Изобретение касается имидазохинолиновых соединений, содержащих тиоэфирную группу в положении 1, и фармацевтических композиций, включающих такие соединения. Изобретение касается также использования этих соединений в качестве иммуномодуляторов для индукции биосинтеза цитокинов у животных и для лечения разнообразных клинических расстройств, включая вирусные и онкологические заболевания.
Первое достоверное сообщение, касающееся 1Н-имидазо[4,5-с]хинолинового кольца (Backman et al., J. Org. Chem. 15, 1278-1284, 1950) описывает синтез 1-(6-метокси-8-хинолинил)-2-метил-1Н-имидазо[4,5-с]хинолина для потенциального использования в качестве лекарственного средства против малярии. В последующем появились сообщения о синтезе различных замещенных 1Н-имидазо[4,5-с]хинолинов. В частности, Jain et al., J. Med. Chem., 11, стр.87-92 (1968), синтезировали соединение 1-[2-(4-пиперидил)этил]-1Н-имидазо[4,5-с]хинолин для применения в качестве противосудорожного и сердечно-сосудистого средства. Другими примерами служат работы Baranov et al., Chem. Abs., 85, 94362 (1976), и Bereneyi et al., J. Heterocyclic Chem., 18, 1537-1540 (1981), в каждой из которых было синтезировано по несколько 2-оксоимидазо[4,5-с]хинолинов.
Позднее было установлено, что некоторые 1Н-имидазо[4,5-с]хинолин-4-амины и их 1- и 2-замещенные производные могут использоваться в качестве проивовирусных средств, бронходилятаторов и иммуномодуляторов. Они описаны, в частности, в Патентах США No 4689338; 4698348; 4929624; 5037986; 5346905 и 5389640.
Интерес к имидазохинолиновому кольцу сохраняется по-прежнему.
Известно несколько Н1-имидазо[4,5-с]нафтиридин-4-аминов, Н1-имидазо[4,5-с]пиридин-4-аминов и Н1-имидазо[4,5-с]хинолин-4-аминов, имеющих замещенную эфирную группировку в положении 1. Они описаны в Патентах США No 5268376; 5389640; 5494916 и WO 99/29693.
Несмотря на все попытки получить соединения, которые можно было бы использовать в качестве иммуномодуляторов, сохраняется потребность в препаратах, изменяющих функцию иммунной системы путем индукции биосинтеза цитокинов или посредством иных механизмов.
Краткое описание изобретения
Мы обнаружили новый класс соединений, индуцирующих биосинтез цитокинов у животных. Таким образом, изобретение касается имидазохинолин-4-аминов и тетрагидроимидазохинолин-4-аминов, содержащих заместитель тиоэфира в положении 1. Эти соединения соответствуют формулам (I) и (II), их подробное описание приведено ниже. Общей структурной формулой этих соединения является
где X, Z, R1, R2 и R даются в дальнейшем для каждого класса соединений, соответствующих формулам (I) и (II).
Соединения, соответствующие формулам (I) и (II), влияют на иммунные реакции благодаря способности индуцировать биосинтез цитокинов или вызывать иные эффекты при введении животным. Это позволяет использовать их для лечения разнообразных патологических состояний, чувствительных к изменениям активности иммунной системы, таких как вирусные заболевания или опухоли.
Изобретение касается также фармацевтических композиций на основе соединений, влияющих на иммунные реакции, и способов индукции биосинтеза цитокинов у животных, терапии вирусных инфекций у животных и/или лечения новообразований у животных посредством введения им того или иного соединения, имеющего формулу (I) или (II).
Кроме того, изобретение относится к способам синтеза указанных соединений.
Подробное описание изобретения
Как указывалось ранее, мы обнаружили несколько соединений, которые индуцируют биосинтез цитокинов и модифицируют иммунные реакции у животных. Эти соединения имеют общие структурные формулы (I) и (II), как описано ниже.
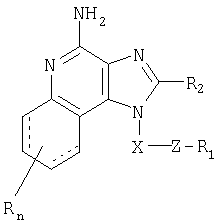
Имидазохинолиновые соединения, содержащие тиоэфирную группировку в положении 1, соответствуют формуле (I)
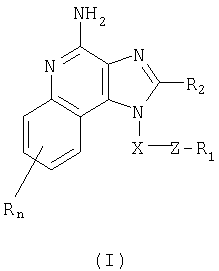
R1 выбран из группы, включающей
- алкил,
- арил, где X: -CHR3-, -CHR3-алкил или -CHR3-алкенил;
Z: -S-, -SO- -SO2-;
- гетероарил,
- гетероциклил,
- алкенил,
- R4-арил,
- R4-гетероарил и
- R4-гетероциклил;
R2 выбран из группы, включающей
- водород,
- алкил,
- алкенил,
- арил,
- гетероарил,
- гетероциклил,
- алкил-Y-алкил,
- алкил-Y-алкенил,
- алкил-Y-арил и
- алкил или алкенил, замещенный одним или несколькими заместителями, выбранными из группы, включающей
- ОН,
- галоген,
- N(R3)2,
- СО-N(R3)2,
- CO-C1-10 алкил,
- CO-O-C1-10 алкил,
- N3,
- арил,
- гетероарил,
- гетероциклил,
- СО-арил и
- СО-гетероарил.
Каждый R3 представляет собой независимо либо Н, либо С1-10 алкил,
R4 представляет собой или алкил или алкенил,
каждый Y представляет собой независимо -О- или -
S(O)0-2-,
n = от 0 до 4 и
каждый R независимо выбран из группы, включающей C1-10 алкил, С1-10 алкокси, гидрокси, галоген и трифторометил или их фармацевтически приемлемую соль.
Изобретение относится также к
тетрагидроимидазохинолинолиновым соединениям, содержащим заместитель тиоэфира в положении 1. Такие тетрагидроимидазохинолинолиновые соединения соответствуют структурной формуле (II)
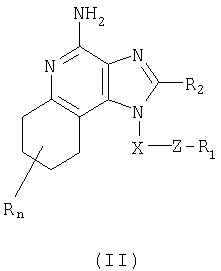
где X: -CHR3-, -CHR3алкил или -CHR3алкенил;
Z: -S-, -SO- -SO2-;
R1 выбран из группы, включающей
- алкил,
- арил,
- гетероарил,
- гетероциклил,
- алкенил,
- R4-арил,
- R4-гетероарил и
- R4-гетероциклил;
R2 выбран из группы, включающей
- водород,
- алкил,
- алкенил,
- арил,
- гетероарил,
- гетероциклил,
- алкил-Y-алкил,
- алкил-Y-алкенил,
- алкил-Y-арил и
- алкил или алкенил, замещенный одним или несколькими заместителями, выбранными из группы, включающей
- ОН,
- галоген,
- N(R3)2,
- СО-N(R3)2,
- CO-C1-10 алкил,
- CO-O-C1-10 алкил,
- N3,
- арил,
- гетероарил,
- гетероциклил,
- СО-арил и
- СО-гетероарил.
Каждый R3 представляет собой независимо либо Н, либо C1-10 алкил,
R4 представляет собой или алкил, или алкенил,
каждый Y представляет собой независимо -О- или -S(O)0-2-,
n = от 0 до 4 и
каждый R независимо выбран из группы, включающей С1-10 алкил, C1-10 алкокси, гидрокси, галоген и трифторометил или их соль, пригодную для фармацевтического использования.
Получение соединений
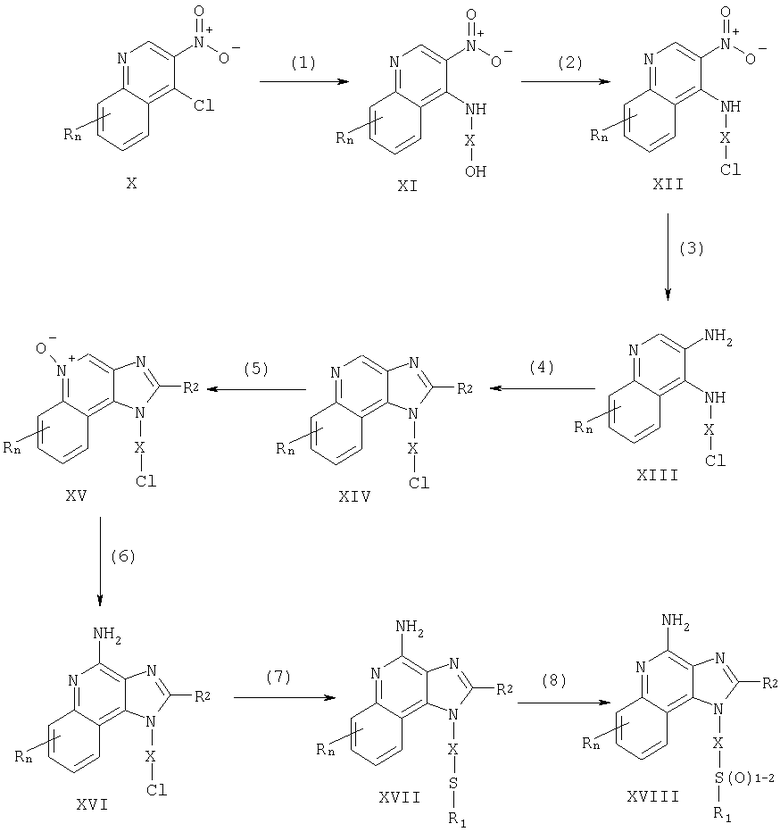
Соединения по данному изобретению могут быть получены посредством реакции по схеме I, где R, R1, R2, Х и n имеют указанные выше значения.
На стадии (1) схемы I 4-хлоро-3-нитрохинолин (X) взаимодействует с амином, имеющим формулу HO-X-NH2, что приводит к образованию 3-нитрохинолин-4-амина (XI). Для проведения этой реакции амин добавляют в раствор соединения (X) в подходящем растворителе, таком как хлороформ или дихлорометан, в присутствии триэтиламина. В случае необходимости полученную смесь можно подогреть. Многие хинолины, имеющие формулу X, хорошо известны (см., например, Патент США No 4689338 и прилагаемый к нему перечень литературы). Многие амины формулы HO-X-NH2 имеются в продаже, другие легко получить, используя известные способы синтеза.
На стадии (2) схемы I 3-нитрохинолин-4-амин (XI) хлорируют для получения 3-нитрохинолин-4-амина, имеющего формулу (XII). Используют обычные хлорирующие реагенты. Реакцию предпочтительно проводят, растворяя соединение (XI) и тионила хлорид в соответствующем растворителе, например в дихлорометане. Реакция протекает при комнатной температуре или в условиях нагревания. По альтернативному способу реагирующие соединения не разводят.
На стадии (3) схемы I 3-нитрохинолин-4-амин (XII) восстанавливают для получения хинолин-3,4-диамина (XIII). Для восстановления предпочтительно используют обычный гетерогенный гидрогенированный катализатор, например платину или уголь. Удобно проводить реакцию в аппарате Парра, в соответствующем растворителе (толуоле).
На стадии (4) схемы I 3-нитрохинолин-4-амин (XIII) реагирует с карбоксильной кислотой или эквивалентным реагентом, что приводит к образованию 1Н-имидазо[4,5-с]хинолина (XIV). К числу соединений, эквивалентных карбоксильной кислоте, относятся ортоэфиры и 1,1-диалкоксиалкил-алканоаты. Карбоксильную кислоту или ее эквивалент выбирают таким образом, чтобы ввести желаемый заместитель R2 в соединение (XIV). В частности, триэтилортоформиат является соединением, в котором R2 представлен водородом, а триметилортовалерат соединением, в котором R2 представлен бутилом. Реакция может проходить в отсутствие растворителя или в таком инертном растворителе, как толуол. Реакционную систему нагревают для упаривания спирта или воды, образующихся в качестве побочных продуктов взаимодействия. В случае необходимости используют катализатор, например пиридина гидрохлорид.
По альтернативной методике стадию (4) осуществляют в два этапа:
(1) смешивают диамин (XIII) с ацильным галидом, имеющим формулу R2C(O)C1 или R2C(O)Br, и (2) проводят циклизацию. На этапе (1) ацильный галид добавляют в раствор диамина в подходящем растворителе, например в пиридине. Реакция протекает при комнатной температуре в присутствии пиридина гидрохлорида.
На стадии (5) схемы I 1Н-имидазо[4,5]хинолин, имеющий общую формулу XIV, окисляют для получения 1Н-имидазо[4,5]хинолин-5N-оксида (XV), используя обычный окислитель, обеспечивающий образование 5N-оксида. Предпочтительно раствор соединения (XV) в подходящем растворителе, таком как хлороформ или дихлорометан, обрабатывают 3-хлоропероксибензойной кислотой при комнатной температуре.
На стадии (6) схемы I 1Н-имидазо[4,5]хинолин-5N-оксид (XV) аминируют для получения 1Н-имидазо[4,5]хинолин-4-амина (XVI). Стадия (6) включает два этапа: (1) реакцию соединения (XV) с ацилирующим реагентом и (2) реакцию полученного продукта с аминирующим реагентом.
На этапе (1) стадии (6) N-оксид (XV) реагирует с ацилирующим реагентом. В качестве последнего используют алкил- или арилсульфонилхлориды (например, бензолсульфонилхлорид, метанолсульфонилхлорид и п-толуолсульфонилхлорид). Предпочтение отдается арилсульфонилахлоридам. Наиболее предпочтителен п-толуолсульфонила хлорид. На этапе (2) стадии (6) продукт, полученный на предыдущем этапе (1), реагирует с избыточным количеством аминирующего соединения. К числу предпочтительных аминирующих реагентов относятся аммиак (в частности, в форме гидроокиси аммония) и соли аммония (например, аммония карбонат, аммония бикарбонат или аммония фосфат). Предпочтение отдается гидроокси аммония. Реакцию предпочтительно проводят, растворяя N-оксид (XV) в инертном растворителе, таком как дихлорометан или хлороформ. При этом в раствор сначала добавляют аминирующее соединение, а затем (медленно) ацилирующий реагент.
На стадии (7) схемы I 1Н-имидазо[4,5]хинолин-4-амин (XVI) взаимодействует с соединением, имеющим формулу R1-SNa, что приводит к образованию 1Н-имидазо[4,5]хинолин-амина формулы XVII, которая является подродом формулы I. Реакцию осуществляют, смешивая соединение (XVI) и соединение, имеющее формулу R1SNa, в подходящем растворителе, таком как N,N-диметилформамид или диметилсульфоксид, при комнатной температуре или при 60-80°С. Продукт реакции или его пригодную для фармацевтического использования соль выделяют обычными способами.
На стадии (8) схемы I 1Н-имидазо[4,5]-4-амин (XVII) окисляют обычным окисляющим реагентом для получения 1Н-имидазо[4,5]-4-амина формулы XVIII, которая является подродом формулы I. Предпочтительно раствор соединения (XVII) в подходящем растворителе, таком как хлороформ или дихлорометан, обрабатывают 3-хлоропероксибензойной кислотой при комнатной температуре. Интенсивность окисления регулируют, подбирая нужное количество 3-хлоропероксибензойной кислоты (примерно один эквивалент дает сульфоксид, приблизительно два эквивалента дают сульфон). Продукт реакции или его пригодную для фармацевтического использования соль выделяют обычными способами.
Реакция по схеме I
Соединения в соответствии с изобретением могут быть получены посредством реакции по схеме II, где R, R1, R2, X и n имеют указанные выше значения.
На стадии (1) схемы II осуществляют реакцию 3-нитрохинолин-4-амина (XII) с соединением, имеющим формулу R1-SNa, используя процедуру (7) схемы I и получая 3-нитрохинолин-4-амин (XIX).
На стадии (3) схемы II осуществляют циклизацию хинолин-3,4-диамина (XX), используя процедуру (4) схемы I и получая 1Н-имидазо[4,5-с]хинолин (XXI).
На стадии (4) схемы II 1Н-имидазо[4,5-с]хинолин (XXI) окисляют для получения 1Н-имидазо[4,5-с]хинолин-5N-оксида (XXII), используя обычный окисляющий реагент. Предпочтительно раствор соединения (XXI) в подходящем растворителе, например хлороформе или дихлорометане, по меньшей мере три раза обрабатывают эквивалентом 3-хлоропероксибензойной кислоты при комнатной температуре.
На стадии (5) схемы II 1Н-имидазо[4,5-с]хинолин-5N-оксид (XXII) аминируют, используя процедуру (6) схемы I, для получения 1Н-имидазо[4,5-с]хинолин-4-амина формулы XVIII, которая является подродом формулы (I). Реакция протекает при комнатной температуре или при 60-80°С. Продукт реакции или его пригодную для фармацевтического использования соль выделяют обычными способами.
Реакция по схеме II
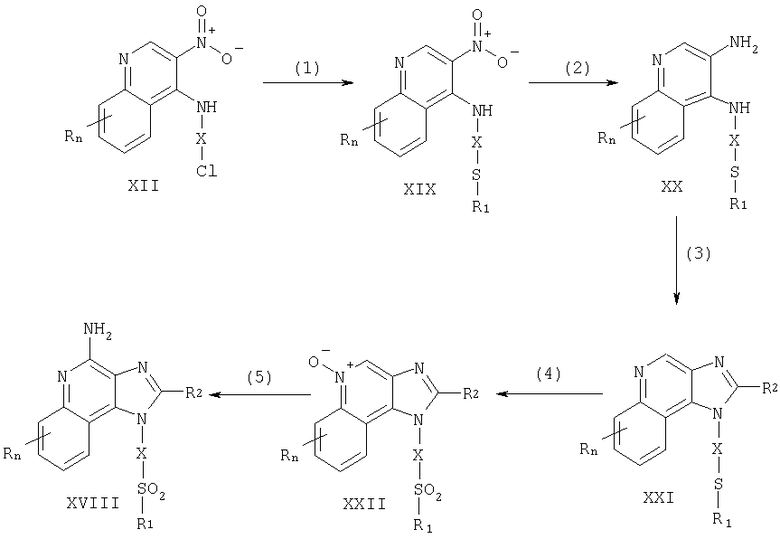
Соединения по данному изобретению могут быть получены посредством реакции по схеме III, где R, R1, R2, X и n имеют указанные выше значения.
На стадии (1) схемы III 3-нитро-4-аминохинолин-1-ил-алкоголь, имеющий формулу XI, защищают трет-бутилдиметилсилильной группой, используя обычную методику. Предпочтительно соединение (XI) вводят вместе с трет-бутилдиметилсилила хлоридом в подходящий растворитель, такой как хлороформ, в присутствии триэтиламина и каталитического количества 4-диметиламинопиридина.
На стадии (2) схемы III защищенный 3-нитро-4-аминохинолин-1-ил-алкоголь (XXIII) восстанавливают, используя процедуру (3) схемы I, для получения защищенного 3,4-диаминохинолин-1-ил-алкоголя, имеющего формулу (XXIV).
На стадии (3) схемы III осуществляют циклизацию защищенного 3,4-диаминохинолин-1-ил-алкоголя (XXIV), используя процедуру (4) схемы 1, для получения 1Н-имидазо[4,5-с]хинолина (XXV).
На стадии (4) схемы III 1Н-имидазо[4,5-с]хинолин (XXV) окисляют для получения 1Н-имидазо[4,5-с]хинолин-5N-оксида (XXVI), используя процедуру (5) схемы I.
На стадии (5) схемы III 1Н-имидазо[4,5-с]хинолин-5N-оксид (XXVI) аминируют используя процедуру (6) схемы I, для получения 1Н-имидазо[4,5-с]хинолин-4-амина (XXVII).
На стадии (6) схемы III из 1Н-имидазо[4,5-с]хинолин-4-амина (XXVII) удаляют защитную группировку и получают 1Н-имидазо[4,5-с]хинолин-4-амин (XXVIII). Предпочтительно раствор соединения (XXVII) в подходящем растворителе, таком как тетрагидрофуран, обрабатывают тетрабутиламмония фторатом. Некоторые соединения, имеющие общую формулу (XXVIII), хорошо известны (см., например, Gerster, U.S. Patent No 4689338, и Gerster et al., U.S. Patent No 5605899).
На стадии (7) схемы III 1Н-имидазо[4,5-с]хинолин-4-амин (XXVIII) хлорируют обычным способом для получения 1Н-имидазо[4,5-с]хинолин-4-амина (XVI). Неразбавленное соединение (XXVIII) можно нагревать в присутствии тионила хлорида. По альтернативному способу в раствор соединения XXVIII в подходящем растворителе, таком как N,N-диметилформамид, добавляют контролируемое количество фосфора оксихлорида вместе с триэтиламином.
Стадии (8) и (9) по схеме III осуществляют таким же образом, как соответственно стадии (7) и (8) по схеме I.
Реакция по схеме III
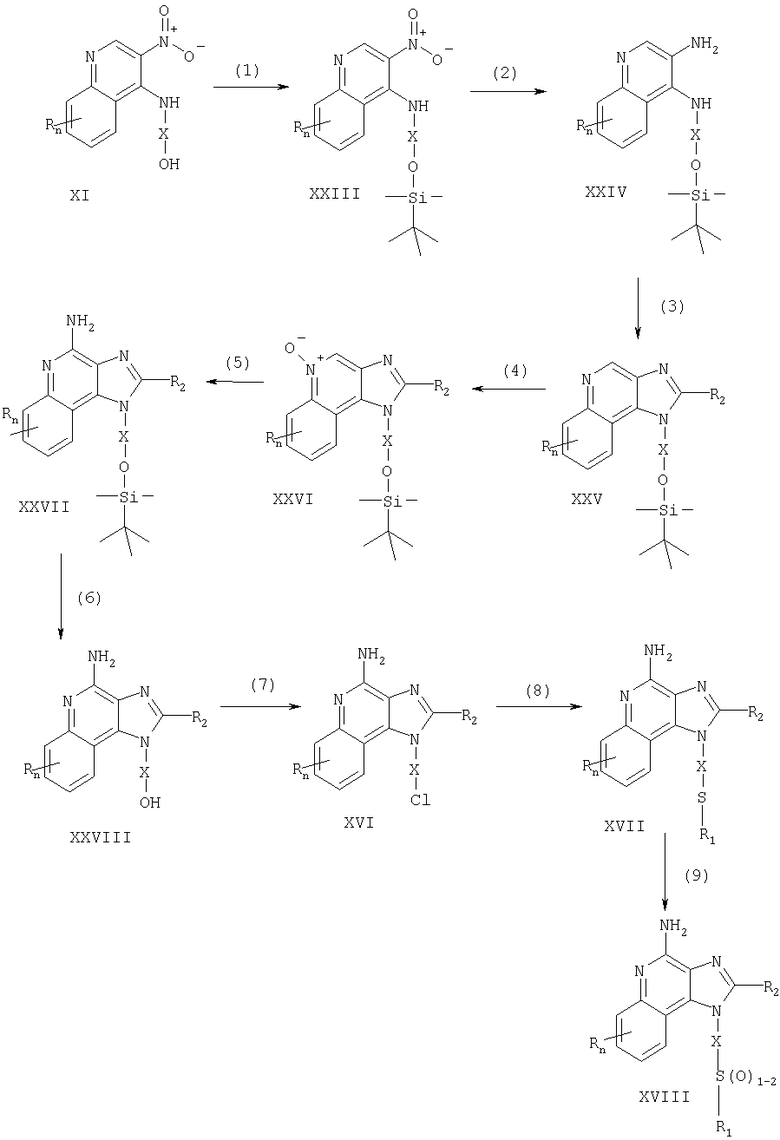
Соединения по данному изобретению могут быть получены посредством реакции по схеме IV где R, R1, R2, X и n имеют указанные выше значения, а ВОС - трет-бутоксикарбонил.
На стадии (1) схемы IV гидроксильную группу 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил-алкоголя (XXIX) защищают трет-бутилдиметилсилильной группой, используя процедуру (1) схемы III. Соединения, имеющие общую формулу (XXIX), описаны ранее и могут быть получены известными способами синтеза (см., например, Nikolaides et al., U.S. Patent No 5352784, и Lindtsrom, U.S. Patent No 5693811, а также перечни литературы, прилагаемые к этим документам).
На стадии (2) схемы IV аминогруппу 1Н-имидазо[4,5-с]хинолин-4-амина (XXX) защищают обычными способами для получения защищенного 1Н-имидазо[4,5-с]хинолина, имеющего формулу (XXXI). Предпочтительно соединение XXX обрабатывают ди-трет-бутилдикарбонатом в подходящем растворителе (например, в тетрагидрофуране) в присутствии триэтиламина и 4-диметиламинопиридина.
Реакция протекает в условиях повышенной температуры (60°С).
На стадии (3) схемы IV из соединения (XXXI) удаляют трет-бутилдиметилсилильную защитную группировку, используя процедуру (6) схемы III, и получают 1Н-имидазо[4,5-с]хинолин-1-ил-алкоголь (XXXII).
На стадии (4) схемы IV 1Н-имидазо[4,5-с]хинолин-1-ил-алкоголь (XXXII) преобразуют в метансульфонат (XXXIII). Предпочтительно раствор соединения (XXXII) в подходящем растворителе, таком как дихлорометан, обрабатывают метансульфонила хлоридом в присутствии триэтиламина.
Реакцию можно проводить при пониженной температуре (-10°С).
На стадии (5) схемы IV осуществляют реакцию метансульфонила (XXXIII) с тиолом, имеющим формулу R1SH, для получения тиоэфира (XXXIV). Предпочтительно раствор соединения (XXXIII) в подходящем растворителе, таком как N,N-диметилформамид, обрабатывают тиолом в присутствии триэтиламина. Реакция протекает в условиях повышенной температуры (80°С).
На стадии (6) схемы IV посредством гидролиза в кислой среде удаляют защитную трет-бутоксикарбонильную группировку и получают 1Н-имидазо[4,5-с]хинолин-4-амин формулы (XXXV), которая является подродом формулы II. Предпочтительно соединение XXXIV в подходящем растворителе, таком как дихлорометан, обрабатывают при комнатной температуре раствором соляной кислоты в диоксане. Продукт реакции или его пригодную для фармацевтического использования соль выделяют обычными способами.
На стадии (7) схемы IV тиоэфир (XXXV) окисляют, используя процедуру (8) схемы I, для получения сульфона или сульфоксида формулы XXXVI, которая является подродом формулы II. Продукт реакции или его пригодную для фармацевтического использования соль выделяют обычными способами.
Реакция по схеме IV
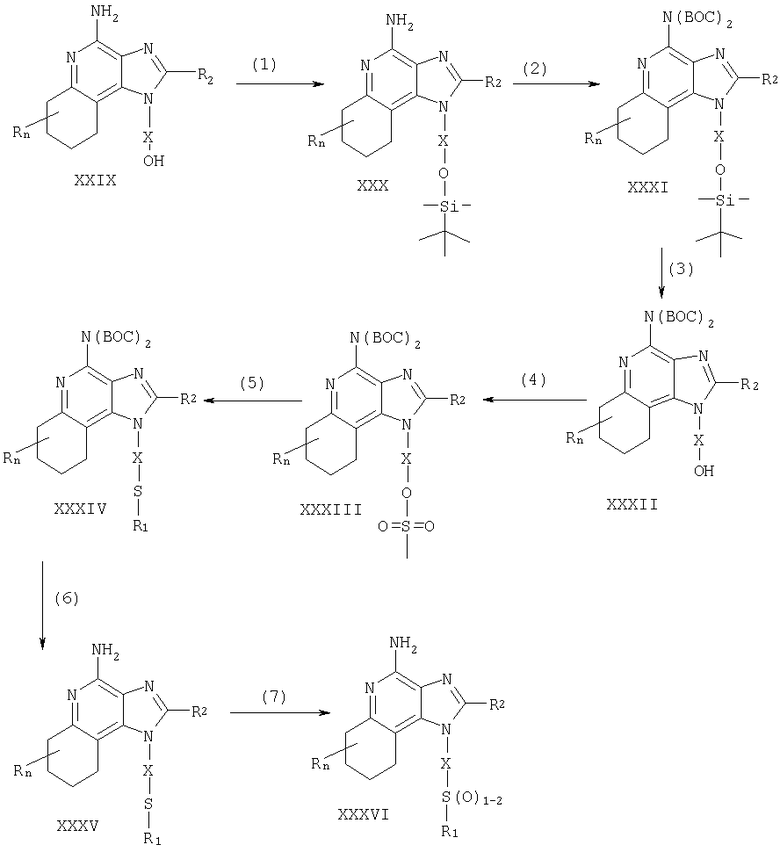
Соединения в соответствии с изобретением могут быть получены посредством реакции по схеме V, где R, R1, R2, X и n имеют указанные выше значения.
На стадии (1) схемы V 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил-алкоголь (XXIX) хлорируют, используя процедуру (7) схемы III, для получения соединения (XXXVII).
На стадии (2) схемы V осуществляют реакцию соединения (XXXVII) с соединением, имеющим формулу R1SNa, используя процедуру (7) схемы 1, для получения тиоэфира формулы XXXV, которая является подродом формулы II. Продукт реакции или его пригодную для фармацевтического использования соль выделяют обычными способами.
На стадии (3) схемы V тиоэфир (XXXV) окисляют, используя процедуру (8) схемы I, для получения сульфона или сульфоксида формулы XXXVI, которая является подродом формулы II. Продукт реакции или его пригодную для фармацевтического использования соль выделяют обычными способами.
Реакция по схеме V
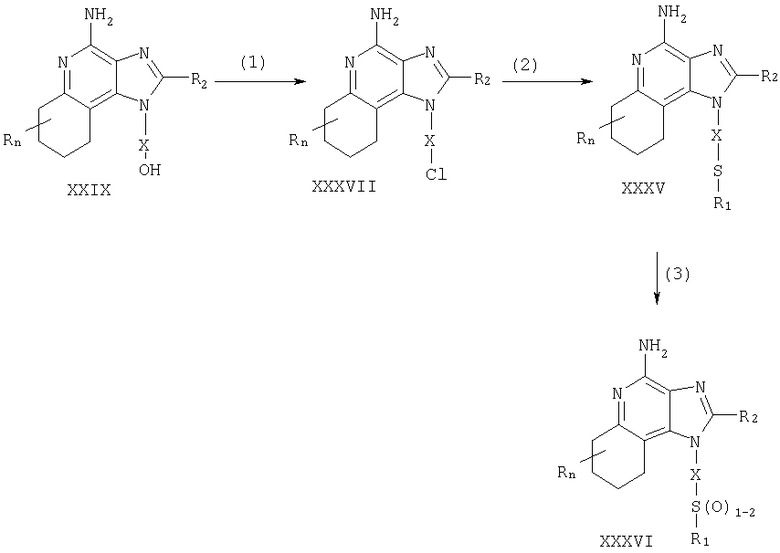
Соединения в соответствии с изобретением могут быть получены посредством реакции по схеме VI, где R, R1, R2, Х и n имеют указанные выше значения.
В реакции по схеме VI 1Н-имидазо[4,5-с]хинолин, имеющий формулу (I) восстанавливают для получения 5,6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолина (II). С этой целью соединение (I) разводят в трифторуксусной кислоте, добавляют в полученный раствор каталитическое количество оксида платины (IV), после чего смесь подвергают давлению водорода. Реакцию удобно проводить в аппарате Парра. Продукт реакции или его пригодную для фармацевтического использования соль выделяют обычными способами.
Реакция по схеме VI
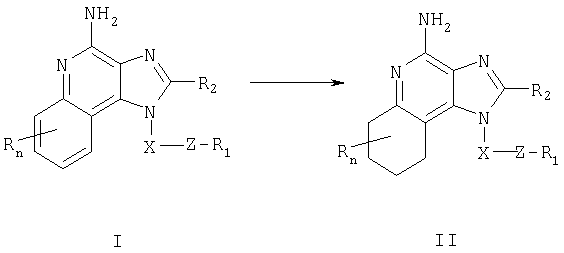
Соединения в соответствии с изобретением могут быть получены посредством реакции по схеме VII, где R, R1, R2, X и n имеют указанные выше значения; Ph - фенил.
