Настоящее изобретение относится к имидазохиолиновым соединениям с сульфонамидным или сульфамидным замещением в положении 1и к фармацевтическим препаратам, содержащим эти соединения. Кроме того, настоящее изобретение относится к использованию этих соединений в качестве иммуномодуляторов, индуцирующих биосинтез цитокинов у животных, и при лечении различных патологий, в том числе вирусных и неоплазических заболеваний.
В первом заслуживающем доверии сообщении о циклической системе 1Н-имидазо[4,5-с]хинолина Бэкман и др. (Backman et al., J.Org.Chem. 15, 1278-1284 (1950)) описывают синтез 1-(6-метокси-8-хинолинил)-2-метил-1Н-имидазо[4,5-с]хинолина для возможного использования в качестве противомалярийного средства. Впоследствии, были описаны синтезы различных замещенных 1Н-имидазо[4,5-с]хинолинов. Например, Джэйн и др. (Jain et al., J.Med.Chem. 11, pp.87-92 (1968)) синтезировали соединение 1-[2-(4-пиперидил)этил]-1Н-имидазо[4,5-с]хинолин для использования в качестве противосудорожного и сердечно-сосудистого средства. Баранов и др. (Baranov et al., Chem.Abs. 85, 94362 (1976)) также сообщали о нескольких 2-оксоимидазо[4,5-с]хинолинах, а Берени и др. (Berenyi et al., J. Heterocyclic Chem., 18, 1537-1540 (1981)) описали некоторые 2-оксоимидазо[4,5-с]хинолины.
Впоследствии выяснилось, что некоторые 1Н-имидазо[4,5-с]хинолин-4-амины и их замещенные в положении 2 производные могут применяться в качестве антивирусных средств, бронхолитических средств и иммуномодуляторов. Они описаны, в том числе, в патентах США №4689338, 4698348, 4929624, 5037986, 5268376, 5346905 и 5389640, которые все инкорпорированы в настоящее изобретение путем отсылки.
Как видно, например, из WO 98/30562, ЕР 894797 и WO 00/09506, интерес к циклическим имидазохинолиновым системам сохраняется. ЕР 894797 описывает амидозамещенные имидазохинолиновые соединения, которые могут быть использованы в качестве иммуномодуляторов, тогда как WO 00/09506 описывает имидазохинолиновые соединения, содержащие сульфонамидный заместитель, в котором сульфонамидный азот является частью насыщенного гетероциклического кольца. Тем не менее, сохраняется потребность в соединениях, способных модулировать иммунный ответ, индуцирующих биосинтез цитокинов или активирующих другие механизмы.
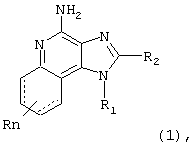
Мы открыли новый класс соединений, которые могут применяться для индукции у животных биосинтеза цитокинов. В соответствии с этим, в настоящем изобретении представлены соединения Формулы I:
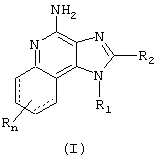
где R, R1 и R2 определены ниже.
Соединения Формулы I могут быть использованы в качестве иммуномодуляторов благодаря их способности индуцировать биосинтез цитокинов или иным способом модулировать иммунный ответ при их введении животным. Это позволяет применять эти соединения для лечения различных патологий, таких как вирусные заболевания и опухоли, чувствительных к таким изменениям иммунного ответа.
Кроме того, настоящее изобретение представляет фармацевтические препараты, содержащие терапевтически эффективные количества соединения Формулы I, и методы индукции биосинтеза цитокинов у животных, лечения вирусной инфекции и/или лечения неоплазических патологий у животных путем введения животному эффективного количества соединения Формулы I.
Помимо этого, представлены методы синтеза соединений Формулы I и промежуточных продуктов, используемых в синтезе этих соединений.
Как сказано выше, в настоящем изобретении представлены соединения Формулы I:
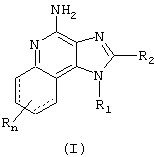
где
связи, изображенные пунктирной линией, могут как присутствовать, так и отсутствовать
R1 - это -алкил-NR3-SO2-Х-R4;
Х - это связь или -NR5-;
R4 - это фенил, нафтил, тиенил, хинолинил, изохинолинил; тетрагидрохинолинил, тиазолил, оксазолил, пиразолил, пиперазинил, пиперидинил, тиоморфолинил, пирролидинил, алкил или алкенил каждый из которых может быть незамещенным или замещенным одним или более заместителями, выбираемыми из группы, в которую входят:
- алкил;
- фенил;
- фенил, замещенный NO2;
- O-алкил;
- СООН;
- СО-O-алкил;
- S(O)0-2-алкил;
- (алкил)0-1-NR3R3;
- (алкил)0-1-NR3СО-O-алкил;
- (алкил)0-1-NR3-СО-алкил;
- (алкил)0-1-NR3-СО-замещенный фенил;
- галоген;
- галогеналкил;
- галогеналкоксил;
- СО-галогеналкоксил;
- NO2;
- CN; и в случае алкила, оксогруппа;
R2 выбирается из группы, в которую входят:
- водород;
- алкил;
- фенил;
- алкил-O-алкил; и
- алкил, замещенный одним или более заместителями, выбираемыми из:
- фенил, замещенный алкоксилом;
каждый из R3 независимо выбирают из группы, в которую входят водород и алкил C1-10;
R5 выбирают из группы, в которую входят водород и алкил C1-10; либо R4 и R5 в совокупности могут образовывать кольцо пиперазина, пиперидина, тиоморфолина или пирролидина;
n - это число 0
или его фармацевтически приемлемая соль.
Получение соединений
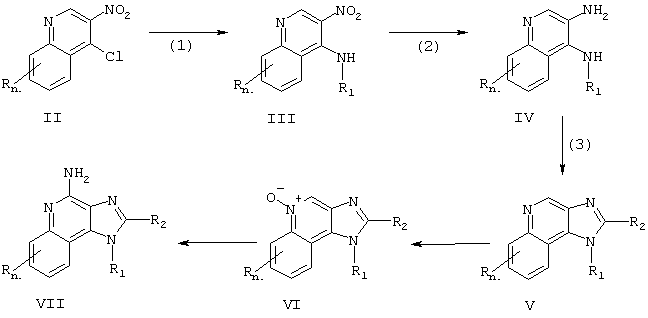
Иминоазохинолины настоящего изобретения можно получить в соответствии со Схемой реакции I, в которой R, R1, R2 и n определены выше.
На стадии (1) Схемы реакции I 4-хлор-3-нитрохинолин Формулы II взаимодействует с амином формулы R1NH2, в которой R1 определен выше, с образованием 3-нитрохинолин-4-амина Формулы III. Реакцию можно осуществить, прибавляя амин к раствору соединения Формулы II в подходящем растворителе, таком как хлороформ или дихлорметан, с возможным нагреванием. Многие хинолины Формулы II - это известные соединения (см., например, патент США №4689338 и ссылки в нем).
На стадии (2) Схемы реакции I 3-нитрохинолин-4-амин Формулы III восстанавливается до хинолин-3,4-диамина Формулы IV. Восстановление предпочтительно проводить на обычном гетерогенном катализаторе гидрирования, таком как платина на угле или палладий на угле. Реакцию удобно осуществлять в аппарате Парра в соответствующем растворителе, таком как изопропиловый спирт или толуол.
На стадии (3) Схемы реакции I хинолин-3,4-диамин Формулы IV взаимодействует с карбоновой кислотой или ее эквивалентом с образованием 1 Н-имидазо[4,5-с]хинолина Формулы V. Эквивалентом карбоновой кислоты вполне могут служить галогениды, простые ортоэфиры и 1,1-диалкоксиалкилалканоаты. Карбоновую кислоту или ее эквивалент выбирают таким образом, чтобы обеспечить желаемый заместитель R2 в Формуле V. Например, триэтилортоформиат позволит получить соединение, в котором R2 - это водород, а использование триэтилортоацетата приводит к соединению, в котором R2 - это метил. Реакция может протекать как в отсутствие растворителя, так и в таком инертном растворителе, как толуол. Реакцию проводят при значительном нагревании с тем, чтобы удалять любые спирты или воду, образующиеся в качестве побочных продуктов реакции.
На стадии (4) Схемы реакции I 1Н-имидазо[4,5-с]хинолин Формулы V под действием обычного окислителя, способного образовывать N-оксиды, окисляется до 1Н-имидазо[4,5-с]хинолин-5N-оксида Формулы VI. Реакцию предпочтительно проводить путем взаимодействия раствора соединения Формулы V в хлороформе с 3-хлорбензойной кислотой при комнатных условиях.
На стадии (5) Схемы реакции I 1Н-имидазо[4,5-с]хинолин-5N-оксид Формулы VI аминируют с образованием 1Н-имидазо[4,5-с]хинолин-4-амина Формулы VII, которая является одним из вариантов Формулы I. Стадия (5) включает в себя (i) взаимодействие соединения Формулы VI с ацилирующим агентом, а затем (ii) взаимодействие продукта этой реакции с аминирующим агентом. Часть (i) стадии (5) включает в себя взаимодействие N-оксида Формулы VI с ацилирующим агентом. В качестве ацилирующих агентов целесообразно использовать алкил- или арилсульфонилхлориды (например, бензолсульфонилхлорид, метансульфонилхлорид, п-толуолсульфонилхлорид). Арилсульфонилхлориды предпочтительны. Наиболее предпочтителен паратолуолсульфонилхлорид. Часть (ii) стадии (5) включает в себя взаимодействие продукта из части (i) с избытком аминирующего агента. В качестве аминирующих агентов целесообразно использовать аммиак (например, в виде гидроокиси аммония) или соли аммония (например, карбонат аммония, бикарбонат аммония, фосфат аммония). Гидроокись аммония предпочтительна. Реакцию предпочтительно осуществлять путем растворения N-оксида Формулы VI в инертном растворителе, таком как дихлорметан, прибавляя к раствору ацилирующий агент, а затем медленно прибавляя аминирующий агент. Продукт или его фармацевтически приемлемую соль можно выделить обычными способами.
По-другому стадию (5) можно провести путем (i) взаимодействия N-оксида Формулы VI с изоцианатом с последующим (ii) гидролизом полученного продукта. Часть (i) включает в себя взаимодействие N-оксида с изоцианатом, в котором изоцианатная группа подсоединена к карбонильной группе. К предпочтительным изоцианатам относятся трихлоацетилизоцианат и ароилизоцианаты, например, бензоилизоцианат. Взаимодействие изоцианата с N-оксидом протекает в безводных условиях путем прибавления изоцианата к раствору N-оксида в таком инертном растворителе, как хлороформ или дихлорметан. Часть (ii) включает в себя гидролиз продукта, полученного в части (i). Гидролиз можно осуществлять обычными методами, такими как нагревание в присутствии воды или низшего спирта, либо в присутствии такого катализатора, как гидроокись щелочного металла или низшего алкоголята.
Схема реакции I
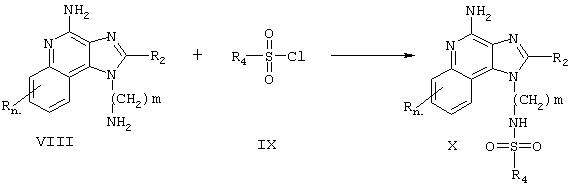
Соединения настоящего изобретения, в которых R1 - это сульфонамид, также могут быть получены в соответствии со Схемой реакции II, где R1, R2, R4 и n определены выше, а m - это число от 1 до 20.
Согласно Схеме реакции II аминоалкилзамещенный 1 Н-имидазо[4,5-с]хинолин-4-амин Формулы VII взаимодействует с сульфонилхлоридом Формулы IX с образованием соединения Формулы X, которая является одним из вариантов Формулы I. Реакцию можно проводить при комнатной температуре в таком инертном растворителе, как дихлорметан, в присутствии такого основания, как пиридин или N,N-диизопропилэтиленамин. Многие 1Н-имидазо[4,5-с]хинолин-4-амины - это известные соединения, см., например, патент США №6069149 (Намба (Namba)); другие легко получаются известными синтетическими методами. Многие сульфонилхлориды Формулы IX являются промышленными продуктами; другие легко получаются известными синтетическими методами. Продукт или его фармацевтически приемлемую соль выделяют с помощью обычных методов.
Схема реакции II
Соединения настоящего изобретения, в которых R1 - это сульфонамид, также могут быть получены в соответствии со Схемой реакции III, где R1, R2, R4 и n определены выше, a m - это число от 1 до 20.
Согласно Схеме реакции III аминоалкилзамещенный 1Н-имидазо[4,5-с]хинолин-4-амин Формулы VIII взаимодействует с ангидридом сульфоновой кислоты Формулы XI с образованием соединения Формулы X, которая является одним из вариантов Формулы I. Реакцию можно проводить при комнатной температуре в таком инертном растворителе, как дихлорметан, в присутствии такого основания, как пиридин или N,N-диизопропилэтиленамин. Реакцию можно проводить также при комнатной температуре в ацетонитриле. Многие ангидриды сульфоновых кислот Формулы XI являются промышленными продуктами; другие легко получаются известными синтетическими методами. Продукт или его фармацевтически приемлемую соль выделяют с помощью обычных методов.
Схема реакции III
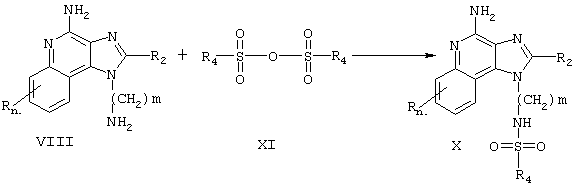
Третичные сульфонамиды настоящего изобретения также могут быть получены в соответствии со Схемой реакции IV, где R1, R2, R3, R4 и n определены выше, a m - это число от 1 до 20.
Согласно Схеме реакции IV аминоалкилзамещенный 1 Н-имидазо[4,5-с]хинолинилсульфонамид Формулы Х взаимодействует с галогенидом Формулы XII с образованием соединения Формулы XIII, которая является одним из вариантов Формулы I. Реакцию можно проводить при комнатной температуре, прибавляя к раствору соединения Формулы Х в N,N-диметилформамиде сначала гидрид натрия, а затем галогенид в таком инертном растворителе, как дихлорметан, в присутствии такого основания, как пиридин или N,N-диизопропилэтиленамин. Многие галогениды Формулы XII являются промышленными продуктами; другие легко получаются известными синтетическими методами. Продукт или его фармацевтически приемлемую соль выделяют с помощью обычных методов.
Схема реакции IV
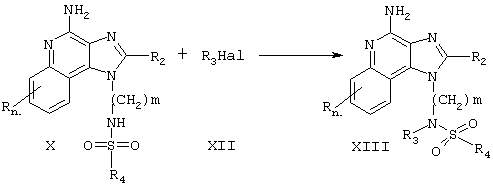
Соединения настоящего изобретения, в которых R1 содержит сульфамидную группу, могут быть получены в соответствии со Схемой реакции V, где R1, R2, R4, R5 и n определены выше, a m - это число от 1 до 20.
На стадии (1) Схемы реакции V аминоалкилзамещенный 1Н-имидазо[4,5-с]хинолин-4-амин Формулы VIII взаимодействует с сульфурилхлоридом с образованием на месте сульфамоилхлорида Формулы XIV. Реакцию можно проводить, прибавляя раствор сульфурилхлорида в дихлорметане к раствору соединения Формулы VIII дихлорметане в присутствии одного эквивалента 4-(диметиламино)пиридина. Реакцию предпочтительно проводить при пониженной температуре (-78°С). По окончании прибавления реакционной смеси можно дать нагреться до комнатной температуры.
На стадии (2) Схемы реакции V амин Формулы R5R4NH взаимодействует с сульфамоилхлоридом Формулы XIV с образованием 1Н-имидазо[4,5-с]хинолинилсульфамида Формулы XV, которая является одним из вариантов Формулы I. Реакцию можно проводить, прибавляя раствор, содержащий 2 эквивалента амина и 2 эквивалента триэтиламина в дихлорметане, к реакционной смеси из стадии (1). Прибавлять раствор предпочтительно при пониженнной температуре (-78°С). По окончании прибавления реакционной смеси можно дать нагреться до комнатной температуры. Продукт или его фармацевтически приемлемую соль выделяют с помощью обычных методов.
Схема реакции V
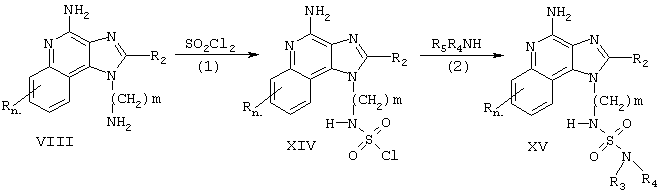
Тетрагидроимидазохинолины настоящего изобретения можно синтезировать в соответствии со Схемой реакции VI, где R2, R3, R4 и R5 определены выше, a m - это число от 1 до 20.
На стадии (1) Схемы реакции VI аминоалкилзамещенный 1Н-имидазо[4,5-с]хинолин-4-амин Формулы XVI восстанавливается до аминоалкилзамещенного 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина Формулы XVII. Реакцию восстановления предпочтительно проводить путем суспендирования или растворения соединения Формулы XVI в трифторуксусной кислоте, добавления каталитических количеств оксида платины (IV) и помещения реакционной смеси под давление водорода. Реакцию удобно проводить в аппарате Парра. Продукт или его соль можно выделить обычными методами.
На стадии (2а) Схемы реакции VI аминоалкилзамещенный 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин Формулы XVII взаимодействует с образованием соединения Формулы XVIII, которая является одним из вариантов Формулы I. Если R3 - это водород, то реакцию можно осуществить в одну стадию с помощью методов, описанных в представленных выше Схемах реакций II и III, используя тетрагидроимидазохинолин Формулы XVII вместо имидазохинолина Формулы VIII. Если R3 - это не водород, то реакцию можно осуществить в две стадии, причем первая стадия проводится с помощью методов, описанных в представленных выше Схемах реакций II и III, а вторая стадия проводится по методу в соответствии со Схемой реакции IV, используя тетрагидроимидазохинолиновый аналог имидазохинолина. Продукт или его фармацевтически приемлемую соль выделяют с помощью обычных методов.
