Изобретение относится к соединениям, обладающим активностью агонистов гормонов-гликопротеинов, в частности, к соединениям-агонистам лютеинизирующего гормона (LH) и фолликулостимулирующего гормона (FSH). Кроме того, изобретение относится к содержащим их фармацевтическим композициям, также как и к применению указанных соединений в медицинской терапии, в частности для регулирования их фертильности.
Гонадотропные гормоны выполняют важные функции в различных функциях организма, включая метаболизм, регуляцию температуры и репродуктивный процесс. Гонадотропные гормоны гипофиза FSH-и LH, например, играют основную роль в стимулировании роста и созревания фолликулов, тогда как LH связаны с индуцированнием процесса овуляции (Sharp, R.M. Clin. Endocrinol 33, 787-807, 1990; Dorrington и Armstwong, Recent Prog. Horm. Res 35, 301-342, 1979; Levy и др. Human Reproduction 15, 2258-2265, 2000).
В настоящее время LH применяют в клинике в комбинации с FSH для стимулирования (деятельности) яичников, то есть гиперстимуляции яичников для оплодотворения in vitro (IVF) и индуцирования овуляции у женщин при бесплодии, связанном с аковуляцией (Insler, V., Int. J.Fertility 33, 85-97, 1988; Navot: и Rosenwaks, J.Vitro Fert. Embryo Transfer 5, 3-13, 1988), также как и при гипогонадизме и мужском бесплодии.
Гонадотропные гормоны действуют на специфические типы гонадных клеток для стимулирования яичников и тестикулярной дифференциации и стероидогенеза. Действие указанных гипофизарных и плацентарных гормонов опосредовано специфическими рецепторами мембраны плазмы, которые относятся к большой группе G-протеинсвязанных рецепторов. Они состоят из отдельных полипептидов с семью трансмембранными доменами и способны взаимодействовать с Gs-протеинами, приводя к активации аденилциклазы.
Гонадотропные гормоны, предназначенные для терапевтических целей, могут быть выделены из мочи человека и имеют низкую степень чистоты (Morse и др., Amer. J. Reproduct. Immunol. and Microbiology 17, 143, 1988). В качестве альтернативы они могут быть получены в виде рекомбинантных гонадотропинов. Дополнительно к указанным белкам рецепторы гонадотропина могут быть активированы или дезактивированы синтетическими соединениями с низкой молекулярной массой. Бициклические гетероароматические соединения, описанные в патенте WO 00/61586, в экспериментах in vitro и in vivo показали, что они применимы в качестве агонистов LH.
В норме у женщин секреция гипофизарных LH и FSH характеризуется всплеском в середине цикла, который предшествует овуляции. Овуляция характеризуется тремя различными физиологическими явлениями, то есть созреванием овоцита, разрывом фолликула и лютеинизацией. В то время как роль выброса LH в развитии in vivo указанных процессов неоспорима, роль выброса FSH менее ясна. Однако, в настоящее время показано, что FSH стимулирует созревание овоцита in vitro, побуждая совокупные клетки продуцировать фактор, который позитивно преодолевает задержку мейоза, индуцированную гипоксантином (Lu и др., Mol. Cell. Endocrinol. 164, 191-196, 2000). Полагают, что указанный фактор является стерином (MAS), активирующим мейоз.
При стимулировании овуляции желательно обеспечить влияние LH как главного компонента. В соответствии с данным изобретением найдены соединения, в частности, с превосходными свойствами при их применении для повышения фертильности. Для указанных соединений LH-активность сопровождается FSH-активностью.
Таким образом, данное изобретение предоставляет соединения с низкой молекулярной массой, которые в дополнение к LH-активности неожиданно проявляют также FSH-активность. В общем указанные соединения являются тиено[2,3-d]пиримидинами, которые в 4-положении пиримидинового кольца замещены фенильной группой, которая в свою очередь замещена в мета-положении.
В данном изобретении представлены производные тиено[2,3-d]пиримидинов в соответствии с общей формулой I
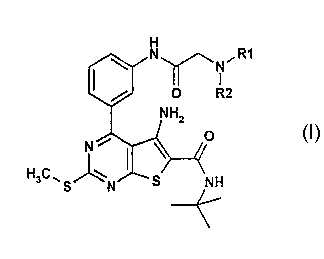
или их фармацевтически приемлемые соли, где N(R1)R2 объединены в (2-6С)гетероциклоалкильное кольцо.
Наиболее предпочтительными соединениями являются: трет-бутил-5-амино-2-метилтио-4-(3-(2-(азетидин-1-ил)-ацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид; трет-бутил-5-амино-2-метилтио-4-(3-(2-(морфолин-4-ил)-ацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид; трет-бутил-5-амино-2-метилтио-4-(3-(2-(тиоморфолин-4-ил)-ацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид; трет-бутил-5-амино-2-метилтио-4-(3-(2-(пиперидин-1-ил)-ацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид; трет-бутил-5-амино-2-метилтио-4-(3-(2-(пирролидин-1-ил)-ацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид и трет-бутил-5-амино-2-метилтио-4-(3-(2-(пиперазин-1-ил)-ацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид.
Термин «объединены в (2-6С)гетероциклоалкильное кольцо» в определении формулы I означает, что R1 и R2 вместе с атомом азота, к которому они присоединены, образуют кольцо, имеющее 2-6 атомов углерода, возможно, содержащее один или более гетероатомов, выбранных из N, О и/или S. Примерами таких колец являются азетидин, пирролидин, пиперидин, пиперазин, морфолин и тиоморфолин.
Показано, что соединения указанной выше формулы I проявляют активность агонистов LH и FSH. В биопробах in vitro, использующих СНО клетки, устойчиво трансфицированные рецепторами LH и FSH человека, соответственно, было найдено, что EC50 по отношению к LH-рецептору составляет менее 5Е-08, тогда как по отношению к FSH-рецептору ЕС50 составляет менее Е-05. Обычно активность FSH колеблется от около 1% активности агонистической стимуляции LH до примерно 10% агонистической стимуляции LH.
Дополнительно изобретение относится к фармацевтической композиции, содержащей производное тиено[2,3-d]пиримидина или его соли общей формулы I.