На стадии (1) схемы VII 2,4-дигидро-3-нитро-6,7,8,9-тетрагидрохинолин, имеющий формулу (XXXVIII), хлорируют, используя обычные хлорирующие реагенты, для получения 2,4-дигидро-3-нитро-6,7,8,9-тетрагидрохинолина (XXXIX).
Предпочтительно осуществляют взаимодействие соединения (XXXVIII) с фосфора оксихлоридом при повышенной температуре. Некоторые соединения, имеющие общую формулу (XXXVIII), описаны ранее и могут быть получены известными способами синтеза (см., например, Nikolaides et al., U.S. Patent No 5352784, и перечень литературы, прилагаемый к этому документу).
На стадии (2) схемы VII осуществляют взаимодействие 2,4-дигидро-3-нитро-6,7,8,9-тетрагидрохинолина (XXXIX) с амином (XXX).
С этой целью амин добавляют в раствор соединения (XXXIX) в подходящем растворителе, таком как N,N-диметилформамид, в присутствии триэтиламина. В случае необходимости реакционную смесь можно нагреть.
На стадии (3) схемы VII осуществляют взаимодействие 2-хлоро-3-нитро-6,7,8,9-тетрагидрохинолина (ХХХХ) с натрия феноксидом для получения 3-нитро-2-фенокси-6,7,8,9-тетрагидрохинолина (XXXXI). С этой целью фенол добавляют к натрия гидриду в подходящем растворителе, таком как 1,2-диметоксиэтан, и получают феноксид. После этого осуществляют реакцию феноксида с соединением (ХХХХ) при повышенной температуре.
На стадии (4) схемы VII 3-нитро-2-фенокси-6,7,8,9-тетрагидрохинолин (XXXXI) хлорируют, используя обычные хлорирующие реагенты, для получения 3-нитро-2-фенокси-6,7,8,9-тетрагидрохинолина (XXXXII).
Предпочтительно осуществляют взаимодействие N-хлоросукцинимида с трифенилфосфином в подходящем растворителе, таком как тетрагидрофуран, и получают фосфинхлорид. После этого осуществляют реакцию фосфинхлорида с соединением (XXXXI).
На стадии (5) схемы VII 3-нитро-2-фенокси-6,7,8,9-тетрагидрохинолин (XXXXII) восстанавливают обычными способами для получения 3-амино-2-фенокси-6,7,8,9-тетрагидрохинолина (XXXXIII). Предпочтительная методика состоит в генерировании Ni2B in situ. Натрия борогидрид добавляют в смесь гексагидрата никеля(II) хлорида и соединения (XXXXII) в метаноле/хлороформе (50:50).
На стадии (6) схемы VII осуществляют циклизацию 3-амино-2-фенокси-6,7,8,9-тетрагидрохинолина (XXXXIII), используя процедуру (4) схемы I, для получения 4-фенокси-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолина формулы (III).
На стадии (7) схемы VII осуществляют взаимодействие 4-фенокси-6,7,8,9-тетрагидро- 1Н-имидазо[4,5-с]хинолина формулы (III) с соединением, имеющим формулу R1SNa, для получения 4-фенокси-6,7,8,9-тетрагидро-6,7,8,9-1Н-имидазо[4,5-с]хинолина формулы (XXXXIV), которая является подродом формулы (IV). Предпочтительно осуществляют реакцию тиола формулы R1SH с натрия гидридом в подходящем растворителе, таком как N,N-диметилформамид, для генерирования аниона, который затем реагирует с соединением формулы (III).
На стадии (8а) схемы VII 4-фенокси-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин (XXXXIV) аминируют для получения 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина формулы XXXV, которая является подродом формулы (II). Реакцию осуществляют, смешивая соединение (XXXXIV) с аммония ацетатом при нагревании примерно до 150°С. В случае необходимости реакцию проводят в сосуде высокого давления. Полученный продукт или его пригодную для фармацевтического использования соль выделяют обычными способами.
На стадии (8б) схемы VII 4-фенокси-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолина формулы (XXXXIV) окисляют, используя процедуру (8) схемы I, для получения 4-фенокси-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолина формулы (XXXXV), которая является подродом формулы (IV).
На стадии (9) схемы VII 4-фенокси-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолина формулы (XXXXV) аминируют, используя процедуру (8а), для получения 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина, имеющего формулу (XXXVI), которая является подродом формулы (II). Полученный продукт или его пригодную для фармацевтического использования соль выделяют обычными способами.
Реакция по схеме VII
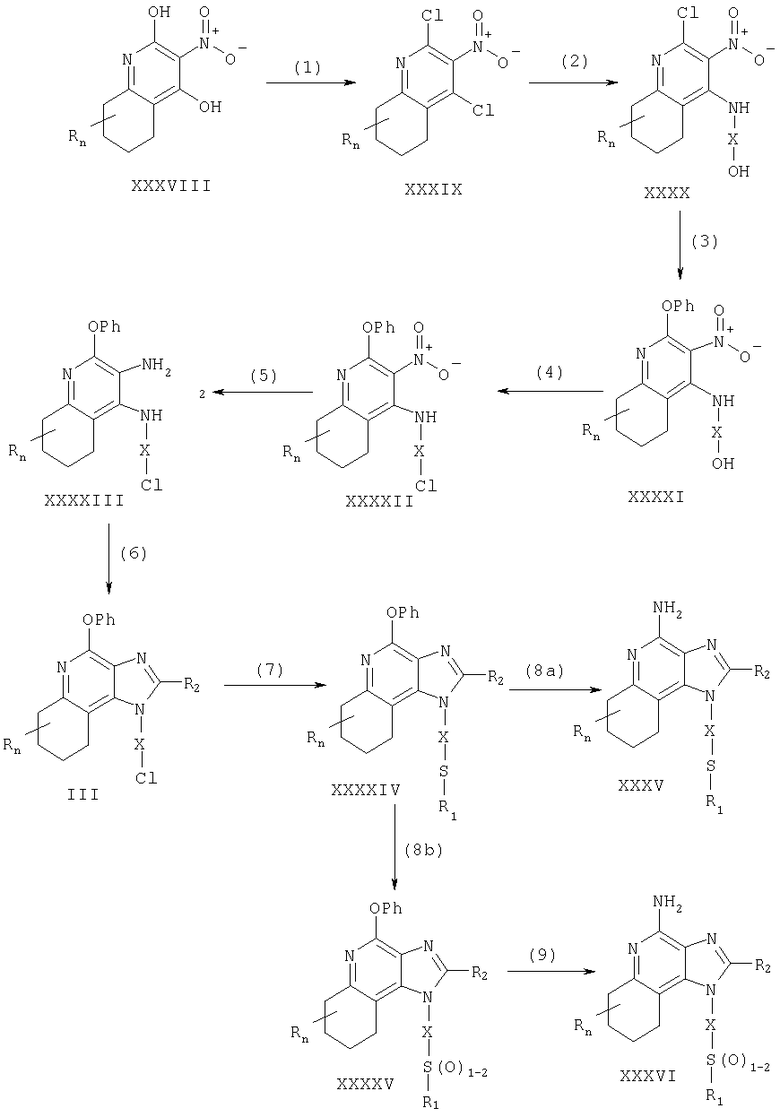
Изобретение относится также к новым соединениям, которые являются промежуточными продуктами при биосинтезе соединений с общей формулой (II). Эти соединения имеют структурные формулы (III) и (IV), более подробная характеристика которых приводится ниже.
Один класс промежуточных продуктов имеет общую формулу III
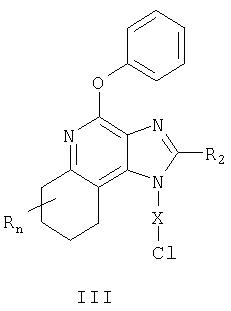
где X: -CHR3-, -CHR3-алкил или -CHR3-алкенил;
R2 выбран из группы, включающей
- водород,
- алкил,
- алкенил,
- арил,
- гетероарил,
- гетероциклил,
- алкил-Y-алкил,
- алкил-Y-алкенил,
- алкил-Y-арил и
- алкил или алкенил, замещенный одним или несколькими заместителями, выбранными из группы, включающей
- ОН,
- галоген,
- N(R3)2,
- СО-N(R2)2,
- CO-C1-10 алкил,
- CO-O-C1-10 алкил,
- N3,
- арил,
- гетероарил,
- гетероциклил,
- СО-арил и
- СО-гетероарил.
Каждый R3 представляет собой независимо либо Н, либо C1-10 алкил,
Y представляет собой независимо -О- или -S(O)0-2-,
n = от 0 до 4 и
каждый R независимо выбран из группы, включающей C1-10 алкил, C1-10 алкокси, гидрокси, галоген и трифторметил или их соль, пригодную для фармацевтического использования.
Другой класс промежуточных соединений имеет общую формулу (IV)

где X: -CHR3-, -CHR3-алкил или -CHR3-алкенил;
Z: -S-, -SO-, -SO2-;
R1 выбран из группы, включающей
- алкил,
- арил,
- гетероарил,
- гетероциклил,
- алкенил,
- R4-арил,
- R4-гетероарил и
- R4-гетероциклил;
R2 выбран из группы, включающей
- водород,
- алкил,
- алкенил,
- арил,
- гетероарил,
- гетероциклил,
- алкил-Y-алкил,
- алкил-Y-алкенил,
- алкил-Y-арил и
- алкил или алкенил, замещенный одним или несколькими заместителями, выбранными из группы, включающей
- ОН,
- галоген,
- N(R3)2,
- СО-N(R3)2,
- CO-C1-10 алкил,
- CO-O-C1-10 алкил,
- N3,
- арил,
- гетероарил,
- гетероциклил,
- СО-арил и
- СО-гетероарил.
Каждый R3 представляет собой независимо либо Н, либо С1-10алкил,
R4 представляет собой или алкил или алкенил,
Y представляет собой независимо -О- или -S(O)0-2-,
n = от 0 до 4 и
каждый R независимо выбран из группы, включающей С1-10 алкил, C1-10 алкокси, гидрокси, галоген и трифторметил или их соль, пригодную для фармацевтического использования.
В данном описании термины "алкил", "алкенил" и приставка "алк-" применяются для обозначения как прямых, так и разветвленных цепей, а также циклических групп, в частности циклоалкильных и циклоалкенильных. Эти группы содержат от 1 до 20 атомов углерода, если не указано иначе. При этом алкенильные группы содержат от 2 до 20 углеродных атомов. Предпочтительные группы содержат в общей сложности до 10 атомов углерода. Циклические группы могут быть моноциклическими или полициклическими; предпочтительны циклические группы, содержащие в кольце от 3 до 10 углеродных атомов. Примерами циклических групп являются циклопропиловая, циклопропилметиловая, циклопентиловая, циклогексиловая и адамантиловая.
Кроме того, алкильные или алкенильные компоненты -Х- групп могут быть незамещенными или замещенными одним либо несколькими заместителями, выбранными из групп, включающих алкил, алкенил, арил, гетероарил, гетероциклил, арилалкил, гетероарилалкил и гетероциклилалкил.
Термин "галоалкил" используется для обозначения групп, замещенных одним или несколькими галогеновыми атомами, в том числе перфторированных групп. То же самое относится к группам, в названиях которых используется приставка "гало-". Примерами галоалкильных групп являются хлорметиловая, трифторметиловая и т.п.
Термин "арил" используется в данном описании для обозначения карбоциклических ароматических колец или их систем.
Примерами арильных группировок являюся фенил, нафтил, бифенил, флюоренил и инденил. Термин "гетероарил" распространяется на ароматические кольца или их системы, содержащие в кольце по меньшей мере один гетероатом (например, О, S, N). Примерами гетероарильных группировок служат фурил, тиенил, пиридил, хинолинил, изохинолинил, индолил, изоиндолил, триазолил, пирролил, тетразолил, имидазолил, пиразолил, оксазолил, тиазолил, бензофуранил, бензотиофенил, карбазолил, бензоксазолил, пиримидинил, бензимидазолил, хиноксалинил, бензотиазолил, нафтиридинил, изоксазолил, изотиазолил, пуринил, хиназолинил и т.д.
Термин "гетероциклил" используется для обозначения неароматических колец или их систем, которые содержат по меньшей мере один гетероатом кольца (например, О, S, N), и всех полностью или частично ненасыщенных производных вышеупомянутых гетероарильных группировок. Примерами гетероцикличных группировок являются пирролидинил, тетрагидрофуранил, морфолинил, тиоморфолинил, пиперидинил, пиперазинил, тиазолидинил, имидазолидинил, изотиазолидинил и т.п.
Арильные, гетероарильные и гетероцикличные группы могут быть незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из группы, включающей алкил, алкокси, метилендиокси, этилендиокси, алкилтио, галоалкил, галоалкилтио, галоген, нитро, гидрокси, меркапто, циано, формил, арил, арилокси, арилтио, арилалкокси, арилалктио, гетероарил, гетероарилокси, гетероарилтио, гетероарилалкокси, гетероарилалкилтио, алкилкарбонил, гетероарилкарбонил, арилоксикарбонил, гетероарилоксикарбонил, арилтиокарбонил, гетероарилтиокарбонил, алканоилокси, алканоилтио, алканоиламино, арилкарбонилокси, арилкарбонилтио, алкиламиносульфонил, алкилсульфонил, арилсульфонил, гетероарилсульфонил, арилдиазинил, алкилсульфониламино, арилсульфониламино, арилалкилсульфониламино, алкилкарбониламино, алкенилкарбониламино, алкенилкарбониламино, арилкарбониламино, арилалкилкарбониламино, гетероарилкарбониламино, гетероарилалкенилкарбониламино, алкилсульфониламино, алкенилсульфониламино, арилсульфониламино, арилалкилсульфониламино, гетероарилсульфониламино, гетероарилалкилсульфониламино, алкиламинокарбониламино, алкениламинокарбониламино, ариламинокарбониламино, арилалкиламинокарбониламино, гетероариламинокарбониламино, гетероалкилкарбониламино и, в случае гетероциклила, оксо. Любые другие группы, идентифицируемые как "замещенные" или "факультативно замещенные", также могут включать в качестве заместителей одно или несколько вышеперечисленных соединений.
Некоторые заместители предпочтительны в принципе. Например, группы Х включают этилен и н-бутилен, предпочтительные группы R1 алкильная и арильная, а в арильной группе - фенильная или замещенная фенильная. Предпочтительно отсутствие R заместителей (т.е. n должно быть равно 0). Предпочтительные R2 группы включают водород, алкильные группы, содержащие от 1 до 4 углеродных атомов (т.е. метиловую, этиловую, пропиловую, изопропиловую, н-бутиловую, втор-бутиловую, изобутиловую и циклопропилметиловую), метилоксиэтиловую и этоксиметиловую. При наличии одного или нескольких таких предпочтительных заместителей они входят в состав соединений по данному изобретению в любой комбинации.
Изобретение касается описанных здесь соединений в любой приемлемой для фармацевтического использования форме, включая изомеры (например, диастереомеры и энантиомеры), соли, сольваты, полиморфные соединения и т.п. В частности, изобретение относится к оптически активным соединениям, включая их энантиомеры, а также рацемические смеси энантиомеров.
Фармацевтические композиции и биологическая активность
Фармацевтические композиции по данному изобретению содержат терапевтически эффективное количество одного из вышеперечисленных соединений в комбинации с фармацевтически приемлемым носителем.
Термин "терапевтически эффективное количество" означает количество того или иного соединения, достаточное для индукции терапевтического эффекта, в частности индукции биосинтеза цитокинов, противоопухолевой активности и/или противовирусной активности. Точное количество активного соединения, входящего в состав фармацевтических композиций по данному изобретению, вариирует (в зависимости от известных на настоящее время факторов, в том числе физической и химической природы соединения, природы носителя и ожидаемых дозировок). Однако любая такая композиция содержит активный ингредиент в количестве, достаточном, чтобы обеспечить ее терапевтическую дозу в диапазоне от 100 нг/кг до 50 мг/кг, предпочтительно от 10 мкг/кг до 5 мг/кг.
Композиции могут входить в состав любых существующих лекарственных форм, таких как таблетки, лепешки, препараты для парентерального введения, сиропы, кремы, мази, аэрозоли, пластыри для нанесения лекарственных средств на кожу и слизистые и т.п.
Соединения по данному изобретению предназначены в качестве лекарственных средств для монотерапии, а также для сочетанного использования друг с другом или с другими активными соединениями, включая иммуномодуляторы иной природы, противовирусные и противобактериальные препараты и т.п.
Соединения по данному изобретению индуцировали продукцию некоторых цитокинов в экспериментах, проводившихся для их тестирования, как описано ниже. Полученные результаты показывают, что эти соединения являются ценными модуляторами иммунных реакций и имеют разные механизмы действия, что позволяет использовать их для лечения разнообразных заболеваний.
К числу цитокинов, продукция которых индуцируется введением соединений в соответствии с изобретением, относятся интерферон-α (ИФН-α) и/или фактор некроза опухолей-α (ФНО-α), а также некоторые интерлейкины (ИЛ). Соединения по данному изобретению индуцируют биосинтез следующих цитокинов: ИФН-α, ФНО-α, ИЛ-1, ИЛ-6, ИЛ-10 и ИЛ-12, а также большого числа цитокинов иных типов. Клинические эффекты этих и других цитокинов включают ингибирование размножения вирусов и роста опухолевых клеток, что позволяет использовать их для лечения вирусных инфекций и новообразований. В связи с этим изобретение относится также к способу индукции биосинтеза цитокинов у животных, который состоит во введении им терапевтически эффективного количества соединения или композиции по данному изобретению.
Установлено, что отдельные соединения по данному изобретению преимущественно индуцируют экспрессию ФНО-α в популяции кроветворных клеток, таких как МКПК (моноядерные клетки периферической крови), содержащую клетки pDC2 (предшественники дендритов 2 типа). При этом они не вызывают сопутствующей продукции больших количеств воспалительных цитокинов.
В дополнение к способности индуцировать биосинтез цитокинов соединения по данному изобретению влияют на другие аспекты врожденной иммунной функции. В частности, они стимулируют активность природных клеток-убийц; это их действие может быть следствием индукции биосинтеза цитокинов. Указанные соединения могут также активировать макрофаги, что, в свою очередь, усиливает секрецию оксида азота и образование дополнительных цитокинов. Кроме того, эти соединения вызывают пролиферацию и дифференцировку В-лимфоцитов.
Соединения в соответствии с изобретением влияют также на приобретенные иммунные реакции. Не оказывая непосредственного действия на Т-клетки или продукцию цитокинов в Т-клетках, они косвенным образом индуцируют образование ИФН-γ в Т-хелперах 1 типа (ТХ1) и ингибируют продукцию ИЛ-4, ИЛ-5 и ИЛ-13 Т-хелперами 2 типа (ТХ2) после введения животным.
Такие проявления активности этих соединений означают, что они могут использоваться для лечения заболеваний, при которых необходимо усилить реакцию ТХ1 и ослабить реакцию ТХ2. В связи со способностью соединений по данному изобретению подавлять иммунные реакции, опосредуемые через ТХ2, они могут оказаться полезными для терапии атопических расстройств, таких как атопический дерматит, астма, аллергия, аллергический ринит и системная красная волчанка, а также в качестве вакцинных адъювантов для клеточного иммунитета и, возможно, для лечения рецидивирующих грибковых заболеваний и хламидиоза.
Модулирующее действие соединений по данному изобретению на иммунные реакции делает их перспективными средствами лечения многочисленных клинических расстройств. Благодаря способности индуцировать продукцию цитокинов, таких как ИФН-α и/или ФНО-α, эти соединения представляют особенно большой интерес в качестве терапевтических средств при вирусных заболеваниях и опухолях. Эта иммуномодулирующая активность предполагает возможность использования соединений по данному изобретению для лечения следующих клинических состояний (перечень которых не ограничивается нижеперечисленными):
вирусные заболевания, включая обычные бородавки, остроконечные кондиломы, подошвенные бородавки; гепатит В и гепатит С;
простой герпес, вызываемый вирусами I и II типа;
контагиозный моллюск; натуральная оспа, особенно в тяжелой форме; ВИЧ-инфекция; инфекции, вызываемые цитомегаловирусом, вирусом ветряной оспы и опоясывающего лишая, риновирусами и аденовирусами; грипп и парагрипп; интраэпителиальные неоплазии, включая опухоли шейки матки; папилломавирусную инфекцию у людей и ассоциирующиеся с ней неоплазии; грибковые заболевания, в частности кандидиоз и аспергиллоз; криптококковый менингит;
новообразования (такие как базально-клеточная карцинома, лейкемический ретикулоэндотелиоз, саркома Капоши, карцинома почечных клеток, плоскоклеточная карцинома, миелогенная лейкемия, множественная миелома, меланома, не-ходжкинская лимфома, лимфома кожных Т-клеток и другие формы рака); паразитарные заболевания (такие как pneumocystis carnii, криптоспоридиоз, гистоплазмоз, токсоплазмоз, трипаносомная инфекция и лейшманиоз) и бактериальные инфекции (в частности, туберкулез и инфекция Mycobacterium avium). К числу других болезней и патологических состояний, которые можно лечить соединениями по данному изобретению, относятся следующие: лучевой кератоз, экзема, эозинофилия, эссенциальная тромбоцитемия, проказа, множественный склероз, синдром Омена, красная дискоидная волчанка, болезнь Боуена и сопутствующий папулез, очаговая алопеция. Кроме того, данные соединения ингибируют формирование келоидов после хирургического вмешательства и других типов послеоперационных шрамов, улучшают или стимулируют заживление ран, в том числе хронических. Эти соединения могут использоваться для лечения оппортунистических инфекций и опухолей, развивающихся вследствие подавления клеточного иммунитета, например, у пациентов, перенесших трансплантацию органа, раковых больных и лиц, страдающих ВИЧ-инфекцией.
Количество соединения, эффективно индуцирующее биосинтез цитокинов, - это количество, достаточное, чтобы повысить продукцию клетками одного или нескольких типов (такими как моноциты, макрофаги, дендриты или В-клетки) продукцию цитокинов (например, ИФН-α, ФНО-α, ИЛ-1, ИЛ-6, ИЛ-10 и ИЛ-12) до уровня, превышающего ее базальную величину. Точное эффективное количество варьирует в зависимости известных на настоящее время факторов, но достаточно, чтобы обеспечить терапевтическую дозу данного соединения в диапазоне от 100 нг/кг до 50 мг/кг, предпочтительно от 10 мкг/кг до 5 мг/кг. Изобретение относится также к способу терапии вирусной инфекции у животных и к способу лечения новообразований у животных, которые включают введение им терапевтически эффективного количества данного соединения или содержащей его композиции. Количество соединения, обеспечивающее эффективное излечение или подавление вирусной инфекции, - это количество, достаточное, чтобы устранить или уменьшить одно или несколько ее клинических последствий, таких как виремия, вызываемые вирусом поражения, интенсивность репродукции вируса или гибель животного, по сравнению с их тяжестью у контрольных особей. Точное количество варьирует в зависимости от известных на настоящее время факторов, достаточно, чтобы обеспечить терапевтическую дозу данного соединения в диапазоне от 100 нг/кг до 50 мг/кг, предпочтительно от 10 мкг/кг до 5 мг/кг. Количество соединения, обеспечивающее эффективное лечение опухоли, - это количество, достаточное, чтобы уменьшить размер новообразования или число очагов неоплазии. Как и в предыдущем случае, точное количество варьирует в зависимости от известных в настоящее время факторов, но достаточно, чтобы обеспечить терапевтически эффективную дозу данного соединения в диапазоне от 100 нг/кг до 50 мг/кг, предпочтительно от 10 мкг/кг до 5 мг/кг.
Изобретение описывается следующими примерами, которые приводятся только в порядке иллюстрации и ни в коей мере не являются ограничивающими.
Пример 1
2-Бутил-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
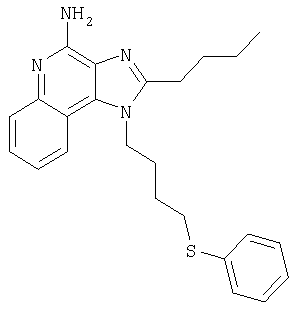
Часть А
Круглодонную колбу помещают на магнитную мешалку и вносят в нее 4-хлоро-3-нитрохинолин (109,70 г, 525,87 ммоля) и дихлорметан (500 мл). К полученному раствору добавляют триэтиламин (79,82 г, 788,81 ммоля) и 4-амино-1-бутанол (46,87 г, 525,87 ммоля). В результата образуется гомогенная жидкость темно-желтого цвета. Реакцию считают завершенной после нагревания раствора в условиях обратного тока на протяжении 30 минут. Раствор охлаждают и разделяют между хлороформом и насыщенным водным раствором аммония хлорида. Образовавшиеся слои отделяют один от другого и однократно экстрагируют водную фракцию хлороформом. Органические фракции объединяют и концентрируют при пониженном давлении, получая 4-[(3-нитрохинолин-4-ил)амино]бутан-1-ол (104,67 г, 400,60 ммоля) в форме твердого вещества темно-желтого цвета. Этот материал в дальнейшем используют без дополнительной очистки.
Часть Б
Круглодонную колбу помещают на магнитную мешалку и вносят в нее 4-[(3-нитрохинолин-4-ил)амино]бутан-1-ол (5,0 г, 19,14 ммоля), триэтиламин (2,91 г, 28,71 ммоля), трет-бутилдиметилсилила хлорид (3,75 г, 24,9 ммоля), 4-диметиламинопиридин (0,10 г) и хлороформ (40 мл), получая раствор темно-желтого цвета. Реакцию считают завершенной после непрерывного перемешивания при комнатной температуре на протяжении 2 часов. Раствор разделяют между этилацетатом и насыщенным водным раствором аммония хлорида. Образовавшиеся слои отделяют один от другого, органическую фракцию промывают насыщенным водным раствором натрия бикарбоната, упаривают над безводным натрия сульфатом, фильтруют и, наконец, концентрируют в условиях пониженного давления, получая N-(4-[трет-бутил(диметил)силил]оксибутил)-3-хинолин-4-амин (6,05 г, 16,11 ммоля), имеющий форму твердого вещества темно-желтого цвета. Этот материал в дальнейшем используют без дополнительной очистки.
Масс-спектрометрия (CI) для С19Н29N3О3Si: m/z 376 (MH+), 342, 210.
Часть В
В сосуд Парра вносят N-(4-[трет-бутил(диметил)силил]оксибутил)-3-хинолин-4-амин (6,05 г, 16,11 ммоля), 5%-ный платиновый реагент на угле (3,0 г) и толуол (32 мл). Сосуд помещают на мешалку Парра и заполняют водородом, создавая давление 50 Пси (3,5 кг/см2). После перемешивания на протяжении 1 часа в реакционную систему вводят дополнительные количества катализатора (3,0 г) и толуола (15 мл). Сосуд снова заполняют водородом до получения давления 50 Пси (3,5 кг/см2) и продолжают перемешивание.
Реакцию считают завершенной по прошествии 1 часа. Катализатор удаляют фильтрованием раствора через фильтровальную бумагу. Оставшийся на фильтре материал дважды промывают толуолом (50 мл), фильтраты объединяют. Летучие компоненты удаляют при пониженном давлении, получая N-(4-[трет-бутил(диметил)силил]оксибутил)хинолин-3,4-диамин (5,57 г, 16,11 ммоля) в форме темной маслянистой жидкости. Этот материал в дальнейшем используют без дополнительной очистки.
Часть Г
Круглодонную колбу помещают на магнитную мешалку и вносят в нее N-(4-[трет-бутил(диметил)силил]оксибутил)хинолин-3,4-диамин (5,57 г, 16,11 ммоля), триметилортовалерат (5,23 г, 32,33 ммоля) и толуол (47 мл). Реакционную систему нагревают таким образом, чтобы достаточно низкая скорость перегонки обеспечивала выведение метанола, образующегося в качестве побочного продукта реакции. Реакцию считают завершенной спустя 15 часов. Реакционную систему охлаждают, а летучие компоненты удаляют в условиях пониженного давлениия, получая 2-бутил-1-(4-[трет-бутил(диметил)силил]оксибутил)-1Н-имидазо[4,5-с]хинолин (4,65 г, 11,30 ммоля), который имеет форму густой маслянистой жидкости темно-желтого цвета. Этот материал в дальнейшем используют без дополнительной очистки.
Масс-спектрометрия (CI) для С24H37N3O3Si: m/z 412 (MH+), 298.
Часть Д
Круглодонную колбу помещают на магнитную мешалку и вносят в нее 2-бутил-1-(4-[трет-бутил(диметил)силил]оксибутил)-1Н-имидазо[4,5-с]хинолин (4,65 г, 11,30 ммоля) и хлороформ (57 мл). В течение 15 минут в полученный раствор отдельными порциями добавляют твердую 3-хлорпербензойную кислоту (2,78 г, 12,43 ммоля), перемешивая реакционную систему при комнатной температуре на протяжении 1 часа.
Затем добавляют еще одну порцию 3-хлорпербензойной кислоты (0,5 г, 2,9 ммоля). Исходные материалы реакции полностью утилизируются спустя 30 минут. Раствор разделяют между хлороформом и насыщенным водным раствором натрия бикарбоната. Образовавшиеся слои отделяют один от другого, органическую фракцию промывают насыщенным водным раствором натрия бикарбоната и солевым раствором, упаривают над безводным натрия сульфатом, фильтруют, а потом концентрируют в условиях пониженного давления, получая 2-бутил-1-(4-[трет-бутил(диметил)силил]оксибутил)-1Н-имидазо[4,5-с]хинолин-5N-оксид (4,83 г, 11,30 ммоля). Этот материал в дальнейшем используют без дополнительной очистки.
Часть Е
Круглодонную колбу помещают на магнитную мешалку и вносят в нее 2-бутил-1-(4-[трет-бутил(диметил)силил]оксибутил)-1Н-имидазо[4,5-с]хинолин-5N-оксид (4,83 г, 11,30 ммоля) и безводный диметилформамид (57 мл) в атмосфере азота. В реакционную систему по каплям добавляют фосфора оксихлорид (1,91 г, 12,43 ммоля), получая, по окончании этой процедуры, гомогенный раствор. Реакцию считают завершенной после его непрерывного перемешивания при комнатной температуре на протяжении 1,5 часа. Раствор разделяют между дихлорметаном и насыщенным водным раствором натрия бикарбоната. Образовавшиеся слои отделяют один от другого и органическую фракцию промывают насыщенным водным раствором натрия бикарбоната и солевым раствором, упаривают над безводным натрия сульфатом, фильтруют, а потом концентрируют в условиях пониженного давления, получая 2-бутил-4-хлоро-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин (3,65 г, 10,42 ммоля).
Масс-спектрометрия (CI) для C18H21Cl2N3: m/z 350 (MH+), 314.
Часть Ж
Круглодонную колбу помещают на магнитную мешалку и вносят в нее 2-бутил-4-хлоро-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин (1,18 г, 3,37 ммоля), бензолтиол (0,56 г, 5,05 ммоля), триэтиламин 0,68 г, 6,74 ммоля) и диметилформамид (15 мл) в атмосфере азота. Реакционную систему нагревают до 80°С, получая гомогенный раствор, который оставляют при 80°С на протяжении 2,5 часов. Анализ с помощью ВЭЖХ свидетельствует об отсутствии в конечном растворе исходных материалов и наличии смеси 2-бутил-4-хлоро-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолина и 2-бутил-4-(фенилтио)-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолина в соотношении 3:1. Раствор охлаждают и разделяют между этилацетатом и насыщенным водным раствором натрия бикарбоната. Образовавшиеся слои отделяют один от другого и органическую фракцию промывают насыщенным водным раствором натрия бикарбоната и солевым раствором, упаривают над безводным натрия сульфатом, фильтруют, а потом концентрируют при пониженном давлении, получая смесь вышеупомянутых соединений в соотношении 3:1 (1,43 г). Этот материал в дальнейшем используют без дополнительной очистки.
