ЗАЯВЛЕНИЕ, ОТНОСЯЩЕЕСЯ К СПИСКУ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Список последовательностей, относящийся к настоящей заявке предоставлен в текстовом формате вместо бумажной копии, и посредством этого введен в настоящее описание с помощью ссылки. Название текстового файла, содержащего список последовательностей, 800059_407WO_SEQUENCE_LISTING.txt. Текстовый файл имеет размер приблизительно 1 KB, был создан 12 декабря 2012 и подан электронно через EFS-Web.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ НАСТОЯЩЕЕ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям, которые связываются с рецептором глюкагонподобного пептида 1 (GLP-1), способам их получения, и способам их терапевтического и/или профилактического применения. Настоящее изобретение относится к соединениям, приспособленным для того, чтобы действовать в качестве модуляторов или потенциаторов GLP-1 рецептора, включая пептиды GLP-1 (7-36) и GLP-l(9-36), а также терапии на основе пептидов, таких как эксенатид и лираглютид.
УРОВЕНЬ ТЕХНИКИ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Рецептор глюкагонподобного пептида 1 (GLP-1R) относится к семейству B1 сопряженных с G-белком рецепторов с семью трансмембранными доменами, и его природный лиганд, являющийся агонистом, представляет собой пептидный гормон, глюкагонподобный пептид-1 (GLP-1). GLP-1 представляет собой пептидный гормон, получаемый его альтернативным ферментативным расщеплением из проглюкагона, прогормонового предшественника для GLP-1, который в значительном количестве экспрессируется в энтероэндокринных клетках кишечного тракта, альфа клетках эндокринной части поджелудочной железы (островках Лангерганса) и мозге (Kieffer T. J. and Habener, J. F. Endocrin. Rev. 20:876-913 (1999); Drucker, D. J., Endocrinology 142:521-7 (2001); Hoist, J. J., Diabetes Metab. Res. Rev. 18:430-41 (2002)). Первоначальное наблюдаемое действие GLP-1 осуществлялось на продуцирующих инсулин клетках островков, где он стимулировал зависящую от глюкозы секрецию инсулина. Впоследствии, обнаружили дополнительное противодиабетическое действие GLP-1, включая стимуляцию роста и ингибирование апоптоза панкреатических бета-клеток (Drucker, D. J., Endocrinology 144:5145-8 (2003); Holz, G. G. and Chepurny O. G., Curr. Med. Chem. 10:2471-83 (2003); List, J. F. and Habener, J. F., Am. J. Physiol. Endocrinol. Metab. 286: E875-81 (2004)).
После активации GLP-1 рецепторы связываются с α-субъединицей G белка, с последующей активацией аденилатциклазы и повышением цАМФ концентрации, посредством этого потенцируя зависящую от глюкозы секрецию инсулина. Следовательно, GLP-1 представляет собой привлекательную терапевтическую мишень для снижения глюкозы в крови и сохранения β-клеток поджелудочной железы пациентов с диабетом. Глюкагон применяют в течение десятилетий в медицинской практике для лечения диабета, и несколько глюкагонподобных пептидов разрабатывают для различных терапевтических показаний. GLP-1 аналоги и производные разрабатывают для лечения пациентов, страдающих от диабета.
СУЩНОСТЬ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, приспособленным для того, чтобы действовать в качестве потенциаторов или модуляторов GLP-1 рецептора; способам их получения и способам их применения, таким как в лечении патологических состояний, опосредованных GLP-1 рецепторной активацией, или когда медицински показано модулирование или потенцирование GLP-1 рецептора.
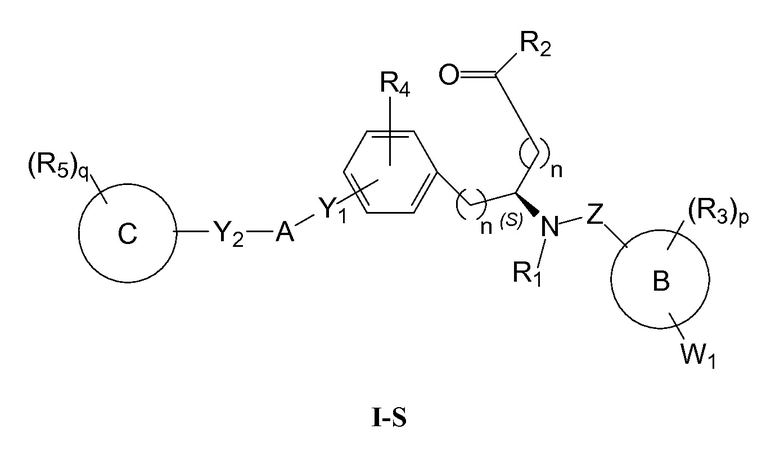
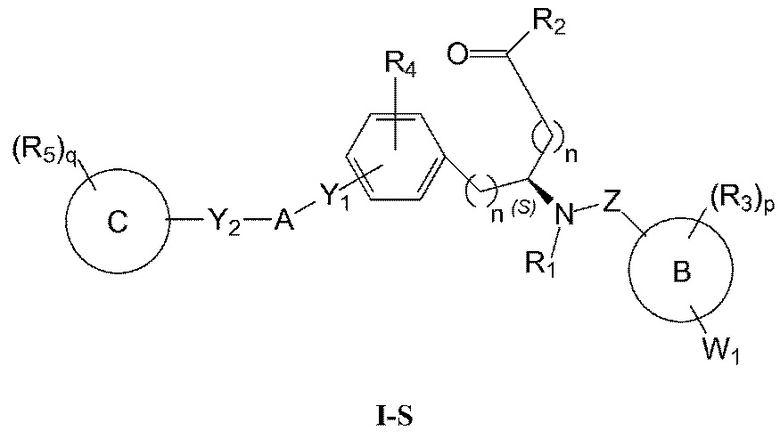
Определенные варианты осуществления настоящего изобретения включают соединение, имеющее структуру формулы I-R или I-S, или его фармацевтически приемлемый изомер, энантиомер, рацемат, соль, изотоп, пролекарство, гидрат или сольват:
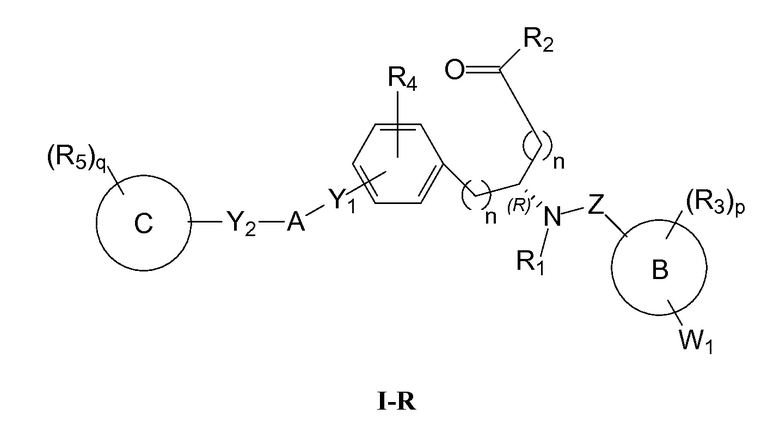
где
A представляет собой 5-, 6- или 7-членный гетероциклил, содержащий один, два или три гетероатома, где каждый данный гетероатом независимо выбран из O, N, и S, и где любой атом кольца такого гетероциклила может быть необязательно замещен одним или более R4;
B представляет собой арил, аралкил, гетероциклил или гетероциклилалкил;
C представляет собой арил, арилалкил, гетероциклил или гетероциклилалкил;
обе Y1 и Y2 являются отсутствующими, или одна из Y1 или Y2 представляет собой -NH- или -O-, и другая Y1 или Y2 является отсутствующей;
Z представляет собой -C(O)- или -S(O)2-;
каждый R1 независимо представляет собой H или C1-4 алкил;
R2 представляет собой -OH, -O-R8, -N(R1)-SO2-R8, -NR41R42, -N(R1)-(CRaRb)m-COOH, -N(R1)-(CRaRb)m-CO-N(R1)-гетероциклил, -N(R1)-(CRaRb)m-CO-N(R1)(R7) или -N(R1)-гетероциклил;
каждый R3 и R4 независимо представляет собой H, галоген, алкил, алкил, замещенный R31, алкокси, галогеналкил, пергалогеналкил, галогеналкокси, пергалогеналкокси, арил, гетероциклил, -OH, -OR8, -CN, -NO2, -NR1R8, -C(O)R8, -C(O)NR1R8, -NR1C(O)R8, -SR8, -S(O)R8, -S(O)2R8, -OS(O)2R8, -S(O)2NR1R8, -NR1S(O)2R8, -(CRaRb)mNR1R8, -(CRaRb)mO(CRaRb)mR8, -(CRaRb)mNR1(CRaRb)mR8 или -(CRaRb)mNR1(CRaRb)mCOOH; или любые две R3 или R4 группы при одном атоме углерода, взятые вместе, образуют оксо;
каждый R31 независимо представляет собой H, галоген, гидроксил, -NR41R42 или алкокси;
каждый R40 независимо представляет собой H или алкил;
каждый R41 и R42 независимо представляют собой R40 или -(CH2)n-COO-R40, -C(O)-R40, арил, гетероарил, или две, взятые вместе с N атомом, с которым они соединены, могут образовывать 3- - 7-членный гетероциклил;
W1 является отсутствующей или -L1-(CRaRb)m-L1-R6;
каждый L1 независимо является, от ближнего к дальнему концу структуры формулы I-R или I-S, отсутствующим, -C(O)O-, -S(O)2-, -S-, -N(R1)-C(O)-N(R1)-, -N(R1)-C(O)-O-, -C(O)- или -S(O)2-NR;
каждый Ra и Rb независимо представляет собой H, алкил, алкокси, арил, арилалкил, гетероциклил или гетероциклилалкил, любой из данных алкила, алкокси, арила, арилалкила, гетероциклила или гетероциклилалкила может быть необязательно (однократно или несколько раз) замещен R7 или -(CH2)mC(O)OR40, -(CH2)mOR40, -(CH2)mSR40, -(CH2)mNR41R42, -(CH2)mC(O)NR41R42; или любые две Ra и Rb, взятые вместе с углеродом, с которым они соединены, образуют циклоалкил или гетероциклил; или R1 и одна из Ra или Rb, взятые вместе, образуют гетероциклил;
R5 представляет собой R7, -(CH2)m-L2-(CH2)m-R7 или -(-L3-(CRaRb)r-)s-L3-R7;
R6 представляет собой H, алкил, циклоалкил, арил, гетероарил, гетероциклил, гетероциклоалкил, любой из которых может быть необязательно однократно или несколько раз замещен R7 или -(CH2)m-L2-(CH2)m-R7;
R7 представляет собой H, галоген, алкил, галогеналкил, пергалогеналкил, алкокси, -OH, -OR8, -CN, -NR1R8, -(CRaRb)mO(CRaRb)mR8, -NR1(CRaRb)mR8, -C(O)R8, -NR1(CRaRb)mCOOH, -NR1C(O)R8, -C(O)NR1R8, -SR8, -S(O)R8, -S(O)2R8, -S(O)2NR1R8, -NR1S(O)2R8; или кольцевую группу, выбранную из циклоалкила, арила, арилалкила, гетероциклила или гетероциклилалкила, где данная кольцевая группа необязательно однократно или несколько раз замещена галогеном, -OH, -CN, алкилом, алкокси, галогеналкилом или пергалогеналкилом;
каждый R8 независимо представляет собой H, алкил, циклоалкил или арил;
L2 независимо является, от ближнего к дальнему концу структуры формулы I-R или I-S, отсутствующей, -O-, -OC(O)-, -NR1-, -C(O)NR1-, -N(R1)-C(O)-, -S(O)2-, -C(O)- или -S(O)2-N(R1)-;
каждый L3 независимо является отсутствующей, -O- или -N(R1)-
каждое m независимо равно 0, 1, 2, 3, 4, 5 или 6;
каждое n независимо равно 0 или 1 или 2;
p равно 0, 1, 2 или 3;
q равно 0, 1, 2 или 3;
каждое r независимо равно 2, 3, или 4; и
каждое s независимо равно 1, 2, 3, или 4.
В определенных вариантах осуществления обеспечивают фармацевтическую композицию, содержащую соединение настоящего изобретения вместе, по меньшей мере, с одним фармацевтически приемлемым носителем, разбавителем или вспомогательным веществом.
В определенных вариантах осуществления обеспечивают способ применения соединения настоящего изобретения, включающий получение лекарственного препарата.
В определенных вариантах осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей соединение настоящего изобретения и второй лекарственный препарат. В различных данных вариантах осуществления второй лекарственный препарат представляет собой агонист или модулятор глюкагонового рецептора, GIP рецептора, GLP-2 рецептора или PTH рецептора или рецептора глюкагонподобного пептида 1 (GLP-1). В различных данных вариантах осуществления второй лекарственный препарат представляет собой эксенатид, лираглутид, таспоглутид, альбиглутид или ликсисенатид или другой регулирующий инсулин пептид. В различных данных вариантах осуществления второй лекарственный препарат представляет собой DPPIV ингибитор. В различных данных вариантах осуществления второй лекарственный препарат медицински показан для лечения диабета II типа.
В определенных вариантах осуществления обеспечивают способ активации, потенцирования или агонизма GLP-1 рецептора, включающий контакт рецептора с соединением, фармацевтической композицией или фармацевтической комбинацией настоящего изобретения.
В определенных вариантах осуществления обеспечивают способ лечения патологического состояния у субъекта, для которого медицински показана активация, потенцирование или агонизм GLP-1 рецептора, где данный способ включает введение данному субъекту соединения, фармацевтической композиции или фармацевтической комбинации настоящего изобретения. В различных данных вариантах осуществления медицински показана селективная активация, потенцирование или агонизм GLP-1 рецептора. В различных данных вариантах осуществления патологическое состояние включает диабет I типа, диабет II типа, гестационный диабет, ожирение, повышенный аппетит, недостаточное насыщение или метаболическое расстройство.
В определенных вариантах осуществления настоящее изобретение относится к способам получения определенных соединений настоящего изобретения. В определенных других вариантах осуществления настоящее изобретение относится к определенным промежуточным соединениям, связанным с данными способами получения.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Определенные варианты осуществления включают соединение, имеющее хиральную структуру формулы I-R или I-S (с хиральностью, как показано), или его фармацевтически приемлемый изомер, энантиомер, рацемат, соль, изотоп, пролекарство, гидрат или сольват:
Определенные варианты осуществления настоящего изобретения включают соединение, имеющее структуру формулы I-R или I-S, или его фармацевтически приемлемый изомер, энантиомер, рацемат, соль, изотоп, пролекарство, гидрат или сольват:
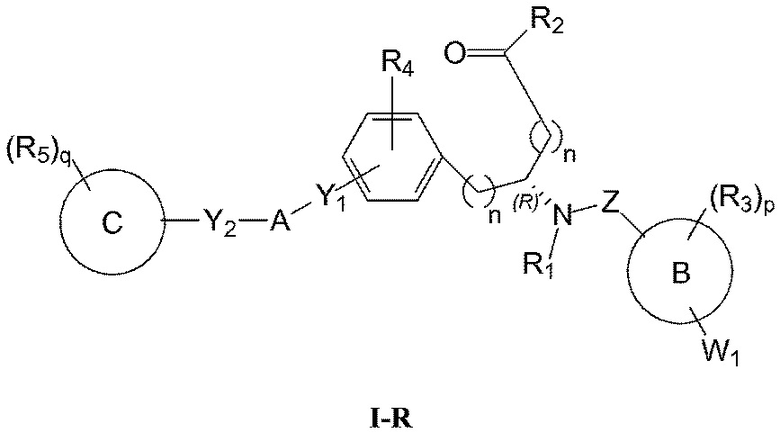
где A, B, C, Y1, Y2, Z, R1, R2, R3, R4, R5, W1, n, p и q представляют собой, как определено выше.
В определенных вариантах осуществления соединения имеют структуру формулы I-R или ее фармацевтически приемлемый изомер, энантиомер, соль, изотоп, пролекарство, гидрат или сольват. В других вариантах осуществления соединения имеют структуру формулы I-S или ее фармацевтически приемлемый изомер, энантиомер, соль, изотоп, пролекарство, гидрат или сольват.
В определенных вариантах осуществления, соединения могут быть по существу энантиомерно чистыми.
В определенных вариантах осуществления настоящее изобретение относится к соединению формулы I-R и/или формулы I-S, где Y1 и Y2 являются отсутствующими, Z представляет собой -C(O)-, и A представляет собой 5- или 6-членную гетероарильную группу. Репрезентативные соединения данного варианта осуществления включают соединения следующих структур (где 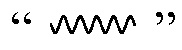 представляет собой одну из или обе R и S формы соединения):
представляет собой одну из или обе R и S формы соединения):
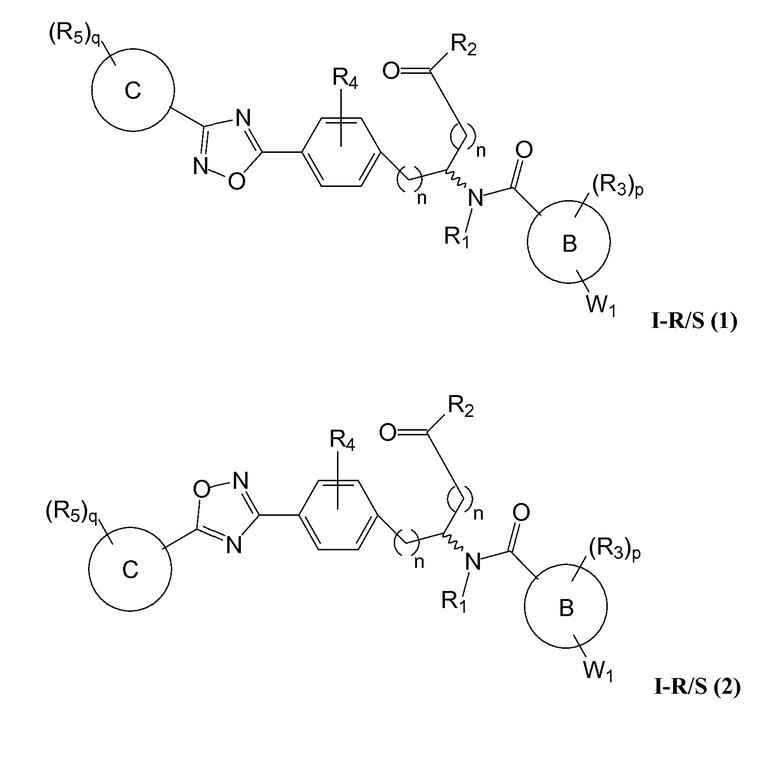
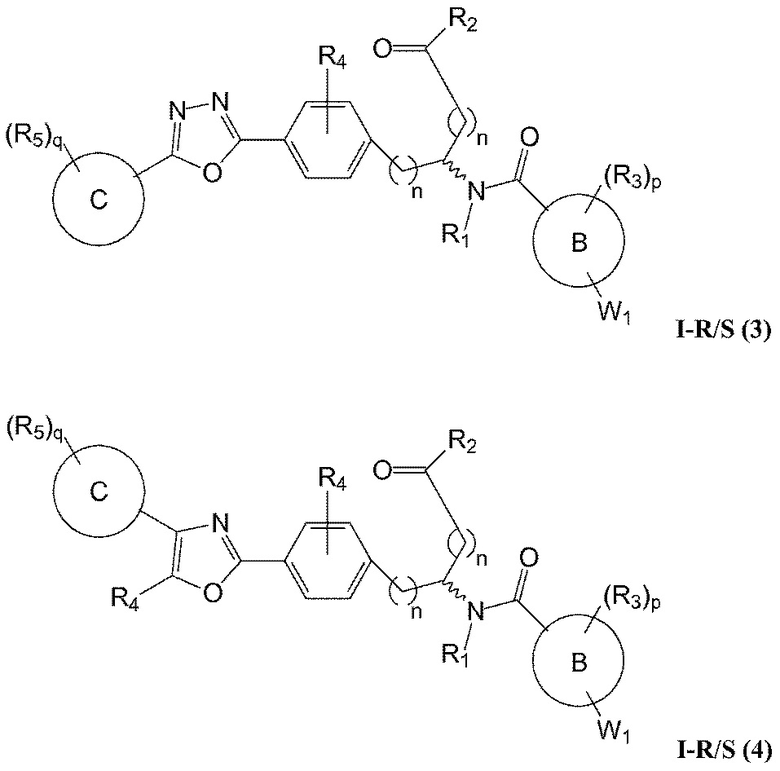

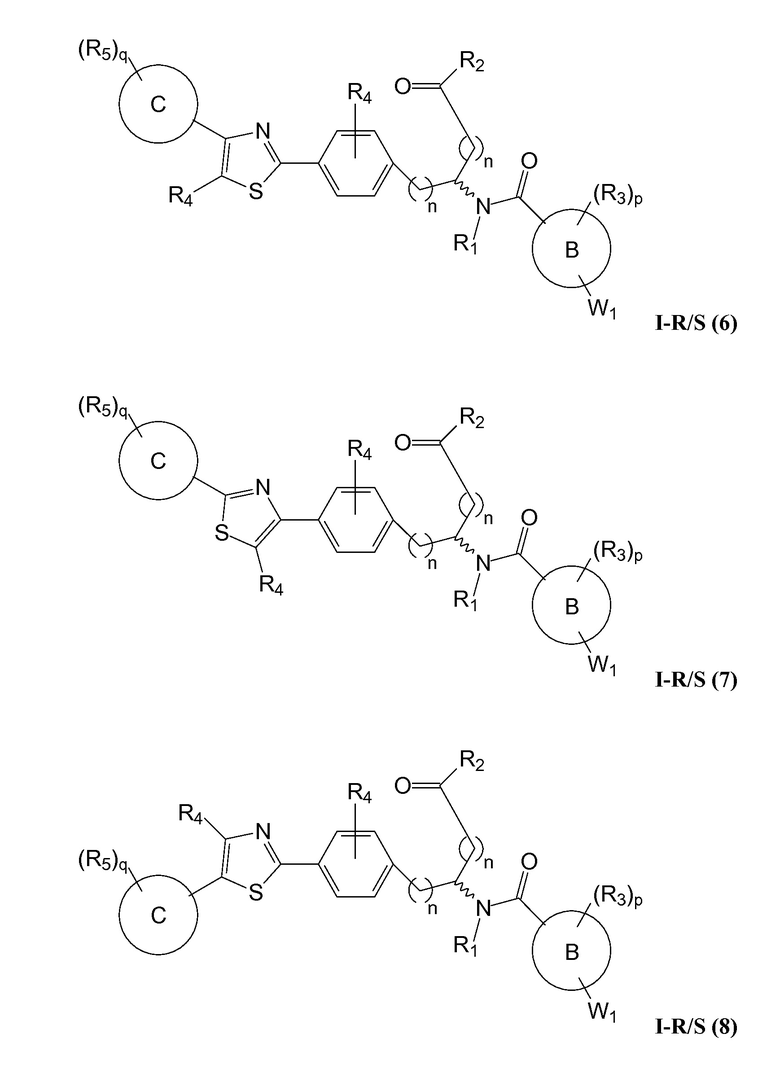
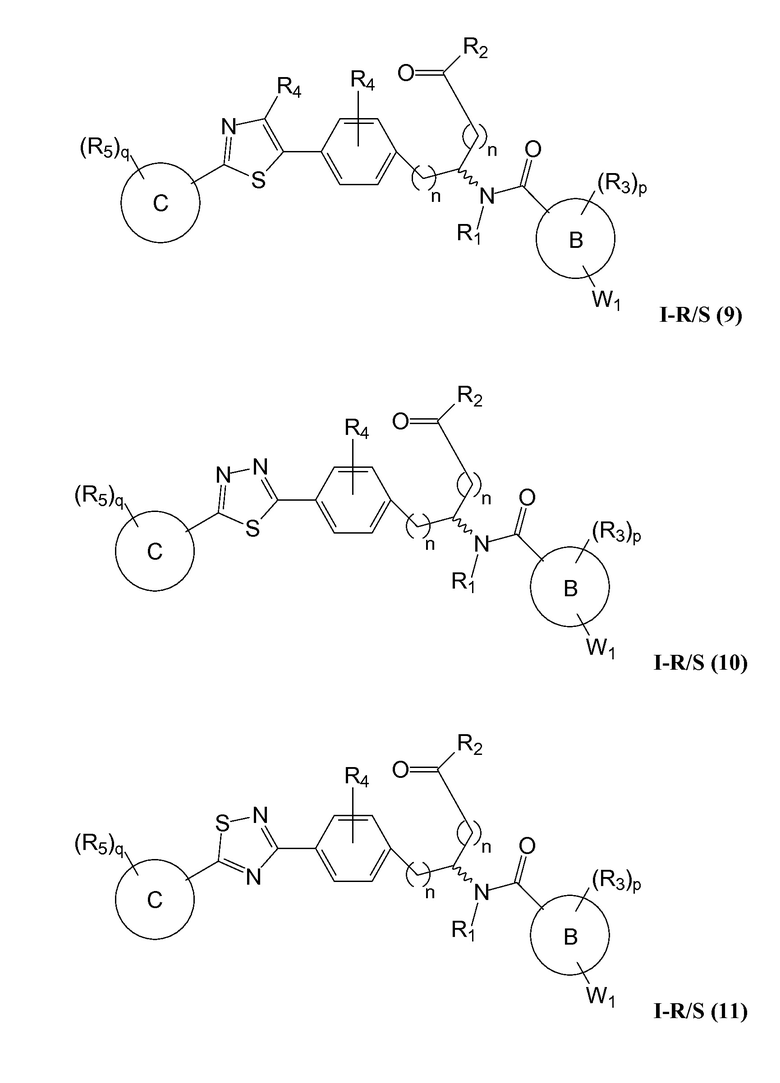
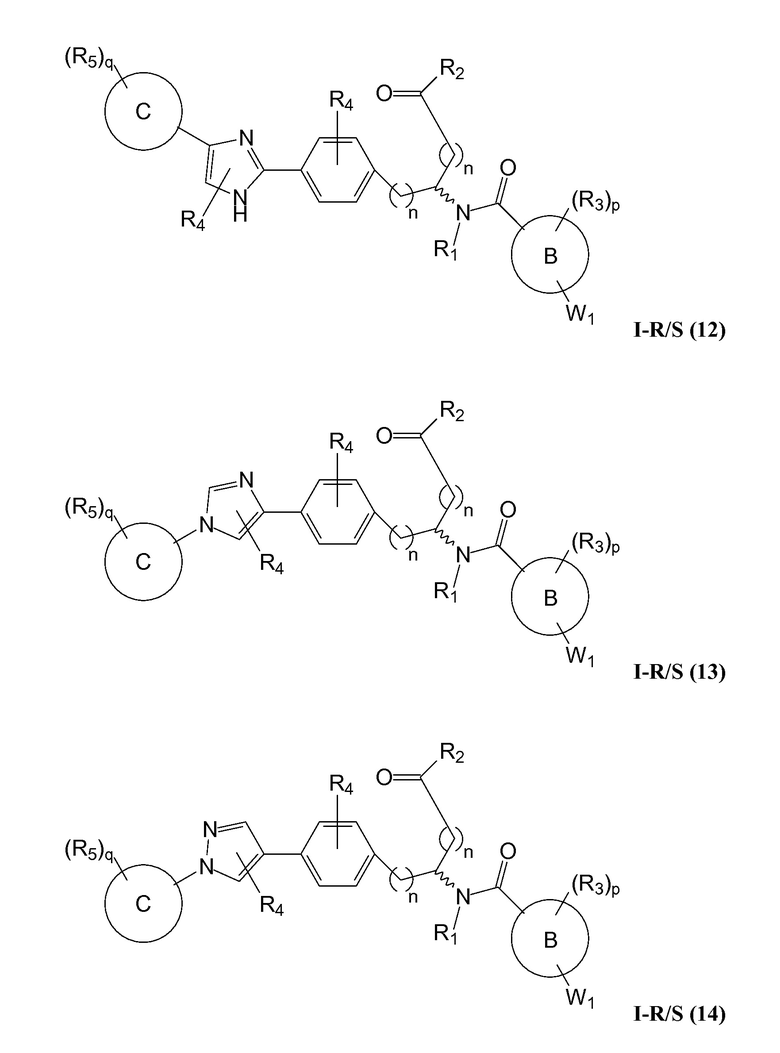
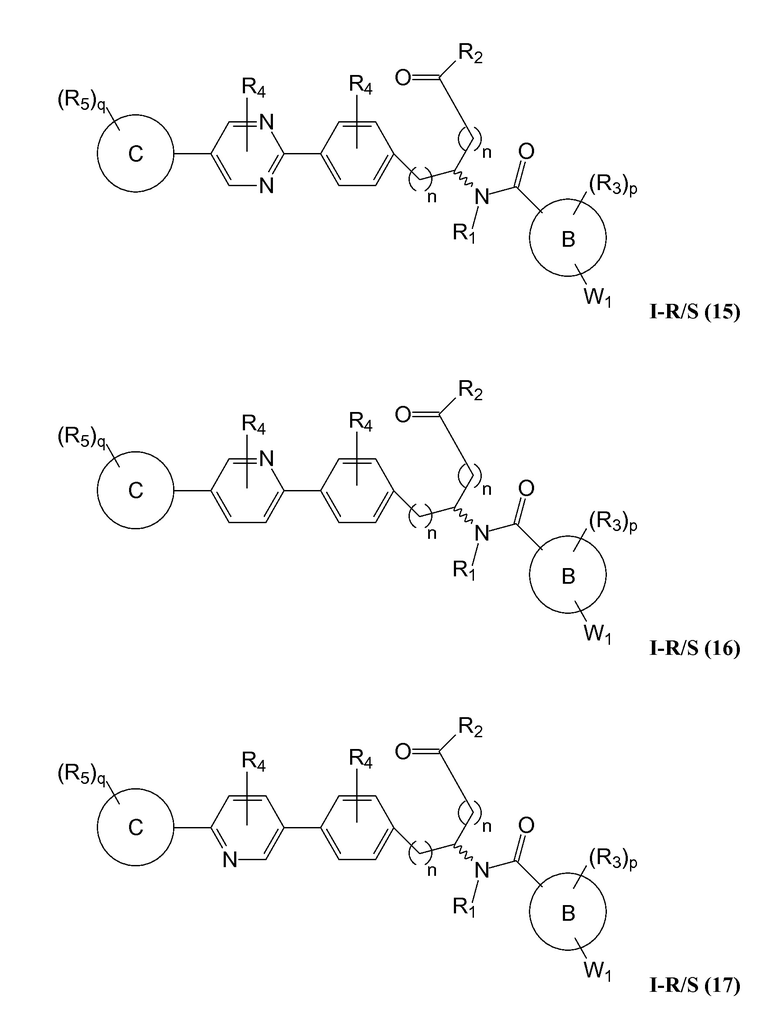
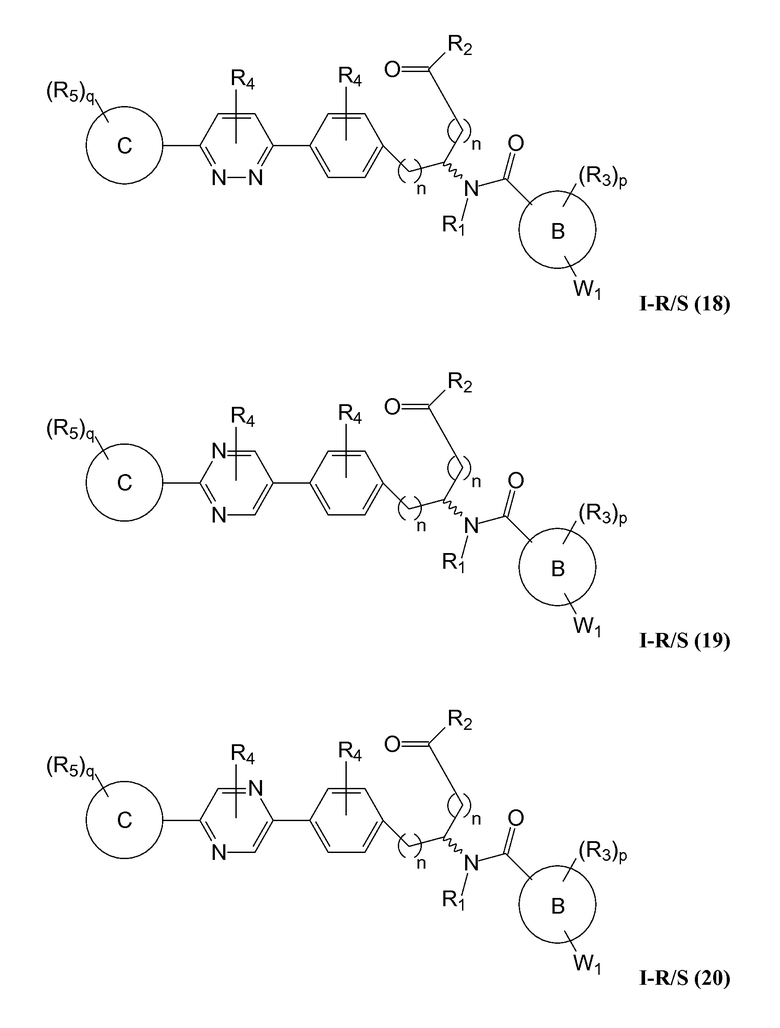
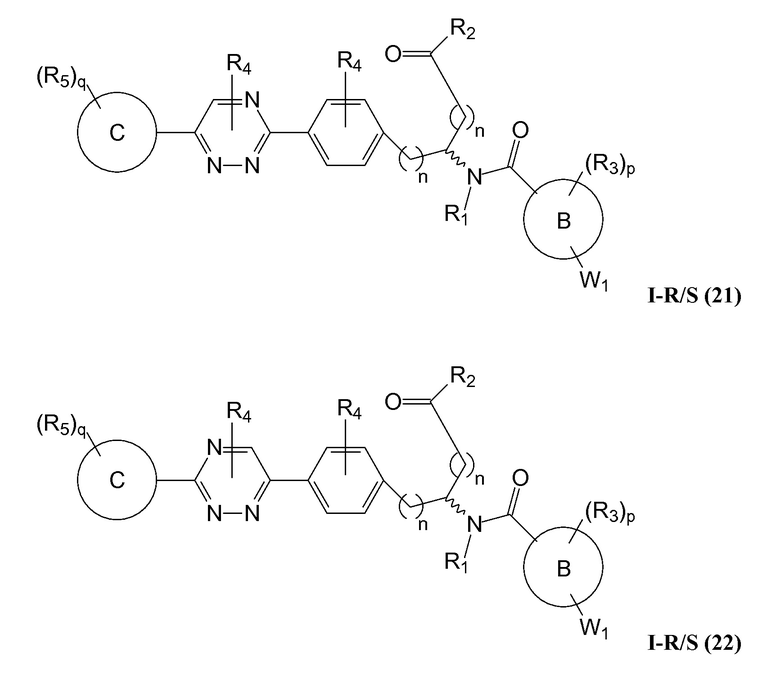
В определенных вариантах осуществления настоящее изобретение относится к соединению, где Y1 и Y2 являются отсутствующими, Z представляет собой -C(O)-, и A представляет собой 5-, 6- или 7-членную неароматическую гетероциклильную группу. Репрезентативные соединения данного варианта осуществления включают соединения следующих структур (где представляет собой одну из или обе R и S формы соединения):
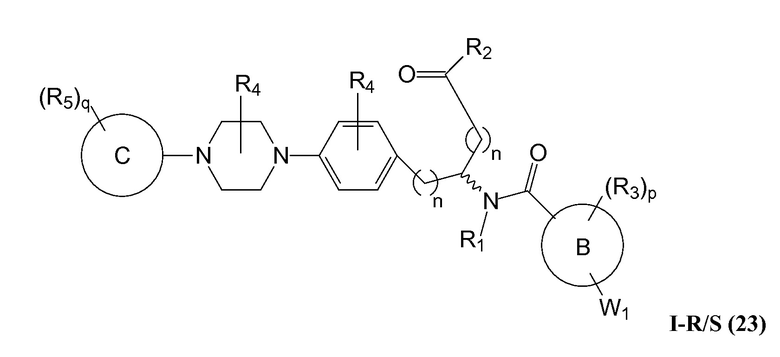
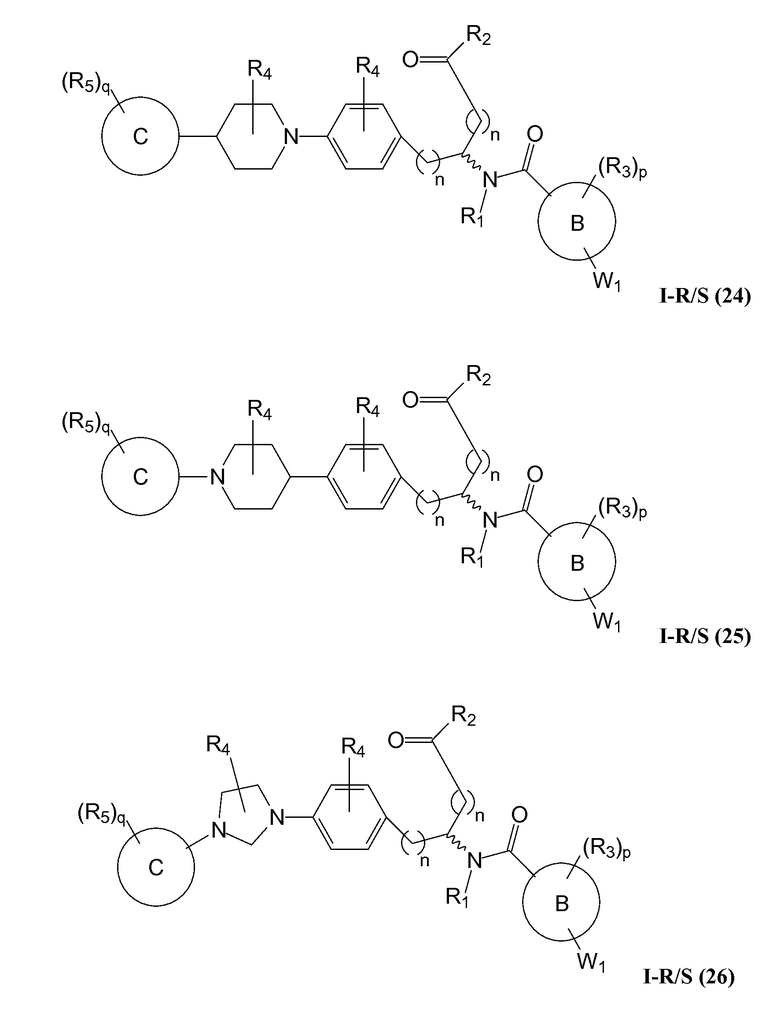
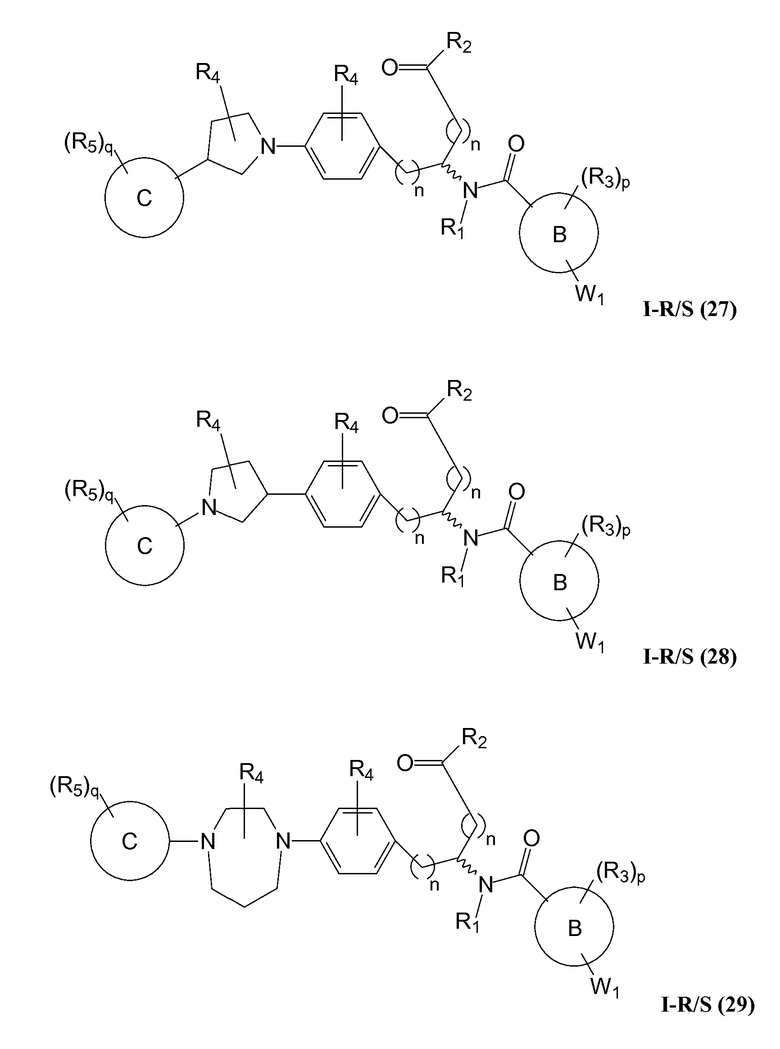
В определенных вариантах осуществления настоящее изобретение относится к соединениям каждой из структур I-R/S(1)-(29), где R4 фенильной группы представляет собой H.