Таким образом, соединения изобретения могут быть использованы в терапии. Дополнительный аспект изобретения состоит в применении производных тиено[2,3-d]пиримидина общей формулы I для приготовления медикамента для регулирования фертильности, более предпочтительно, для стимулирования овуляции. Указанные соединения применимы для активации рецепторов как LH, так и FSH. Соединения данного изобретения поэтому могут быть использованы в способах лечения женщин с проблемами бесплодия (фертильности).
Терапевтически применимыми солями соединений формулы I являются такие соли, в которых противоион фармацевтически приемлем. Однако аддитивные соли с кислотами оснований, соответствующих формуле I, могут найти применение, например, при получении или очистке фармацевтически приемлемых соединений. Все соли, являются они фармацевтически приемлемыми или нет, включены в область изобретения.
Примерами аддитивных солей являются соли, производные таких минеральных кислот, как соляная кислота, фосфорная кислота, серная кислота, предпочтительно соляная кислота, и органических кислот, таких как лимонная кислота, винная кислота, уксусная кислота, молочная кислота, малеиновая кислота, малоновая кислота, фумаровая кислота, гликолевая кислота, янтарная кислота и подобные.
Подходящими способами введения соединений формулы I или их фармацевтически приемлемых солей, также относящимися к применению их в качестве активных ингредиентов, являются внутримышечные инъекции, подкожные инъекции, внутривенные вливания или внутрибрюшинные инъекции, пероральное и интраназальное введение. Предпочтительно соединения могут быть введены перорально. Точная доза и режим введения активного ингредиента или его фармацевтической композиции безусловно должны зависеть от терапевтического эффекта, который должен быть дистигнут (лечение бесплодия, контрацепция), и могут варьироваться в зависимости от конкретного соединения, способа введения, возраста и состояния отдельного субъекта, которому медикамент должен быть введен.
Обычно парентеральное введение требует меньших доз, чем другие способы введения, которые больше зависят от адсорбции. Однако доза для человека предпочтительно содержит 0,0001-25 мг на кг веса тела. Желаемая доза может представлять собой разовую дозу или множество субдоз, вводимых через соответствующие интервалы в течение дня. В случае реципиента-женщины дозы могут вводиться в соответствующие интервалы дней менструального цикла для поддержки фолликул или в виде разовой дозы для вызывания овуляции. Дозы, также как и режим введения, могут быть различными для реципиентов женщин и мужчин.
В случае введения in vitro или ex vivo подобно IVF-введению соединения изобретения должны быть использованы в инкубационной среде в концентрации примерно 0,01-5 мкг/мл.
Данное изобретение относится также к фармацевтическим композициям, содержащим производные тиено[2,3-d]пиримидина формулы I в смеси с фармацевтически приемлемыми вспомогательными веществами и, возможно, другими терапевтическими агентами. Вспомогательные вещества должны быть «приемлемыми» в смысле совместимости с другими компонентами композиции и не быть вредными для реципиента.
Фармацевтические композиции включают такие, которые подходят для перорального, ректального, назального, местного (включая чрезкожное, буккальное и сублингвальное), вагинального или парентерального (включая подкожное, трансдермальное, внутримышечное, внутривенное и чрезкожное) введение. Композиции могут быть получены любым способом, хорошо известным в области фармации, например, с применением таких способов, как описанные в Gennaro и др., Remington's Pharmaceutical Sciences (18-е изд. Mack Publishing company, 1990, см. особенно часть 8: Pharmaceutical Preparations and Their Manufacture).
Указанные способы включают стадию объединения активного компонента с любым вспомогательным агентом. Вспомогательными агентами, также называемыми дополнительными компонентами, являются такие обычные компоненты (Gennaro, supra), как наполнители, связующие, разбавители, дезинтегранты, лубриканты, красители, отдушки и смачивающие агенты.
Фармацевтические композиции, подходящие для перорального введения, могут быть представлены в форме дискретных дозированных единиц, таких как пилюли, таблетки или капсулы, или в форме порошка или гранул, или в виде раствора или суспензии. Активный компонент может также быть представлен в виде болюса или пасты. Композиции могут также быть приготовлены в виде суппозитории или клизмы для ректального введения.
Для парентерального введения подходящими композициями являются водные и неводные стерильные инъекционные растворы. Композиции могут быть представлены в виде упаковки для однократного приема или для многократного приема, например, в виде запаянных пузырьков и ампул и могут храниться в лиофилизированном состоянии, требуя перед употреблением только добавления стерильного жидкого носителя, например воды.
Композициями или препаратами, подходящими для назальной ингаляции, могут быть тонкоизмельченные порошки или аэрозоли, которые могут быть получены путем распыления отмеренной дозы аэрозоля под давлением, при помощи распылителя или путем вдувания.
Производные тиено[2,3-d]пиримидина изобретения могут также быть введены в форме имплантируемых фармацевтических устройств, состоящих из ядра активного материала, упакованного в мембрану, высвобождающую его с регулируемой скоростью. Указанные имплантанты применяют подкожно или местно и они способны высвобождать активный компонент примерно с постоянной скоростью в течение относительно длительного промежутка времени, например, от недель до лет. Способы получения имплантируемых фармацевтических устройств, такие, как известные в современном уровне техники, описаны, например в Европейском патенте 0303306 (AKZO N.V.).
Таким образом, соединения согласно данному изобретению могут быть использованы для тех же клинических целей, что и природный LH, с тем преимуществом, что они обладают также FSH-активностью, проявляют измененные свойства стабильности и могут вводиться различными способами.
Соединения данного изобретения, представленные формулой (I), обычно могут быть получены нуклеофильным замещением галогенидов (II), где Q=Cl или Br, с (циклическими) вторичными аминами формулы (III) в соответствующем растворителе, таком как N,N-диметилформамид или ТГФ (тетрагидрофуран) при комнатной температуре в присутствии третичного основания, такого как N,N-диизопропилэтиламин (DIPEA).