Часть З
Смесь 2-бутил-4-хлоро-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолина и 2-бутил-4-(фенилтио)-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолина в соотношении 3:1 (1,38 г) и 7%-ный раствор аммиака в метаноле (30 мл) объединяют в баллоне и нагревают до 150°С. Реакцию считают завершенной по истечении 5 часов. Летучие соединения удаляют упариванием при пониженном давлении, а остаток перемешивают в воде и добавлением натрия карбоната доводят рН смеси до 10. Затем водную смесь трижды экстрагируют хлороформом. Объединенные органические фракции промывают насыщенным водным раствором натрия бикарбоната и солевым раствором, упаривают над безводным натрия сульфатом, фильтруют, а потом концентрируют в условиях пониженного давления, получая твердое кристаллическое вещество желтого цвета. Его навеску (0,8 г) разводят в этилацетате (50 мл) и перегоняют в обратном токе. Добавляют активированный уголь (0,4 г), полученную смесь нагревают в условиях обратного тока на протяжении 5 минут, а затем удаляют уголь фильтрованием через гофрированную фильтровальную бумагу, после которой остается бесцветная жидкость. Эту жидкость концентрируют при пониженном давлении, получая твердый материал. Последний подвергают перекристаллизации из этилацетата и гексана до 2-бутил-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с] хинолин-4-амина (0,51 г, 1,25 ммоля), который имеет форму игольчатых кристаллов белого цвета с температурой плавления 118-120°С. Результаты анализа. Расчетное содержание в C24H28N4S: % С 71,25; % Н 6,98; % N 13,85, найденное содержание: % С 71,12; % Н 6,81;% N 13,62.
1H-ЯМР (300 МГц, ДМСО): δ 8,02 (d, J=8,3 Hz, 1Н), δ 7,61 (d, J=8,3 Hz, 1Н), δ 7,41 (t, J=8,3 Hz, 1Н), δ 7,16-7,30 (m, 6H), δ 6,46 (bs, 2H), δ 4,52 (t, J=7,6 Hz, 2 Н), δ 3,02 (t, J=7,3 Hz, 2 Н), δ 2,89 (t, J=7,8 Hz, 2 Н), δ 1,95 (m, 2 Н), δ 1,75 (m, 4 Н), δ 1,43 (секстет, J=7,3 Hz, 2 Н), δ 0,94 (t, J=7,3 Hz, 3 Н). Масс-спектрометрия (CI) для C24H28N4S: m/z 405 (MH+), 282, 241.
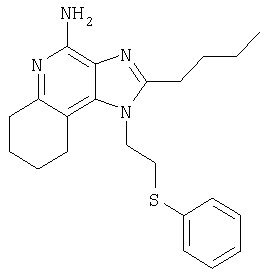
Пример 2
2-Бутил-1-[2-(фенилтио)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина гидрохлорид
Часть А
Круглодонную колбу помещают на магнитную мешалку и вносят в нее 2-(4-амино-2-бутил-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил)этанол (1,0 г, 3,47 ммоля), трет-бутилметилсилилхлорид (1,62 г, 10,75 ммоля), триэтиламин (1,58 г, 15,62 ммоля), 4-диметиламинопиридин (0,1 г) и хлороформ (30 мл). Реакцию считают завершенной после непрерывного перемешивания при 60°С на протяжении 2 часов. Раствор разделяют между этилацетатом и насыщенным водным раствором аммония хлорида. Образовавшиеся слои отделяют один от другого и органическую фракцию промывают насыщенным водным раствором натрия бикарбоната и солевым раствором, упаривают над безводным натрия сульфатом, фильтруют, а потом концентрируют при пониженном давлении, получая смесь 2-бутил-1-(2-[трет-бутил(диметил)силил]оксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина и 2-бутил-N-[трет-бутил(диметил)силил]-1-(2-[трет-бутил(диметил)силил] оксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-аминав соотношении 3:1, в форме темной маслянистой жидкости. Этот материал в дальнейшем используют без дополнительной очистки.
Часть Б
Круглодонную колбу помещают на магнитную мешалку и вносят в нее смесь 2-бутил-1-(2-[трет-бутил(диметил)силил]оксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина и 2-бутил-N-[трет-бутил(диметил)силил]-1-(2-[третбутил(диметил)силил]оксиэтил)-6,7,8,9-тетрагидро- 1Н-имидазо[4,5-с]хинолин-4-амина в соотношении 3:1 (1,6 г) и 1 М уксусную кислоту в дихлорметане (85 мл), получая гомогенный раствор. Реакцию считают завершенной после непрерывного перемешивания при комнатной температуре на протяжении 30 минут. Материал разделяют между хлороформом и солевым раствором. Образовавшиеся слои отделяют один от другого, органическую фракцию промывают насыщенным водным раствором натрия бикарбоната и солевым раствором, упаривают над безводным натрия сульфатом, фильтруют и затем концентрируют при пониженном давлении, получая масло темно-бурого цвета. Полученный таким образом материал подвергают очистке посредством хроматографии на силикагеле, используя для элюции смесь дихлорметана, метанола и гидроокиси аммония [14,8 М в воде] в соотношении 95:4:1. В результате получают 2-бутил-1-(2-[третбутил(диметил)силил]оксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин, который имеет форму бесцветного масла (1,24 г, 3,10 ммоля).
Часть В
Круглодонную колбу помещают на магнитную мешалку и вносят в нее 2-бутил-1-(2-[трет-бутил(диметил)силил]оксиэтил)-6,7,8,9-тетрагидро1Н-имидазо[4,5-с]хинолин-4-амин (0,83 г, 2,06 ммоля), ди-трет-бутила дикарбонат (1,79 г, 8,24 ммоля), триэтиламин (0,52 г, 5,15 ммоля), 4-диметиламинопиридин (0,1 г) и безводный тетрагидрофуран (21 мл) в атмосфере азота. Реакционную систему нагревают до 60°С, получая гомогенный раствор, который оставляют при указанной температуре на протяжении 2,5 часов. По истечении этого времени реакцию считают завершенной. Полученный материал охлаждают до комнатной температуры и добавляют в него 1 М раствор тетрабутиламмония фтората в тетрагидрофуране (2,27 мл, 2,27 ммоля). Эта реакция завершается после непрерывного перемешивания при комнатной температуре в течение 30 минут. Раствор разделяют между этилацетатом и насыщенным водным раствором аммония хлорида. Образовавшиеся слои отделяют один от другого, органическую фракцию промывают насыщенным водным раствором натрия бикарбоната и солевым раствором, упаривают над безводным натрия сульфатом, фильтруют и затем концентрируют в условиях пониженного давления, получая твердое вещество светло-желтого цвета. Этот материал очищают посредством хроматографии на силикагеле, используя для элюции смесь дихлорметана, метанола и гидроокиси аммония [14,8 М в воде] в соотношении 95:4:1. В результате получают ди(трет-бутил)-2-бутил-1-(2-гидроксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-илимидодикарбонат в форме прозрачной смолы (0,55 г, 1,13 ммоля).
Часть Г
Круглодонную колбу помещают на магнитную мешалку и вносят в нее ди(трет-бутил)-2-бутил-1-(2-гидроксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-илимидодикарбонат (0,55 г, 1,13 ммоля) и безводный дихлорметан (11 мл) в атмосфере азота.
Полученный гомогенный раствор охлаждают до -10°С в низкотемпературной бане с метанолом. К охлажденному раствору добавляют триэтиламин (0,23 г, 2,26 ммоля) и метансульфонила хлорид (0,19 г, 1,70 ммоля). Реакцию считают завершенной после непрерывного перемешивания при -15°С на протяжении 15 минут. Раствор разделяют между этилацетатом и насыщенным водным раствором аммония хлорида. Образовавшиеся слои отделяют один от другого, органическую фракцию промывают насыщенным водным раствором натрия бикарбоната и солевым раствором, упаривают над безводным натрия сульфатом, фильтруют и затем концентрируют при пониженном давлении, получая 2-4-[бис(трет-бутоксикарбонил)амино]-2-бутил-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-илэтилметансульфонат в форме твердого смолообразного вещества желтого цвета (0,61 г, 1,08 ммоля). Этот материал в дальнейшем используют без дополнительной очистки. Масс-спектрометрия (CI) для C27H42N4O7S: m/z 567 (МН+), 467, 367, 271.
Часть Д
Круглодонную колбу помещают на магнитную мешалку и вносят в нее 2-4-[бис(трет-бутоксикарбонил)амино]-2-бутил-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-илэтилметансульфонат (0,61 г, 1,08 ммоля), бензолтиол (0,21 г, 1,88 ммоля), триэтиламин (0,25 г, 2,43 ммоля) и безводный диметила формамид (11 мл) в атмосфере азота.
Реакционную систему нагревают до 80°С, получая гомогенный раствор желтого цвета, который оставляют при 80°С на протяжении 2,5 часов. По истечении этого времени реакцию считают завершенной. Полученный материал разделяют между этилацетатом и насыщенным водным раствором натрия бикарбоната. Образовавшиеся слои отделяют один от другого, органическую фракцию промывают насыщенным водным раствором натрия бикарбоната и солевым раствором, упаривают над безводным натрия сульфатом, фильтруют и затем концентрируют при пониженном давлении, что приводит к образованию вещества в форме желтого масла. Этот материал подвергают очистке посредством хроматографии на силикагеле в смеси дихлорметана и метанола (в соотношении 95:5), получая ди(трет-бутил)-2-бутил-1-[2-(фенилтио)этил]- 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-илимидодикарбонат в форме маслянистой жидкости светло-желтого цвета (0,54 г, 0,93 ммоля). Масс-спектрометрия (CI) для С32Н44Н4O48: m/z 581 (МН+), 481, 381, 245.
Часть Е
Круглодонную колбу помещают на магнитную мешалку и вносят в нее ди(трет-бутил)-2-бутил-1-[2-(фенилтио)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-илимидодикарбонат (0,50 г, 0,86 ммоля), 4 М раствор соляной кислоты в диоксане (5 мл) и дихлорметан (5 мл). Реакцию считают завершенной после непрерывного перемешивания реакционной смеси при комнатной температуре на протяжении 2 часов. Летучие соединения упаривают при пониженном давлении, получая в остатке твердое вещество белого цвета. Этот материала подвергают перекристаллизации из ацетонитрила, в результате которой образуются 2-бутил-1-[2-(фенилтио)этил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-илимидодикарбонат в форме рыхлых игольчатых кристаллов белого цвета с температурой плавления 237-238°С. Результаты анализа. Расчетное содержание в C22H28N4S·(H2O)1/4·(HCl)2: % С 57,70; % Н 6,71; % N 12,23, найденное содержание: % С 57,62; % Н 6,57; % N 12,41.
1Н-ЯМР (300 МГц, ДМСО): δ 7,81 (bs, 2H), δ 7,22-7,39 (m, 5H), 3 4,64 (t, J=6,8 Hz, 2Н), δ 2,75 (m, 6H), δ 1,71 (m, 6H), δ 1,34 (секстет, J=7,3 Hz, 2Н), δ 0,89 (t, J=7,3, Hz, 3 Н). Масс-спектрометрия (Cl) для C22H28N4S·(H2O)1/4·(HCl)2: m/z 381 (МН+), 245, 137.
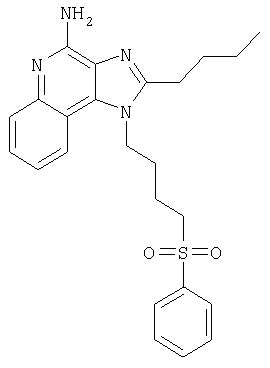
Пример 3
2-Бутил-1-[4-(фенилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
С помощью общего способа, описанного в примере 1, часть Д, 2-бутил-1-(4-[трет-бутил(диметил)силил]оксибутил)-1Н-имидазо[4,5-с]хинолин (16,0 г, 38,87 ммоля) окисляли до 2-бутил-1-(4-[трет-бутил(диметил)силил]оксибутил)-1Н-имидазо[4,5-с]хинолин-5N-оксида (16,61 г, 38,87 ммоля), который выделяли без дополнительной очистки в форме твердого вещества желтовато-коричневого цвета.
Часть Б
Круглодонную колбу помещали на магнитную мешалку и вносили в нее 2-бутил-1-(4-[трет-бутил(диметил)силил]оксибутил)-1Н-имидазо[4,5-с]хинолин-5N-оксида (16,0 г, 38,87 ммоля), 14,8 М раствор гидроокиси аммония в воде (75 мл) и хлороформ (200 мл). В полученную систему при энергичном перемешивании добавляли отдельными порциями п-толуола хлорид (8,15 г, 42,76 ммоля). Эта процедура сопровождалась слабой экзотермической реакцией. Ее считали завершенной после непрерывного перемешивания при комнатной температуре на протяжении 10 минут. Раствор разделяли между хлороформом и насыщенным водным раствором натрия бикарбоната. Образовавшиеся слои отделяли один от другого, органическую фракцию промывали насыщенным водным раствором натрия бикарбоната и солевым раствором, упаривали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая твердое вещество беловатого цвета. Этот материал мелко растирали в этиловом эфире, а затем отфильтровывали, получая 2-бутил-1-(4-[трет-бутил(диметил)силил]оксибутил)-1Н-имидазо[4,5-с]хинолин-4-амина (9,3 г, 21,80 ммоля), который имеет форму мелкого порошка белого цвета. Этот материал в дальнейшем использовали без дополнительной очистки.
Часть В
Круглодонную колбу помещали на магнитную мешалку и вносили в нее смесь 2-бутил-1-(4-[трет-бутил(диметил)силил]оксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амин (9,2 г, 21,56 ммоля), 1 М раствор тетрабутиламмония фтората (23,72 мл, 23,72 ммоля) и безводный тетрагидрофуран (100 мл). В результате получали гомогенный раствор светло-оранжевого цвета. Реакцию считали завершенной после перемешивания при комнатной температуре на протяжении 1 часа.
Добавление воды (100 мл) в условиях непрерывного перемешивания вызывало слабую экзотермическую реакцию. Летучие компоненты удаляли упариванием при пониженном давлении до выпадения твердого осадка.
Последний собирали посредством фильтрования и промывали водой (20 мл) и ацетоном (20 мл), получая твердое вещество белого цвета. Этот материал растирали в порошок в этиловом эфире (50 мл), а затем отфильтровывали.
Остаток представлял собой 4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутанол (6,12 г, 19,59 ммоля), имеющий форму твердого вещества белого цвета с температурой плавления 184-186°С.
Результаты анализа. Расчетное содержание в С18Н24N4О: % С 69,20; % Н 7,74; % N 17,93, найденное содержание: % С 69,05; % Н 8,02; % N 18,03. Масс-спектрометрия (CI) для C18H24N4): m/z 313 (MH+).
Часть Г
Круглодонную колбу помещали на магнитную мешалку и вносили в нее 4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутанол (7,3 г, 23,37 ммоля), триэтиламин (3,55 г, 35,06 ммоля) и безводный диметилформамид (93 мл) в атмосфере азота. В полученный раствор при непрерывном перемешивании добавляли по каплям фосфора оксихлорид (3,94 г, 25,70 ммоля). Эта процедура индуцировала слабую экзотермическую реакцию, которая приводила к образованию гетерогенной смеси темно-желтого цвета. Нагревание реакционной смеси до 60°С давало гомогенный раствор, который оставляли при указанной температуре на срок до 5 часов. В течение этого периода происходила полная утилизация исходных материалов реакции. Летучие компоненты удаляли упариванием при пониженном давлении до образования масла темно-бурого цвета.
Полученный материал разделяли между хлороформом и насыщенным водным раствором натрия бикарбоната. Образовавшиеся слои отделяли один от другого и водную фракцию однократно экстрагировали хлороформом. Органические фракции объединяли и удаляли из них летучие соединения упариванием при пониженном давлении. В результате получали смесь N'-[2-бутил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-ил]-N,N-диметилимидоформамида и 2-бутил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-амина (7,70 г) в форме твердого вещества беловатого цвета. Этот материал в дальнейшем использовали без дополнительной очистки.
Часть Д
Круглодонную колбу помещали на магнитную мешалку и вносили в нее смесь N'-[2-бутил-1-(4-хлоробутил-1Н-имидазо[4,5-с]хинолин-4-ил]-N,N-диметилимидоформамида и 2-бутил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-амина (1,3 г), натриевую соль бензолсульфиновой кислоты (1,67 г, 10,11 ммоля) и безводный диметилформамид (15 мл) в атмосфере азота. Полученный таким образом раствор нагревали до 100°С, что приводило к образованию гомогенной жидкости. Ее оставляли при указанной температуре на протяжении 90 часов. В течение этого периода происходила полная утилизация исходных материалов реакции. Полученный раствор разделяли между хлороформом и водой. Образовавшиеся слои отделяли один от другого, органическую фракцию промывали насыщенным водным раствором натрия бикарбоната и солевым раствором, упаривали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая смолообразное вещество темно-желтого цвета. Этот материал растворяли в метаноле (20 мл) и 4 М растворе соляной кислоты в диоксане (3,02 мл, 12,1 ммоля). Полученную жидкость светло-оранжевого цвета непрерывно перемешивали при комнатной температуре на протяжении 12 часов, после чего считали реакцию завершенной. Летучие соединения удаляли упариванием при пониженном давлении, получая смолистое вещество светло-желтого цвета. Этот материал разделяли между хлороформом и насыщенным водным раствором натрия бикарбоната.
Образовавшиеся слои отделяли один от другого и водную фракцию однократно экстрагировали хлороформом. Органические фракции объединяли и промывали солевым раствором, упаривали над безводным натрия сульфатом, фильтровали и затем концентрировали при пониженном давлении, получая твердое вещество светло-желтого цвета. Полученный таким образом материал очищали посредством хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 95:5. В результате получали твердое вещество беловатого цвета. Его навеску (0,63 г) разводили в этилацетате (50 мл) и перегоняли в обратном токе. Добавляли активированный древесный уголь (0,6 г) и нагревали полученную смесь в условиях обратного тока. Уголь отделяли фильтрованием через гофрированную фильтровальную бумагу. Фильтрат представлял собой бесцветную жидкость. Ее концентрировали при пониженном давлении, а твердый остаток подвергали перекристаллизации из этилацетата и гексана. Конечный материал состоял из 2-бутил-1-[4-(фенилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина (0,37 г, 0,85 ммоля) в форме рыхлого твердого вещества белого цвета, имеющего температуру плавления 179-180°С.
Результаты анализа. Расчетное содержание в C24H28N4O2S: % C 66,03; % Н 6,46; % N 12,83, полученное содержание: % C 65,88; % Н 6,49; % N 12,76.
1H-ЯМР (300 МГц, ДМСО): δ 7,98 (d, J=8,3, Hz, 1H), δ 7,82 (m, 2H), δ 7,73 (d, J=6,8 Hz, 1 H), δ 7,62 (m, 3 H), δ 7,41 (t, J=7,6 Hz, 1 H), δ 7,22 (t, J=7,6 Hz, 1 H), δ 6,45 (bs, 2 H), δ 4,51 (t, J=7,3 Hz, 2 H), δ 3,90 (t, J=7,8 Hz, 2 H), δ 2,86 (t, J=7,6 Hz, 3H), δ 1,69-1,90 (m, 6 H), δ 1,43 (секстет, J=7,3 Hz, 2 H), δ 0,95 (t, J=7,3, Hz, 3 H). Масс-спектрометрия (CI) для C24H28N4O2S: m/z 437 (MH+), 295.
Пример 4
2-Бутил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин

Часть А
Круглодонную колбу помещали на магнитную мешалку и вносили в нее смесь N'-[2-бутил-1-(4-хлоробутил-1Н-имидазо[4,5-с]хинолин-4-ил]-N,N-диметилимидоформамида и 2-бутил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-амина в соотношении 2:1 (6,17 г), 4 M раствор соляной кислоты в диоксане (21,15 мл, 84,56 ммоля) и метанол (200 л), получая раствор светло-оранжевого цвета. Реакцию считали завершенной после непрерывного перемешивания раствора при комнатной температуре на протяжении 43 часов. Летучие компоненты удаляли упариванием при пониженном давлении, а образовавшийся твердый остаток светло-желтого цвета разделяли между хлороформом и насыщенным водным раствором натрия бикарбоната. Образовавшиеся слои отделяли один от другого и водную фракцию однократно экстрагировали хлороформом. Органические фракции объединяли, высушивали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая 2-бутил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-амин (4,65 г, 14,05 ммоля), который имел форму твердого вещества беловатого цвета. Этот материал в дальнейшем использовали без дополнительной очистки. Масс-спектрометрия (CI) для С18H23ClN4: m/z 331 (МН+), 295.
Часть Б
Круглодонную колбу помещали на магнитную мешалку и вносили в нее 2-бутил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-амин (1,5 г, 4,53 ммоля), натрия тиометоксид (0,48 г, 6,80 ммоля) и безводный диметилформамид (18 мл) в атмосфере азота. Реакционную систему нагревали при 60°С на протяжении 16 часов. В течение этого времени происходила полная утилизация исходных материалов реакции. Раствор охлаждали и разделяли между хлороформом и водой. Образовавшиеся слои отделяли один от другого и органическую фракцию промывали насыщенным водным раствором натрия бикарбоната. Объединенные водные фракции однократно экстрагировали хлороформом. Органические фракции объединяли и промывали солевым раствором, упаривали над безводным натрия сульфатом, фильтровали, а затем концентрировали в условиях пониженного давления, получая масло темно-бурого цвета. Полученный таким образом материал очищали посредством хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 90:10. В результате получали твердое вещество светло-желтого цвета. Его подвергали перекристаллизации из диметилформамида и воды с образованием 2-бутил-1-[(4-метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина (0,83 г, 2,42 ммоля), который имел форму игольчатых кристаллов светло-желтого цвета с температурой плавления 127-130°С.
Результаты анализа. Расчетное содержание в C19H26N4S: % С 66,63; % Н 7,65; % N 16,36, полученное содержание: % С 66,88; % Н 7,53; % N 16,35.
1Н-ЯМР (500 МГц, ДМСО): δ 8,04 (d, J=8,3, Hz, 1H), δ 7,61 (m, 2H), (t, J=8,3 Hz, 1Н), δ 7,25 (t, J=8,3 Hz, 1Н), δ 6,43 (bs, 2 Н), δ 4,52 (t, J=7,6 Hz, 2 Н), δ 2,92 (t, J=7,8 Hz, 2 Н), δ 2,53 (t, J=7,6 Hz, 3 Н), δ 2,01 (s, 3 Н), δ 1, 90 (m, 2 Н), δ 1,80 (p, J=7,8 Hz, 2 Н), δ 1,71 (p, J=7,3 Hz, 2 Н), δ 1,46 (секстет, J=7,3 Hz, 2 Н), δ 0,96 (t, J=7,3, Hz, 3 Н). Масс-спектрометрия (CI) для C19H26N4O2S: m/z 343 (MH+), 295, 241.
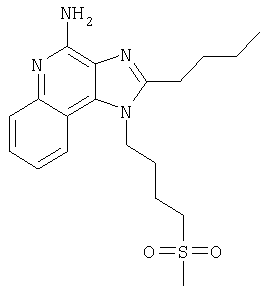
Пример 5
2-Бутил-1-[(4-метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
Круглодонную колбу помещали на магнитную мешалку и вносили в нее 2-бутил-1-[(4-метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин (1,2 г, 3,50 ммоля) и хлороформ (18 мл). В полученный раствор отдельными порциями на протяжении 15 минут добавляли твердую 3-хлорпербензойную кислоту (1,72 г, 7,71 ммоля). Реакцию считали завершенной после непрерывного перемешивания раствора при комнатной температуре в течение 5 минут. Раствор разделяли между хлороформом и 1%-ным водным раствором натрия карбоната. Образовавшиеся слои отделяли один от другого, органическую фракцию промывали солевым раствором, высушивали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая твердое вещество светло-коричневого цвета. Полученный таким образом материал подвергали очистке посредством хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 90:10. В результате получали твердое вещество беловатого цвета. Его подвергали перекристаллизации из ацетонитрила и воды с образованием 2-бутил-1-[(4-метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина (0,61 г, 1,63 ммоля), который имел форму иглообразных кристаллов беловатого цвета, имеющих температуру плавления 164-165°С.
Результаты анализа. Расчетное содержание в C19H26N4O2S: % С 60,94; % Н 7,00; % N 14,96, полученное содержание: % С 60,70; % Н 6,94; % N 14,94.
1H-ЯМР (300 МГц, ДМСО): δ 8,03 (d, J=8,3, Hz, 1Н), δ 7,61 (d, J=8,3 Hz, 1Н), 7,42 (t, J=8,3 Hz, 1Н), δ 7,26 (t, J=8,3 Hz, 1Н), δ 6,46 (bs, 2 Н), δ 4,56 (t, J=7,6 Hz, 2 Н), δ 3,21 (t, J=7,3 Hz, 2 Н), δ 2,96 (s, 3 Н), δ 2, 93 (t, J=7,8 Hz, 2 Н), δ 1,91 (m, 4 Н), δ 1,81 (p, J=7,3 Hz, 2 Н), δ 1,45 (секстет, J=7,3 Hz, 2 Н), δ 0,96 (t, J=7,3 Hz, 3 Н). Масс-спектрометрия (CI) для C19H26N4O2S: m/z 375 (MH+), 295.
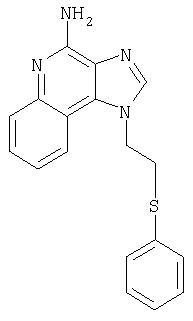
Пример 6
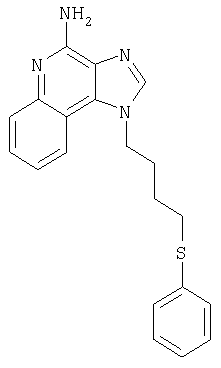
1-[2-(Фенилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
Круглодонную колбу помещали на магнитную мешалку и вносили в нее 2-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)этанол (8,46 г, 37,06 ммоля) и тионила хлорид (68,99 г, 57,99 ммоля) в атмосфере азота. Реакционную систему нагревали до 80°С, получая гетерогенный раствор, который оставляли при указанной температуре на протяжении 2 часов. В течение этого времени происходила полная утилизация исходных материалов реакции. Раствор охлаждали и останавливали реакцию добавлением воды (400 мл). Затем в раствор при непрерывном перемешивании добавляли твердый натрия карбонат, доводя его рН до 10,0. При этом выпадал твердый осадок. Последний собирали посредством фильтрования. Он состоит из 1-(2-хлороэтил)-1Н-имидазо[4,5-с]хинолин-4-амина (7,86 г, 31,86 ммоля) в форме твердого вещества беловатого цвета. Полученный таким образом материал в дальнейшем использовали без дополнительной очистки.
Часть Б
Круглодонную колбу помещали на магнитную мешалку и вносили в нее 1-(2-хлороэтил)-1Н-имидазо[4,5-с]хинолин-4-амин (2,0 г, 8,11 ммоля), натрия бензолтиолат (1,79 г, 12,16 ммоля) и безводный диметилсульфоксид (40 мл) в атмосфере азота. Реакционную систему нагревали до 100°С, получая гомогенный раствор, который оставляли при указанной температуре на протяжении 30 мин. В течение этого времени происходила полная утилизация исходных материалов реакции. Горячий раствор переливали в воду (300 мл) при энергичном перемешивании, что вызывало выпадение твердого осадка. Последний собирали посредством фильтрования. Он состоял из 1-[2-(фенилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амина (2,08 г, 6,49 ммоля) в форме порошка беловатого цвета, имеющего температуру плавления 233-235°С.
Результаты анализа. Расчетное содержание в C18H16N4S: % С 67,47; % Н 5,03; % N 17,49, полученное содержание: % С 67,20; % Н 4,95;% N 17,52.
1Н-ЯМР (300 МГц, ДМСО): δ 8,14 (s, 1Н), δ 7,76 (d, J=8,3 Hz, 1Н), δ 7,60 (t, J=8,3 Hz, 1Н), δ 7,28-7,44 (m, 6 Н), δ 7,12 (t, J=8,3 Hz, 1Н), δ 6,58 (bs, 2 Н), δ 7,49 (t, J=6,8 Hz, 2 Н), δ 3,48 (t, J=6,8 Hz, 2 Н). Масс-спектрометрия (CI) для C18H16N4S: m/z 321 (MH+), 185, 137.
Пример 7
1-[4-(Фенилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
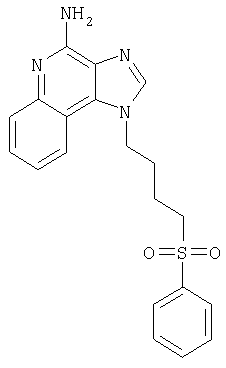
Часть А
Круглодонную колбу помещали на магнитную мешалку и вносили в нее N,N-дибензил-1Н-имидазо[4,5-с]хинолин-4-амин (20,0 г, 55,04 ммоля), натрия гидрид (3,3 г 60%-ной дисперсии, 82,56 ммоля) и безводный диметилформамид (275 мл) в атмосфере азота. После непрерывного перемешивания реакционной смеси при комнатной температуре на протяжении 2 часов в раствор добавляли 4-хлоро-1-йодобутират (19,23 г, 88,06 ммоля), а полученную гомогенную жидкость снова перемешивали при комнатной температуре в течение 48 часов. При этом происходила полная утилизация исходных материалов реакции. Раствор разделяли между этилацетатом и водой. Образовавшиеся слои отделяли один от другого, органическую фракцию промывали насыщенным водным раствором натрия бикарбоната и солевым раствором, высушивали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая твердое вещество светло-желтого цвета. Полученный таким образом материал подвергали перекристаллизации из этилацетата и гексана, которая приводила к образованию N,N-дибензил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-амина (20,7 г, 45,49 ммоля), который имел форму игольчатых кристаллов белого цвета. Масс-спектрометрия (CI) для C28H27ClN4: m/z 455 (МН+), 365, 329, 239.
Часть Б
Круглодонную колбу помещали на магнитную мешалку и вносили в нее N,N-дибензил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-амина (7,0 г, 15,38 ммоля), натрия бензолтиолат (3,46 г, 26,15 ммоля) и безводный диметилформамид (77 мл) в атмосфере азота. Реакционную систему нагревали до 60°С, получая гетерогенную смесь, которую оставляли при указанной температуре на протяжении 4 часов. В течение этого времени происходила полная утилизация исходных материалов реакции. Охлажденный раствор разделяли между этилацетатом и водой. Образовавшиеся слои отделяли один от другого, органическую фракцию промывали насыщенным водным раствором натрия бикарбоната и солевым раствором, высушивали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая конечный продукт, который имел форму бесцветного масла. Этот материал подвергали очистке посредством хроматографии на силикагеле в дихлорметане, которая приводила к выходу N,N-дибензил-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]-4-амина (7,5 г, 14,19 ммоля) в форме бесцветного масла. Масс-спектрометрия (CI) для С34Н32N4S: m/z 529 (МН+), 439, 349.
Часть В
Круглодонную колбу помещали на магнитную мешалку и вносили в нее N,N-дибензил-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин (3,64 г, 6,88 ммоля) и хлороформ (34 мл). В полученный раствор отдельными порциями, на протяжении 5 минут, добавляли твердую 3-хлоропербензойную кислоту (3,39 г, 1,14 ммоля). Реакцию считали завершенной после непрерывного перемешивания раствора при комнатной температуре в течение 5 минут. Раствор разделяли между хлороформом и 1%-ным водным раствором натрия карбоната. Образовавшиеся слои отделяли один от другого, органическую фракцию промывали солевым раствором, высушивали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая смолообразное вещество красного цвета. Полученный таким образом материал подвергали очистке посредством хроматографии на силикагеле, используя для элюции дихлорметан. В результате получали N,N-дибензил-1-[4-(фенилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин (2,85 г, 5,08 ммоля) в форме смолообразного вещества розового цвета. Масс-спектрометрия (CI) для C34H32N4O2S: m/z 561 (МН+), 471, 381.