На стадии (2b) Схемы реакции VI аминоалкилзамещенный 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин Формулы XVII взаимодействует с образованием соединения Формулы XIX, которая является одним из вариантов Формулы I. Реакцию можно осуществить методом, описанным в Схеме реакции V, используя тетрагидроимидазохинолин Формулы XVII вместо имидазохинолина Формулы VIII. Продукт или его фармацевтически приемлемую соль выделяют с помощью обычных методов.
Схема реакции VI
Тетрагидроимидазохинолины настоящего изобретения также можно синтезировать в соответствии со Схемой реакции VII, где R, R2, R3, R4, R5 и n определены выше, a m - это число от 1 до 20.
На стадии (1) Схемы реакции VII 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолинил-трет-бутилкарбамат Формулы XX гидролизуется до аминоалкилзамещенного 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина Формулы XXI. Реакцию можно осуществить путем растворения соединения Формулы XX в смеси трифторуксусной кислоты и ацетонитрила и перемешивания при комнатной температуре. Помимо этого, соединение Формулы XX можно смешать с разбавленной соляной кислотой и нагреть на паровой бане. Тетрагидро-1Н-имидазо[4,5-с]хинолинил-трет-бутилкарбаматы Формулы XX можно получить синтетическим методом, описанным в патенте США №5352784 (Николаидис (Nikolaides)). Продукт или его соль выделяют обычными методами.
Стадии (2а) и (2b) можно провести способом, аналогичным представленному в Схеме реакции VI.
Схема реакции VII
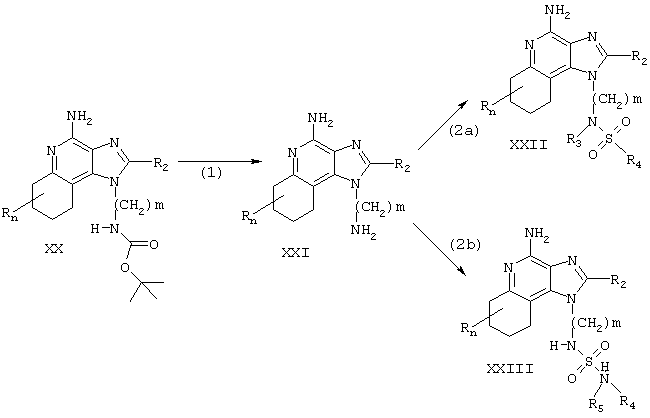
Некоторые соединения Формулы 1 легко можно получить из других соединений Формулы I. Например, соединения, в которых заместитель R4 содержит хлоралкильную группу, могут взаимодействовать с амином с образованием заместителя R4, замещенного вторичной или третичной аминогруппой; соединения, в которых заместитель R4 содержит нитрогруппу, могут восстанавливаться до соединения, в которых заместитель R4 содержит первичный амин.
В рамках настоящего изобретения, термиными “алкил”, “алкенил”, “алкинил” и приставка “-алк” обозначают как линейные и разветвленные, так и циклические группы, т.е. циклоалкильные и циклоалкенильные группы. Если не указано иное, то эти группы содержат от 1 до 20 атомов углерода, тогда как алкенильные и алкинильные группы содержат от 2 до 20 атомов углерода.
Предпочтительны группы, содержащие до 10 атомов углерода. Циклические группы могут быть моноциклическими или полициклическими и предпочтительно содержат от 3 до 10 атомов углерода, входящих в циклы. Примерами циклических групп могут служить циклопропил, циклогексил и адамантил.
Термином “галогеналкил” обозначают группы, замещенные одним или более галогенным атомами, в том числе группы, в которых все доступные атомы водорода замещены на атомы галогена. Это также относится к группам, в названии которых содержится приставка “галогеналк-”. Примерами приемлемых галогеналкильных групп могут служить хлорметил, трифторметил и т.п.
В рамках настоящего изобретения, термином “арил” обозначают ароматические кольца и циклические системы. Примерами арильных групп могут служить фенил, нафтил, бифенил, флуоренил и инденил. Термином “гетероарил” обозначают ароматические кольца и циклические системы, содержащие в кольце по крайней мере один гетероатом (например, О, S, N). К приемлемым гетероарильным группам относятся фурил, тиенил, пиридил, хинолинил, тетразолил, пиразолил, тиазолил, оксазолил и т.п.
Термином “гетероциклил” обозначают неароматические кольца и циклические системы, содержащие в кольце по крайней мере один гетероатом (например, О, S, N). Примерами гетероциклических групп могут служить пирролидинил, тетрагидрофуранил, морфолинил, тиоморфолинил, пиперидинил, пиперазинил, фурил, тиазолидинил, имидазолинил и т.п.
Если не указано иное, термины “замещенный циклоалкил”, “замещенный арил”, “замещенный гетероарил” и “замещенный гетероциклил” подразумевают, что кольца или циклические системы дополнительно замещены одним или более заместителями, независимо отбираемыми из группы, в которую входят алкил, алкоксил, алкилтио, гидроксил, галоген, галогеналкил, галогеналкилкарбонил, галогеналкоксил (например, трифторметоксил), нитрогруппа, алкилкарбонил, алкенилкарбонил, арилкарбонил, гетероарилкарбонил, арил, арилалкил, гетероарил, гетероарилалкил, гетероциклил, гетероциклоалкил, нитрил, алкоксикарбонил, алканоилоксил, алканоилтио и, в случае циклоалкила и гетероциклила, кетогруппа.
В структурных формулах, представляющих соединения настоящего изобретения, некоторые связи изображены пунктирными линиями. Такие линии означают, что связи, изображенные пунктирной линией могут как присутствовать, так и отсутствовать. Соответственно, Формулой I могут быть описаны как имидазохинолиновые соединения, так и тетрагидроимидазохинолиновые соединения.
Настоящее изобретение включает в себя описанные в нем соединения в любой из их фармацевтически приемлемых форм, в том числе такие изомеры, как диастереомеры, энантиомеры, соли, сольваты, полиморфы и т.п.
Фармацевтические препараты и биологическая активность
Фармацевтические препараты настоящего изобретения содержат терапевтически эффективное количество соединения Формулы I в сочетании с фармацевтически приемлемым носителем.
В рамках настоящего изобретения, термин “терапевтически эффективное количество” означает количество соединения, достаточное для проявления такого терапевтического эффекта, как индукция цитокинов, противоопухолевая активность и/или противовирусная активность. Хотя точное количество активного соединения в фармацевтическом препарате настоящего изобретения может меняться в зависимости от таких известных специалистам в данной области факторов, как физическая и химическая природа соединения и природа носителя, а также схема приема препарата, следует понимать, что препараты настоящего изобретения должны содержать активного ингредиента достаточно для того, чтобы организм получил дозу соединения от 100 нг/кг до 50 мг/кг, а предпочтительно - от 10 мг/кг до 5 мг/кг. Могут быть использованы любые обычные лекарственные формы, такие как таблетки, лепешки, парентеральные препараты, сиропы, кремы, мази, аэрозольные препараты, черескожные пластыри, пластыри на слизистую и т.п.
В опытах, проведенных по описанным ниже методикам, было показано, что соединения настоящего изобретения индуцируют синтез некоторых цитокинов. Эти результаты показывают, что такие соединения могут быть использованы в качестве иммуномодуляторов, способных несколькими разными способами модифицировать иммунный ответ, что позволяет применять их для лечения различных отклонений.
К цитокинам, которые могут быть индуцированы вследствие введения соединений настоящего изобретения, вообще говоря, относятся α-интерферон (α-ИФН), α-фактор некроза опухоли (α-ФНО) и некоторые интерлейкины (ИЛ). Цитокины, биосинтез которых могут индуцировать соединения настоящего изобретения, включают α-ИФН, α-ФНО, ИЛ-1, 6, 10, 12 и различные другие цитокины. Среди прочих эффектов, цитокины ингибируют размножение вирусов и рост опухолевых клеток, что делает их эффективным средством для лечения вирусных заболеваний и опухолей.
Помимо индуцирования синтеза цитокинов, соединения настоящего изобретения оказывают воздействие и на другие аспекты врожденного иммунного ответа. Например, под действием вырабатываемых цитокинов, скорее всего, повышается активность клеток естественных киллеров. Эти соединения также могут активировать макрофаги, что, в свою очередь, стимулирует выделение окиси азота и продукцию дополнительных цитокинов. Кроме того, эти соединения могут способствовать пролиферации и дифференцировке В-лимфоцитов.
Соединения настоящего изобретения также могут повлиять на приобретенный иммунный ответ. Например, хотя считается, что не существует какого-либо воздействия на Т-лимфоциты или прямой индукции цитокинов Т-лимфоцитов, введение этих соединений косвенно индуцирует продукцию цитокина γ-ИФН Т-хелпера 1-го типа (Тh-1) и ингибирует продукцию цитокинов ИЛ-4, ИЛ-5 и ИЛ-13 Т-хелпера 2-го типа (Тh-2). Такая активность указывает на то, что эти соединения являются эффективным средством при лечении таких заболеваний, где требуется активизация Тh-1 ответа и/или подавление Тh-2 ответа, то есть, ожидается, что эти соединения будут полезны в лечении, например, таких атопических заболеваний, как атопический дерматит, астма и аллергический ринит, как системная красная волчанка; а также в качестве адъювантной вакцины для клеточно опосредованного иммунитета; и возможно для лечения рецидивирующих грибковых заболеваний и хламидий.
Способность этих соединений модифицировать иммунный ответ делает их полезными в лечении широкого спектра отклонений. Благодаря их способности индуцировать выработку таких цитокинов, как α-ИФН и/или α-ФНО, эти соединения особенно эффективны при лечении вирусных заболеваний и опухолей. Такая иммуномодулирующая активность предполагает, что соединения настоящего изобретения эффективны при лечении таких и не только таких заболеваний, как вирусные заболевания, к которым относятся остроконечная бородавка; обычная бородавка; подошвенная бородавка; гепатит В, гепатит С, вирус простого герпеса типа I и типа II; кантагиозный моллюск; ВИЧ; цитомегаловирус; вирус ветряной оспы; интраэпителиальные неоплазии, такие как цервикальная интраэпителиальная неоплазия, папилломавирус человека (ПВЧ) и связанные с ним неоплазии; грибковые заболевания, например, кандидозный, аспергиллезный и криптококковый менингит; неоплазические патологии, например, базально-клеточная карцинома; лейкемический ретикулез, саркома Капоши, почечно-клеточная карцинома, плоскоклеточная карцинома, миелогенная лейкемия, множественная миелома, меланома, не-ходжкинская лейкома, кожная Т-клеточная лимфома, другие виды рака; паразитические заболевания, например, pneumocystis carnii, криптоспоридиоз, гистоплазмоз, токсоплазмоз, трипаносомная инфекция, лейшманиоз; а также бактериальные инфекции, например, туберкулез, mycobacterium avium. Кроме того, с помощью соединений настоящего изобретения можно лечить такие заболевания, как экзема; эозинофилия; врожденная тромбоцитэмия; лепра; рассеянный склероз; синдром Омена; болезнь Боуэна; боуэновидный папулез. Эти соединения также способствуют заживлению ран, в том числе хронических язв, и стимулируют его.
Таким образом, настоящее изобретение представляет способ индукции биосинтеза цитокинов в организме животного путем введения в него эффективного количества соединения Формулы I. Количество соединения, эффективное для индукции биосинтеза цитокинов - это количество, достаточное для того, чтобы один или несколько типов клеток, таких как моноциты, макрофаги, дендритные клетки и В-клетки, стали продуцировать один или несколько таких цитокинов, как, например α-ИФН, α-ФНО, ИЛ-1, 6, 10 и 12, которое превысило бы базовый уровень этих цитокинов. Точное количество может меняться, в зависимости от факторов, известных специалистам в данной области, однако, скорее всего, это будет доза от 100 нг/кг до 50 мг/кг, а предпочтительно от 10 мкг/кг до 5 мг/кг. Настоящее изобретение также представляет способ лечения вирусных инфекций у животных и способ лечения неоплазических состояний у животных, включающий в себя введение в организм эффективного количества соединения Формулы I. Количество, эффективное для лечения или подавления вирусного заболевания, это такое количество, которое вызывает ослабление одного или более признаков инфекции, таких как вирусное поражение, вирусная нагрузка, уровень продукции вируса и смертность, по сравнению с контрольной группой животных, не подвергавшихся воздействию соединения. Точное количество может меняться в зависимости от факторов, известных специалистам в данной области, однако можно предположить, что доза составит от 100 нг/кг до 50 мг/кг, а предпочтительно от 10 мкг/кг до 5 мг/кг. Количество соединения, эффективное для лечения неоплазического состояния, это такое количество, которое приводит к уменьшению размера опухоли или количества очагов опухолей. Опять же, точное количество может меняться в зависимости от факторов, известных специалистам в данной области, однако можно предположить, что доза составит от 100 нг/кг до 50 мг/кг, а предпочтительно от 10 мкг/кг до 5 мг/кг.
Далее настоящее изобретение описывается на примерах, которые предназначены только для иллюстрации, а вовсе не для ограничения.
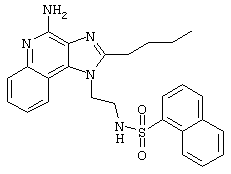
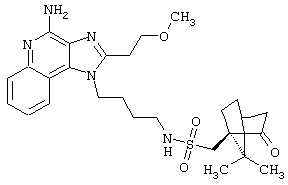
Пример 1
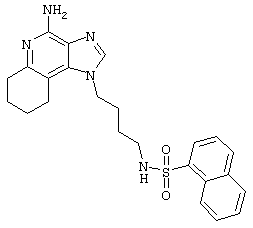
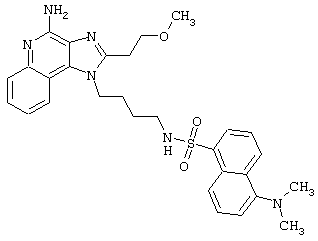
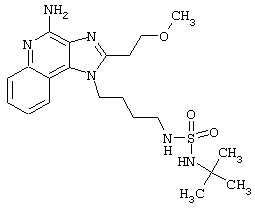
N1-[4-(4-Aминo-2-бyтил-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-5-(диметиламино)-1-нафталинсульфонамид
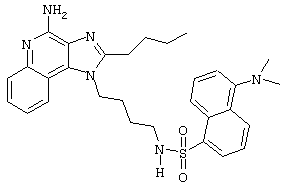
5-Диметиламино-1-нафталинсульфонилхлорид (1,82 г, 6,74 ммоль) прибавляют к смеси N,N-диизопропилэтиламина (1,23 мл, 7,06 ммоль), дихлорметана (15 мл) и 1-(4-аминобутил)-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амина (2,0 г, 6,42 ммоль). Реакционную смесь оставляют на ночь перемешиваться при комнатной температуре. К смеси прибавляют метанол до получения прозрачного раствора. К реакционной смеси прибавляют силикагель, а затем удаляют растворители. Силикагель помещают в колонку, а затем элюируют хлороформом с пошаговым градиентом до соотношения хлороформ / метанол 9:1. Полученный продукт перекристаллизовывают из N,N-диметилформамида и деионизированной воды, выделяя 2,5 г N1-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-5-(диметиламино)-1-нафталинсульфонамида в виде желтого кристаллического вещества с температурой плавления 223-224°С. Элементный анализ: Теоретически рассчитано для С30Н36N6О2S: %С 66,15; %Н 6,66; %N 15,43; Экспериментально найдено: %С 66,36; %Н 6,34; %N 15,23.
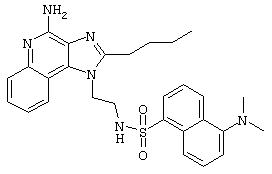
Пример 2
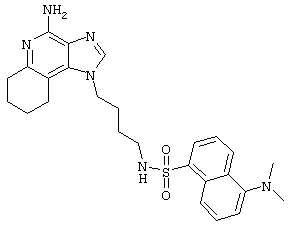
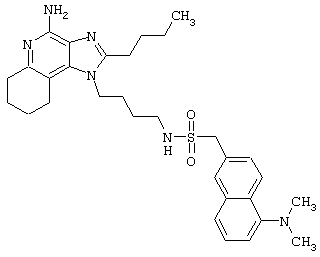
N1-[4-(4-Амино-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-5-(диметиламино)-1-нафталинсульфонамид
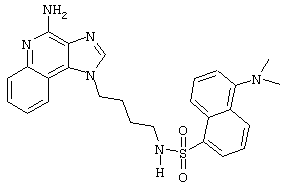
Суспензию 1-(4-аминобутил)-1Н-имидазо[4,5-с]хинолин-4-амина (0,5 г, 2,0 ммоль) в пиридине (250 мл) нагревают до 60°С до растворения амина.
Раствору дают остыть до приблизительно 30°С, а затем медленно прибавляют 5-диметиламино-1-нафталинсульфонилхлорид (0,5 г, 1,8 ммоль). Через 1 час прибавляют еще 0,3 г 5-диметиламино-1-нафталинсульфонилхлорида. Реакционную смесь нагревают до 60°С и при этой температуре оставляют на ночь. Реакционную смесь концентрируют под вакуумом. Остаток перекристаллизовывают из пропилацетата. Выход: твердый N1-[4-(4-амино-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-5-(диметиламино)-1-нафталинсульфонамид с температурой плавления 200-201°С.
Пример 3
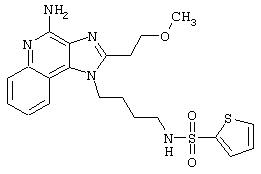
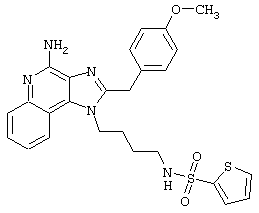
N2-[4-(4-Aминo-2-бyтил-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-2-тиофенсульфонамид

