил)пентил]-1H-имидазо[4,5-с]хинолин-4-амина. Полученный твердый продукт подвергают хроматографической очистке на силикагеле (95/5 дихлорметан/метанол) и выделяют клейкое твердое вещество, которое после перекристаллизации из ацетонитрила дает 0,54 г (1,38 ммоля) целевого продукта в виде беловатого порошка. Температура плавления этого продукта составляет 158-160°С.
Анализ. Рассчитано для C19H26N4O3S: %С 58,44; %Н 6,71; %N 14,35. Найдено: %С 58,24; %Н 6,76; %N 14,70.
1H ЯМР (300 МГц, ДМСО) δ 8,02 (дублет, J=8,3 Гц, 1Н); δ 7,62 (дублет, J=8,3 Гц, 1Н); δ 7,42 (триплет, J=8,3 Гц, 1Н); δ 7,26 (триплет, J=8,3 Гц, 1Н); δ 6,50 (широкий синглет, 2Н); δ 4,53 (триплет, J=7,6 Гц, 2Н); δ 3,83 (триплет, J=6,8 Гц, 2Н); δ 3,30 (синглет, 3Н); δ 3,19 (триплет, J=6,8 Гц, 2Н); δ 3,11 (триплет, J=7,8 Гц, 2Н); δ 2,93 (синглет, 3Н), δ 1,85 (мультиплет, 2Н); δ 1,76 (мультиплет, 2Н); δ 1,57 (мультиплет, 2Н).
MS (Cl) для С19Н26N4O3S m/z 391 (МН+), 359.
Пример 17
2-Бутил-1-[5-(метилтио)пентил]-1H-имидазо[4,5-с]хинолин-4-амин

Часть А
Используя основной способ, применяемый в примере 1, часть С, проводят восстановление 2,0 г (6,80 ммоля) N-(5-хлорпентил)-3-нитрохинолин-4-амина до 1,79 г (6,80 ммоля) N4-(5-хлорпентил)-хинолин-3,4-диамина, который выделяют в виде масла коричневого цвета и используют без последующей очистки.
Часть В
Используя основной способ, применяемый в примере 1, часть D, под действием 2,55 г (15,72 ммоля) триметилортовалериата и 0,079 г солянокислого пиридина, проводят циклизацию 1,79 г (6,80 ммоля) N4-(5-хлорпентил)-хинолин-3,4-диамина до 2-бутил-1-(5-хлорпентил)-1H-имидазо[4,5-с]хинолина. Продукт (1,95 г, 5,91 ммоля) выделяют в виде беловатого твердого вещества и используют без дополнительной очистки.
Часть С
Используя основной способ, применяемый в примере 1, часть Е, 1,95 г (5,91 ммоля) 2-бутил-1-(5-хлорпентил)-1H-имидазо[4,5-с]хинолина окисляют до 2,04 г (5,91 ммоля) 2-бутил-1-(5-хлорпентил)-1H-имидазо[4,5-с]хинолин-5N-оксида, который выделяют из реакционной смеси в виде желтовато-коричневого твердого продукта и используют без дополнительной очистки.
Часть D
Используя основной способ, применяемый в примере 3, часть В, проводят аминирование 2,04 г (5,91 ммоля) 2-бутил-1-(5-хлорпентил)-1H-имидазо[4,5-с]хинолин-5N-оксида до 2-бутил-1-(5-хлорпентил)-1H-имидазо[4,5-с]хинолин-4-амина. После перекристаллизации образующегося продукта из этанола получают 0,85 г (2,46 ммоля) целевого амина в виде белого мелкого порошка, температура плавления которого составляет 144-146°С.
Анализ. Рассчитано для С19Н25CIN4: %С 66,17; %Н 7,31; %N 16,24. Найдено: %С 66,44; %Н 7,55; %N 16,29.
MS (Cl) для С19Н25CIN4 m/z 345 (МН+), 309.
Часть Е
По способу, приведенному в примере 6, часть В, но при температуре 80°С и при использовании тиометилата натрия (0,68 г, 8,70 ммоля) вместо бензолтиолата натрия 2-бутил-1-(5-хлорпентил)-1H-имидазо[4,5-с]хинолин-4-амин (2,0 г, 5,80 ммоля) превращают в 2-бутил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин-4 амин. Образующийся твердый продукт распределяют между хлороформом и насыщенным водным раствором бикарбоната натрия. Водный и органический слои отделяют друг от друга и органический слой промывают рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения твердого белого вещества. После перекристаллизации этого вещества из ацетонитрила получают 1,91 г (5,36 ммоля) целевого продукта в виде мелкого порошка. Температура плавления этого продукта 112-114°С.