Часть Г
Круглодонную колбу помещали на магнитную мешалку и вносили в нее N,N-дибензил-1-[4-(фенилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин (10,0 г, 1,78 ммоля), трифловую кислоту (2,68 г, 17,83 ммоля) и безводный дихлорметан (14 мл) в атмосфере азота. Реакцию считали завершенной после непрерывного перемешивания раствора при комнатной температуре в течение 24 часов.
Раствор разделяли между хлороформом и избыточным количеством 20%-ного водного раствора гидроокиси натрия. Образовавшиеся слои отделяли один от другого, водную фракцию трижды экстрагировали хлороформом.
Органические фракции объединяли и концентрировали при пониженном давлении, получая твердое вещество светло-коричневого цвета.
Полученный таким образом материал очищали посредством хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 90:10. В результате получали мелкий порошок белого цвета.
Продукт его перекристаллизации из ацетонитрила состоял из 1-[4-(фенилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина (0,32 г, 0,84 ммоля), который имел форму игольчатых кристаллов белого цвета с температурой плавления 175-177°С.
Результаты анализа. Расчетное содержание в C20H20N4O2S: % С 63,14; % Н 5,30; % N 14,73, полученное содержание: % С 63,14; % Н 5,24; % N 14,77.
1H-ЯМР (300 МГц, ДМСО): δ 8,15 (s, 1Н), δ 8,01 (d, J=8,3 Hz, 1Н), δ 7,80 (m, 2 Н), 7,7 (m, 1Н), δ 7,60 (m, 3 Н), δ 7,44 (t, J=8,3 Hz, 1H), δ 7,24 (m, 6 H), (t, J=8,3 Hz, 1 H), δ 6,59 (bs, 2 H), δ 4,59 (t, J=6,8 Hz, 2 H), δ 3,38 (t, J=7,8 Hz, 2 H), δ 1,93 (m, 2 H), δ 1,58 (m, 2 H).
Масс-спектрометрия (CI) для C20H20N4O2S: m/z 381 (MH+), 239.
Пример 8
1-[4-(Метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
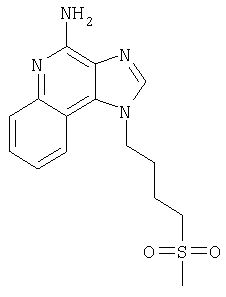
Часть А
С помощью общего способа, описанного в примере 7, часть Б, используя натрия тиометоксид (1,16 г, 16,48 ммоля), N,N-дибензил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-амин (5,0 г, 10,99 ммоля) преобразовывали в N,N-дибензил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин. Полученный материал подвергали очистке посредством хроматографии на силикагеле в системе гексан-этилацетат в соотношении 80:20. Продуктом хроматографии являлось бесцветное масло (4,91 г, 10,52 ммоля). Масс-спектрометрия (CI) для C29H30N4S: m/z 467 (МН+), 377, 287, 185.
Часть Б
С помощью общего способа, описанного в примере 7, часть В, N,N-дибензил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин (4,91 г, 15,52 ммоля) окисляли до N,N-дибензил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина, который подвергали очистке посредством хроматографии на силикагеле в системе гексан-этилацетат в соотношении 80:20. Продуктом хроматографии являлось твердое вещество светло-оранжевого цвета (4,53 г, 9,08 ммоля). Масс-спектрометрия (CI) для С29Н30N4O2S: m/z 499 (МН+), 409, 319.
Часть В
С помощью общего способа, описанного в примере 7, часть Г, N,N-дибензил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин (4,53 г, 9,08 ммоля) преобразовывали в 1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин (1,16 г, 16,48 ммоля). Полученный материал подвергали перекристаллизации из метанола и воды, которая приводила к образованию 1-[4-(метилсульфонил)бутил]-1Н-имидазо [4,5-с]хинолин-4-амина, имевшего форму иглообразных кристаллов белого цвета (1,33 г, 4,18 ммоля), имеющих температуру плавления 203-204°С.
Результаты анализа. Расчетное содержание в C15H18N4O2S: % С 56,58; % Н 5,70; % N 17,60, полученное содержание: % С 56,33; % Н 5,63;% N 17,41.
1H-ЯМР (300 МГц, ДМСО): δ 8,22 (s, 1Н), δ 8,06 (d, J=8,3, Hz, 1Н), δ 7,62 (d, J=8,3 Hz, 1Н), δ 7,45 (t, J=8,3 Hz, 1H), δ 7,27 (t, J=8,3 Hz, 1Н), δ 6,59 (bs, 2 Н), δ 4,65 (t, J=6,8 Hz, 2 Н), δ 3,19 (t, J=7,8 Hz, 2 Н), δ 2,93 (s, 3 Н), δ 1,99 (m, 2 Н), δ 1,74 (m, 2 Н).
Масс-спектрометрия (CI) для C15H18O2S: m/z 319 (MH+), 239.
Пример 9
1-[4-(Фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
С помощью общего способа, описанного в примере 1, часть Г, используя триэтилортоформиат (65,11 г, 439,35 ммоля), осуществляли циклизацию N-(4-[трет-бутил(диметил)силил]оксибутил)хинолин-3,4-диамина (101,21 г, 292,90 ммоля) до 1-(4-[трет-бутил(диметил)силил]оксибутил-1Н-имидазо [4,5-с]хинолина.
Полученный продукт (65,11 г, 439,35 ммоля) выделяли в форме масла бурого цвета и в дальнейшем использовали без дополнительной очистки.
Часть Б
С помощью общего способа, описанного в примере 1, часть Д, 1-(4-[трет-бутил(диметил)силил]оксибутил-1Н-имидазо[4,5-с]хинолин (42,2 г, 118,69 ммоля) окисляли до 1-(4-[трет-бутил(диметил)силил]оксибутил-1Н-имидазо[4,5-с]хинолин-5N-оксида (44,10 г, 118,69 ммоля), который выделяли в форме твердого вещества желто-коричневого цвета и в дальнейшем использовали без дополнительной очистки.
Часть В
С помощью общего способа, описанного в примере 3, часть Б, 1-(4-[трет-бутил(диметил)силил]оксибутил-1Н-имидазо[4,5-с]хинолин 5N-оксид (44,10 г, 118,69 ммоля) аминировали до 1-(4-[трет-бутил(диметил)силил]оксибутил-1Н-имидазо[4,5-с]хинолин-4-амина.
Полученный материал растирали в порошок в этиловом эфире и собирали посредством фильтрования в виде твердого вещества светло-коричневого цвета (21,54 г, 58,12 ммоля), которое в последующем использовали без дополнительной очистки.
Часть Г
С помощью общего способа, описанного в примере 3, часть В, 1-(4-[трет-бутил(диметил)силил]оксибутил-1Н-имидазо[4,5-с]хинолин-4-амин (21,5 г, 58,02 ммоля) преобразовывали в 4-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)бутан-1-ол. Полученный материал растирали в порошок в холодном метаноле и собирали посредством фильтрования в виде твердого вещества (13,92 г, 54,30 ммоля), которое в последующем использовали без дополнительной очистки. Масс-спектрометрия (CI) для C14H16N4O: m/z 257 (MH+), 185.
Часть Д
С помощью общего способа, описанного в примере 6, часть А, 4-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)бутан-1-ол (5,0 г, 19,51 ммоля) хлорировали для получения 1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-амина (4,92 г, 17,91 ммоля), который выделяли без дополнительной очистки в форме твердого вещества беловатого цвета.
Часть Е
С помощью общего способа, описанного в примере 6, часть Б (но понизив температуру до 80°С), 1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-амин (1,5 г, 5,46 ммоля) преобразовывали в 1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин. Полученное твердое вещество (1,53 г) разводили в ацетонитриле (90 мл) и перегоняли в обратном токе. Добавляли активированный древесный уголь (0,9 г) и в течение 5 минут нагревали полученную смесь в условиях обратного тока. Уголь отделяли фильтрованием через гофрированную фильтровальную бумагу. Фильтрат представлял собой бесцветную жидкость, содержавшую 1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин (0,86 г, 2,47 ммоля) в форме игольчатых кристаллов белого цвета с температурой плавления 158-160°С.
Пример 10
1-[4-(Метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
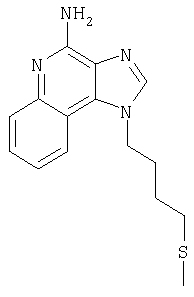
Часть А
С помощью общего способа, описанного в примере 6, часть Б (но понизив температуру до 80°С), 1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-амин (1,5 г, 5,46 ммоля) преобразовывали в 1-[4-[(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин, используя для этой цели реакцию с натрия тиометоксидом (0,88 г, 12,56 ммоля) вместо натрия бензолтиолата. Полученное твердое вещество (1,26 г) разводили в ацетонитриле (40 мл) и перегоняли в обратном токе. Добавляли активированный древесный уголь (0,7 г) и в течение 5 минут нагревали полученную смесь в условиях обратного тока.
Уголь отделяли фильтрованием через гофрированную фильтровальную бумагу. Фильтрат представлял собой бесцветную жидкость, которую концентрировали при пониженном давлении, получая твердый остаток.
Последний подвергали перекристаллизации из ацетонитрила. Искомое соединение, т.е. 1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин (0,66 г, 2,30 ммоля), выделяли в форме игольчатых кристаллов белого цвета с температурой плавления 163-164°С.
Результаты анализа. Расчетное содержание в C15H18N4S: % С 62,91; % Н 6,34; % N 19,56, полученное содержание: % С 62,70; % Н 6,19; % N 19,45.
1Н-ЯМР (300 МГц, ДМСО): δ 8,212 (s, 1Н), δ 8,06 (d, J=8,3 Hz, 1Н), δ 7,62 (d, J=8,3 Hz, 1Н), δ 7,44 (t, J=8,3 Hz, 1Н), δ 7,26 (t, J=8,3 Hz, 1Н), δ 6,59 (bs, 2 Н), δ 4,62 (t, J=7,6 Hz, 2 Н), δ 2,50 (t, J=6,8 Hz, 2 Н), δ 1,99 (s, 3 Н), δ 1,95 (p, J=7,3 Hz, 2 Н), δ 1,59 (p, J=7,3 Hz, 2 Н). Масс-спектрометрия (CI) для C15H18N4S: m/z 287 (MH+), 185.
Пример 11
2-Бутил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин
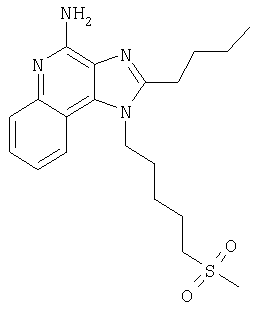
Часть А
С помощью общего способа, описанного в примере 1, часть А, 4-хлоро-3-нитрохинолин (107,7 г, 525,87 ммоля) преобразовывали в 5-[3-(нитрохинолин-4-ил)амино]пентан-1-ол, используя для это цели амино-1-пентанол (79,82 г, 788,81 ммоля) вместо 4-аминобутанола. Полученное твердое вещество темно-желтого цвета (117,22 г, 425,77 ммоля) в дальнейшем использовали без дополнительной очистки.
Масс-спектрометрия (CI) для C14H17N4O3: m/z 276(MH+), 224.
Часть Б
Круглодонную колбу помещали на магнитную мешалку и вносили в нее 5-[(3-нитрохинолин-4-ил)амино]пентан-1-ол (5,0 г, 18,16 ммоля) и тионила хлорид (40,78 г, 0,34 ммоля) в атмосфере азота. Полученный таким образом раствор нагревали до 80°С, что приводило к образованию гомогенной жидкости. Ее оставляли при указанной температуре на протяжении 1 часа. В течение этого периода происходила полная утилизация исходных материалов реакции. Летучие компоненты удаляли упариванием при пониженном давлении, а к оставшейся маслянистой жидкости добавляли при непрерывном перемешивании твердый натрия карбонат, доводя ее рН до 10. Конечный твердый продукт, представлявший собой N-(5-хлорофенил)-3-нитрохинолин-4-амин, собирали посредством фильтрования (4,80 г, 16,34 ммоля) и в дальнейшем использовали без дополнительной очистки.
Часть В
С помощью общего способа, описанного в примере 6, часть Б (но понизив температуру реакции до 80°С), N-(5-хлорофенил)-3-нитрохинолин-4-амин (4,75 г, 16,17 ммоля) преобразовывали в N-[5-(метилтио)пентил]-3-нитрохинолин-4-амин, используя для этой цели натрия тиометоксид (1,43 г, 19,40 ммоля) вместо натрия бензолтиолата. Полученное твердое вещество светло-желтого цвета (3,28 г, 10,74 ммоля) в дальнейшем использовали без дополнительной очистки. Масс-спектрометрия (CI) для C15H19N3O2S: m/z 306 (MH+), 272, 117.
Часть Г
С помощью общего способа, описанного в примере 1, часть В, N-[5-(метилтио)пентил]-3-нитрохинолин-4-амин (3,20 г, 10,48 ммоля) восстанавливали до N4-[5-(метилтио)пентил]хинолин-3,4-диамина (2,89 г, 10,48 ммоля), который выделяли без дополнительной очистки в форме масла бурого цвета.
Часть Д
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-[5-(метилтио)пентил]-3-хинолин-3,4-диамина (2,89 г, 10,48 ммоля) для получения 2-бутил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолина. Полученный материал очищали посредством хроматографии на силикагеле, используя для элюции этилацетат. Конечный продукт представлял собой масло светло-желтого цвета (2,10 г, 6,15 ммоля).
Часть Е
Круглодонную колбу помещали на магнитную мешалку и вносили в нее 2-бутил-1- [5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолина (2,1 г, 6,15 ммоля) и хлороформ (31 мл). В полученный раствор отдельными порциями на протяжении 10 минут добавляли твердую 3-хлорпербензойную кислоту (4,41 г, 19,68 ммоля) и реакционную смесь непрерывно перемешивали при комнатной температуре в течение 30 мин. Эта процедура обеспечивала полную утилизацию исходных материалов реакции. Раствор разделяли между хлороформом и насыщенным водным раствором натрия бикарбоната. Образовавшиеся слои отделяли один от другого, органическую фракцию промывали насыщенным водным раствором натрия бикарбоната и солевым раствором, высушивали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая 2-бутил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-5N-оксид (2,40 г, 6,15 ммоля), который имел форму смолообразного вещества желто-коричневого цвета. Полученный таким образом материал в дальнейшем использовали без дополнительной очистки.
Часть Ж
С помощью общего способа, описанного в примере 3, часть Б, 2-бутил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-5N-оксид (2,40 г, 6,15 ммоля) аминировали для получения 2-бутил-1-[5-(метилсульфонило)пентил]-1Н-имидазо[4,5-с]хинолин-4-амина.
Полученный твердый материал (2,24 г) разводили в ацетонитриле (40 мл) и перегоняли в обратном токе. Добавляли активированный уголь (1 г), полученную смесь нагревали в условиях обратного тока на протяжении 5 минут, а затем удаляли уголь фильтрованием через гофрированную фильтровальную бумагу, после которой оставалась жидкость светло-коричневого цвета. После ее охлаждения выделяли 2-бутил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин (0,90 г, 2,32 ммоля) в форме иглообразных кристаллов белого цвета с температурой плавления 173-175°С.
Результаты анализа. Расчетное содержание в C20H28N4O2S: % С 61,83; % Н 7,26; % N 14,42, найденное содержание: % С 61,58; % Н 7,27; % N 14,36.
1H-ЯМР (300 МГц, ДМСО): δ 8,01 (d, J=8,3 Hz, 1Н), δ 7,61 (d, J=8,3 Hz, 1Н), δ 7,41 (t, J=8,3 Hz, 1Н), δ 7,26 (t, J=8,3 HZ, 1Н), δ 6,45 (bs, 2 Н), δ 4,51 (t, J=7,6 Hz, 2 Н), δ 3,10 (t, J=7,8 Hz, 2 Н), δ 2,92 (t, J=7,3 Hz, 2 Н), δ 1,76 (m, 6 Н), δ 1,54 (m, 2 Н), δ 1,46 (секстет, J=7,3 Hz, 2 H), δ 0,99 (t, J=7,3 Hz, 3H).
Масс-спектрометрия (CI) для C20H28N4O2S: m/z 389 (MH+).
Пример 12
2-Метил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин
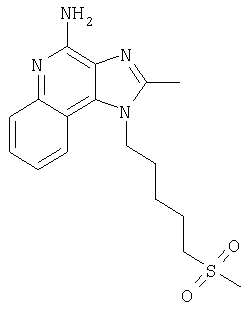
Часть А
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-[5-(метилтио)пентил]-3-хинолин-3,4-диамина (4,53 г, 16,37 ммоля) для получения 2-метил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолина, используя для этой цели обработку триметоксиэтаном (2,95 г, 24,6 ммоля) в присутствии пиридина гидрохлорида (0,1 г). Полученный таким образом материал растирали в порошок в ацетонитриле и собирали посредством фильтрования в форме твердого вещества светло-коричневого цвета (3,78 г, 12,62 ммоля). В дальнейшем его использовали без дополнительной очистки.
Часть Б
С помощью общего способа, описанного в примере 11, часть Е, 2-метил-1- [5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин (3,78 г, 12,62 ммоля) окисляли до 2-метил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолина-5N-оксида (4,38 г, 12,62 ммоля), который выделяли в форме твердого вещества желтовато-коричневого цвета. В дальнейшем его использовали без дополнительной очистки.
Часть В
С помощью общего способа, описанного в примере 3, часть Б, 2-метил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-5N-оксид (4,38 г, 12,62 ммоля) аминировали для получения 2-метил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амина. Полученный твердый материал (2,24 г) растирали в порошок в ацетонитриле (40 мл), после чего искомое соединение (2-метил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин) собирали посредством фильтрования (0,8 г, 2,31 ммоля) в форме твердого вещества беловатого цвета с температурой плавления 235-240°С.
Результаты анализа. Расчетное содержание в C17H22N4O2S: % С 58,94; % Н 6,40; % N 16,17, найденное содержание: % С 58,77; % Н 6,34; % N 16,39.
1Н-ЯМР (300 МГц, ДМСО): δ 8,02 (d, J=8,3 Hz, 1Н), δ 7,60 (d, J=8,3 Hz, 1Н), δ 7,41 (t, J=8,3 Hz, 1Н), δ 7,25 (t, J=8,3 Hz, 1Н) δ 6,49 (bs, 2 Н), δ 4,50 (t, J=7,3 Hz, 2 Н), δ 3,12 (t, J=7,8 Hz, 2 Н), δ 2,92 (s, 3 Н), δ 2,61 (s, 3 Н), (t, J=7,3 Hz, 2 Н), δ 1,76 (m, 6H), δ 1,86 (m, 2 Н), δ 1,74 (m, 2H), δ1,53 (m,2H).
Масс-спектрометрия (CI) для C17H22N4O2S: m/z 347 (MH+), 267.
Пример 13
2-Этил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин
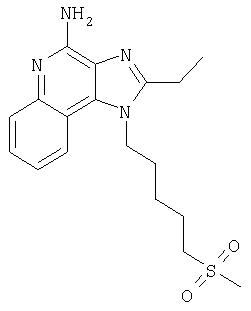
Часть А
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-[5-(метилтио)пентил]-3-хинолин-3,4-диамина (4,53 г, 16,37 ммоля) для получения 2-этил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолина, используя для этой цели реакцию с триэтила ортопропионатом (4,3 г, 24,56 ммоля) в присутствии пиридина гидрохлорида (0,1 г). Полученный таким образом материал мелко растирали в этиловом эфире и собирали посредством фильтрования в форме порошка беловатого цвета (3,25 г, 10,37 ммоля). В дальнейшем его использовали без дополнительной очистки.
Часть Б
С помощью общего способа, описанного в примере 11, часть Е, 2-этил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин (3,25 г, 10,37 ммоля) окисляли до 2-этил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолина-5N-оксида (3,75 г, 10,37 ммоля), который выделяли в форме твердого вещества желтовато-коричневого цвета. В дальнейшем его использовали без дополнительной очистки.
Часть В
С помощью общего способа, описанного в примере 3, часть Б, 2-этил-1- [5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-5N-оксид (3,75 г, 10,37 ммоля) аминировали для получения 2-этил-1-[5-(метилсульфонило)пентил]-1Н-имидазо [4,5-с]хинолин-4-амина.
Полученный твердый материал подвергали последовательной перекристаллизации из этанола и ацетонитрила, которая приводила к образованию искомого соединения, имевшего форму иглообразных кристаллов беловатого цвета, имеющих температуру плавления 189-191°С.
Результаты анализа. Расчетное содержание в C18H24N4O2S: % С 59,98; % Н 6,71; % N 15,54, найденное содержание: % С 59,71; % Н 6,68; % N 15,64.
1H-ЯМР (300 МГц, ДМСО): δ 8,01 (d, J=8,3 Hz, 1Н), δ 7,61 (d, J=8,3 Hz, 1Н), δ 7,42 (t, J=8,3 Hz, 1Н), δ 7,26 (t, J=8,3 Hz, 1Н), δ 6,45 (bs, 2 Н), δ 4,50 (t, J=7,6 Hz, 2 Н), δ 3,10 (t, J=7,8 Hz, 2 Н), δ 2,95 (t, J=7,3 Hz, 2 Н), δ 2,92 (s, 3 Н), δ 1,85 (m, 2 Н), δ 1,74 (m, 2 Н), δ 1,55 (m, 2 Н), δ 1,38 (t, J=7,3 Hz, 3H).
Масс-спектрометрия (CI) для C18H24N4O2S: m/z 361 (MH+), 281.
Пример 14
1-[5-(Метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин
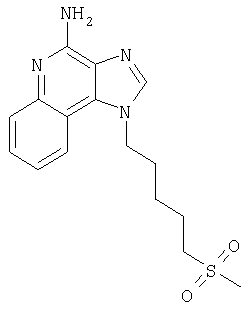
Часть А
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-[5-(метилтио)пентил]-3-хинолин-3,4-диамина (4,53 г, 16,37 ммоля) для получения 1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолина, используя для этой цели реакцию с триэтила ортопропионатом (3,64 г, 24,56 ммоля) в присутствии пиридина гидрохлорида (0,1 г).
Полученный таким образом (4,05 г, 14,19 ммоля) материал, имевший вид масла бурого цвета, выделяли и использовали без дополнительной очистки.
Часть Б
С помощью общего способа, описанного в примере 11, часть Е, 2-этил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин (14,05 г, 14,19 ммоля) окисляли до 1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолина-5N-оксида (4,05 г, 14,19 ммоля), который выделяли в форме твердого вещества желтовато-коричневого цвета. В дальнейшем его использовали без дополнительной очистки.
Часть В
С помощью общего способа, описанного в примере 3, часть Б, 1 [5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-5N-оксид (4,73 г, 14,19 ммоля) аминировали для получения 1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амина.
Полученный материал подвергали хроматографической очистке на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 95:5. Конечный продукт имел форму твердого вещества светло-желтого цвета. Его перекристаллизовывали из диметилформамида и получали искомое соединение, т.е. 1-[5-(метилсульфонил)пентил] -1Н-имидазо[4,5-с]хинолин-4-амин, в виде твердого гранулированного материала светло-желтого цвета с температурой плавления 199-201°С.
Результаты анализа. Расчетное содержание в C16H20N4O2S: % С 57,70; % Н 6,06; % N 16,58, найденное содержание: % С 57,01; % Н 6,06; % N 16,70.
1Н-ЯМР (300 МГц, ДМСО): δ 8,20 (s, 1 H), δ 8,04 (d, J=8,3 Hz, 1 H), δ 7,62 (d, J=8,3 Hz, 1 H), δ 7,44 (t, J=8,3 Hz, 1 H), δ 7,27 (t, J=8,3 Hz, 1 H) δ 6,57 (bs, 2 H), δ 4,61 (t, J=6,8 Hz, 2 H), δ 3,09 (t, J=7,8 Hz, 2 H), δ 2,92 (s, 3 H), δ 1,91 (p, J=7,6 Hz, 2 H), δ 1,73 (m, 2 H), δ 1,45 (m, 2 H).
Масс-спектрометрия (CI) для C16H20N4O2S: m/z 333 (MH+).
Пример 15
2-Гексил-1-[5-(метилсульфонил)пентил]-1Н-имидазо [4,5-с]хинолин-4-амин
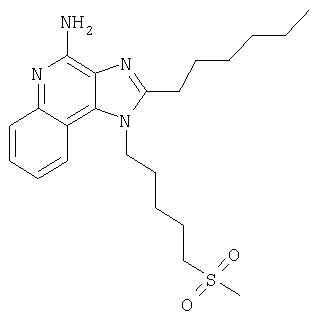
Часть А
Круглодонную колбу помещали на магнитную мешалку и вносили в нее N4-[5-(метилтио)пентил]хинолин-3,4-диамин (3,17 г, 11,46 ммоля) и безводный пиридин (46 мл) в атмосфере азота. Полученный гомогенный раствор охлаждали на водяной бане со льдом до 0°С. В охлажденную жидкость добавляли неразведенный гептаноила хлорид (1,87 г, 12,61 ммоля).
Реакцию считали завершенной после непрерывного перемешивания раствора при комнатной температуре на протяжении 1 часа. Летучие компоненты удаляли упариванием при пониженном давлении, а полученный материал, который имел форму масла, разделяли между хлороформом и водой.
Образовавшиеся слои отделяли один от другого и органическую фракцию промывали насыщенным водным раствором натрия бикарбоната и солевым раствором, высушивали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая N-(4-[5-(метилтио)пентил]аминохинолин-3-ил)гептанамид (4,44 г, 11,46 ммоля), имевший форму масла бурого цвета, в дальнейшем использовался без дополнительной очистки.
Часть Б
Круглодонную колбу помещали на магнитную мешалку и вносили в нее N-(4-[5-(метилтио)пентил]аминохинолин-3-ил)гептанамид (4,44 г, 11,46 ммоля), пиридина гидрохлорид (0,13 г, 1,15 ммоля) и безводный пиридин (50 мл) в атмосфере азота. Реакцию считали завершенной после непрерывного перемешивания раствора в условиях перегонки на протяжении 1,5 часа. Раствор охлаждали и разделяли между этилацетатом и водой.
Образовавшиеся слои отделяли один от другого и органическую фракцию промывали насыщенным водным раствором натрия бикарбоната и солевым раствором, высушивали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая 2-гексил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин (4,0 г, 10,82 ммоля), который имел форму масла бурого цвета и в дальнейшем использовался без дополнительной очистки.
Часть В
С помощью общего способа, описанного в примере 1, часть Е, 2-гексил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин (4,0 г, 10,82 ммоля) окисляли до 2-гексил-1-[5-(метилсулфонил)пентил]-1Н-имидазо[4,5-с]хинолин-5N-оксида (4,52 г, 10,82 ммоля), который выделяли в форме твердого вещества желтовато-коричневого цвета и в последующем использовали без дополнительной очистки.
Часть Г
С помощью общего способа, описанного в примере 3, часть Б, 2-гексил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин-5N-оксид (4,0 г, 10,82 ммоля) аминировали для получения 2-гексил-1-[5-(метилсулфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амина.
Полученный материал подвергали перекристаллизации из ацетонитрила, которая приводила к образованию искомого соединения (2-гексил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амина) (2,25 г, 5,40 ммоля), имевшего форму иглообразных кристаллов беловатого цвета с температурой плавления 168-171°С.
Результаты анализа. Расчетное содержание в C22H32N4O2S: % С 63,43; % Н 7,74; % N 13,45, найденное содержание: % С 63,06; % Н 7,66; % N 13,81.
1H-ЯМР (300 МГц, ДМСО): δ 8,01 (d, J=8,3 Hz, 1Н), δ 7,62 (d, J=8,3 Hz, 1Н), δ 7,42 (t, J=8,3 Hz, 1Н), δ 7,26 (t, J=8,3 Hz, 1Н) δ 6,51 (bs, 2 Н), δ 4,51 (t, J=7,3 Hz, 2 Н), δ 3,10 (t, J=7,8 Hz, 2 Н), δ 2,93 (s, 3 Н), δ 2,93 (t, J=7,3 Hz, 2 Н), δ 1,71-1,87 (m, 6 Н), δ 1,54 (m, 2 Н), δ 1,44 (m, 2 Н), δ 1,33 (m, 4 Н), δ 0,89 (t, J=7,3 Hz, 3 Н).
Масс-спектрометрия (CI) для C22H32N4O2S: m/z 417 (MH+), 337.
Пример 16
2-(2-Метоксиэтил)-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин
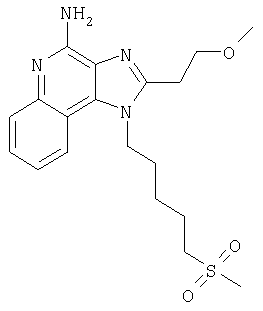
Часть А
Круглодонную колбу помещали на магнитную мешалку и вносили в нее N4-[5-(метилтио)пентил]хинолин-3,4-диамин (3,56 г, 12,93 ммоля) и безводный пиридин (52 мл) в атмосфере азота. Полученный гомогенный раствор охлаждали на водяной бане со льдом до 0°С. В охлажденную жидкость добавляли неразведенный 3-метоксипропионила хлорид (2,74 г, 22,36 ммоля). После добавления кислого хлорида реакционную систему нагревали в условиях обратного тока на протяжении 14 часов. В течение этого времени происходила полная утилизация исходных материалов реакции. Раствор разделяли между хлороформом и насыщенным водным раствором аммония хлорида. Образовавшиеся слои отделяли один от другого, органическую фракцию промывали насыщенным водным раствором натрия бикарбоната, высушивали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая 2-(2-метоксиэтил)-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин (3,0 г, 8,73 ммоля), который имел форму масла бурого цвета и в дальнейшем использовался без дополнительной очистки.
Часть Б
С помощью общего способа, описанного в примере 11, часть Е, 2-(2-метоксиэтил)-1-[5-(метилтио)пентил]-1Н-имидазо [4,5-с]хинолин (3,0 г, 8,73 ммоля) окисляли до 2-(2-метоксиэтил)-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин-5N-оксида (3,41 г, 8,73 ммоля), который выделяли в форме твердого вещества желтовато-оранжевого цвета и в дальнейшем использовали без дополнительной очистки.
Часть В
С помощью общего способа, описанного в примере 3, часть Б, 2-(2-метоксиэтил)-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин-5N-оксид (3,41 г, 8,73 ммоля) аминировали для получения 2-(2-метоксиэтил)-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин-4-амина. Полученный твердый материал подвергали хроматографической очистке на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 95:5. В результате получали смолообразное вещество, которое подвергали перекристаллизации из ацетонитрила, которая приводила к образованию искомого соединения (2-(2-метоксиэтил)-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амина), который имел форму порошка беловатого цвета с температурой плавления 158-160°С.
Результаты анализа. Расчетное содержание в C19H26N4O3S: % С 58,44; % Н 6,71; % N 14,35, найденное содержание: % С 58,24; % Н 6,76; % N 14,70.
1Н-ЯМР (300 МГц, ДМСО): δ 8,02 (d, J=8,3 Hz, 1Н), δ 7,62 (d, J=8,3 Hz, 1Н), δ 7,42 (t, J=8,3 Hz, 1Н), δ 7,26 (t, J=8,3 Hz, 1Н), δ 6,50 (bs, 2 Н), δ 4,53 (t, J=7,6 Hz, 2 Н), δ 3,83 (t, J=6,8 Hz, 2 Н), δ 3,30 (s, 3 Н), δ 3,19 (t, J=6,8 Hz, 2 Н), δ 3,11 (t, J=7,8 Hz, 2 Н), δ 2,93 (s, 3 Н), δ 1,85 (m, 2 Н), δ 1,76 (m, 2 Н), δ 1,57 (m, 2 Н).