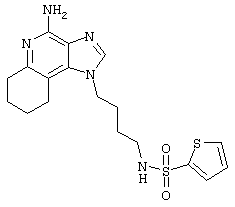
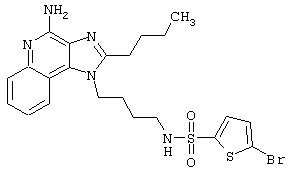
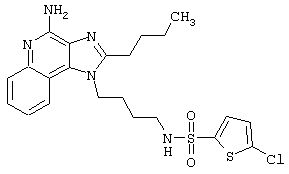
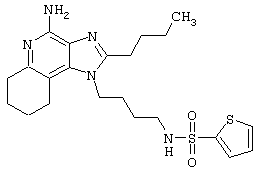
2-Тиофенсульфонилхлорид (0,3 г в 10 мл дихлорметана, 1,6 ммоль) при перемешивании прикапывают к раствору 1-(4-аминобутил)-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амина (0,5 г, 1,6 ммоль) в дихлорметане (40 мл) и пиридине (0,8 мл). В течение нескольких часов реакционную смесь выдерживают при комнатной температуре, а затем прибавляют дополнительную порцию 2-тиофенсульфонилхлорида (0,1 г, 0,6 ммоль). Реакционную смесь оставляют на ночь, а затем концентрируют под вакуумом. Полученный остаток очищают колоночной хроматографией под давлением (силикагель, 9/1 дихлорметан / метанол), а фракции, содержащие продукт, промывают насыщенным водным раствором бикарбоната натрия. Органический слой сушат (МgSO4), фильтруют, и после концентрации получают 0,2 г N2-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-2-тиофенсульфонамида в виде белого порошка с температурой плавления 137,5-141,5°С. ЯМР 1H (300 МГц, ДМСО-D6) δ 8,00 (дублет, J=8,0 Гц, 1Н), 7,89 (двойной дублет, J=5,0, 1,3 Гц, 1Н), 7,83 (шир. с., 1Н), 7,61 (двойной дублет, J=8,3, 1,1 Гц, 1Н), 7,54 (двойной дублет, J=3,7, 1,3 Гц, 1Н), 7,42 (триплет, J=7,2 Гц, 1Н), 7,25 (ср., 1Н), 7,15 (ср., 1Н), 6,44 (шир. с., 2Н), 4,47 (триплет, J=7,4 Гц, 2Н), 2,87 (ср., 4Н), 1,80 (ср., 4Н), 1,58-1,38 (ср., 4Н), 0,96 (триплет, J=7,4 Гц, 3Н); ИК (KBr) 3467, 3361, 3167, 3091, 2957, 2933, 2870, 1644, 1617, 1585, 1533, 1478, 1405, 1336, 1154, 1095, 1014, 854, 761, 733 см-1; Масс-спектрометрия (электронный удар) m/e 457,1606 (457,1606 рассчитано для C22H27N5O2S2); Элементный анализ: теор. для C22H27N5O2S2: С - 57,74; Н - 5,95; N - 15,30; эксп.: С - 57,50; Н - 5,98; N - 15,15.
Пример 4
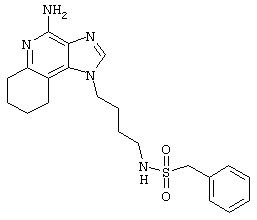
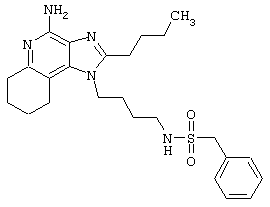
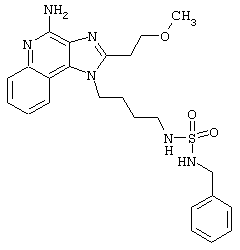
N-[4-(4-Амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-фенилметансульфонамид
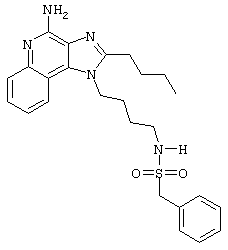
2-Толуолсульфонилхлорид (0,5 г в 10 мл дихлорметана, 2,7 ммоль) при перемешивании прикапывают к раствору 1-(4-аминобутил)-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амина (0,75 г, 2,4 ммоль) в дихлорметане (115 мл) и пиридине (1 мл). В течение 4 часов реакционную смесь выдерживают при комнатной температуре, а затем концентрируют под вакуумом. Полученный остаток очищают колоночной хроматографией под давлением (силикагель, 9/1 дихлорметан / метанол, Rf 0,16). Фракции, содержащие продукт, соединяют и промывают насыщенным водным раствором бикарбоната натрия. Органический слой сушат (МgSO4), фильтруют и концентрируют. После конечной перекристаллизации из смеси дихлорметана с диэтиловым эфиром получают 0,65 г N-[4-(4-амино-2-бутил-1 Н-имидазо[4,5-с]хинолин-1-ил)бутил]-фенилметансульфонамида в виде белого порошка с температурой плавления 197,0-199,5°С. ЯМР 1H (300 МГц, ДМСО-D6) δ 8,02 (дублет, J=7,6 Гц, 1Н), 7,62 (двойной дублет, J=8,3, 1,1 Гц, 1Н), 7,42 (двойной триплет, J=7,5, 1,1 Гц, 1Н), 7,35-7,23 (ср., 1Н), 7,12 (триплет, J=5,4 Гц, 1Н), 6,46 (шир. с., 2Н), 4,49 (триплет, J=7,5 Гц, 2Н), 4,29 (с., 2Н), 2,91 (ср., 4Н), 1,83-1,42 (ср., 8Н), 0,96 (триплет, J=7,4 Гц, 3Н); ИК (KBr) 3460, 3293, 3226, 3158, 2955, 2931, 2867, 1632, 1586, 1534, 1482, 1437, 1389, 1331, 1152, 1094, 752, 700 см-1; Масс-спектрометрия (электронный удар) m/e 465, 2204 (465, 2198 рассчитано для С26Н31N5O2S); Элементный анализ: теор. для C25H31N5O2S: С - 64,49; Н - 6,71; N - 15,04; эксп.: С - 64,15; Н - 6,71; N - 15,00.
Пример 5
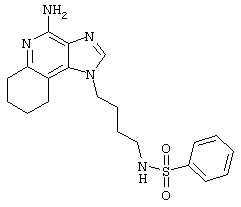
N1-[4-(4-Амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-бензолсульфонамид
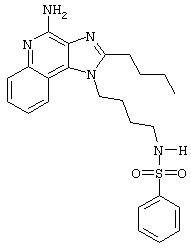
2-Бензолсульфонилхлорид (0,45 мл в 10 мл дихлорметана, 3,5 ммоль) при перемешивании прикапывают к раствору 1-(4-аминобутил)-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амина (1,0 г, 3,2 ммоль) в дихлорметане (140 мл) и пиридине (0,8 мл). В течение 4 часов реакционную смесь выдерживают при комнатной температуре, а затем концентрируют под вакуумом. Полученный остаток очищают колоночной хроматографией под давлением (силикагель, 9/1 дихлорметан / метанол, Rf 0,28) с последующей перекристаллизацией из смеси дихлорметана с диэтиловым эфиром. Выход: 1,14 г N1-[4-(4-амино-2-бутил-1H-имидазо[4,5-с]хинолин-1-ил)бутил]-1-бензолсульфонамида в виде белого порошка с температурой плавления 75,5-79,0°С. ЯМР 1H (300 МГц, ДМСО-D6) δ 7,99 (дублет, J=7,7 Гц, 1Н), 7,76 (дублет, J=7,2 Гц, 1Н), 7,63-7,53 (ср., 5Н), 7,42 (ср., 5Н), 7,25 (ср., 1Н), 6,43 (шир. с., 2Н), 4,45 (триплет, J=7,6 Гц, 2Н), 2,87 (триплет, J=7,7 Гц, 2Н), 2,78 (ср., 2Н), 1,79 (ср., 4Н), 1,55-1,40 (ср., 4Н), 0,95 (триплет, J=7,4 Гц, 3Н); Масс-спектрометрия (электронный удар) m/e 451,2036 (451,2042 рассчитано для C24H29N5O2S); Элементный анализ: теор. для C24H29N5O2S: С - 63,83; Н - 6,47; N - 15,51; эксп.: С - 63,89; Н - 6,42; N - 15,30.
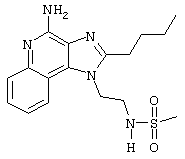
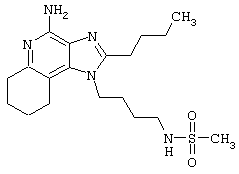
Пример 6
N-[4-(4-Амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]метансульфонамид
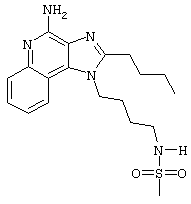
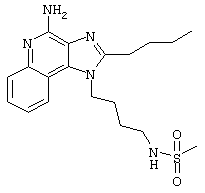
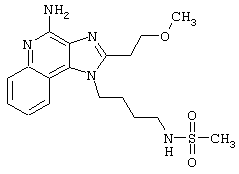
Метансульфоновый ангидрид (0,6 г, 3,4 ммоль) при перемешивании прикапывают к раствору 1-(4-аминобутил)-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амина (1,0 г, 3,2 ммоль) в ацетонитриле (200 мл). Через несколько минут образуется осадок. Растворитель удаляют под вакуумом, а остаток разделяют между дихлорметаном и насыщенным водным раствором бикарбоната натрия. Фракции отделяют, и органическую фракцию сушат (MgSO4), фильтруют и концентрируют. Получается сырой твердый продукт белого цвета. После перекристаллизации из метилацетата получают белый кристаллический N-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]метансульфонамид с температурой плавления 195,1-196,0°С. ЯМР 1H (300 МГц, ДМСО-D6) δ 8,04 (дублет, J=7,4 Гц, 1Н), 7,61 (двойной дублет, J=8,3, 1,2 Гц, 1Н), 7,50 (двойной триплет, J=7,5, 1,1 Гц, 1Н), 7,26 (двойной триплет, J=7,5, 1,2 Гц, 1Н), 6,99 (триплет, J=5,7 Гц, 1Н), 6,44 (шир. с., 2Н), 4,52 (триплет, J=7,5 Гц, 2Н), 3.02-2,86 (ср., 7Н), 1,82 (ср., 4Н), 1,62 (ср., 2Н), 1,46 (квадруплет, J=7,4 Гц, 2Н), 0,96 (триплет, J=7,4 Гц, 3Н); ИК (KBr) 3348, 3299, 3152, 2952, 2931, 2869, 1642, 1584, 1530, 1480, 1323, 1155, 1142, 1094, 982, 765 см-1; Масс-спектрометрия (электронный удар) m/e 389, 1889 (389, 1885 рассчитано для C19H27N5O2S);
Элементный анализ: теор. для C19H27N5O2S: С - 58,59; Н - 6,99; N - 17,98; эксп. С - 58,26; Н-6,64; N-17,69.
Пример 7
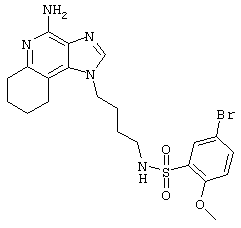
N1-[4-(4-Амино-2-бутил-1Н-имидазо[4,5-c]хинолин-1-ил)бутил]-3-нитро-1-бензолсульфонамид гидрохлорид
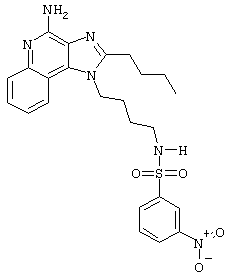
В соответствии с общей методикой, описанной в Примере 5, 3-нитробензолсульфонилхлорид соединяют с 1-(4-аминобутил)-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амином. N1-[4-(4-Амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-3-нитро-1-бензолсульфонамид выделяют в виде хлоргидратной соли (белое твердое вещество) с температурой плавления 176,0-178,2°С. ЯМР 1H (300 МГц, ДМСО-D6) δ 8.70 (оч. шир., с., 2Н), 8,49-8,42 (ср., 2Н), 8,21-8,17 (ср., 2Н), 8,06 (триплет, J=5,7 Гц, 1Н), 7,88-7,81 (ср., 2Н), 7,71 (триплет, J=7,7 Гц, 1Н), 7,57 (триплет, J=7,7 Гц, 1Н), 4,56 (триплет, J=7,3 Гц, 2Н), 2,94 (триплет, J=7,7 Гц, 2Н), 2,86 (ср., 2Н), 1,81 (ср., 4Н), 1,60-1,42 (ср., 4Н), 0,96 (триплет, J=7,3 Гц, 3Н); ИК (KBr) 3096, 2954, 2771, 1671, 1607, 1528, 1351, 1335, 1163, 1128, 1083, 879, 758, 735, 672, 661 см-1; Масс-спектрометрия (электронный удар) m/e 496,1897 (496,1893 рассчитано для C24H28N6O4S); Элементный анализ: теор. для C24H28N6O4S·HCl·H2O: С - 52.31; Н - 5.67; N - 15,25; эксп.: С - 52.26; Н - 5.46; N - 15,09.
Пример 8
N1-[4-(4-Амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-3-амино-1-бензолсульфонамид гидрохлорид
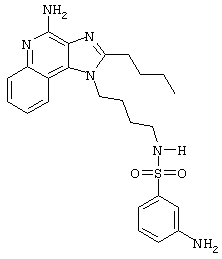
В раствор N1-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-3-нитро-1-бензолсульфонамида гидрохлорида (0,4 г) в метаноле (250 мл) загружают каталитическое количество 10% палладия на угле (0,085 г). Реакционную смесь помещают в атмосферу водорода (50 фунт/кв.дюйм; 3,44·105 Па) и в течение 2 часов встряхивают в аппарате Парра. Затем реакционную смесь фильтруют и растворитель удаляют под вакуумом. После перекристаллизации твердого продукта из пропанола-2 получают 0,18 г желтовато-белого кристаллического N1-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-3-амино-1-бензолсульфонамида гидрохлорида с температурой плавления 110,2°С (разл.). ЯМР 1H (300 МГц, ДМСО-D6) δ 8,70 (оч. шир., с., 2Н), 8,22 (дублет, J=8,2 Гц, 1Н), 7,83 (дублет, J=7,8 Гц, 1Н), 7,72 (триплет, J=7,6 Гц, 1Н), 7,59 (триплет, J=7,7 Гц, 1Н), 7,43 (триплет, J=5,9 Гц, 1Н), 7,15 (триплет, J=7,9 Гц, 1Н), 6,95 (триплет, J=1,9 Гц, 1Н),6,84 (дублет, J=7,7 Гц, 1Н), 6,73 (двойной дублет, J=8,0, 1,5 Гц, 1Н), 5,63 (шир., с., 2Н), 4,56 (триплет, J=7,5 Гц, 2Н), 2,77 (квадруплет, J=6,3 Гц, 2Н), 1,83 (ср., 4Н), 1,60-1,40 (ср., 4Н), 0,97 (триплет, J=7,3 Гц, 3Н); ИК (KBr) 3313, 3135, 2957, 2870, 2782, 1671, 1599, 1485, 1454, 1313, 1155, 1084, 754, 735, 686 см-1;
Масс-спектрометрия (электронный удар) m/е 466,2150 (466, 2151 рассчитано для C24H30N6O2S); Элементный анализ: теор. для C24H30N6O2S·HCl·0,25H2O: С - 56,79; Н - 6,26; N - 16,56, Cl - 6,98; эксп.: С - 56,87; Н - 6,22; N - 16,19, Cl - 7,22.
Пример 9
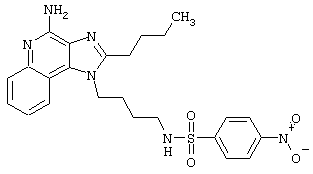
N1-[4-(4-Амино-2-бутил-1H-имидазо[4,5-с]хинолин-1-ил)бутил]-4-нитро-1-бензолсульфонамид гидрохлорид
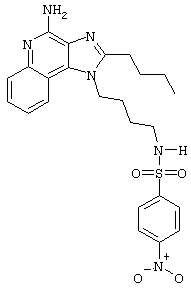
В соответствии с общей методикой, описанной в Примере 5, 4-нитробензолсульфонилхлорид соединяют с 1-(4-аминобутил)-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амином. N1-[4-(4-Амино-2-бутил-1H-имидазо[4,5-с]хинолин-1-ил)бутил]-4-нитро-1-бензолсульфонамид выделяют в виде хлоргидратной соли (белое твердое вещество) с температурой плавления 96,0°С (разл.). ЯМР 1H (300 МГц, ДМСО-D6) δ 8,70 (оч. шир., с., 2Н), 8,38-8,34 (ср., 2Н), 8,19 (дублет, J=8,2 Гц, 1Н), 8,09 (триплет, J=5,6 Гц, 1Н), 8,03-7,99 (ср., 2Н), 7,80 (дублет, J=7,4 Гц, 1Н), 7,68 (триплет, J=7,4 Гц, 1Н), 7,54 (триплет, J=7,2 Гц, 1Н), 4,55 (триплет, J=7,4 Гц, 2Н), 2,94 (триплет, J=7,7 Гц, 2Н), 2,86 (квадруплет, J=6,2 Гц, 2Н), 1,80 (ср., 4Н), 1,58 (ср., 4Н), 1,45 (квадруплет, J=7,5 Гц, 2Н), 0,96 (триплет, J=7,3 Гц, 3Н); ИК (KBr) 3283, 3100, 2957, 2870, 2782, 1670, 1606, 1528, 1347, 1311, 1162, 1092, 854, 746, 737, 686 см-1; Масс-спектрометрия (электронный удар) m/e 496, 1902 (496, 1893 рассчитано для C24H28N6O4S); Элементный анализ: теор. для С24Н28N6O4S·НСl·0,85Н2O: С - 52.57; Н - 5.64; N - 15,33; эксп.: С - 52.57; Н - 5.46; N - 15,33.
Пример 10
N1-[4-(4-Aминo-2-бyтил-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-4-амино-1-бензолсульфонамид гидрохлорид
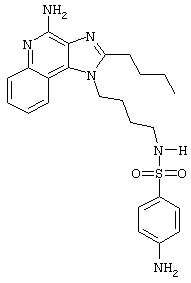
В раствор N1-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-4-нитро-1-бензолсульфонамида гидрохлорида (0,38 г) в метаноле (250 мл) загружают каталитическое количество 10% палладия на угле (0,085 г). Реакционную смесь помещают в атмосферу водорода (50 фунт/кв.дюйм; 3,44·105 Па) и в течение 2 часов встряхивают в аппарате Парра. Реакционную смесь фильтруют и растворитель удаляют под вакуумом. После перекристаллизации твердого продукта из пропанола-2 получают 0,34 г желтовато-белого порошка N1-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-4-амино-1-бензолсульфонамида гидрохлорида с температурой плавления 203,1-205,0°С. ЯМР 1H (300 МГц, ДМСО-D6) δ 8,65 (оч. шир, с., 2Н), 8,21 (дублет, J=8,0 Гц, 1Н), 7,82 (ср., 1Н), 7,71 (триплет, J=7,7 Гц, 1Н), 7,58 (триплет, J=7,7 Гц, 1Н), 7,38 (дублет, J=8,7 Гц, 1Н), 7,13 (триплет, J=5,9 Гц, 1Н), 6,60 (дублет, J=8,7 Гц, 1Н), 5,92 (шир., с., 2Н), 4,55 (триплет, J=7,6 Гц, 2Н), 2,96 (триплет, J=7,6 Гц, 2Н), 2,70 (квадруплет, J=6,4 Гц, 2Н), 1,81 (ср., 4Н), 1,58-1,43 (ср., 4Н), 0,96 (триплет, J=7,4 Гц, 3Н); ИК (KBr) 3430, 3316, 3215, 3046, 2955, 2868, 2679, 1671, 1594, 1334, 1157, 1091, 851, 776, 759 см-1; Масс-спектрометрия (электронный удар) m/e 466,2145 (466,2151 рассчитано для C24H30N6O2S); Элементный анализ: теор. для C24H30N6O2S·HCl: С - 57,30; Н - 6,21; N - 16,71; эксп.: С - 57,36; Н - 6,31;N - 16,21.
Пример 11
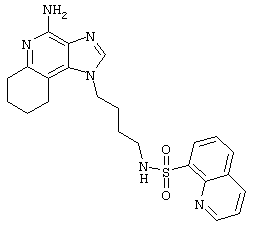
N5-[4-(4-Aминo-2-бyтил-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-5-изохинолинсульфонамид
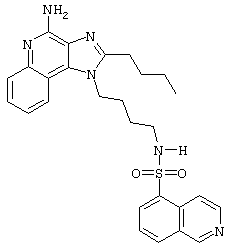
Суспензию изохинолин-5-сульфонилхлорида гидрохлорида (0,83 г в 50 мл пиридина, 3,1 ммоль) при перемешивании прикапывают к раствору 1-(4-аминобутил)-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амина (1,0 г, 3,2 ммоль) в дихлорметане (175 мл). Раствор приобретает ярко-желтый цвет, и в течение 4 часов его выдерживают при комнатной температуре. Прибавляют еще 0,18 г изохинолин-5-сульфонилхлорида гидрохлорида и поддерживают реакцию в течение еще 60 часов. Желтый раствор концентрируют под вакуумом, растворяют в дихлорметане и последовательно промывают насыщенным водным раствором бикарбоната натрия и водой. Органическую фракцию сушат (МgSO4), фильтруют и концентрируют под вакуумом. Полученный остаток очищают колоночной хроматографией под давлением (силикагель, 9/1 дихлорметан / метанол). Выход: 0,7 г N5-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-5-изохинолинсульфонамида в виде белого порошка с температурой плавления 96,0°С (разл.). ЯМР 1H (300 МГц, ДМСО-D6) δ 9,44 (дублет, J=0,7 Гц, 1Н), 8,64 (дублет, J 6,1 Гц, 1Н), 8,41-8,35 (ср., 2Н), 8,30 (двойной дублет, J=7,4, 1,2 Гц, 1Н), 8,11 (триплет, J=5,6 Гц, 1Н) 7,92 (дублет, J=7,6 Гц, 1Н), 7,75 (дублет, J=7,7 Гц, 1Н), 7,61 (двойной дублет, J=8,3, 1,2 Гц, 1Н), 7,41 (двойной триплет, J=7,7, 1,2 Гц, 1Н), 7,22 (двойной триплет, J=7,6, 1,2 Гц, 1Н), 6,47 (шир. с., 2Н), 4,38 (триплет, J=7,5 Гц, 2Н), 2,86-2,74 (ср., 4Н), 1,78-1,63 (ср., 4Н), 1,50-1,34 (ср., 4Н), 0,94 (триплет, J=7,4 Гц, 3Н); Масс-спектрометрия (электронный удар) m/е 502,2151 (502,2151 рассчитано для С27Н30N6O2S); Элементный анализ: теор. для C27H30N6O2S: С - 64,52; Н - 6,02; N - 16,72; эксп.: С - 64,03; Н - 6,03; N - 16,55.
Пример 12
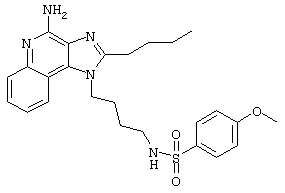
N-[4-(4-Амино-2-(4-метоксибензил)-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]метансульфонамид