Анализ. Рассчитано для C20H28N4S: %С 67,38; %Н 7,92; %N 15,71. Найдено: %С 67,26; %Н 8,08; %N 15,74.
1H ЯМР (300 МГц, ДМСО) δ 8,01 (дублет, J=8,3 Гц, 1Н); δ 7,61 (дублет, J=8,3 Гц, 1Н); δ 7,41 (триплет, J=8,3 Гц, 1Н); δ 7,25 (триплет, J=8,3 Гц, 1Н); δ 6,45 (широкий синглет, 2Н); δ 4,50 (триплет, J=7,8 Гц, 2Н); δ 3,83 (триплет, J=6,8 Гц, 2Н); δ 3,30 (синглет, 3Н); δ 3,19 (триплет, J=6,8 Гц, 2Н); δ 2,92 (триплет, J=7,6 Гц, 2Н); δ 2,46 (триплет, J=7,3 Гц, 2Н); δ 2,01 (синглет, 3Н), δ 1,80 (мультиплет, 4Н); δ 1,42 - 1,61 (мультиплет, 6Н); δ 0,96 (триплет, J=7,3 Гц, 3Н)
MS (Cl) для C20H28N4S m/z 357 (MH+), 309.
Пример 18
2-Бутил-1-[5-(метилсульфинил)пентил]-1H-имидазо[4,5-с]хинолин-4-амин
В круглодонную колбу, снабженную магнитной мешалкой, загружают 1,0 г (2,80 ммоля) 2-бутил-1-[5-(метилтио)пентил]-1H-имидазо[4,5-с]хинолин-4-амина и 14 мл хлороформа. К раствору порциями в течение 5 минут добавляют 0,69 г (3,09 ммоля) твердой 3-хлорпербензойной кислоты и смесь для завершения реакции перемешивают при температуре окружающей среды в течение 20 минут. Полученный раствор распределяют между хлороформом и насыщенным водным раствором бикарбоната натрия. Водный и органический слои отделяют друг от друга и органический слой промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения беловатого твердого вещества, которое согласно данным 1H-ЯМР-спектроскопии представляет собой хлорбензойную соль целевого продукта. Это вещество перемешивают в воде и раствор делают щелочным (рН 10), добавляя к нему твердый карбонат натрия.
Образующееся при этом свободное основание отфильтровывают и перекристаллизовывают из ацетонитрила. В результате получают 0,40 г (1,07 ммоля) 2-бутил-1-[5-(метилсульфинил)пентил]-1H-имидазо[4,5-с]хинолин-4-амина в виде белого порошка. Температура плавления этого продукта 119-121°С. Анализ. Рассчитано для C20H28N4OS(H2O)1: %С 61,51; %Н 7,74; %N 14,35. Найдено: %С 61,64; %Н 7,82; %N 14,32.
1H ЯМР (300 МГц, ДМСО) δ 8,01 (дублет, J=8,3 Гц, 1Н); δ 7,60 (дублет, J=8,3 Гц, 1Н); δ 7,26 (триплет, J=8,3 Гц, 1Н); δ 7,26 (триплет, J=8,3 Гц, 1Н); δ 6,44 (широкий синглет, 2Н); δ 4,51 (триплет, J=7,6 Гц, 2Н); δ 2,92 (триплет, J=7,8 Гц, 2Н); δ 2,57 -2,74 (мультиплет, 2Н); δ 2,50 (синглет, 3Н), δ 1,80 (мультиплет, 4Н); δ 1,66 (мультиплет, 2Н); δ 1,55 (мультиплет, 2Н); δ 1,48 (мультиплет, 2H); δ 0,96 (триплет, J=7,3 Гц, 3Н.
MS (Cl) для C20H28N4OS(H2O)1: m/z 373 (МН+), 309, 253.