В определенных вариантах осуществления настоящее изобретение относится к соединениям каждой из структур I-R/S(1)-(29), где A группа (т.е., 5-, 6- или 7-членный гетероциклил) не замещена R4, или замещена R4, когда R4 представляет собой алкил, галогеналкил, алкокси, -NR41R42, когда R41 и R42 независимо представляют собой водород или алкил, или замещена двумя R4 группами, которые, взятые вместе, образуют оксо.
В определенных вариантах осуществления настоящее изобретение относится к соединению формулы I-R и/или формулы I-S, где Y1 и Y2 являются отсутствующими, Z представляет собой -C(O)-, и A представляет собой арил. Репрезентативные соединения данного варианта осуществления включают соединения следующих структур (где представляет собой одну из или обе R и S формы соединения):
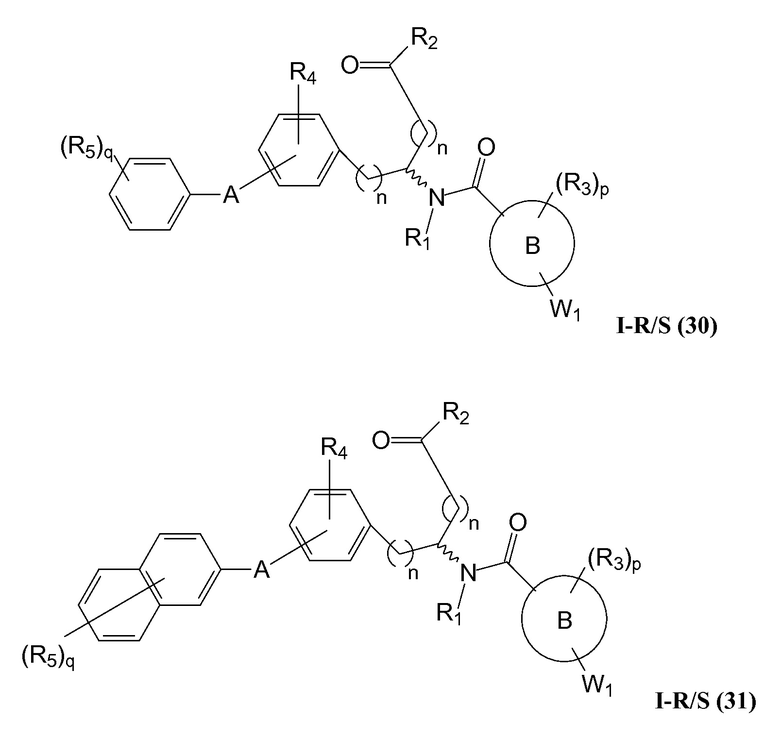
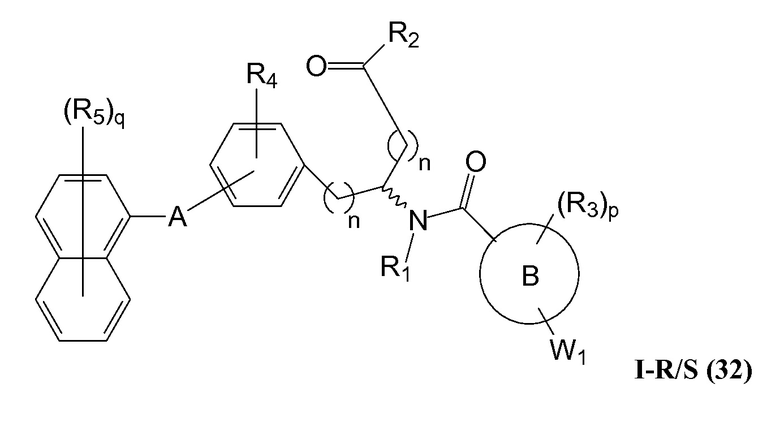
В определенных вариантах осуществления настоящее изобретение относится к соединениям каждой из структур I-R/S(30)-(32), где q равно нулю.
В определенных вариантах осуществления настоящее изобретение относится к соединениям каждой из структур I-R/S(30)-(32), где q равно одному, двум или трем.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(30), где q равно одному, и R5 представляет собой -(CH2)m-L2-(CH2)m-R7 или -(-L3-(CRaRb)r-)s-L3-R7. Репрезентативные соединения данного варианта осуществления включают соединения следующей структуры (где " представляет собой одну из или обе R и S формы соединения):
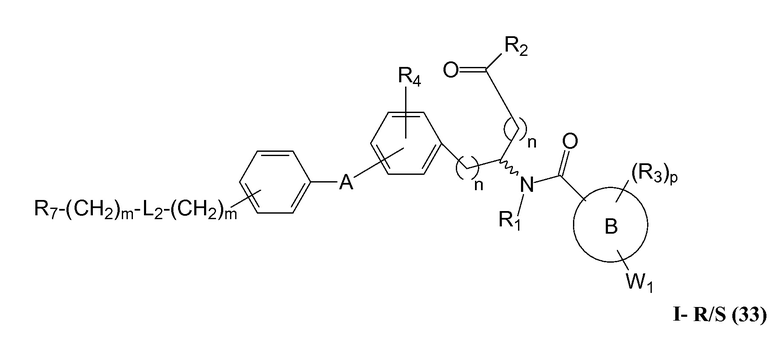
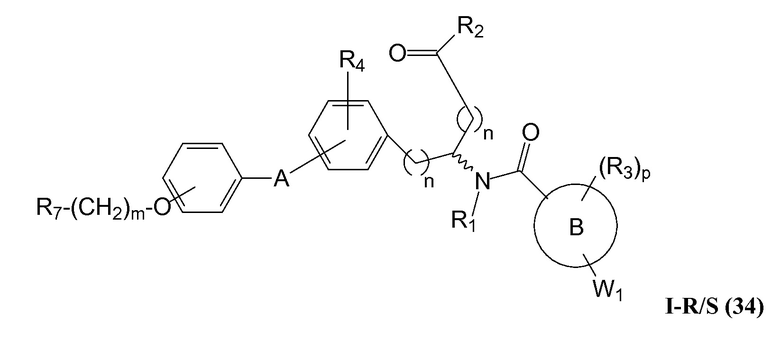
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(33), где R7 представляет собой H или алкил, и L2 представляет собой O. Репрезентативные соединения данного варианта осуществления включают соединения следующей структуры (где " представляет собой одну из или обе R и S формы соединения):
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(30), где R5 представляет собой R7. Репрезентативные соединения данного варианта осуществления включают соединения следующей структуры (где представляет собой одну из или обе R и S формы соединения):
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(35), где R7 представляет собой галоген, алкил, галогеналкил, пергалогеналкил, алкокси, -OH, -OR8, -CN, -R1R8, -(CRaRb)mO(CRaRb)mR8, -NR1(CRaRb)mR8, -C(O)R8, -NR1(CRaRb)mCOOH, -NR1C(O)R8, -C(O)NR1R8, -SR8, -S(O)R8, -S(O)2R8, -S(O)2NR1R8 или -NR1S(O)2R8.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(35), где R7 представляет собой кольцевую группу, выбранную из циклоалкила, арила, арилалкила, гетероциклила или гетероциклилалкила, где каждая кольцевая группа необязательно (однократно или несколько раз) замещена галогеном, -OH, -CN, алкилом, алкокси, галогеналкилом или пергалогеналкилом.
В определенных вариантах осуществления настоящее изобретение относится к соединению формулы I-R и/или формулы I-S, где Y1 и Y2 являются отсутствующими, Z представляет собой -C(O)-, и A представляет собой гетероциклил. Репрезентативные соединения данного варианта осуществления включают соединения следующих структур (где представляет собой одну из или обе R и S формы соединения):

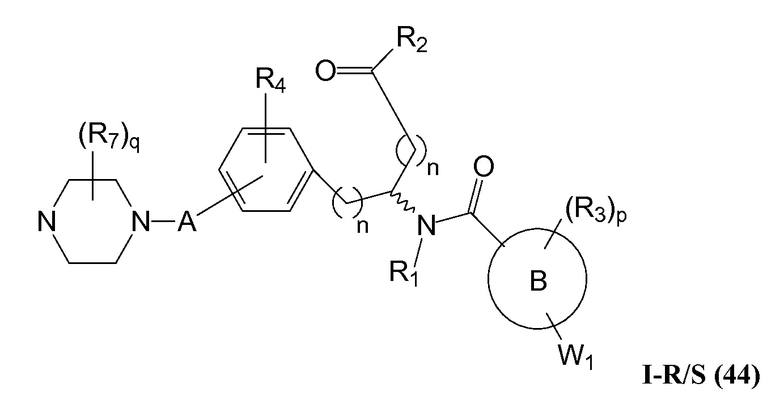
В определенных вариантах осуществления настоящее изобретение относится к соединениям каждой из структур I-R/S(36)-(44), где R7 представляет собой галоген, алкил, галогеналкил, пергалогеналкил, алкокси, -OH, -OR8, -CN, -NR1R8, -(CRaRb)mO(CRaRb)mR8, -NR1(CRaRb)mR8, -C(O)R8, -NR1(CRaRb)mCOOH, -NR1C(O)R8, -C(O)NR1R8, -SR8, -S(O)R8, -S(O)2R8, -S(O)2NR1R8 или -NR1S(O)2R8.
В определенных вариантах осуществления настоящее изобретение относится к соединениям каждой из структур I-R/S(36)-(44), где R7 представляет собой кольцевую группу, выбранную из циклоалкила, арила, арилалкила, гетероциклила или гетероциклилалкила, где данная кольцевая группа необязательно (однократно или несколько раз) замещена галогеном, -OH, -CN, алкилом, алкокси, галогеналкилом или пергалогеналкилом.
В определенных вариантах осуществления настоящее изобретение относится к соединению формулы I-R и/или формулы I-S, где Y1 и Y2 являются отсутствующими, Z представляет собой -C(O)-, и B представляет собой арил или арилалкил. Репрезентативные соединения данного варианта осуществления включают соединения следующих структур (где представляет собой одну из или обе R и S формы соединения):
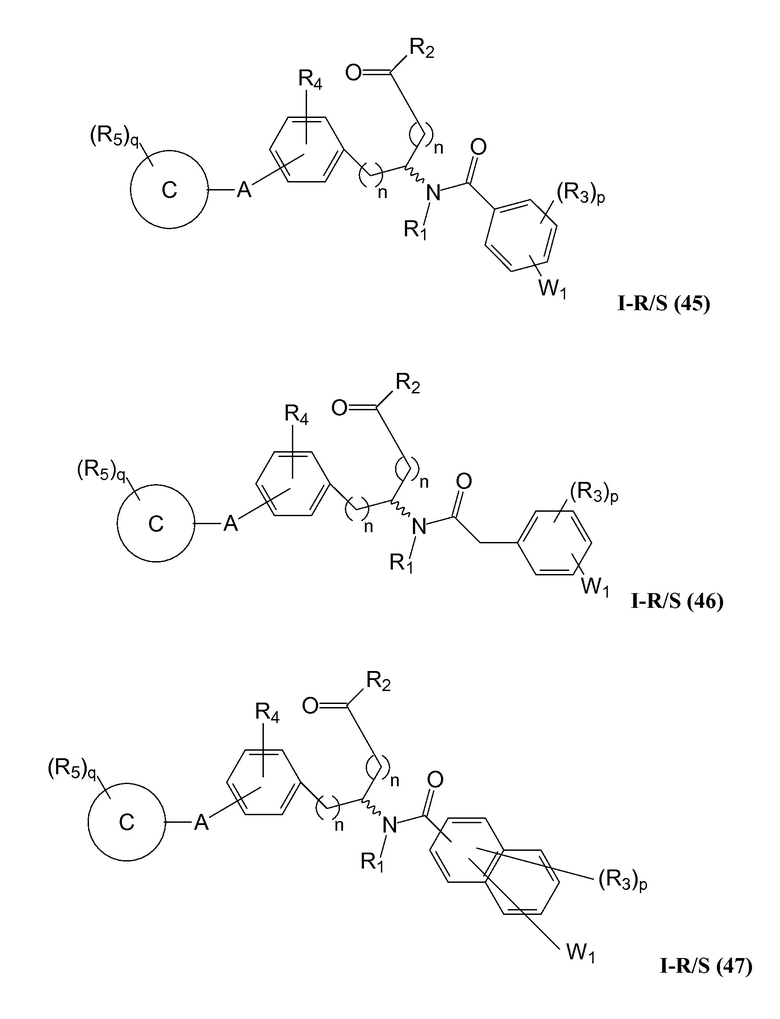
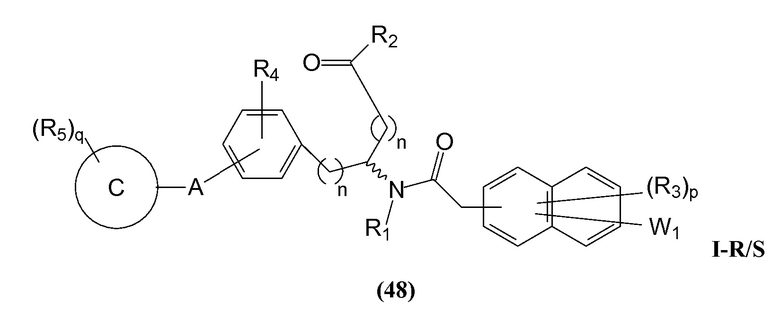
В определенных вариантах осуществления настоящее изобретение относится к соединениям каждой из структур I-R/S (45)-(48), где W1 является отсутствующей.
Репрезентативные соединения данного варианта осуществления включают соединения следующей структуры (где представляет собой одну из или обе R и S формы соединения):
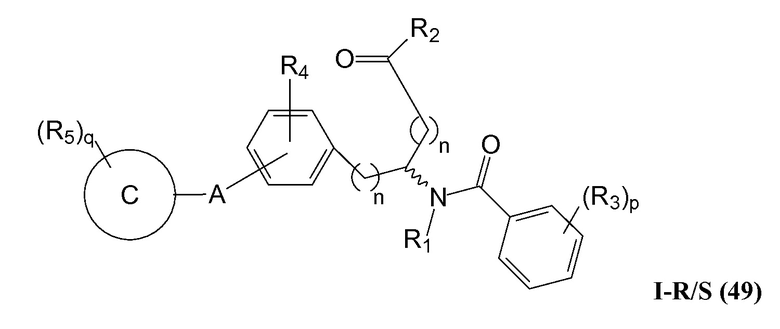
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(49), где R3 представляет собой галоген, алкил, алкокси, галогеналкил, пергалогеналкил, галогеналкокси, пергалогеналкокси, -OH, -OR8, -CN, -NR1R8, -C(O)R8, -C(O)NR1R8, -NR1C(O)R8, -SR8, -S(O)R8, -S(O)2R8, -OS(O)2R8, -S(O)2NR1R8, -NR1S(O)2R8, -(CRaRb)mNR1R8 или -(CRaRb)mO(CRaRb)mR8.
В определенных вариантах осуществления настоящее изобретение относится к соединениям каждой из структур I-R/S (45)-(49), где R3 представляет собой алкил.
В определенных вариантах осуществления настоящее изобретение относится к соединению формулы I-R и/или формулы I-S, где Y1 и Y2 являются отсутствующими, Z представляет собой -C(O)-, и B представляет собой гетероциклил или гетероциклилалкил. Репрезентативные соединения данного варианта осуществления включают соединения следующих структур (где представляет собой одну из или обе R и S формы соединения):

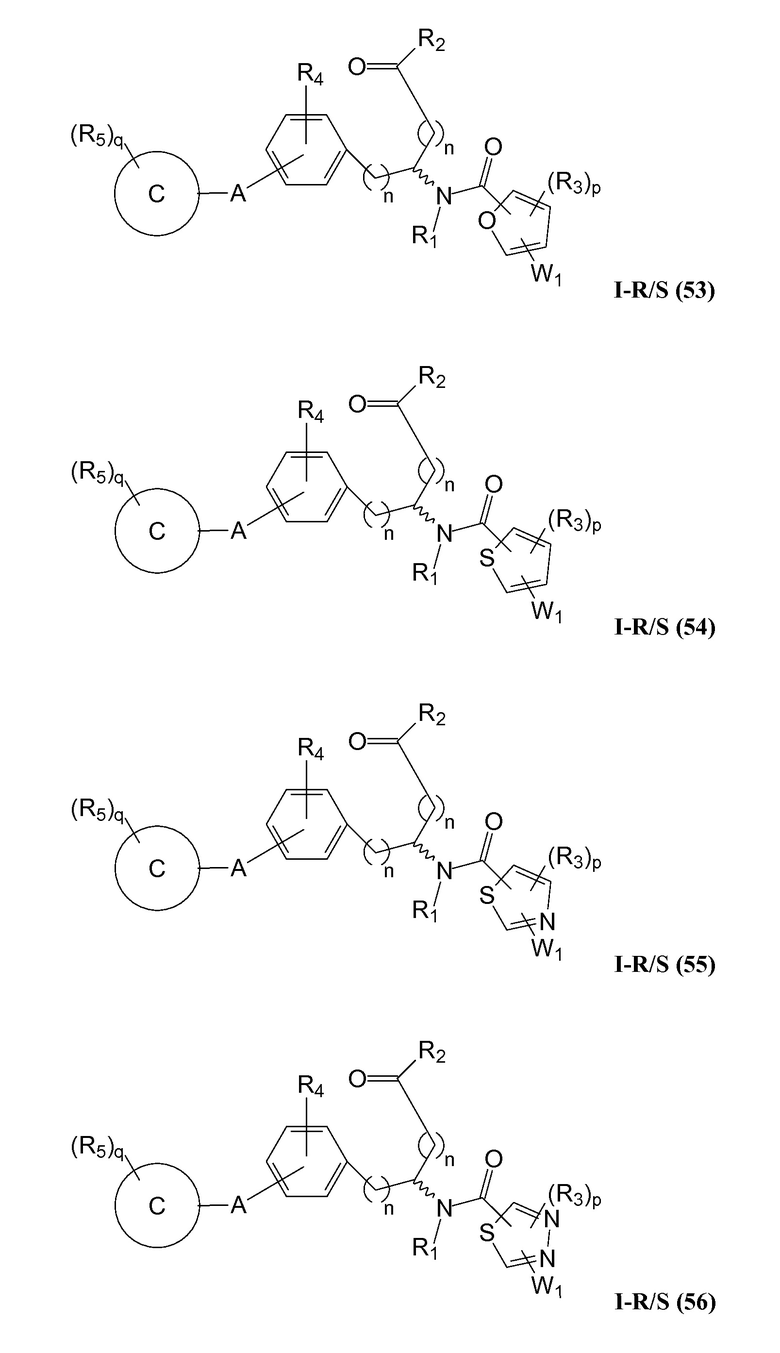
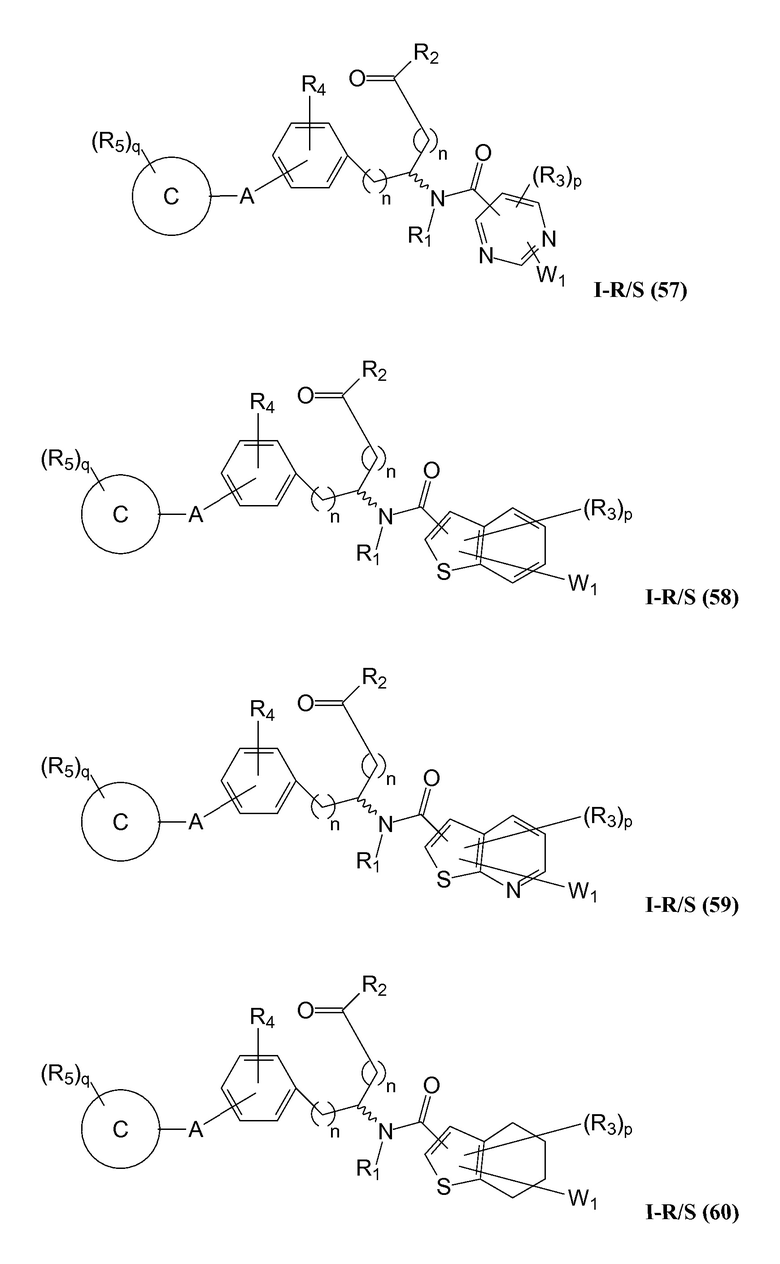
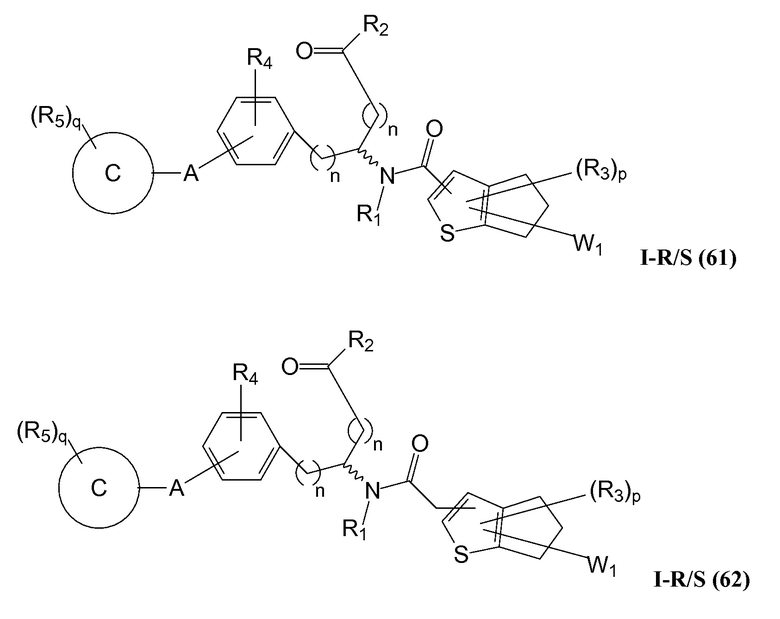
В определенных вариантах осуществления настоящее изобретение относится к соединениям каждой из структур I-R/S(50)-(62), где W1 является отсутствующей.
В определенных вариантах осуществления настоящее изобретение относится к соединениям каждой из структур I-R/S(50)-(62), где W1 является отсутствующей, и R3 представляет собой галоген, алкил, алкокси, галогеналкил, пергалогеналкил, галогеналкокси, пергалогеналкокси, -OH, -OR8, -CN, -NR1R8, -C(O)R8, -C(O)NR1R8, -NR1C(O)R8, -SR8, -S(O)R8, -S(O)2R8, -OS(O)2R8, -S(O)2NR1R8, -NR1S(O)2R8, -(CRaRb)mNR1R8 или -(CRaRb)mO(CRaRb)mR8.
В определенных вариантах осуществления настоящее изобретение относится к соединениям каждой из структур I-R/S(50)-(62), где W1 является отсутствующей, p равно 1, и R3 представляет собой алкил.
В определенных вариантах осуществления настоящее изобретение относится к соединениям каждой из структур I-R/S(1)-(62), где R2 представляет собой -OH, -N(R1)-(CRaRb)m-COOH или -N(R1)-SO2-R8; где R1 представляет собой H; где Ra и Rb независимо представляют собой H, алкил, алкокси, -(CH2)mC(O)NR41R42, -(CH2)mC(O)OR40, -(CH2)mNR41R42, -(CH2)mSR40, -N(R1)-гетероциклил, арил, необязательно замещенный R7, или где R1 и один из Ra или Rb, взятые вместе, образуют гетероциклил; R8 представляет собой алкил; и m равно 1 или 2.
В определенных вариантах осуществления настоящее изобретение относится к соединениям следующих структур (где представляет собой одну из или обе R и S формы соединения):
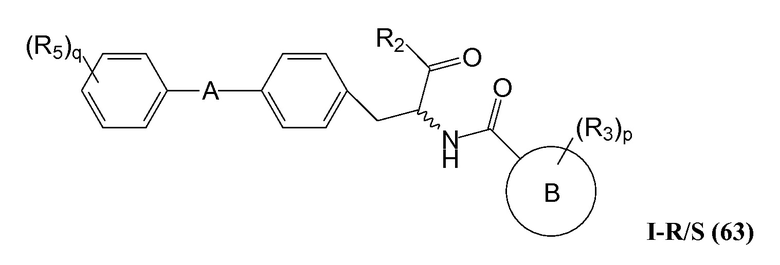
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где A представляет собой 5-членный гетероарил.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где A представляет собой 6-членный гетероарил.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где A представляет собой 6-членный гетероарил, содержащий один или два атома азота.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где A представляет собой пиримидинил.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где A представляет собой пиридинил.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где B представляет собой арил.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где B представляет собой фенил.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где B представляет собой гетероарил.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где B представляет собой тиофенил.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где R2 представляет собой -OH.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где R2 представляет собой -NH(CRaRb)mCOOH.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где R2 представляет собой -NHSO2R8.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где R2 представляет собой -NHCH2COOH.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где R2 представляет собой -NH(CHRb)COOH, где Rb представляет собой алкил, -(CH2)mOR40, -(CH2)mSR40, -(CH2)mC(O)OR40, -(CH2)mNR41R42 или -(CH2)mC(O)NR41R42.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где R2 представляет собой -NH(CRaRb)mCOOH, где Ra и Rb независимо представляют собой H, алкил, -(CH2)mOR40, -(CH2)mSR40, -(CH2)mC(O)OR40, -(CH2)mNR41R42 или -(CH2)mC(O)NR41R42.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где R2 представляет собой -NR1(CHRb)COOH, где R1 и Rb, взятые вместе, образуют гетероциклил.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где R2 представляет собой -NR1(CRaRb)mCOOH, где R1 и один из Rb, взятые вместе, образуют гетероциклил.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где любые две Ra и Rb, взятые вместе с углеродом, с которым они соединены, образуют циклоалкил.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где R2 представляет собой -NH(CRaRb)mCOOH, где один из Ra и Rb представляет собой H, и другой Ra и Rb представляет собой арил, замещенный R7.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где p равно 1 или 2, и каждый R3 независимо представляет собой алкил, алкокси, -OH, пергалогеналкил или -C(O)R8.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где p равно 1, и каждый R3 представляет собой алкил.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где q равно 1, и R5 представляет собой -(CH2)m-L2-(CH2)m-R7.