Метансульфоновый ангидрид (0,19 г, 1,1 ммоль) при перемешивании прикапывают к раствору 1-(4-аминобутил)-2-(4-метоксибензил)-1Н-имидазо[4,5-с]хинолин-4-амина (1,0 г, 3,2 ммоль) в дихлорметане (75 мл) и ацетонитриле (100 мл). Реакцию поддерживают в течение 60 часов при комнатной температуре. Растворитель удаляют под вакуумом, а остаток очищают колоночной хроматографией под давлением (силикагель, 9/1 дихлорметан / метанол). Фракции, содержащие продукт, соединяют и промывают насыщенным водным раствором бикарбоната натрия, сушат (МgSO4), фильтруют и концентрируют. Выход: N-[4-(4-амино-2-(4-метоксибензил)-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]метансульфонамида в виде белого твердого вещества с температурой плавления 78,1-79,5°С. ЯМР 1H (300 МГц, ДМСО-D6) δ 7,99 (дублет, J=7,6 Гц, 1Н), 7,62 (двойной дублет, J=8,3, 1,2 Гц, 1Н), 7,42 (ср., 1Н), 7,27-7,21 (ср., 3Н), 6,98 (триплет, J=5,7 Гц, 1Н), 6,89 (дублет, J=8,7 Гц, 1Н), 6,58 (шир. с, 2Н), 4,45 (шир. с., 2Н), 4,33 (с., 2Н), 3,72 (с., 3Н), 2,87 (ср., 5Н), 1,55 (шир. с., 2Н; Масс-спектрометрия (химическая ионизация (ХИ)) m/е 454 (М+Н).
Пример 13
N-[4-(4-Амино-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-1-бутансульфонамид
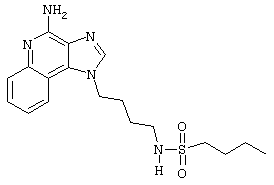
Раствор 1-(4-аминобутил)-2-(4-метоксибензил)-1Н-имидазо[4,5-с]хинолин-4-амина (9,3, 36 мкмоль) в 10 мл дихлорметана в пробирке с завинчивающейся крышкой охлаждают до -5°С. Прибавляют бутансульфонилхлорид (45 мкмоль) в виде 0,3 М раствора в дихлорметане; в ходе прибавления и в течение 15 секунд после его окончания через смесь пропускают аргон. Смесь оставляют на ночь при -5°С. Прибавляют аминометиловую полистирольную смолу (приблизительно 90 мг, 0,62 мг-экв/г, 100-200 меш, ф. “Бахем” (Bachem)) и смесь нагревают до кипения и встряхивают при 600 об/мин в течение 3 часов. Для того, чтобы удалить смолу, смесь фильтруют через колонку Poly-Prep (Bio-Rad №731-1550). Растворитель удаляют под вакуумом, а остаток очищают методом полупрепаративной жидкостной хроматографии на системе Джилсона (Gilson) (колонка Rainin Microsorb C18, 21,4×250 мм, размер частиц 8 мкм, поры 60 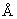 , 10 мл/мин, градиентное элюирование от 2 до 95% Б в течение 25 минут, 5-минутная задержка на 95% Б, где А=0,1% трифторуксусной кислоты в воде, а Б=0,1% трифторуксусной кислоты в ацетонитриле, включение сбора фракций детектируется по пику 254 нм). Фракции, полученные полупрепаративной жидкостной хроматографией, анализируют на хромато-масс-спектрометре ЖХ-ХИАД/МС и соответствующие фракции соединяют и лиофилизуют. Твердое вещество растворяют приблизительно в 3 мл смеси дихлорметан / метанол 2/1 и для того, чтобы выделить свободный амин, в течение 2 часов встряхивают с 80 мг (300 мкмоль) диизопропиламинометил-полистирольной смолой (Argonaut PS-DIEA, 3,86 ммоль/г), а затем фильтруют, и твердый продукт высушивают под вакуумом. Масс-спектрометрия (химическая ионизация при атмосферном давлении (ХИАД)) m/е 376,16 (М+Н).
, 10 мл/мин, градиентное элюирование от 2 до 95% Б в течение 25 минут, 5-минутная задержка на 95% Б, где А=0,1% трифторуксусной кислоты в воде, а Б=0,1% трифторуксусной кислоты в ацетонитриле, включение сбора фракций детектируется по пику 254 нм). Фракции, полученные полупрепаративной жидкостной хроматографией, анализируют на хромато-масс-спектрометре ЖХ-ХИАД/МС и соответствующие фракции соединяют и лиофилизуют. Твердое вещество растворяют приблизительно в 3 мл смеси дихлорметан / метанол 2/1 и для того, чтобы выделить свободный амин, в течение 2 часов встряхивают с 80 мг (300 мкмоль) диизопропиламинометил-полистирольной смолой (Argonaut PS-DIEA, 3,86 ммоль/г), а затем фильтруют, и твердый продукт высушивают под вакуумом. Масс-спектрометрия (химическая ионизация при атмосферном давлении (ХИАД)) m/е 376,16 (М+Н).
Пример 14
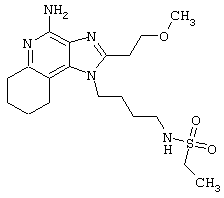
N1-{4-[4-Амино-2-(2-метоксиэтил)-6,7,8,9-тетратдро-1Н-имидазо[4,5-с]хинолин-1-ил]бутил}-4-фтор-1-бензолсульфонамид
В соответствии с общей методикой, описанной в Примере 5, 1-(4-аминобутил)-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин соединяют с фторбензолсульфонилхлоридом. После перекристаллизации из смеси н-пропилацетат / метанол 4/1 получают белый кристаллический N1-{4-[4-амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-1-ил]бутил}-4-фтор-1-бензолсульфонамид с температурой плавления 191,0-193,0°С. ЯМР 1H (300 МГц, ДМСО-D6) δ 7,86-7,81 (ср., 2Н), 7,45-7,39 (ср., 2Н), 5,65 (шир. с., 2Н), 4,15 (ср., 2Н), 3,76 (триплет, J=6,7 Гц, 2Н), 3,27 (ср., 3Н), 3,00 (триплет, J=6,7 Гц, 2Н), 2,90 (шир. с., 2Н), 2,78 (ср., 2Н), 2,65 (шир. с., 2Н), 1,75 (шир. с., 4Н), 1,61 (ср., 2Н), 1,43 (ср., 2Н); Масс-спектрометрия (ХИ) m/е 476 (М+Н); Элементный анализ: теор. для С23Н30FN5O3S: %С 58,09; %Н 6,36; %N 14,73; эксп.: %С 58,37; %Н 6,35; %N 14,60.
Пример 15
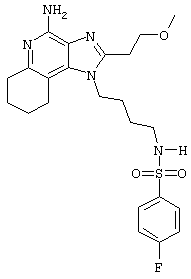
N1-[4-(4-Aминo-2-фeнил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-4-фтор-1-бензолсульфонамид
Часть А
Раствор бензоилхлорида (5,3 г, 37,7 ммоль) в дихлорметане (100 мл) при комнатной температуре медленно прибавляют к раствору трет-бутил-N-{4-[(3-аминохинолин-4-ил)амино]бутил}карбамата (12,5 г, 37,7 ммоль) в дихлорметане (250 мл). Реакционную смесь оставляют на ночь при комнатной температуре. Полученный осадок отфильтровывают и высушивают. Получают трет-бутил-N-{4-[(3-(бензоиламино)хинолин-4-ил)амино]бутил}карбамат гидрохлорид в виде твердого белого вещества.
Часть Б
Триэтиламин (7,26 г, 71,7 ммоль) прибавляют к раствору материала, полученного в Части А, в этаноле и в течение 2 дней кипятят с обратным холодильником. Реакционную смесь концентрируют и получают сиропообразную жидкость оранжевого цвета. Хромато-масс-спектрометрический анализ этой жидкости выявил в ней содержание как целевого продукта, так и исходного материала. Жидкость растворяют в дихлорметане (100 мл) и охлаждают на ледяной бане. Добавляют триметиламин (5 мл) и бензоилхлорид (1,9 мл). Реакционную смесь выдерживают при комнатной температуре в течение 2 суток, и анализ, проведенный в этот момент методом высокоэффективной жидкостной хроматографии (ВЭЖХ) показал, что реакция не завершена. Реакционную смесь концентрируют под вакуумом. Остаток растворяют в изопропиловом спирте (150 мл). Добавляют триметиламин (5 мл), и оставляют реакционную на ночь кипеть с обратным холодильником. Реакционную смесь концентрируют под вакуумом. Остаток очищают колоночной хроматографией под давлением (силикагель; элюирование 10% метанолом в дихлорметане). Содержащие продукт фракции соединяют и концентрируют под вакуумом. После перекристаллизации остатка из ацетонитрила получают 6,7 г твердого трет-бутил-N-[4-(2-фенил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]карбамата с температурой плавления 158-159°С.
Часть В
Хлорпероксибензойную кислоту (1,05 эквивалентов 65%) медленно небольшими порциями прибавляют к раствору трет-бутил-N-[4-(2-фенил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]карбамата (6,56 г, 15,75 ммоль) в дихлорметане (120 мл). Через 3 часа реакцию останавливают 1%-ным водным раствором бикарбоната натрия (200 мл). Разделяют слои. Водный слой экстрагируют дихлорметаном (2 раза по 50 мл). Органические фракции соединяют, сушат сульфатом магния и концентрируют под вакуумом. Получается сироп светло-оранжевого цвета. Этот сироп перетирают с диэтиловым эфиром в порошок. Получается 6,8 г светло-коричневго твердого трет-бутил-N-[4-(2-фенил-1Н-имидазо[4,5-с]хинолин-5N-оксида с температурой плавления 178-181°С.
Часть Г
Раствор трет-бутил-N-[4-(2-фен ил-1Н-имидазо[4,5-с]хинолин-5N-оксида (6,8 г, 15,75 ммоль) в дихлорметане (100 мл) охлаждают на ледяной бане. Прибавляют концентрированную гидроокись аммония (30 мл). В течение 30 минут небольшими порциями прибавляют тозилхлорид (3,0 г, 15,75 ммоль).
Реакционную смесь оставляют на ночь отогреваться до комнатной температуры. Реакцию останавливают водой (350 мл). Разделяют слои. Водный слой экстрагируют дихлорметаном (2 раза по 50 мл). Органические фракции соединяют, сушат сульфатом магния и концентрируют под вакуумом с образованием твердого вещества желтовато-коричневого цвета. Это вещество очищают колоночной хроматографией под давлением (силикагель; элюирование 10% метанолом в дихлорметане). Выход продукта 4,8 г. Основную часть материала переносят в следующую стадию. Небольшую часть перекристаллизовывают из толуола, и получается твердый трет-бутил-N-[4-(4-амино-2-фенил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]карбамат с температурой плавления 182-183°С. Элементный анализ: теор. для C25H29N5O2: %С 69,58; %Н 6,77; %N 16,22; эксп.: %С 69,86; %Н 6,95; %N 15,80.
Часть Д
Материал из Части Г растворяют в метаноле (15 мл) и 1Н соляной кислоте (100 мл), а затем нагревают и в течение 2 часов кипятят с обратным холодильником. Реакционную смесь концентрируют под вакуумом до объема около 50 мл. При добавлении концентрированной гидроокиси аммония до рН 12 осадка не образуется. С помощью 1Н соляной кислоты рН доводят до 7. Смесь экстрагируют дихлорметаном, а затем этилацетатом. Водный слой упаривают досуха. Остаток растворяют в воде (50 мл), а затем в течение 36 часов непрерывно экстрагируют кипящим хлороформом. После упаривания хлороформенного экстракта под вакуумом образуется твердый продукт светло-коричневого цвета. После перекристаллизации этого продукта из ацетонитрила получается 2,5 г 1-(4-амино)-2-фенил-1Н-имидазо[4,5-с]хинолин-4-амина в виде желтовато-белого вещества с температурой плавления 175-177°С. Элементный анализ: теор. для C20H21N5: %С 72,48; %Н 6,39; %N 21,13; эксп.: %С 72,72; %Н 6,32; %N 20,71.
Часть Е
1-(4-Амино)-2-фенил-1Н-имидазо[4,5-с]хинолин-4-амин (0,331 г, 1,0 ммоль) растворяют в безводном ацетонитриле (35 мл) и раствор остужают до 4°С. Медленно прибавляют раствор 4-фторбензолсульфонилхлорида (0,194 г, 1,0 ммоль) в безводном дихлорметане (10 мл). Реакционную смесь оставляют на выходные дни медленно нагреваться до комнатной температуры. Реакцию останавливают, прибавляя насыщенный водный раствор бикарбоната натрия. Слои разделяют и после упаривания органического слоя получают светло-желтое твердое вещество. Этот материал перекристаллизовывают из изопропилового спирта, а затем очищают колоночной хроматографией под давлением (силикагель; элюирование 10% метанолом в дихлорметане). Чистые фракции соединяют и концентрируют. После перекристаллизации остатка из изопропилового спирта получают 0,2 г светло-желтого твердого N-[4-(4-амино-2-фенил-1 Н-имидазо[4,5-с]хинолин-1-ил)бутил]-4-фтор-1-бензолсульфоамида с температурой плавления 214-216°С. Элементный анализ: теор. для C26H24FN5O2S: %С: 63,79; %Н 4,94; %N 14,30; эксп.: %С: 63,19; %Н 4,85; %N 13,90; Масс-спектрометрия М+1=490,2.
Пример 16
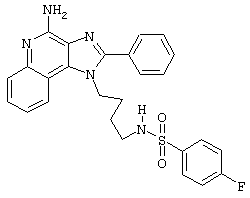
N-[4-(4-Амино-2-фенил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]метансульфонамид
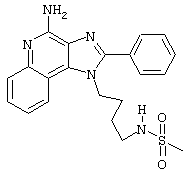
В соответствии с общей методикой, описанной в Части Е Примера 15, 1-(4-амино)-2-фенил-1Н-имидазо[4,5-с]хинолин-4-амин (0,331 г, 1,0 ммоль) взаимодействует с метансульфоновым ангидридом с образованием белого твердого N-[4-(4-амино-2-фенил-1 Н-имидазо[4,5-с]хинолин-1-ил)бутил]метансульфонамида с температурой плавления 234-235°С. Масс-спектрометрия М+1=410,2.
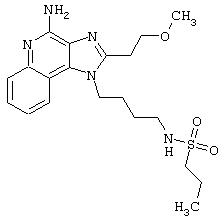
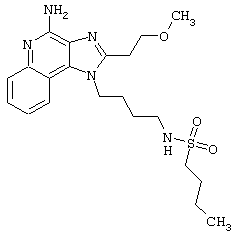
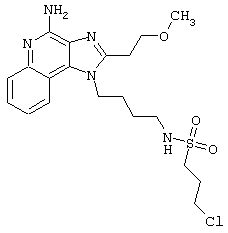
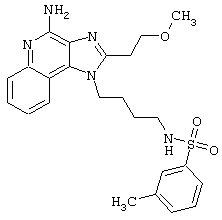
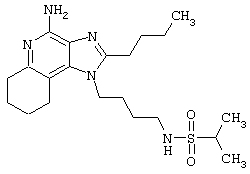
Примеры 17-33
Соединения, представленные ниже в Таблице, получают в соответствии с общей методикой синтеза, описанной выше в Схеме реакции II.
1-(2-Аминоэтил)-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амин (25 мг) помещают в ампулу на 2 драхмы (7,4 мл). По очереди прибавляют диизопропилэтиламин (11 мкл, 1,2 эквивалента), дихлорметан (1 мл) и сульфонилхлорид (1,1 эквивалента). На 2 часа ампулу помещают во встряхиватель, а затем на 0,5 часа в ультразвуковую камеру. Реакционную смесь оставляют на ночь при комнатной температуре, а затем для того, чтобы подтвердить образование целевого продукта, анализируют на хромато-масс-спектрометре. Растворитель удаляют, а остаток очищают полупрепаративной ЖХВД (колонка Capcell Pak C18, 35 мм×20 мм, размер частиц 5 мкм, 20 мл/мин, градиентное элюирование от 5 до 95% Б в течение 10 минут, 2-минутная задержка на 95% Б, где А=0,1% трифторуксусной кислоты в воде, а Б=0,1% трифторуксусной кислоты в ацетонитриле, включение сбора фракций детектируется по пику 254 нм). Фракции, полученные полупрепаративной жидкостной хроматографией, анализируют на хромато-масс-спектрометре ЖХ-ХИАД/МС и соответствующие фракции соединяют и лиофилизуют. Образуется трифторацетатная соль нужного сульфонамида (см. табл. 1).
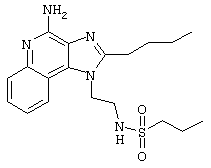
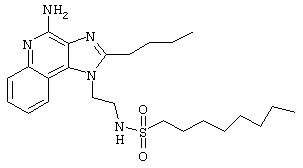