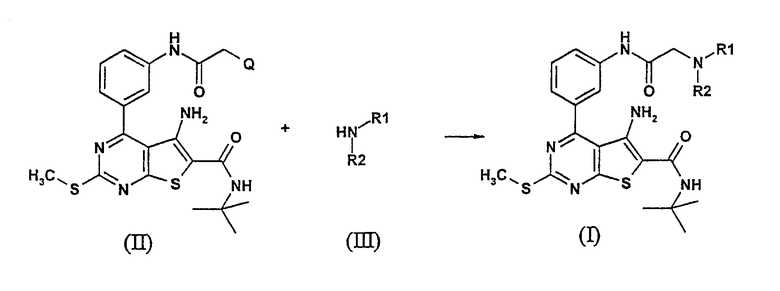
Производные формулы (II), где Q=Cl или Br, могут быть получены региоселективным ацилированием производного мета-анилина (V) ацилхлоридом типа (IV), где Q=Cl или Br, в присутствии третичного основания, такого как N,N-диизопропилэтиламин, в подходящем растворителе, таком как дихлорметан или ТГФ.
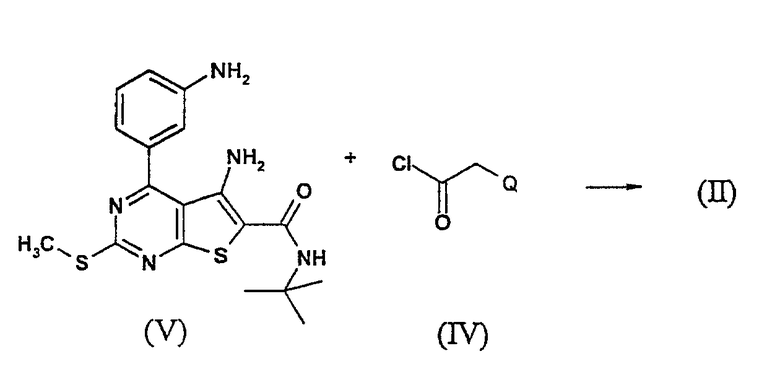
Соединение (V) может быть получено известным в уровне техники восстановлением нитрофункции в производном (VI) с применением соответствующего восстановителя, такого как хлорид олова(II), в протонном растворителе, таком как этанол, в присутствии соляной кислоты при повышенной температуре (J. Heilbron, J.Chem. Soc., 1279 (1940)).
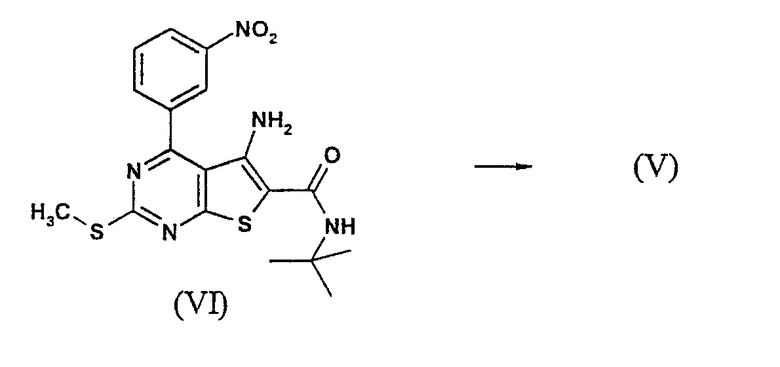
Тиенопиримидины (VI) могут быть получены конденсацией карбоновой кислоты (VII) с трет-бутиламином под действием агента сочетания, такого как тетрафторборат О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU) или гексафторфосфат бромтрипирролидинофосфония (PyBrOP) и третичного основания, например, N,N-диизопропилэтиламина.
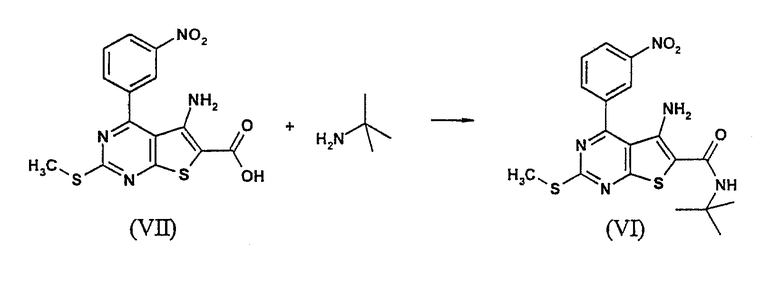
Омыление сложного этилового эфира (VIII) до соответствующей карбоновой кислоты (VII) протекает в присутствии основания, такого как гидроксид лития, гидроксид калия или гидроксид натрия, в водном диоксане при повышенной температуре (от 80°С до кипения).
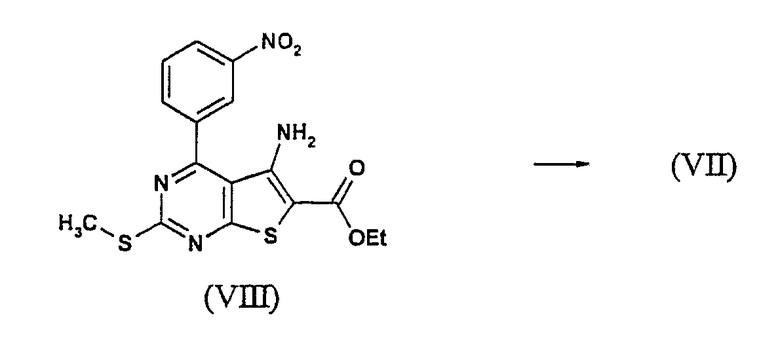
Бициклическое соединение (VIII) образовано замещением хлорида (X) этилмеркаптоацетатом под действием N,N-диизопропилэтиламина с последующим замыканием цикла промежуточного простого тиоэфира (IX) катализируемым основанием. Указанный тип образования тиено[2,3-d]пиримидинового кольца описан в: S.A. Abdel-Hady, M.A. Badawy, Y.A. Ibrahim, Sulfur Lett. 9, 101 (1989) и S. Tumkevicius, Liebigs Ann., 1703 (1995).