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(63), где q равно 1, и R5 представляет собой алкокси.
В определенных вариантах осуществления настоящее изобретение относится к соединению формулы I-R и/или формулы I-S, где Y1 и Y2 являются отсутствующими, и Z представляет собой -S(O)2-. Репрезентативные соединения данного варианта осуществления включают соединения следующих структур (где представляет собой одну из или обе R и S формы соединения):
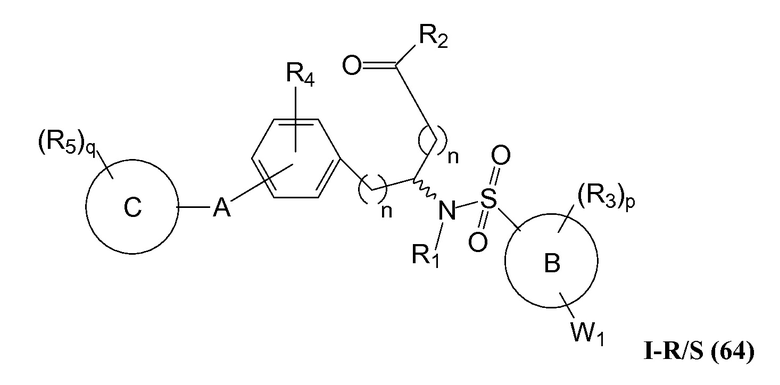
В определенных вариантах осуществления настоящее изобретение относится к соединениям структуры I-R/S(64), где A представляет собой пиримидинил, B представляет собой фенил, и C представляет собой фенил. Репрезентативные соединения данного варианта осуществления включают соединения следующей структуры (где представляет собой одну из или обе R и S формы соединения):
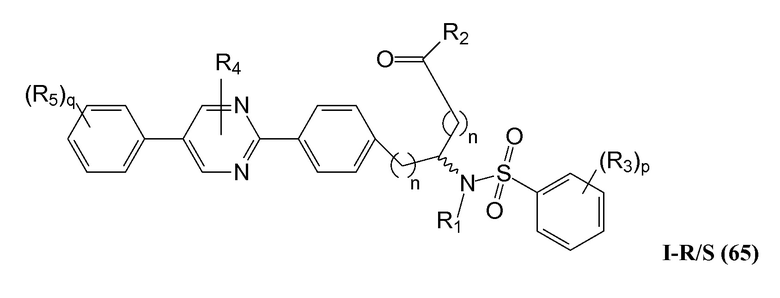
В определенных вариантах осуществления настоящее изобретение относится к соединению формулы I-R и/или формулы I-S, где Y1 является отсутствующей, Y2 представляет собой -O-, и Z представляет собой -C(O)-. Репрезентативные соединения данного варианта осуществления включают соединения следующих структур (где представляет собой одну из или обе R и S формы соединения):
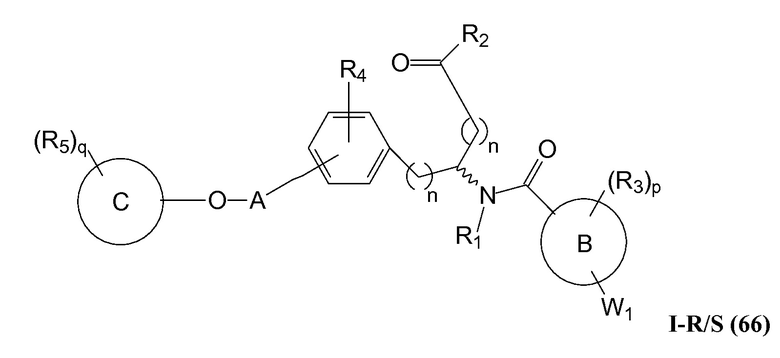
В определенных вариантах осуществления настоящее изобретение относится к соединению формулы I-R и/или формулы I-S, где Y1 представляет собой NH, Y2 является отсутствующей, и Z представляет собой -C(O)-. Репрезентативные соединения данного варианта осуществления включают соединения следующих структур (где представляет собой одну из или обе R и S формы соединения):
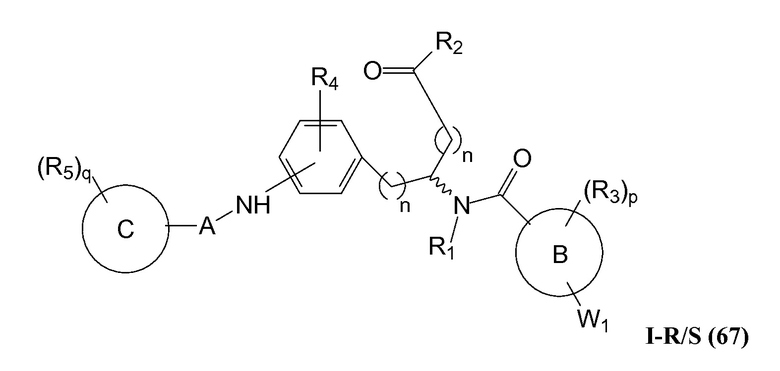
В определенных вариантах осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение настоящего изобретения вместе, по меньшей мере, с одним фармацевтически приемлемым носителем, разбавителем или вспомогательным веществом.
В определенных вариантах осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение настоящего изобретения и второй лекарственный препарат. В некоторых из данных вариантов осуществления, второй лекарственный препарат представляет собой GLP-1 агонист или DPPIV ингибитор.
В определенных вариантах осуществления настоящее изобретение относится к способу применения соединений настоящего изобретения для получения лекарственного препарата.
В определенных вариантах осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей соединение настоящего изобретения и второй лекарственный препарат. В различных данных вариантах осуществления второй лекарственный препарат представляет собой агонист или модулятор глюкагонового рецептора, GIP рецептора, GLP-2 рецептора или PTH рецептора или рецептора глюкагонподобного пептида 1 (GLP-1). В различных данных вариантах осуществления второй лекарственный препарат представляет собой эксенатид, лираглутид, таспоглутид, альбиглутид или ликсисенатид, или другой регулирующий инсулин пептид. В различных данных вариантах осуществления второй лекарственный препарат представляет собой DPPIV ингибитор. В различных данных вариантах осуществления второй лекарственный препарат медицински показан для лечения диабета II типа.
В определенных вариантах осуществления обеспечивают способ активации, потенцирования или агонизма глюкагонподобного пептида, включающий контакт рецептора с эффективным количеством соединения, фармацевтической композиции или фармацевтической комбинации настоящего изобретения.
В следующих вариантах осуществления обеспечивают способ активации или агонизма GLP-1 рецептора контактом рецептора с эффективным количеством соединения настоящего изобретения и GLP-1 пептидов GLP-1 (9-36) и GLP-1 (7-36), фармацевтической композиции или фармацевтической комбинации, где GLP-1 рецептор находится в живом млекопитающем; в определенных вариантах осуществления данное млекопитающее представляет собой человека.
В определенных вариантах осуществления обеспечивают способ лечения патологического состояния у субъекта, для которого медицински показана активация, потенцирование или агонизм GLP-1 рецептора, введением эффективного количества соединения настоящего изобретения субъекту при частоте и в течение периода времени, достаточного для обеспечения полезного эффекта пациенту. В еще следующих вариантах осуществления обеспечивают способ лечения патологического состояния у пациента, для которого медицински показана активация, потенцирование или агонизм GLP-1 рецептора, введением эффективного количества соединения настоящего изобретения пациенту при частоте и в течение периода времени, достаточного для обеспечения полезного эффекта пациенту, где патологическое состояние включает диабет I типа, диабет II типа, гестационный диабет, ожирение, повышенный аппетит, недостаточное насыщение или метаболическое расстройство. В определенных вариантах осуществления субъект представляет собой пациента или человека. В определенных вариантах осуществления человек подвержен или находится в группе риска развития заболевания или состояния, выбранного из группы, состоящей из диабета I типа, диабета II типа, гестационного диабета, ожирения, повышенного аппетита, недостаточного насыщения и метаболического расстройства. В некоторых из данных вариантов осуществления, указанное заболевание представляет собой диабет I типа или диабет II типа.
В определенных вариантах осуществления настоящее изобретение относится к способам получения определенных соединений, включая соединения настоящего изобретения, как более подробно проиллюстрировано в настоящем изобретении. В определенных других вариантах осуществления настоящее изобретение относится к определенным промежуточным соединениям, относящимся к данным способам получения, как показано в настоящем изобретении.
В определенных вариантах осуществления обеспечивают способы применения соединения настоящего изобретения в получении лекарственного препарата, приспособленного для лечения заболевания или патологического состояния, где медицински показана активация или ингибирование GLP-1 рецептора. В определенных вариантах осуществления патологическое состояние включает диабет I типа, диабет II типа, гестационный диабет, ожирение, повышенный аппетит, недостаточное насыщение и метаболическое расстройство. Предпочтительно указанное заболевание представляет собой диабет I типа или диабет II типа.
В определенных вариантах осуществления способ дополнительно включает введение субъекту второго лекарственного препарата, выбранного из группы пептидных GLP-1 агонистов и DPPIV ингибиторов, где данный второй лекарственный препарат представляет собой или компонент фармацевтической композиции или вторую фармацевтическую композицию. В некоторых из данных вариантов осуществления второй лекарственный препарат может представлять собой эксенатид или ситаглиптин.
Как применяют в описании и прилагаемой формуле изобретения, единичные формы "a," "an" и "the" включают множественные референты, если контекст явно не указывает иначе.
Как применяют в настоящем изобретении, "индивид" (как в субъекте лечения) обозначает и млекопитающих и немлекопитающих. Млекопитающие включают, например, людей; приматов, отличных от человека, например, человекообразных обезьян и нечеловекообразных обезьян; крупный рогатый скот; лошадей; овец; и коз.
Немлекопитающие включают, например, рыб и птиц.
"Рецептор", как хорошо известно в данной области техники, представляет собой биомолекулу, обычно включающую белок, которая специфически связывается со структурным классом лигандов или одним нативным лигандом в живом организме, связывание которого вызывает преобразование рецептором сигнала связывания в другой тип биологического действия, такой как сигнал клетке, что связывание осуществилось, которое вызывает изменение функционирования клетки некоторым способом. Пример преобразования представляет собой рецепторное связывание лиганда, вызывающее изменение активности "G-белка" в цитоплазме живой клетки. Любую молекулу, имеющуюся в природе или нет, которая связывается с рецептором и активирует его для преобразования сигнала, называют "агонистом" или "активатором". Любую молекулу, имеющуюся в природе или нет, которая связывается с рецептором, но не вызывает преобразование сигнала, и которая может блокировать связывание агониста и его последующее преобразование сигнала, называют "антагонистом". Определенные молекулы связываются с рецепторами на участках, отличных от сайтов связывания их природных лигандов, и данные аллостерические связывающие молекулы могут потенцировать, активировать или агонизировать рецептор и могут усилить эффект природного лиганда или совместно вводимого лиганда.
"GLP-1 соединение" или "GLP-1 агонист" или "GLP-1 активатор" или "GLP-1 ингибитор" или "GLP-1 антагонист" или "GLP-1 потенциатор" или "GLP-1 модулятор" в качестве терминов применяют в настоящем изобретении для ссылки на соединения, которые взаимодействуют некоторым способом с GLP-1 рецептором. Они могут представлять собой агонисты, потенциаторы или активаторы, или они могут представлять собой антагонисты или ингибиторы. "GLP-1 соединение" настоящего изобретения может быть селективным для действия на GLP-1 рецепторное семейство.
"По существу" в качестве термина, применяемого в настоящем изобретении, обозначает полностью или почти полностью; например, композиция, которая "по существу не содержит" компонента, или не содержит компонента или содержит такое следовое количество, что любое соответствующее функциональное свойство композиции не подвергается воздействию наличия следового количества, или соединение является "по существу чистым", то есть присутствуют только ничтожные количества примесей.
По существу энантиомерно или диастереомерно чистое обозначает степень энантиомерного или диастереомерного обогащения одного энантиомера относительно другого энантиомера или диастереомера, по меньшей мере, 90%, 95%, 98%, 99%, 99,5% или 99,9%.
"Лечение" в пределах значения в настоящем изобретении относится к облегчению симптомов, связанных с расстройством или заболеванием, или ингибированию дальнейшего развития или ухудшения данных симптомов, или предотвращению или профилактике заболевания или расстройства.
Выражение "эффективное количество" при применении для описания применения соединения настоящего изобретения для осуществления терапии пациента, страдающего от расстройства или патологического состояния, опосредованного GLP-1, относится к количеству соединения настоящего изобретения, которое является эффективным для связывания в качестве агониста или антагониста с GLP-1 рецептором в тканях индивида, где GLP-1 вовлечен в заболевание, где данное связывание протекает до степени, достаточной для оказания полезного терапевтического эффекта на пациента. Аналогично, как применяют в настоящем изобретении, "эффективное количество" или "терапевтически эффективное количество" соединения настоящего изобретения относится к количеству соединения, которое облегчает, полностью или частично, симптомы, связанные с расстройством или заболеванием, или останавливает или замедляет дальнейшее развитие или ухудшение данных симптомов, или предотвращает или обеспечивает профилактику расстройства или заболевания. В частности, "терапевтически эффективное количество" относится к количеству, эффективному, при дозах и в течение требуемого периода времени, для достижения требуемого терапевтического результата действием в качестве агониста GLP-1 активности. Терапевтически эффективное количество также представляет собой количество, при котором любые токсические или вредные эффекты соединений настоящего изобретения перевешиваются терапевтически полезными эффектами. Например, в контексте лечения патологического состояния, опосредованного активацией GLP-1 рецептора, терапевтически эффективное количество GLP-1 рецепторного агониста настоящего изобретения представляет собой количество, достаточное для контроля патологического состояния, ослабления развития патологического состояния или облегчения симптомов патологического состояния.
Примеры патологических состояний, которые можно лечить данным способом, включают, но не ограничиваются, диабет II типа.
Предполагаются все хиральные, диастереомерные, рацемические формы структуры, если специально не показана конкретная стереохимия или изомерная форма. Соединения, применяемые в настоящем изобретении, могут включать обогащенные или разделенные оптические изомеры при любом или всех асимметрических атомах, как ясно из изображения, при любой степени обогащения. И рацемические и диастереомерные смеси, а также отдельные оптические изомеры можно получить так, чтобы они были по существу свободными от их энантиомерных или диастереомерных партнеров, и все из них включены в объем определенных вариантов осуществления настоящего изобретения.
Изомеры, являющиеся результатом наличия, хирального центра, включают пару неналожимых изомеров, которые называют "энантиомерами". Отдельные энантиомеры чистого соединения являются оптически активными, т.е., они способны вращать плоскость плоскополяризованного света. Отдельные энантиомеры обозначают согласно системе Кана-Ингольда-Прелога. После определения приоритетности четырех групп, молекулу ориентируют так, чтобы группа с наименьшим приоритетом располагалась от наблюдателя. Затем, если уменьшение приоритета других групп осуществляется по часовой стрелке, молекулу обозначают как (R), и если уменьшение приоритета других групп осуществляется против часовой стрелки, молекулу обозначают (S). В примере на схеме 14, приоритетность по Кану-Ингольду-Прелогу представляет собой A > B > C > D. Атом с наименьшим приоритетом, D, ориентируют от наблюдателя.
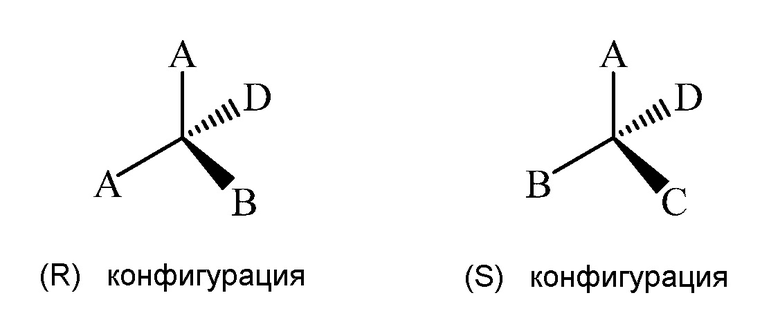
"Выделенный оптический изомер" обозначает соединение, которое по существу очищено от соответствующего оптического изомера (изомеров) той же формулы. Предпочтительно, выделенный изомер имеет, по меньшей мере, приблизительно 80%, более предпочтительно, по меньшей мере, 90% чистоту, даже более предпочтительно, по меньшей мере, 98% чистоту, самое предпочтительное, по меньшей мере, приблизительно 99% чистоту, по весу.
Энантиомеры иногда называют оптическими изомерами, поскольку чистый энантиомер вращает плоскополяризованный свет в конкретном направлении. Если свет вращается по часовой стрелке, то данный энантиомер обозначают "(+)" или "d" для правовращающего, его партнер будет вращать свет против часовой стрелки, и его обозначают "(-)" или "l" для левовращающего.
Термины "рацемат" и "рацемическая смесь" часто применяют взаимозаменяемо. Рацемат представляет собой равную смесь двух энантиомеров. Рацемат обозначают "(±)", поскольку он не является оптически активным (т.е., не будет вращать плоскополяризованный свет в любом из двух направлений, поскольку составляющие его энантиомеры уравновешивают друг друга).
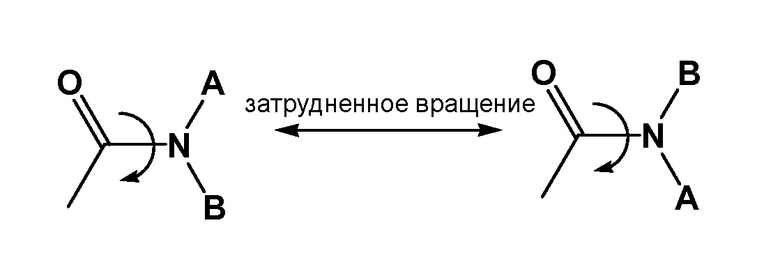
Ясно, что из-за химических свойств (т.е., резонанс, предающий двойной связи характер C-N связи) ограниченного вращения вокруг амидной связи (как показано ниже) можно наблюдать отдельные ротамеры и даже, в некоторых обстоятельствах, выделить данные молекулы, пример показан ниже. Кроме того ясно, что определенные структурные элементы, включая объемные группы или заместители при амидном азоте, могут повышать стабильность ротамера до такой степени, что можно выделить соединение, и оно стабильно, в виде единичного стабильного ротамера. Настоящее изобретение, следовательно, включает все возможные стабильные ротамеры соединений настоящего изобретения, которые являются биологически активными при лечении диабета I типа, диабета II типа, гестационного диабета, ожирения, повышенного аппетита, недостаточного насыщения или метаболического расстройства.
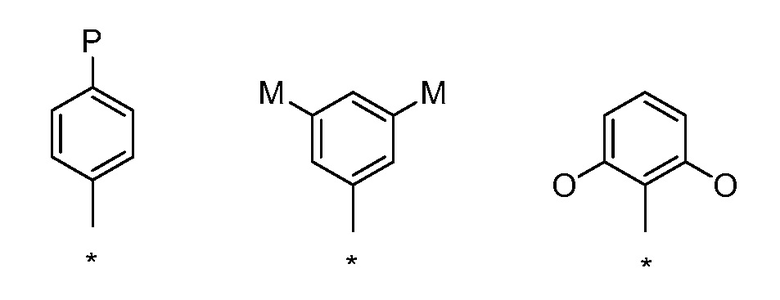
Предпочтительные соединения настоящего изобретения имеют конкретное пространственное расположение заместителей в ароматических кольцах, которое связано с соотношением структура-активность, демонстрируемым данным классом соединений. Часто данное расположение заместителей обозначают системой нумерации; однако, системы нумерации часто не согласуются для различных кольцевых систем. В шестичленных ароматических системах, пространственное расположение показывают стандартной номенклатурой "пара" для 1,4-замещения, "мета" для 1,3-замещения и "орто" для 1,2-замещения, как показано ниже.
Все структуры, включенные в какой-либо пункт формулы изобретения, являются "химически возможными", под этим понимают то, что предполагается, что структура, показанная любой комбинацией или подкомбинацией необязательных заместителей, определена пунктом формулы изобретения, физически возможно ее существование, по меньшей мере, с некоторой стабильностью, как можно определить законами структурной химии и экспериментированием. Структуры, которые не являются химически возможными, не включены в заявленный набор соединений. Кроме того, изотопы показанных атомов (такие как дейтерий и тритий в случае водорода) включены в объем настоящего изобретения.
В общем, "замещенный" относится к органической группе, как определено в настоящем изобретении, в которой одна или более связей с атомом водорода, содержащемся в нем, заменены одной или более связями с атомом, отличным от водорода, таким как, но не ограничиваясь, галоген (т.е., F, Cl, Br, и I); атом кислорода в группах, таких как гидроксильная группа, алкокси группы, арилокси группы, аралкилокси группы, оксо(карбонильные) группы, карбоксильные группы, включая карбоновые кислоты, карбоксилаты и эфиры карбоновых кислот; атом серы в группах, таких как тиольные группы, алкильные и арилсульфидные группы, сульфоксидные группы, сульфогруппы, сульфонильные группы и сульфамидные группы; атом азота в группах, таких как амины, гидроксиламины, нитрилы, нитро группы, N-оксиды, гидразиды, азиды и енамины; и другие гетероатомы в различных других группах. Неограничивающие примеры заместителей, которые могут быть соединены с замещенным атомом углерода (или другим атомом), включают F, Cl, Br, I, OR', OC(O)N(R')2, CN, CF3, OCF3, R', O, S, C(O), S(O), метилендиокси, этилендиокси, N(R')2, SR', SOR', SO2R', SO2N(R')2, SO3R', C(O)R', C(O)C(O)R', C(O)CH2C(O)R', C(S)R', C(O)OR', OC(O)R', C(O)N(R')2, OC(O)N(R')2, C(S)N(R')2, (CH2)0-2NHC(O)R', (CH2)0-2N(R')N(R')2, N(R')N(R')C(O)R', N(R')N(R')C(O)OR', N(R')N(R')CON(R')2, N(R')SO2R', N(R')SO2N(R')2, N(R')C(O)OR', N(R')C(O)R', N(R')C(S)R', N(R')C(O)N(R')2, N(R')C(S)N(R')2, N(COR')COR', N(OR')R', C(=NH)N(R')2, C(O)N(OR')R' или C(=NOR')R', где R' может представлять собой водород или группу на основе углерода, и где группа на основе углерода может быть сама дополнительно замещена.
Замещенные алкильные, алкенильные, алкинильные, циклоалкильные и циклоалкенильные группы, а также другие замещенные группы также включают группы, в которых одна или более связей с атомом водорода заменена одной или более связями, включая двойные или тройные связи, с атомом углерода или с гетероатомом, таким как, но не ограничиваясь, кислород в карбонильной (оксо), карбоксильной, эфирной, амидной, имидной, уретановой и карбамидной группах; и азот в иминах, гидроксииминах, оксимах, гидразонах, амидинах, гуанидинах и нитрилах.
Замещенные кольцевые группы включают замещенные арильные, гетероциклильные и гетероарильные группы. Замещенные кольцевые группы можно заместить одним или более заместителями при любом доступном кольцевом положении. В некоторых вариантах осуществления два заместителя в замещенной кольцевой группе можно брать вместе с кольцом, с которым они соединены, получая такое кольцо, что два кольца конденсированы вместе. Например, бензодиоксолил представляет собой конденсированную кольцевую систему, образованную двумя заместителями, взятыми вместе при фенильной группе.
Данные замещенные кольцевые группы также включают кольца и конденсированные кольцевые системы, в которых связь с атомом водорода заменена связью с атомом углерода. Следовательно, замещенные арильные, гетероциклильные и гетероарильные группы можно также замещать алкильными, алкенильными, циклоалкильными, арильными, гетероарильными и алкинильными группами, как определено в настоящем изобретении, которые могут быть сами дополнительно замещены.
Линкерные группы (например, L1 и L2) формулы I-R или I-S представляют собой частичные структуры, которые можно представить формулой, например, -N(R1)-C(O)-, которая читается слева направо. Соответственно, атом азота -N(R1)-C(O)-линкера будет соединен с ближайшим концом структуры формулы I-R или I-S, и карбонильный атом углерода -N(R1)-C(O)-линкера будет соединен с дальним концом структуры формулы I-R или I-S.
Термин "гетероатомы", как применяют в настоящем изобретении, относится к атомам, отличным от водорода и углерода, способным образовывать ковалентные связи с углеродом, и иначе они не ограничены. Типичные гетероатомы представляют собой N, O и S. Что касается серы (S), ясно, что сера может иметь любую из степеней окисления, в которых ее обнаруживают, таким образом, включая сульфоксиды (R-S(O)-R') и сульфоны (R-S(O)2-R'), если не указано степени окисления; таким образом, термин "сульфон" включает только сульфоновую форму серы; термин "сульфид" включает только сульфидную (R-S-R') форму серы. Когда применяют фразы, такие как "гетероатомы, выбранные из группы, состоящей из O, NH, NR' и S", или "[переменная] представляет собой O, S …", понятно, что они включают все из сульфидных, сульфоксидных и сульфоновых степеней окисления серы.
Алкильные группы включают алкильные группы и циклоалкильные группы с нормальной или разветвленной цепью, содержащие от 1 до приблизительно 20 атомов углерода и обычно 1-12 углеродов (C1-C12 алкил), или, в некоторых вариантах осуществления, 1-8 атомов углерода (C1-C8 алкил), или, в некоторых вариантах осуществления, 1-4 атомов углерода (C1-C4 алкил). Примеры алкильных групп с нормальной цепью включают, но не ограничиваются, метильную, этильную, н-пропильную, н-бутильную, н-пентильную, н-гексильную, н-гептильную и н-октильную группы. Примеры разветвленных алкильных групп включают, но не ограничиваются, изопропильную, изобутильную, втор-бутильную, трет-бутильную, неопентильную, изопентильную и 2,2-диметилпропильную группы. Алкильные группы, как применяют в настоящем изобретении, могут необязательно содержать одну или более дополнительных замещающих групп. Репрезентативные замещенные алкильные группы можно заместить один или более раз любой из групп, перечисленных выше, например, амино, гидрокси, циано, карбокси, нитро, тио, алкокси и галогеновыми группами.
Циклоалкильные группы представляют собой алкильные группы, образованные кольцевой структурой, которая может быть замещенной или незамещенной, где кольцо является или полностью насыщенным, частично ненасыщенным или полностью ненасыщенным, где, если имеется ненасыщенность, сопряжение пи-электронов в кольце не приводит к ароматичности. Примеры циклоалкила включают, но не ограничиваются, циклопропильную, циклобутильную, циклопентильную, циклогексильную, циклогептильную и циклооктильную группы. В некоторых вариантах осуществления циклоалкильная группа содержит 3-8 кольцевых членов, тогда как в других вариантах осуществления количество кольцевых атомов углерода находится в диапазоне 3-5, 3-6 или 3-7. Циклоалкильные группы дополнительно включают полициклические циклоалкильные группы, такие как, но не ограничиваясь, норборнильную, адамантильную, борнильную, камфенильную, изокамфенильную и каренильную группы, и конденсированные кольца, такие как, но не ограничиваясь, декалинил и подобные. Циклоалкильные группы также включают кольца, которые замещены алкильными группами с нормальной или разветвленной цепью, как определено выше. Репрезентативные замещенные циклоалкильные группы могут быть монозамещенными или замещенными один или более раз любой из групп, перечисленных выше, например, но не ограничиваясь, амино, гидрокси, циано, карбокси, нитро, тио, алкокси и галогеновыми группами.
Термины "карбоциклический" и "карбоцикл" обозначает кольцевую структуру, где атомы кольца представляют собой углерод. В некоторых вариантах осуществления карбоцикл содержит 3-8 кольцевых членов, тогда как в других вариантах осуществления количество кольцевых атомов углерода составляет 4, 5, 6 или 7. Если специально не указано обратное, карбоциклическое кольцо можно заместить вплоть до N заместителями, где N представляет собой размер карбоциклического кольца, например, амино, гидрокси, циано, карбокси, нитро, тио, алкокси и галогеновыми группами.
(Циклоалкил)алкильные группы, также обозначаемые как циклоалкилалкил, представляют собой алкильные группы, как определено выше, в которых водородная или углеродная связь алкильной группы заменена связью с циклоалкильной группой, как определено выше.
Алкенильные группы включают алкильные группы с нормальной и разветвленной цепью и циклические алкильные группы, как определено выше, за исключение того, что, по меньшей мере, одна двойная связь присутствует между двумя атомами углерода. Таким образом, алкенильные группы содержат от 2 до приблизительно 20 атомов углерода и обычно от 2 до 12 углеродов или, в некоторых вариантах осуществления, от 2 до 8 атомов углерода. Примеры включают среди прочих, но не ограничиваются, -CH=CH(CH3), -CH=C(CH3)2, -C(CH3)=CH2, -C(CH3)=CH(CH3), -C(CH2CH3)=CH2, винил, циклогексенил, циклопентенил, циклогексадиенил, бутадиенил, пентадиенил и гексадиенил.
Термин "циклоалкенил" отдельно или в комбинации обозначает циклическую алкенильную группу, в которой, по меньшей мере, одна двойная связь присутствует в кольцевой структуре.
Циклоалкенильные группы включают циклоалкильные группы, содержащие, по меньшей мере, одну двойную связь между двумя соседними атомами углерода. Таким образом, например, циклоалкенильные группы включают, но не ограничиваются циклогексенильную, циклопентенильную и циклогексадиенильную группы.
(Циклоалкенил)алкильные группы представляют собой алкильные группы, как определено выше, в которых водородная или углеродная связь алкильной группы замещена связью с циклоалкенильной группой, как определено выше.
Алкинильные группы включают алкильные группы с нормальной и разветвленной цепью, за исключение того, что, по меньшей мере, одна тройная связь присутствует между двумя атомами углерода. Таким образом, алкинильные группы содержат от 2 до приблизительно 20 атомов углерода и обычно от 2 до 12 углеродов или, в некоторых вариантах осуществления, от 2 до 8 атомов углерода. Примеры включают среди прочих, но не ограничиваются, -C≡CH, -C≡C(CH3), -C≡C(CH2CH3), -CH2C≡CH, -CH2C≡C(CH3) и -CH2C≡C(CH2CH3).
Арильные группы представляют собой циклические ароматические углеводороды, которые не содержат гетероатомов. Таким образом, арильные группы включают, но не ограничиваются, фенильную, азуленильную, гепталенильную, бифенильную, индаценильную, флуоренильную, фенантренильную, трифениленильную, пиренильную, нафтаценильную, кризенильную, бифениленильную, антраценильную и нафтильную группы. В некоторых вариантах осуществления, арильные группы содержат 6-14 углеродов в кольцевой части группы. Фраза "арильные группы" включает группы, содержащие конденсированные кольца, такие как конденсированные ароматические алифатические кольцевые системы (например, инданил, тетрагидронафтил и подобные), и также включает замещенные арильные группы, которые содержат другие группы, включая, но не ограничиваясь, алкильные, галогеновые, амино, гидрокси, циано, карбокси, нитро, тио или алкокси группы, соединенные с одним из кольцевых атомов. Репрезентативные замещенные арильные группы могут быть монозамещенными или замещенными более одного раза, такими как, но не ограничиваясь, 2-, 3-, 4-, 5- или 6-замещенные фенильные или нафтильные группы, которые можно заместить группами, включающими, но не ограничивающимися, группы, перечисленные выше.
Аралкильные группы представляют собой алкильные группы, как определено выше, в которых атом водорода алкильной группы замещен арильной группой, как определено выше. Репрезентативные аралкильные группы включают бензильную и фенилэтильную группы и конденсированные (циклоалкиларил)алкильные группы, такие как 4-этилинданил. Арильная группа или алкильная группа или обе необязательно замещены другими группами, включая, но не ограничиваясь, алкильную, галогеновую, амино, гидрокси, циано, карбокси, нитро, тио или алкокси группы. Аралкенильная группа представляет собой алкенильные группы, как определено выше, в которой водородная или углеродная связь алкильной группы заменена связью с арильной группой, как определено выше.
Гетероциклильные или гетероциклические группы включают ароматические и неароматические кольцевые системы, содержащие 3 или более кольцевых членов, из которых один или более представляет собой гетероатом, такой как, но не ограничиваясь, N, O, S или P. В некоторых вариантах осуществления гетероциклильные группы содержат 3-20 кольцевых членов, тогда как другие данные группы содержат 3-15 кольцевых членов, включая, например, системы с одним кольцом, содержащие 5, 6 или 7 кольцевых членов. По меньшей мере, одно кольцо содержит гетероатом, но необязательно, чтобы каждое кольцо в полициклической системе содержало гетероатом. Например, обе из диоксоланильной кольцевой и бенздиоксоланильной кольцевой систем (метилендиоксифенильная кольцевая система) представляют собой гетероциклильные группы, включенные в данное значение в настоящем изобретении. Гетероциклильная группа, обозначенная как C2-гетероциклил, может представлять собой 5-членное кольцо с двумя атомами углерода и тремя гетероатомами, 6-членное кольцо с двумя атомами углерода и четырьмя гетероатомами, и так далее. Аналогично, C4-гетероциклил может представлять собой 5-членное кольцо с одним гетероатомом, 6-членное кольцо с двумя гетероатомами, и так далее. Количество атомов углерода плюс количество гетероатомов в сумме равно суммарному количеству кольцевых атомов.