Пример 19
2-Бутил-1-[3-(метилсульфонил)пропил]-1H-имидазо[4,5-с]хинолин-4-амин

Часть А
В круглодонную колбу, снабженную магнитной мешалкой, загружают 20,75 г (83,93 ммоля) 3-[(3-нитрохинолин-4-ил)амино]пропан-1-ола, 15,0 г (125,89 ммоля) тионилхлорида и 420 мл дихлорметана. Полученный ярко-желтый раствор перемешивают при температуре окружающей среды в течение 2 часов; за это время происходит полное исчерпание исходного материала. Летучие продукты отгоняют при пониженном давлении и оставшийся твердый продукт перемешивают с 400 мл воды и подщелачивают раствор до рН 10, добавляя к нему твердый карбонат натрия. Из раствора отфильтровывают 21,63 г (81,41 ммоля) ярко-желтого твердого N-(3-хлорпропил)-3-нитрохинолин-4-амина, который используют без дополнительной очистки.
Часть В
Используя основной способ, применяемый в примере 1, часть С, проводят восстановление 10,0 г (37,63 ммоля) N-(3-хлорпропил)-3-нитрохинолин-4-амина до 8,87 г (37,63 ммоля) N4-(3-хлорпропил)хинолин-3,4-диамина, который выделяют в виде масла коричневого цвета и используют без последующей очистки.
Часть С
Используя основной способ, применяемый в примере 1, часть D, под действием 7,33 г (45,16 ммоля) триметилортовалериата и 0,43 г солянокислого пиридина проводят циклизацию 8,87 г (37,63 ммоля) N4-(3-хлорпропил)хинолин-3,4-диамина до 2-бутил-1-(3-хлорпропил)-1H-имидазо[4,5-с]хинолина. Продукт обрабатывают этиловым эфиром и после фильтрации выделяют 9,00 г, (29,82 ммоля) беловатого твердого вещества, которое используют без дополнительной очистки.
Часть D
Используя основной способ, применяемый в примере 1, часть Е, 9,0 г (29,82 ммоля) 2-бутил-1-(3-хлорпропил)-1H-имидазо[4,5-с]хинолина окисляют до 9,48 г (29,82 ммоля) 2-бутил-1-(3-хлорпропил)-1H-имидазо[4,5-с]хинолин-5N-оксида, который выделяют из реакционной смеси в виде желтовато-коричневого твердого продукта и используют без дополнительной очистки.
Часть Е
Используя основной способ, применяемый в примере 3, часть В, проводят аминирование 9,48 г (29,82 ммоля) 2-бутил-1-(3-хлорпропил)-1Н-имидазо[4,5-с]хинолин-5N-оксида до 2-бутил-1-(3-хлорпропил)-1Н-имидазо[4,5-с]хинолин-4-амина. Образующийся твердый продукт подвергают хроматографической очистке на силикагеле (95/5 дихлорметан/метанол) и получают 6,4 г (20,20 ммоля) целевого амина в виде желтовато-коричневого твердого вещества.
Часть F
По способу, приведенному в примере 6, часть В, но при температуре 80°С и при использовании тиометилата натрия (0,74 г, 9,47 ммоля) вместо бензолтиолата натрия, 2-бутил-1-(3-хлорпропил)-1Н-имидазо[4,5-с]хинолин-4-амин (2,0 г, 6,31 ммоля) превращают в 2-бутил-1-[3-(метилтио)пропил]-1H-имидазо[4,5-с]хинолин-4-амин. Образующийся твердый продукт распределяют между хлороформом и насыщенным водным раствором бикарбоната натрия. Водный и органический слои отделяют друг от друга и органический слой промывают рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения 2,0 г (6,09 ммоля) твердого белого вещества, которое используют в дальнейшем без дополнительной очистки.
Часть G
Используя основной способ, применяемый в примере 5, часть А, проводят окисление 2,0 г (6,09 ммоля) 2-бутил-1-[3-(метилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амина до 2-бутил-1-[3-(метилсульфонил)пропил]-1H-имидазо[4,5-с]хинолин-4-амина. Полученный твердый продукт промывают метанолом и отфильтровывают. В результате получают 0,96 г (2,66 ммоля) беловатого порошка 2-бутил-1-[3-(метилсульфонил)пропил]-1H-имидазо[4,5-с]хинолин-4 амина, температура плавления которого составляет 233-236°С.
Анализ. Рассчитано для C18H24N4O2S: %С 59,98; %Н 6,71; %N 15,54. Найдено: %С 59,71; %Н 6,65; %N 15,43.