Масс-спектрометрия (CI) для C19H26N4O2S: m/z 391 (MH+), 359.
Пример 17
2-Бутил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин
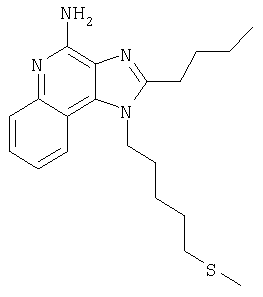
Часть А
С помощью общего способа, описанного в примере 1, часть В, N-(5-хлоропентил)-3-нитрохинолин-4-амин (2,0 г, 6,80 ммоля) восстанавливали для получения N4-(5-хлоропентил)хинолин-3,4-диамина (1,79 г, 6,80 ммоля), который выделяли в форме масла бурого цвета и в последующем использовали без дополнительной очистки.
Часть Б
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-(5-хлоропентил)хинолин-3,4-диамина (1,79 г, 6,80 ммоля) для получения 2-бутил-1-(5-хлоропентил)-1Н-имидазо[4,5-с]хинолина, используя для этой цели реакцию с триметилортовалератом (2,55 г, 15,72 ммоля) в присутствии пиридина гидрохлорида (0,079 г). Полученный продукт (1,95 г, 5,91 ммоля) выделяли в форме твердого вещества беловатого цвета и в дальнейшем использовали без дополнительной очистки.
Часть В
С помощью общего способа, описанного в примере 1, часть Д, 2-бутил-1-(5-хлоропентил)-1Н-имидазо[4,5-с]хинолин (1,95 г, 5,91 ммоля) окисляли для получения 2-бутил-1-(5-хлоропентил)-1Н-имидазо[4,5-с]хинолин-5N-оксида (2,04 г, 5,91 ммоля), который выделяли в форме твердого вещества желтовато-коричневого цвета и в дальнейшем использовали без дополнительной очистки.
Часть Г
С помощью общего способа, описанного в примере 3, часть Б, 2-бутил-1-(5-хлоропентил)-1Н-имидазо[4,5-с]хинолин-5N-оксид (2,04 г, 5,91 ммоля) аминировали для получения 2-бутил-1-(5-хлоропентил)-1Н-имидазо[4,5-с]хинолин-4-амина, который подвергали перекристаллизации из этанола, получая искомый продукт (0,85 г, 2,46 ммоля), который имел форму мелкого порошка белого цвета с температурой плавления 144-146°С.
Результаты анализа. Расчетное содержание в С19Н25ClN4: % С 66,17; % Н 7,31; % N 16,24, найденное содержание: % С 66,44; % Н 7,55; % N 16,29.
Масс-спектрометрия (CI) для C19H25ClN4: m/z 345 (MH+), 399.
Часть Д
С помощью общего способа, описанного в примере 6, часть Б (но понизив температуру реакции до 80°С), осуществляли преобразование 2-бутил-1-(5-хлоропентил)-1Н-имидазо[4,5-с]хинолин-4-амина (2,04 г, 5,91 ммоля) для получения 2-бутил-1-(5-метилтио)-1Н-имидазо[4,5-с]хинолин-4-амина, используя для этой цели натрия тиометоксид (0,68 г, 8,70 ммоля) вместо натрия бензолтиолата. Полученный материал разделяли между хлороформом и насыщенным водным раствором натрия бикарбоната. Образовавшиеся слои отделяли один от другого, органическую фракцию промывали солевым раствором, высушивали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая твердое вещество. Его подвергали перекристаллизации из ацетонитрила, которая приводила к образованию искомого соединения, т.е. 2-бутил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин-4-амина (1,91 г, 5,36 ммоля), имевшего вид мелкого порошка белого цвета с температурой плавления 112-114°С.
Результаты анализа. Расчетное содержание в C20H28N4S: % С 67,38; % Н 7,92; % N 15,17, найденное содержание: % С 67,26; % Н 8,08;% N15,74.
1Н-ЯМР (300 МГц, ДМСО): δ 8,01 (d, J=8,3 Hz, 1Н), δ 7,61 (d, J=8,3 Hz, 1Н), δ 7,41 (t, J=8,3 Hz, 1Н), δ 7,25 (t, J=8,3 Hz, 1Н) δ 6,45 (bs, 2 Н), δ 4,50 (t, J=7,8 Hz, 2 Н), δ 2,92 (t, J=7,6 Hz, 2 Н), δ 2,46 (t, J=7,3 Hz, 2 Н), δ 2,01 (s, 3 Н), δ 1,80 (m, 4 Н), δ 1,42-1,61 (m, 6 Н), δ 0,96 (t, J=7,3 Hz, 3 Н).
Масс-спектрометрия (CI) для С20Н28N4S: m/z 357 (MH+), 309.
Пример 18
2-Бутил-1-[5-(метилсульфинил)пентил]-1Н-имидазо[4,5-с]хинолинамин
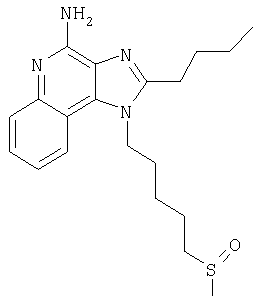
Часть А
Круглодонную колбу помещали на магнитную мешалку и вносили в нее 2-бутил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин (1,0 г, 2,80 ммоля) и хлороформ (14 мл). В полученный раствор на протяжении 5 минут отдельными порциями добавляли твердую 3-хлорпербензойную кислоту (0,69 г, 3,09 ммоля), перемешивая реакционную смесь при комнатной температуре в течение 20 минут. За это время происходила полная утилизация исходных материалов реакции. Полученный раствор разделяли между хлороформом и насыщенным водным раствором натрия бикарбоната. Образовавшиеся слои отделяли один от другого, органическую фракцию промывали насыщенным водным раствором натрия бикарбоната и солевым раствором, упаривали над безводным натрия сульфатом, фильтровали, а потом концентрировали при пониженном давлении, получая твердое вещество беловатого цвета. Способом 1Н-ЯМР оно было идентифицировано как соль 3-хлоропербензойной кислоты искомого продукта. Этот материал смешивали с водой и добавлением твердого натрия карбоната доводили рН полученной смеси до 10,0.
Полученное таким образом свободное основание собирали посредством фильтрования в форме твердого вещества белого цвета. Последнее подвергали перекристаллизации из ацетонитрила, в результате которой образовывался белый порошок, представлявший собой 2-бутил-1-[5-метилсульфинил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин (0,40 г, 1,07 ммоля) с температурой плавления 119-121°С.
Результаты анализа. Расчетное содержание в C20H28N4OS(H2O)1: % С 61,51; % Н 7,74; % N 13,45, найденное содержание: % С 61,64; % Н 7,82; % N 14,32.
1Н-ЯМР (300 МГц, ДМСО): δ 8,01 (d, J=8,3 Hz, 1Н), δ 7,60 (d, J=8,3 Hz, 1Н), δ 7,41 (t, J=8,3 Hz, 1Н), δ 7,26 (t, J=8,3 Hz, 1Н), δ 6,44 (bs, 2 Н), δ 4,51 (t, J=7,6 Hz, 2 Н), δ 2,92 (t, J=7,8 Hz, 2 Н), δ 2,93 (t, J=7,8 Hz, 2 Н), δ 2,57-2,74 (m, 2 Н), δ 2,50 (s, 3 Н), δ 1,80 (m, 4 Н), δ 1,66 (m, 2 Н), δ 1,55 (m, 2 Н), δ 1,48 (m, 2 Н), δ 0,96 (t, J=7,3 Hz, 3 Н).
Масс-спектрометрия (CI) для C20H28N4OS(H2O)1: m/z 373 (MH+), 309, 253.
Пример 19
2-Бутил-1-[3-(метилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолинамин
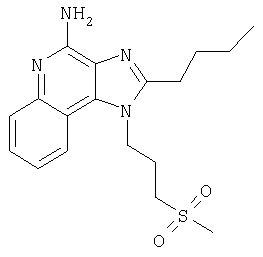
Часть А
Круглодонную колбу помещали на магнитную мешалку и вносили в нее 3-[(3-нитрохинолин-4-ил)амино]пропан-1-ол (20,75 г, 83,93 ммоля), тионила хлорид (15,0 г, 125,89 ммоля) и дихлорметан (420 мл).
Полученный гомогенный раствор ярко-желтого цвета непрерывно перемешивали при комнатной температуре в течение 20 минут. На протяжении этого времени происходила полная утилизация исходных материалов реакции. Летучие соединения удаляли упариванием в условиях пониженного давления, а оставшийся твердый остаток смешивали с водой (400 мл) и добавлением твердого натрия карбоната доводили рН полученной смеси до 10,0. Полученное таким образом твердое вещество ярко-желтого цвета собирали посредством фильтрования и идентифицировали как N-(3-хлоропропил)-3-нитрохинолин-4-амин (21,63 г, 81,41 ммоля), который в дальнейшем использовали без дополнительной очистки.
Часть Б
С помощью общего способа, описанного в примере 1, часть В, N-(3-хлоропропил)-3-нитрохинолин-4-амин (10,0 г, 37,63 ммоля) восстанавливали для получения N4-(3-хлоропропил)хинолин-3,4-диамина (8,87 г, 37,63 ммоля), который выделяли в форме масла бурого цвета и в дальнейшем использовали без дополнительной очистки.
Часть В
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-(3-хлоропропил)хинолин-3,4-диамина (8,87 г, 37,63 ммоля) для получения 2-бутил-1-(3-хлоропропил)-1Н-имидазо[4,5-с]хинолина, используя для этой цели обработку триметилортовалератом (7,33, 45,16 ммоля) в присутствии пиридина гидрохлорида (0,43 г). Полученный твердый материал растирали в порошок в этиловом эфире и собирали посредством фильтрования в форме твердого вещества беловатого цвета (9,00 г, 29,82 ммоля). В дальнейшем этот материал использовали без дополнительной очистки.
Часть Г
С помощью общего способа, описанного в примере 1, часть Д, 2-бутил-1-(3-хлоропропил)-1Н-имидазо[4,5-с]хинолин (9,0 г, 29,82 ммоля) окисляли до 2-бутил-1-(3-хлоропропил)-1Н-имидазо[4,5-с]хинолин-5N-оксида (9,48 г, 29,82 ммоля), который выделяли в форме твердого вещества желтовато-коричневого цвета и в дальнейшем использовали без дополнительной очистки.
Часть Д
С помощью общего способа, описанного в примере 3, часть Б, 2-бутил-1-(3-хлоропропил)-1Н-имидазо[4,5-с]хинолин-5N-оксид (9,48 г, 29,82 ммоля) аминировали для получения 2-бутил-1-(3-хлоропропил)-1Н-имидазо[4,5-с]хинолин-4-амина. Полученный твердый материал подвергали очистке посредством хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 95:5. Конечный продукт (6,4 г, 20,20 ммоля) представлял собой твердое вещество желтовато-коричневого цвета.
Часть Е
С помощью общего способа, описанного в примере 6, часть Б, осуществляли преобразование 2-бутил-1-(3-хлоропропил)-1Н-имидазо[4,5-с]хинолин-4-амина (2,0 г, 6,31 ммоля) в 2-бутил-1-(3-метилтио)-1Н-имидазо[4,5-с]хинолин-4-амин, используя для этой цели тиометоксид (0,74 г, 9,47 ммоля) вместо натрия бензолтиолата. Полученное таким образом твердое вещество разделяли между хлороформом и насыщенным водным раствором натрия бикарбоната.
Образовавшиеся слои отделяли один от другого, органическую фракцию промывали солевым раствором, высушивали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая искомое соединение 2-бутил-1-[3-(метилсульфонил)пропил]-1Н-имидазо [4,5-с]хинолин-4-амин (2,0, 6,09 ммоля), имевшее форму твердого вещества белого цвета. Этот материал в дальнейшем использовался без дополнительной очистки.
Часть Ж
С помощью общего способа, описанного в примере 5, часть А, 2-бутил-1-[3-(метилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин (2,0 г, 6,09 ммоля) окисляли до 2-бутил-1-[3-(метилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолина-4-амина.
Полученное твердое вещество мелко растирали в метаноле и собирали посредством фильтрования в форме порошка беловатого цвета, представлявшего собой искомый 2-бутил-1-[3-(метилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолинамин (0,96 г, 2,66 ммоля) с температурой плавления 233-236°С.
Результаты анализа. Расчетное содержание в C18H24N4O2S: % С 59,98; % Н 6,71; % N 15,54, найденное содержание: % С 59,71; % Н 6,65; % N 15,43.
1H-ЯМР (300 МГц, ДМСО): δ 8,10 (d, J=8,3 Hz, 1Н), δ 7,61 (d, J=8,3 Hz, 1Н), δ 7,42 (t, J=8,3 Hz, 1Н), δ 7,25 (t, J=8,3 Hz, 1Н), δ 6,47 (bs, 2 Н), δ 4,66 (t, J=7,8 Hz, 2 Н), δ 3,40 (t, J=7,3 Hz, 2 Н), δ 3,01 (s, 3 Н), 2,94 (t, J=7,8 Hz, 2 Н), δ 2,22 (m, 2 Н), δ 1,80 (m, 2 Н), δ 1,46 (секстет J=7,3 Hz, 2 Н), δ 0,96 (t, J=7,3 Hz, 3 Н).
Масс-спектрометрия (CI) для C18H24N4OS: m/z 361 (MH+), 281, 235.
Пример 20
2-Бутил-1-[3-(фенилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин
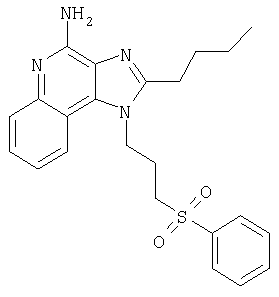
Часть А
Круглодонную колбу помещали на магнитную мешалку и вносили в нее бензолтиол (0,68 г, 6,21 ммоля), натрия гидрид (9,25 г 60%-ной дисперсии, 6,21 ммоля) и безводный диметилформамид (28 мл) в атмосфере азота. После непрерывного перемешивания реакционной смеси при комнатной температуре на протяжении 30 минут в нее добавляли 2-бутил-1-(3-хлоропропил)-1Н-имидазо[4,5-с]хинолин-4-амин (1,64 г, 5,18 ммоля), а образовавшийся мутный раствор нагревали до 80°С и оставляли при этой температуре в течение 2,5 часов. За это время происходила полная утилизация исходных материалов реакции. Горячий раствор переливали в воду (200 мл) при энергичном перемешивании смеси, которую затем дважды экстрагировали хлороформом. Объединенные органические фракции промывали насыщенным водным раствором натрия бикарбоната и солевым раствором, высушивали над безводным натрия сульфатом, фильтровали, а затем концентрировали при пониженном давлении, получая масло светло-желтого цвета. Полученный материал подвергали очистке с помощью хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 95:5. В результате получали 2-бутил-1-[3-(фенилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин (1,38 г, 3,53 ммоля) в форме твердого вещества белого цвета.
Часть Б
С помощью общего способа, описанного в примере 5, часть А, 2-бутил-1-[3-(фенилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин (1,38, 3,53 ммоля) окисляли до 2-бутил-1-[3-(фенилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амина.
Полученный материал подвергали перекристаллизации из этанола, которая приводила к образованию исходного соединения 2-бутил-1-[3-(фенилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амина (0,85 г, 3,53 ммоля) в форме порошка белого цвета с температурой плавления 224-227°С.
Результаты анализа. Расчетное содержание в C23H26N4O2S: % С 65,38; % Н 6,20; % N 13,26, найденное содержание: % С 65,25; % Н 6,23; % N 13,20.
1Н-ЯМР (300 МГц, ДМСО): δ 7,96 (d, J=8,3 Hz, 1 H), δ 7,89 (m, 2 H), 3 7,73 (m, 1 H), δ 7,63 (m, 3 H), δ 7,40 (d, J=8,3 Hz, 1 H), δ 7,17 (t, J=8,3 Hz, 1 H), δ 6,46 (9bs, 2 H), δ 4,60 (t, J=7,8 Hz, 1 H), δ 3,66 (t, J=7,3 Hz, 2 H), δ 2,86 (t, J=7,8 Hz, 2 H), δ 2,04 (m, 2 H), δ 1,73 (p, J=7,6 Hz, 2 H), δ 1,93 (секстет, J=7,3 Hz, 2 H), δ 0,92 (t, J=7,3 Hz, 3 H).
Масс-спектрометрия (CI) для С23Н26N4O2S: m/z 423 (MH+), 322, 281.
Пример 21
1-[5-(Метилсульфонил)пентил]-2-пропил-1Н-имидазо[4,5-с]хинолин-4-амин
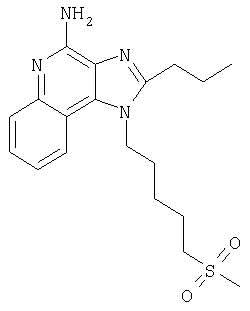
Часть А
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-(5-хлоропентил)хинолин-3,4-диамина (примерно 20,4 ммоля), используя для этой цели реакцию с триметилортобутиратом (3,6 г, 24,5 ммоля) в присутствии пиридина гидрохлорида (приблизительно 0,1 г).
Первичный продукт этой реакции очищали с помощью колоночной хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 95:5. В результате получали 3,9 г 1-(5-хлоропентил)-2-пропил-1Н-имидазо[4,5-с]хинолина, представлявшего собой твердое вещество светло-зеленого цвета.
Часть Б
С помощью общего способа, описанного в примере 1, часть Д, 1-(5-хлоропентил)-2-пропил-1Н-имидазо[4,5-с]хинолин (3,9 г, 12,36 ммоля) окисляли до 1-(5-хлоропентил-2-пропил)-1Н-имидазо[4,5-с]хинолин-5N-оксида, который получали в форме масла темно-оранжевого цвета.
Часть В
С помощью общего способа, описанного в примере 3, часть Б, материал, полученный в части Б, аминировали до 1-(5-хлоропентил)-2-пропил-1Н-имидазо[4,5-с]хинолин-4-амина. Это вещество смешивали с диэтилэфиром, отфильтровывали, промывали диэтилэфиром и высушивали, получая 3,42 г порошка белого цвета.
Часть Г
Суспензию 1-(5-хлоропентил)-2-пропил-1Н-имидазо[4,5-с]хинолин-4-амина (2,5 г, 7,56 ммоля) в безводном N,N-диметилформамиде (38 мл) нагревали до 80°С, получая раствор светло-желтого цвета. Затем в него за один прием вносили навеску натрия тиометоксида (0,67 г 95%-ного материала, 9,07 ммоля) и продолжали нагревать на протяжении еще 110 минут. Образовавшуюся суспензию светло-коричневого цвета переливали в воду (300 мл) при энергичном перемешивании. В результате выпадал осадок белого цвета.
После охлаждения суспензии до комнатной температуры в нее добавляли несколько порций твердого натрия карбоната. Затем суспензию помещали на водяную баню со льдом и перемешивали на протяжении 1 часа. Твердый материал отделяли посредством фильтрования, отмывали холодной водой и высушивали, получая 2,3 г 1-[5-(метилсульфонил)пентил]-2-пропил-1Н-имидазо[4,5-с]хинолин-4-амина, имевшего форму порошка белого цвета.
Часть Д
С помощью общего способа, описанного в примере 5, часть Г, материал, полученный в части Г, окисляли, а первичный продукт реакции подвергали очистке, получая 0,88 г 1-[5-(метилсульфонил)пентил]-2-пропил-1Н-имидазо[4,5-с]хинолин-4-амина в форме порошка белого цвета. Это вещество смешивали с диэтилэфиром, отфильтровывали, промывали диэтилэфиром и высушивали, получая 3,42 г порошка белого цвета с температурой плавления 179-181°С.
Результаты анализа. Расчетное содержание в C19H26N4O2S: % С 60,94; % Н 7,00; % N 14,96, найденное содержание: % С 60,60; % Н 7,03; % N 14,84.
1H-ЯМР (300 МГц, ДМСО-d6): δ 8,0 (d, J=7,8 Hz, 1Н), δ 7,61 (d J=7,5 Hz, 1Н), 7,41 (d, J=8,4 Hz, 1Н), δ 7,25 (dt, J=8,1; 1,2, Hz, 1Н), δ 6,43 (s, 2 Н), δ 4,60 (t, J=7,8 Hz, 1Н), δ 3,66 (t, J=7,3 Hz, 2 Н), δ 4,50 (t, J=7,5 Hz, 2 Н), δ 4,50 (t, J=7,5 Hz, 2 Н), δ 3,10 (t, J=8,1 Hz, 2 Н), δ 2,92 (s, 3 Н), δ 2,90 (m, 2 Н), δ 1,84 (квинтет, J=7,5 Hz, 4 Н), δ 1,74 (m, 2 Н), δ 1,54 (квинтет, J=8,1 Hz, 2 Н), δ 1,04 (t, J=7,5 Hz, 3 Н).
Масс-спектрометрия (CI) для C19H26N4O2S: m/z 375 (МН+).
Пример 22
2-Метил-1-[3-(метилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин
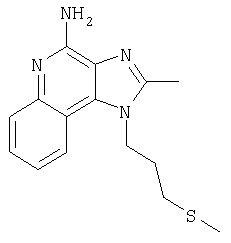
Часть А
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-(3-хлоропропил)хинолин-3,4-диамина (приблизительно 37,6 ммоля), используя для этой цели обработку 1,1,1-триметоксиэтаном (5,43 г, 45,2 ммоля) в присутствии пиридина гидрохлорида (0,43 г). В результате получали 7,6 г 1-(3-хлоропропил)-2-метил-1Н-имидазо[4,5-с]хинолина, представлявшего собой твердое вещество светло-желтого цвета.
Часть Б
С помощью общего способа, описанного в примере 1, часть Д, 1-(3-хлоропропил)-2-метил-1Н-имидазо[4,5-с]хинолин (7,53 г, 29,0 ммоля) окисляли до 1-(3-хлоропропил)-2-метил)-1Н-имидазо[4,5-с]хинолин-5N-оксида, который получали в форме порошка светло-желтого цвета.
Часть В
С помощью общего способа, описанного в примере 3, часть Б, аминировали материал, полученный в части Б. Первичный продукт этой реакции смешивали с диэтилэфиром и перекристаллизовывали из изопропанола, получая 3,07 г 1-(3-хлоропропил)-2-метил-1Н-имидазо[4,5-с]хинолин-4-амина в форме порошка светло-желтого цвета.
Часть Г
С помощью общего способа, описанного в примере 21, часть В, осуществляли реакцию материала, полученного в части В, с натрия тиометоксидом. Первичный продукт этой реакции перекристаллизовывали из ацетонитрила и мелко растирали в диэтилэфире, получая 3,07 г 1-(3-хлоропропил)-2-метил-1Н-имидазо[4,5-с]хинолин-4-амина, который имел форму игольчатых кристаллов золотистого цвета с температурой плавления 199-202°С.
Результаты анализа. Расчетное содержание в C15H18N4S: % С 62,91; % Н 6,34; % N 19,56, найденное содержание: % С 62,74; % Н 6,20;% N 19,47.
1H-ЯМР (300 МГц, ДМСО-d6): δ 8,13 (d, J=7,5 Hz, 1Н), δ 7,61 (d J=7,5 Hz, 1Н), 7,42 (d, J=7,2 Hz, 1Н), δ 7,24 (t, J=7,2 Hz, 1Н), δ 6,51 (s, 2 Н), δ 4,58 (t, J=7,5 Hz, 2 Н), δ 3,66 (t, J=7,3 Hz, 2 Н), δ 4,58 (t, J=7,5 Hz, 2 Н), δ 2,67-2,61 (m, 5 Н), δ 2,09 (m, 5 Н).
Масс-спектрометрия (CI) для C15H18N4S: m/z 287(MH+).
Пример 23
2-Метил-1-[3-(метилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин
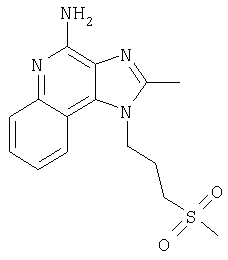
С помощью общего способа, описанного в примере 3, окисляли 2-метил-1-[3-(метилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин (1,8 г, 6,28 ммоля), очищали первичный продукт реакции, получая 0,91 г 2-сульфонил-1-[3-(метилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амина в форме твердого вещества белого цвета, имеющего температуру плавления 225-228°С.
Результаты анализа. Расчетное содержание в C15H18N4O2S: % С 56,59; % Н 5,70; % N 17,60, найденное содержание: % С 56,60; % Н 5,68; % N 17,61.
1H-ЯМР (300 МГц, ДМСО-d6): δ 8,04 (d, J=8,1 Hz, 1Н), δ 7,43 (d J=7,2 Hz, 1Н), 7,25 (dt, J=6,9; 1,2 Hz, 1Н), δ 6,56 (s, 2 Н), δ 4,65 (t, J=7,8 Hz, 2 H), δ 3,38 (t, J=7,8 Hz, 2 H), δ 3,01 (s, 3 H), δ 2,24 (квинтет, J=7,5 Hz, 2 H).
Масс-спектрометрия (CI) для C15H18N4O2S: m/z 319 (MH+).
Пример 24
2-Этил-1-[3-(метилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин
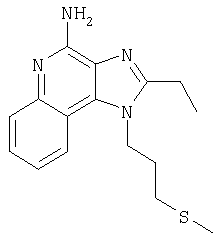
Часть А
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-(3-хлоропропил)хинолин-3,4-диамина (приблизительно 37,6 ммоля), используя для этой цели реакцию с 1,1,1-триэтилортопропионатом (7,69 г, 45,2 ммоля) в присутствии пиридина гидрохлорида (0,43 г).
Первичный продукт реакции подвергали очистке посредством хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 95:5. В результате получали 7,33 г 1-(3-хлоропропил)-2-этил-1Н-имидазо[4,5-с]хинолина, представлявшего собой твердое вещество белого цвета.
Часть Б
С помощью общего способа, описанного в примере 1, часть Д, 1-(3-хлоропропил)-2-этил-1Н-имидазо[4,5-с]хинолин (7,33 г, 26,8 ммоля) окисляли до 1-(3-хлоропропил)-2-этил)-1Н-имидазо[4,5-с]хинолин-5N-оксида, который представлял собой твердое вещество.
Часть В
С помощью общего способа, описанного в примере 3, часть Б, аминировали материал, полученный в части Б. Первичный продукт этой реакции смешивали с диэтилэфиром, получая 6,2 г 1-(3-хлоропропил)-2-этил-1Н-имидазо[4,5-с]хинолин-4-амина в форме порошка белого цвета.
Часть Г
С помощью общего способа, описанного в примере 21, часть В, осуществляли реакцию 1-(3-хлоропропил)-2-этил)-1Н-имидазо[4,5-с]хинолин-4-амина (4,0, 13,85 ммоля) с натрия тиометоксидом (1,53 г, 20,78 ммоля). Первичный продукт этой реакции мелко растирали в диэтилэфире, получая 3,65 г белого порошка.
Навеску этого материала (1,5 г) очищали посредством хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 95:5. В результате получали 1 г 2-этил-1-[3-(метилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амина, представлявшего собой порошок белого цвета, имеющий температуру плавления 210-212°С.
Результаты анализа. Расчетное содержание в C16H20N4S: % С 63,97; % Н 6,71; % N 18,65, найденное содержание: % С 63,70; % Н 5,69;% N 18,62.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 8,14 (d, J=8,7 Hz, 1Н), δ 7,62 (dd J=6,9; 1,2 Hz, 1Н), 7,42 (dt, J=7,2; 1,2 Hz, 1Н), δ 7,25 (dt, J=6,9; 1,2), δ 6,48 (s, 2 Н), δ 4,58 (t, J=7,5 Hz, 2 Н), δ 2,97 (квартет, J=7,5 Hz, 2 Н), δ 2,65 (t, J=6,9 Hz, 2 Н), δ 2,12-2,02 (m, 5 Н), δ 1,38 (t, J=7,5 Hz, 3 Н).
Масс-спектрометрия (CI) для C16H20N4S: m/z 301 (MH+).
Пример 25
2-Этил-1-[3-(метилсульфонил)пропил]-1Н-имидазо [4,5-с]хинолин-4-амин
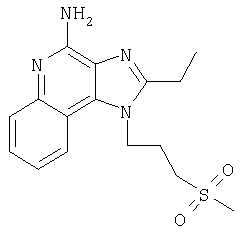
С помощью общего способа, описанного в примере 5, окисляли 2-этил-1-[3-(метилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин (2,1 г, 6,99 ммоля), получая 1,7 г 2-этил-1-[3-(метилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амина, который имел форму мелкого порошка белого цвета с температурой плавления выше 250°С.
Результаты анализа. Расчетное содержание в C16H20N4O2S: % С 57,81; % Н 6,06; % N 16,85, найденное содержание: % С 57,81; % Н 5,88; % N 16,78.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 8,12 (d, J=8,1 Hz, 1Н), δ 7,62 (d J=8,1 Hz, 1Н), 7,43 (t, J=8,1 Hz, 1Н), δ 7,25 (t, J=8,4 Hz, 1Н), δ 6,45 (s, 2 Н), δ 4,65 (t, J=7,8 Hz, 2 Н), δ 3,39 (t, J=7,6 Hz, 2 Н), 3,00 (s, 3 Н), δ 2,96 (квартет, J=7,2 Hz, 2 Н), δ 2,22 (квинтет, J=7,8 Hz, 2 Н), δ 1,38 (t, J=7,2 Hz, 3 Н).
Масс-спектрометрия (CI) для C16H20N4O2S: m/z 333 (MH+).
Пример 26
2-Метил-1-[4-(метитио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин
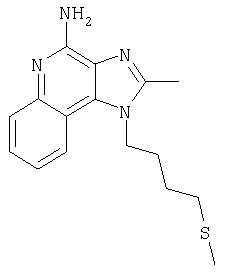
Часть А
С помощью общего способа, описанного в примере 19, часть А, хлорировали 4-[(3-нитрохинолин-4-ил)амино]бутан-1-ол (120 г, 0,459 ммоля), используя для этой цели реакцию с тионила хлоридом (109 г, 0,919 ммоля). В результате получали 127,9 г N-(4-хлоробутил)-3-нитрохинолин-4-амина, который имел форму порошка желтого цвета.
Часть Б
С помощью общего способа, описанного в примере 1, часть В, N-(4-хлоробутил)-3-нитрохинолин-4-амин восстанавливали до N4-(4-хлоробутил)хинолин-3,4-диамина, имевшего форму масла бурого цвета.
Часть В
С помощью общего способа, описанного в примере 1, часть В, осуществляли циклизацию материала, полученного в части Б, используя для этой цели реакцию с 1,1,1-триметоксиэтаном (3,6 г, 30,12 ммоля) в присутствии пиридина хлорида (0,29 г). В результате получали 7,6 г 1-(4-хлоробутил)-2-метил-1Н-имидазо[4,5-с]хинолина, который выделяли в форме масла бурого цвета.
Часть Г
С помощью общего способа, описанного в примере 1, часть Д, 1-(4-хлоробутил)-2-метил-1Н-имидазо[4,5-с]хинолин (5,8 г материала, полученного в части В) окисляли до 1-(4-хлоробутил)-2-метил-1Н-имидазо[4,5-с]хинолин-5N-оксида (приблизительно 6,33 г), который выделяли в форме масла янтарного цвета.
Часть Д
С помощью общего способа, описанного в примере 3, часть Б, материал, полученный в части Г, аминировали и подвергали очистке, получая 1,84 г 1-(4-хлоробутил)-2-метил-1Н-имидазо[4,5-с]хинолин-4-амина, который имел форму рыхлых кристаллов беловатого цвета.