Термин "гетероциклил" включает конденсированные кольцевые системы, включая системы, содержащие конденсированные ароматические и неароматические группы. Фраза также включает полициклические кольцевые системы, содержащие гетероатом, такие как, но не ограничиваясь, хинуклидил, и также включает гетероциклильные группы, которые содержат заместители, включая, но не ограничиваясь, алкильные, галогеновые, амино, гидрокси, циано, карбокси, нитро, тио или алкокси группы, соединенные с одним из кольцевых членов. Гетероциклильная группа, как определено в настоящем изобретении, может представлять собой гетероарильную группу или частично или полностью насыщенную циклическую группу, включая, по меньшей мере, один кольцевой гетероатом. Гетероциклильные группы включают, но не ограничиваются, пиразинильную, пиримидинильную, пиридазинильную, тиадиазолильную, оксадиазолильную, имидазолинильную, гексагидропиримидинильную, диазепанильную, триазинильную, имидазолильную, пирролидинильную, фуранильную, тетрагидрофуранильную, тетрагидро-2H-пиранильную, диоксоланильную, пиперидинильную, пиперазинильную, морфолинильную, пирролильную, пиразолильную, триазолильную, тетразолильную, оксазолильную, изооксазолильную, тиазолильную, пиридинильную, тиофенильную, бензотиофенильную, бензофуранильную, дигидробензофуранильную, индолильную, дигидроиндолильную, азаиндолильную, индазолильную, бензимидазолильную, азабензимидазолильную, бензоксазолильную, бензотиазолильную, бензотиадиазолильную, имидазопиридинильную, изоксазолопиридинильную, тианафталенильную, пуринильную, ксантинильную, аденинильную, гуанинильную, хинолинильную, изохинолинильную, тетрагидрохинолинильную, хиноксалинильную и хиназолинильную группы.
Гетероциклильные группы могут быть замещенными. Репрезентативные замещенные гетероциклильные группы могут быть монозамещенными или замещенными более одного раза, включая, но не ограничиваясь, кольца, содержащие, по меньшей мере, один гетероатом, который является моно, ди, три, тетра, пента, гекса или более замещенным заместителями, таким как заместители, перечисленные выше, включая, но не ограничиваясь алкильные, галогеновые, амино, гидрокси, циано, карбокси, нитро, тио и алкокси группы.
Гетероарильные группы представляют собой ароматические кольцевые системы, содержащие 5 или более кольцевых членов, из которых, один или более представляет собой гетероатом, такой как, но не ограничиваясь, N, O и S. Гетероарильная группа, обозначенная как C2-гетероарил, может представлять собой 5-членное кольцо с двумя атомами углерода и тремя гетероатомами, 6-членное кольцо с двумя атомами углерода и четырьмя гетероатомами и так далее. Аналогично C4-гетероарил может представлять собой 5-членное кольцо с одним гетероатомом, 6-членное кольцо с двумя гетероатомами, и так далее. Количество атомов углерода плюс количество гетероатомов в сумме равно суммарному количеству кольцевых атомов. Гетероарильные группы включают, но не ограничиваются, группы, такие как пирролильная, пиразолильная, пиридинильная, пиридазинильная, пиримидильная, пиразильная, пиразинильная, пиримидинильная, тиадиазолильная, имидазолильная, оксадиазолильная, тиенильная, триазолильная, тетразолильная, триазинильная, тиазолильная, тиофенильная, оксазолильная, изооксазолильная, бензотиофенильная, бензофуранильная, индолильная, азаиндолильная, индазолильная, бензимидазолильная, азабензимидазолильная, бензоксазолильная, бензотиазолильная, бензотиадиазолильная, имидазопиридинильная, изоксазолопиридинильная, тианафталенильная, пуринильная, ксантинильная, аденинильная, гуанинильная, хинолинильная, изохинолинильная, тетрагидрохинолинильная, тетрагидроизохинолинильная, хиноксалинильная и хиназолинильная группы. Термины "гетероарил" и "гетероарильные группы" включают конденсированные кольцевые соединения, такие как, в которых, по меньшей мере, одно кольцо, но не обязательно все кольца, является ароматическим, включая тетрагидрохинолинил, тетрагидроизохинолинил, индолил и 2,3-дигидроиндолил. Термин также включает гетероарильные группы, которые содержат другие группы, соединенные с одним из кольцевых членов, включая, но не ограничиваясь алкильные, галогеновые, амино, гидрокси, циано, карбокси, нитро, тио или алкокси группы. Репрезентативные замещенные гетероарильные группы могут быть замещены один или более раз группами, таким как группы, перечисленные выше.
Дополнительные примеры арильных и гетероарильных групп включают, но не ограничиваются, фенил, бифенил, инденил, нафтил (1-нафтил, 2-нафтил), N-гидрокситетразолил, N-гидрокситриазолил, N-гидроксиимидазолил, антраценил (1-антраценил, 2-антраценил, 3-антраценил), тиофенил (2-тиенил, 3-тиенил), фурил (2-фурил, 3-фурил), индолил, оксадиазолил (1,2,4-оксадиазолил, 1,3,4-оксадиазолил), тиадиазолил (1,2,4-тиадиазолил, 1,3,4-тиадиазолил), изооксазолил, хиназолинил, флуоренил, ксантенил, изоинданил, бензгидрил, акридинил, тиазолил, пирролил (2-пирролил), пиразолил (3-пиразолил), имидазолил (1-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил), триазолил (1,2,3-триазол-1-ил, 1,2,3-триазол-2-ил, 1,2,3-триазол-4-ил, 1,2,4-триазол-3-ил), оксазолил (2-оксазолил, 4-оксазолил, 5-оксазолил), тиазолил (2-тиазолил, 4-тиазолил, 5-тиазолил), пиридил (2-пиридил, 3-пиридил, 4-пиридил), пиримидинил (2-пиримидинил, 4-пиримидинил, 5-пиримидинил, 6-пиримидинил), пиразинил, пиридазинил (3-пиридазинил, 4-пиридазинил, 5 -пиридазинил), пиразоло[1,5-a]пиридинил, хинолил (2-хинолил, 3-хинолил, 4-хинолил, 5-хинолил, 6-хинолил, 7-хинолил, 8-хинолил), изохинолил (1-изохинолил, 3-изохинолил, 4-изохинолил, 5-изохинолил, 6-изохинолил, 7-изохинолил, 8-изохинолил), бензо[b]фуранил (2-бензо[b]фуранил, 3-бензо[b]фуранил, 4-бензо[b]фуранил, 5-бензо[b]фуранил, 6-бензо[b]фуранил, 7-бензо[b]фуранил), изобензофуранил, 2,3-дигидробензо[b]фуранил (2-(2,3-дигидробензо[b]фуранил), 3-(2,3-дигидробензо[b]фуранил), 4-(2,3-дигидробензо[b]фуранил), 5-(2,3-дигидробензо[b]фуранил), 6-(2,3-дигидробензо[b]фуранил), 7-(2,3-дигидробензо[b]фуранил), бензо[b]тиофенил (2-бензо[b]тиофенил, 3-бензо[b]тиофенил, 4-бензо[b]тиофенил, 5-бензо[b]тиофенил, 6-бензо[b]тиофенил, 7-бензо[b]тиофенил), 2,3-дигидробензо[b]тиофенил, (2-(2,3-дигидробензо[b]тиофенил), 3-(2,3-дигидробензо[b]тиофенил), 4-(2,3-дигидробензо[b]тиофенил), 5-(2,3-дигидробензо[b]тиофенил), 6-(2,3-дигидробензо[b]тиофенил), 7-(2,3-дигидробензо[b]тиофенил), индолил (1-индолил, 2-индолил, 3-индолил, 4-индолил, 5-индолил, 6-индолил, 7-индолил), индазол (1-индазолил, 3-индазолил, 4-индазолил, 5-индазолил, 6-индазолил, 7-индазолил), бензимидазолил (1-бензимидазолил, 2-бензимидазолил, 4-бензимидазолил, 5-бензимидазолил, 6-бензимидазолил, 7-бензимидазолил, 8-бензимидазолил), бензоксазолил (1-бензоксазолил, 2-бензоксазолил), бензотиазолил (1-бензотиазолил, 2-бензотиазолил, 4-бензотиазолил, 5-бензотиазолил, 6-бензотиазолил, 7-бензотиазолил), бензо[d]изоксазолил, карбазолил (1-карбазолил, 2-карбазолил, 3-карбазолил, 4-карбазолил), 5H-дибенз[b,f]азепин (5H-дибенз[b,f]азепин-1-ил, 5H-дибенз[b,f]азепин-2-ил, 5H-дибенз[b,f]азепин-3-ил, 5H-дибенз[b,f]азепин-4-ил, 5H-дибенз[b,f]азепин-5-ил), 10,11-дигидро-5H-дибенз[b,f]азепин (10,11-дигидро-5H-дибенз[b,f]азепин-1-ил, 10,11-дигидро-5H-дибенз[b,f]азепин-2-ил, 10,11-дигидро-5H-дибенз[b,f]азепин-3-ил, 10,11-дигидро-5H-дибенз[b,f]азепин-4-ил, 10,11-дигидро-5H-дибенз[b,f]азепин-5-ил) и подобные.
Гетероциклилалкильные группы представляют собой алкильные группы, как определено выше, в которых водородная или углеродная связь алкильной группы заменена связью с гетероциклильной группой, как определено выше. Репрезентативные гетероциклилалкильные группы включают, но не ограничиваются, фуран-2-ил метил, фуран-3-ил метил, пиридин-2-илметил (α-пиколил), пиридин-3-илметил (β-пиколил), пиридин-4-илметил (γ-пиколил), тетрагидрофуран-2-илэтил и индол-2-илпропил. Гетероциклилалкильные группы могут быть замещены по гетероциклильной группе, алкильной группе или обеим.
Гетероарилалкильные группы представляют собой алкильные группы, как определено выше, в которых водородная или углеродная связь алкильной группы заменена связью с гетероарильной группой, как определено выше. Гетероарилалкильные группы могут быть замещены по гетероарильной группе, алкильной группе, или обеим.
При применении "кольцевой системы" в качестве термина в настоящем изобретении, она обозначает группу, содержащую одно, два, три или более колец, которые могут быть замещены некольцевыми группами или другими кольцевыми системами, или обеими, которые могут быть полностью насыщенными, частично ненасыщенными, полностью ненасыщенными или ароматическими, и где кольцевая система содержит более одного кольца, кольцо может быть конденсировано, соединено мостиковой связью или быть спироциклическим. Под "спироциклическим" понимают класс структур, где два кольца конденсированы при одном тетраэдрическом атоме углерода, как это хорошо известно в данной области техники.
"Моноциклическое, бициклическое или полициклическое, ароматическое или частично ароматическое кольцо" в качестве термина, применяемого в настоящем изобретении, относится к кольцевой системе, содержащей ненасыщенное кольцо, содержащее 4n+2 пи-электронов, или его частично восстановленную (гидрированную) форму. Ароматическое или частично ароматическое кольцо может содержать дополнительные конденсированные, мостиковые или спирокольца, которые сами не являются ароматическими или частично ароматическими. Например, и нафталин и тетрагидронафталин представляют собой "моноциклическое, бициклическое или полициклическое, ароматическое или частично ароматическое кольцо" в пределах данного значения в настоящем изобретении. Кроме того, например, бензо[2,2,2]бициклооктан также представляет собой "моноциклическое, бициклическое или полициклическое, ароматическое или частично ароматическое кольцо" в пределах данного значения в настоящем изобретении, содержащее фенильное кольцо, конденсированное с мостиковой бициклической системой. Полностью насыщенное кольцо не содержит в себе двойных связей, и является карбоциклическим или гетероциклическим, в зависимости от наличия гетероатомов, в пределах данного значения в настоящем изобретении.
Считают, что когда две "R" группы соединены вместе или берутся вместе, образуя кольцо, это подразумевает, что вместе с атомом углерода или атомом, отличным от углерода (например, атомом азота), с которым они соединены, они могут образовывать кольцевую систему. В общем, они соединены друг с другом, образуя 3- - 7-членное кольцо или 5- - 7-членное кольцо.
Неограничивающие конкретные примеры представляют собой циклопентил, циклогексил, циклогептил, пиперидинил, пиперазинил, пирролидинил, пирролил, пиридинил.
Термин "алкокси" относится к атому кислорода, соединенному с алкильной группой, включая циклоалкильную группу, как определено выше. Примеры линейных алкокси групп включают, но не ограничиваются, метокси, этокси, н-пропокси, н-бутокси, н-пентилокси, н-гексилокси, н-гептилокси, н-октилокси, н-нонилокси и подобные. Примеры разветвленных алкокси включают, но не ограничиваются, изопропокси, втор-бутокси, трет-бутокси, изопентилокси, изогексилокси и подобные. Примеры циклических алкокси включают, но не ограничиваются, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и подобные.
Термины "арилокси" и "арилалкокси" относятся, соответственно, к арильной группе, соединенной с атомом кислорода, и аралкильной группе, соединенной с атомом кислорода, по алкильной группе. Примеры включают, но не ограничиваются, фенокси, нафтилокси и бензилокси.
"Ацильная" группа в качестве термина, применяемого в настоящем изобретении, относится к группе, содержащей карбонильную группу, где группа присоединена через карбонильный атом углерода. Карбонильный атом углерода также соединен с другим атомом углерода, который может быть частью алкильной, арильной, аралкильной, циклоалкильной, циклоалкилалкильной, гетероциклильной, гетероциклилалкильной, гетероарильной, гетероарилалкильной группы или подобных. В конкретном случае, где карбонильный атом углерода соединен с водородом, группа представляет собой "формильную" группу, ацильную группу, как данный термин определяют в настоящем изобретении. Ацильная группа может содержать от 0 до приблизительно 12-20 дополнительных атомов углерода, соединенных с карбонильной группой. Ацильная группа может содержать двойные или тройные связи в пределах данного значения в настоящем изобретении. Акрилоильная группа представляет собой пример ацильной группы. Ацильная группа может также содержать гетероатомы в пределах значения в настоящем изобретении. Никотиноильная (пиридил-3-карбонильная группа) представляет собой пример ацильной группы в пределах данного значения в настоящем изобретении. Другие примеры включают ацетильную, бензоильную, фенилацетильную, пиридилацетильную, циннамоильную и акрилоильную группы и подобные. Когда группа, содержащая атом углерода, который соединен с карбонильным атомом углерода, содержит галоген, группу называют "галогенацильной" группой. Пример представляет собой трифторацетильную группу.
Термин "амин" включает первичные, вторичные и третичные амины, имеющие, например, формулу N(группа)3, где каждая группа может независимо представлять собой H или не H, такую как алкил, арил и подобную. Амины включают, но не ограничиваются, R-NH2, например, алкиламины, ариламины, алкилариламины; R2NH, где каждый R выбран независимо, такие как диалкиламины, диариламины, аралкиламины, гетероциклиламины и подобные; и R3N, где каждый R выбран независимо, такие как триалкиламины, диалкилариламины, алкилдиариламины, триариламины и подобные. Термин "амин" также включает аммониевые ионы, как применяют в настоящем изобретении.
"Амино" группа представляет собой заместитель вида -NH2, -NHR, -NR2, -NR3+, где каждый R выбран независимо, и протонированные формы каждой из них.
Соответственно, любое соединение, замещенное группой, может рассматриваться как амин.
"Аммониевый" ион включает незамещенный аммониевый ион NH4+, но если не указано иначе, он также включает любые протонированные или кватернизованные формы аминов. Таким образом, и гидрохлорид триметиламмония и хлорид тетраметиламмония представляют собой аммониевые ионы и амины, в пределах данного значения в настоящем изобретении.
Термин "амид" (или "амидо") включает C- и N-амидные группы, т.е., -C(O)NR2 и -NRC(O)R группы, соответственно. Амидные группы, следовательно, включают, но не ограничиваются, карбамоильные группы (-C(O)NH2) и формамидные группы (-NHC(O)H). "Карбоксамидо" группа представляет собой группу формулы C(O)NR2, где R может представлять собой H, алкил, арил и т.д.
Термин "карбонил" относится к -C(O)- группе.
"Галоген" и "галид" включает фтор, хлор, бром и йод.
Термин "пергалогеналкил" относится к алкильной группе, где все из атомов водорода заменены атомами галогена. Пергалогеналкильные группы включают, но не ограничиваются, -CF3 и -C(CF3)3. Термин "галогеналкил" относится к алкильной группе, где некоторые, но не обязательно все из атомов водорода, заменены атомами галогена. Галогеналкильные группы включают, но не ограничиваются, -CHF2 и -CH2F.
Термин "пергалогеналкокси" относится к алкокси группе, где все из атомов водорода заменены атомами галогена. Пергалогеналкокси группы включают, но не ограничиваются, -OCF3 и -OC(CF3)3. Термин "галогеналкокси" относится к алкокси группе, где некоторые, но не обязательно все из атомов водорода, заменены атомами галогена. Галогеналкокси группы включают, но не ограничиваются, -OCHF2 и -OCH2F.
Термины "содержащий", "включающий", "имеющий", "состоящий из" представляют собой неограничивающие термины, как применяют в настоящем изобретении, и не исключают наличия дополнительных элементов или компонентов. В элементах пунктов формулы изобретения применение форм "содержащий", "включающий", "имеющий", "состоящий из" обозначает то, что содержится, включен, имеется или составляет какой-либо элемент, необязательно только элемент, включенный предметом пункта, который содержит данное слово.
"Соль", как хорошо известно в данной области техники, включает органические соединения, такие как карбоновая кислота, сульфокислота или амин, в ионной форме, в комбинации с противоионом. Например, кислоты в их анионной форме могут образовывать соли с катионами, такими как катионы металлов, например, натрия, калия и подобных; аммониевые соли, такие как NH4+, или с катионами различных аминов, включая тетраалкиламмониевые соли, такие как тетраметиламмоний, или другими катионами, такими как триметилсульфоний, и подобными. "Фармацевтически приемлемая" или "фармакологически приемлемая" соль представляет собой соль, образованную ионом, который одобрен для потребления человеком и обычно является нетоксичным, такую как хлоридная соль или натриевая соль. "Цвиттерион" представляет собой внутреннюю соль, такую как соль, которая может образовываться в молекуле, которая содержит, по меньшей мере, две ионизируемые группы, причем одна образует анион и другая катион, которые служат для уравновешивания друг друга. Например, аминокислоты, такие как глицин, могут существовать в цвиттерионной форме. "Цвиттерион" представляет собой соль в пределах данного значения в настоящем изобретении. Соединения настоящего изобретения могут принимать форму солей. Термин "соли" включают соли присоединения свободных кислот или свободных оснований, которые представляют собой соединения настоящего изобретения. Соли могут представлять собой "фармацевтически приемлемые соли". Термин "фармацевтически приемлемая соль" относится к солям, которые имеют профиль токсичности в диапазоне, который обеспечивает применимость для фармацевтических нужд. Фармацевтически неприемлемые соли могут, тем не менее, обладать свойствами, такими как кристалличность, которые применимы на практике настоящего изобретения, такой как, например, применение в получении, очистке или формулировании соединений настоящего изобретения.
Подходящие фармацевтически приемлемые соли присоединения кислот можно получить из неорганических кислот или из органических кислот. Примеры неорганических кислот включают хлористоводородную, бромистоводородную, йодистоводородную, азотную, угольную, серную и фосфорную кислоты. Подходящие органические кислоты можно выбрать из алифатических, циклоалифатических, ароматических, аралифатических, гетероциклических, карбоновых и сульфоновых классов органических кислот, примеры которых включают муравьиную, уксусную, пропионовую, янтарную, гликолевую, глюконовую, молочную, яблочную, винную, лимонную, аскорбиновую, глюкуроновую, малеиновую, фумаровую, пировиноградную, аспарагиновую, глутаминовую, бензойную, антраниловую, 4-гидроксибензойную, фенилуксусную, миндальную, эмбоновую (памовую), метансульфоновую, этансульфоновую, бензолсульфоновую, пантотеновую, трифторметансульфоновую, 2- гидроксиэтансульфоновую, п-толуолсульфоновую, сульфаниловую, циклогексиламиносульфоновую, стеариновую, альгиновую, β-гидроксимасляную, салициловую, галактаровую и галактуроновую кислоты. Примеры фармацевтически неприемлемых солей присоединения кислот включают, например, перхлораты и тетрафторбораты.
Подходящие фармацевтически приемлемые соли присоединения оснований соединений настоящего изобретения включают, например, металлические соли, включая соли щелочных, щелочноземельных и переходных металлов, такие как, например, кальциевые, магниевые, калиевые, натриевые и цинковые соли. Фармацевтически приемлемые соли присоединения оснований также включают органические соли, полученные из основных аминов, таких как, например, N,N'-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, меглумин (N-метилглюкамин) и прокаин. Примеры фармацевтически неприемлемых солей присоединения оснований включают литиевые соли и цианатные соли. Хотя фармацевтически неприемлемые соли обычно являются непригодными в качестве лекарственных препаратов, данные соли могут быть пригодными, например, в качестве промежуточных соединений в получении соединений формулы I, например, в их очистке перекристаллизацией. Все из данных солей можно получить общепринятыми способами из соответствующего соединения согласно формуле I реакцией, например, подходящей кислоты или основания с соединением согласно формуле I. Термин "фармацевтически приемлемые соли" относится к нетоксичным солям присоединения неорганических или органических кислот и/или оснований, смотри, например, Lit et al, SAlt Selection for Basic Drugs (1986), IntJ. Pharm., 33, 201-217, включенную в настоящее изобретение с помощью ссылки.
"Гидрат" представляет собой соединение, которое существует в композиции с молекулами воды. Композиция может включать воду в стехиометрических количествах, такая как моногидрат или дигидрат, или может содержать воду в произвольном количестве. В качестве термина, применяемого в настоящем изобретении, "гидрат" относится к твердой форме, т.е., соединение в водном растворе, притом, что оно может быть гидратировано, не является гидратом в значении термина, применяемого в настоящем изобретении.
"Сольват" представляет собой аналогичную композицию, за исключением того, что растворитель, отличный от воды, заменяет воду. Например, метанол или этанол могут образовывать "алкоголяты", которые могут быть снова стехиометрическими или нестехиометрическими. В качестве термина, применяемого в настоящем изобретении, "сольват" относится к твердой форме, т.е., соединение в растворе в растворителе, притом, что оно может быть сольватировано, не является сольватом в значении термина, применяемого в настоящем изобретении.
"Пролекарство", как хорошо известно в данной области техники, представляет собой вещество, которое можно вводить пациенту, где вещество превращается in vivo под действием биохимических веществ в теле человека, таких как ферменты, в активный фармацевтический ингредиент. Примеры пролекарств включают эфиры карбоксильных групп, которые могут гидролизоваться эндогенными эстеразами, которые присутствуют в кровотоке людей и других млекопитающих.
"Изотопы" являются хорошо известными в данной области техники и относятся к атомам с тем же количеством протонов, но отличным количеством нейтронов. Например, углерод 12, самая распространенная форма углерода, содержит шесть протонов и шесть нейтронов, тогда как углерод 14 содержит шесть протонов и восемь нейтронов.
Кроме того, где признаки или аспекты настоящего изобретения описаны в терминах группы Маркуша, специалисту в данной области техники ясно, что настоящее изобретение также описано, посредством этого, в терминах любого отдельного члена или подгруппы членов группы Маркуша. Например, если X описывают как выбранную из группы, состоящей из брома, хлора и йода, пункты для X, представляющего собой бром, и пункты для X, представляющего собой бром и хлор, описаны полностью. Более того, где признаки или аспекты настоящего изобретения описаны в терминах группы Маркуша, специалисту в данной области техники ясно, что настоящее изобретение также описано, посредством этого, в терминах любой комбинации отдельных членов или подгрупп членов группы Маркуша. Таким образом, например, если X описывают как выбранную из группы, состоящей из брома, хлора и йода, и Y описывают как выбранную из группы, состоящей из метила, этила и пропила, пункты для X, являющейся бромом, и Y, являющейся метилом, описаны полностью.
КОМПОЗИЦИИ И КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ
GLP-1 соединения, их фармацевтически приемлемые соли или гидролизуемые эфиры настоящего изобретения можно комбинировать с фармацевтически приемлемым носителем, получая фармацевтические композиции, пригодные для лечения биологических состояний или расстройств, указанных в настоящем изобретении, у различных видов млекопитающих, и более предпочтительно, у людей. Конкретный носитель, применяемый в данных фармацевтических композициях, может изменяться в зависимости от типа требуемого введения (например, внутривенно, перорально, местно, суппозиторием или парентерально).
Для получения композиций в виде пероральных жидких дозированных форм (например, суспензий, эликсиров и растворов), можно применять стандартную фармацевтическую среду, такую как вода, гликоли, масла, спирты, ароматизаторы, консерванты, красители и подобные. Аналогично, при получении пероральных твердых дозированных форм (например, порошков, таблеток и капсул), можно применять носители, такие как крахмалы, сахара, разбавители, гранулирующие агенты, лубриканты, связующие, дезинтегрирующие агенты и подобные.
Другой аспект варианта осуществления настоящего изобретения относится к композициям соединений настоящего изобретения, отдельно или в комбинации с другим GLP-1 агонистом или другим типом терапевтического агента, или обоими. Неограничивающие примеры GLP-1 рецепторных агонистов включают эксенатид, лираглутид, таспоглутид, альбиглутид, ликсисенатид и их смеси.
В одном варианте осуществления GLP-1 агонист представляет собой эксенатид (Byetta®) или Byetta LAR®. Эксенатид описан, например, в патентах США № 5424286; 6902744; 7297761 и других, содержание каждого из которых включено в настоящее изобретение с помощью ссылки во всей своей полноте.
В одном варианте осуществления GLP-1 агонист представляет собой лираглутид (VICTOZA®) (также называемый NN-2211 и [Arg34, Lys26]-(N-эпсилон-(гамма-Glu(N-альфа-гексадеканоил))-GLP-1 (7-37)), содержащий последовательность HAEGTFTSDVSSYLEGQAAKEFIAWKVRGRG (SEQ ID NO: 1) и имеющийся в продаже у Novo Nordisk (Denmark) или Scios (Fremont, Calif. USA). Смотри, например, Elbrond et al, 2002, Diabetes Care. August; 25(8):1398404; Agerso et al, 2002, Diabetologia. February; 45(2): 195-202).
В одном варианте осуществления GLP-1 агонист представляет собой таспоглутид (CAS регистрационный No. 275371-94-3) и имеется в продаже у Hoffman La-Roche. Смотри, например, патент США No. 7368427, содержание которого включено в настоящее изобретение с помощью ссылки во всей своей полноте.
В одном варианте осуществления GLP-1 агонист представляет собой альбиглутид (SYNCRIA® от Glaxo SmithKline).
В другом варианте осуществления GLP-1 агонист представляет собой ликсисенатид (Lyxumia® от Sanofi-Aventis/Zealand Pharma).
Как указано в настоящем изобретении, соединения настоящего изобретения включают стереоизомеры, таутомеры, сольваты, гидраты, соли, включая фармацевтически приемлемые соли, и их смеси. Композиции, содержащие соединение настоящего изобретения, можно получить общепринятыми способами, например, как описано в Remington: The Science and Practice of Pharmacy, 19th Ed., 1995, включенную в настоящее изобретение с помощью ссылки. Композиции могут находиться в общепринятых формах, например, капсулы, таблетки, аэрозоли, растворы, суспензии, или формах для местного применения.
Типичные композиции содержат соединение настоящего изобретения и фармацевтически приемлемый эксципиент, который может представлять собой носитель или разбавитель. Например, активное соединение будут обычно смешивать с носителем, или разбавлять в носителе, или включать в носитель, который может быть в виде ампулы, капсулы, саше, бумаги или другого контейнера. Когда активное соединение смешивают с носителем, или когда носитель служит в качестве разбавителя, он может представлять собой твердый, полутвердый или жидкий материал, который действует как носитель, эксципиент или среда для активного соединения. Активное соединение можно абсорбировать на гранулированном твердом носителе, например, содержащемся в саше. Некоторые примеры подходящих носителей представляют собой воду, солевые растворы, спирты, полиэтиленгликоли, полигидроксиэтоксилированное касторовое масло, арахисовое масло, оливковое масло, желатин, лактозу, каолин, сахарозу, декстрин, карбонат магния, сахар, циклодекстрин, амилоза, стеарат магния, тальк, желатин, агар, пектин, аравийскую камедь, стеариновую кислоту или низшие алкиловые эфиры целлюлозы, кремниевую кислоту, жирные кислоты, амины жирных кислот, моноглицериды и диглицериды жирных кислот, сложные эфиры пентаэритритола и жирных кислот, полиоксиэтилен, гидроксиметилцеллюлозу и поливинилпирролидон. Аналогично, носитель или разбавитель может включать любой материал, замедляющий высвобождение, известный в данной области техники, такой как моностеарат глицерина или дистеарат глицерина, отдельно или смешанный с воском.
Составы можно смешивать с вспомогательными агентами, которые не реагируют вредоносно с активными соединениями. Данные добавки могут включать смачивающие агенты, эмульгирующие и суспендирующие агенты, соль для регулирования осмотического давления, буферы и/или красители, консерванты, подслащивающие агенты или ароматизаторы. Композиции можно также стерилизовать, при необходимости.
Путь введения может представлять собой любой путь, который эффективно переносит активное соединение настоящего изобретения в подходящее или требуемое место действия, такой как пероральный, назальный, пульмональный, буккальный, подкожный, внутрикожный, чрескожный или парентеральный, например, ректальный, депо, подкожный, внутривенный, внутриуретральный, внутримышечный, интраназальный, в виде офтальмологического раствора или мази, причем пероральный путь является предпочтительным.
Что касается парентерального введения, носитель обычно будет содержать стерильную воду, хотя можно также включать другие ингредиенты, которые способствуют растворимости или служат в качестве консервантов. Более того, можно также получать инъецируемые суспензии, в данном случае можно применять подходящие жидкие носители, суспендирующие агенты и подобные.
Что касается местного введения, соединения настоящего изобретения можно формулировать, применяя мягкие, увлажняющие основы, такие как мази или крема.
Если твердый носитель применяют для перорального введения, препарат можно таблетировать, помещать в твердые желатиновые капсулы в виде порошка или пеллетов, или он может быть в виде пастилки или пастилки для рассасывания. Если применяют жидкий носитель, препарат может быть в виде сиропа, эмульсии, мягкой желатиновой капсулы или стерильной инъецируемой жидкости, такой как водная или неводная жидкая суспензия или раствор.
Инъецируемые дозированные формы обычно включают водные суспензии или масляные суспензии, которые можно получить, применяя подходящие диспергаторы или смачивающие агенты и суспендирующие агенты. Инъецируемые формы могут быть в водной фазе или в виде суспензии, которую получают с растворителем или разбавителем. Приемлемые растворители или среды включают стерилизованную воду, раствор Рингера или изотонический водный соляной раствор. Альтернативно, можно применять стерильные масла в качестве растворителей или суспендирующих агентов.
Предпочтительно, масло или жирная кислота является нелетучей, включая природные или синтетические масла, жирные кислоты, моно-, ди- или триглицериды.
Что касается инъекции, состав может также представлять собой порошок, подходящий для растворения до подходящего раствора, как описано выше. Его примеры включают, но не ограничиваются, лиофилизированные, высушенные на роторе или высушенные распылением порошки, аморфные порошки, гранулы, осадки или частицы. Что касается инъекции, составы могут необязательно содержать агенты, повышающие растворимость, pH модификаторы, поверхностно-активные вещества, агенты, изменяющие биодоступность, и их комбинации. Соединения можно формулировать для парентерального введения инъекцией, такой как болюсная инъекция или непрерывное вливание. Единичная дозированная форма для инъекции может быть в виде ампул или в виде контейнеров для нескольких доз.
Составы настоящего изобретения можно разрабатывать так, чтобы получить быстрое, замедленное или отсроченное высвобождение активного ингредиента после введения пациенту применением способов, хорошо известных в данной области техники. Таким образом, составы можно также формулировать для контролируемого высвобождения или для медленного высвобождения.
Композиции, предполагаемые в настоящем изобретении, могут включать, например, мицеллы или липосомы, или какие-либо другие инкапсулированные формы, или их можно вводить в форме с длительным высвобождением, обеспечивая длительное хранение и/или эффект доставки. Следовательно, составы можно прессовать до пеллетов или цилиндров и имплантировать внутримышечно или подкожно в виде депо-инъекций. В данных имплантатах можно применять известные инертные материалы, такие как силиконы и биоразлагаемые полимеры, например, полиактид-полигликолид. Примеры других биоразлагаемых полимеров включают поли(ортоэфиры) и поли(ангидриды).
Что касается назального введения, препарат может содержать соединение настоящего изобретения, растворенное или суспендирванное в жидком носителе, предпочтительно водном носителе, для аэрозольного нанесения. Носитель может содержать добавки, такие как агенты, повышающие растворимость, например, пропиленгликоль, поверхностно-активные вещества, усилители поглощения, такие как лецитин (фосфатидилхолин) или циклодекстрин, или консерванты, такие как парабены.
Что касается парентерального применения, особенно подходящими являются инъецируемые растворы или суспензии, предпочтительно водные растворы с активным соединением, растворенным в полигидроксилированном касторовом масле.
Дозированные формы можно вводить ежедневно или более одного раза в день, например, два или три раза в день. Альтернативно, дозированные формы можно вводить менее часто, чем ежедневно, например раз в два дня или раз в неделю, если лечащий врач считает это уместным.
Один вариант осуществления настоящего изобретения также включает пролекарства соединения настоящего изобретения, которые после введения подвергаются химическому превращению метаболическими или другими физиологическими способами перед тем, как превратиться в активные фармакологические вещества. Превращение метаболическими или другими физиологическими способами включает без ограничения ферментативное (например, специфически катализируемое ферментативно) и неферментативное (например, общее или специфическое вызванное кислотой или основанием) химическое превращение пролекарства в активное фармакологическое вещество. В общем, данные пролекарства будут представлять собой функциональные производные соединения настоящего изобретения, которые можно легко превратить in vivo в соединение настоящего изобретения. Общепринятые способы выбора и получения подходящих пролекарственных производных описаны, например, в Design of Prodrugs, ed. H. Bundgaard, Elsevier, 1985.