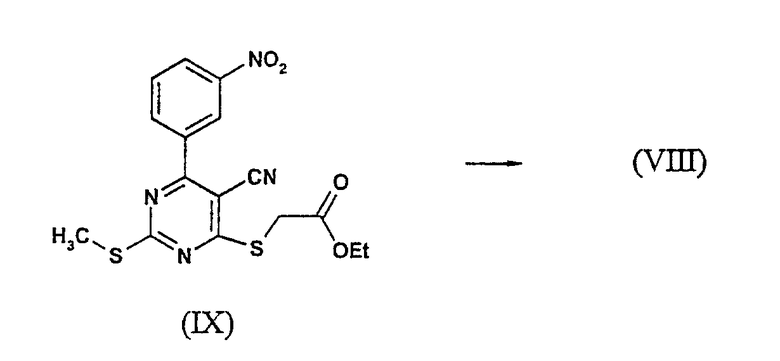
Подходящими условиями реакции циклизации являются этилат натрия в этаноле или N,N-диизопропилэтиламин в толуоле/этаноле (1/1, об./об.) при температуре кипения.
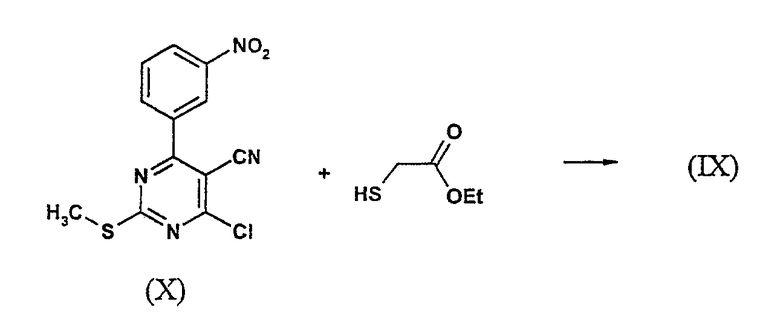
Необходимый хлоримин (Х) может быть синтезирован следующим известным из литературы способом, например, как описано A.A. Santilli, D.H. Kim и S.V.Wanser, J.Heterocycl. Chem. 8, 445, 1971. Согласно указанному способу лактам (XI) обрабатывают POCl3 при повышенной температуре (от 80°С до кипения) с образованием хлорида (Х). Добавление соответствующего растворителя, например диоксана, и/или добавление либо PCl5, либо N,N-диметиланилина к реакционной смеси может привести к сокращению времени реакции и более высоким выходам хлорида (Х).
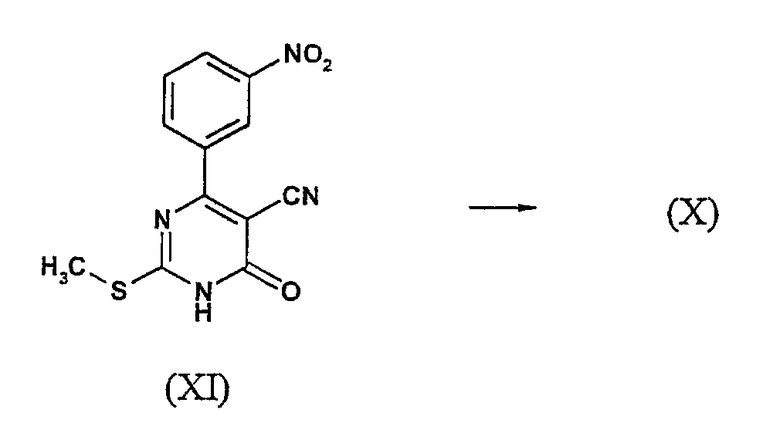
Соответствующий путь к лактаму (XI) предусматривает многокомпонентную конденсацию этилцианоацетата с 3-нитро-бензальдегидом и S-метилизотиомочевиной в этаноле под действием основания, такого как карбонат калия, при повышенной температуре (60°С).
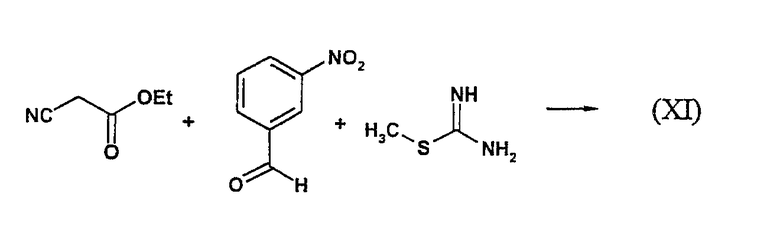
Родственные способы предложены в S. Kambe, K. Saito и H. Kishi, Synthesis, 287 (1979); A.M. Abd-Elfattah, S.M. Hussain и A.M. El-Reedy, Tetrahedron 39, 3197 (1983); S.M. Hussain, A.A. El-Barbary и S.A. Mansour, J.Heterocycl. Chem. 22, 169 (1985).
Способы определения связывания рецептора в пробах как in vitro, так и in vivo, для определения биологической активности гонадотропных гормонов хорошо известны. Обычно экспрессированный рецептор вводят в контакт с соединением, которое должно быть испытано, и измеряют связывание или стимулирование, или ингибирование функциональной реакции.
Для измерения функциональной реакции выделенную ДНК, кодирущую ген рецептора LH или FSH, предпочтительно рецептора человека, экспрессируют в подходящие клетки хозяина. Такой клеткой может быть клетка яичника китайского хомяка, но подходят также и другие клетки. Предпочтительны клетки млекопитающих (Jia и др., Mol. Endocrin., 5, 759-776, 1991).
Способы создания рекомбинантных клеточных линий, экспрессирующих LH или FSH, хорошо известны в современном уровне техники (Sambrook и др., Molecular Cloning: a Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, последнее издание). Экспрессию рецептора достигают экспрессией ДНК, кодирующей целевой белок. Методики сайт-направленного мутагенеза, лигирования дополнительных последовательностей, ПЦР (PCR), и создания подходящих систем экспрессии хорошо известны в уровне техники. Часть или вся ДНК, кодирующая желаемый белок, может быть получена синтетически с использованием стандартной твердофазной методики, предпочтительно для включения сайтов рестрикции для облегчения лигирования. Подходящие контрольные элементы для транскрипции и трансляции включенных кодирующих последовательностей могут быть обеспечены для кодирующих последовательностей ДНК. Как хорошо известно, теперь доступны системы экспрессии, которые совместимы с широким разнообразием хозяев, включая прокариотических хозяев, таких как бактерии, и эукариотических хозяев, таких как дрожжи, клетки растений, клетки насекомых, клетки млекопитающих, клетки птиц и подобные.