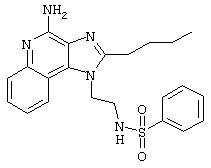
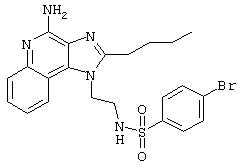
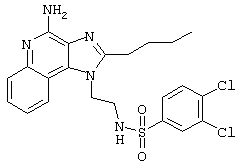
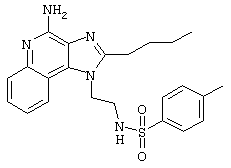
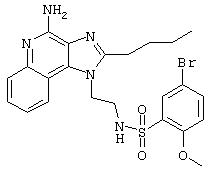
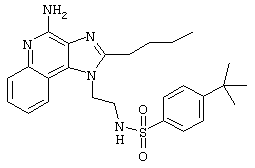
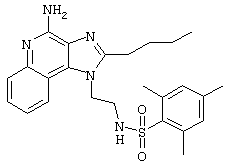

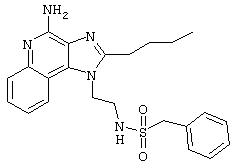
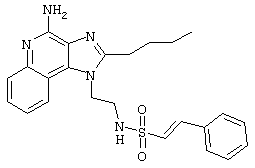
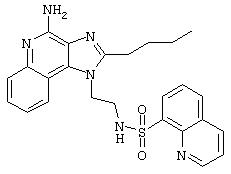

Пример 34
N-[4-(4-Амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)этил]метансульфонамид трифторацетат
Это соединение получают в соответствии с общей методикой, описанной для примеров 17-33, за исключением того, что вместо сульфонилхлорида используют 1,1 эквивалента метансульфонового ангидрида. (Экспериментально найденная молекулярная масса=362,2).
Пример 35
N-[4-(4-Амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)этил] трифторметансульфонамид трифторацетат
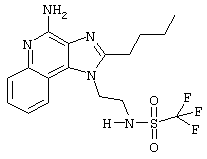
Это соединение получают в соответствии с общей методикой, описанной для примеров 17-33, за исключением того, что, вместо сульфонилхлорида, используют 1,1 эквивалента трифторметансульфонового ангидрида. (Экспериментально найденная молекулярная масса=416,1).
Примеры 36-40
Соединения, представленные ниже в Таблице, получают в соответствии с общей методикой синтеза, описанной выше в Схеме реакции II.
1-(2-Аминобутил)-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амин (25 мг) помещают в ампулу на 2 драхмы (7,4 мл). По очереди прибавляют диизопропилэтиламин (14 мкл, 1,0 эквивалент), дихлорметан (1 мл) и сульфонилхлорид (1,0 эквивалент). На 0,5 часа ампулу помещают во встряхиватель, и за это время почти все переходит в раствор. Несколько позже образуется осадок. Добавляют немного метанола, и осадок растворяется. Реакционную смесь еще на один час помещают во встряхиватель, а затем на 0,5 часа в ультразвуковую камеру. Для того, чтобы подтвердить образование целевого продукта, реакционную смесь анализируют на хромато-масс-спектрометре. Растворитель удаляют, а остаток очищают полупрепаративной ЖХВД (колонка Capcell Pak C18, 35 мм×20 мм, размер частиц 5 мкм, 20 мл/мин, градиентное элюирование от 5 до 95% Б в течение 10 минут, 2-минутная задержка на 95% Б, где А=0,1% трифторуксусной кислоты в воде, а Б=0,1% трифторуксусной кислоты в ацетонитриле, включение сбора фракций детектируется по пику 254 нм). Фракции, полученные полупрепаративной жидкостной хроматографией, анализируют на хромато-масс-спектрометре ЖХ-ХИАД/МС и соответствующие фракции соединяют и лиофилизуют. Образуется трифторацетатная соль нужного сульфонамида (см. табл.2).

Примеры 41-52
Соединения, представленные ниже в Таблице, получают в соответствии с общей методикой синтеза, описанной выше в Схеме реакции II.
1-(2-Аминобутил)-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амин (25 мг) помещают в ампулу на 2 драхмы (7,4 мл). По очереди прибавляют диизопропилэтиламин (14 мкл, 1,0 эквивалент), дихлорметан (1 мл) и сульфонилхлорид (1,0 эквивалент). Примерно на 60 минут ампулу помещают в ультразвуковую камеру при комнатной температуре. Для того, чтобы подтвердить образование целевого продукта, реакционную смесь анализируют на хромато-масс-спектрометре. Растворитель удаляют, а остаток очищают полупрепаративной ВЭЖХ (колонка Capcell Pak C18, 35 мм×20 мм, размер частиц 5 мкм, 20 мл/мин, градиентное элюирование от 5 до 95% Б в течение 10 минут, 2-минутная задержка на 95% Б, где А=0,1% трифторуксусной кислоты в воде, а Б=0,1% трифторуксусной кислоты в ацетонитриле, включение сбора фракций детектируется по пику 254 нм). Фракции, полученные полупрепаративной жидкостной хроматографией, анализируют на хромато-масс-спектрометре ЖХ-ХИАД/МС и соответствующие фракции соединяют и лиофилизуют. Образуется трифторацетатная соль нужного сульфонамида (см. табл.3).
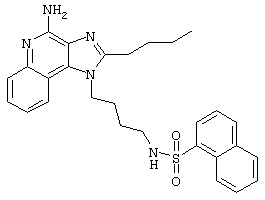
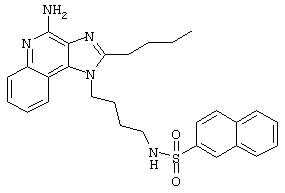
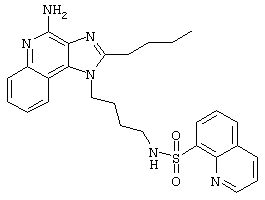
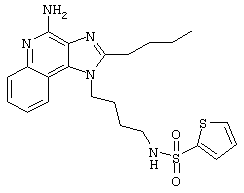
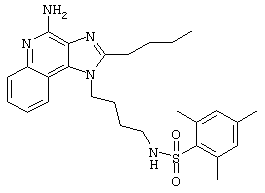
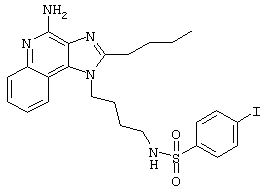
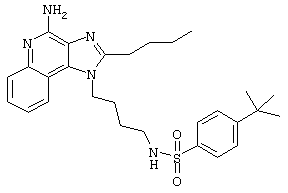
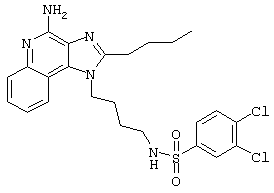
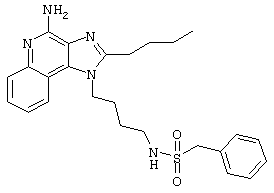
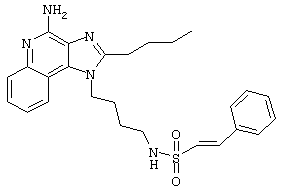
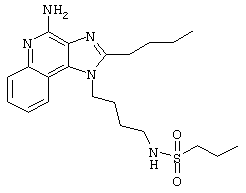
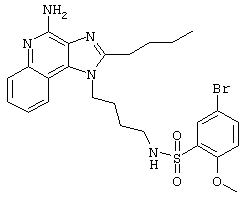
Пример 53
N-[4-(4-Амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]трифторметансульфонамидтрифторацетат
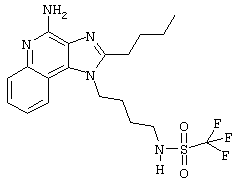
Это соединение получают в соответствии с общей методикой, описанной для примеров 41-52, за исключением того, что, вместо сульфонилхлорида, используют 1,0 эквивалент трифторметансульфонового ангидрида. (Экспериментально найденная молекулярная масса=444,1).
Примеры 54-71
Соединения, представленные ниже в Таблице, получают в соответствии с общей методикой синтеза, описанной выше в Схеме реакции IV.
Часть А
Каталитическое количество оксида платины (IV) прибавляют к раствору 1-(4-аминобутил)-1Н-имидазо[4,5-с]хинолин-4-амина (2,75 г, 10,8 ммоль) в трифторуксусной кислоте (150 мл). Реакционную смесь помещают в атмосферу водорода под давлением 50 фунтов на кв. дюйм (3,44×105 Па). Проведенный через 1 неделю масс-спектрометрический анализ показывает присутствие в реакционной смеси как исходного материала, так и тетрагидрированного продукта. К реакционной смеси добавляют свежий катализатор и продолжают гидрирование при 50 фунтов на кв. дюйм (3,44×105 Па). Через 2 недели для того, чтобы удалить катализатор, смесь фильтруют. Фильтрат концентрируют под вакуумом. Остаток растворяют в 1Н соляной кислоте (120 мл), и в течение 1 часа перемешивают раствор при комнатной температуре. Раствор расщелачивают 50%-ным едким натром до рН 10 и экстрагируют дихлорметаном (5 раз по 100 мл). Экстракты соединяют и концентрируют под вакуумом. Выход: 2,08 г твердого белого 1-(4-аминобутил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амина.
Часть Б
1-(2-Аминобутил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин (25 мг) помещают в ампулу на 2 драхмы (7,4 мл). По очереди прибавляют диизопропилэтиламин (11 мкл, 1,2 эквивалента), дихлорметан (1 мл) и сульфонилхлорид (1,1 эквивалента). Примерно на 6 часов ампулу помещают во встряхиватель. Реакционную смесь оставляют на ночь при комнатной температуре и для того, чтобы подтвердить образование целевого продукта, анализируют ее на хромато-масс-спектрометре. Растворитель удаляют, а остаток очищают полупрепаративной ВЭЖХ (колонка Capcell Pak C18, 35 мм×20 мм, размер частиц 5 мкм, 20 мл/мин, градиентное элюирование от 5 до 95% Б в течение 10 минут, 2-минутная задержка на 95% Б, где А=0,1% трифторуксусной кислоты в воде, а Б=0,1% трифторуксусной кислоты в ацетонитриле, включение сбора фракций детектируется по пику 254 нм). Фракции, полученные полупрепаративной жидкостной хроматографией, анализируют на хромато-масс-спектрометре ЖХ-ХИАД/МС и соответствующие фракции соединяют и лиофилизуют. Образуется трифторацетатная соль нужного сульфонамида (см. табл.4).
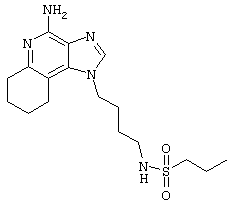
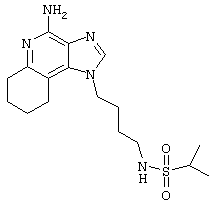
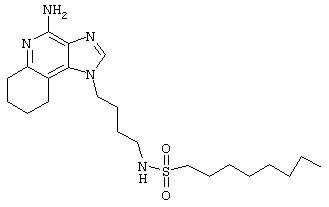