В другом варианте осуществления обеспечивают способы получения композиции соединения, описанного в настоящем изобретении, включающие формулирование соединения настоящего изобретения с фармацевтически приемлемым носителем или разбавителем. В некоторых вариантах осуществления фармацевтически приемлемый носитель или разбавитель является пригодным для перорального введения. В некоторых данных вариантах осуществления способы могут дополнительно включать стадию формулирования композиции в виде таблетки или капсулы. В других вариантах осуществления фармацевтически приемлемый носитель или разбавитель является пригодным для парентерального введения. В некоторых данных вариантах осуществления способы дополнительно включают стадию лиофилизации композиции, получая лиофилизированный препарат.
Соединения настоящего изобретения можно применять терапевтически в комбинации с i) одним или более другими GLP-1 модуляторами и/или ii) одним или более другими типами терапевтических агентов, которые можно вводить перорально в той же дозированной форме, в виде отдельной пероральной дозированной формы (например, последовательно или непоследовательно) или инъекцией вместе или раздельно (например, последовательно или непоследовательно). Примеры комбинированных терапевтических агентов включают, ситаглиптин (MK-0431, Januvia), пероральный гипогликемический препарат (противодиабетическое лекарственное средство) класса ингибиторов дипептидилпептидазы-4 (DPP-4) и эксенатид (Byetta), инкретиномиметик.
Комбинации настоящего изобретения включают смеси соединений из (a) и (b) в виде единого состава и соединений из (a) и (b) в виде отдельных составов. Некоторые комбинации настоящего изобретения можно упаковывать в виде отдельных составов в наборе. В некоторых вариантах осуществления два или более соединений из (b) формулируют вместе, тогда как соединение настоящего изобретения формулируют отдельно.
Дозы и составы для других агентов, которые будут применять, где применимо, будут такими, как это указано в последнем издании настольного справочника врача, включенного в настоящее изобретение с помощью ссылки.
СПОСОБЫ ЛЕЧЕНИЯ
В определенных вариантах осуществления настоящее изобретение включает соединения, которые связываются с высоким сродством и специфичностью с GLP-1 рецептором агонистическим способом или в качестве активатора или потенциатора. В определенных вариантах осуществления соединение настоящего изобретения действует как положительный аллостерический модулятор GLP-1 рецептора.
В определенных вариантах осуществления настоящее изобретение относится к способу активации, потенцирования или агонизирования (т.е., обладает агонистическим эффектом, действуя как агонист) GLP-1 рецептора, соединением настоящего изобретения. Способ включает контактирование рецептора с подходящей концентрацией соединения настоящего изобретения, приводя к активации рецептора. Контактирование может протекать in vitro, например, при осуществлении анализа для определения активности GLP-1 рецепторной активации соединения настоящего изобретения, подвергаемого эксперименту, связанного с подачей на рассмотрение для разрешения контролирующим органом.
В определенных вариантах осуществления способ активации GLP-1 рецептора можно также осуществлять in vivo, то есть, в живом теле млекопитающего, такого как пациент, являющийся человеком, или испытуемого животного. Соединение настоящего изобретения можно поставлять в живой организм одним из путей, как описано выше, например, перорально, или можно обеспечивать местно в ткани тела. В присутствии соединения настоящего изобретения протекает активация рецептора, и можно исследовать его эффект.
Один вариант осуществления настоящего изобретения относится к способу лечения патологического состояния у пациента, для которого медицински показана активация GLP-1 рецептора, где пациенту вводят соединение настоящего изобретения в дозе, частоте и в течение некоторого периода времени, получая полезный для пациента эффект. Соединение настоящего изобретения можно вводить подходящими способами, примеры которых описаны выше.
В определенных вариантах осуществления настоящее изобретение относится к соединениям, приспособленным для того, чтобы действовать в качестве модуляторов или потенциаторов GPCR класса B. Данные соединения могут обладать активностью сами по себе или в присутствии рецепторных лигандов. Рецепторы включают инкретиновые пептиды, включая GLP-1 (7-36) и GLP-1 (9-36).
Способы лечения, относящиеся к настоящему изобретению, включают введение соединения настоящего изобретения, отдельно или в комбинации с другим фармакологически активным агентом субъекту или пациенту, страдающему от патологического состояния, для которого медицински показана активация, потенцирование или агонизм рецептора глюкагонподобного пептида 1, такого как диабет I типа, диабет II типа, гестационный диабет, ожирение, повышенный аппетит, недостаточное насыщение или метаболическое расстройство.
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ ОПРЕДЕЛЕННЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Общие способы получения соединений
Молекулярные варианты осуществления настоящего изобретения можно получить, применяя синтетические способы, известные специалисту в данной области техники. Соединения настоящего изобретения можно получить, применяя общие способы получения, показанные на схемах 1-21.
Схема 1:
Реагенты: PG1 и PG2 представляют собой защитные группы: (i) если Z = CO, то амидная конденсация с хлорангидридом карбоновой кислоты: DIEA, DCM или амидная конденсация с кислотой: EDC, HOBt, DMF или HATU, DMF; если Z=SO2, то конденсация с сульфонилхлоридом: DIEA или NEt3, DCM или DMF; (ii) деблокирование PG1, например, деблокирование метилового эфира: LiOH, диоксан, вода.
Другой энантиомер можно получить аналогичным способом, применяя схему 1.
Схема 2:

Реагенты: (i) Zn(CN)2, Pd(PPh3)4, NMP; (ii) NH2OH HCl, TEA, EtOH.
Схема 3:
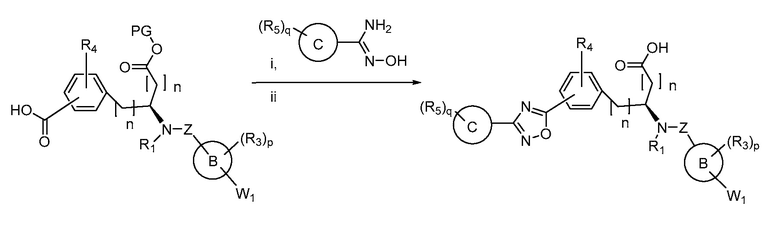
Реагенты: PG представляет собой защитную группу (i) EDC, HOBt, DMF, затем нагревание; (ii) деблокирование, например, деблокирование метилового эфира: NaOH, MeOH, вода.
Другой энантиомер можно получить аналогичным способом, применяя схему 3.
Схема 4:

Реагенты: PG представляет собой защитную группу (i) NH2OH, TEA, вода или EtOH; (ii) EDC, HOBt, DMF, затем нагревание; (iii) деблокирование, например, деблокирование метилового эфира: NaOH, MeOH, вода.
Другой энантиомер можно получить аналогичным способом, применяя схему 4.
Схема 5:
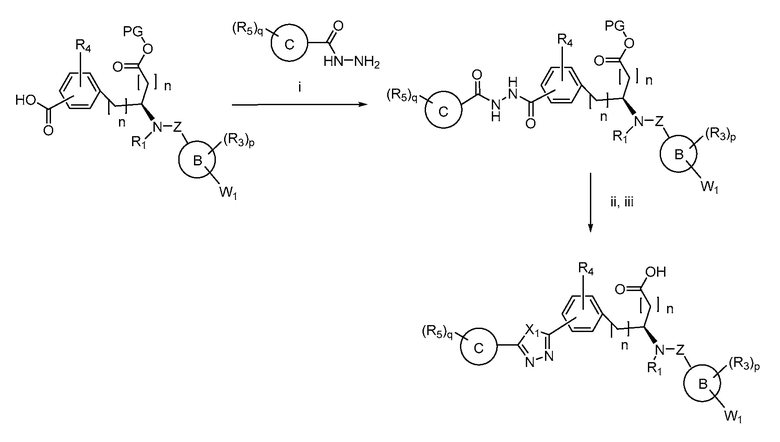
Реагенты: X1 = O или S; (i) N-метилморфолин, изобутилхлорформиат, THF, DMF; (ii) если X1 = кислород, то 2-хлор-1,3-диметилимидазолинийхлорид, TEA, DCM; если X1 = сера, то 2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан 2,4-дисульфид, THF; (iii) деблокирование, например, деблокирование метилового эфира: NaOH, MeOH, вода.
Другой энантиомер можно получить аналогичным способом, применяя схему 5.
Схема 6:
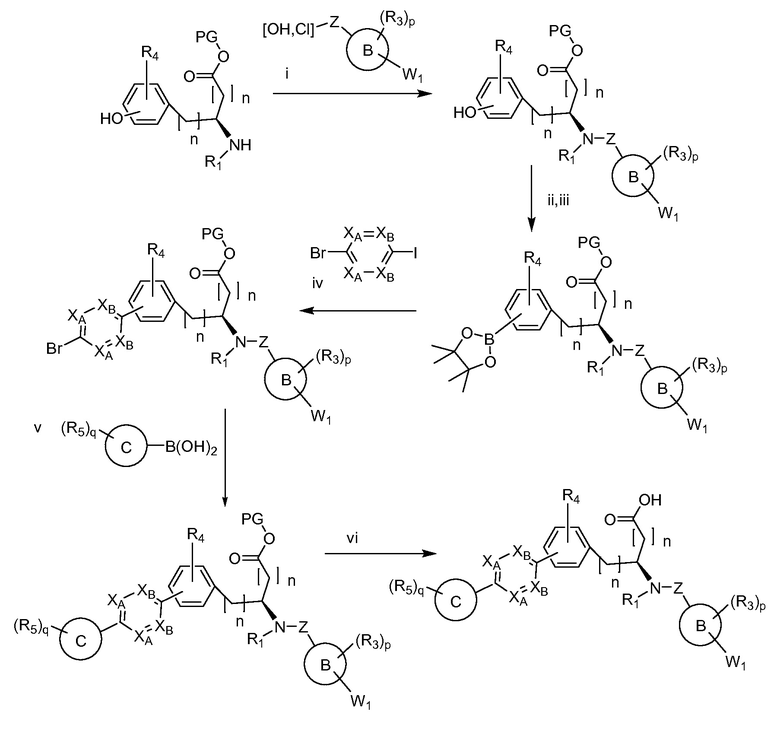
Реагенты: PG представляет собой защитную группу (i) если Z=CO, то амидная конденсация с хлорангидридом карбоновой кислоты: DIEA, DCM или амидная конденсация с кислотой: EDC, HOBt, DMF или HATU, DMF; если Z=SO2, то конденсация с сульфонилхлоридом, DIEA или NEt3, DCM или DMF (ii) DIEA, 1,1,1-трифтор-N-фенил-N-((трифторметил)сульфонил)метансульфамид, DCM; (iii) KOAc, бис-пинаколатоборан, PdCl2(dppf) или Pd(dppf)Cl2, Na2CO3, THF, MeCN, вода; (iv) Pd(dppf)Cl2, Na2CO3, THF, MeCN, вода; (v) Pd(dppf)Cl2, Na2CO3, THF, MeCN, вода; (vi) деблокирование, например, деблокирование метилового эфира: NaOH, MeOH, вода. Каждый случай XA и XB независимо представляет собой CR4 или N.
Другой энантиомер можно получить аналогичным способом, применяя схему 6.
Схема 7:
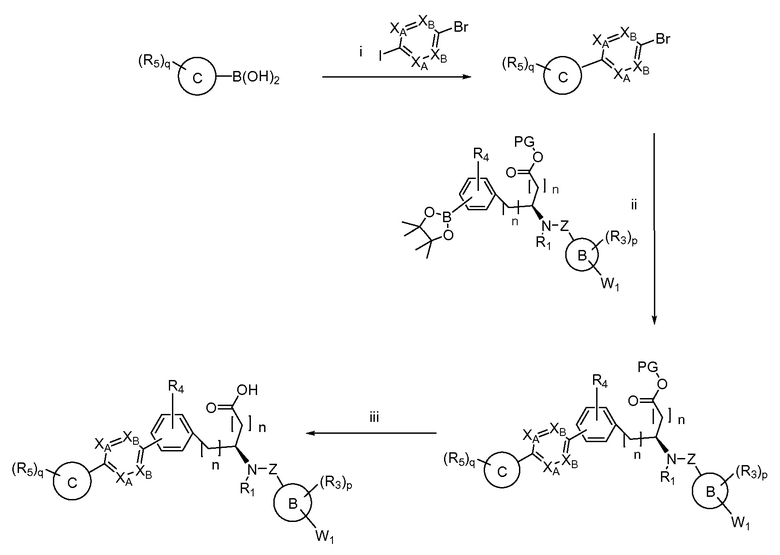
Реагенты: PG представляет собой защитную группу; (i) Pd(dppf)Cl2, Na2CO3, THF, MeCN, вода; (ii) Pd(dppf)Cl2, Na2CO3, THF, MeCN, вода; (iii) деблокирование, например, деблокирование метилового эфира: NaOH, MeOH, вода. Каждый случай XA и XB независимо представляет собой CR4 или N.
Другой энантиомер можно получить аналогичным способом, применяя схему 7.
Схема 8:
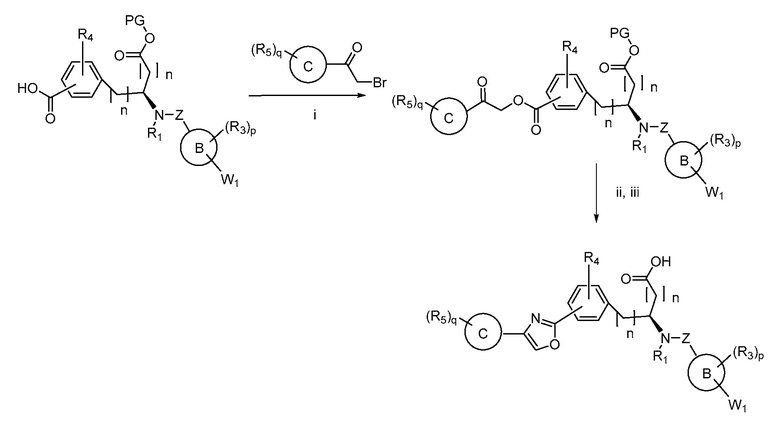
Реагенты: PG представляет собой защитную группу (i) DIEA или TEA, ацетонитрил; (ii) ацетамид, эфират трехфтористого бора, DCM; (iii) деблокирование, например, деблокирование метилового эфира: NaOH, MeOH, вода.
Другой энантиомер можно получить аналогичным способом, применяя схему 8.
Схема 9:
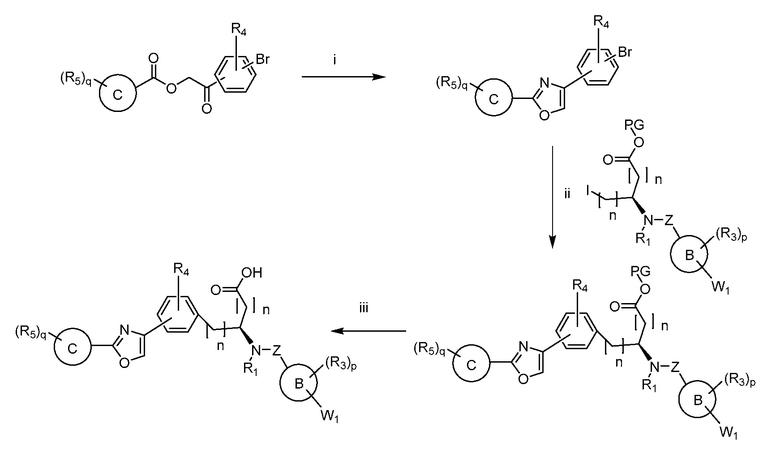
Реагенты: PG представляет собой защитную группу (i) эфират трехфтористого бора, ацетамид, DCM; (ii) Zn, I2, Pd2(dba)3, дициклогексил(2',6'-диметокси-[1,1'-бифенил]-2-ил)фосфин, DMF; (iii) деблокирование, например, деблокирование метилового эфира: NaOH, MeOH, вода.
Другой энантиомер можно получить аналогичным способом, применяя схему 9.
Схема 10:
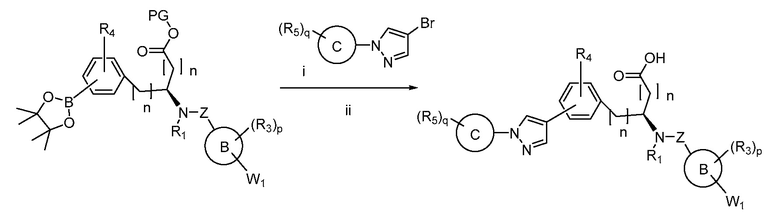
Реагенты: PG представляет собой защитную группу (i) Pd(dppf)Cl2, Na2CO3, THF, MeCN, вода; (ii) деблокирование, например, деблокирование метилового эфира: NaOH, MeOH, вода.
Другой энантиомер можно получить аналогичным способом, применяя схему 10.
Схема 11:
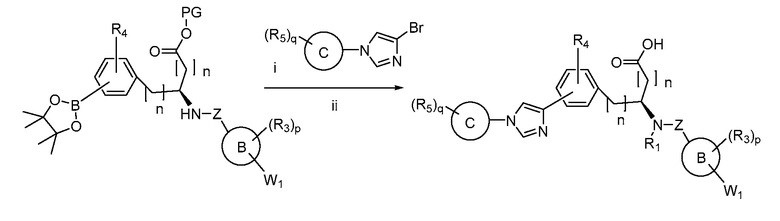
Реагенты: PG представляет собой защитную группу (i) Pd(dppf)Cl2, Na2CO3, THF, MeCN, вода; (ii) деблокирование, например, деблокирование трет-бутилового эфира: DCM, TFA
Другой энантиомер можно получить аналогичным способом, применяя схему 11.
Схема 12:
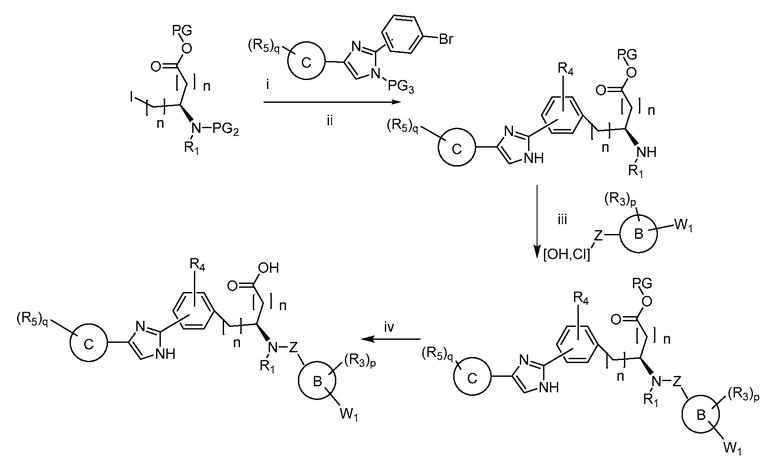
Реагенты: PG1, PG2 и PG3 представляют собой защитные группы (i) Zn, I2, Pd2(dba)3, дициклогексил(2',6'-диметокси-[1,1'-бифенил]-2-ил)фосфин, DMF; (ii) деблокирование PG2, например, трет-бутилкарбоната, и PG3, например, SEM деблокирование: DCM, TFA; (iii) если Z=CO, то конденсация с кислотой:основанием (DIEA, TEA или NMM), конденсирующие реагенты (EDC, HOBt или DCC, HOBt, или DCC, DMAP или HATU), растворитель (DMF или DCM); если Z=SO2, то конденсация с сульфонилхлоридом: DIEA или TEA, DCM или DMF; (iv) деблокирование PG, например, деблокирование трет-бутилового эфира: DCM, TFA
Другой энантиомер можно получить аналогичным способом, применяя схему 12.
Схема 13:
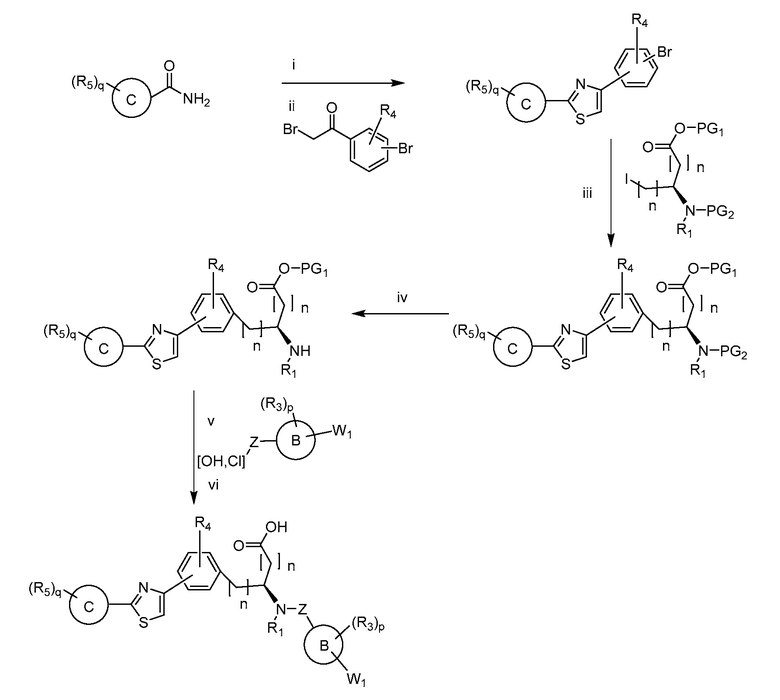
Реагенты: PG1 и PG2 представляют собой защитные группы (i) 2,4-бис(4-феноксифенил)-1,3,2,4-дитиадифосфетан 2,4-дисульфид, DME, THF; (ii) изопропанол; (iii) Zn, I2, Pd2(dba)3, дициклогексил(2',6'-диметокси-[1,1'-бифенил]-2-ил)фосфин, DMF; (iv) деблокирование PG2, например, деблокирование трет-бутилового эфира: DCM, TFA; (v) если Z=CO, то конденсация с кислотой:основанием (DIEA, TEA, или NMM), конденсирующие реагенты (EDC, HOBt или DCC, HOBt, или DCC, DMAP или HATU), растворитель (DMF или DCM); если Z=SO2, то конденсация с сульфонил-Cl: DIEA или TEA, DCM или DMF; (vi) деблокирование PG1, например, деблокирование метилового эфира: NaOH, MeOH, вода.
Другой энантиомер можно получить аналогичным способом, применяя схему 13.
Схема 14:
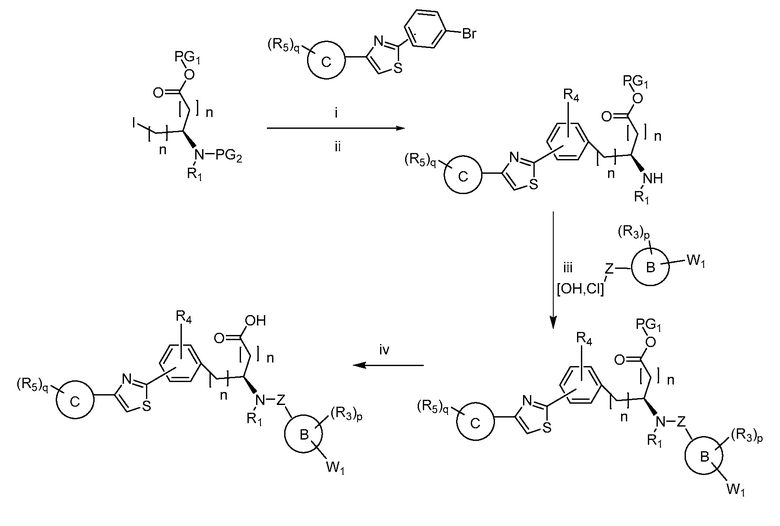
Реагенты: PG1 и PG2 представляют собой защитные группы: (i) Zn, I2, Pd2(dba)3, дициклогексил(2',6'-диметокси-[1,1'-бифенил]-2-ил)фосфин, DMF; (ii) деблокирование PG2, например, деблокирование трет-бутилкарбоната: DCM, TFA; (iii) если Z=CO, то конденсация с кислотой:основанием (DIEA, TEA, или NMM), конденсирующие реагенты (EDC, HOBt или DCC, HOBt, или DCC, DMAP или HATU), растворитель (DMF или DCM); если Z=SO2, то конденсация с сульфонил-Cl: DIEA или TEA, DCM или DMF; (iv) деблокирование PG1, например, деблокирование трет-бутилового эфира: DCM, TFA.
Другой энантиомер можно получить аналогичным способом, применяя схему 14.
Схема 15:
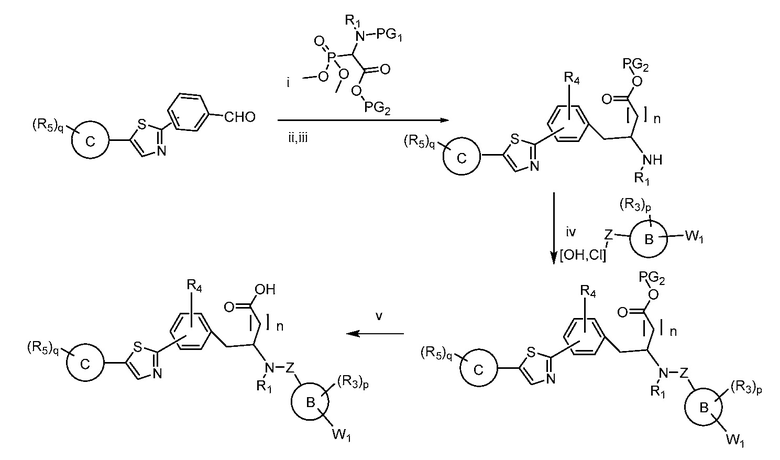
Реагенты: PG1 и PG2 представляют собой защитную группу (i) 1,1,3,3-тетраметилгуанидин, THF; (ii) H2, диоксан; (iii) деблокирование PG1, например, деблокирование boc-амина: DCM, TFA; (iv) если Z=CO, то конденсация с кислотой:основанием (DIEA, TEA, или NMM), конденсирующие реагенты (EDC, HOBt или DCC, HOBt, или DCC, DMAP или HATU), растворитель (DMF или DCM); если Z=SO2, то конденсация с сульфонил-Cl: DIEA или TEA, DCM или DMF; (v) деблокирование PG2, например, деблокирование трет-бутилового эфира: DCM, TFA.
Схема 16:
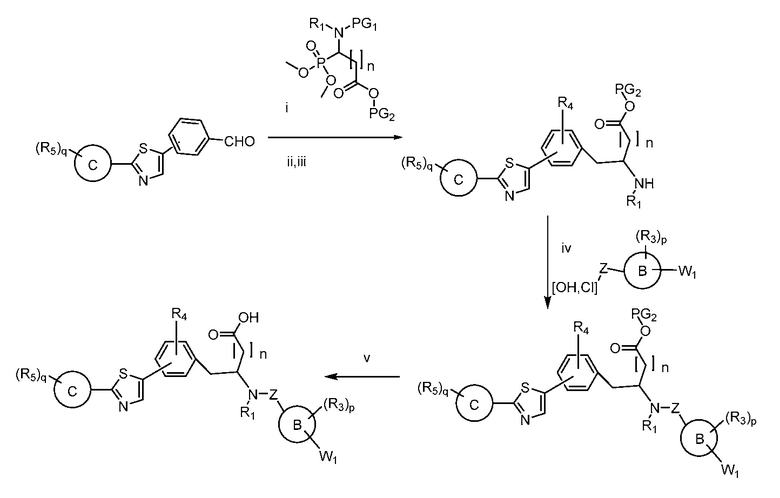
Реагенты: PG1 и PG2 представляют собой защитные группы (i) 1,1,3,3-тетраметилгуанидин, THF; (ii) H2, диоксан; (iii) деблокирование PG1, например, деблокирование boc-амина: DCM, TFA; (iv) Если Z=CO, то конденсация с кислотой:основанием (DIEA, TEA, или NMM), конденсирующие реагенты (EDC, HOBt или DCC, HOBt, или DCC, DMAP или HATU), растворитель (DMF или DCM); Если Z=SO2, то конденсация с сульфонил-Cl: DIEA или TEA, DCM или DMF; (v) деблокирование PG2, например, деблокирование трет-бутилового эфира: DCM, TFA.
Схема 17:
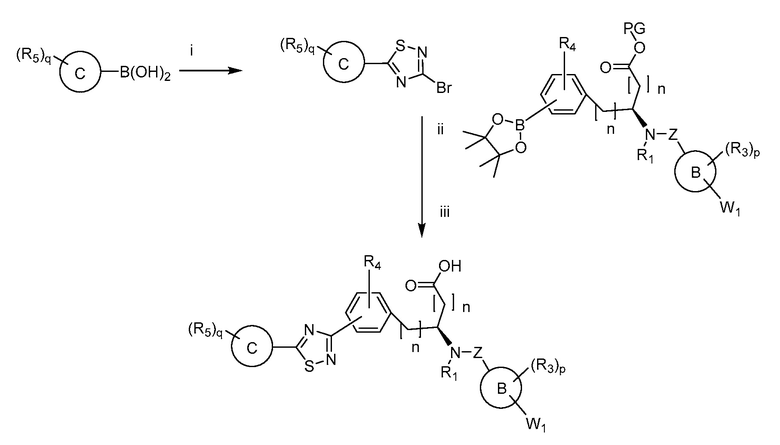
Реагенты: PG представляет собой защитную группу (i) 3-бром-5-хлор-1,2,4-тиадиазол, NaHCO3, Pd(dppf)Cl2, вода и THF, ACN или диоксан; (ii) NaHCO3, Pd(dppf)Cl2, вода и THF, ACN или диоксан; (iii) деблокирование PG, например, деблокирование трет-бутилового эфира: DCM, TFA.
Другой энантиомер можно получить аналогичным способом, применяя схему 17.
Схема 18:
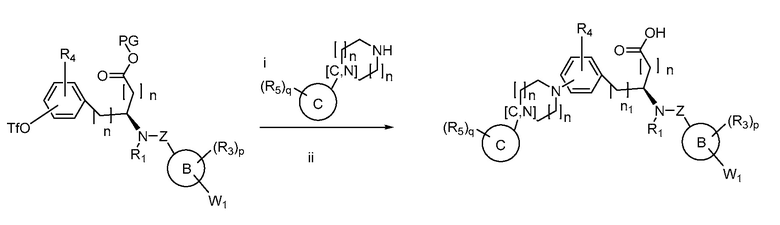
Реагенты: PG представляет собой защитную группу (i) NaO'Bu или Cs2CO3, Pd(dppf)Cl2 или Pd2(dba)3, 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил, вода и THF, ACN или диоксан; (ii) деблокирование PG, например, деблокирование трет-бутилового эфира: DCM, TFA.
Другой энантиомер можно получить аналогичным способом, применяя схему 18.
Схема 19:
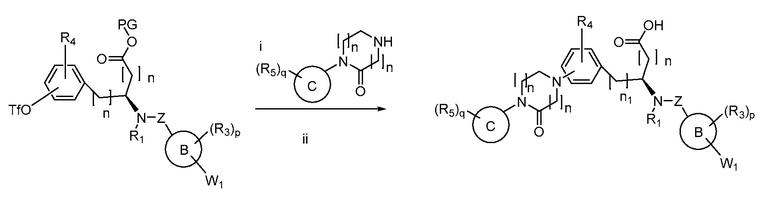
Реагенты: PG представляет собой защитную группу (i) NaO'Bu или Cs2CO3, Pd2(dppf)Cl2 или Pd2(dba)3, 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил, вода и THF, ACN или диоксан; (ii) деблокирование PG, например, деблокирование трет-бутилового эфира: DCM, TFA.
Другой энантиомер можно получить аналогичным способом, применяя схему 19.
Схема 20:
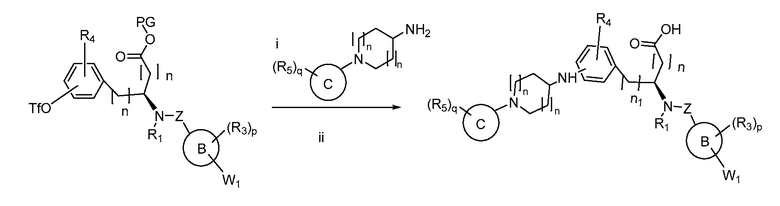
Реагенты: PG представляет собой защитную группу (i) NaO'Bu или Cs2CO3, Pd(dppf)Cl2 или Pd2(dba)3, 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил, вода и THF, ACN или диоксан; (ii) деблокирование PG, например, деблокирование трет-бутилового эфира: DCM, TFA.
Другой энантиомер можно получить аналогичным способом, применяя схему 20.
Схема 21:
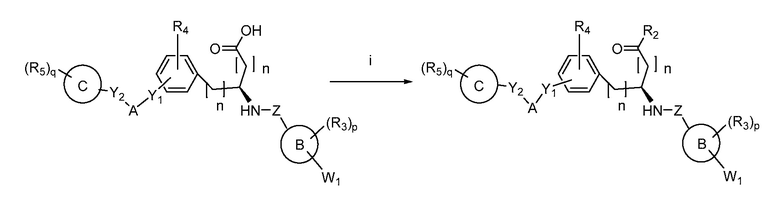
Реагенты: PG представляет собой защитную группу (i) (a) где R2 представляет собой NH-(CRaRb)m-COOH: NH2-(CRaRb)m-COOPG, HATU, DMF, затем деблокирование, например, деблокирование трет-бутилового эфира: DCM, TFA;(b) где R2 представляет собой NH-SO2-R8: R8SO2NH2, DCC, DMAP, DCM (c) где R2 представляет собой NR41R42:HNR41R42, HATU, DMF, затем деблокирование, например, деблокирование трет-бутилового эфира: DCM, TFA: (d) где R2 представляет собой N(R1)-(CRaRb)m-CO-N(R1)-гетероциклил: HN(R1)-(CRaRb)m-CO-N(R1)-гетероциклил, HATU, DMF, затем деблокирование, например, деблокирование трет-бутилового эфира: DCM, TFA; (e) где R2 представляет собой -N(R1)-(CRaRb)m-CO-N(R1)(R7): NH2-(CRaRb)m-COOPG, HATU, DMF, затем деблокирование, например, деблокирование трет-бутилового эфира: DCM, TFA, затем HN(R1)(R7), HATU, DMAP, DCM (f), где R2 представляет собой N(R1)-гетероциклил: HN(R1)-гетероциклил, HATU, DMF, затем деблокирование, например, деблокирование трет-бутилового эфира: DCM, TFA.