1H ЯМР (300 МГц, ДМСО) δ 8,10 (дублет, J=8,3 Гц, 1Н); δ 7,61 (дублет, J=8,3 Гц, 1Н); δ 7,42 (триплет, J=8,3 Гц, 1Н); δ 7,25 (триплет, J=8,3 Гц, 1Н); δ 6,47 (широкий синглет, 2Н); δ 4,66 (триплет, J=7,8 Гц, 2Н); δ 3,40 (триплет, J=7,3 Гц, 2Н); δ 3,01 (синглет, 3Н); δ 2,94 (триплет, J=7,8 Гц, 2Н); δ 2,22 (мультиплет, 2Н); δ 1,80 (мультиплет, 2Н); δ 1,46 (секстет, J=7,3 Гц, 2Н); δ 0,96 (триплет, J=7,3 Гц, 3Н).
MS (Cl) для C18H24N4O2S: m/z 361 (МН*), 281, 235.
Пример 20
2-Бутил-1-[3-(фенилсульфонил)пропил]-1H-имидазо[4,5-с]хинолин-4-амин
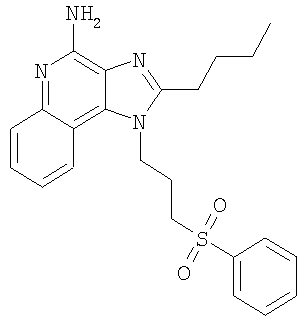
Часть А
В круглодонную колбу, снабженную магнитной мешалкой, в атмосфере азота загружают 0,68 г (6,21 ммоля) бензолтиола, 0,25 г 60%-ной дисперсии (6,21 ммоля) гидрида натрия и 28 мл безводного диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение 30 мин, добавляют к ней 1,64 г (5,18 ммоля) 2-бутил-1-(3-хлорпропил)-1Н-имидазо[4,5-с]хинолин-4-амина, полученный мутный раствор нагревают до 80°С и для завершения реакции перемешивают эту смесь при 80°С в течение 2,5 часов. Горячий раствор при быстром перемешивании выливают в 200 мл воды, и смесь дважды экстрагируют хлороформом. Органические фракции соединяют вместе, промывают насыщенным водным раствором бикарбоната натрия и рассолом, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении до получения светло-желтого масла. Хроматографическая очистка этого вещества на силикагеле (95/5 дихлорметан/метанол) позволяет получить 1,38 г (3,53 ммоля) твердого белого 2-бутил-1-[3-(фенилтио)пропил]-1H-имидазо[4,5-с]хинолин-4 амина.
Часть В
Используя основной способ, применяемый в примере 5, часть А, проводят окисление 1,38 г (3,53 ммоля) 2-бутил-1-[3-(фенилтио)пропил]-1H-имидазо[4,5-с]хинолин-4 амина до 2-бутил-1-[3-(фенилсульфонил)пропил]-1H-имидазо[4,5-с]хинолин-4-амина. Полученный твердый продукт перекристаллизовывают из этанола и в результате получают 0,85 г (2,01 ммоля) беловатого порошка 2-бутил-1-[3-(фенилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амина, температура плавления которого составляет 224-227°С.
Анализ. Рассчитано для С23Н26N4O3S: %С 65,38; %Н 6,20; %N 13,26. Найдено: %С 65,25; %Н 6,23; %N 13,20.
1H ЯМР (300 МГц, ДМСО) δ 7,96 (дублет, J=8,3 Гц, 1Н); δ 7,89 (мультиплет, 2Н); δ 7,73 (мультиплет, 1Н); δ 7,63 (мультиплет, 3Н); δ 7,40 (триплет, J=8,3 Гц, 1Н); δ 7,17 (триплет, J=8,3 Гц, 1Н); δ 6,46 (широкий синглет, 2Н); δ 4,60 (триплет, J=7,8 Гц, 2Н); δ 3,66 (триплет, J=7,3 Гц, 2Н); δ 2,86 (триплет, J=7,8 Гц, 2Н); δ 2,04 (мультиплет, 2Н); δ 1,73 (квинтет, J=7,6 Гц, 2Н); δ 1,39 (секстет, J=7,3 Гц, 2Н); δ 0,92 (триплет, J=7,3 Гц, 3Н).
MS (Cl) для С23Н26N4O3S m/z 423 (МН+), 322, 281.