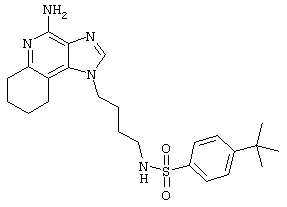
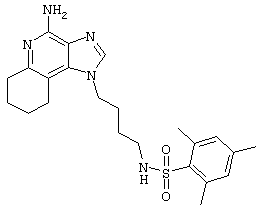
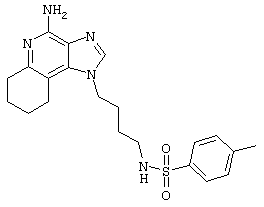
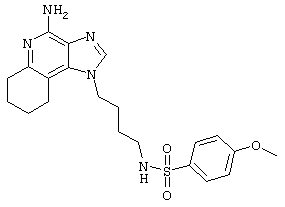
Пример 72
N-[4-(4-Амино-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]метансульфонамид трифторацетат

Это соединение получают в соответствии с общей методикой, описанной для примеров 54-71, за исключением того, что, вместо сульфонилхлорида, используют 1,1 эквивалента метансульфонового ангидрида. (Экспериментально найденная молекулярная масса=338,2).
Примеры 73-201
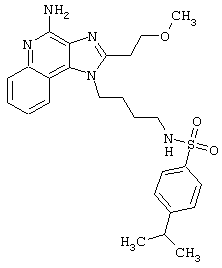
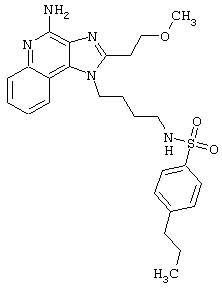
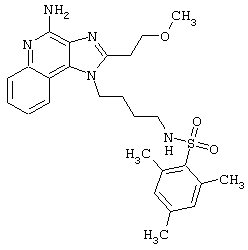
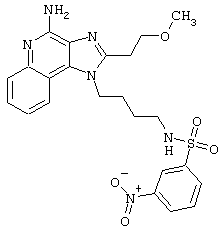
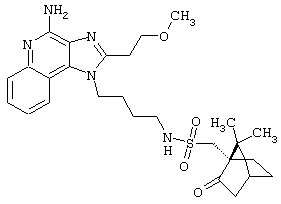
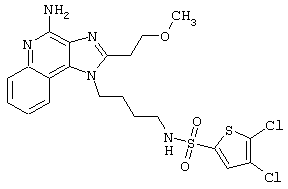
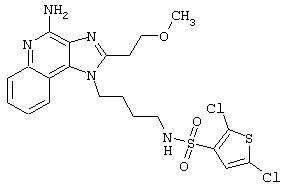
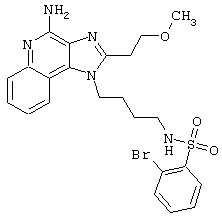
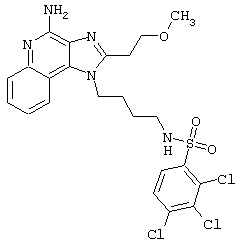
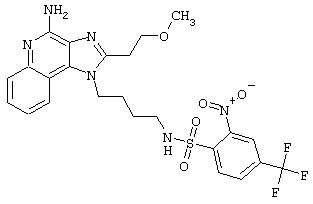
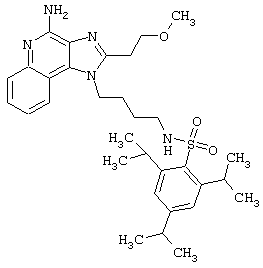
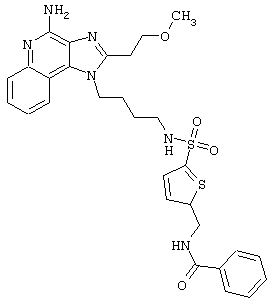
Соединения, представленные ниже в Таблице, получают в соответствии с методом, описанной выше в Схеме реакции II, с помощью следующей общей методики.
1Н-Имидазо[4,5-с]хинолин-4-амин (50 мг) помещают в ампулу на 2 драхмы (7,4 мл). Прибавляют диизопропилэтиламин (1,2 эквивалента) в дихлорметане (1 мл). Прибавляют раствор, содержащий сульфонилхлорид (1,1 эквивалента) в дихлорметане (~1 мл). Примерно на 2-16 часов (обычно на 2 часа) ампулу помещают во встряхиватель при комнатной температуре. Для того, чтобы подтвердить образование целевого продукта, реакционную смесь анализируют на хромато-масс-спектрометре. Растворитель удаляют, а остаток очищают полупрепаративной ВЭЖХ (колонка Capcell Рак С18, 35 мм×20 мм, размер частиц 5 мкм, 20 мл/мин, градиентное элюирование от 5 до 95% Б в течение 10 минут, 2-минутная задержка на 95% Б, где А=0,1% трифторуксусной кислоты в воде, а Б=0,1% трифторуксусной кислоты в ацетонитриле, включение сбора фракций детектируется по пику 254 нм). Фракции, полученные полупрепаративной жидкостной хроматографией, анализируют на хромато-масс-спектрометре ЖХ-ХИАД/МС и соответствующие фракции соединяют и лиофилизуют. Образуется трифторацетатная соль нужного сульфонамида (см. таблицу 5).
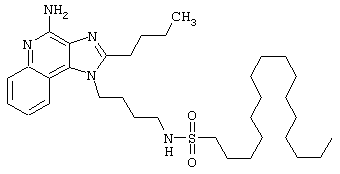
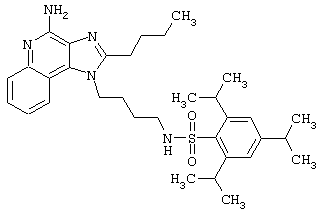

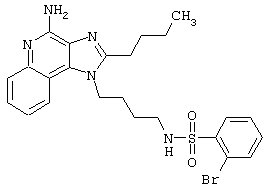
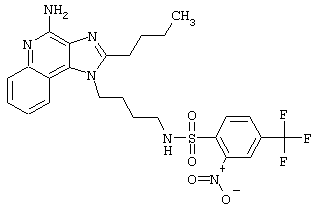
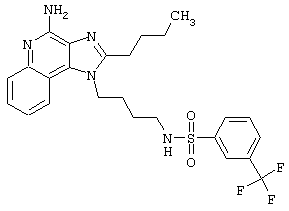
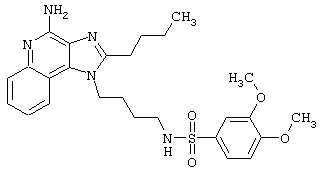
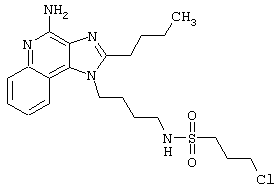
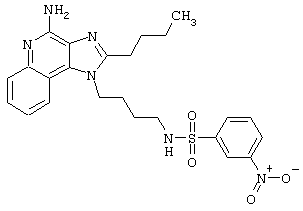
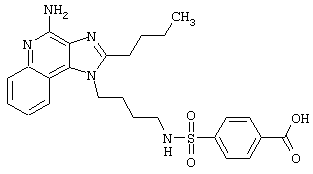
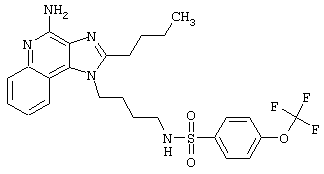
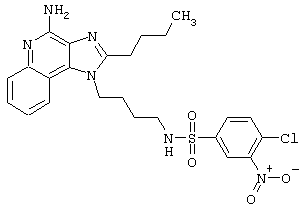
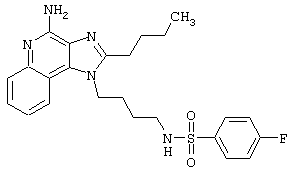



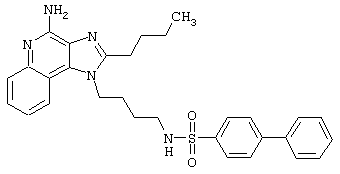
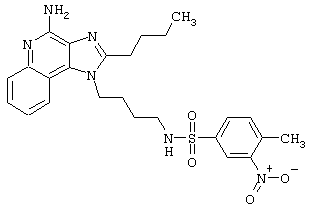
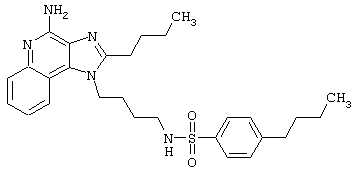
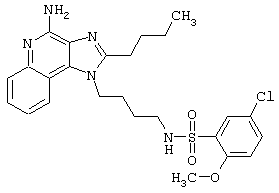
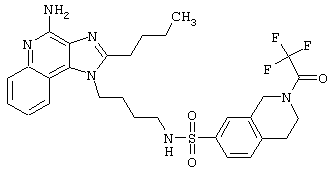
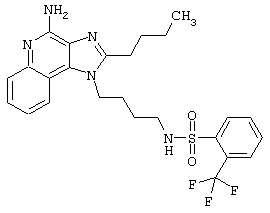
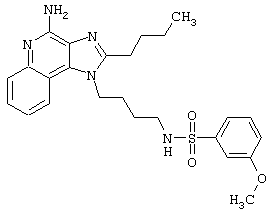
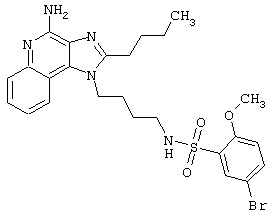
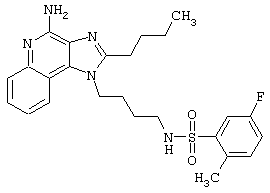
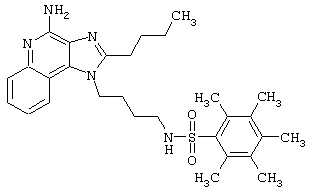
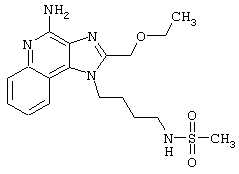
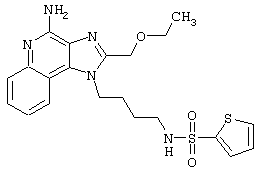
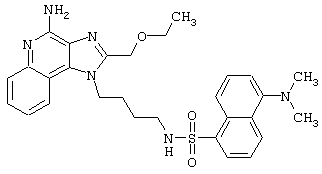

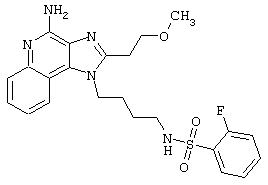
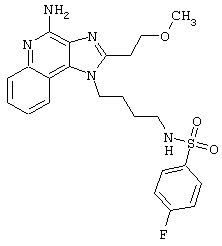
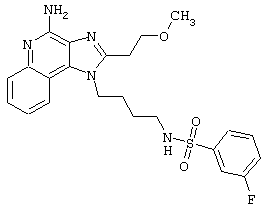
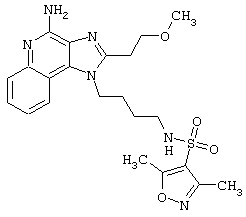
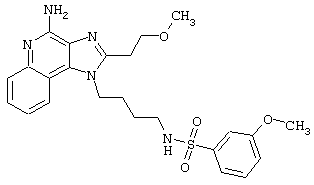
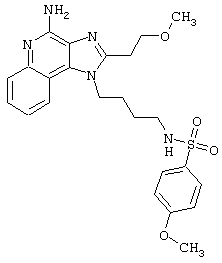
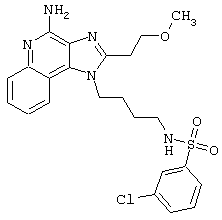
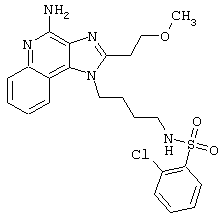
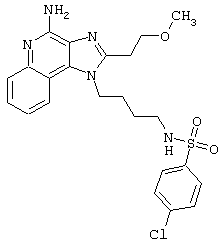
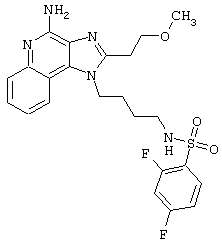
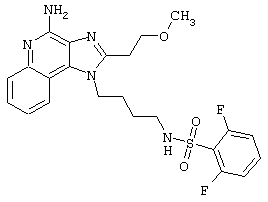
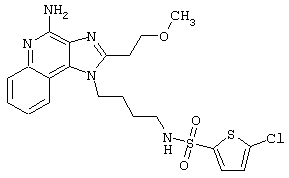
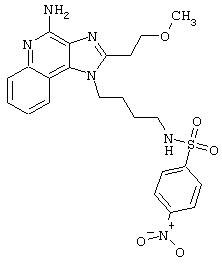
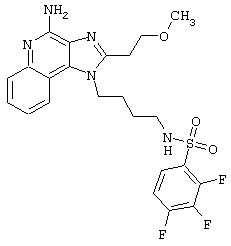
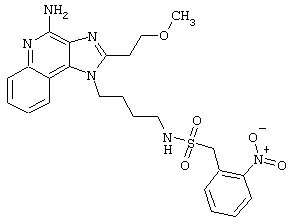
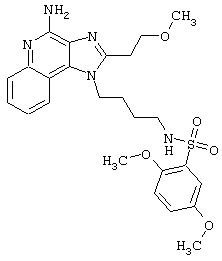
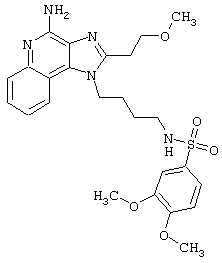
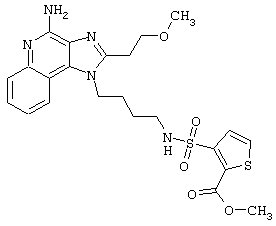
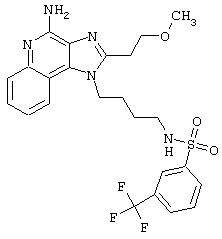
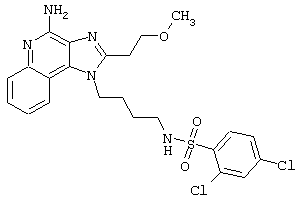
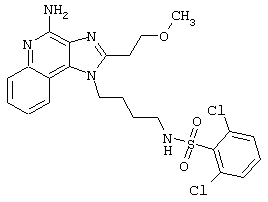
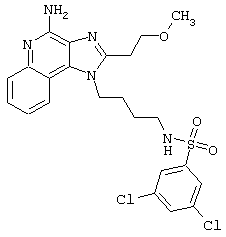
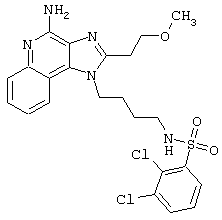
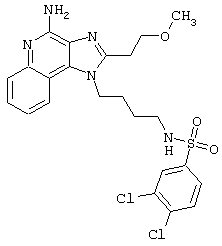
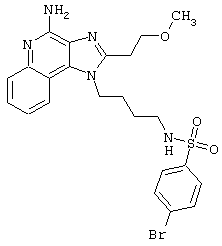
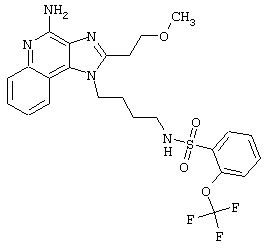
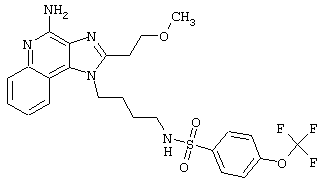
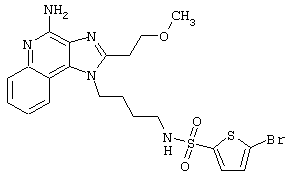
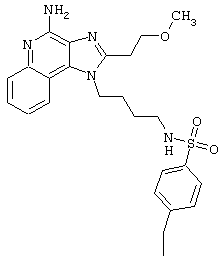

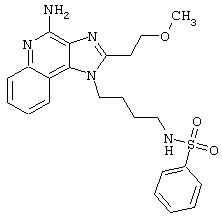
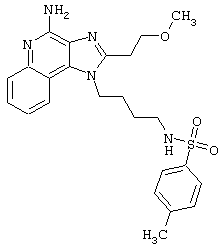
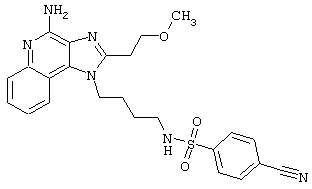
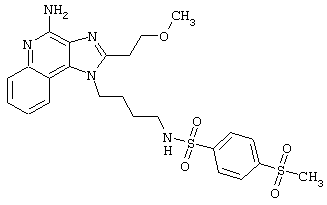