Другой энантиомер и диастереоизомер можно получить аналогичным способом, применяя схему 21.
Схема 22:
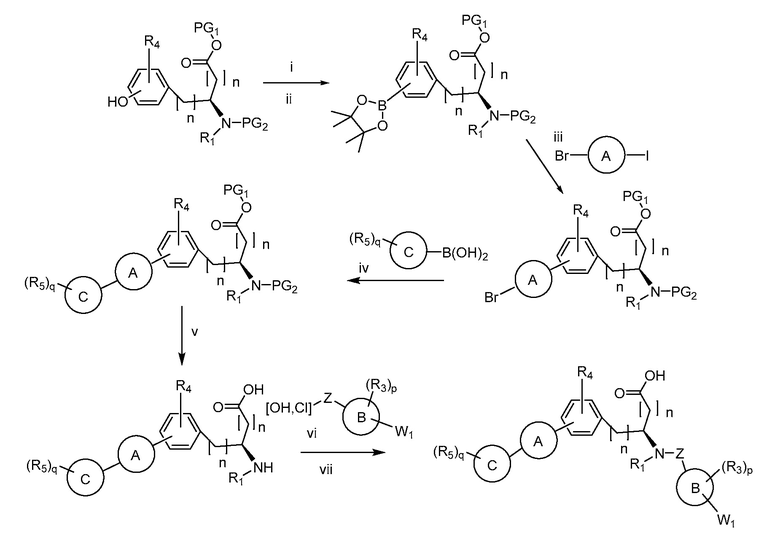
Реагенты: PG1 и PG2 представляют собой защитные группы (i) DIEA, 1,1,1-трифтор-N-фенил-N-((трифторметил)сульфонил)метансульфамид, DCM; (ii) KOAc, бис-пинаколатоборан, PdCl2(dppf); (iii) Pd(dppf)Cl2, Na2CO3, THF, MeCN, вода; (iv) Pd(dppf)Cl2, Na2CO3, THF, MeCN, вода; (v) деблокирование PG2, например, CBZ: Pd/C, H2, EA; (vi) Если Z= CO, то амидная конденсация с хлорангидридом карбоновой кислоты: DIEA, DCM или амидная конденсация с кислотой: EDC, HOBt, DMF или HATU, DMF; если Z=SO2, то конденсация с сульфонилхлоридом: DIEA или NEt3, DCM или DMF; (vii) деблокирование PG1, например, деблокирование трет-бутилового эфира: DCM, TFA.
Другой энантиомер можно получить аналогичным способом, применяя схему 22.
Схема 23:
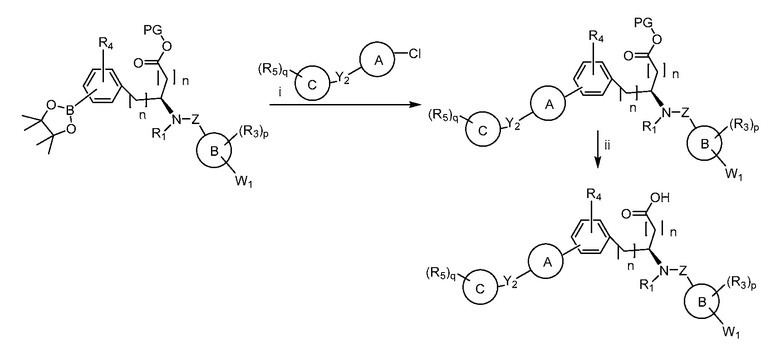
Реагенты: PG представляет собой защитную группу (i) Pd(dppf)Cl2, Na2CO3, THF, MeCN, вода; (ii) деблокирование PG, например, деблокирование трет-бутилового эфира: DCM, TFA.
Другой энантиомер можно получить аналогичным способом, применяя схему 23.
Схема 24:
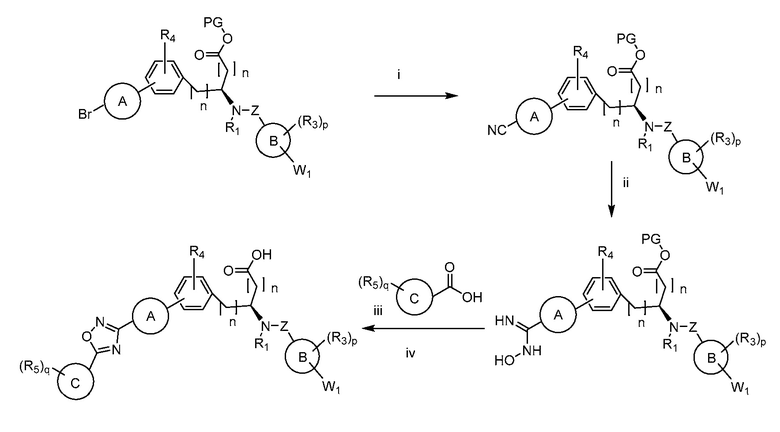
Реагенты: PG представляет собой защитную группу (i) Zn(CN)2, Pd(Ph3)4, NMP; (ii) гидроксиламин, NEt3, EtOH; (iii) EDC, HOBt, DMF, затем нагревание; (iv) деблокирование PG, например, деблокирование трет-бутилового эфира: DCM, TFA.
Другой энантиомер можно получить аналогичным способом, применяя схему 24.
Схема 25:

Реагенты: PG представляет собой защитную группу (i) NH4Cl, NaN3, DMF; (ii) CsCO3 или K2CO3, DMF, ацетон или ацетонитрил; (iii) деблокирование PG, например, деблокирование трет-бутилового эфира: DCM, TFA.
Другой энантиомер можно получить аналогичным способом, применяя схему 25.
Схема 26:
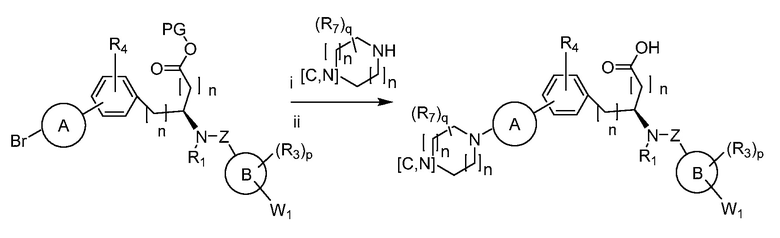
Реагенты: PG представляет собой защитную группу (i) трет-бутоксид натрия, Pd2(dba)3, диоксан; (ii) деблокирование PG, например, деблокирование трет-бутилового эфира: DCM, TFA.
Другой энантиомер можно получить аналогичным способом, применяя схему 26.
Схема 27:
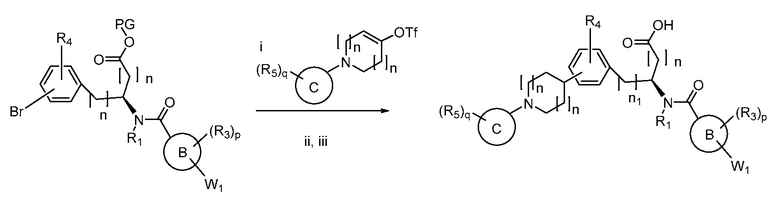
Реагенты: PG представляет собой защитную группу (i) NaO'Bu или Cs2CO3, Pd2(dppf)Cl2 или Pd2(dba)3, 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил, вода и THF, ACN или диоксан; (ii) Pd/C, H2, EtOH, (iii) деблокирование PG, например, деблокирование трет-бутилового эфира: DCM, TFA.
Другой энантиомер можно получить аналогичным способом, применяя схему 27.
ПРИМЕРЫ
Настоящее изобретение далее проиллюстрировано следующими примерами. Примеры ниже являются неограничивающими и просто репрезентативными различных аспектов настоящего изобретения.
Общие способы
ЯМР спектры
1H NMR (400 МГц) и 13C NMR (100 МГц) получали в растворе дейтерохлороформа (CDCl3) или диметилсульфоксида (d6-DMSO). ЯМР спектры обрабатывали, применяя MestReNova 6.0.3-5604.
LCMS данные
Масс-спектры (LCMS) получали, применяя одну из 5 систем. Система 1: Agilent 1100/6110 ВЭЖХ система, снабженная Thompson ODS-A, 100A, 5 мкм (50×4,6 мм) колонкой, применяя воду с 0,1% муравьиной кислотой в качестве подвижной фазы A, и ацетонитрил с 0,1% муравьиной кислотой в качестве подвижной фазы B. Способ 1: 20-100% подвижной фазы B в течение 2,5 минут, затем выдерживание при 100% в течение 2,5 минут со скоростью потока 1 мл/мин. Способ 2: 5% подвижной фазы B в течение 1 минуты, 5-95% в течение 9 минут, затем выдерживание при 95% в течение 10 минут, со скоростью потока 1 мл/мин. Способ 3: 20-100% подвижной фазы B в течение 2,5 минут, затем выдерживание при 100% в течение 4,5 минут со скоростью потока 1 мл/мин. Система 2: Agilent 1200 LCMS, снабженная Agilent Zorbax Extend RRHT 1,8 мкм (4,6×30 мм) колонкой, применяя воду с 0,1% муравьиной кислотой в качестве подвижной фазы A и ацетонитрил с 0,1% муравьиной кислотой в качестве подвижной фазы B. Способ 4: 5-95% подвижной фазы B в течение 3,0 минут со скоростью потока 2,5 мл/мин, затем выдерживание при 95% в течение 0,5 минут со скоростью потока 4,5 мл/мин. Способ 5: 5-95% подвижной фазы B в течение 14 минут со скоростью потока 2,5 мл/мин, затем выдерживание при 95% в течение 0,5 минут со скоростью потока 4,5 мл/мин. Система 3: Waters Fractionlynx LCMS система, снабженная Agilent Zorbax Extend RRHT 1,8 мкм (4,6×30 мм) колонкой, применяя воду с 0,1% муравьиной кислотой в качестве подвижной фазы A и ацетонитрил с 0,1% муравьиной кислотой в качестве подвижной фазы B. Способ 6: 5-95% подвижной фазы B в течение 3,0 минут со скоростью потока 2,5 мл/мин, затем выдерживание при 95% в течение 0,5 минут со скоростью потока 4,5 мл/мин. Способ 7: 5-95% подвижной фазы B в течение 14 минут со скоростью потока 2,5 мл/мин, затем выдерживание при 95% в течение 0,5 минут со скоростью потока 4,5 мл/мин. Система 4: Agilent 1260 LCMS, снабженная Agilent Zorbax Extend RRHT 1,8 мкм (4,6×30 мм) колонкой, применяя воду с 0,1% муравьиной кислотой в качестве подвижной фазы A и ацетонитрил с 0,1% муравьиной кислотой в качестве подвижной фазы B. Способ 8: 5-95% подвижной фазы B в течение 3,0 минут со скоростью потока 2,5 мл/мин, затем выдерживание при 95% в течение 0,5 минут со скоростью потока 4,5 мл/мин. Способ 9: 5-95% подвижной фазы B в течение 14 минут со скоростью потока 2,5 мл/мин, затем выдерживание при 95% в течение 0,5 минут со скоростью потока 4,5 мл/мин. Система 5: Agilent 1260 LCMS, снабженная Waters Xselect CSH C18 3,5 мкм (4,6×50 мм) колонкой, применяя воду с 0,1% муравьиной кислотой в качестве подвижной фазы A и ацетонитрил с 0,1% муравьиной кислотой в качестве подвижной фазы B. Способ 10: Градиент представлял собой 5-95% подвижной фазы B в течение 13,0 минут со скоростью потока 2,5 мл/мин, затем выдерживание при 95% в течение 1,0 минуты со скоростью потока 4,5 мл/мин. Способ 11: Градиент представлял собой 5-95% подвижной фазы B в течение 3,0 минут со скоростью потока 2,5 мл/мин, затем выдерживание при 95% в течение 0,6 минут со скоростью потока 4,5 мл/мин.
Условия реакций и сокращения
Пиридин, дихлорметан (DCM), тетрагидрофуран (THF) и толуол, применяемые в способах, получали из Aldrich Sure-Seal емкостей или Acros AcroSeal сухого растворителя и хранили в атмосфере азота (N2). Все реакционные смеси перемешивали механически, и температуры представляли собой внешние температуры реакции. Соединения с центрами, способными образовывать соль, считали солями трифторуксусной кислоты (TFA). Применяли следующие сокращения: этилацетат (EA), 1-метил-2-пирролидинон (NMP), триэтиламин (TEA), N-гидроксибензотриазол (HOBt), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодимида (EDC), N,N-диметилформамид (DMF), диметилацетамид (DMA), ди-трет-бутилдикарбонат (Boc2O), N,N-диизопропилэтиламин (DIEA), уксусная кислота (AcOH), хлористоводородная кислота (HCl), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU), 4-диметиламинопиридин (DMAP), трет-бутанол (трет-BuOH), гидрид натрия (NaH), триацетоксиборгидрид натрия (Na(OAc)3BH), этанол (EtOH), метанол (MeOH), ацетонитрил (ACN).
Очистка
Хроматографию осуществляли, применяя или Combiflash Rf систему для быстрой очистки (Teledyne Isco), снабженную Redisep (Teledyne Isco), Telos (Kinesis) или GraceResolv (Grace Davison Discovery Sciences) силикагельными (SiO2) колонками. Препаративную ВЭЖХ очистку осуществляли, применяя одну из двух систем. Система 1: Varian Pro Star/Prep Star система, снабженная Waters SunFire Prep C18 OBD, 5 мкм (19×150 мм) колонкой, применяя воду, содержащую 0,05% трифторуксусной кислоты в качестве подвижной фазы A, и ацетонитрил с 0,05% трифторуксусной кислоты в качестве подвижной фазы B. Градиент представлял собой 40-95% подвижной фазы B в течение 10 минут, выдерживание при 95% в течение 5-10 минут, и затем возвращение 40% в течение 2 минут со скоростью потока 18 мл/мин. Фракции собирали, применяя Varian Prostar коллектор фракций при УФ-детектировании при 254 нм и упаривали, применяя Savant SpeedVac Plus вакуумный насос или Genevac EZ-2. Система 2: Waters Fractionlynx система, снабженная Agilent Prep-C18, 5 мкм (21,2×50 мм) колонкой, применяя воду, содержащую 0,1% муравьиной кислоты в качестве подвижной фазы A и ацетонитрил с 0,1% муравьиной кислоты в качестве подвижной фазы B. Градиент представлял собой 45-95% подвижной фазы B в течение 7,5 минут, выдерживание при 95% в течение 1 минуты, и затем возвращение до 45% в течение 1,5 минут со скоростью потока 28 мл/мин. Фракции собирали при УФ-детектировании при 254 нм или масс и упаривали, применяя Genevac EZ-2.
Хиральные способы
Энантиомерный избыток определяли интегрированием пиков, которые разделялись на Diacel Chiralpak IA, 4,6×250 мм колонке, 5 мкм размер частиц. Применяемыми растворителями были "растворитель A": 4:1 (гексан с 0,2% TFA): DCM, и "растворитель B": EtOH. Скорость потока поддерживали равной 1,0 мл/минута со следующим градиентом: повышение количества растворителя B 2-10% в течение 30 минут, выдерживание растворителя B при 10% в течение 15 минут.
Экспериментальные способы
Общие способы
Общий способ 1: Получение нитрилов.
Перемешиваемый раствор бромида или трифлата (1 экв), цианида цинка (2 экв.) и тетракис(трифенилфосфин)палладия (1-5 моль %) в сухом NMP (0,5-1 M) дегазировали N2. Реакцию нагревали до 100°C в течение 18 часов при перемешивании в атмосфере N2. Реакционную смесь охлаждали и выливали в воду и DCM. Твердый материал удаляли фильтрованием, и фильтрат экстрагировали водой. Органический слой сушили над MgSO4 и концентрировали. Неочищенный продукт очищали хроматографией.
Общий способ 2: получение амидоксимов.
К перемешиваемому раствору нитрила (1 экв.) в EtOH добавляли гидроксиламин (50% раствор в H2O, 5 экв.) и TEA (1,1 экв.). Смесь нагревали в течение 2-12 часов при 80-85°C, затем концентрировали. Полученный в результате твердый остаток растворяли в EA, промывали водой, затем сушили Na2SO4, концентрировали и применяли без дополнительной очистки. Альтернативно, к перемешиваемому раствору нитрила (1 экв.) и TEA (2-3 экв.) в DMF или EtOH добавляли гидрохлорид гидроксиламина (2-3 экв.). Смесь перемешивали при комнатной температуре вплоть до 80°C в течение вплоть до 24 часов, затем концентрировали. Полученный в результате твердый остаток растворяли в EA или DCM, промывали водой или соляным раствором, затем сушили Na2SO4, концентрировали и применяли без дополнительной очистки.
Общий способ 3: получение амидов через хлорангидриды карбоновых кислот.
Раствор амина (1 экв.) и основания (или DIEPA или TEA) (2-3 экв.) в DCM (0,06-0,30 M) обрабатывали подходящим хлорангидридом карбоновой кислоты (1,0-1,5 экв.). Реакционную смесь перемешивали до завершения реакции. Реакцию разбавляли DCM и промывали насыщенным водным NaHCO3. Органический слой сушили над MgSO4 и концентрировали. Продукт очищали хроматографией. Альтернативно, неочищенную реакционную смесь применяли в следующей стадии без дополнительной очистки.
Общий способ 4: гидролиз эфиров до кислот.
К перемешиваемому раствору эфира (1 экв.) в THF или диоксане и воде добавляли NaOH или LiOH (1-3 экв.). Реакционную смесь перемешивали при вплоть до 60°C в течение вплоть до 18 часов. Реакционную смесь нейтрализовали AcOH или HCl, и разбавляли водой или концентрировали. Если реакционную смесь разбавляли водой, то HCl добавляли для подкисления реакционной смеси до pH приблизительно 2. Полученный в результате остаток выделяли фильтрованием, получая продукт, который можно очищать хроматографией, препаративной ВЭЖХ или применять без очистки. Если реакционную смесь концентрировали, неочищенный материал разбавляли DCM или EA и промывали соляным раствором. Органический слой концентрировали и очищали хроматографией или препаративной ВЭЖХ, получая конечный продукт. Альтернативно, неочищенный материал можно применять далее без очистки.
Общий способ 5: получение оксадиазолов из кислот или хлорангидридов карбоновых кислот.
Оксадиазолы из кислот:
К раствору кислоты (1 экв.) в DMF добавляли HOBt (2 экв.) и EDC (2 экв.). После перемешивания в течение 2 часов добавляли амидоксим (2 экв.), и смесь перемешивали при комнатной температуре в течение вплоть до 12 часов. Затем, реакционную смесь нагревали при 100°C в течение вплоть до 12 часов.
Альтернативно, после перемешивания при комнатной температуре, реакционную смесь разбавляли DCM, промывали NaHCO3, затем сушили Na2SO4 и концентрировали. Полученный в результате остаток растворяли в EtOH и грели в микроволновой печи в течение 35 минут при 110°C. Растворитель удаляли, и конечный продукт очищали препаративной ВЭЖХ.
Оксадиазолы из хлорангидридов карбоновых кислот:
Для получения оксадиазолов из хлорангидридов карбоновых кислот, диоксаны и DIEA (1,5 экв.) добавляли к перемешиваемому раствору амидоксима (1 экв.), с последующим добавлением хлорангидрида карбоновой кислоты (1,1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, затем при 120°C в течение вплоть до 6 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли EA и промывали соляным раствором. Органические слои концентрировали, и остаток очищали хроматографией.
Общий способ 6: удаление трет-бутилкарбамата.
Раствор трет-бутилкарбамата (1 экв.) в DCM (0,06 M) обрабатывали TFA (0,16-0,33 M). Реакционную смесь перемешивали или при комнатной температуре или при 30°C до завершения. Растворитель удаляли, и продукт очищали хроматографией или препаративной ВЭЖХ.
Общий способ 7: Получение амидов пептидной конденсацией.
Раствор амина (1,0 экв.) и основания (DIEA, TEA или NMM) (0-3,0 экв.) в DCM или DMF (0,08-0,10 M) обрабатывали подходящей карбоновой кислотой (1,0-1,5 экв.). К данной смеси добавляли конденсирующий реагент. Конденсирующий реагент мог представлять собой HATU (1,05-2,5 экв.), EDC (1,5 экв.) с HOBt (1,5 экв.), DCC (1,1 экв.) с HOBt (1,1 экв.) или DCC (1,5 экв.) с DMAP (2,0 экв.). Реакционную смесь перемешивали до завершения реакции. Реакцию разбавляли EA и промывали насыщенным водным NaHCO3. Органический слой сушили над MgSO4 и концентрировали. Продукт очищали хроматографией или альтернативно применяли в следующей стадии без дополнительной очистки.
Общий способ 8: деблокирование трет-бутиловых эфиров до кислот или деблокирование Boc-аминов
Раствор трет-бутилового эфир или Boc-амина (1,00 экв.) в DCM (0,06 M) обрабатывали TFA (0,16-0,33 M). Реакционную смесь перемешивали или при комнатной температуре или при 30°C до завершения. Растворитель удаляли, и продукт очищали хроматографией или препаративной ВЭЖХ.
Общий способ 9: получение трифлата.
Раствор фенола (1,0 экв.) в DCM (0,25 M) обрабатывали 1,1-трифтор-N-фенил-N-((трифторметил)сульфонил)метансульфамидом (1,1 экв.). Реакционную смесь перемешивали при комнатной температуре до завершения. Реакцию перемешивали с водой и насыщенным водным NaHCO3. Органические слои сушили и концентрировали. Материал очищали хроматографией или альтернативно применяли без очистки.
Способ 10: катализируемые палладием реакции конденсации.
Раствор бороновой кислоты или боронатного эфира (1,0-1,3 экв.), галогенида (1,0-1,3 экв.), бикарбоната натрия или декагидрата карбоната натрия (2,0-2,5 экв.), и Pd(dppf)Cl2 смешивали в THF, ацетонитриле или диоксане (0,1-0,2 M) и воде (0,25-0,50 M). Реакцию нагревали при 80-100°C до завершения. Реакцию разбавляли EA и промывали насыщенным водным NaHCO3. Органический слой сушили над MgSO4 и концентрировали. Продукт можно очищать хроматографией, препаративной ВЭЖХ или применять в следующей стадии без дополнительной очистки.
Общий способ 11: катализируемое палладием арильное амидирование.
Раствор арилбромида или трифлата (1,00 экв.), трет-бутоксида натрия или карбоната цезия (1-2 экв.) и амина (1,0-1,5 экв.) в диоксане или THF (0,05M) дегазировали, применяя барботирование N2 в течение 10 минут. Добавляли Pd2(dba)3 (0,10 экв.) и 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил (0,15 экв.), и реакционную смесь нагревали в течение 45-60 минут при 100-120°C в микроволновом реакторе или вплоть до 80°C общепринятым нагреванием в течение вплоть до 18 часов. Реакцию разбавляли EA и промывали насыщенным водным NaHCO3. Органический слой сушили над Na2SO4 и концентрировали. Продукт можно очищать хроматографией, препаративной ВЭЖХ, или применять в следующей стадии без дополнительной очистки.
Общий способ 12: алкилирование фенолов, имидазолов и лактамов.
К раствору фенольного промежуточного соединения в DMF, ацетоне или ACN (0,1 M) добавляли подходящий бромалкан (1,5 экв.), или CsCO3 (1,5 -2,0 экв.) или K2CO3 (1,5 -2,0 экв.). Реакционную смесь грели при 40-70°C в течение 18 часов, затем разбавляли DCM и промывали H2O. Органический слой сушили над Na2SO4 и концентрировали. Продукт можно очищать хроматографией, препаративной ВЭЖХ, или применять в следующей стадии без дополнительной очистки.
Общий способ 13: получение сульфоната или сульфамида.
К раствору спирта или амина в DCM (0,02 M) добавляли сульфонилхлорид (2 экв.) и триэтиламин (3 экв.). Реакцию перемешивали при комнатной температуре до завершения. Реакцию разбавляли DCM и промывали насыщенным водным NaHCO3. Органический слой сушили над MgSO4 и концентрировали. Продукт можно очищать хроматографией, препаративной ВЭЖХ, или применять в следующей стадии без дополнительной очистки.
Общий способ 14: восстановление арилнитро до ариламина.
К перемешиваемому раствору арилнитро (1 экв.) в THF, продутому N2, добавляли палладий на угле. Реакционную смесь помещали в атмосферу H2 на вплоть до 4 часов. Реакционную смесь можно фильтровать через слой целлита, и растворитель концентрировали. Неочищенный материал применяли в следующей стадии без дополнительной очистки.
Общий способ 15: Получение вторичного или третичного амина восстановительным аминированием.
К перемешиваемому раствору альдегида или кетона (0,9-1,0 экв.) в DCM или 1,2-дихлорэтане или THF добавляли амин (0,9-1,1 экв.). После перемешивания при комнатной температуре в течение вплоть до 2 часов, добавляли одну каплю уксусной кислоты (необязательно), с последующим добавлением триацетоксиборгидрида натрия (1,5-2,0 экв.), и реакционную смесь перемешивали в течение ночи. В некоторых случаях необходимо фильтровать реакционную смесь, перерастворять и добавлять дополнительное количество восстанавливающего агент для доведения реакции до завершения. Неочищенную реакционную смесь гасили NaHCO3 и перемешивали в течение 5 минут. Водный слой экстрагировали DCM, и органический слой сушили над MgSO4 и концентрировали. Конечный продукт выделяли хроматографией.
Общий способ 16: Получение 2-йодпиримидинов
К перемешиваемому раствору 2-хлорпиримидина (1 экв.) в 57% водной йодистоводородной кислоте (1 мл) добавляли йодид натрия (2 экв.). Реакционную смесь перемешивали при температуре окружающей среды до потребления исходного соединения. Реакционную смесь гасили NaHCO3 (5 мл), затем экстрагировали EA (3×5 мл). Объединенный органический слой промывали соляным раствором (10 мл), сушили над MgSO4 и концентрировали. Неочищенный продукт применяли в следующей стадии без очистки.
Общий способ 17. Получение 2-йодпиридинов
К перемешиваемому раствору 2-хлорпиридина (1 экв.) в ацетонитриле (2 мл) добавляли йодид натрия (6 экв.). Реакционную смесь нагревали до 40°C и добавляли ацетилхлорид (0,6 экв.). Реакционную смесь перемешивали до потребления исходного соединения. Реакцию гасили NaHCO3 (5 мл) и экстрагировали EA (3×5 мл). Объединенный органический слой промывали соляным раствором (10 мл), сушили над MgSO4 и концентрировали. Неочищенный продукт применяли в следующей стадии без очистки.
Получение репрезентативных соединений
4-(гептилокси)бензонитрил
Получали, применяя общий способ 1: перемешиваемый раствор 1-бром-4-(гептилокси)бензола (2,0 г, 7,37 ммоль), цианида цинка (1,73 г, 14,74 ммоль) и тетракис(трифенилфосфин)палладия (76,12 мг, 0,07 моль) в сухом NMP (20 мл) дегазировали N2. Реакцию нагревали при 100°C в течение 18 часов при перемешивании в атмосфере азота. Реакционную смесь охлаждали и выливали в воду (100 мл) и DCM (20 мл). Твердый материал удаляли фильтрованием, и фильтрат экстрагировали водой (3×20 мл). Органический слой сушили над MgSO4 и концентрировали. Неочищенный продукт очищали хроматографией (EA/гексан), получая 1,15 г (73%) 4-(гептилокси)бензонитрила в виде светло-желтого твердого остатка. LCMS-ESI (m/z) рассчитанная для C14H19NO: 217,1; найденная 218,1 [M+H]+, tR = 11,14 минут (способ 2). 1H ЯМР (400 МГц, CDCl3) δ 7,64-7,50 (м, 2H), 7,05-6,83 (м, 2H), 3,99 (т, J=6,5 Гц, 2H), 1,89-1,69 (м, 2H), 1,58-1,12 (м, 8H), 0,90 (дд, J=9,1, 4,5 Гц, 3H). 13C ЯМР (101 МГц CDCL3) δ 162,47, 133,91, 132,78, 132,12, 129,13, 119,31, 115,18, 103,58, 68,41, 31,73, 28,98, 25,89, 22,58, 14,07.
(Z)-4-(гептилокси)-N'-гидроксибензимидамид
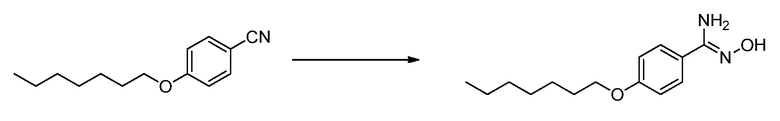
Получали, применяя общий способ 2: к перемешиваемому раствору 4-(гептилокси)бензонитрила (1,0 г, 4,6 ммоль) в EtOH (15 мл) добавляли гидрохлорид гидроксиламина (0,96 г, 13,8 ммоль) и TEA (2,22 г, 23,0 ммоль). Реакцию нагревали при 85°C в течение 2 часов. Растворитель удаляли при пониженном давлении, и остаток разбавляли водой (20 мл) и экстрагировали DCM (3×10 мл). Объединенные органические слои концентрировали при пониженном давлении. Неочищенный материал кристаллизовали из изопропанола (20 мл), получая 1,05 г (91%) (Z)-4-(гептилокси)-N'-гидроксибензимидамида в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C14H22N2O2: 250,2; найденная 251,3 [M+H]+, tR = 1,70 минут (способ 1). 1H ЯМР (400 МГц, CDCI3) δ 9,45 (с, 1H), 7,59 (д, J=8,6 Гц, 2H), 6,93 (т, J=14,7 Гц, 2H), 5,82-5,48 (м, 2H), 3,97 (т, J=6,5 Гц, 2H), 1,83-1,55 (м, 2H), 1,56-1,05 (м, 8H), 0,87 (т, J=6,7 Гц, 3H). 13C ЯМР (101 МГц CDCL3) δ 159,19, 150,53, 126,64, 125,55, 113,87, 67,40, 31,21, 28,62, 28,40, 25,44, 22,02, 13,92.
(S)-метил 4-(2-амино-3-метокси-3-оксопропил)бензоат
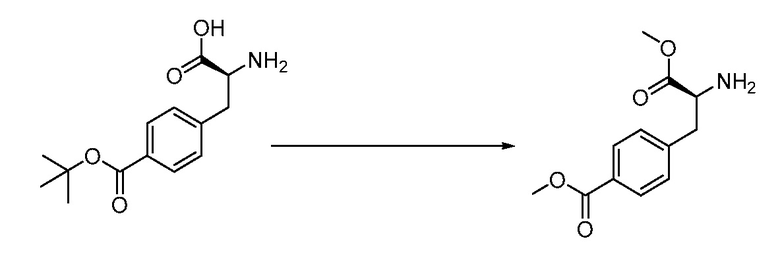
К раствору (S)-2-амино-3-(4-(трет-бутоксикарбонил)фенил)пропионовой кислоты (500,0 мг, 1,88 ммоль) в MeOH (20 мл) при 0°C медленно добавляли тионилхлорид (447,64 мг, 3,77 ммоль). Реакцию перемешивали в течение 1 часа при 0°C, затем нагревали до комнатной температуры и перемешивали в течение 1 часа. Растворитель удаляли при пониженном давлении. Реакционную смесь промывали насыщенным водным NaHCO3 (20 мл) и экстрагировали DCM (3×10 мл). Органический слой сушили над MgSO4 и концентрировали. Неочищенный продукт очищали хроматографией (EA/гексан), получая 425 мг (95%) (S)-метил 4-(2-амино-3-метокси-3-оксопропил)бензоата в виде HCl соли. LCMS-ESI (m/z) рассчитанная для C12H15NO4: 237,1; найденная 238,0 [M+H]+, tR =1,01 минут (способ 1). 1H ЯМР (400 МГц, CDCL3) δ 8,55 (с, 3H), 7,94 (д, J=8,3 Гц, 2H), 7,41 (д, J=8,3 Гц, 2H), 4,37 (т, J=6,8 Гц, 1H), 3,86 (с, 3H), 3,68 (с, 3H), 3,20 (дд, J=11,8, 6,8 Гц, 2H).
(S)-метил 4-(2-(4-(трет-бутил)бензамидо)-3-метокси-3-оксопропил)бензоат
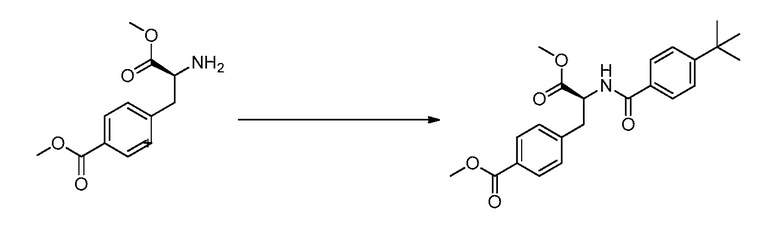
Получали, применяя общий способ 3: к раствору (S)-метил 4-(2-амино-3-метокси-3-оксопропил)бензоата (425,0 мг, 1,79 ммоль) в DCM (10 мл) и DIEA (463,0 мг, 3,58 ммоль) добавляли 4-(трет-бутил)бензоилхлорид (556,6 мг, 2,83 ммоль) при комнатной температуре. Реакцию перемешивали в течение 2 часов, и реакцию распределяли между DCM и насыщенным водным NaHCO3. Органический слой сушили над MgSO4 и концентрировали. Неочищенный продукт очищали хроматографией (EA/гексан), получая 317 мг (45%) (S)-метил 4-(2-(4-(трет-бутил)бензамидо)-3-метокси-3-оксопропил)бензоата. LCMS-ESI (m/z) рассчитанная для C23H27NO5: 397,2; найденная 398,1 [M+H]+, tR =2,31 минут (способ 1). 1H ЯМР (400 МГц, CDCL3) δ 7,97-7,75 (м, 2H), 7,67-7,51 (м, 2H), 7,46-7,26 (м, 2H), 7,14 (д, J=8,3 Гц, 2H), 6,60 (д, J=7,4 Гц, 1H), 5,03 (дт, J=7,4, 5,7 Гц, 1H), 3,82 (с, 3H), 3,68 (с, 3H), 3,28 (дд, J=13,7, 5,8 Гц, 1H), 3,18 (дд, J=13,7, 5,5 Гц, 1H), 1,24 (с, 9H).