Часть Е
С помощью общего способа, описанного в примере 21, часть Г, осуществляли реакцию материала, полученного в части Д, с натрия тиометоксидом. Первичный продукт этой реакции подвергали перекристаллизации из 1,2-дихлорэтана, после чего мелко растирали в диэтилэфире, получая 1,21 г 2-метил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина в форме порошка белого цвета, имеющего температуру плавления 190-193°С.
Результаты анализа. Расчетное содержание в C16H20N4S: % С 63,97; % Н 6,71; % N 18,65, найденное содержание: % С 63,70; % Н 6,55; % N 18,55.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 8,05 (d, J=8,1 Hz, 1Н), δ 7,69 (d, J=9,3 Hz, 1Н), 7,41 (t, J=8,4 Hz, 1H), 7,24 (t, J=8,4 Hz, 1Н), δ 6,48 (s, 2 Н), δ 5,42 (t, J=7,4 Hz, 2 Н) δ 2,60 (s, 3 Н), δ 2,53 (m, 2 Н), δ 2,02 (s, 3 Н), δ 1,91 (квинтет, J=7,5 Hz, 2 Н), δ 1,69 (квинтет, J=7,5 Hz, 2 Н).
Масс-спектрометрия (CI) для C16H20N4S: m/z 301 (M+Н).
Пример 27
2-Метил-1-[4-(метилсульфинил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
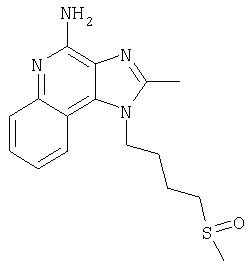
С помощью общего способа, описанного в примере 18, окисляли 2-метил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин (2,15 г, 7,16 ммоля), получая неочищенный сульфоксид. Этот материал последовательно подвергали очистке посредством рекристаллизации из ацетонитрила, хроматографии на силикагеле (используя для элюции смесь дихлорметана и метанола в соотношении 90:10) и растирания в диэтилэфире. В результате получали 0,7 г 2-метил-1-[4-(метилсульфинил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин (2,15 г, 7,16 ммоля), который имел форму белого порошка с температурой плавления 184-187°С.
Результаты анализа. Расчетное содержание в C16H20N4OS: % С 60,73; % Н 6,37;% N 17,71, найденное содержание: % С 60,37;% Н 6,38; % N 17,52.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 8,04 (d, J=8,1 Hz, 1 Н), δ 7,61 (d J=8,1 Hz, 1 Н), 7,42 (dt, J=6,9; 1,3 Hz, 1 Н), δ 7,26 (dt, J=6,9; 1,3 Hz, 1 Н), δ 6,53 (s, 2 Н), δ 4,25 (t, J=7,2 Hz, 2 Н), 2,87-2,66 (m, 2 Н), δ 2,67 (s, 3 Н), δ 2,51 (s, 3 Н), δ 1,95 (m, 2 Н), δ 1,81 (квинтет, J=7,5 Hz, 2 Н).
Масс-спектрометрия (CI) для C16H20N4OS: m/z 317 (МН+).
Пример 28
2-Этил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
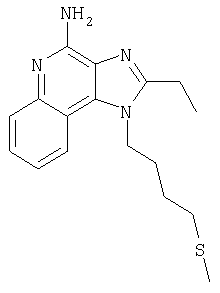
Часть А
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-(3-хлоробутил)хинолин-3,4-диамина (приблизительно 35,75 ммоля), используя для этой цели реакцию с триэтилортопропионатом (7,56 г, 42,9 ммоля) в присутствии пиридина гидрохлорида (0,41 г). Первичный продукт этой реакции подвергали очистке посредством хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 95:5. В результате получали 7,5 г 1-(4-хлоробутил)-2-этил-1Н-имидазо[4,5-с]хинолина в форме белого порошка.
Часть Б
С помощью общего способа, описанного в примере 1, часть В, материал, полученный в части А, окисляли, получая 1-(4-хлоробутил)-2-этил-1Н-имидазо[4,5-с]хинолин-5N-оксид в форме твердого вещества желтовато-коричневого цвета.
Часть В
С помощью общего способа, описанного в примере 3, часть Б, материал, полученный в части Б, подвергали аминированияю и последующей очистке для получения 7,0 г 1-(4-хлоробутил)-2-этил-1Н-имидазо[4,5-с]хинолин-4-амина, который имел форму белого порошка.
Часть Г
С помощью общего способа, описанного в примере 21, часть Г, осуществляли реакцию материала, полученного в части В, с натрия триметоксидом. Первичный продукт этой реакции подвергали рекристаллизации из изопропанола с последующим растиранием в диэтилэфире. В результате получали 1,55 г 2-этил-1-[3-(метилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амина, который имел форму порошка белого цвета с температурой плавления 183-186°С.
Результаты анализа. Расчетное содержание в C17H22N4S: % С 64,93; % Н 7,05; % N 17,82, найденное содержание: % С 65,07; % Н 7,17; % N 17,66.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 8,04 (d, J=7,5 Hz, 1Н), δ 7,61 (dd, J=9,3; 1,5 Hz, 1Н), 7,41 (dt, J=7,8; 1,4 Hz, 1Н), δ 6,44 (s, 2 Н), δ 4,52 (t, J=7,50 Hz, 2 Н), δ 2,95 (квартет, J=7,5 Hz, 2 Н), 2,55 (m, 2 Н), δ 2,02 (s, 3 Н), δ 1,90 (m, 2 Н), δ 1,71 (m, 2 Н), δ 1,38 (t, J=7,2 Hz, 2 Н).
Масс-спектрометрия (CI) для C17H22N4S: m/z 315 (M+Н).
Пример 29
2-Этил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
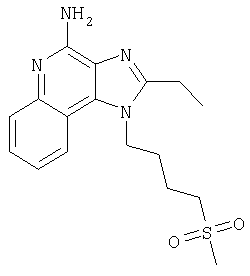
С помощью общего способа, описанного в примере 5, окисляли 2-этил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин (2,3 г, 7,31 ммоля). Полученный неочищенный материал последовательно очищали посредством растирания в порошок в диэтилэфире, хроматографии на силикагеле (используя для элюции смесь дихлорметана и метанола в соотношении 90:10), рекристаллизации из этанола и растирания в диэтилэфире. В результате получали 1,18 г 2-этил-1-[4-(метилсульфонил)бутил]-1Н-имидазо [4,5-с]хинолин-4-амина (2,15 г, 7,16 ммоля), имевшего форму белого порошка с температурой плавления 182-185°С.
Результаты анализа. Расчетное содержание в C17H22N4O2S: % С 58,94; % Н 6,40; % N 16,17, найденное содержание: % С 58,89; % Н 6,51; % N 16,13.
1H-ЯМР (300 МГц, ДМСО-d6): δ 8,04 (d, J=8,1 Hz, 1Н), δ 7,62 (dd, J=8,1; 1,5 Hz, 1Н), 7,42 (dt, J=6,9; 1,2 Hz, 1Н), δ 7,26 (t, J=7,4 Hz, 1Н), δ 6,45 (s, 2 Н), δ 4,55 (t, J=7,05 Hz, 2 Н), 3,21 (t, J=7,2 Hz, 2 Н), δ 2,96 (m, 5 Н), δ 1,91 (m, 4 Н), δ 1,38 (t, J=7,2 Hz, 3 Н).
Масс-спектрометрия (CI) для C17H22N4O2S: m/z 347(MH+).
Пример 30
1-[4-(Метилсульфонил)бутил]-2-пропил-1Н-имидазо[4,5-с]хинолин-4-амин
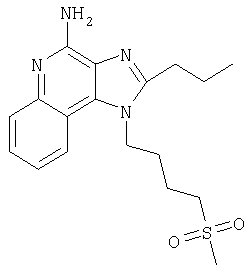
Часть А
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-(3-хлоробутил)хинолин-3,4-диамина (приблизительно 21,45 ммоля), используя для этой цели реакцию с триэтилортобутиратом (3,8 г, 25,74 ммоля) в присутствии пиридина гидрохлорида (0,1 г). Первичный продукт этой реакции подвергали очистке посредством хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 95:5. В результате получали 3,6 г 1-(4-хлоробутил)-2-пропил-1Н-имидазо[4,5-с]хинолина в форме медленно отверждающегося масла зеленого цвета.
Часть Б
С помощью общего способа, описанного в примере 1, часть В, материал, полученный в части А, окисляли, получая 1-(4-хлоробутил)-2-пропил-1Н-имидазо[4,5-с]хинолин-5N-оксид в форме масла светло-оранжевого цвета.
Часть В
С помощью общего способа, описанного в примере 3, часть Б, материал, полученный в части Б, подвергали аминированию и последующей очистке для получения 3,0 г 1-(4-хлоробутил)-2-пропил-1Н-имидазо[4,5-с]хинолин-4-амина, который имел форму беловатого порошка.
Часть Г
С помощью общего способа, описанного в примере 21, часть Г, осуществляли реакцию материала, полученного в части В, с натрия триметоксидом, получая 2,52 г 1-[4-(метилтио)бутил]-2-пропил-1Н-имидазо[4,5-с]хинолин-4-амина в форме твердого вещества.
Часть Д
С помощью общего способа, описанного в примере 5, часть Г, окисляли материал, полученный в части Г. Первичный продукт этой реакции последовательно очищали посредством хроматографии на силикагеле (используя для элюции смесь дихлорметана и метанола в соотношении 95:5), рекристаллизации из этанола и растирания в порошок в диэтилэфире. В результате получали 1,15 г твердого вещества, которое разводили в N,N-диметилформамиде (6 мл), и переливали конечный раствор в воду (100 мл). При этом выпадал осадок, который собирали посредством фильтрования, промывали водой и высушивали, получая 1,0 г 1-[4-(метилсульфонил)бутил]-2-пропил-1Н-имидазо[4,5-с]хинолин-4-амина, который имел форму белого порошка с температурой плавления 202-204°С.
Результаты анализа. Расчетное содержание в C18H24N4O2S: % С 59,98; % Н 6,71; % N 15,54, найденное содержание: % С 59,71; % Н 6,69; % N 15,41.
1H-ЯМР (300 МГц, ДМСО-d6): δ 8,03 (d, J=8,1 Hz, 1Н), δ 7,61 (dd, J=1,8; 1,2 Hz, 1Н), 7,42 (dt, J=7,2; 1,2 Hz, 1Н), δ 7,26 (t, J=7,50 Hz, 2 Н), δ 6,44 (s, 2 Н), δ 4,55 (t, J=6,5 Hz, 2 Н), δ 3,21 (t, J=7,2 Hz, 2 Н), δ 2,96 (s, 3 Н), δ 2,02 (s, 3 Н), δ 2,91 (t, J=7,5 Hz, 2 Н), δ 1,92-1,79 (m, 6 Н), δ 1,04 (t, J=7,5 Hz, 3H).
Масс-спектрометрия (CI) для C18H24N4O2S: m/z 361 (M+Н).
Пример 31
2-Бутил-1-[4-(метилсульфинил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
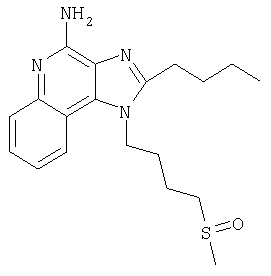
С помощью общего способа, описанного в примере 18, окисляли 2-бутил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин (2,5 г, 7,30 ммоля). После очистки получали 1,5 г 2-бутил-1-[4-(метилсульфинил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин, имевший форму твердого вещества белого цвета с температурой плавления 126-128°С.
Результаты анализа. Расчетное содержание в C19H26N4OS: % С 62,87; % Н 7,36; % N 1,43, найденное содержание: % С 62,57; % Н 7,34; % N 15,47.
1H-ЯМР (300 МГц, ДМСО-d6): δ 8,03 (d, J=8,1 Hz, 1Н), δ 7,61 (d, J=9,0 Hz, 1Н), 7,41 (t, J=6,9, Hz, 1Н), δ 7,25 (t, J=7,1 Hz, 1Н), δ 6,46 (s, 2 Н), δ 4,56 (t, J=7,3 Hz, 2 Н), 2,93 (t, J=7,8 Hz, 2 Н), δ 2,87-2,66 (m, 5 Н), δ 2,51 (s, 3 Н), δ 1,93-1,75 (m, 6 Н), δ 1,46 (секстет, J=7,5 Hz, 3 Н), δ 0,96 (t, J=7,4 Hz, 3 Н).
Масс-спектрометрия (CI) для C19H26N4OS: m/z 359 (M+Н).
Пример 32
2-Метил-1-[2-(метилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
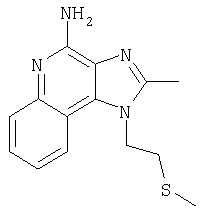
Часть А
С помощью общего способа, описанного в примере 6, часть А (дополнительно в качестве растворителя использовали 55 мл 1,2-дихлорэтана), хлорировали 2-(4-амино-2-метил-1Н-имидазо[4,5-с]хинолин-1-ил)этанол, используя для этой цели реакцию с тионила хлоридом (2,41 мл, 33,02 ммоля). В результате получали 3,9 г 1-(2-хлороэтил)-2-метил-1Н-имидазо[4,5-с]хинолин-4-амина, который имел форму мелкого порошка белого цвета.
Часть Б
С помощью общего способа, описанного в примере 21, часть Г, осуществляли реакцию 1-(2-хлороэтил)-2-метил-1Н-имидазо[4,5-с]хинолин-4-амина (3,75 г, 21,57 ммоля) с натрия тиометоксидом (1,6 г, 21,57 ммоля), получая 3,2 г тиоэфира в форме твердого вещества беловатого цвета. Навеску этого материала (1,4 г) подвергали рекристаллизации из этанола, а потом мелко растирали в диэтилэфире. В результате получали 0,9 г 2-метил-1-[2-(метилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амина, который имел форму мелкого белого порошка с температурой плавления 193-195°С.
Результаты анализа. Расчетное содержание в C14H16N4S: % С 61,74; % Н 5,92; % N 20,57, найденное содержание: % С 61,64; % Н 5,97; % N 20,66.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 8,00 (dd, J=7,8; 1,5 Hz, 1Н), δ 7,61 (dd, J=8,1; 1,4 Hz, 1Н), 7,19 (dt, J=7,8; 1,2, Hz, 1Н), δ 7,25 (dt, J=7,5; 1,5 Hz, 1Н), δ 6,51 (s, 2 Н), δ 4,72 (t, J=6,9 Hz, 2 Н), 2,99 (t, J=6,8 Hz, 2 Н), δ 2,66 (m, 3 Н), δ 2,08 (s, 3 Н).
Масс-спектрометрия (CI) для C14H16N4S: m/z 273 (M+Н).
Пример 33
2-Метил-1-[2-(метилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
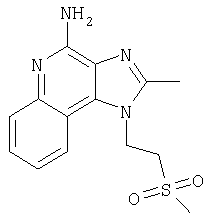
В суспензию 2-метил-1-[4-(метилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амина (1,8 г, 6,61 ммоля) в хлороформе добавляли 3-хлорпербензойную кислоту (33,5 г 75%-ной кислоты, 14,54 ммоля). Добавление примерно одного эквивалента окисляющего реагента вызывало образование осадка. После этого в реакционную систему дополнительно вводили хлороформ, перемешивали смесь при комнатной температуре на протяжении 1 часа и охлаждали на водяной бане со льдом. Твердый осадок белого цвета отделяли посредством фильтрования, после чего промывали его холодным дихлорметаном. Твердый материал суспендировали в воде (100 мл) и добавляли в нее твердый натрия карбонат, доводя рН суспензии до 10,0. После этого ее перемешивали при комнатной температуре на протяжении нескольких часов, твердый материал собирали посредством фильтрации и промывали его водой, получая примерно 1,5 г вещества белого цвета. Анализ с помощью 1H-ЯМР выявил присутствие сульфоксида. Этот материал суспендировали в дихлорметане (23 мл) и отдельными порциями добавляли в суспензию 3-хлорпербензойной кислоты (0,25 г). Спустя приблизительно 15 минут в реакционную систему вводили дополнительное количество окисляющего реагента. Смесь концентрировали при пониженном давлении, а остаток переносили в воду (100 мл), доводя ее рН до 10,0 добавлением твердого натрия карбоната. Твердый материал бурого цвета отделяли посредством фильтрования, промывали водой и подвергали очистке с помощью хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 95:5. Полученный после хроматографии материал мелко растирали в диэтилэфире. В результате получали 0,56 г 1-метил-1-[2-(метилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амина, который имел форму мелкого белого порошка, имеющего температуру плавления 242-245°С.
Результаты анализа. Расчетное содержание в С14Н16N4O2S: % С 55,25; % Н 5,30; % N 18,41, найденное содержание: % С 54,92; % Н 5,19; % N 18,29.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 8,05 (d, J=6,6 Hz, 1Н), δ 7,63 (d, J=6,6 Hz, 1Н), δ 7,43 (t, J=7,2 Hz, 2 Н), δ 7,25 (t, J=6,9 Hz, 2 Н), δ 6,31 (s, 2 Н), δ 4,95 (t, J=7,2 Hz, 2 Н), δ 3,77 (t, J=7,2 Hz, 2 Н), 3,08 (s, 3 Н), 2,66 (s, 3 Н).
Масс-спектрометрия (CI) для C14H16N4O2S: m/z 305 (M+Н).
Пример 34
2-Метил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин
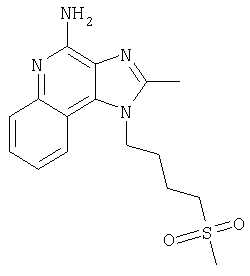
Часть А
В раствор 1-(4-(хлоробутил)-2-метил-1Н-имидазо[4,5-с]хинолина (5,0 г, 18,26 ммоля) в безводном N,N-диметилформамиде (91 мл) за один прием вносили навеску натрия диметоксида (2,02 г, 27,40 ммоля). Спустя 30 минут реакционную смесь переливали в воду (500 мл) при непрерывном перемешивании. Полученный таким образом раствор дважды экстрагировали хлороформом (по 200 мл). Органические фракции объединяли, промывали насыщенным водным раствором натрия бикарбоната (100 мл) и солевым раствором (100 мл), высушивали над безводным натрия сульфатом, фильтровали, а потом концентрировали в условиях пониженного давления, получая 5,0 г 2-метил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолина в форме маслянистой жидкости светло-желтого цвета.
Часть Б
С помощью общего способа, описанного в примере 11, часть А, материал, полученный в части А, окисляли до 2-метил-1-[4-(метилсульфонил)бутил]-1Н-имидазо [4,5-с]хинолин-5N-оксида, который имел форму твердого вещества светло-оранжевого цвета.
Часть В
С помощью общего способа, описанного в примере 3, часть Б (но используя в качестве растворителя дихлорметан вместо хлороформа), аминировали материал, полученный в части Б. Первичный продукт этой реакции подвергали очистке с помощью хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 90:10. Полученный после хроматографии материал мелко растирали в диэтилэфире. В результате получали 1,67 г 2-метил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина, имевшего форму мелкого белого порошка с температурой плавления 206-209°С.
Результаты анализа. Расчетное содержание в C16H20N4O2S: % С 57,81; % Н 6,06; % N 16,85, найденное содержание: % С 57,70; % Н 6,10; % N 16,64.
1H-ЯМР (300 МГц, ДМСО-d6): δ 8,05 (d, J=8,1 Hz, 1Н), δ 7,61 (d J=8,7 Hz, 1Н), 7,42 (d, J=7,8 Hz, 1Н), δ 7,25 (t, J=7,6 Hz, 2 Н), δ 6,51 (s, 2 Н), δ 4,55 (t, J=7,2 Hz, 2 Н), δ 3,20 (t, J=7,4 Hz, 2 Н), 2,96 (s, 3 Н), 2,61 (s,3H), 1,91 (m, 4H).
Macc-спектрометрия (CI) для C16H20N4O2S: m/z 333 (M+Н).
Пример 35
2-Этил-1-[4-(метилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
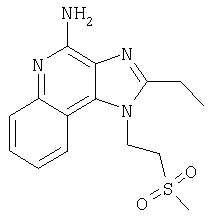
Часть А
С помощью общего способа, описанного в примере 1, часть А, осуществляли реакцию 4-хлоро-3-нитрохинолина (15,0 г, 71,90 ммоля) с 2-хлороэтиламина моногидрохлоридом (8,3 г, 71,90 ммоля). Первичный продукт этой реакции суспендировали в воде (300 мл) и добавлением твердого натрия карбоната доводили рН суспензии до 10,0. Суспензию перемешивали в течение ночи, после чего охлаждали на водяной бане со льдом.
Твердый материал изолировали фильтрованием и промывали холодной водой, получая 15,88 г N-(2-хлороэтил)-3-нитрохинолин-4-амина в форме рыхлого твердого вещества ярко-желтого цвета.
Часть Б
С помощью общего способа, описанного в примере 21, часть Г, осуществляли реакцию N-(2-хлороэтил)-3-нитрохинолин-4-амина (6,0 г, 23,84 ммоля) с натрия тиометоксидом (2,11 г 95%-ного продукта, 28,61 ммоля), получая 4,95 г N-[2-(метилтио)этил]-3-нитрохинолин-4-амина, который имел форму твердого вещества тусклого желтого цвета.
Часть В
С помощью общего способа, описанного в примере 1, часть В, N-[2-(метилтио)этил]-3-нитрохинолин-4-амин (4,71 г, 17,89 ммоля) восстанавливали для получения N4-[2-(метилтио)этил]хинолин-3,4-диамина в форме масла светло-коричневого цвета.
Часть Г
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию материала, полученного в части В. Для этого использовали реакцию с триэтилортофосфатом. Первичный продукт реакции подвергали очистке с помощью хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола. В результате получали 3,1 г 2-этил-1-[2-(метилтио)этил]-1Н-имидазо[4,5-с]хинолина, который имел форму твердого вещества беловатого цвета.
Часть Д
С помощью общего способа, описанного в примере 11, часть Е, осуществляли окисление материала, полученного в части Г, до 2-этил-1-[2-(метилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-5N-оксида, который имел форму твердого вещества беловатого цвета.
Часть Е
С помощью общего способа, описанного в примере 3, часть Б, аминировали материал, полученный в части Д. Продукт этой реакции очищали, получая 0,2 г 2-этил-1-[2-(метилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амина, имевшего форму твердого вещества с температурой плавления 222-225°С.
Результаты анализа. Расчетное содержание в C15H18N4O2S: % С 56,59; % Н 5,70; % N 17,60, найденное содержание: % С 56,37; % Н 5,59; % N 17,34.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 8,06 (d, J=8,7 Hz, 1Н), δ 7,63 (d J=8,1 Hz, 1Н), δ 7,26 (d, J=8,1 Hz, 1Н), δ 6,50 (s, 2 Н), δ 4,95 (t, J=7,2 Hz, 2 Н), δ 3,78 (t, J=7,2 Hz, 2 Н), δ 3,12 (s, 3 Н), δ 3,02 (квартет, J=7,5 Hz, 2 Н), 1,39 (t, J=7,6 Hz, 2 Н), 1,39 (t, J=7,2 Hz, 3 Н).
Масс-спектрометрия (CI) для C15H18N4O2S: m/z 319 (М+Н).
Пример 36
1-[2-(Метилсульфонил)этил]-2-пропил-1Н-имидазо[4,5-с]хинолин-4-амин
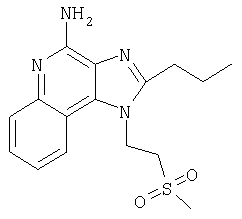
Часть А
В сосуд высокого давления помещали N4-[2-(метилтио)этил]хинолин-3,4-диамин (4,2 г, 19,2 ммоля), триметилортобутират (2,86 г, 19,2 ммоля), пиридина гидрохлорид (каталитическое количество) и толуол. Реакционную систему нагревали при 140°С на протяжении 1 часа. Затем реакционную систему охлаждали и концентрировали при пониженном давлении, получая 4,9 г 1-[2-(метилсульфонил)этил]-2-пропил-1Н-имидазо[4,5-с]хинолина.
Часть Б
Полученный в части А материал растворяли в хлороформе (100 мл) и добавляли в раствор отдельными порциями 3-хлорпербензойную кислоту (14,12 г 65%-ной кислоты, 53,2 ммоля). Спустя примерно 30 минут реакционную систему последовательно промывали водным раствором натрия карбоната, водой и солевым раствором. Органическую фракцию объединяли с избыточным количеством гидроокиси аммония. В этот материал при энергичном перемешивании отдельными порциями вводили п-толуолсульфонила хлорид (3,6 г, 18,9 ммоля). Через 1 час реакционную систему разбавляли хлороформом (100 мл) и водой (100 мл). Отделяли органический слой, промывали его водой и концентрировали при пониженном давлении. Полученную в результате этих операций маслянистую жидкость подвергали очистке с помощью хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 98:2. Полученный после хроматографии материал перекристаллизовывали из дихлорметана и отделяли посредством фильтрования. В результате получали 0,9 г 1-[2-(метилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амина, который представлял собой твердое вещество с температурой плавления 212-214°С.
Результаты анализа. Расчетное содержание в C16H20N4O2S: % С 56,94; % Н 5,99; % N 16,52, найденное содержание: % С 56,95; % Н 5,91; % N 16,59.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 8,06 (d, J=7,5 Hz, 1Н), δ 7,64 (d J=7,2 Hz, 1Н), δ 7,43 (d, J=7,2 Hz, 1Н), δ 7,25 (t, J=6,9 Hz, 2 Н), δ 6,47 (s, 2 Н), δ 4,94 (t, J=7,2 Hz, 2 Н), δ 3,76 (t, J=7,2 Hz, 2 Н), δ 3,11 (s, 3 Н), δ 2,97 (t, J=7,5 Hz, 2 Н), δ 1,87 (секстет, J=7,2 Hz, 2 Н), δ 1,04 (t, J=7,2 Hz, 3 Н).
Масс-спектрометрия (CI) для C16H20N4O2S: m/z 333 (M+Н).
Пример 37
2-Бутил-1-4-[2,4-(дифторофенил)тио]бутил-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-(4-хлоробутил)хинолина-3,4-диамина (119,1 г, 0,48 ммоля) посредством его обработки триэтилортовалератом (93 г, 0,57 ммоля) в присутствии пиридина гидрохлорида (1,1 г, 0,0095 моля).
В результате получали 120 г 2-бутил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолина в форме порошка цвета слоновой кости.
Часть Б
В раствор 2-бутил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолина (118 г, 0,037 моля) в дихлорметане (1700 мл) на протяжении 30 минут отдельными порциями добавляли 3-хлорпербензойную кислоту (110 г 77%-ной кислоты, 0,45 моля). Спустя примерно 90 минут реакционную систему разбавляли дополнительным количеством дихлорметана, трижды промывали 10%-ным водным раствором натрия карбоната, а затем солевым раствором, после чего высушивали, получая 2-бутил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-5N-оксид.
Часть В
2-Бутил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолина-5N-оксид, полученный в части Б, растворяли в дихлорметане и добавляли в раствор концентрированную гидроокись аммония (1100 мл). Затем в эту систему при энергичном перемешивании отдельными порциями вводили тозила хлорид (68 г, 0,36 моля). Спустя 30 минут разделяли слои, органическую фракцию разбавляли дихлорметаном, дважды промывали 10%-ной гидроокисью натрия, а затем солевым раствором, высушивали и концентрировали в условиях пониженного давления, получая твердое вещество желтовато-коричневого цвета. Этот материал подвергали перекристаллизации из ацетонитрила (30 мл/г) и получали 91,6 г 2-бутил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-амина, представлявшего собой иглообразные кристаллы желтовато-коричневого цвета.
Результаты анализа. Расчетное содержание в С18Н23ClN4: % С 65,34%; Н 7,01; % N 16,93, найденное содержание: % С 65,32; % Н 7,09; % N 16,94.
Часть Г
В суспензию натрия гидрида (0,65 г 60%-ного материала, 16,5 ммоля) в безводном N,N-диметилформамиде (30 мл) добавляли 2,4-дифторобензолтиол (2 г, 13,7 ммоля). По завершении этой процедуры реакционную систему перемешивали при комнатной температуре на протяжении примерно 30 минут. Затем в нее за один прием вводили навеску 2-бутил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолин-4-амина (4,5 г, 13,6 ммоля). Смесь перемешивали при комнатной температуре в течение примерно 30 минут, а затем переливали ее в ледяную воду и снова перемешивали. Водную фракцию пятикратно экстрагировали дихлорметаном (по 75 мл). Объединенную органическую фракцию трижды промывали водой (по 100 мл), а затем солевым раствором. Конечный материал концентрировали при пониженном давлении, получая 6,7 г твердого вещества. Последнее подвергали перекристаллизации из этанола. Навеску полученного материала (1,1 г) высушивали в горячей вакуумной печи до получения твердого вещества, которое представляло собой 2-бутил-1-4-[2,4-(дифторофенил)тио]бутил-1Н-имидазо[4,5-с]хинолин-4-амин, имевший температуру плавления 122-126°С.
Результаты анализа. Расчетное содержание в C24H26F2N4S: % С 65,43; % Н 5,95; % N 12,72, найденное содержание: % С 65,41; % Н 5,98: % N 12,80.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 8,02 (d, J=7,5 Hz, 1 H), δ 7,62 (d, J=7,2 Hz, 1 H), δ 7,42 (m, 2 H), δ 7,25 (m, 2 H), δ 7,05 (t, J=6,0 Hz, 1 H), δ 6,46 (s, 2 H), δ 4,50 (t, J=7,5 Hz, 2 H), δ 2,97 (t, J=6,6 Hz, 2 H), δ 2,87 (t, J=7,2 Hz, 2 H), δ 1,92 (квинтет, J=7,8 Hz, 2 H), δ 1,76 (квинтет, J=7,5 Hz, 2 H), δ 1,42 (секстет, J=7,5 Hz, 2 H), δ 0,94 (t, J=7,2 Hz, 3 H).
Масс-спектрометрия (CI) для С24Н26F2N4S: m/z 441 (M+H).
Пример 38
2-Бутил-1-4-[2,4-(дифторофенил)сульфонил]бутил-1Н-имидазо[4,5-с]хинолин-4-амин
В раствор 2-бутил-1-4-[2,4-(дифторофенил)тио]бутил-1Н-имидазо[4,5-с]хинолин-4-амина (5,0 г, 11,3 ммоля) в дихлорметане (50 мл) отдельными порциями добавляли 3-хлорпербензойную кислоту (6,025 г 65%-ной кислоты, 22,6 моля). По завершении этой процедуры реакционную систему перемешивали на протяжении примерно 30 минут. После этого ее разделяли между дихлорметаном и водным раствором натрия карбоната. Объединенные органические фракции трижды экстрагировали дихлорметаном (по 500 мл). Полученный материал пятикратно промывали водой (по 100 мл), а затем солевым раствором, после чего концентрировали при пониженном давлении. Остаток (6,1 г) подвергали перекристаллизации из этанола и получали 91,6 г 2-бутил-1-4-[2,4-(дифторофенил)сульфонил]бутил-1Н-имидазо[4,5-с]хинолин-4-амина, который представлял собой твердое вещество, имевшее температуру плавления 190-193°С.
Результаты анализа. Расчетное содержание в C24H26F2N4O2S: % С 6,00; % Н 5,55; % N 11,86, найденное содержание: % С 61,33; % Н 5,38; % N 11,70.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 8,00 (d, J=8,1 Hz, 1Н), δ 7,83 (d, J=8,7 Hz, 1Н), δ 7,62 (m, 2 Н), δ 7,41 (m, 2 Н), δ 7,22 (t, J=6,9 Hz, 1Н), δ 6,46 (s, 2 Н), δ 4,51 (t, J=6,9 Hz, 2Н), δ 3,49 (t, J=7,5 Hz, 2 Н), δ 2,87 (t, J=7,5 Hz, 2 Н), δ 1,90 (m, 2 Н), δ 1,77 (m, 4 Н), δ 1,43 (секстет, J=7,5 Hz, 2 Н), δ 0,95 (t, J=7,5 Hz, 3 Н).