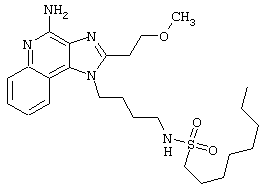
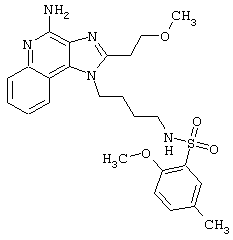
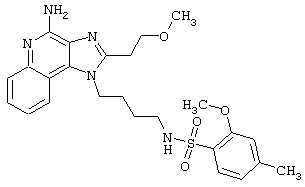
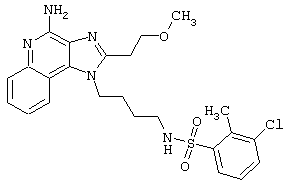
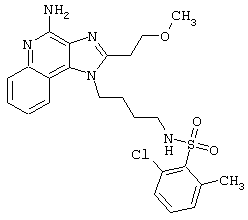
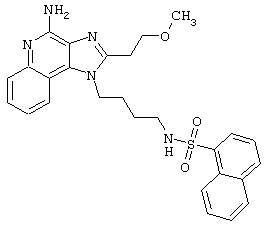
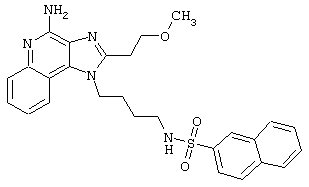
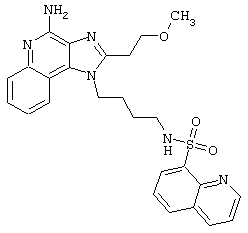
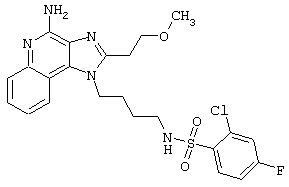
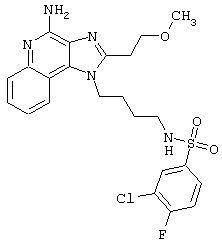
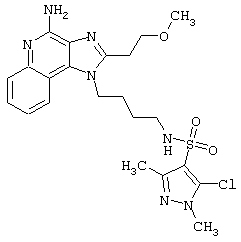
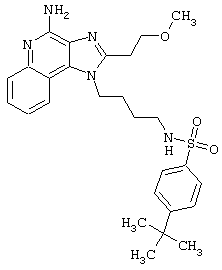
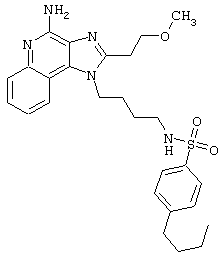
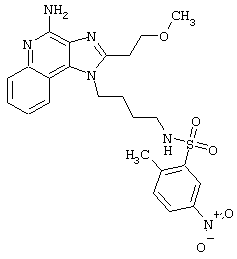
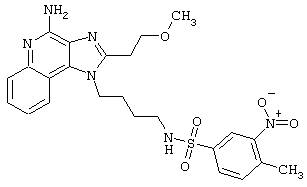
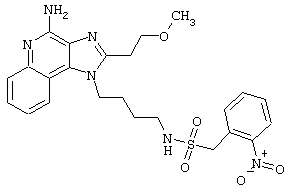
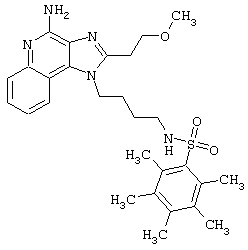
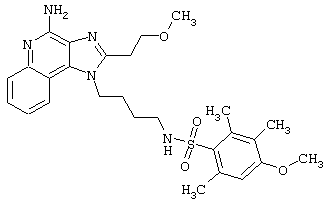
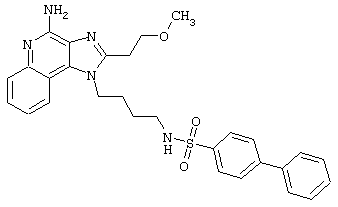
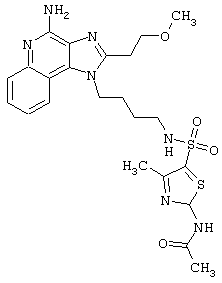
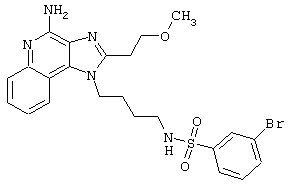
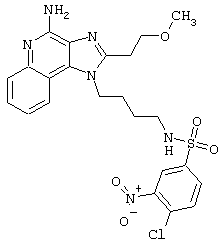
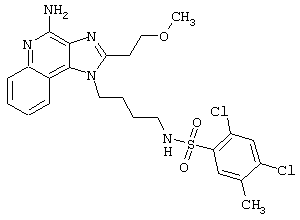
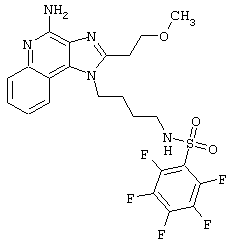
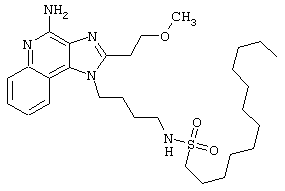
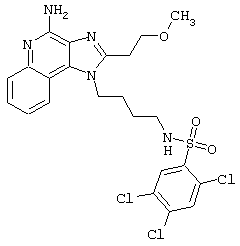
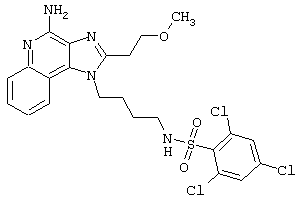

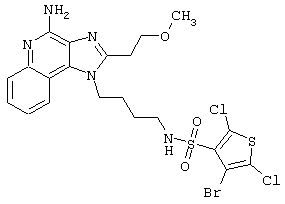
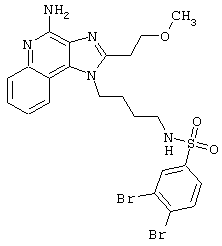
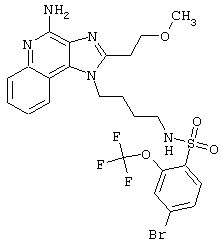
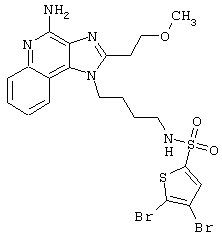
Примеры 202-213
Соединения, представленные ниже в Таблице, получают в соответствии с общей методикой синтеза, описанной выше в Схеме реакции VI.
Часть А
Исходные тетрагидрохинолинамины получают следующим образом. Каталитическое количество оксида платины (IV) прибавляют к раствору 1-(4-аминобутил)-2-бутил-1Н-имидазо[4,5-с]хинолин-4-амина (2,2 г, 7,06 ммоль) в трифторуксусной кислоте (200 мл). В течение 6 суток реакционную смесь гидрируют в аппарате Парра под давлением 50 фунтов на кв. дюйм (3,44×105 Па). Для того, чтобы удалить катализатор, смесь фильтруют и фильтрат концентрируют под вакуумом. Остаток соединяют с 1Н соляной кислотой (100 мл) и в течение 2 часов нагревают на паровой бане. Смесь охлаждают, расщелачивают гидроокисью аммония, а затем экстрагируют дихлорметаном. Экстракт концентрируют под вакуумом и получают твердый 1-(4-аминобутил)-2-бутил-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин с температурой плавления 63-67°С.
Каталитическое количество оксида платины (IV) прибавляют к раствору 1-(4-аминобутил)-2-метоксиэтил-1Н-имидазо[4,5-с]хинолин-4-амина (7,7 г, 24,5 ммоль) в трифторуксусной кислоте (250 мл). Реакционную смесь гидрируют в аппарате Парра под давлением 50 фунтов на кв. дюйм (3,44×105 Па). За ходом реакции следят по хромато-масс-спектрометру. Через 7, 11 и 17 дней после начала реакции добавляют дополнительные количества катализатора. Через 25 дней реакция завершается. Для того, чтобы удалить катализатор, смесь фильтруют через слой ускорителя фильтрования целит (Celite®) и фильтрат концентрируют под вакуумом. Остаток соединяют с 1Н соляной кислотой (100 мл) и оставляют на ночь перемешиваться. Смесь расщелачивают гидроокисью аммония до рН 11, а затем экстрагируют дихлорметаном (3 раза по 300 мл). Экстракты соединяют и концентрируют под вакуумом. Выход: 3,5 г твердого 1-(4-аминобутил)-6,7,8,9-тетрагидро-2-метоксиэтил-1Н-имидазо[4,5-с]хинолин-4-амина.
Часть Б
С помощью методики, описанной для примеров 73-201, тетрагидроимидазохинолинамины из части А взаимодействуют с соответствующим сульфонилхлоридом с образованием нужного сульфонамида (см. табл.6).
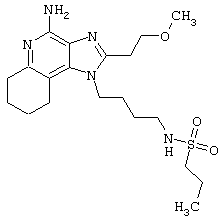
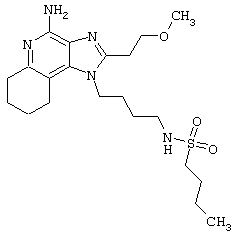
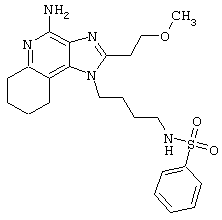
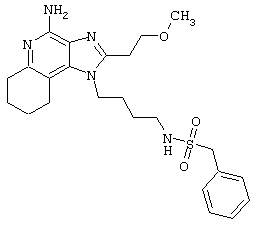
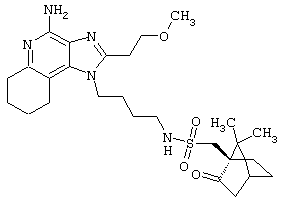
Пример 214
N-[4-(4-Амино-6,7,8,9-тетрагидро-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]метансульфонамидтрифторацетат
Это соединение получают в соответствии с методикой, описанной для примеров 202-213, за исключением того, что вместо сульфонилхлорида используют метансульфоновый ангидрид.
Пример 215
N-[4-(4-Амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-N-метил-3,5-диметилизооксазоло-4-сульфонамидтрифторацетат
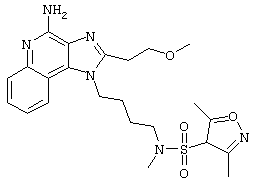
Часть А
В соответствии с общей методикой из примера DC001, 1-(4-аминобутил)-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амин взаимодействует с 3,5-диметилоксазол-4-сульфонилхлоридом с образованием N-[4-(4-Амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-N-метил-3,5-диметилизооксазоло-4-сульфонамида трифторацетата.
Часть Б
Гидрид натрия (5,8 мг) прибавляют к раствору материала из части А (25,4 мг) в диметилформамиде. К реакционной смеси прибавляют иодометан (3,2 мкл) и в течение 2 часов встряхивают реакционную смесь при комнатной температуре. Для того, чтобы подтвердить образование целевого продукта, реакционную смесь анализируют на хромато-масс-спектрометре. Растворитель удаляют, а остаток очищают полупрепаративной ВЭЖХ (колонка Capcell Рак С18, 35 мм×20 мм, размер частиц 5 мкм, 20 мл/мин, градиентное элюирование от 5 до 95% Б в течение 10 минут, 2-минутная задержка на 95% Б, где А=0,1% трифторуксусной кислоты в воде, а Б=0,1% трифторуксусной кислоты в ацетонитриле, включение сбора фракций детектируется по пику 254 нм). Фракции, полученные полупрепаративной жидкостной хроматографией, анализируют на хромато-масс-спектрометре ЖХ-ХИАД/МС и соответствующие фракции соединяют и лиофилизуют. Лиофилизованный материал второй раз очищают полупрепаративной ВЭЖХ при тех же условиях за исключением того, что градиентное элюирование от 5 до 95% Б протекает не за 10 минут, а за 60 минут. Фракции, полученные полупрепаративной жидкостной хроматографией, анализируют на хромато-масс-спектрометре ЖХ-ХИАД/МС и соответствующие фракции соединяют и лиофилизуют. Образуется трифторацетатная соль нужного амида.
Пример 216
N-[4-(4-Амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-N-метилтрифторметансульфонамидтрифторацетат
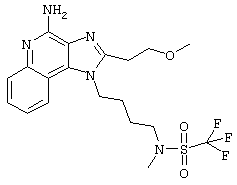
Это соединение получают в соответствии с общей методикой, описанной выше в примере 215, за исключением того, что вместо сульфонилхлорида в части А используют трифторметансульфоновый ангидрид.
Примеры 217-221
Соединения в примерах, представленных в приведенной ниже таблице, получают в соответствии со следующей общей методикой.
1 Н-Имидазо[4,5-с]хинолин-4-амин или 6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-4-амин (50 мг) помещают в ампулу на 2 драхмы (7,4 мл). Прибавляют дихлорметан (2 мл) и диизопропилэтиламин (1,2 эквивалента). Прибавляют диметилсульфамоилхлорид (1,1 эквивалента). Примерно на 2-4 часа ампулу помещают во встряхиватель при комнатной температуре. Для того, чтобы подтвердить образование целевого продукта, реакционную смесь анализируют на хромато-масс-спектрометре. Растворитель удаляют, а остаток очищают полупрепаративной ЖХВД (колонка Capcell Pak C18, 35 мм×20 мм, размер частиц 5 мкм, 20 мл/мин, градиентное элюирование от 5 до 95% Б в течение 10 минут, 2-минутная задержка на 95% Б, где А=0,1% трифторуксусной кислоты в воде, а Б=0,1% трифторуксусной кислоты в ацетонитриле, включение сбора фракций детектируется по пику 254 нм). Фракции, полученные полупрепаративной жидкостной хроматографией, анализируют на хромато-масс-спектрометре ЖХ-ХИАД/МС и соответствующие фракции соединяют и лиофилизуют. Образуется трифторацетатная соль нужного сульфамида (см. табл.7).

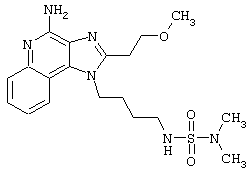
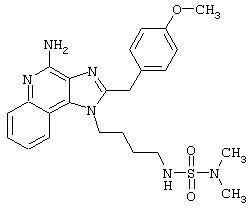
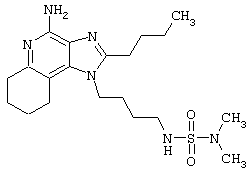
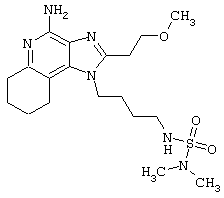
Примеры 222-228
Соединения, представленные ниже в Таблице, получают в соответствии с общей методикой синтеза, описанной выше в Схеме реакции V.
1-(4-Аминобутил)-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амин (50 мг) помещают в ампулу на 2 драхмы (7,4 мл). Прибавляют 4-(диметиламино)пиридин (19 мг, 1,0 эквивалент) в дихлорметане (800 мкл). Ампулу запаивают и охлаждают до -78°С на бане сухой лед / ацетон. Прибавляют сульфурилхлорид (186 мкл 1М раствора в дихлорметане). Примерно на 30 минут ампулу помещают во встряхиватель, а затем снова охлаждают до -78°С. В отдельную ампулу загружают амин общей формулы R4R5NН (2,0 эквивалента), триэтиламин (2,0 эквивалента) и дихлорметан (1 мл) и охлаждают до -78°С. Раствор амина / триэтиламина прибавляют в первую ампулу. Примерно на 1 час ампулу помещают во встряхиватель при комнатной температуре. Для того, чтобы подтвердить образование целевого продукта, реакционную смесь анализируют на хромато-масс-спектрометре. Растворитель удаляют, а остаток очищают полупрепаративной ЖХВД (колонка Capcell Pak С18, 35 мм×20 мм, размер частиц 5 мкм, 20 мл/мин, градиентное элюирование от 5 до 95% Б в течение 10 минут, 2-минутная задержка на 95% Б, где А=0,1% трифторуксусной кислоты в воде, а Б=0,1% трифторуксусной кислоты в ацетонитриле, включение сбора фракций детектируется по пику 254 нм). Фракции, полученные полупрепаративной жидкостной хроматографией, анализируют на хромато-масс-спектрометре ЖХ-ХИАД/МС и соответствующие фракции соединяют и лиофилизуют. Образуется трифторацетатная соль нужного сульфамида (см. табл.8).
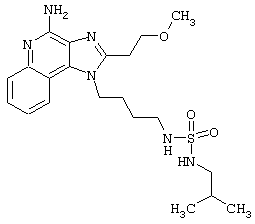
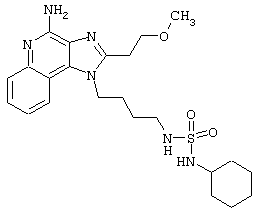
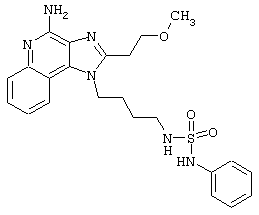
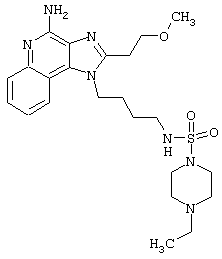
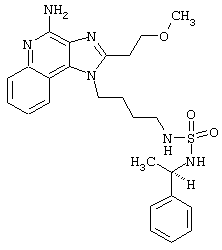
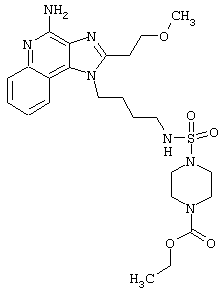
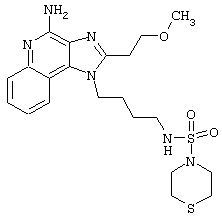
Примеры 229-231
Соединения, представленные ниже в Таблице, получают в соответствии с методом, описанным в примерах 222-228, за исключением того, что амин общей формулы R4R5NH взаимодействует с сульфурилхлоридом с образованием промежуточного сульфамоилхлорида, который затем реагирует с 2 эквивалентами 1-(4-аминобутил)-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-4-амина (см. табл.9).