Клетки, экспрессирующие рецептор, затем контактируют с испытываемым соединением для наблюдения связывания или стимулирования, или ингибирования функциональной реакции.
В качестве альтернативы для измерения связывания соединения могут быть использованы выделенные клеточные мембраны, содержащие экспрессированный рецептор.
Для измерения связывания могут быть использованы радиоактивно или флуоресцентно меченные соединения. В качестве эталонного соединения могут быть использованы рекомбинантные LH или FSH человека. В качестве альтернативы могут быть проведены также тесты конкурентного связывания.
Другой тест заключается в скрининге соединений агонистов LH или FSH путем определения стимуляции рецептора, опосредованной аккумуляцией сАМР. Таким образом, указанный способ предусматривает экспрессию рецептора на поверхности клетки хозяина и воздействие на клетку испытуемого соединения. Затем измеряют количество сАМР. Уровень сАМР может быть снижен или увеличен в зависимости от ингибирующего или стимулирующего действия испытуемого соединения в результате связывания c рецептором.
В дополнение к непосредственному измерению, например, уровня сАМР в подвергнутой действию клетке могут быть использованы клетки, которые в добавление к трансфекции рецептором, кодирующим ДНК, трансфицированы также второй ДНК, кодирующей ген-репортер, экспрессия которого зависит от уровня сАМР. Указанные гены-репортеры могут быть индуцируемыми или могут быть сконструированы таким образом, чтобы они были связаны в новыми элементами, чувствительными к сАМР. Обычно экспрессия гена-репортера может контролироваться любым чувствительным элементом, реагирующим на изменение уровня сАМР. Подходящими генами-репортерами являются, например, LacZ, щелочная фосфатаза, люцифераза светляка и флуоресцирущий зеленым белок. Принципы указанной пробы трансактивации хорошо известны в уровне техники и описаны Stratowa, Ch, Himmler, A и Czernilofsky, A.P., Curr.Opin.Biotechnol. 6, 574 (1995).
Для отбора активных соединений для рецепторов LH и FSH, испытания при 10-5 М должны показать активность более 20% от максимальной активности, когда LH и FSH использованы в качестве эталона. Другим критерием может быть значение ЕС50, которое должно быть <10-5 M, предпочтительно <10-7 M.
Квалифицированный специалист определит, что желаемое значение ЕС50 зависит от испытуемого соединения. Например, соединение с ЕС50 менее 10-5 М обычно рассматривают в качестве кандидата для отбора в качестве лекарственного средства. Предпочтительно указанное значение ниже 10-7 М. Однако соединение, которое имеет более высокое ЕС50, но является селективным для конкретного рецептора, может быть даже лучшим кандидатом.
Скрининг соединений агонистов рецепторов LH может также осуществляться путем использования биопробы клетки мыши Leydig (Van Damme, M., Robersen, D. И Diczfalusy, E., Acta Endocrinol. 77: 655-671 (1974), Mannaerts, B., Kloosterboer, H., и Schuurs, A., Neuroendocrinology of reproduction. R.Rolland и др. Eds., Elsevier Science Publishers B.V., 49-58 (1987)). В указанном тесте рецептор LH, опосредованный образованием тестостерона, может быть измерен в клетке Leydig, выделенной из самца (мужской особи) мыши.
Активность агониста FSH соединений может быть также определена на моделях ex vivo с применением культивированных мышиных фолликул согласно Nayudu, P и Osborn, S. (J. Reproduction and Fertility 95, 349-362 (1992)). Для этого фолликулы яичников мыши выделяли и культивировали в присутствии соединений агонистов FSH для индуцирования роста фолликул. Измерение диаметра фолликул и эстрадиола в культуральной среде показывает рост фолликул.
Для измерения LH-активности соединений in vivo может быть изучено индуцирование овуляции у неполовозрелых мышей. В указанном тесте неполовозрелой женской особи мыши сначала вводили FSH, выделенный из мочи, и примерно через 48 часов соединение-агонист LH. Животных умерщвляли после введения агониста LH и под микроскопом оценивали число яйцеклеток в маточных трубах.
Для измерения FSH-активности соединений in vivo неполовозрелые самки мыши обрабатывали через 0, 8, 24 и 32 часа соединением-агонистом FSH, чтобы вызвать рост фолликул. Через 52 часа после начала эксперимента животным впрыскивали hCG, чтобы вызвать овуляцию. Животных убивали через 72 часа после начала эксперимента и под микроскопом оценивали число яйцеклеток в маточных трубах. Дополнительно определяли массу яичника.
Соединения данного изобретения могут быть использованы клинически в тех схемах (лечения), где теперь применяют LH или hCG. Они предусматривают замещение LH у пациента с гипогонадным гипогонадизмом как у мужчин, так и у женщин, введение в середине цикла для индуцирования овуляции (OI) или регулирования гиперстимуляции (СОН) или стимуляции желтого тела.
Следующие примеры иллюстрируют изобретение и не должны быть интерпретированы как ограничение изобретения.
ПРИМЕРЫ
Пример 1
Трет-бутил-5-амино-2-метилтио-4-(3-(2-(азетидин-1-ил)-ацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид
(а). 5-Циано-4-(3-нитрофенил)-2-метилтио-6-оксопиримидин
Смесь сульфата S-метилизотиомочевины (69,0 г), 3-нитробензальдегида (75,0 г), этилцианоацетата (56,0 мл) и карбоната калия (72,5 г) в абс.EtOH (1500 мл) перемешивали при 60°С 16 ч. Реакционную смесь охлаждали до 0°С на ледяной бане. Получаемый осадок отфильтровывали, промывали абс.EtOH и растворяли в горячей воде (100°С). Раствор охлаждали до комнатной температуры, подкисляли 2 н. HCl до рН 2 и охлаждали до 0°С на ледяной бане. Получаемый осадок отфильтровывали и промывали ледяной водой. Остаточную воду из осадка удаляли соиспарением с 1,4-диоксаном.
Выход: 54,0 мг
MS-ESI (масс-спектр-ионизация электрораспылением): [M+H]+=289,0
ТСХ (тонкослойная хроматография): Rf=0,3, силикагель, DCM (дихлорметан)/МеОН=9/1 (об./об.).