Масс-спектрометрия (CI) для C24H26F2N4O2S: m/z 473 (M+Н).
Примеры 39-42
Тиоэфиры, перечисленные в нижеследующей таблице 1, были получены посредством реакции 2-бутил-1-(4-хлоробутил)-1Н-имидазо[4,5-с]хинолинамина с соответствующим тиолом по способу, описанному в примере 37, часть Г. Сульфоны получали посредством окисления соответствующего тиоэфира по способу, описанному в примере 38.
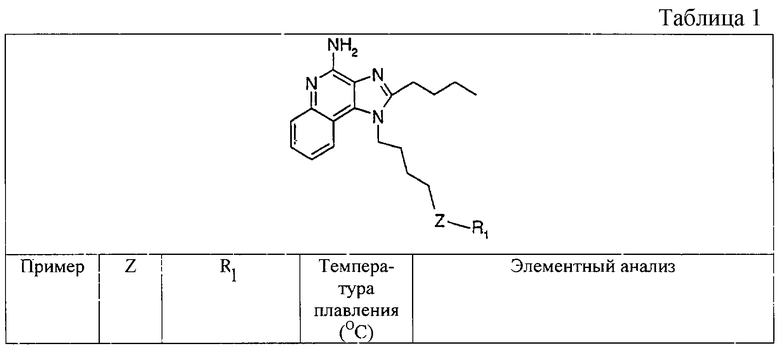

Примеры 43-55
Часть А
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-(4-хлоробутил)хинолин-3,4-диамина (30 г, 0,12 моля), используя для этой цели обработку триметила ортопропионатом (23,3 г, 0,13 моля) в присутствии каталитического количества пиридина гидрохлорида, и получали 25,1 г 1-(4-хлоробутил)-2-этил-1Н-имидазо[4,5-с]хинолина, который представлял собой твердое вещество.
Часть Б
3-Хлорбензойную кислоту (20,1 г 60% кислоты, 0,117 молей) отдельными порциями добавляли в раствор 1-(4-хлоробутил)-2-этил-1Н-имидазо[4,5-с]хинолина (24 г, 0,084 моля) в дихлорметане. Реакционную смесь разбавляли 5% натрия карбонатом в количестве, достаточном для поддержания рН водного слоя на уровне 9-10.
Образовавшиеся слои разделяли, органическую фракцию промывали последовательно дополнительным количеством натрия карбоната, водой (250 мл) и солевым раствором, после чего концентрировали в условиях пониженного давления. Образовавшийся остаток разводили в хлороформе (350 мл). В полученный раствор добавляли гидроокись аммония (250 мл), энергично перемешивая смесь до получения эмульсии. Затем, продолжая перемешивание, отдельными порциями добавляли тозила хлорид (19,2 г, 0,10 моля). Реакционную смесь дважды промывали водой (по 100 мл), 5% натрия карбонатом (дважды по 200 мл) и солевым раствором. Затем ее высушивали над натрия карбонатом и концентрировали при пониженном давлении. Остаток объединяли с диэтилэфиром и перемешивали в течение ночи. Образовавшийся твердый материал отделяли посредством фильтрации и высушивали на воздухе, получая 19,3 г 1-(4-хлоробутил)-2-этил-1Н-имидазо[4,5-с]хинолин-4-амина.
Часть С
Тиоэфиры, включенные в приводимую ниже таблицу 2, получали посредством реакции 1-(4-хлоробутил)-2-этил-1Н-имидазо[4,5-с]хинолин-4-амина с соответствующим тиолом, с помощью методики, описанной в примере 37, часть Г. Сульфоны получали посредством окисления соответствующего тиоэфира по способу, описанному в примере 38.
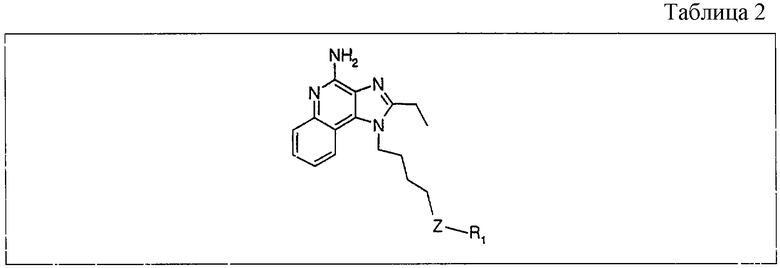
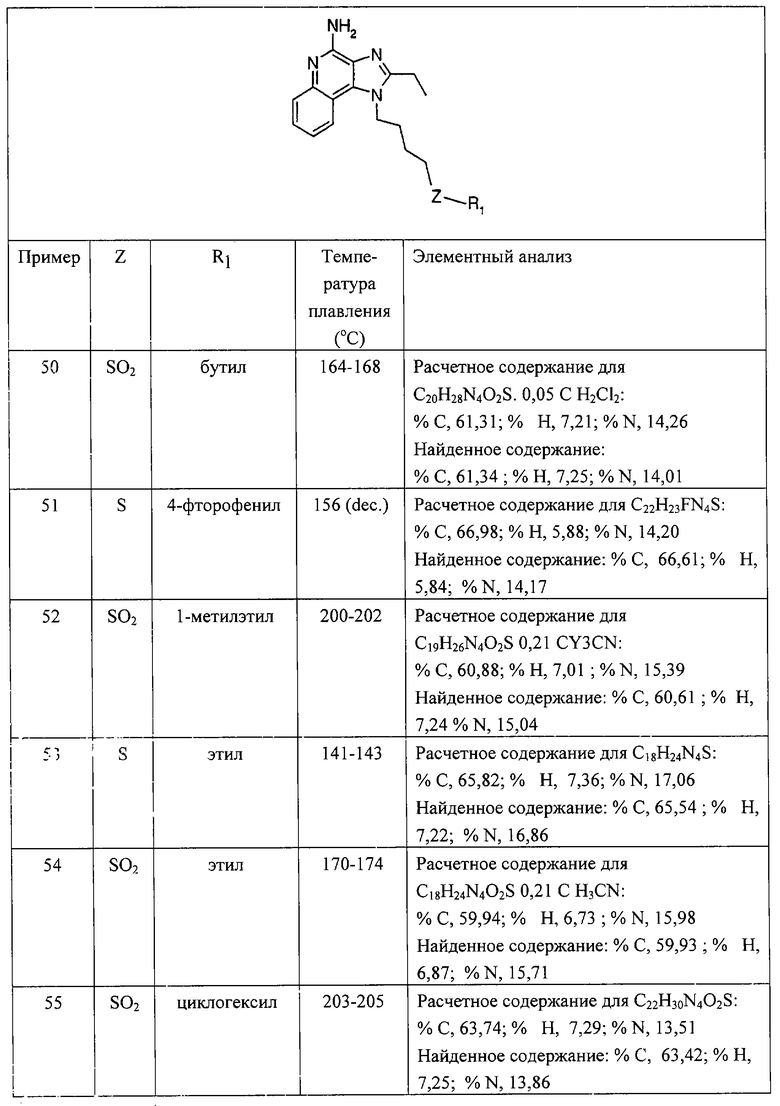


Примеры 56-66
Часть А
В раствор 1-(4-хлоробутил)-2-этил-1Н-имидазо[4,5-с]хинолина (3,7 г, 13 ммолей) в толуоле, содержавшем каталитическое количество N,N-диметилформамида, добавляли тионила хлорид (3,8 г, 32 ммоля). Реакционную смесь нагревали в условиях обратного тока, а затем укупоривали содержащий ее сосуд. После того как данные анализа способом высокоэффективной жидкостной хроматографии указывали на завершение реакции, реакционную смесь концентрировали в условиях пониженного давления. Остаток разводили в подогретом метаноле и объединяли полученный материал с концентрированным раствором аммония хлорида (5 мл). Смесь охлаждали. Выпавший осадок отделяли посредством фильтрации, промывали холодным метанолом, а затем высушивали в вакууме на протяжении ночи, получая 3,01 г 2-бутил-1-(2-хлороэтил)-1Н-имидазо[4,5-с]хинолин-4-амина в форме твердого вещества бурого цвета.
Часть Б
Тиоэфиры, включенные в приводимую ниже таблицу 3, получали посредством реакции 2-бутил-1-(2-хлороэтил)-1Н-имидазо[4,5-с]хинолин-4-амина с соответствующим тиолом, с помощью методики, описанной в примере 37, часть Г. Сульфоны получали посредством окисления соответствующего тиоэфира по способу, описанному в примере 38.

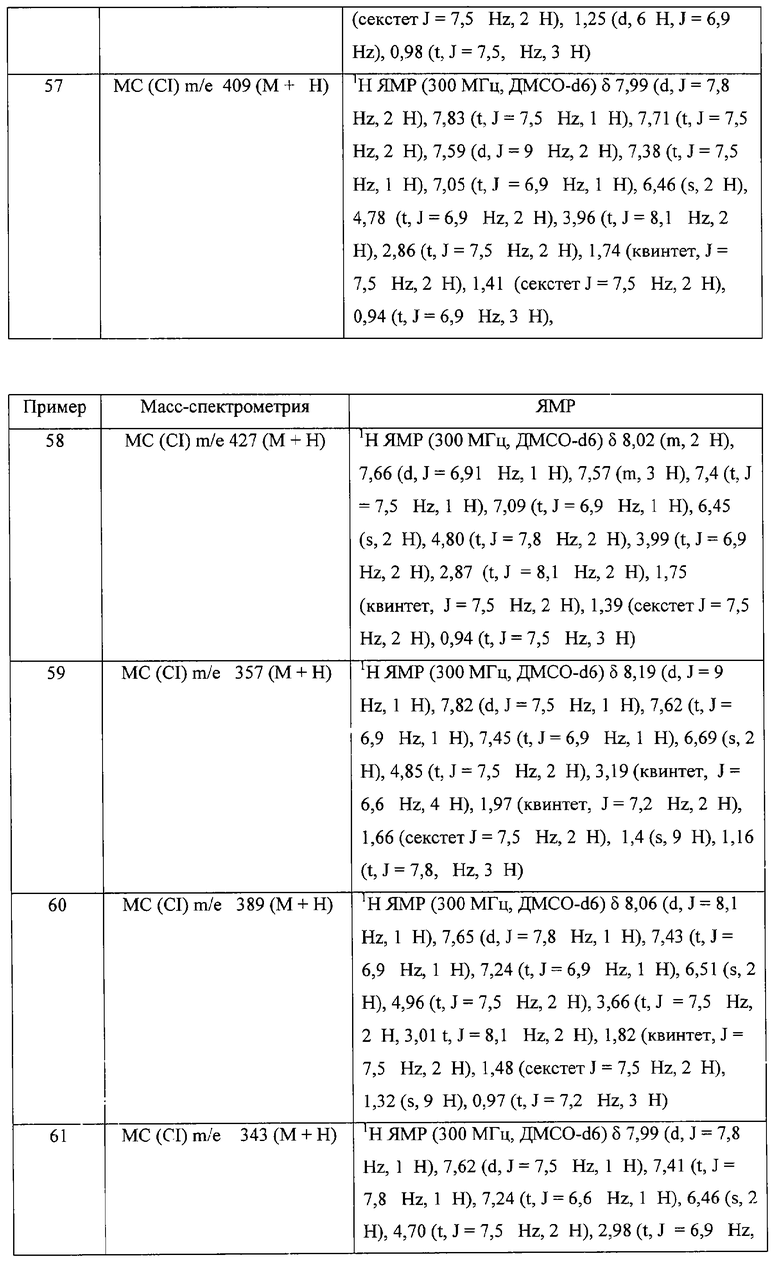
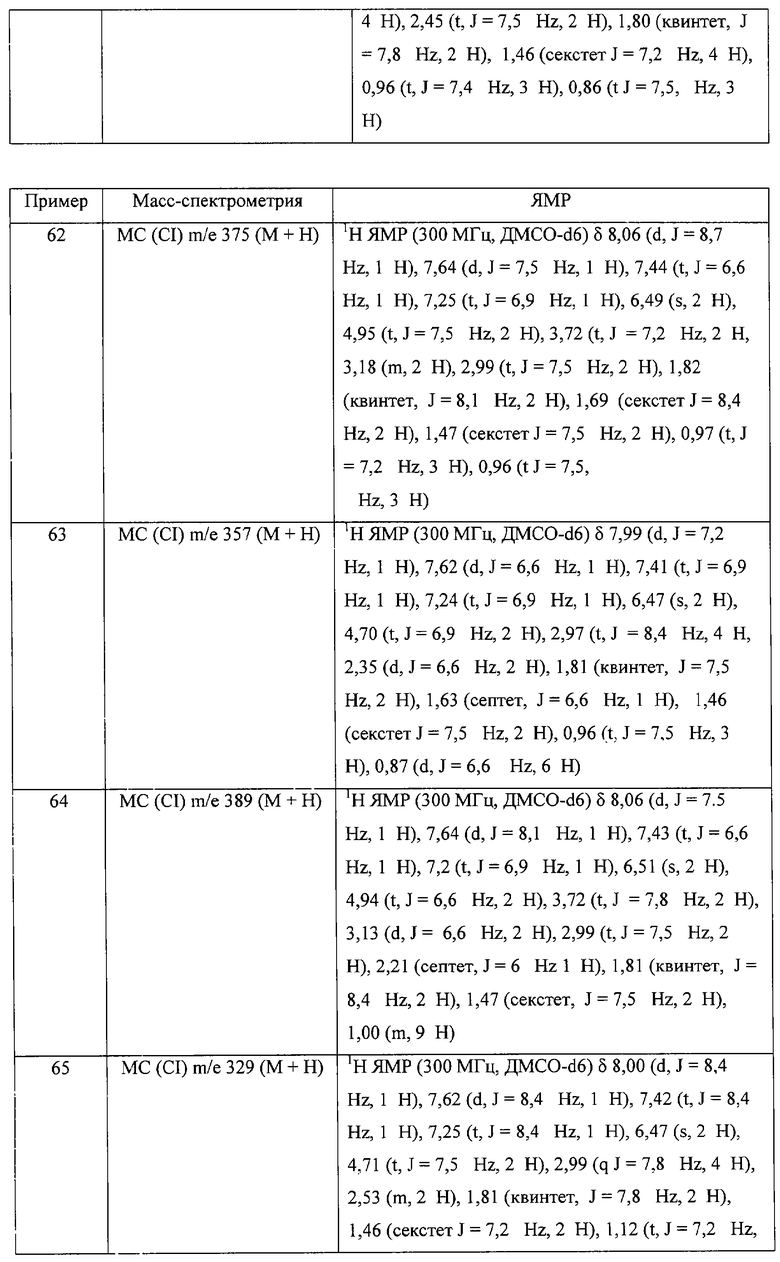

Пример 67
2-Бутил-1-[2-(метилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
С помощью общего способа, описанного в примере 21, часть Г, осуществляли реакцию 2-бутил-1-(2-хлороэтил)-1Н-имидазо[4,5-с]хинолин-4-амина (1,44 г, 4,76 ммоля) с натрия метоксидом (0,42 г 95% вещества, 5,71 ммоля) для получения 1,4 г 2-бутил-1-[2-(метилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в форме порошка белого цвета.
Часть Б
С помощью общего способа, описанного в примере 5, производили окисление 2-бутил-1-[2-(метилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амина (1,35 г, 4,29 моля). Первичный продукт окисления очищали с помощью хроматографии на силикагеле, используя для элюции смесь дихлорметана и метанола в соотношении 95:5. Затем этот материал растирали в диэтилэфире и получали 0,5 г 2-бутил-1-[2-(метилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амина в форме порошка белого цвета, который имел температуру плавления 226-228°С.
Результаты анализа. Расчетное содержание для C17H22N4О2S: % С 58,94; % Н 6,40; % N 16,17; найденное содержание % С 58,91; % Н 6,27; % N 16,13.
1Н ЯМР (300 МГц, ДМСО-d6): δ 8,05 (d, J=8,1 Hz, 1 Н), δ 7,63 (d, J=8,1 Hz, 1 Н), δ 7,44 (t, J=8,4 Hz, 1 Н), δ 7,26 (t, J=8,1 Hz, 1 Н), δ 6,48 (s, 2 Н), δ 4,95 (t, J=7,2, Hz, 2 Н), δ 3,77 (t, J=7,2 Hz, 2 Н), δ 3,11 (s, 3 Н), δ 2,99 (t, J=δ7,8 Hz, 2 Н), 8 1,83 (квинтет, J=7,6 Hz, 2 Н), 6 1,48 (секстет, J=7,4 Hz, 2 Н), δ 0,97 (t, J=7,5 Hz, 3 Н).
Масс-спектрометрия (CI) для C17H22N4O2S: m/e 347 (N+Н).
Пример 68
2-Метил-1-[6-(метилсульфонил)гексил]-2-имидазо[4,5-с]хинолин-4-амин
Часть А
В раствор N-(6-гидроксигексил)-3-нитрохинолин-4-амина (14,9 г, 51,5 ммоля) в дихлорметане (200 мл) медленно добавляли раствор тионила хлорида (6,74 г, 56,6 ммоля) в дихлорметане (50 мл). После этого реакционную систему перемешивали в течение примерно 1 часа, а затем концентрировали при пониженном давлении. Полученный остаток суспендировали в воде, перемешивали в течение примерно 1 часа, отделяли посредством фильтрования, промывали водой, а затем высушивали, получая 14,0 г N-(6-хлорогексил)-3-нитрохинолин-4-амина, имевшего форму твердого вещества.
Часть Б
С помощью общего способа, описанного в примере 1, часть В, 4-(6-хлорогексил)-3-нитрохинолин-4-амин (6 г, 19 ммолей) восстанавливали для получения N4-(6-хлорогексил)хинолин-3,4-диамина.
Часть В
В сосуд высокого давления вводили N4-(6-хлорогексил)хинолин-3,4-диамин (5 г, 18 ммолей), триэтила ортоацетат (2,92 г, 18 ммолей), толуол (75 мл) и каталитическое количество пиридина гидрохлорида. Смесь нагревали до 140°С. Спустя приблизительно 1,5 часа реакционную смесь охлаждали, а затем концентрировали при пониженном давлении, получая 3,8 г 1-(6-хлорогексил)-2-метил-1Н-имидазо[4,5-с]хинолина, который имел форму масла темно-оранжевого цвета.
Часть Г
С помощью общего способа, описанного в примерах 43-55, часть В, 4-(6-хлорогексил)-2-метил-1Н-имидазо[4,5-с]хинолин (3,8 г, 13 молей) сначала окисляли, а затем аминировали, получая 2,5 г 2-метил-1Н-имидазо[4,5-с]хинолин-4-амина.
Часть Д
Объединяли 1-(6-хлорогексил)-2-метил-1Н-имидазо[4,5-с]хинолин-4-амин (2,5 г, 8 ммолей), натрия тиометоксид (1,13 г, 15,5 ммоля) и N,N-диметилформамид (15 мл). Полученную смесь нагревали при 160°С на протяжении 3 часов. Реакцию останавливали добавлением воды. Полученный осадок содержал 1,5 г 2-метил-1-[6-(метилтио)гексил]-1Н-имидазо[4,5-с]хинолин-4-амина.
Часть Е
С помощью общего способа, описанного в примере 38, материл, полученный в части Д, окисляли, получая 0,80 г 2-метил-1-[6-(метилтио)гексил]-1Н-имидазо[4,5-с]хинолин-4-амина с температурой плавления 202-206°С.
Результаты анализа. Расчетное содержание для C18H24N4О2S·0,02 EtOH: % С 59,96; % Н 6,73; % N 15,50; найденное содержание: % С 59,74; % Н 6,81; % N 15,30.
ЯМР (300 МГц, ДМСО-d6): δ 8,02 (d, J=8,1 Hz, 1H), δ 7,62 (d, J=7, 2 Hz, 1Н), δ 7,41 (t, J=8,1 Hz, 1Н), δ 7,25 (t, J=7,2 Hz, 1Н), δ 6,48 (s, 2 Н), δ 4,48 (t, J=7,2 Hz, 2 Н), δ 3,08 (t, J=8,4 Hz, 2 Н), δ 2,92 (s, 3 Н), δ 2,60 (s, 3 Н), δ 1,82 (m, 2 Н), δ 1,68 (m, 2 Н), δ 1,44 (m, 4 Н).
Масс-спектрометрия (CI) для C18H24N4O2S·0,02 EtOH: m/e 361 (M+Н).
Пример 69
1-[5-(Фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин
Часть А
С помощью общего способа, описанного в примере 37, часть Г, осуществляли реакцию N-(5-хлорофенил)-3-нитрохинолин-4-амина (10 г, 34 ммоля) с бензолтиолом (1,1 экв.), получая 12,6 г N-[5-(фенилтио)пентил]хинолин-4-амина.
Часть Б
С помощью общего способа, описанного в примере 1, часть В, материал, полученный, как описано в части А, восстанавливали для получения N4-[5-(фенилтио)пентил]хинолин-3,4-диамина в форме твердого кристаллического вещества бурого цвета.
Часть В
С помощью общего способа, описанного в примере 1, часть Г, осуществляли циклизацию N4-[5-(фенилтио)пентил]хинолин-3,4-диамина (5,1 г, 15,1 ммоля), используя для этой цели реакцию с триэтилортоформиатом (2,46 г, 16,6 ммоля), в присутствии каталитического количества пиридина гидрохлорида. В результате получали 1-[5-(фенилтио)пентил]хинолин-1Н-имидазо[4,5-с]хинолин, имевший форму твердого вещества желтого цвета.
Часть Г
С помощью общего способа, описанного в примере 11, часть Е (но используя в качестве растворителя дихлорметан вместо хлороформа), производили окисление 1-[5-(фенилтио)пентил]-1Н-имидазо[4,5-с]хинолина (5 г, 13,2 ммоля) и получали 4,7 г 1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-5N-оксида в форме масла.
Часть Д
В раствор 1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-5N-оксида (3,6 г, 9,1 ммоля) в дихлорметане (40 мл) медленно добавляли трихлорацетила изоцианат (1,91 г, 10 ммолей). Реакционную смесь концентрировали при пониженном давлении.
Полученный таким образом остаток объединяли примерно с двумя эквивалентами натрия метоксида.
Спустя несколько минут выпадал осадок, который отделяли посредством фильтрации, затем перекристаллизовывали из этанола и очищали с помощью хроматографии на силикагеле, используя для элюции 4% раствор метанола в дихлорметане. В результате этих процедур получали 0,3 г 1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин в форме твердого вещества с температурой плавления 171-172°С.
Результаты анализа. Расчетное содержание для C21H22N4O2S: % С 63,94; % Н 5,62; % N 14,20; найденное содержание: % С 63,7; % Н 5,64;% N 14,07.
ЯМР (300 МГц, ДМСО-d6): δ 8,15 (s, 1Н), δ 7,99 (d, J=8,1 Hz, 1Н), δ 7,86 (m, 2 Н), δ 7,71 (m, 1Н), δ 7,64 (m, 3 Н), δ 7,43 (t, J=6,6 Hz, 1Н), δ 7,22 (t, J=6,9 Hz, 1Н), δ 6,56 (s, 2 Н), δ 4,54 (t, J=7,5 Hz, 2 Н), δ 3,30 (m, 2 Н), δ 1,84 (квинтет, J=7,5 Hz, 2 Н), δ 1,57 (квинтет, J=6,9 Hz, 2 Н), δ 1,40 (квинтет, J=6,9 Hz, 2 Н).
Масс-спектрометрия (CI) для C21H22N4O2S: m/e 395 (M+Н).
Пример 70
2-(2-Метоксиэтил)-1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин
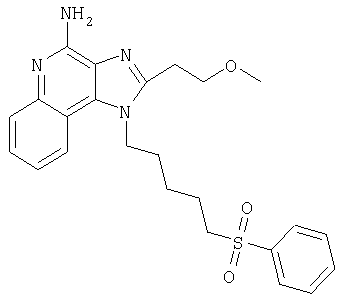
Часть А
В раствор N4-[5-(фенилтио)пентил]хинолин-3,4-диамина (5,1 г, 15,2 ммоля) в пиридине медленно добавляли охлажденный до 0°С раствор 3-метоксипропаноила хлорида (2,04 г, 16,6 ммоля) в пиридине (20 мл). После повышения температуры реакционной системы до комнатной в нее добавляли дополнительное количество кислого хлорида (1 г) и нагревали в условиях обратного тока жидкости в течение ночи. Затем реакционную смесь концентрировали при пониженном давлении, получая 3-метокси-N-(4-{[5-(фенилтио)пентил]амино}хинолин-3-ил)пропанамид, который имел форму клейкого твердого вещества бурого цвета.
Часть Б
Материал, полученный в части А, объединяли с пиридином и полученную смесь перегоняли на протяжении нескольких часов. Затем реакционную смесь концентрировали в условиях пониженного давления. Остаток разделяли между дихлорметаном и водой. Органический слой трижды промывали водой (по 100 мл), фильтровали через целитовый фильтр и снова концентрировали при пониженном давлении. Остаток очищали посредством хроматографии, используя для элюции смесь дихлорметана и метанола в соотношении 4:1, и получали 3,7 г 2-(2-метоксиэтил)-1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолина.
Часть В
С помощью общего способа, описанного в примере 37, часть Б, материал, полученный, как описано в части Б, окисляли для получения 2,09 г 2-(2-метоксиэтил)-1-[5-(фенилсульфонил)пентил]хинолин-3,4-диамин-5N-оксида.
Часть Г
С помощью общего способа, описанного в примере 37, часть В, осуществляли аминирование материала, полученного в части В. Первичный продукт этой реакции подвергали перекристаллизации из этанола и получали 0,26 г 2-(2-метоксиэтил)-1-4-[5-(фенилсульфонил)пентил]хинолин-4-амина, имевшего температуру плавления 172-175°С.
Результаты анализа. Расчетное содержание для С24Н28N4O3S: % С 63,69; % Н 6,24; % N 12,38; найденное содержание: % С 63,40; % H 5,95; % N 12,08.
ЯМР (300 МГц, ДМСО-d6): δ 7,96 (d, J=6,6 Hz, 1 Н), δ 7,87 (d, J=5,4 Hz, 2 H), δ 7,73 (m, 1 Н), δ 7,41 (m, 3 Н), δ 7,43 (t, J=7,8 Hz, 1 Н), δ 7,22 (t, J=7,8 Hz, 1 Н), δ 6,47 (s, 2 Н), δ 4,46 (t, J=7,5 Hz, 2 Н), δ 3,80 (t, J=6,9 Hz, 2 Н), δ 3,27 (s, 2 Н), δ 3,14 (t, J=6 Hz, 2 H), δ 1,76 (m, 2 H), δ 1,53 (m, 4 H).
Масс-спектрометрия (CI) для C24H28N4O3S: m/e 473 (M+Н).
Пример 71
1-[5-(Метилсульфонил)пентил]-2-(трифторметил)-1H-имидазо[4,5-с]хинолин-4-амин
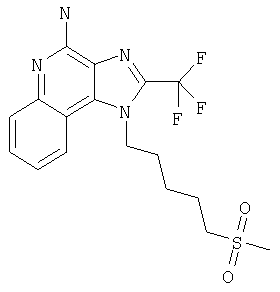
Часть А
В раствор N4-[5-(метилтио)пентил]хинолин-3,4-диамина (6 г, 23,1 ммоля) в смеси толуола и пиридина медленно добавляли охлажденный раствор фторацетила хлорида (3,5 г, 26,5 ммоля) в толуоле. В результате образовывался тяжелый осадок желтого цвета. Реакционную смесь оставляли на выходные дни в условиях непрерывного перемешивания, после чего концентрировали при пониженном давлении, получая 13,2 г неочищенного 2,2,2-трифторо-N-(4-{[5-(метилтио)пентил]амино}хинолин-3-ил)ацетамида.
Часть Б
Материал, полученный в части А, объединяли с толуолом (150 мл), полученную смесь помещали в сосуд высокого давления и нагревали при 140°С на протяжении примерно 30 минут. Затем реакционную смесь концентрировали при пониженном давлении. Остаток разделяли между дихлорметаном и 5%-ным натрия карбонатом. Органический слой промывали водой (по 100 мл), высушивали над магния сульфатом и снова концентрировали в условиях пониженного давления, получая 7,9 г 1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолина, который имел форму твердого вещества.
Часть В
С помощью общего способа, описанного в примере 11, часть Е, производили окисление материала, полученного в части Б, в результате чего происходило образование 7,5 г 1-[5-(метилсульфонил)пентил]-2-(трифторометил)-1Н-имидазо [4,5-с]хинолин-5N-оксида.
Часть Г
С помощью общего способа, описанного в примере 37, часть В, производили аминирование материала, полученного в части В, и получали 2,5 г 1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амина, имевшего форму твердого вещества с температурой плавления 192-195°С.
Результаты анализа. Расчетное содержание для C17H19F3N4O2S·0,05 CH2Cl2: % С 50,61; % Н 4,76; % N 13,84; найденное содержание: % С 50,60; % Н 4,76; % N 13,77.
ЯМР (300 МГц, ДМСО-d6): δ 8,08 (d, J=7,2 Hz, 1 H), δ 7,67 (d, J=7,5 Hz, 1 H), δ 7,54 (t, J=6,9 Hz, 2 H), δ 7,34 (t, J=7,8 Hz, 1 H), δ 6,91 (s, 2 H), δ 4,67 (t, J=8,4 Hz, 1 H), δ 3,11 (t, J=7,5 Hz, 2 H), δ 2,93 (s, 2 H), δ 1,91 (квинтет, J=7,2 Hz, 2 H), δ 1,744 (квинтет, J=8,1 Hz, 2 H), δ 1,58 (квинтет, J=6,9 Hz, 2 H).
Масс-спектрометрия (CI) для C17H19F3N4O2S·0,05 СН2Cl2: m/e 401 (M+H).
Пример 72
2-Этил-1-[4-(пиримидин-2-илтио)бутил]-1H-имидазо [4,5-с]хинолин-4-амин
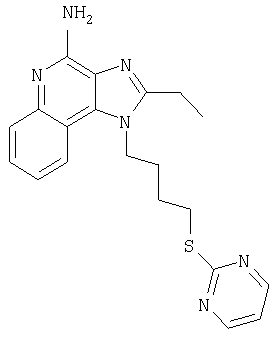
С помощью общего способа, описанного в примере 20, часть А, осуществляли реакцию 1-(4-хлоробутил)-2-этил-1H-имидазо[4,5-с]хинолин-4-амина (1,0 г, 3,30 ммоля) с 2-меркаптопиримидином (0,59 г, 5,3 ммоля) и получали 1,0 г 2-этил-1-[4-(пиримидин-2-илтио)бутил]-1H-имидазо[4,5-с]хинолин-4-амина в форме белого порошка, который имел температуру плавления 182-185°С.
Результаты анализа. Расчетное содержание для С20Н22N6S·0,25 Н2O: % С 62,72, % Н 5,92, % N 21,94, найденное содержание: % С 63,00, % Н 5,88, % N 22,21.
Пример 73
2-Этил-1-[4-(пиримидин-2-илсульфонил)бутил]-1H-имидазо[4,5-с]хинолин-4-амин
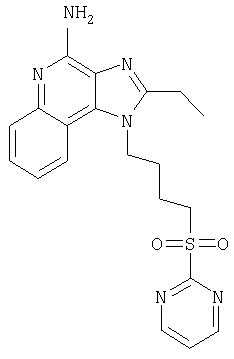
С помощью общего способа, описанного в примере 5, часть А, 2-этил-1-[4-(пиримидин-2-илтио)бутил)]-1Н-имидазо[4,5-с]хинолин-4-амина (0,3 г) окисляли для получения 10 мг 2-этил-1-[4-(пиримидин-2-илсульфонил)бутил]-1H-имидазо[4,5-с]хинолин-4-амина в форме твердого вещества персикового цвета с температурой плавления 172-175°С.