ИНДУКЦИЯ ЦИТОКИНОВ В КЛЕТКАХ ЧЕЛОВЕКА
Для изучения индукции цитокинов соединениями настоящего изобретения была использована система клеток крови человека in vitro. Метод основан на измерении уровня (а) интерферона и фактора некроза опухоли (ИФН и ФНО, соответственно), секретируемых в культуральную среду, как описано Тестерманом и др (Testerman et al, "Cytokine induction by Immunomodulators Imiquimod and S-27609", Journal of Leukocyte Biology, 58, 365-372 (September, 1995))
Приготовление клеток крови для культуры
Цельную кровь забирают из вены здорового человека и помещают в пробирки. Мононуклеарные клетки периферической крови (МКПК) выделяют из цельной крови в градиенте плотности путем центрифугирования с использованием прибора Histipaque®-1077 (ф. Sigma Chemicals, St.Louis, МО). Мононуклеары периферической крови суспендируют в концентрации 3-4×106 клеток/мл в среде RPMI 1640, содержащей 10% фетальной бычьей сыворотки, 2 мМ L-глютамина и 1% смеси пеницилина и стрептомицина (полная среда RPMI). Суспензию МКПК вносят в 48-луночные плоскодонные стерильные планшеты для культуры тканей (ф. Costar, Cambridge, МА, или ф. Becton Dickinson Labware, Lincoln Park, NJ), в которых находятся равные объемы полной среды RPMI, содержащей тестируемый препарат.
Приготовление препарата
Препарат солюбилизируют в диметилсульфоксиде (ДМСО). Конечная концентрация ДМСО для добавления в культуральные лунки не должна превышать 1%.
Инкубация
Раствор тестируемого препарата в концентрации 60 мкМ вносят в первую лунку, содержащую полную среду RPMI, затем делают серию последовательных разведении (3- или 10-кратных). Суспензию МКПК затем добавляют в лунки в равных объемах, что позволяет добиться необходимого спектра концентраций тестируемого препарата. Конечная концентрация суспензии МКПК составляет 1,5-2×106 клеток/мл. Плашки закрывают стерильными пластиковыми крышками и подвергают аккуратному перемешиванию и далее инкубируют в течение 18-24 часов при температуре 37°С в атмосфере с 5%-ной концентрацией двуокиси углерода.
Выделение
Плашки центрифугируют в течение 5-10 минут в режиме 1000 об/мин (~200×g) при температуре 4°С. Супернатант извлекают из клеточной культуры стерильной полипропиленовой пипеткой и переносят в стерильные полипропиленовые пробирки. Полученные пробы хранят до анализа при температуре от -30 до -70°С. Затем пробы тестируют на содержание α-интерферона и α-фактора некроза опухоли методом твердофазного иммуноферментного анализа (ТИФА) [ELISA].
Анализ методом ТИФА (ELISA) на содержание α-интерферона и α-фактора некроза опухоли.
Метод ТИФА позволяет определить концентрацию α-интерферона с помощью набора - Human Multi-Species kit (ф. PBL Biomedical Laboratories, New Brunswick, NJ).
Концентрация α-фактора некроза опухоли определяют с помощью набора для ТИФА от ф. Genzyme, Cambridge, МА; ф. R&D Systems, Minneapolis, MN; или ф. Pharmigen, San Diego, CA)
Таблица, приведенная ниже, демонстрирует найденную наименьшую концентрацию каждого препарата для индукции фактора некроза опухоли. “**” - отражает отсутствие индукции в какой-либо из тестированных концентраций (0,12, 0,37, 1,11, 3,33, 10 и 30 мкМ); “***” - отражает отсутствие индукции в какой-либо из тестированных концентраций (0,0001, 0,001, 0,01, 0,1, 1 и 10 мкм) (см. табл.10).
примера
В описании настоящего изобретения рассмотрены некоторые примеры его осуществления. Приведенные выше подробное описание и примеры представлены здесь только для ясности понимания, и из них не следует каких-либо ограничений. Специалистам в данной области понятно, что возможны многочисленные изменения в описанных примерах осуществления, не меняющие сущности и объема настоящего изобретения. Таким образом, объем настоящего изобретения ограничен не конкретными деталями описанных здесь составов и структур, а лишь формулировками нижеследующей формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОЧЕВИНОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2265020C2 |
| АМИДОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2000 |
|
RU2295343C2 |
| АРИЛЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ВИРУСНОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ОПУХОЛЕВОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2308456C2 |
| ТИОЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2001 |
|
RU2315049C2 |
| N-{2-[4-АМИНО-2-(ЭТОКСИМЕТИЛ)-1Н-ИМИДАЗО-[4,5-с]-ХИНОЛИН-1-ИЛ]-1,1-ДИМЕТИЛЭТИЛ}-МЕТАНСУЛЬФОНАМИД И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2004 |
|
RU2374246C2 |
| ГЕТЕРОЦИКЛИЛЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2001 |
|
RU2351598C2 |
| КАРБАМИДЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНОВЫЕ ЭФИРЫ | 2001 |
|
RU2302418C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНЫ | 2001 |
|
RU2294934C2 |
| Способ получения имидазохинолиновых соединений или их фармацевтически приемлемых солей | 1987 |
|
SU1470192A3 |
| ТИОЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2304143C2 |
Изобретение относится к новым имидазохинолинам формулы 1:
где R, R1, R2 и n имеют значения, указанные в описании, обладающим действием иммуномодуляторов, индуцирующих биосинтез цитокинов у животных при лечении различных патологий, в том числе вирусных и неоплазических заболеваний. Изобретение также относится к фармацевтическому препарату для индукции интерферона-α или α-фактора некроза опухоли, способу индукции биосинтеза цитокинов у животных и способам лечения вирусных заболеваний и неоплазменных патологий у животных. Технический результат - получение новых биологически активных соединений. 7 с. и 16 з.п. ф-лы, 10 табл.
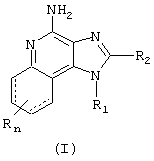
где
связи, изображенные пунктирной линией, могут как присутствовать, так и отсутствовать,
R1 - это -алкил-NR3-SO2-Х-R4;
Х - это связь или -NR5-;
R4 - это фенил, нафтил, тиенил, хинолинил, изохинолинил; тетрагидрохинолинил, тиазолил, оксазолил, пиразолил, пиперазинил, пиперидинил, тиоморфолинил, пирролидинил, алкил или алкенил, каждый из которых может быть незамещенным или замещенным одним или более заместителями, выбираемыми из группы, в которую входят:
- алкил;
- фенил;
- фенил, замещенный NO2;
- O-алкил;
- СООН;
- СО-O-алкил;
- S(O)0-2-алкил;
- (алкил)0-1-NR3R3;
- (алкил)0-1-NR3СО-О-алкил;
- (алкил)0-1-NR3-СО-алкил;
- (алкил)0-1-NR3-СО- фенил;
- галоген;
- галогеналкил;
- галогеналкоксил;
- СО-галогеналкоксил;
- NO2;
- CN; и в случае алкила, оксогруппа;
R2 выбирается из группы, в которую входят:
- водород;
- алкил;
- фенил;
- алкил-O-алкил; и
- алкил, замещенный одним или более заместителями, выбираемыми из:
- фенил, замещенный алкоксилом;
каждый из R3 независимо выбирают из группы, в которую входят водород и алкил C1-10;
R5 выбирают из группы, в которую входят водород и алкил C1-10; либо R4 и R5 в совокупности могут образовывать кольцо пиперазина, пиперидина, тиоморфолина или пирролидина;
n - это число 0,
или его фармацевтически приемлемая соль.
- алкил;
- фенил;
- фенил, замещенный NO2;
- O-алкил;
- СООН;
- СО-O-алкил;
- S(O)0-2-алкил;
- (алкил)0-1-NR3R3;
- (алкил)0-1-NR3СО-О-алкил;
- (алкил)0-1-NR3-СО-алкил;
- (алкил)0-1-NR3-СО-фенил;
- галоген;
- галогеналкил;
- галогеналкоксил;
- СО-галогеналкоксил;
- NO2;
- CN; и в случае алкила, оксогруппа;
N2-[2-(4-амино-2-бутил-1H-имидазо[4,5-с]хинолин-1-ил)этил]-2-тиофенсульфонамид;
N1-[2-(4-амино-2-бутил-1H-имидазо[4,5-с]хинолин-1-ил)этил]-1-бензолсульфонамид;
N8-[2-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)этил]-8-хинолинсульфонамид;
N1-[2-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)этил]-5-(диметиламино)-1-нафталинсульфонамид;
N-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]метансульфонамид;
N1-[4-(4-aминo-2-бyтил-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-1-бензолсульфонамид;
N8-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-8-хинолинсульфонамид;
N2-[4-(4-aминo-2-бyтил-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-2-тиофенсульфонамид;
N2-[4-(4-aминo-6,7,8,9-тeтpaгидpo-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-2-тиофенсульфонамид;
N1-[4-(4-aминo-6,7,8,9-тeтpaгидpo-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-1-бензолсульфонамид;
N8-[4-(4-амино-6,7,8,9-тетрагидро-1Н-имидазо[4,5-c]хинолин-1-ил)бутил]-8-хинолинсульфонамид;
N1-[4-(4-aминo-6,7,8,9-тeтpaгидpo-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-5-(диметиламино)-1-нафталинсульфонамид;
N1-[4-(4-aминo-6,7,8,9-тeтpaгидpo-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-4-фтор-1-бензолсульфонамид;
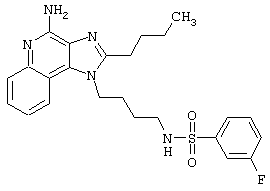
N1-[4-(4-амино-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-3-фтор-1-бензолсульфонамид;
N-{2-[4-амино-2-(этоксиметил)-1Н-имидазо[4,5-с]хинолин-1-ил)этил}метансульфонамид;
N2-{2-[4-aминo-2-(этoкcимeтил)-1H-имидaзo[4,5-c]xинoлин-1-ил]этил}-2-тиофенсульфонамид;
N1-{2-[4-aминo-2-(этoкcимeтил)-1H-имидaзo[4,5-c]xинoлин-1-ил]этил}-1-нафталинсульфонамид;
N-{2-[4-амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил]бутил}метансульфонамид;
N2-{2-[4-aминo-2-(2-мeтoкcиэтил)-1H-имидaзo[4,5-c]xинoлин-1-ил]бyтил}-2-тиофенсульфонамид;
N1-{4-[4-aминo-2-(2-мeтoкcиэтил)-1H-имидaзo[4,5-c]xинoлин-1-ил]бyтил}-5-(диметиламино)-1-нафталинсульфонамид;
N1-{4-[4-aминo-2-(2-мeтoкcиэтил)-1H-имидaзo[4,5-c]xинoлин-1-ил]бyтил}-4-фтор-1-бензолсульфонамид;
N1-{4-[4-aминo-2-(2-мeтoкcиэтил)-1H-имидaзo[4,5-c]xинoлин-1-ил]бyтил}-3-фтор-1-бензолсульфонамид;
N1-{4-[4-aминo-2-(2-мeтoкcиэтил)-1H-имидaзo[4,5-c]xинoлин-1-ил]бyтил}-1-бензолсульфонамид;
N8-{4-[4-амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-c]хинолин-1-ил]бутил}-8-хинолинсульфонамид;
N2-{4-[4-амино-2-(4-метоксибензил)-1Н-имидазо[4,5-c]хинолин-1-ил]бутил}-8-тиофенсульфонамид;
N-[4-(4-aмино-2-бутил-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]метансульфонамид;
N2-[4-(4-амино-2-бутил-6,7,8,9-тетрагидро-1H-имидазо[4,5-c]хинолин-1-ил)бутил]-2-тиофенсульфонамид;
N1-[4-(4-амино-2-бутил-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-5-(диметиламино)-1-нафталинсульфонамид;
N1-{4-[4-амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил]бутил}-1-бензолсульфонамид;
N1-{4-[4-амино-2-(метоксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил]бутил}-5-(диметиламино)-1-нафталинсульфонамид;
N'-{2-[4-aминo-2-(этoкcимeтил)-1H-имидaзo[4,5-c]xинoлин-1-ил]этил}-N,N-диметилульфамид;
N'-{2-[4-амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил]бутил}-N,N-диметилульфамид;
N'-{2-{4-амино-2-(4-метоксибензил)-1Н-имидазо[4,5-с]хинолин-1-ил]бутил}-N,N-диметилульфамид;
N'-[4-(4-aминo-2-бyтил-6,7,8,9-тeтpaгидpo-1H-имидaзo[4,5-c]xинoлин-1-ил)бутил]-N,N-диметилульфамид;
N'-[4-(4-амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-N,N-диметилульфамид;
N4-{4-[4-амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-c]хинолин-1-ил]бутил}-4-тиоморфолинсульфонамид;
N1-{4-[4-амино-2-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил]бутил}-1-пирролидинсульфонамид;
N1-[4-(4-амино-2-бутил-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-4-фтор-1-бензолсульфонамид;
N-[4-(4-амино-2-бутил-(2-метоксиэтил)-6,7,8,9-тетрагидро-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]метансульфонамид; и N-[4-(4-амино-2-бутил-(2-метоксиэтил)-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]фенилметансульфонамид;
N1-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-5-(диметиламино)-1-нафталинсульфонамид;
N1-[4-(4-aминo-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-5-(димeтилaминo)-1-нафталинсульфонамид;
N2-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-2-тиофенсульфонамид;
N-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-фенилметансульфонамид;
N1-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-1-бензолсульфонамид;
N1-[4-(4-aминo-2-бyтил-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-3-нитpo-1-бензолсульфонамид;
N1-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-3-амино-1-бензолсульфонамид;
N1-[4-(4-aминo-2-бyтил-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-4-нитpo-1-бензолсульфонамид;
N1-[4-(4-амино-2-бутил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-4-амино-1-бензолсульфонамид;
N5-[4-(4-aминo-2-бyтил-1H-имидaзo[4,5-c]xинoлин-1-ил)бyтил]-5-изохинолинсульфонамид;
N-[4-(4-амино-2-(4-метоксибензил)-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]-метансульфонамид;
N1-[4-(4-амино-1H-имидазо[4,5-с]хинолин-1-ил)бутил]-1-бутансульфонамид;
N1-{4-[4-амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-с]хинолин-1-ил]бутил}-4-фтор-1-бензолсульфонамид;
N1-[4-(4-амино-2-фенил-1Н-имидазо[4,5-c]хинолин-1-ил)бутил]-4-фтор-1-бензолсульфонамид; и
N-[4-(4-амино-2-фенил-1Н-имидазо[4,5-с]хинолин-1-ил)бутил]метансульфонамид.
Приоритет от 10.06.1999
По пунктам 1, 2, 3, 5, 6, 8, 10, 14, 15, 16, 17, 20, 21, 22, 23 в случае, если радикал R4 представляет фенил; -O-алкил; -СО-O-алкил; -S-(O)0-2-алкил; оксогруппу; галоген; NO2; а радикал R2 выбирают из группы, в которую входят: водород; -алкил; -фенил; и по п.19, в случае когда соединение представляет N1-[4-(амино-1H-имидазо[4,5-с]хинолин-1-ил)бутил]-1-бутансульфамид.
От 07.06.2000
По пунктам 1, 2, 3, 5, 6, 8, 10, 14, 15, 16, 17, 20, 21, 22, 23 при всех остальных значениях указанных радикалов и других признаков и по пунктам 4; 7; 9; 11; 12; 13; 18.
| JP 9208584 А2, 12.08.1997 | |||
| WO 9830562 А, 16.07.1998 | |||
| ИМИДАЗО-АННЕЛИРОВАННЫЕ ИЗО- И ГЕТЕРОЦИКЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2076105C1 |
| ПРОИЗВОДНОЕ 1,3-ДИГИДРО-2H-ИМИДАЗО/4,5-B/ХИНОЛИН-2-ОНА В КАЧЕСТВЕ ИНГИБИТОРА ФОСФОДИЭСТЕРАЗЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ ДЛЯ НЕГО, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1992 |
|
RU2127273C1 |