(b). 6-Хлор-5-циано-4-(3-нитрофенил)-2-метилтиопиримидин
POCl3 (100 мл) прибавляли к перемешиваемому раствору 5-циано-4-(3-нитрофенил)-2-метилтио-6-оксопиримидина (пример 1(а), 25,0 г) в сухом 1,4-диоксане (300 мл). Через 3 ч при 90°С смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток растворяли в 1,4-диоксане (100 мл) и получаемый раствор охлаждали до 0°С: осторожно прибавляли ледяную воду. Получаемый осадок отфильтровывали и промывали водой. Остаточную воду из осадка удаляли соиспарением с 1,4-диоксаном.
Выход: 26,0 г
MS-ESI: [M+H]+=307,0
ТСХ: Rf=0,5, силикагель, гептан/EtOAc=3/2 (об./об.).
(с). Этил-5-циано-4-(3-нитрофенил)-2-метилтио-6-(этоксикарбонилметилтио)-пиримидин
DIPEA (15,7 мл) прибавляли к перемешиваемому раствору этил-2-меркаптоацетата (9,3 мл) и 6-хлор-5-циано-4-(3-нитрофенил)-2-метилтиопиримидина (пример 1(b), 26,0 г) в смеси EtOH и DCM (250 мл). Через 1 ч при комнатной температуре к смеси прибавляли 0,1 н. водн. HCl (500 мл) и затем экстрагировали DCM (3*500 мл), сушили (MgSO4) и концентрировали при пониженном давлении.
Выход: 28,0 г
MS-ESI: [M+H]+=390,4
ТСХ: Rf=0,5, силикагель, гептан/EtOAc=3/2 (об./об.).
(d). Этиловый эфир 5-амино-4-(3-нитрофенил)-2-метилтио-тиено[2,3-d]пиримидин-6-карбоновой кислоты
Смесь этил-5-циано-4-(3-нитрофенил)-2-метилтио-6-(этоксикарбонилметилтио)-пиримидина (пример 1(с), 28,0 г) и DIPEA (30 мл) в смеси толуола (150 мл) и EtOH (150 мл) перемешивали при температуре кипения (100°С) 16 ч. Смесь затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаточный DIPEA удаляли соиспарением с толуолом.
Выход: 28,0 г
MS-ESI: [M+H]+=391,2
ТСХ: Rf=0,6, силикагель, гептан/EtOAc=3/2 (об./об.).
(е). Этиловый эфир 5-амино-4-(3-аминофенил)-2-метилтио-тиено[2,3-d]пиримидин-6-карбоновой кислоты
EtOH (440 мл) прибавляли к смеси этилового эфира 5-амино-4-(3-нитрофенил)-2-метилтио-тиено[2,3-d]пиримидин-6-карбоновой кислоты (пример 1(d), 28,0 г), концентрированной водн. HCl (15 мл) и хлорида олова(II) (41,0 г) в 1,4-диоксане (400 мл). Смесь перемешивали при 90°С 16 ч. Смесь затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток суспендировали в EtOAc (1000 мл) и добавляли 4 н. водн. NaOH до рН 10-11. Смесь энергично перемешивали, органический слой отделяли, сушили (MgSO4) и концентрировали при пониженном давлении.
Выход: 21,0 г
MS-ESI: [M+H]+=361,0
ТСХ: Rf=0,6, силикагель, гептан/EtOAc=3/2 (об./об.).
(f). 5-Амино-4-(3-аминофенил)-2-метилтио-тиено[2,3-d]пиримидин-6-карбоновая кислота
Гидроксид калия (32,4 г) прибавляли к раствору этилового эфира 5-амино-4-(3-аминофенил)-2-метилтио-тиено[2,3-d]пиримидин-6-карбоновой кислоты (пример 1(е), 21,0 г) в смеси 1,4-диоксана (300 мл) и воды (100 мл). Через 16 ч при 90°С смесь охлаждали до 10°С и при энергичном перемешивании прибавляли 2 н. водн. лимонную кислоту (300 мл). Получаемый осадок отфильтровывали, промывали водой (180 мл) и сушили в вакууме.
Выход: 14,0 г
MS-ESI: [M+H]+=333,0
ТСХ: Rf=0,5, силикагель, DCM/MeOH=9/1 (об./об.).
(g). Трет-бутил-5-амино-4-(3-аминофенил)-2-метилтио-тиено[2,3-d]пиримидин-6-карбоксамид
TBTU (16,1 г) прибавляли к раствору 5-амино-4-(3-аминофенил)-2-метилтио-тиено[2,3-d]пиримидин-6-карбоновой кислоты (пример 1(f), 14,0 г), DIPEA (17,4 мл) и трет-бутиламина (7,3 г) в DCM/ДМФА (диметилформамид) (1/1, об./об., 250 мл). Через 3 ч при комнатной температуре смесь промывали насыщ. водн. NaHCO3 (3*100 мл), 0,1 н. водн. HCl (100 мл) и водой (100 мл). Органический слой концентрировали при пониженном давлении. Сырой продукт очищали кристаллизацией из теплого абс.EtOH (300 мл).
Выход: 10,5 г
MS-ESI: [M+H]+=388,2
ВЭЖХ (высокоэффективная жидкостная хроматография): Rt=30,72 мин, Luna C-18(2), 5 мкм, 250*2,0 мм, детектирование в УФ=210 нм, температура колонки=40°С, расход 0,25 мл/мин, элюент вода/ACN/МеОН=от 90/9,5/0,5 до 0/95/5, время пробега=50 мин.
(h). Трет-бутил-5-амино-2-метилтио-4-(3-(2-бромацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид
Бромацетилхлорид (615 мг) прибавляли к раствору трет-бутил-5-амино-2-метилтио-4-(3-аминофенил)-тиено[2,3-d]пиримидин-6-карбоксамида (пример 1(g), 1,08 г) и DIPEA (2,43 мл) в сухом DCM (20 мл). Через 3 ч при комнатной температуре смесь разбавляли DCM, промывали насыщ. водн. NaHCO3, сушили (MgSO4) и концентрировали при пониженном давлении. Сырой продукт очищали хроматографированием на силикагеле, используя в качестве элюента гептан/EtOAc=3/2 (об./об.).