ОБРАЗОВАНИЕ ЦИТОКИНА В КЛЕТКАХ ЧЕЛОВЕКА
Определение активности полученных в изобретении соединений для стимулирования образования цитокина проводили с использованием клеток крови человека in vitro. Активность оценивали, исходя из количества интерферона и фактора-α некроза опухоли (IFN и TNF соответственно), определенного по методике, приведенной в статье Тестермана с сотр. «Индукция кинина с помощью иммуномодуляторов Imiquimod и S-27609», опубликованной в журнале «Journal of Leukocyte Biology», 58, 365-372 (Сентябрь, 1995).
Подготовка культуры на основе клеток крови
Цельную кровь собирали венепункцией от здоровых доноров в специальные вакуумные контейнеры, соответствующие требованиям EDTA. Одноядерные клетки периферийной крови (РВМС) отделяли от цельной крови с помощью способа градиентного центрифугирования с использованием аппарата Histopaque®-1077. РВМС дважды промывали раствором сбалансированных солей Hank (Ханка) и затем суспендировали в среде RPMI. Концентрации клеток крови в такой суспензии составляла (3-4)×106 клеток на 1 мл суспензии. Суспензию РВМС помещали на плоские стерильные планшеты с 48 лунками (поставляемые компанией Costar, Кэмбридж, шт. Массачусетс или Becton Dickinson Labware, Линкольн Парк, шт. Нью-Джерси), в которых находились равные объемы среды RPMI, содержащей исследуемое соединение.
Подготовка соединения
Соединения солюбилизировали в диметилсульфоксиде (ДМСО). Конечная концентрация ДМСО в приготовленных образцах, используемых для испытания, не должна превышать 1%. Первоначально соединения обычно испытывали при концентрации, находящейся в пределах от 0,12 до 30 мкмоль/л. Соединения, обладающие активностью при концентрации 0,12 мкмоль/л, могли быть испытаны затем и при более низкой концентрации.
Инкубация
Раствор применяемого для испытания соединения с концентрацией 60 мкмоль/л добавляли сначала в первую лунку, в которой находилась полная среда RPMI, а в остальные лунки вводят растворы после серийных трехкратных разбавлений. После этого добавляли суспензию РМВС в таком количестве, чтобы концентрация исследуемого вещества находилась в заданных пределах (0,12-30 мкмоль/л). Конечная концентрация суспензии РМВС составляла (1,5-2)×106 клеток/мл. Лунки закрывали стерильными пластмассовыми крышками, аккуратно перемешивали образцы и выдерживали их в течение 18-24 часов при 37°С в атмосфере, содержащей 5% углекислого газа.
Отделение
После окончания инкубационного периода образцы подвергали центрифугированию в течение 5-10 минут при скорости вращения центрифуги 1000 об/мин (приблизительно 200 g) и температуре 4°С. Очищенный раствор, не содержащий клеток культуры, отбирали с помощью стерильной полипропиленовой пипетки и переносили в стерильные полипропиленовые ампулы. Образцы до проведения анализа хранили при температуре от -30 до -70°С. Образцы анализировали на интерферон-α и фактор-α некроза опухоли с помощью способа ELISA.
Анализ интерферона (α) и фактора-α некроза опухоли по способу ELISA
Концентрацию интерферона (α) определяли по способу ELISA, используя набор человеческих мультичастиц, полученный в PBL биомедицинской лаборатории, Нью-Брунсвик, шт. Нью-Джерси. Этот способ анализа позволяет определить содержание интерферона в единицах пг/мл.
Концентрацию фактора-α некроза опухоли (TNF) определяли с помощью набора ELISA, поставляемого фирмой Genzyme, Кэмбридж, шт. Массачусетс; R&D Systems, Миннеаполис, шт. Миннесота, или фирмой Pharmingen, Сан-Диего, шт. Калифорния. Этот способ анализа позволяет определить содержание интерферона в единицах пг/мл.