Результаты анализа. Расчетное содержание для С20Н22N6O2·0,25 Н2О: % С 57,88, % Н 5,46, % N 20,25, найденное содержание: % С 57,76, % Н 5,48, % N 19,88.
Пример 74
2-Метил-1-[4-(метилсульфонил)бутил]-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амин
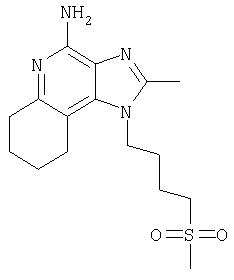
В гидрогенизационный сосуд Парра вносили раствор 2-метил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амина (1,0 г) в трифторуксусной кислоте (11 мл) и добавляли катализатор (0,2 г платины оксида). Полученная смесь на протяжении примерно 90 часов находилась под давлением водорода (50 Пси, 3,5 кг/см2). По истечении этого времени реакционную смесь фильтровали через целитовый фильтр, предварительно промытый трифторуксусной кислотой (приблизительно 125 мл). Оставшийся на фильтре остаток промывали примерно 100 мл трифторуксусной кислоты, а фильтрат концентрировали при пониженном давлении. Полученную в результате этих процедур маслянистую жидкость разводили в 1 N соляной кислоте (20 мл). Спустя несколько минут выпадал белый осадок. Его рН доводили до 14,0 добавлением 50%-ной водной гидроокиси натрия. После этого осадок разводили, получая раствор желтого цвета. Из него вскоре также выпадал осадок. Конечную суспензию оставляли на ночь при комнатной температуре в условиях непрерывного перемешивания. После этого ее в течение 2 часов охлаждали на водяной бане со льдом, а затем фильтровали, получая порошок белого цвета (1,0 г). Этот материал очищали перекристаллизацией из метанола с последующей колоночной хроматографией на силикагеле (используя для элюции смесь дихлорметана и метанола в соотношении 9:1). Очищенный продукт в количестве 0,44 г имел форму твердого вещества белого цвета и представлял собой 2-метил-1-[4-(метилсульфонил)бутил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин с температурой плавления 213-216°С.
Результаты анализа. Расчетное содержание для C16H24N6O2S: % С 57,12, % Н 7,19, % N 16,65, найденное содержание: % С 56,86, % Н 7,09,% N,16,61.
Пример 75
2-Метил-1-[5-(метилсульфонил)пентил]-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амин
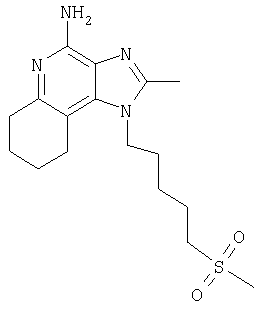
С помощью общего способа, описанного в примере 74, восстанавливали 2-метил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин (1,3 г), продукт реакции подвергали очистке и получали 0,6 г 2-метил-1-[5-(метилсульфонил)пентил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина, имевшего форму иглообразных кристаллов белого цвета с температурой плавления 172-174°С.
Результаты анализа. Расчетное содержание для C17H26N4O2S: % С 58,26, % Н 7,48, % N 15,99, найденное содержание: % С 58,22, % Н 7,54,% N 16,12.
Пример 76
2-Метил-1-{4-[(1-метилэтил)сульфонил]бутил}-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амин
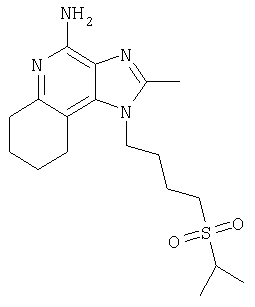
Часть А
Суспензию 2,4-дигидрокси-3-нитро-6,7,8,9-тетрагидрохинолина (10,56 г, 50,3 ммоля) в фосфора оксихлориде (60 мл) нагревали при 50-60°С на протяжении 48 часов. После этого реакционную смесь охлаждали до комнатной температуры и медленно, при энергичном перемешивании, переливали ее в охлажденную на водяной бане со льдом двухфазную смесь дихлорметана (300 мл) и 20%-ного водного карбоната (500 мл). Добавлением твердого натрия карбоната рН смеси доводили до 8,0 и разделяли образовавшиеся слои. Водную фракцию дважды экстрагировали дихлорметаном (по 125 мл), а объединенные органические фракции высушивали над магния сульфатом, после чего концентрировали в условиях пониженного давления, получая 11,9 г 2,4-дихлоро-3-нитро-6,7,8,9-тетрагидрохинолина в форме твердого вещества светло-коричневого цвета.
Часть Б
В раствор 2,4-дихлоро-3-нитро-6,7,8,9-тетрагидрохинолина (9,0 г, 36,4 ммоля, 1 эквивалент) в N,N-диметилформамиде (60 мл) добавляли триэтиламин (6,1 мл, 1,2 эквивалента). Затем в реакционную систему вносили 4-амино-1-бутанол (3,7 мл, 1,1 эквивалента) и нагревали ее приблизительно при 50°С на протяжении 5 часов. По завершении нагревания смесь оставляли для охлаждения до комнатной температуры и концентрировали в условиях пониженного давления. В результате получали масло красного цвета. Масло разбавляли хлороформом (500 мл), трижды промывали водой (по 200 мл) и однократно солевым раствором (200 мл). Затем материал высушивали над магния сульфатом и концентрировали при пониженном давлении, получая 11,9 г масла красного цвета. Масло растирали с диэтилэфиром (40 мл). Образовывавшееся твердое вещество изолировали с помощью фильтрования, затем трижды промывали его диэтилэфиром (по 10 мл) и получали 5,76 г 4-[(2-хлоро-3-нитро-6,7,8,9-тетрагидрохинолин-4-ил)амино]бутан-1-ола, имевшего форму твердого вещества светло-желтого цвета.
Часть В
В суспензию натрия гидрида (1,15 г 60%-ного материала, 1,5 эквивалента) в диглиме (34 мл) на протяжении 15 минут добавляли фенол (2,71 г, 1,5 эквивалента). Реакционную смесь перемешивали в течение еще 30 минут, а затем вносили в нее 5,75 г (19,16 ммоля, 1,0 эквивалент) твердого 4-[(2-хлоро-3-нитро-6,7,8,9-тетрагидрохинолин-4-ил)амино] бутан-1-ола. Эту систему нагревали при 85°С на протяжении 24 часов, а потом оставляли на ночь для охлаждения при комнатной температуре. Охлажденную реакционную смесь концентрировали упариванием при пониженном давлении до объема примерно 10 мл. Концентрат разбавляли хлороформом (400 мл), высушивали над магния сульфатом и снова концентрировал при пониженном давлении. Остаток разводили в ацетонитриле (200 мл), дважды промывали гексаном (пл 100 мл) и концентрировали в условиях пониженного давления, получая 5,31 г 4-[(3-нитро-2-фенокси-6,7,8,9-тетрагидрохинолин-4-ил)амино]бутан-1-ола, имевшего форму темного масла.
Часть Г
В раствор трифенилфосфина (4,68 г, 1,2 эквивалента) в тетрагидрофуране (30 мл) добавляли N-хлорсукцинимид (2,38 г, 1,2 эквивалента). Реакционную систему перемешивали на протяжении 20 минут, после чего вносили в нее раствор полученного в части В материала в тетрагидрофуране (30 мл). Смесь перемешивали в течение еще 75 минут, а затем концентрировали в условиях пониженного давления. Остаток разводили в хлороформе (350 мл), дважды промывали водой (по 250 мл), высушивали над магния сульфатом и снова концентрировали в условиях пониженного давления. Остаток очищали посредством хроматографии на силикагеле, используя для элюции хлороформ, и получали 4,88 г N4-(4-хлоробутил)-3-нитро-6,7,8,9-тетрагидрохинолин-4-амина, который имел форму твердого вещества желтого цвета.
Часть Д
В суспензию N4-(4-хлоробутил)-3-нитро-2-фенокси-6,7,8,9-тетрагидрохинолин-4-амина (4,78 г, 12,71 ммоля, 1,0 эквивалент) в 120 мл смеси метанола с хлороформом (в соотношении 1:1) вносили гексагидрат никеля(II) хлорида (303 мг, 0,1 эквивалента). После охлаждения этой реакционной системы до 0°С в нее добавляли натрия борогидрид (1,92 г, 4 эквивалента) четырьмя равными порциями на протяжении 50 минут. Полученную смесь перемешивали в течение еще 30 минут, а затем концентрировали в условиях пониженного давления. Остаток разводили в хлороформе (300 мл), трижды промывали водой (по 100 мл), высушивали над магния сульфатом и снова концентрировали при пониженном давлении, получая 5,01 г N4-(4-хлоробутил)-2-фенокси-6,7,8,9-тетрагидрохинолин-3,4-диамина, имевшего форму густого масла.
Часть Е
Материал, полученный, как описано в части Д, растворяли в толуоле (40 мл) и добавляли к нему триметилортоацетат (2,0 мл, 1,2 эквивалента), а затем пиридина гидрохлорид (150 мл, 0,1 эквивалента). Реакционную смесь нагревали при 100°С на протяжении часа, потом охлаждали до комнатной температуры и концентрировали в условиях пониженного давления. Полученный остаток разводили в хлороформе (300 мл), дважды промывали водой (по 75 мл), высушивали над магния сульфатом, разбавляли ацетонитрилом (40 мл) и снова концентрировали при пониженном давлении, получая 4,5 г полужидкого вещества темно-красного цвета. Этот материал подвергали очистке посредством хроматографии на силикагеле, используя для элюции смесь метанола и хлороформа в соотношении 2:98. В результате получали масло красного цвета. Его разбавляли изопропанолом (50 мл), концентрировали, а полученный остаток растирали до порошкообразного состояния в диэтилэфире. Образовавшееся твердое вещество отделяли с помощью фильтрования и промывали диэтилэфиром. Конечный продут в количестве 2,77 г представлял собой твердое вещество белого цвета, состоявшее из 1-(4-хлоробутил)-2-метил-4-фенокси-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолина.
Часть Ж
В суспензию натрия гидрида (25 мг 60%-ного материала, 1,2 эквивалента) в N,N-диметилформамиде (1 мл) по каплям добавляли 1-метилэтилтиол (57 мкл, 1,2 эквивалента). Реакционную смесь перемешивали на протяжении 30 минут, после чего доливали в нее раствор 1-(4-хлоробутил)-2-метил-4-фенокси-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолина (189 мг, 0,51 ммоля, 1,0 эквивалент) в N,N-диметилформамиде (1,5 мл). Смесь снова перемешивали в течение 3 часов, разводили хлороформом (50 мл), по одному разу промывали 5%-ным водным натрия карбонатом (50 мл) и водой (25 мл), высушивали над магния сульфатом и концентрировали в условиях пониженного давления. В результате получали 176 мг 1-{[4-(1-метилэтил)тио]бутил}-2-метил-4-фенокси-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолина, имевшего форму масла светло-коричневого цвета.
Часть З
Материал, полученный, как описано в части Ж, разводили в хлороформе (2,2 мл) и добавляли в него 3-хлорпербензойную кислоту (218 мг, 2,2 эквивалента при титре 75%). Реакционную смесь перемешивали при 0°С на протяжении 20 минут, разбавляли хлороформом (50 мл), дважды промывали насыщенным водным раствором натрия бикарбоната (по 25 мл), высушивали над магния сульфатом и концентрировали в условиях пониженного давления, получая 202 мг 1-{[4-(1-метилэтил)сульфонил]бутил}-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин в форме полужидкого вещества желтого цвета.
Часть И
Смесь 1-{[4-(1-метилэтил)сульфонил]бутил} -6,7,8,9-тетрагидро-1H-имидазо [4,5-с]хинолина (200 мг) и твердого уксуснокислого аммония (2,1 г) нагревали при 145°С в плотно укупоренной пробирке в течение 24 часов. После этого реакционную систему охлаждали до комнатной температуры, разбавляли хлороформом (40 мл) и дважды промывали 10%-ной водной гидроокисью натрия (по 20 мл). Водный слой дважды экстрагировали хлороформом (по 20 мл), а объединенные органические фракции высушивали над магния сульфатом и концентрировали в условиях пониженного давления, получая 204 г желтого масла. Растирание этого материала в ацетонитриле до порошкообразного состояния приводило к образованию 48 г твердого вещества белого цвета. Маточный раствор концентрировали, а остаток очищали посредством хроматографирования на силикагеле, используя для элюции смесь метанола и хлороформа в соотношении 10:90. Растирание полученного материала в ацетонитриле давало 18 мг твердого вещества. Оба твердых продукта объединяли, рехроматографировали и перекристаллизовывали из этанола. Образовавшееся вещество в форме призматических кристаллов концентрировали из метанола и получали 40 мг 2-метил-1-{4-[(1-метилэтил)сульфонил]бутил}-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амин, имевший форму порошка белого цвета с температурой плавления 177-178°С.
Результаты анализа. Расчетное содержание для C18H28N4O2S: % С 59,31, % Н 7,74, % N 15,37, найденное содержание: % С 59,27, % H 7,82,% N 15,19.
1H ЯМР (300 Гц, ДМСО-d6): δ 5,64 (s, 2 H); δ 4,21 (m, 2 H); δ 3,21 (септет, 1 H, J=6,9); δ 3,14 (m, 2 H); δ 2,94 (m, 2 H); δ 2,65 (m, 5 H); δ 2,47 (s, 3 H); δ 1,76 (br. m, 8 H); δ 1,23 (d, 6 H, J=6,6 Hz).
EI-масс-спектрометрия для C18H28N4O2S (m/z): 365 (M+1).
Пример 77
2-Метил-1-{4-[(4-фторофенил)сульфонил]бутил}-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амин
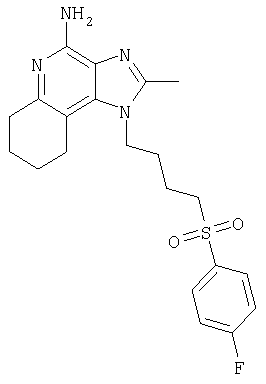
Часть А
С помощью общего способа, описанного в примере 76, часть Ж, осуществляли реакцию 1-(4-хлоробутил)-2-метил-4-фенокси-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолина (740 г, 2,00 ммоля, 1,0 эквивалент) с 4-фторобензолтиолом (260 мкл, 1,2 эквивалента) для получения 0,91 г 1-{[4-(4-фторофенил)тио]бутил}-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолина, имевшего форму твердого вещества светло-желтого цвета.
Часть Б
С помощью общего способа, описанного в примере 76, часть З, полученный в части А материал окисляли до 1-{[4-(4-фторофенил)тио] бутил) -6,7,8,9-тетрагидро-1Н-имидазо [4,5-с]хинолина (1,02 г), имевшего форму белой пены.
Часть В
С помощью общего способа, описанного в примере 76, часть И, полученный в части Б материал аминировали до 2-метил-1-{[4-(4-(фторофенил)сульфонил]бутил}-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина (148 г), который имел форму твердого вещества белого цвета, которое размягчалось при температуре 137-144°С, а затем плавилось при 159-162°С.
Результаты анализа. Расчетное содержание для C21H25FN4O2S·H2O: % С 58,05; % Н 6,26; % N 12,98; найденное содержание: % С 57,78; % Н 5,93; % N 12,72.
ЯМР (300 МГц, ДМСО-d6): δ 7,93 (m, 2 Н), δ 7,50 (m, 2 Н), δ 5,67 (s, 2 Н), δ 4,15 (m, 2 Н), δ 2,87 (br m, 2 Н), δ 2,64 (br m, 2H), δ 2,41 (s, 3 Н), δ 1,74 (br m, 8 H).
EI-масс-спектрометрия для C21H25FN4O2S·H2O: (CI) m/z 417 (M+1).
Пример 78
2-Метил-1-{4-[(1,1-(диметилэтил)сульфонил]бутил}-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амин
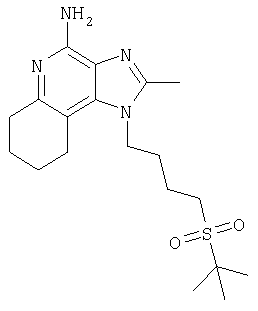
Часть А
С помощью общего способа, описанного в примере 76, часть Ж, осуществляли реакцию 1-(4-хлоробутил)-2-метил-4-фенокси-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолина (750 г, 2,03 ммоля, 1,0 эквивалент) с 1,1-диметилэтилтиолом (275 мкл, 1,2 эквивалента) для получения 0,91 г 1-{[4-(1,1-диметилэтил)тио]бутил}-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолина в форме масла, которое кристаллизовалось при длительном стоянии.
Часть Б
С помощью общего способа, описанного в примере 76, часть З, полученный в части А материал окисляли до 1-{[4-(1,1-диметилэтил)сульфонил]бутил}-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолина (1,0 г), имевшего форму белой пены.
Часть В
С помощью общего способа, описанного в примере 76, часть И, полученный в части Б материал аминировали до 2-метил-1-{4-[(1,1-диметилэтил)сульфонил]бутил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-4-амина (460 мг), который имел форму твердого вещества белого цвета с температурой плавления 208-210°С.
Результаты анализа. Расчетное содержание для C19H30FN4O2S: % С 60,29; % Н 7,99; % N 14,80; найденное содержание: % С 60,26; % Н 7,88; % N 14,89.
ЯМР (300 МГц, ДМСО-d6): δ 5,65 (m, 2 Н), δ 4,23 (m, 2 Н), δ 3,13 (m, 2 Н), δ 2,95 (br m, 2 Н), δ 2,65 (br m, 2H), δ 2,47 (s, 3 Н), δ 1,75 (br m, 8 Н).
EI-масс-спектрометрия для C19H30FN4O2S (CI) m/z: 379 (M+1).
Пример 79
2-Этоксиметил-1-(4-метансульфонилбутил)-6,7,8,9-тетрагидро-1H-имидазо [4,5-с]хинолин-4 амин
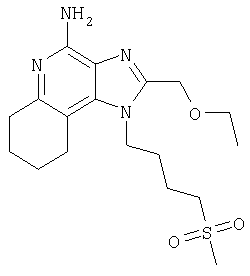
Часть А
1-(4-Хлоробутил)-2-метил-4-фенокси-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолина (1,00 г, 2,66 ммоля, 1,0 эквивалент) разводили в диметилформамиде. В полученный раствор добавляли раствор натрия тиометоксида (205 мг, 1,1 эквивалента). Реакционную смесь перемешивали на протяжении 1 часа, а затем концентрировали в условиях пониженного давления. Остаток растворяли в метилена хлориде (110 мл), один раз промывали водой (30 мл), высушивали над магния сульфатом и концентрировали при пониженном давлении, получая 0,97 г (4-метилсульфонилбутил)-(3-нитро-2-фенокси-5,6,7,8-тетрагидрохинолин-4-ил)амина, который имел форму твердого вещества желтого цвета.
Часть Б
С помощью общего способа, описанного в примере 76, часть Д, полученный в части А материал восстанавливали до N4-(4-метилсульфонилбутил)-2-фенокси-5,6,7,8-тетрагидрохинолин-3,4-диамина (0,89 г), имевшего форму бесцветного масла.
Часть В
Материал, полученный, как описано в части Б, растворяли в пиридине (10 мл) и добавляли к нему этоксиацетила хлорид. После перемешивания реакционной системы при комнатной температуре на протяжении 1 часа ее нагревали при 95°С в течение 1 часа, а затем при 105°С на протяжении еще 4 часов. После этого реакционную смесь охлаждали до комнатной температуры и концентрировали в условиях пониженного давления. Полученный остаток разводили в метилена хлориде (100 мл), промывали насыщенным водным раствором натрия бикарбоната, высушивали над магния сульфатом и снова концентрировали при пониженном давлении. В результате получали 0,89 г 2-этоксиметил-1-(4-метилсульфонилбутил)-4-фенокси-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолина, имевшего форму прозрачного бесцветного масла.
Часть Г
С помощью общего способа, описанного в примере 76, часть З, материал, полученный, как описано в части В, окисляли до получения 0,60 г 2-этоксиметил-1-(4-метилсульфонилбутил)-4-фенокси-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолина, который имел форму пены бурого цвета.
Часть Д
С помощью общего способа, описанного в примере 76, часть И, материал, полученный, как описано в части Г, аминировали для получения 355 мг г 2-этоксиметил-1-(4-метилсульфонилбутил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина (0,60 г), имевшего форму твердого вещества белого цвета, который имел температуру плавления 170-171°С.
Результаты анализа. Расчетное содержание для С24Н31N3О3S: % С 56,82, % Н 7,42% N 17,72, найденное содержание: % С 56,64, % Н 7,32,% N 14,47.
1H ЯМР (300 Гц, ДМСО-d6): δ 5,89 (br s, 2 Н); δ 4,64 (s, 2 Н); δ 4,29 (m, 2 Н); δ 3,51 (q, 2 Н, J=7,0 Hz); δ 3,17 (m, 2 Н); δ 2,96 (br s, 5 Н); δ 2,67 (m, 2 Н); δ 1,80 (m, 8 Н), δ 1,15 (t, 3 Н, J=7,0 Hz).
EI-масс-спектрометрия для С24Н31N3О3S (m/z): 381 (M+1).
Дополнительные соединения, которые могут быть получены с использованием вышеописанных способов, включают следующие:
2-бутил-1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[2-(фенилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[4-(фенилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(фенилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(фенилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(фенилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-гексил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метокси-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[5-(метилтио)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[3-(метилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[3-(фенилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-пропил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[3-(метилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-пропил-1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-(2-циклопропилэтил)-1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-(2-циклопропилэтил)-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-циклопропилметил-1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-циклопропилметил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метоксиэтил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метоксиэтил-1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этоксиметил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-пропил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[3-(метилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[3-(метилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[3-(метилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-пропил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[2-(метилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[2-(метилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-пропил-1-[2-(метилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{4-[(2,4-дифторофенил)тио]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{4-[(2,4-дифторофенил)сульфонил]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[4-(этилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[4-(трет-бутилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{4-[(4-фторофенил)тио]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{4-[(4-фторофенил)сульфонил]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
4-амино-2-метил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-{4-[(1-метилэтил)тио]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-{4-[(3,5-дихлорофенил)тио]бутил)-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(циклопентилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-{4-[(3,5-дихлорофенил)сульфонил]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-циклопентилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-4-[(3,5-дихлорофенил)сульфонил]бутил-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(пропилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-{4-[(4-хлорофенил)тио]бутил)-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(бутилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-{4-[(4-фторофенил)тио]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-{4-[4-(хлорофенил)сульфонил]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(этилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(этилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(циклогексилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{2-[1-(метилэтил)сульфонил]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{2-[4-(фторофенил)сульфонил]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{2-[1,1-(диметилэтил)сульфонил]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{2-[4-(фторофенил)тио]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[2-(пропилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-2-[(2-метилропил)сульфонил]этил-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[2-(этилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[2-(этилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[2-(метилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[6-(метилсульфонил)гексил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-трифторометил-1-[5-(метилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метоксиэтил-1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(пиримидин-2-илтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(пиримидин-2-илсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[4-(метилсульфонил)бутил]-6,7,8,9-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[5-(метилсульфонил)пентил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-{5-[(1-метилэтил)сульфонил]пентил}-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-{4-[(4-фторофенил)сульфонил]бутил}-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-{4-[(1,1-диметилэтил)сульфонил]бутил}-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этоксиметил-1-[3-(пиримидин-2-илсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-пропил-1-[4-(пиримидин-2-илсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-пропил-1-[3-(пиримидин-2-илсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-пропил-1-[5-(пиримидин-2-илсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этоксиметил-1-[3-(пиримидин-2-илсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этоксиметил-1-[5-(пиримидин-2-илсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин и их пригодные для фармацевтического применения соли.
ИНДУКЦИЯ ЦИТОКИНОВ В ЧЕЛОВЕЧЕСКИХ КЛЕТКАХ
Для характеристики индукции цитокинов используется культивирование человеческих клеток in vitro. Индукция оценивается на основании результатов измерения активности интерферона и фактора некроза опухолей-α (соответственно ИФН и ФНО), которые секретируются клетками в культуральную среду, с помощью способа, см. Testerman с соавторами "Cytokine Induction by the Immunomodulators Imiquimod and S-27609", Journal of Cytokine Biology, 58, 365-372 (September, 1995).
Подготовка клеток крови для культивирования
Цельную донорскую кровь здоровых людей, полученную посредством венепункции, собирают в вакуумные пробирки, содержащие ЭДТА. С помощью центрифугирования в градиенте плотности, используя Histopaque-1077, из периферической крови выделяют моноядерные клетки (МКПК). Для этого кровь разбавляют фосфатным буферным раствором Дюльбекко (ДФБР) или солевым раствором Хэнкса (ХСР). Собирают содержащий МКПК слой и дважды промывают его ДФБР или ХСР, а затем ресуспендируют клетки в полной питательной среде (RPMI) до концентрации 4×106 клеток/мл. Суспензию МКПК распределяют между 48 ячейками плоскодонной платы для стерильного культивирования тканей (Costar, Cambridge, МА, США, или Becton Dickinson Labware, Lincoln Park, NJ, США). Ячейки предварительно заполняют равными объемами полной питательной среды (RPMI), содержащей тестируемое соединение.
Подготовка соединений
Тестируемые соединения растворяют в диметилсульфоксиде (ДМСО). Концентрация последнего не должна превышать конечную концентрацию 1% раствора для внесения в ячейки платы. Индивидуальные соединения обычно тестируют в концентрациях от 30 до 0,014 мкмолей.
Инкубация
Раствор тестируемого соединения в концентрации 60 мкмолей вносят в первую ячейку платы, содержащую полную питательную среду (RPMI). Для остальных ячеек готовят серийные трехкратные разведения раствора. После этого в ячейки вносят равные объемы суспензии МКПК, доводя концентрацию тестируемого соединения до желаемой величины в диапазоне от 30 до 0,014 мкмолей.
Конечная концентрация МКПК составляет 2×106 клеток/мл. Плату накрывают стерильной пластмассовой крышкой, слегка встряхивают для перемешивания содержимого ячеек и оставляют для инкубации при 37°С в атмосфере углекислого газа на протяжении от 18 до 24 часов.
Разделение
По окончании инкубации плату центрифугируют при 1000 об/мин (примерно 200 g) при 4°С в течение 10 минут. Бесклеточный супернатант переносят стерильной полипропиленовой пипеткой в стерильные пробирки из того же материала. До проведения анализа пробы хранят при температуре от -30 до -70°С. Анализы на интерферон-α проводят с помощью твердофазного иммуноферментативного способа (ELISA), а анализы на фактор некроза опухолей-α - с помощью иммунологических способов ELISA или IGEN.
Анализ интерферона-α и фактора некроза опухолей-α с помощью ELISA
Концентрацию интерферона-α определяют с помощью твердофазного иммуноферментативного способа, используя многовидовые аналитические наборы медицинского назначения производства PBL Biomedical Laboratories, New Brunswick, NJ, США. Результаты измерений выражают в пг/мл.
Концентрацию фактора некроза опухолей-α определяют с помощью твердофазного иммуноферментативного способа, используя аналитические наборы производства Biosource International, Camarillo, СА, США. Вместо этого для определения концентрации ФНО-α можно использовать иммунологическую методику Origen M-Series Jmmunoassay в сочетании с анализатором IGEN M-8 (IGEN International, Gaithersburg, MD, США). Методика основана на захвате человеческого ФНО-α и определении антител с помощью наборов Biosource International, Camarillo, СА, США. Результаты измерений выражают в пг/мл.
В приводимой ниже таблице 4 перечислены минимальные концентрации индивидуальных соединений, вызывавшие индукцию интерферона и фактора некроза опухолей. Звездочкой (*) отмечены случаи отсутствия индукции при любых тестировавшихся концентрациях данного соединения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТИОЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2001 |
|
RU2315049C2 |
| КАРБАМИДЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНОВЫЕ ЭФИРЫ | 2001 |
|
RU2302418C2 |
| N-{2-[4-АМИНО-2-(ЭТОКСИМЕТИЛ)-1Н-ИМИДАЗО-[4,5-с]-ХИНОЛИН-1-ИЛ]-1,1-ДИМЕТИЛЭТИЛ}-МЕТАНСУЛЬФОНАМИД И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2004 |
|
RU2374246C2 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ | 2021 |
|
RU2780103C1 |
| ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2248975C2 |
| АМИДОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2000 |
|
RU2295343C2 |
| АРИЛЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ВИРУСНОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ОПУХОЛЕВОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2308456C2 |
| МОЧЕВИНОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2265020C2 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ-ИНФЕКЦИИ | 2020 |
|
RU2780101C2 |
| ГЕТЕРОЦИКЛИЛЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2001 |
|
RU2351598C2 |
Описываются новые тиоэфирзамещенные имидазохинолины и их фармацевтически приемлемые соли. Соединения способны индуцировать биосинтез цитокинов в клетках человека, что позволяет использовать их в медицине. Описывается также фармацевтическая композиция на их основе. 2 н.п. ф-лы, 4 табл.
1-[5-(метилсульфонил)пентил]-2-пропил-1Н-имидазо[4,5-с]хинолин4-амин;
2-метил-1-[3-(метилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[3-(метилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[3-(метилтио)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[3-(метилсульфонил)пропил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[4-(метилсульфинил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(метилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(метилсульфонил)бутил]-2-пропил-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[4-(метилсульфинил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[2-(метилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[2-(метилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[4-(метилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[2-(метилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[2-(метилсульфонил)этил]-2-пропил-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{4-[(2,4-дифторофенил)тио]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{4-[(2,4-дифторофенил)сульфонил]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[4-(этилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{4-[(1,1-диметилэтил)тио] бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{4-[(4-фторофенил)тио]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{4-[(4-фторофенил)сульфонил]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-{4-[(1-метилэтил)тио]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
1-{4-[(3,5-дихлорофенил)тио]бутил}-2-этил-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(циклопентилсульфонил)бутил]-2-этил-1Н-имидазо[4,5-с]хинолин-4-амин;
1-{4-[(3,5-дихлорофенил)сульфонил]бутил}-2-этил-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(циклогексилтио)бутил]-2-этил-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(бутилтио)бутил]-2-этил-1Н-имидазо[4,5-с]хинолин-4-амин;
1-{4-[(4-хлорофенил)тио]бутил}-2-этил-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(бутилсульфонил)бутил]-2-этил-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-{4-[(4-фторофенил)тио]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-{4-[(1-метилэтил)сульфонил]бутил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(этилтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(этилсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[4-(циклогексилсульфонил)бутил]-2-этил-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{2-[(1-метилэтил)сульфонил]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[2-(фенилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{2-[(4-фторофенил)сульфонил]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{2-[(1,1-диметилэтил)сульфонил]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{2-[(1,1-диметилэтил)тио]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[2-(пропилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[2-(пропилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{2-[(2-метилпропил)сульфонил]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-{2-[(2-метилпропил)тио]этил}-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[2-(этилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[2-(этилтио)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-бутил-1-[2-(метилсульфонил)этил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[6-(метилсульфонил)гексил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
1-[5-(метилсульфонил)пентил]-2-(трифторометил)-1Н-имидазо[4,5-с]хинолин-4-амин;
2-(2-метоксиэтил)-1-[5-(фенилсульфонил)пентил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(пиримидин-2-илтио)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-этил-1-[4-(пиримидин-2-илсульфонил)бутил]-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[4-(метилсульфонил)бутил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-[5-(метилсульфонил)пентил]-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-{4-[(1-метилэтил)сульфонил]бутил}-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-{4-[(4-фторофенил)сульфонил]бутил}-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин;
2-метил-1-{4-[(1,1-диметилэтил)сульфонил]бутил}-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин;
или их фармацевтически приемлемые соли.
| WO 9215582 A1, 17.09.1992 | |||
| WO 9502598 A1, 26.01.1995 | |||
| Способ получения производных 1,3-дигидро-2Н-имидазо/4,5-в/хинолин-2-онов или их фармацевтически приемлемых солей | 1986 |
|
SU1450746A3 |