Выход: 910 мг
MS-ESI: [M+H]+=510,2
ТСХ: Rf=0,3, силикагель, гептан/EtOAc=3/2 (об./об.)
(i). Трет-бутил-5-амино-2-метилтио-4-(3-(2-(азетидин-1-ил)-ацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид
Трет-бутил-5-амино-4-(3-(2-бромацетамидо)-фенил)-2-метилтио-тиено[2,3-d]пиримидин-6-карбоксамид (пример 1(h), 91 мг) прибавляли к раствору гидрохлорида азетидина (120 мг) и N,N-диизопропилэтиламина (0,25 мл) в DCM (5 мл). Через 16 ч при комнатной температуре смесь промывали насыщ. водн. NaHCO3, сушили (MgSO4) и концентрировали при пониженном давлении. Сырой продукт очищали ВЭЖХ, используя колонну Luna C-18 с градиентом элюирования: 0,1% водн. TFA (трифторуксусная кислота)+10% водн. ACN/ACN=от 90/10 до 10/90 за 30 мин. Соединение, указанное в заглавии, затем лиофилизировали от воды 0,1% TFA.
Выход: 56 мг (соль с TFA)
MS-ESI: [M+H]+=485,2
ВЭЖХ: Rt=13,45 мин, колонка Luna C-18(2), 3 мкм, 100*2,0 мм, детектирование в УФ=210 нм, температура колонки=40°С, расход 0,25 мл/мин, элюент фосфатный буферный раствор 50 мМ рН 2,1/вода/ACN=от 10/70/20 до 10/10/80 (об./об.), время пробега=20 мин.
Пример 2
Трет-бутил-5-амино-2-метилтио-4-(3-(2-(морфолин-4-ил)-ацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид
Морфолин (5,0 мл) прибавляли к раствору трет-бутил-5-амино-4-(3-(2-бромацетамидо)фенил)-2-метилтио-тиено[2,3-d]пиримидин-6-карбоксамида (пример 1(h), 1,0 г) в ТГФ (50 мл). Через 16 ч при комнатной температуре смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, используя DCM/MeOH=9/1 в качестве элюента. Сырой продукт дополнительно очищали ВЭЖХ, используя колонку Luna C-18 с градиентом элюирования: 0,1% водн.TFA/вода/ACN=от 3/97/0 до 3/7/90 за 30 мин. Чистое соединение, указанное в заглавии, лиофилизировали из смеси 0,1% водн. TFA и воды.
Выход: 215 мг (соль с TFA)
MS-ESI: [M+H]+=515,2
ВЭЖХ: Rt=20,62 мин, Luna C-18(2), 5 мкм, 150*2 мм, детектирование в УФ=210 нм, температура колонки=40°С, расход=0,25 мл/мин, элюент фосфатный буферный раствор 50 мМ рН 2,1/вода/ACN/MeOH=от 10/72/17/1 до 10/18/68/4 (об./об.), время пробега=40 мин.
Пример 3
Трет-бутил-5-амино-2-метилтио-4-(3-(2-(тиоморфолин-4-ил)-ацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид
Тиоморфолин (2,16 мл) прибавляли к раствору трет-бутил-5-амино-2-метилтио-4-(3-(2-бромацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамида (пример 1(h), 1,09 г) в DCM (50 мл). Через 16 ч при комнатной температуре смесь разбавляли DCM, промывали насыщ. водн. NaHCO3, сушили (MgSO4) и концентрировали при пониженном давлении. Сырой продукт очищали ВЭЖХ, используя колонку Luna C-18 с градиентом элюирования: 0,1% водн.TFA+10% водн. ACN/ACN=от 100/0 до 10/90 за 30 мин. Чистое соединение, указанное в заглавии, лиофилизировали из воды, подкисленной 1 н. водн. HCl.
Выход: 816 мг
MS-ESI: [M+H]+=531,2ВЭЖХ: Rt=14,72 мин., колонка Luna C-18(2), 3 мкм, 100*2 мм, детектирование в УФ=210 нм+254 нм, температура колонки=40°С, расход=0,25 мл/мин, элюент фосфатный буферный раствор 50 мМ рН 2,1/вода/ACN/MeOH=от 10/72/17/1 до 10/18/68/4 (об./об.), время пробега=20 мин.
Пример 4
Трет-бутил-5-амино-2-метилтио-4-(3-(2-(пиперидин-1-ил)-ацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид
Пиперидин (3,0 мл) прибавляли к раствору трет-бутил-5-амино-4-(3-(2-бромацетамидо)-фенил)-2-метилтио-тиено[2,3-d]пиримидин-6-карбоксамида (пример 1(h), 1,0 г) в CH2Cl2 (50 мл). Через 16 ч при комнатной температуре смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, используя DCM/MeOH=9/1 в качестве элюента. Сырой продукт дополнительно очищали ВЭЖХ, используя колонку Luna C-18 с градиентом элюирования: 0,1% водн.TFA/ACN=от 100/0 до 10/90 за 30 мин. Чистое соединение, указанное в заглавии, лиофилизировали из смеси 0,1% водн. TFA и воды.
Выход: 851 мг (соль с TFA)
MS-ESI: [M+H]+=513,2
ВЭЖХ: Rt=37,3 мин, Luna C-18(2), 5 мкм, 150*2 мм, детектирование в УФ=210 нм, температура колонки=40°С, расход=0,25 мл/мин, элюент фосфатный буферный раствор 50 мМ рН 2,1/вода/ACN=от 20/60/20 до 20/0/80 (об./об.), время пробега=40 мин.
Пример 5
Трет-бутил-5-амино-2-метилтио-4-(3-(2-(пирролидин-1-ил)-ацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид
Пирролидин (3,0 мл) прибавляли к раствору трет-бутил-5-амино-4-(3-(2-бромацетамидо)-фенил)-2-метилтио-тиено[2,3-d]пиримидин-6-карбоксамида (пример 1(h), 1,0 г) в СН2Cl2 (50 мл). Через 16 ч при комнатной температуре смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, используя DCM/MeOH=9/1 в качестве элюента. Сырой продукт дополнительно очищали ВЭЖХ, используя колонку Luna C-18 с градиентом элюирования: 0,1% водн.TFA/ACN=от 100/0 до 10/90 за 30 мин. Чистое соединение, указанное в заглавии, лиофилизировали из смеси 0,1% водн. TFA и воды.