(S)-4-(2-(4-(трет-бутил)бензамидо)-3-метокси-3-оксопропил)бензойная кислота (INT-1)
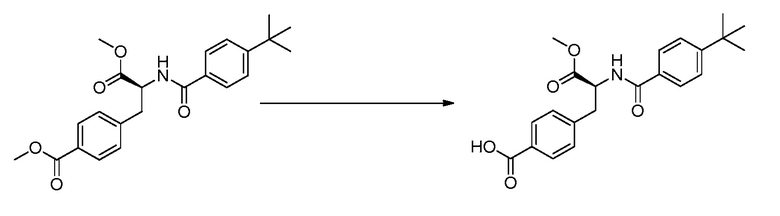
Получали, применяя общий способ 4: к перемешиваемому раствору (S)-метил 4-(2-(4-(трет-бутил)бензамидо)-3-метокси-3-оксопропил)бензоата (316,6 мг, 0,79 ммоль) в диоксане (15 мл) и воде (1 мл) при 0°C добавляли моногидрат гидроксида лития (93,52 мг, 2,23 ммоль). Через 2 часа, раствор нейтрализовали 1 M HCl до pH 7,0. Смесь распределяли между DCM (15 мл) и насыщенным водным NaHCO3 (10 мл). Органический слой промывали насыщенным водным NaHCO3 (3×10 мл) и соляным раствором (10 мл). Органический слой сушили над MgSO4 и концентрировали, получая 208 мг (69%) (S)-4-(2-(4-(трет-бутил)бензамидо)-3-метокси-3-оксопропил)бензойной кислоты INT-1. LCMS-ESI (m/z) рассчитанная для C22H25NO5: 383,2; найденная 384,1 [M+H]+, tR = 2,13 минут (способ 1). 1H ЯМР (400 МГц, DMSO) δ 12,86 (с, 1H), 8,80 (д, J=8,0 Гц, 1H), 7,87-7,78 (м, 2H), 7,75-7,65 (м, 2H), 7,50-7,35 (м, 4H), 4,72 (ддд, J=10,3, 8,0, 5,1 Гц, 1H), 3,65 (с, 3H), 3,28-3,05 (м, 2H), 1,29 (с, 9H). 13C ЯМР (101 МГц, DMSO) δ 173,00, 167,21, 166,29, 154,39, 143,10, 130,85, 129,34, 129,27, 129,21, 129,03, 127,21, 125,39, 125,10, 53,75, 52,04, 34,64, 30,92, 30,88.
(S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(3-(4-(гептилокси)фенил)-1,2,4-оксадиазол-5-ил)фенил)пропаноат
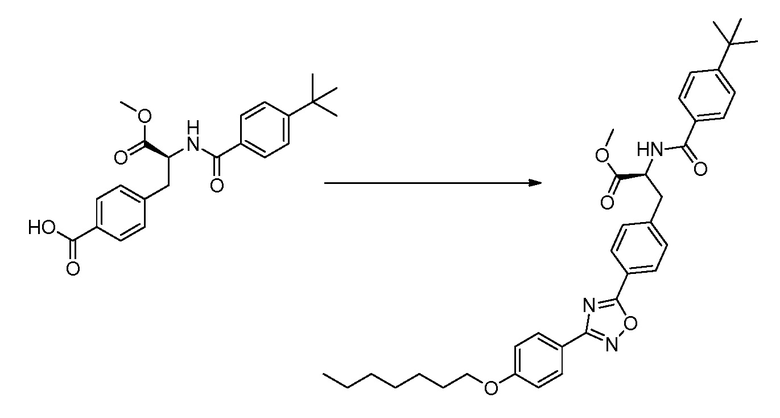
Получали, применяя общий способ 5: к раствору (S)-4-(2-(4-(трет-бутил)бензамидо)-3-метокси-3-оксопропил)бензойной кислоты, INT-1 (10,0 мг, 0,026 ммоль) в безводном DMF (1 мл) добавляли HOBt (5,27 мг, 0,39 ммоль) и EDC (7,48 мг, 0,39 ммоль). После перемешивания в течение 2 часов добавляли (Z)-4-(гептилокси)-N'-гидроксибензимидамид (9,76 мг, 0,39 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, распределяли между насыщенным водным NaHCO3 (5 мл) и EA (5 мл), и концентрировали при пониженном давлении, получая промежуточное соединение, (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(((4-(гептилокси)бензимидамидо)окси)карбонил)фенил)пропаноат.
Промежуточное соединения растворяли в DMF (1 мл) и нагревали при 100°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры и распределяли между EA (5 мл) и насыщенным водным NaHCO3 (5 мл). Органический слой экстрагировали водой (2×5 мл) и соляным раствором (5 мл). Органический слой сушили над MgSO4 и концентрировали. Коричневое масло очищали препаративной ВЭЖХ, получая 4,5 мг (29%) (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(3-(4-(гептилокси)фенил)-1,2,4-оксадиазол-5-ил)фенил)пропаноата. LCMS-ESI (m/z) рассчитанная для C36H43N3O5: 597,3; m/z не наблюдали, tR = 12,75 минут (способ 2). 1H ЯМР (400 МГц, DMSO) δ 8,85 (д, J=8,0 Гц, 1H), 8,09 (д, J=8,3 Гц, 2H), 8,00 (д, J=8,9 Гц, 2H), 7,74 (д, J=8,5 Гц, 2H), 7,59 (д, J=8,4 Гц, 2H), 7,48 (д, J=8,6 Гц, 2H), 7,12 (д, J=8,9 Гц, 2H), 4,87-4,56 (м, 1H), 4,06 (т, J=6,5 Гц, 2H), 3,67 (с, 3H), 3,32-3,13 (м, 4H), 1,74 (дд, J=14,2, 6,5 Гц, 2H), 1,51-1,37 (м, 2H), 1,33 (с, 4H), 1,26 (д, J=20,2 Гц, 9H), 0,88 (т, J=6,9 Гц, 3H). 13C ЯМР (101 МГц, DMSO) δ 175,00, 171,91, 167,89, 166,27, 161,21, 154,37, 143,68, 130,78, 130,30, 128,76, 127,80, 127,18, 125,07, 121,69, 118,21, 115,07, 67,72, 53,61, 52,05, 36,15, 34,60, 31,20, 30,87, 28,54, 28,39, 25,40, 22,02, 13,93.
(S)-2-(4-(трет-бутил)бензамидо)-3-(4-(3-(4-(гептилокси)фенил)-1,2,4-оксадиазол-5-ил)фенил)пропионовая кислота (соединение 1)
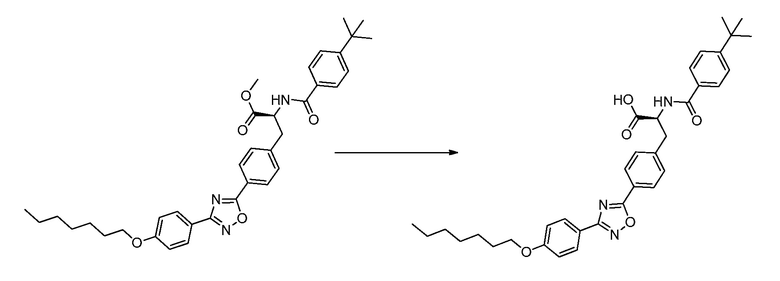
Получали, применяя общий способ 4: к раствору (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(3-(4-(гептилокси)фенил)-1,2,4-оксадиазол-5-ил)фенил)пропаноата (4,52 мг, 0,008 ммоль) в MeOH (2 мл) добавляли 1н NaOH (1 мл). Реакционную смесь перемешивали при 50°C в течение 3 часов. Полученную в результате смесь очищали препаративной ВЭЖХ, получая 0,36 мг (8%) (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(3-(4-(гептилокси)фенил)-1,2,4-оксадиазол-5-ил)фенил)пропионовой кислоты. LCMS-ESI (m/z) рассчитанная для C35H41N3O5: 583,7; m/z не наблюдали, tR = 12,59 минут (способ 2).
(S)-метил 2-амино-3-(4-цианофенил)пропаноат
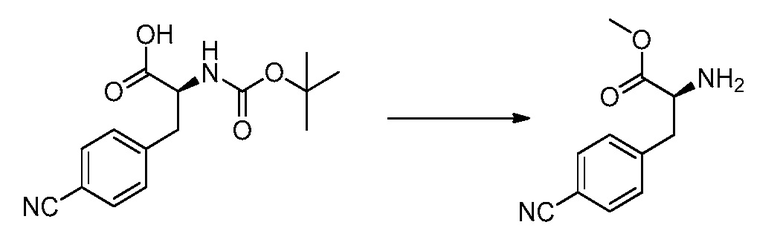
К раствору (S)-2-((трет-бутоксикарбонил)амино)-3-(4-цианофенил)пропионовой кислоты (1,0 г, 3,44 ммоль) в MeOH (20 мл) при 0°C медленно добавляли тионилхлорид (818,1 мг, 6,89 ммоль) в течение 1 часа. Реакцию нагревали до комнатной температуры и перемешивали в течение 1 часа. Растворитель удаляли при пониженном давлении. Реакционную смесь промывали насыщенным водным NaHCO3 (20 мл) и экстрагировали DCM (3×10 мл). Органический слой сушили над MgSO4 и концентрировали. Неочищенный продукт очищали хроматографией (EA/гексан), получая 789 мг (97%) (S)-метил 2-амино-3-(4-цианофенил)пропаноата в виде HCl соли. LCMS-ESI (m/z) рассчитанная для C11H12N2O2: 204,1; найденная 205,0 [M+H]+, tR = 3,25 минут (способ 1). 1H ЯМР (400 МГц, CDCL3) δ 8,69 (с, 3H), 7,83 (д, J=8,3 Гц, 2H), 7,51 (т, J=8,8 Гц, 2H), 4,37 (т, J=6,7 Гц, 1H), 3,68 (с, 3H), 3,23 (кв.д, J=14,4, 7,7 Гц, 2H).
(S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-цианофенил)пропаноат
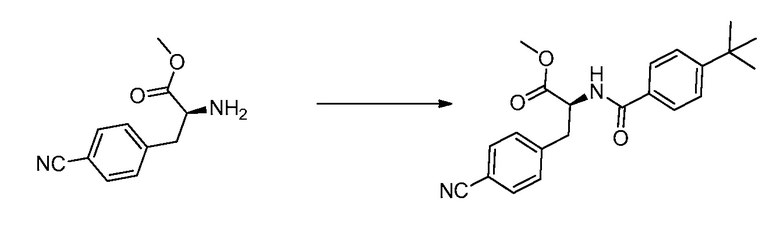
Получали, применяя общий способ 3: к раствору (S)-метил 2-амино-3-(4-цианофенил)пропаноата (789,2 мг, 3,32 ммоль) в DCM (15 мл) и DIEA (1,29 г, 9,96 ммоль) добавляли 4-(трет-бутил)бензоилхлорид (981,3 мг, 4,99 ммоль) при комнатной температуре. Реакцию перемешивали в течение 2 часов, и реакцию распределяли между DCM и насыщенным водным NaHCO3. Органический слой сушили над MgSO4 и концентрировали. Неочищенный продукт очищали хроматографией (EA/гексан), получая 1,06 г (88%) (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-цианофенил)пропаноата. LCMS-ESI (m/z) рассчитанная для C22H24N2O3: 364,2; найденная 365,3 [M+H]+, tR = 3,55 минут (способ 1). 1H ЯМР (400 МГц, CDCL3) δ 8,81 (д, J=8,0 Гц, 1H), 7,85-7,60 (м, 4H), 7,49 (дд, J=15,1, 8,4 Гц, 4H), 4,85-4,60 (м, 1H), 3,65 (с, 3H), 3,30-3,23 (м, 1H), 3,18 (дд, J=13,7, 10,6 Гц, 1H), 1,29 (с, 9H).
(S,Z)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(N'-гидроксикарбамимидоил)фенил)пропаноат (INT-2)

Получали, применяя общий способ 2: к перемешиваемому раствору (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-цианофенил)пропаноата (1,0 г, 2,74 ммоль) в EtOH (15 мл) добавляли гидрохлорид гидроксиламина (572,2 мг, 8,22 ммоль) и TEA (1,38 г, 13,7 ммоль). Реакцию нагревали при 85°C в течение 2 часов. Растворитель удаляли при пониженном давлении, и остаток разбавляли водой (20 мл) и экстрагировали DCM (3×10 мл). Объединенные органические слои концентрировали при пониженном давлении. Неочищенный материал кристаллизовали из изопропанола (20 мл), получая 1,04 г (95%) (S,Z)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(N'-гидроксикарбамимидоил)фенил)пропаноата INT-2 в виде белого твердого остатка. LCMS-ESI (m/z): рассчитанный для: C22H27N3O4, 397,2; найденная 398,1 [M+1]+, tR = 2,26 минут (способ 1). 1H ЯМР (400 МГц, DMSO) δ 10,19 (с, 1H), 9,57 (с, 1H), 8,78 (д, J=7,9 Гц, 1H), 7,74 (д, J=8,4 Гц, 2H), 7,58 (д, J=8,2 Гц, 2H), 7,48 (д, J=8,4 Гц, 2H), 7,30 (д, J=8,3 Гц, 2H), 4,79-4,49 (м, 1H), 3,65 (с, 3H), 3,15 (дт, J=13,6, 6,0 Гц, 2H), 1,75 (д, J=13,6 Гц, 1H), 1,29 (с, 9H).
(S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,2,4-оксадиазол-3-ил)фенил)пропаноат
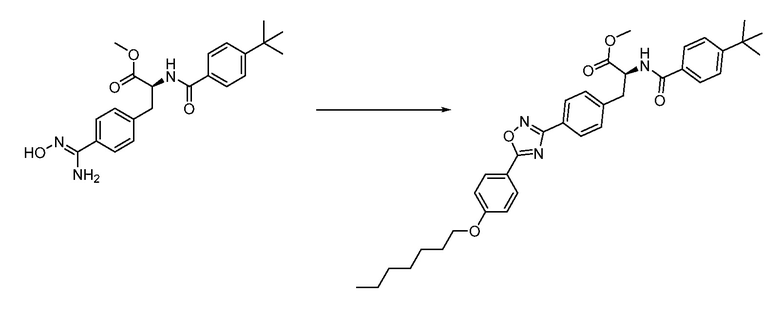
Получали, применяя общий способ 5: к раствору 4-(гептилокси)бензойной кислоты (400,0 мг, 1,54 ммоль) в безводном DMF (6 мл) добавляли HOBt (312,3 мг, 2,31 ммоль) и EDC (442,75 мг, 2,31 ммоль). После перемешивания в течение 2 часов добавляли (S,Z)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(N'-гидроксикарбамимидоил)фенил)пропаноат, INT-2 (673,3 мг, 1,69 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, распределяли между насыщенным водным NaHCO3 (15 мл) и EA (15 мл), и концентрировали при пониженном давлении, получая промежуточное соединение (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(N-((4-(гептилокси)бензоил)окси)карбамимидоил)фенил)пропаноат. Промежуточное соединение растворяли в DMF (10 мл) и нагревали при 100°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры и распределяли между EA (10 мл) и насыщенным водным NaHCO3 (50 мл). Органический слой экстрагировали водой (2×10 мл) и соляным раствором (10 мл). Органический слой сушили над MgSO4 и концентрировали. Коричневое масло очищали хроматографией (EA/гексан), получая 710 мг (77%) (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,2,4-оксадиазол-3-ил)фенил)пропаноата в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C36H43N3O5: 597,3; m/z не наблюдали, tR = 12,80 минут (способ 2). 1H ЯМР (400 МГц, DMSO) δ 8,84 (д, J=8,0 Гц, 1H), 8,08 (т, J=17,2 Гц, 2H), 7,97 (дд, J=18,2, 8,5 Гц, 2H), 7,74 (д, J=8,5 Гц, 2H), 7,50 (дд, J=18,6, 8,3 Гц, 4H), 7,18 (д, J=8,9 Гц, 2H), 4,85-4,63 (м, 1H), 4,09 (дд, J=13,8, 7,3 Гц, 2H), 3,67 (с, 3H), 3,24 (ддд, J=23,8, 15,7, 7,3 Гц, 4H), 2,08 (с, 4H), 1,74 (дд, J=14,1, 6,9 Гц, 2H), 1,42 (дд, J=13,6, 6,3 Гц, 2H), 1,30 (д, J=14,5 Гц, 9H), 0,88 (т, J=6,8 Гц, 3H). 13C ЯМР (101 МГц, DMSO) δ 174,05, 170,87, 133,81, 165,14, 161,43, 153,21, 140,51, 129,70, 128,85, 128,78, 126,06, 125,84, 123,93, 123,39, 114,36, 114,25, 66,86, 52,66, 50,88, 34,32, 33,47, 30,06, 29,74, 27,33, 27,24, 24,23, 20,89, 12,80.
(S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,2,4-оксадиазол-3-ил)фенил)пропионовая кислота (соединение 2)
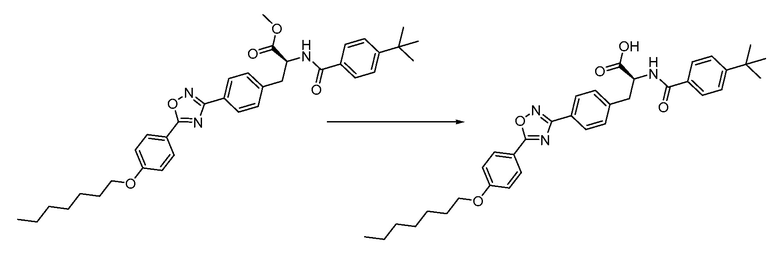
Получали, применяя общий способ 4: к раствору (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,2,4-оксадиазол-3-ил)фенил)пропаноата (710,0 мг, 1,19 ммоль) в MeOH (20 мл) добавляли 1н NaOH (10 мл). Реакционную смесь перемешивали при 50°C в течение 3 часов. Полученную в результате смесь очищали хроматографией (DCM/MeOH), получая 218 мг (31%) (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,2,4-оксадиазол-3-ил)фенил)пропионовой кислоты в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C35H41N3O5: 583,3; m/z не наблюдали, tR = 12,16 минут (способ 2). 1H ЯМР (400 МГц, DMSO) δ 8,69 (д, J=8,3 Гц, 1H), 8,16-8,02 (м, 2H), 7,98 (д, J=8,3 Гц, 2H), 7,74 (д, J=8,5 Гц, 2H), 7,53 (д, J=8,3 Гц, 2H), 7,47 (д, J=8,5 Гц, 2H), 7,18 (д, J=9,0 Гц, 2H), 4,70 (ддд, J=10,8, 8,4, 4,5 Гц, 1H), 4,09 (т, J=6,5 Гц, 2H), 3,30 (дд, J=13,8, 4,2 Гц, 1H), 3,17 (дд, J=13,8, 10,7 Гц, 1H), 1,74 (дд, J=14,5, 6,7 Гц, 2H), 1,42 (дд, J=13,8, 6,1 Гц, 2H), 1,37-1,14 (м, 14H), 0,87 (т, J=6,9 Гц, 3H). 13C ЯМР (101 МГц, DMSO) δ 175,16, 173,00, 167,96, 166,19, 162,55, 154,18, 142,11, 131,08, 129,95, 129,89, 127,14, 126,92, 125,01, 124,39, 115,49, 115,37, 67,98, 53,72, 36,19, 34,58, 31,19, 30,89, 28,46, 28,37, 25,36, 22,01, 13,92.
Соединения 3-11 и 13-61 получали из (S,Z)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(N'-гидроксикарбамимидоил)фенил)пропаноата INT-2, применяя последовательно общие способы 5 и 4.
Соединения 62-66 получали из (S,Z)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(N'-гидроксикарбамимидоил)фенил)пропаноата INT-2, применяя последовательно общие способы 5, 6 и 4.
(S)-2-(2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,2,4-оксадиазол-3-ил)фенил)пропанамидо)уксусная кислота (соединение 67)
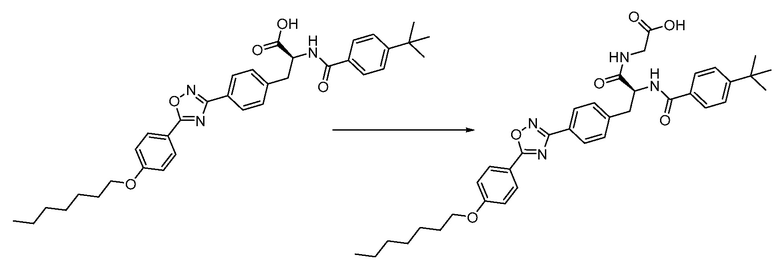
Получали, применяя общие способы 7 и 8: к раствору (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,2,4-оксадиазол-3-ил)фенил)пропионовой кислоты, соединения 2 (10,0 мг, 0,017 ммоль), в безводном DMF (1 мл) добавляли HOBt (3,52 мг, 0,027 ммоль) и EDCI (4,88 мг, 0,027 ммоль) при комнатной температуре. Через 2 часа добавляли трет-бутил 2-аминоацетат (3,49 мг, 0,027 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. LCMS анализ показал полное превращение в промежуточное соединение. Реакционную смесь распределяли между водным NaHCO3 (5 мл) и DCM (1 мл), органический слой собирали и концентрировали в вакууме, и затем перерастворяли в 1 мл DCM и 0,1 мл TFA. Смесь нагревали при 30°C в течение 3 часов. Конечное соединение очищали ВЭЖХ, получая 9,6 мг (88%) (S)-2-(2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,2,4-оксадиазол-3-ил)фенил)пропанамидо)уксусной кислоты. LCMS-ESI (m/z) рассчитанная для C37H44N4O6: 640,3; m/z не наблюдали, tR = 11,51 минут (способ 2). 1H ЯМР (400 МГц, DMSO) δ: 8,60 (д, J=8,4 Гц, 1H), 8,47 (т, J=5,7 Гц, 1H), 8,10 (д, J=8,8 Гц, 2H), 7,96 (д, J=8,2 Гц, 2H), 7,75 (д, J=8,4 Гц, 2H), 7,57 (д, J=8,0 Гц, 2H), 7,45 (д, J=8,4 Гц, 2H), 7,17 (д, J=8,8 Гц, 2H), 4,83 (д, J=8,1 Гц, 1H), 4,09 (т, J=6,4 Гц, 2H), 3,94-3,69 (м, 2H), 3,34 (с, 2H), 3,26 (д, J=13,5 Гц, 1H), 3,15-3,01 (м, 1H), 1,83-1,65 (м, 2H), 1,50-1,15 (м, 16H), 0,87 (т, J=6,7 Гц, 3H). 13C ЯМР (101 МГц, DMSO) δ: 175,12, 171,58, 171,13, 167,99, 166,02, 162,54, 154,10, 142,44, 131,16, 130,02, 129,89, 127,23, 126,81, 124,91, 124,25, 115,50, 115,36, 67,97, 54,23, 40,10, 37,12, 34,57, 31,19, 30,88, 28,46, 28,37, 25,36, 22,02, 13,93.
Соединение 68 получали из соединения 5, применяя последовательно общие способы 7 и 8.
Соединения 69 и 70 получали из (S,Z)-метил 2-(4-(трет-бутил)бензамидо)-3 -(4-(N'-гидроксикарбамимидоил)фенил)пропаноата, соединения 2, применяя последовательно общие способы 7 и 8.
Соединения 71 и 72 получали из гидрохлорида метил 2-амино-2-(4-бромфенил)ацетата, применяя последовательно общие способы 7, 1, 2, 5 и 4.
Соединения 73 и 74 получали из гидробромида (S)-метил 2-амино-4-(4-гидроксифенил)бутаноата, применяя последовательно общие способы 7, 9, 1, 2, 5 и 4.
Соединение 75 получали из гидрохлорида (S)-метил 3-амино-4-(4-гидроксифенил)бутаноата, применяя последовательно общие способы 7, 9, 1, 2, 5 и 4.
4-(гептилокси)бензогидразид
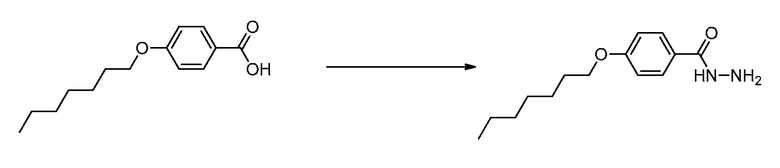
К перемешиваемому раствору 4-(гептилокси)бензойной кислоты (679 мг, 2,87 ммоль) в THF (5 мл) добавляли 1,1'-карбонилдиимидазол (559 мг, 3,45 ммоль). После перемешивания при комнатной температуре в течение 2 часов, раствор добавляли к перемешиваемой смеси гидразингидрата (0,729 мл, 5,75 ммоль) в THF (2 мл) и перемешивали дополнительные 2 часа. Реакционную смесь выливали в воду (20 мл) и перемешивали в течение 30 минут. Полученный в результате остаток собирали фильтрованием, промывали водой (2×10 мл), затем ацетонитрилом (3 мл), получая 0,54 г (71%) 4-(гептилокси)бензогидразида в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C14H22N2O2: 250,3; найденная 251,0 [M+H]+, tR = 2,05 минут (способ 4).
(S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(2-(4-(гептилокси)бензоил)гидразинкарбонил)фенил)пропаноат (INT-3)
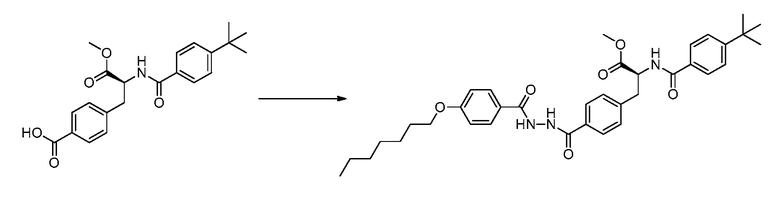
К перемешиваемому раствору (S)-4-(2-(4-(трет-бутил)бензамидо)-3-метокси-3-оксопропил)бензойной кислоты INT-1 (260 мг, 0,68 ммоль) в THF (5 мл) добавляли 4-метилморфолин (0,15 мл, 1,36 ммоль) и изобутилкарбонохлоридат (0,09 мл, 0,71 ммоль). После перемешивания при комнатной температуре в течение 2 часов добавляли 4-(гептилокси)бензогидразид (187 мг, 0,75 ммоль), и перемешивание продолжали в течение следующих 2 часов. Реакционную смесь выливали в NaHCO3 (50 мл) и экстрагировали DCM (3×20 мл). Объединенные органические слои сушили над MgSO4 и упаривали. Неочищенный продукт очищали колоночной хроматографией (100% EA в изогексане), получая 297 мг (71%) (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(2-(4- (гептилокси)бензоил)гидразинкарбонил)фенил)пропаноата INT-3 в виде грязно-белой пены. LCMS-ESI (m/z) рассчитанная для C36H45N306: 615,8 найденная 616,0 [M+H]+, tR = 2,89 минут (способ 4).
(S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,3,4-оксадиазол-2-ил)фенил)пропаноат
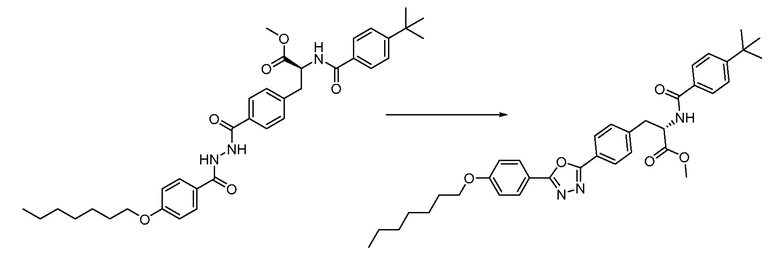
К перемешиваемому раствору (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(2-(4-(гептилокси)бензоил)гидразинкарбонил)фенил)пропаноата INT-3 (127 мг, 0,21 ммоль) и TEA (0,09 мл, 0,62 ммоль) в DCM (4 мл) добавляли хлорид 2-хлор-1,3-диметилимидазолидиния (41,8 мг, 0,25 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем нагревали при 40°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, разбавляли NaHCO3 (15 мл), встряхивали, разделяли через гидрофобную порошковую керамику и упаривали, получая 120 мг (95%) (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,3,4-оксадиазол-2-ил)фенил)пропаноат в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C36H43N305: 597,8; найденная 598,0 [M+H]+, tR = 3,25 минут (способ 4).
Соединение 76 получали, применяя (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,3,4-оксадиазол-2-ил)фенил)пропаноат и общий способ 4.
2-бром-1-(4-(гептилокси)фенил)этанон (INT-4)
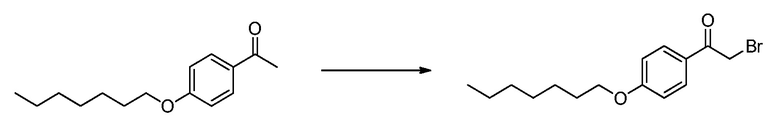
К перемешиваемому раствору 1-(4-(гептилокси)фенил)этанона (500 мг, 2,13 ммоль) в THF (8,5 мл) в атмосфере азота добавляли трибромид фенилтриметиламмония (842 мг, 2,24 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, фильтровали в вакууме, и полученный твердый остаток промывали THF. Объединенные маточные растворы концентрировали, получая 919 мг (100%) 2-бром-1-(4-(гептилокси)фенил)этанона INT-4 в виде желтого масла. LCMS-ESI (m/z) рассчитанная для C15H21BrO2: 313,2; найденная 313,0 [M+H]+, tR = 2,12 минут (способ 4).
(S)-2-(4-(гептилокси)фенил)-2-оксоэтил 4-(2-(4-(трет-бутил)бензамидо)-3-метокси-3-оксопропил)бензоат
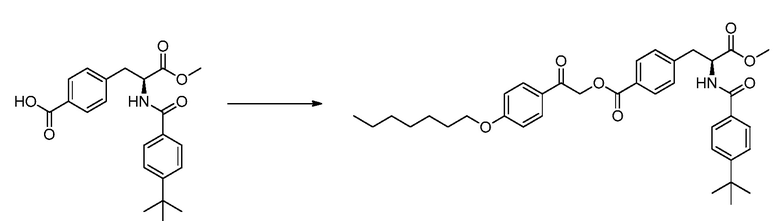
Раствор 2-бром-1-(4-(гептилокси)фенил)этанона, INT-4 (166 мг, 0,45 ммоль) в ацетонитриле (1 мл) добавляли к раствору (S)-4-(2-(4-(трет-бутил)бензамидо)-3-метокси-3-оксопропил)бензойной кислоты INT-1 (190 мг, 0,50 ммоль) и TEA (75,0 мкл, 0,54 ммоль) в ацетонитриле (4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем выливали в 0,5 M лимонную кислоту (30 мл) и экстрагировали EA (3×25 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток растирали с Et2O (10 мл), и фильтрат концентрировали, получая 159 мг (49%) (S)-2-(4-(гептилокси)фенил)-2-оксоэтил 4-(2-(4-(трет-бутил)бензамидо)-3-метокси-3-оксопропил)бензоата в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C37H45NO7: 615,8; найденная 616,0 [M+H]+, tR = 2,76 минут (способ 4).
(S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(4-(4-(гептилокси)фенил)оксазол-2-ил)фенил)пропаноат
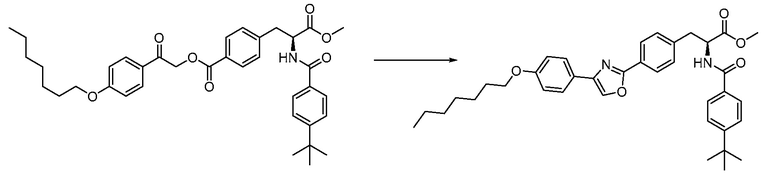
К диэтилэферату трифторида бора (33,3 мкл, 0,27 ммоль) добавляли смесь ацетамида (763 мг, 12,9 ммоль) и (S)-2-(4-(гептилокси)фенил)-2-оксоэтил 4-(2-(4-(трет-бутил)бензамидо)-3-метокси-3-оксопропил)бензоата (159 мг, 0,26 ммоль). Реакционную смесь перемешивали при 140°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, разбавляли EA (15 мл) и экстрагировали NaHCO3 (3×15 мл) и соляным раствором (15 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток перекристаллизовывали из Et2O (5 мл), фильтровали и промывали Et2O. Фильтрат концентрировали, получая 55 мг (16%) (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(4-(4-(гептилокси)фенил)оксазол-2-ил)фенил)пропаноата в виде оранжевого масла. LCMS-ESI (m/z) рассчитанная для C37H44N2O5: 596,8; найденная 597,0 [M+H]+, tR = 3,11 минут (способ 4).
Соединение 77 получали из (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(4-(4-(гептилокси)фенил)оксазол-2-ил)фенил)пропаноата, применяя общий способ 4.
2-(4-бромфенил)-2-оксоэтил 4-(гептилокси)бензоат
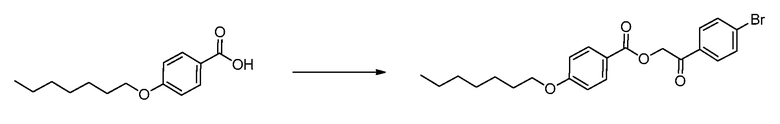
К перемешиваемой смеси 4-(гептилокси)бензойной кислоты (2,0 г, 8,46 ммоль) в ацетонитриле (30 мл) при комнатной температуре добавляли по каплям TEA (1,24 мл, 8,87 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, выливали в 0,05 M лимонную кислоту (100 мл) и EA (10 мл), затем перемешивали в течение 10 минут. Осадок выделяли фильтрованием, промывали водой (30 мл) и изогексаном (2×10 мл), затем сушили на воздухе, получая 3,8 г (98%) 2-(4-бромфенил)-2-оксоэтил 4-(гептилокси)бензоата. LCMS-ESI (m/z) рассчитанная для C22H25BrO4: 433,3; найденная 455,0/457,0 [M+Na]+, tR = 3,21 минут (способ 4).