В нижеследующей таблице приведены минимальные концентрации соединений, при которых наблюдается образование интерферона и фактора некроза опухоли. Знак «*» означает, что при любой исследованной концентрации добавленного соединения (0,12, 0,37, 1,11, 3,33, 10 и 30 мкмоль/л) не наблюдалось образования интерферона и фактора некроза опухоли.
| название | год | авторы | номер документа |
|---|---|---|---|
| АРИЛЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ВИРУСНОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ОПУХОЛЕВОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2308456C2 |
| ТИОЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2304143C2 |
| АМИДОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2000 |
|
RU2295343C2 |
| N-{2-[4-АМИНО-2-(ЭТОКСИМЕТИЛ)-1Н-ИМИДАЗО-[4,5-с]-ХИНОЛИН-1-ИЛ]-1,1-ДИМЕТИЛЭТИЛ}-МЕТАНСУЛЬФОНАМИД И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2004 |
|
RU2374246C2 |
| ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2248975C2 |
| ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ НА ОСНОВЕ КЕТОАМИДА | 2021 |
|
RU2819346C1 |
| МОЧЕВИНОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2265020C2 |
| КАРБАМИДЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНОВЫЕ ЭФИРЫ | 2001 |
|
RU2302418C2 |
| СОЕДИНЕНИЕ, ИМЕЮЩЕЕ ИНГИБИРУЮЩУЮ АКТИВНОСТЬ В ОТНОШЕНИИ КИНАЗ | 2021 |
|
RU2831403C1 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНЫ | 2001 |
|
RU2294934C2 |
Описываются имидазохинолиновые производные формулы (I)
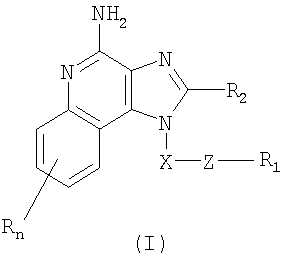
где Х представляет собой алкил, -CHR3-; Z представляет собой -S-, -SO- или -SO2- группу; R1 выбран из группы, включающей алкил и незамещенный фенил; R2 выбран из группы, включающей атом водорода; алкил; алкил-Y-алкил; каждый R3 представляет собой атом водорода; Y представляет собой -О-; n равен 0. Описывается также тетраимидазохинолиновое производное, фармацевтические композиции на их основе, способ стимулирования биосинтеза цитокина в организме животного. Предлагаемые в изобретении соединения и композиции могут вызывать биосинтез различных цитокинов и в связи с этим использоваться для лечения различных болезней, включая вирусные и опухолевые заболевания. 9 н. и 11 з.п. ф-лы, 1 табл.
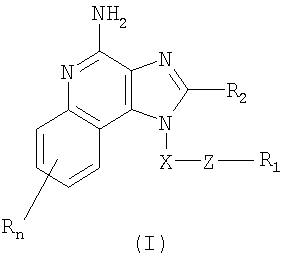
где X представляет собой алкил, -CHR3-;
Z представляет собой -S-, -SO- или -SO2- группу;
R1 выбран из группы, включающей
алкил; и
незамещенный фенил;
R2 выбран из группы, включающей
атом водорода;
алкил;
алкил-Y-алкил;
каждый R3 представляет собой атом водорода;
Y представляет собой -О-;
n равен 0.
2-бутил-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[2-(фенилтио)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[4-(фенилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(фенилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(фенилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-гексил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-(2-метоксиэтил)-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[5-(метилсульфинил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[3-(метилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин; и
2-бутил-1-[3-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
или соль фармацевтического качества на их основе.
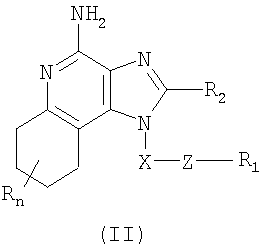
где X представляет собой -CHR3-;
Z представляет собой -S- группу;
R1 выбран из группы, включающей незамещенный фенил;
R2 выбран из группы, включающей алкил;
R3 представляет собой атом водорода;
n равен 0.
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| US 5389640 A, 14.02.1995 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2-ХЛОР-3-НИТРОХИНОЛИНАМИНОВ, СПОСОБ ПОЛУЧЕНИЯ 2-ХЛОР-3,4-ДИАМИНОХИНОЛИНОВ, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 4-ХЛОР-1H-ИМИДАЗО (4,5-C)-ХИНОЛИНОВ, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 1H-ИМИДАЗО (4,5-C)ХИНОЛИНОВ, 2-ХЛОР-3-НИТРОАМИНОХИНОЛИН, 2-ХЛОР-3,4-ДИАМИНОХИНОЛИН | 1990 |
|
RU2083563C1 |