Выход: 616 мг (соль с TFA)
MS-ESI: [M+H]+=499,2
ВЭЖХ: Rt=37,5 мин, Luna C-18(2), 5 мкм, 150*2 мм, детектирование в УФ=210 нм, температура колонки=40°С, расход=0,25 мл/мин, элюент фосфатный буферный раствор 50 мМ рН 2,1/вода/ACN=от 20/60/20 до 20/0/80 (об./об.), время пробега=40 мин.
Пример 6
Трет-бутил-5-амино-2-метилтио-4-(3-(2-(пиперидин-1-ил)-ацетамидо)-фенил)-тиено[2,3-d]пиримидин-6-карбоксамид
Пиперазин (2,5 г) прибавляли к раствору трет-бутил-5-амино-4-(3-(2-бромацетамидо)-фенил)-2-метилтио-тиено[2,3-d]пиримидин-6-карбоксамида (пример 1(h), 1,0 г) в CH2Cl2 (50 мл). Через 16 ч при комнатной температуре смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, используя DCM/MeOH=7/1 в качестве элюента. Сырой продукт дополнительно очищали ВЭЖХ, используя колонку Luna C-18 с градиентом элюирования: 0,1% водн.TFA/ACN=от 100/0 до 10/90 за 30 мин. Чистое соединение, указанное в заглавии, лиофилизировали из смеси 0,1% водн. TFA и воды.
Выход: 766 мг (соль с TFA)
MS-ESI: [M+H]+=514,4
ВЭЖХ: Rt=33,7 мин, Luna C-18(2), 5 мкм, 150*2 мм, детектирование в УФ=210 нм, температура колонки=40°С, расход=0,25 мл/мин, элюент фосфатный буферный раствор 50 мМ рН 2,1/вода/ACN=от 20/60/20 до 20/0/80 (об./об.), время пробега=40 мин.
Пример 7
Биологическая активность СНО-LH и СНО-FSH in vitro
LH-агонистическую активность соединений определяли на клетках яичников китайского хомяка (СНО), устойчиво трансфицированных рецепторами человека и сотрансфицированных чувствительным элементом сАМР (CRE)/промотором, направляющим экспрессию гена-репортера люциферазы светляка. Связывание лиганда с Gs-связанным LH-рецептором приводит к повышению сАМР, что, в свою очередь, индуцирует повышенную трансактивацию конструкции репортера люциферазы. Сигнал люциферазы определяли количественно с применением люминесценции. Для испытуемых соединений вычисляли значение ЕС50 (концентрация испытуемого соединения, вызывающая 50% максимального стимулирования). Для указанной цели применяли программное обеспечение GraphPad PRISM, версия 3,0 (GraphPad software Inc., SanDiego).
Подобным образом была испытана FSH-агонистическая активность соединений на клетках СНО, трансфицированных геном-репортером люциферазы и рецептором FSH человека. Результаты показаны в таблице.
Биологическая активность in vivo
Для измерения активности соединений агонистов рецептора LH/FSH изучали индуцирование овуляции у неполовозрелых мышей. В указанной пробе неполовозрелым самкам мышей вводили FSH, выделенный из мочи (Humegon 12,5 IU/животное). Примерно через 48 часов животным вводили соединение-агонист LH/FSH в дозе 50 мг/кг. Животных убивали через 24 часа после введения агониста LH/FSH и под микроскопом определяли число яйцеклеток в маточных трубах. Результаты приведены в таблице.
LHR (M)
FSHR (M)
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЛИЦИН-ЗАМЕЩЕННЫЕ ТИЕНО[2,3-D]ПИРИМИДИНЫ С ОБЪЕДИНЕННОЙ LH И FSH АГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2002 |
|
RU2294331C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОАРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ | 2001 |
|
RU2271360C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОАРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ LH АГОНИСТОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2248979C2 |
| НОВЫЕ МОДУЛЯТОРЫ РЕЦЕПТОРА GLP-1 | 2012 |
|
RU2634896C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОХИНОЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ РЕГУЛЯТОРОВ ФЕРТИЛЬНОСТИ | 2003 |
|
RU2328488C2 |
| НЕКОТОРЫЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ | 2015 |
|
RU2671494C2 |
| ПРОИЗВОДНЫЕ ДИГИДРОПИРИДИНА | 2005 |
|
RU2372337C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛЫ, АКТИВНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2013 |
|
RU2666538C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ IRAK II ТИПА И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2810338C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2769696C2 |
Настоящее изобретение относится к производным тиено[2,3-d]пиримидина общей формулы I или их фармацевтически приемлемым солям
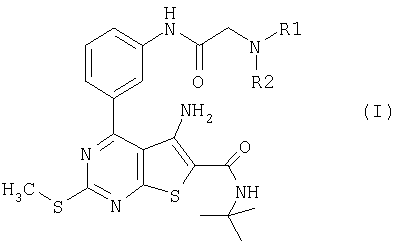
где R1 и R2 вместе с атомом азота, к которому они присоединены, образуют кольцо, имеющее от 2 до 6 атомов углерода, необязательно содержащее один или более гетероатомов, выбранных из N, О и/или S. Соединения обладают способностью активировать рецепторы как LH, так и FSH, и могут быть использованы в терапии для регулирования фертильности. Описана также фармацевтическая композиция на основе соединений формулы I. 5 н.п. ф-лы, 1 табл.
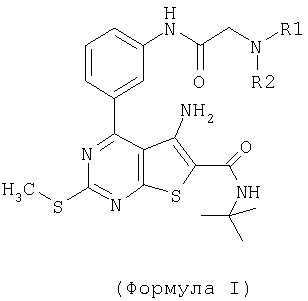
или их фармацевтически приемлемые соли, где R1 и R2 вместе с атомом азота, к которому они присоединены, образуют кольцо, имеющее от 2 до 6 атомов углерода, необязательно содержащее один или более гетероатомов, выбранных из N, О и/или S.
| WO 00/61586 А, 19.10.2000 | |||
| RU 99104796 A1, 20.01.2001. |