4-(4-бромфенил)-2-(4-(гептилокси)фенил)оксазол

К эфирату трехфтористого бора (0,322 мл, 2,5 ммоль) добавляли 2-(4-бромфенил)-2-оксоэтил 4-(гептилокси)бензоат (1,0 г, 2,3 ммоль) и ацетамид (4,91 г, 83,0 ммоль) в DCM (10 мл). Реакционную смесь нагревали до 50°C, затем при 140°C в течение 16 часов, DCM отгоняли. Реакционную смесь охлаждали, разбавляли ацетонитрилом и перемешивали при комнатной температуре в течение 1 часа. Осадок выделяли фильтрованием, получая 273 мг (23%) 4-(4-бромфенил)-2-(4-(гептилокси)фенил)оксазола в виде коричневого твердого остатка. LCMS-ESI (m/z) рассчитанная для C22H24BrNO2: 414,3; найденная 414,0 [M+H]+, tR = 3,00 минут (способ 4).
(S)-метил 2-((трет-бутоксикарбонил)амино)-3-(4-(2-(4-(гептилокси)фенил)оксазол-4-ил)фенил)пропаноат

К цинку (104 мг, 1,59 ммоль), пермешиваемому в DMF (1,5 мл), добавляли йод (20,2 мг, 0,08 ммоль). После исчезновения цвета, добавляли (R)-метил 2-((трет-бутоксикарбонил)амино)-3-йодпропаноат (175 мг, 0,53 ммоль) и дополнительное количество йода (20,2 мг, 0,08 ммоль). Через 30 минут смесь дегазировали барботированием N2, затем обрабатывали 4-(4-бромфенил)-2-(4-(гептилокси)фенил)оксазолом (220 мг, 0,53 ммоль), Pd2dba3 (12,2 мг, 0,01 ммоль) и дициклогексил(2',6'-диметокси-[1,1'-бифенил]-2-ил)фосфином (10,9 мг, 0,03 ммоль), с последующим добавлением THF (1 мл). Реакционную смесь нагревали до 50°C в течение 2 часов, охлаждали до комнатной температуры и очищали колоночной хроматографией (градиент 15-95% EA в изогексане), получая 188 мг (65%) (S)-метил 2-((трет-бутоксикарбонил)амино)-3-(4-(2-(4-(гептилокси)фенил)оксазол-4-ил)фенил)пропаноата. LCMS-ESI (m/z) рассчитанная для C31H40N206: 536,6; найденная 537,0 [M+H]+, tR = 3,72 минут (способ 11).
Соединение 78 получали из (S)-метил 2-((трет-бутоксикарбонил)амино)-3-(4-(2-(4-(гептилокси)фенил)оксазол-4-ил)фенил)пропаноата и 4-(трет-бутил)бензойной кислоты, применяя общие способы 8, 7, затем 4.
2-(4-бромфенил)-4-(4-(гептилокси)фенил)тиазол

К перемешиваемому раствору 2-бром-1-(4-(гептилокси)фенил)этанона INT-4 (1,37 г, 4,38 ммоль) в EtOH (10 мл) добавляли 4-бромбензотиоамид (0,95 г, 4,38 ммоль) и изопропанол (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Твердый остаток выделяли фильтрованием, промывали EtOH (5 мл), затем растворяли в DCM (10 мл) и NaHCO3 (20 мл) и перемешивали в течение 1 часа при комнатной температуре. Твердый остаток выделяли фильтрованием, промывали водой (2×10 мл) и ацетонитрилом (2×4 мл), затем сушили, получая 1,02 г (52%) 2-(4-бромфенил)-4-(4-(гептилокси)фенил)тиазола в виде белого микрокристаллического остатка. LCMS-ESI (m/z) рассчитанная для C22H24BrNOS: 429,1; найденная 430,0 [M+H]+, tR = 3,20 минут (способ 4).
(S)-метил 2-((трет-бутоксикарбонил)амино)-3-(4-(4-(4-(гептилокси)фенил)тиазол-2-ил)фенил)пропаноат
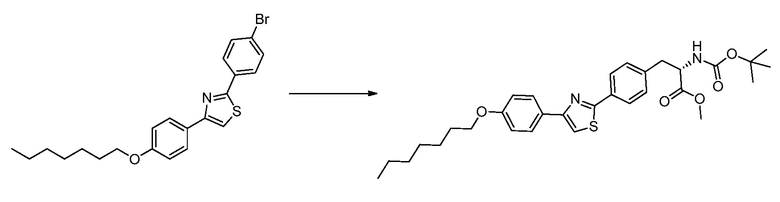
К перемешиваемой суспензии цинка (228 мг, 3,49 ммоль) в DMF (2 мл) добавляли дийод (44 мг, 0,17 ммоль). Когда цвет исчезал, добавляли (R)-метил 2-((трет-бутоксикарбонил)амино)-3-йодпропаноат (382 мг, 1,16 ммоль) и дополнительное количество дийода (44,2 мг, 0,17 ммоль). После перемешивания при комнатной температуре в течение 30 минут, реакционную смесь дегазировали барботированием N2, затем добавляли 2-(4-бромфенил)-4-(4-(гептилокси)фенил)тиазол (500 мг, 1,16 ммоль), дициклогексил(2',6'-диметокси-[1,1'-бифенил]-2-ил)фосфин (23,8 мг, 0,06 ммоль), Pd2dba3 (26 мг, 0,03 ммоль) и DMF (2 мл). Реакционную смесь нагревали при 50°C в течение 3 часов, охлаждали и очищали колоночной хроматографией (10-80% EA в изогексане), получая 620 мг (96%) (S)-метил 2-((трет-бутоксикарбонил)амино)-3-(4-(4-(4-(гептилокси)фенил)тиазол-2-ил)фенилпропаноата. LCMS-ESI (m/z) рассчитанная для C31H40N2O5S: 552,3; ион не наблюдали, tR = 3,37 минут (способ 4).
Соединение 79 получали из (S)-метил 2-((трет-бутоксикарбонил)амино)-3-(4-(4-(4-(гептилокси)фенил)тиазол-2-ил)фенилпропаноата и 4-(трет-бутил)бензойной кислоты, применяя общие способы 8, 7, затем 4.
4-(гептилокси)бензотиоамид
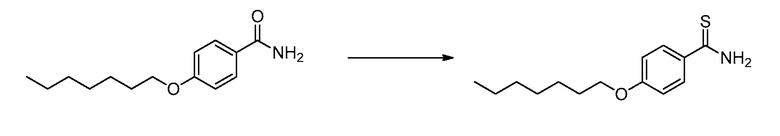
К перемешиваемой суспензии 4-(гептилокси)бензамида (1,24 г, 5,29 ммоль) в DME (20 мл) и THF (10 мл) добавляли 2,4-бис(4-феноксифенил)-1,3,2,4-дитиадифосфетан 2,4-дисульфид (2,80 г, 5,29 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали на силикагеле и очищали колоночной хроматографией (0-60% EA в изогексане), получая 1,4 г (62%) 4-(гептилокси)бензотиоамида в виде желтого восковидного остатка. LCMS-ESI (m/z) рассчитанная для C14H21NOS: 251,4; найденная 252,0 [M+H]+, tR = 3,13 минут (способ 6).
4-(4-бромфенил)-2-(4-(гептилокси)фенил)тиазол
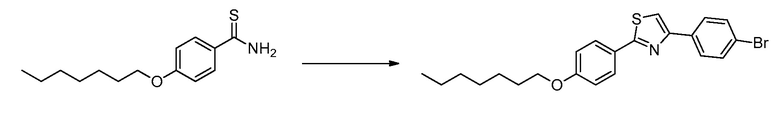
К перемешиваемой смеси 4-(гептилокси)бензотиоамида (1,30 г, 5,17 ммоль) в изопропаноле (20 мл) добавляли 2-бром-1-(4-бромфенил)этанон (1,44 г, 5,17 ммоль). Осадок собирали фильтрованием и промывали EtOH (2×5 мл). Остаток на фильтре суспендировали с NaHCO3 (2×20 мл), водой (2×20 мл), затем EtOH (2×5 мл) и сушили, получая 926 мг (41%) 4-(4-бромфенил)-2-(4-(гептилокси)фенил)тиазола в виде бледно-желтого порошка. LCMS-ESI (m/z) рассчитанная для C22H24BrNOS: 429,1; найденная 430,0 [M+H]+, tR = 3,41 минут (способ 4).
(S)-метил 2-((трет-бутоксикарбонил)амино)-3-(4-(2-(4-(гептилокси)фенил)тиазол-4-ил)фенил)пропаноат
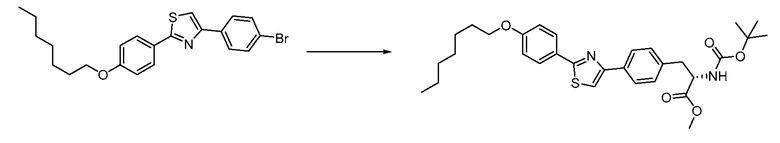
К перемешиваемой смеси цинка (182 мг, 2,79 ммоль) в DMF (2 мл) добавляли дийод (35,4 мг, 0,14 ммоль). Когда цвет исчезал, добавляли дополнительное количество дийода (35,4 мг, 0,14 ммоль) и (R)-метил 2-((трет-бутоксикарбонил)амино)-3-йодпропаноата (306 мг, 0,93 ммоль). Через 30 минут добавляли DMF (1 мл), и смесь дегазировали барботированием N2. К реакционной смеси добавляли 4-(4-бромфенил)-2-(4-(гептилокси)фенил)тиазол (400 мг, 0,93 ммоль), Pd2dba3 (21 мг, 0,02 ммоль) и дициклогексил(2',6'-диметокси-[1,1'-бифенил]-2-ил)фосфин (19 мг, 0,05 ммоль), смесь дополнительно дегазировали, затем нагревали при 50 °C в течение 3 часов. Реакционную смесь охлаждали и очищали колоночной хроматографией (10-80% EA в изогексане). Полученный продукт растворяли в DCM (4 мл) и промывали водой (20 мл) и сушили через гидрофобный керамический порошок. Органические слои суспендировали в ACN (4 мл) и концентрировали, получая 432 мг (83%) (S)-метил 2-((трет-бутоксикарбонил)амино)-3-(4-(2-(4-(гептилокси)фенил)тиазол-4-ил)фенил)пропаноата в виде желтой пены. LCMS-ESI (m/z) рассчитанная для C31H40N2O5S: 552,7; ион не наблюдали, tR = 3,36 минут (способ 4).
Соединение 80 получали из (S)-метил 2-((трет-бутоксикарбонил)амино)-3-(4-(2-(4-(гептилокси)фенил)тиазол-4-ил)фенил)пропаноата и 4-(трет-бутил)бензойной кислоты, применяя общие способы 8, 7, затем 4.
4-(5-(4-(гептилокси)фенил)тиазол-2-ил)бензальдегид
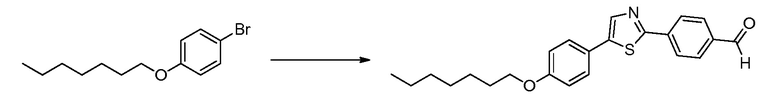
К перемешиваемой суспензии 4-(тиазол-2-ил)бензальдегида (349 мг, 1,84 ммоль), трициклогексилфосфина (27 мг, 0,07 ммоль), пивалиновой кислоты (64,2 мкл, 0,55 ммоль), карбоната калия (382 мг, 2,77 ммоль) и ацетата палладий (II) (8 мг, 0,04 ммоль) в DMA (5,15 мл) в атмосфере азота добавляли раствор 1-бром-4-(гептилокси)бензола (500 мг, 1,84 ммоль) в DMA (1 мл). Реакционную смесь вакуумировали и продували азотом 3 раза, затем нагревали при 100°C в течение 6 часов. После охлаждения реакционную смесь разбавляли EA (40 мл), промывали водой (3×40 мл) и соляным раствором (40 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали в вакууме, получая коричнево-зеленый твердый остаток. Неочищенный продукт очищали хроматографией (0-50% EA в гексане), получая 270 мг (37%) 4-(5-(4-(гептилокси)фенил)тиазол-2-ил)бензальдегида в виде переливающегося желтого твердого остатка. LCMS-ESI (m/z) рассчитанная для C23H25NO2S: 379,5; найденная 380,0 [M+H]+, tR = 2,99 минут (способ 8).
Метил 2-((трет-бутоксикарбонил)амино)-3-(4-(5-(4-(гептилокси)фенил)тиазол-2-ил)фенил)акрилат
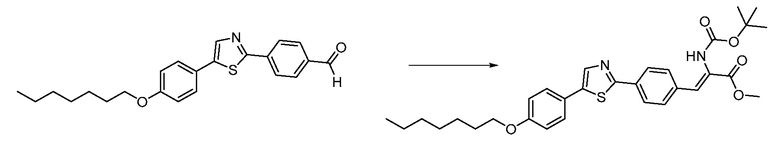
Перемешиваемую смесь 1,1,3,3-тетраметилгуанидина (86 мкл, 0,69 ммоль) добавляли к суспензии 4-(5-(4-(гептилокси)фенил)тиазол-2-ил)бензальдегида (260 мг, 0,685 ммоль) и метил 2-((трет-бутоксикарбонил)амино)-2-(диметоксифосфорил)ацетата (185 мг, 0,62 ммоль) в безводном THF (10 мл) в атмосфере азота при -70°C. Реакционную смесь перемешивали при -70°C в течение 1 часа, затем при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли DCM (50 мл), промывали водой (50 мл), пропускали через картридж для разделения фаз, и органическую фазу концентрировали в вакууме, получая желтый твердый остаток. Твердый остаток растирали с EA/EtOH (20 мл), и собранный твердый остаток промывали EtOH (10 мл) и Et2O, получая 284 мг (79%) метил 2-((трет-бутоксикарбонил)амино)-3-(4-(5-(4-(гептилокси)фенил)тиазол-2-ил)фенил)акрилата в виде желтого твердого остатка. LCMS-ESI (m/z) рассчитанная для C31H38N2O5S: 550,7; найденная 551,0 [M+H]+, tR = 3,11 минут (способ 8).
Метил 2-((трет-бутоксикарбонил)амино)-3-(4-(5-(4-(гептилокси)фенил)тиазол-2-ил)фенил)пропаноат

Перемешиваемую смесь метил 2-((трет-бутоксикарбонил)амино)-3-(4-(5-(4-(гептилокси)фенил)тиазол-2-ил)фенил)акрилата (50 мг, 0,091 ммоль), растворенного в диоксане (5 мл), гидрировали, применяя H-Cube гидрогенизатор (10% Pd/C, 30×4 мм, полностью водород, 40°C, 1 мл/мин). Реакционную смесь концентрировали в вакууме, получая 21 мг (29%) метил 2-((трет-бутоксикарбонил)амино)-3-(4-(5-(4-(гептилокси)фенил)тиазол-2-ил)фенил)пропаноата в виде желтого твердого остатка. LCMS-ESI (m/z) рассчитанная для C31H40N2O5S: 552,7; найденная 553,0 [M+H]+, tR = 1,85 минут (способ 8).
Соединение 81 получали из метил 2-((трет-бутоксикарбонил)амино)-3-(4-(5-(4-(гептилокси)фенил)тиазол-2-ил)фенил)пропаноата и 4-(трет-бутил)бензоилхлорида, применяя общие способы 8, 3, затем 4.
Соединение 82 получали способом, аналогичным способу получения соединения 81, применяя 4-(2-(4-(гептилокси)фенил)тиазол-5-ил)бензальдегид вместо 4-(5-(4-(гептилокси)фенил)тиазол-2-ил)бензальдегида.
(S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,3,4-тиадиазол-2-ил)фенил)пропаноат

Получали, применяя INT-3: к перемешиваемому раствору 2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан 2,4-дисульфида (65,7 мг, 0,16 ммоль) в THF (3 мл) добавляли (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(2-(4-(гептилокси)бензоил)гидразинкарбонил)фенил)пропаноат INT-3 (100,0 мг, 0,16 ммоль), и смесь нагревали до 65°C. Через 1 час, реакционную смесь концентрировали и очищали колоночной хроматографией (10-100% EA в изогексане), получая 37,0 мг (29%) (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,3,4-тиадиазол-2-ил)фенил)пропаноата в виде желтого твердого остатка. LCMS-ESI (m/z) рассчитанная для C36H43N3O4S: 613,8; ион не наблюдали, tR = 3,31 минут (способ 4).
Соединение 83 получали из (S)-метил 2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-1,3,4-тиадиазол-2-ил)фенил)пропаноата, применяя общий способ 4.
Соединение 84 получали, применяя 3-бром-5-хлор-1,2,4-тиадиазол, (4-(гептилокси)фенил)бороновую кислоту и INT-13, применяя последовательно общие способы 10, 10 и 8.
(S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(((трифторметил)сульфонил)окси)фенил)пропаноат (INT-5)
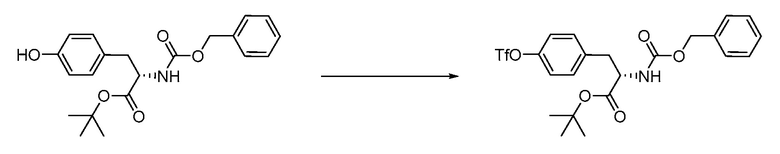
Получали, применяя общий способ 9: перемешиваемый раствор гидрата (S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-гидроксифенил)пропаноата (25 г, 64,2 ммоль) в DCM (100 мл) обрабатывали сульфатом магния (4,01 г, 33,7 ммоль). Через 15 минут смесь фильтровали и промывали DCM (2×20 мл). Органические слои обрабатывали N-этил-N-изопропилпропан-2-амином (17,41 г, 134,7 ммоль) и перемешивали. Данный раствор обрабатывали 1,1,1-трифтор-N-фенил-N-((трифторметил)сульфонил)метансульфамидом (26,44 г, 74,01 ммоль), и смесь перемешивали в течение ночи при комнатной температуре. Смесь обрабатывали водой (50 мл) и насыщенным водным NaHCO3 (20 мл) и интенсивно перемешивали в течение 10 минут. Слои разделяли, и органический слой дополнительно промывали насыщенным водным NaHCO3 (2×50 мл), водой (50 мл) и насыщенным водным NaHCO3 (50 мл) и концентрировали. Соединение очищали хроматографией (EA/гексан), получая 26,85 г (79%) (S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(((трифторметил)сульфонил)окси)фенил)пропаноата INT-5. LCMS-ESI (m/z) рассчитанная для C22H24F3NO7S: 503,1; найденная 526,1 [M + Na]+, tR = 4,12 минут (способ 3).
(S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноат (INT-6)
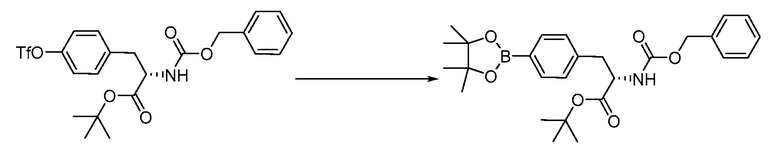
Раствор (S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(((трифторметил)сульфонил)окси)фенил)пропаноата INT-5 (26,85 г, 53,4 ммоль), ацетата калия (15,71 г, 160,1 ммоль), бис-пинаколатоборана (27,1 г, 106,7 ммоль) и DMSO (100 мл) дегазировали постоянным потоком газообразного азота в течение 5 минут. К данному раствору добавляли PdCl2(dppf) (1,95 г, 2,67 ммоль), и раствор дополнительно дегазировали и выдерживали в атмосфере азота. Смесь нагревали при 100°C в течение 18 часов, затем охлаждали до комнатной температуры и разбавляли EA (50 мл) и промывали насыщенным водным NaHCO3 (20 мл), водой (3×30 мл), сушили над MgSO4, фильтровали, и растворитель удаляли при пониженном давлении. Соединение очищали колоночной хроматографией, получая 11,10 г (41 %) (S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноата INT-6 в виде масла. LCMS-ESI (m/z) рассчитанная для C27H36BNO6: 481,3; найденная 504,3 [M+Na]+, tR = 4,21 минут (способ 3). 1H ЯМР (400 МГц, DMSO) δ 7,72 (д, J=8,3 Гц, 1H), 7,60 (д, J=8,0 Гц, 2H), 7,42-7,11 (м, 6H), 4,98 (с, 2H), 4,22-4,08 (м, 1H), 3,03 (дд, J=13,7, 5,2 Гц, 1H), 2,85 (дд, J=13,6, 10,1 Гц, 1H), 1,36 (с, 6H), 1,30 (с, 9H), 1,22-1,13 (м, 6H).
(S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(5-бромпиримидин-2-ил)фенил)пропаноат (INT-7)
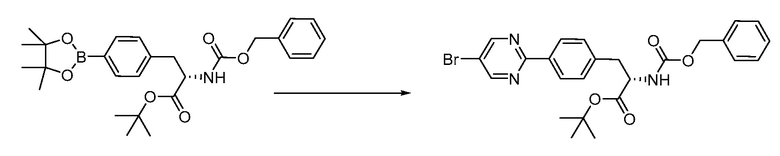
Получали, применяя общий способ 10: дегазировали перемешиваемую смесь (S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноата INT-6 (21,7 г, 45,0 ммоль) и 5-бром-2-йодпиримидина (15,4 г, 54,0 ммоль) в диоксане (400 мл) с декагидратом карбоната натрия (25,7 г, 90 ммоль) в воде (100 мл). Добавляли PdCl2(dppf) (0,99 г, 1,4 ммоль), и смесь дополнительно дегазировали, затем кипятили с обратным холодильником в течение 5 часов. Смесь охлаждали при перемешивании в течение ночи. Смесь выливали в воду (1 л) и EA (300 мл) и перемешивали в течение 30 минут. Смесь фильтровали, и слои разделяли. Водный слой дополнительно экстрагировали EA (2×200 мл), и объединенные органические слои промывали водой (2×100 мл), затем соляным раствором (50 мл), сушили над MgSO4 и концентрировали. Колоночная хроматография (EA/гексан) давала 14,84 г (63 %) (S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(5-бромпиримидин-2-ил)фенил)пропаноата INT-7. LCMS-ESI (m/z) рассчитанная для C25H26BrN3O4 511,1; найденная 534,0 [M + Na]+, tR = 2,97 минут (способ 11).
(S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(5-(4- гептилокси)фенил)пиримидин-2-ил)фенил)пропаноат (INT-8)
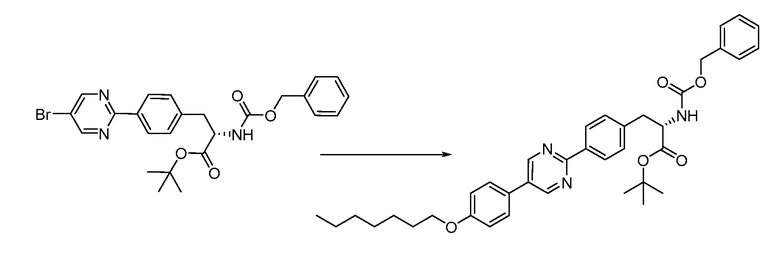
Получали, применяя общий способ 10: перемешиваемый раствор (S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(5-бромпиримидин-2-ил)фенил)пропаноата INT-7 (759 мг, 1,48 ммоль), (4-(гептилокси)фенил)бороновой кислоты (455 мг, 1,93 ммоль) и бикарбоната натрия (311 мг, 3,70 ммоль) в ацетонитриле (5 мл), THF (5 мл) и воде (4 мл) дегазировали N2 в течение 5 минут. Добавляли Pd(dppf)Cl2 (108 мг, 0,15 ммоль), и реакцию нагревали при 110°C в микроволновой печи в течение 50 минут. Реакцию разбавляли EA и водой, затем фильтровали. Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле (EA/гексан), получая 591 мг (62 %) (S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропаноата INT-8 в виде желтого твердого остатка. LCMS-ESI (m/z) рассчитанная для C38H45N3O5: 623,8; m/z не наблюдали, tR = 3,42 минут (способ 8).
(S)-трет-бутил 2-амино-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропаноат (INT-9)
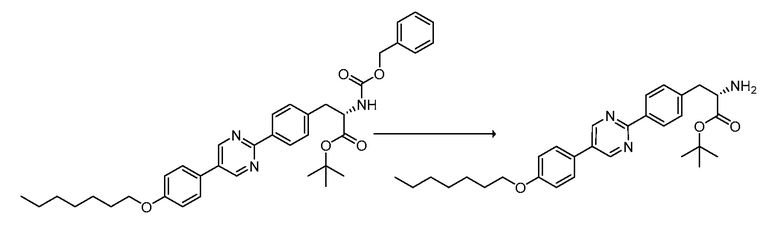
К перемешиваемому раствору (S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропаноата INT-8 (591 мг, 0,95 ммоль) в EA (25 мл) добавляли Pd/C (101 мг, 0,09 ммоль), и суспензию дегазировали H2. Смесь интенсивно перемешивали в атмосфере H2 в течение ночи, затем фильтровали через целит, и фильтрат концентрировали, получая 405 мг (83%) (S)-трет-бутил 2-амино-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропаноата INT-9. LCMS-ESI (m/z) рассчитанная для C30H39N3O3: 489,3; найденная: 490,2 [M+H]+, tR = 2,35 минут (способ 8).
(S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропионовая кислота (соединение 85)
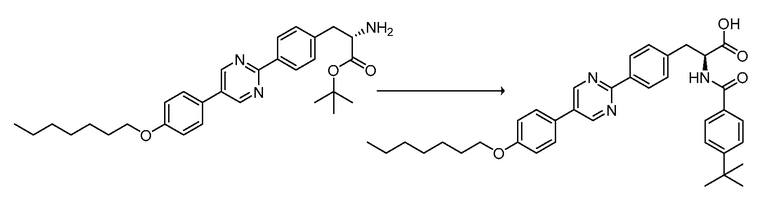
Перемешиваемый раствор (S)-трет-бутил 2-амино-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропаноата INT-9 (1,34 г, 2,74 ммоль) и 4-(трет-бутил)бензойной кислоты (0,54 г, 3,01 ммоль) в DMF (5 мл) и N-этил-N-изопропилпропан-2-амина (1,01 мл, 5,47 ммоль) обрабатывали HATU (1,09 г, 2,87 ммоль). После перемешивания в течение 1 часа, смесь обрабатывали водой (60 мл) и изогексаном (20 мл) и перемешивали в течение 1 часа. Продукт собирали фильтрованием, промывали водой (3×10 мл), затем изогексаном (10 мл), и сушили в вакуумном сушильном шкафу. Эфир растворяли в DCM (5 мл) и обрабатывали TFA (5 мл). Через 2 часа смесь обрабатывали толуолом (5 мл) и упаривали. Остаток растворяли в DMSO (6 мл), затем обрабатывали водой (20 мл) и перемешивали в течение 1 часа. Продукт собирали фильтрованием, промывали водой (3×15 мл), затем ацетонитрилом (2×5 мл) и сушили в вакуумном сушильном шкафу, получая 1,40 г (85 %) (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропионовой кислоты, соединения 85, в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C37H43N3O4: 593,3; найденная: 594,0 [M+H]+, tR = 11,18 минут (способ 9) и 97% e.e. (хиральный способ). 1H ЯМР (400 МГц, DMSO-d6) δ 12,79 (шир.с, 1H), 9,16 (с, 2H), 8,66 (д, J=8,2 Гц, 1H), 8,45-8,27 (м, 2H), 7,89-7,69 (м, 4H), 7,57-7,38 (м, 4H), 7,18-7,02 (м, 2H), 4,77-4,62 (м, 1H), 4,03 (т, J=6,5 Гц, 2H), 3,30-3,24 (м, 1H), 3,22-3,12 (м, 1H), 1,80-1,68 (м, 2H), 1,48-1,20 (м, 17H), 0,96-0,82 (м, 3H).
Соединения 86-102, 104-158 и 296 получали из (S)-трет-бутил 2-амино-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропаноата INT-8, применяя общие способы 3 или 7, с последующим 4 или 8.
(S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(5-(4-(трет-бутил)фенил)пиримидин-2-ил)фенил)пропаноат (INT-10)
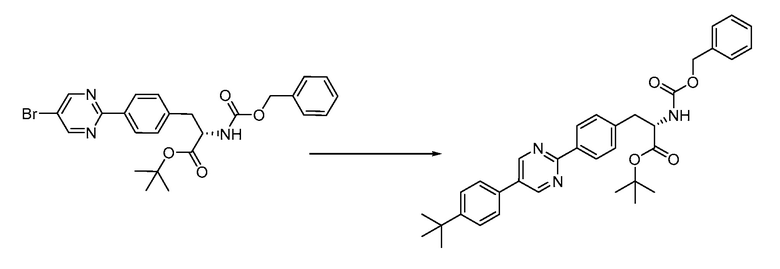
Получали, применяя общий способ 10: перемешиваемый раствор (S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(5-бромпиримидин-2-ил)фенил)пропаноата INT-7 (0,96 г, 1,86 ммоль), (4-(трет-бутил)фенил)бороновой кислоты (0,43 г, 2,42 ммоль) и бикарбоната натрия (0,39 г, 4,66 ммоль) в ацетонитриле (5 мл), THF (5 мл) и воде (5 мл) дегазировали N2 в течение 5 минут. Добавляли Pd(dppf)Cl2 (0,136 г, 0,186 ммоль), и реакцию нагревали при 110°C в микроволновой печи в течение 45 минут. Реакцию разбавляли EA (50 мл) и фильтровали через целит. Органическую фазу промывали водой (100 мл) и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле (EA/изогексан), получая 757 мг (70 %) (S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(5-(4-(трет-бутил)фенил)пиримидин-2-ил)фенил)пропаноат INT-10 в виде белого порошка. LCMS-ESI (m/z) рассчитанная для C35H39N3O4: 565,3; m/z не наблюдали, tR= 3,39 минут (способ 8).
(S)-трет-бутил 2-амино-3-(4-(5-(4-(трет-бутил)фенил)пиримидин-2-ил)фенил)пропаноат (INT-11)
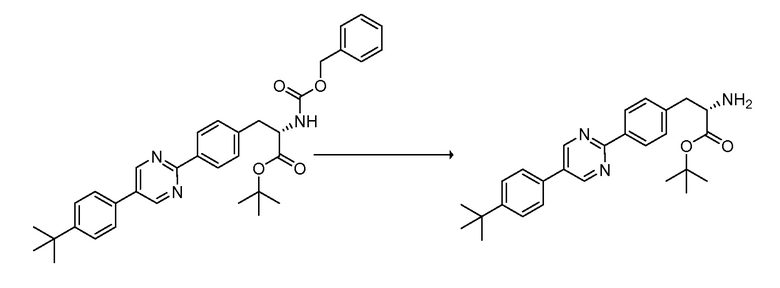
К перемешиваемому раствору (S)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-(5-(4-(трет-бутил)фенил)пиримидин-2-ил)фенил)пропаноата INT-10 (757 мг, 1,34 ммоль) в EA (100 мл) добавляли Pd/C (142 мг, 0,13 ммоль), и суспензию дегазировали H2. Смесь интенсивно перемешивали в атмосфере H2 в течение ночи, затем фильтровали через целит, и фильтрат концентрировали, получая 532 мг (88%) (S)-трет-бутил 2-амино-3-(4-(5-(4-(трет-бутил)фенил)пиримидин-2-ил)фенил)пропаноата INT-11. LCMS-ESI (m/z) рассчитанная для C27H33N3O2: 431,3; найденная: 432,0 [M+H]+, tR = 2,01 минут (способ 4).
Соединения 159-181 получали из (S)-трет-бутил 2-амино-3-(4-(5-(4-(трет-бутил)фенил)пиримидин-2-ил)фенил)пропаноата INT-11, применяя общие способы 3 или 7, с последующим 4 или 8.
Соединение 182 можно получить способом, аналогичным способу для 165, исходя из (R)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-гидроксифенил)пропаноата.
Соединение 183 получали из (S)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)-2-(4-гидроксибензамидо)пропионовой кислоты, соединения 114, применяя общий способ 13.
Соединения 184-190 получали из (S)-трет-бутил 2-амино-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропаноата INT-9, применяя последовательно общие способы 13 и 8.
Соединение 191 можно получить способом, аналогичным способу получения 85, исходя из (R)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-гидроксифенил)пропаноата.
(S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропионовая кислота (соединение 192)
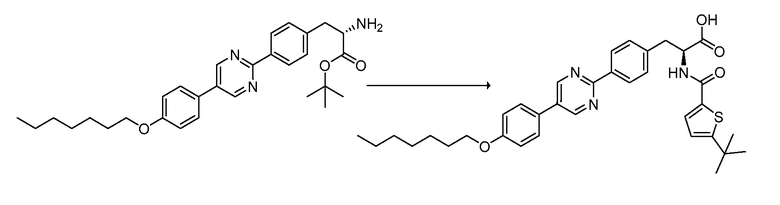
Перемешиваемый раствор (S)-трет-бутил 2-амино-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропаноата INT-9 (5,50 г, 11,23 ммоль) и 5-(трет-бутил)тиофен-2-карбоновой кислоты (2,13 г, 11,57 ммоль) в DMF (50 мл) и DIEA (6,22 мл, 33,70 ммоль) обрабатывали порциями HATU (4,48 г, 11,79 ммоль). После перемешивания в течение 1 часа, смесь обрабатывали водой (200 мл) и изогексаном (20 мл) и перемешивали в течение 10 минут. Продукт собирали фильтрованием, промывали изогексаном (2×30 мл), водой (2×50 мл), затем MeOH (20 мл) и изогексаном (30 мл). Эфир растворяли в DCM (50 мл) и обрабатывали TFA (10 мл). Через 1 час добавляли дополнительное количество TFA (15 мл). Через следующие 5 часов смесь обрабатывали толуолом (20 мл) и концентрировали. Остаток промывали ацетонитрилом (25 мл), затем растворяли в DMSO (20 мл), затем обрабатывали водой (100 мл) и перемешивали в течение 1 часа. Продукт собирали фильтрованием, промывали водой (4×50 мл), затем ацетонитрилом (3×30 мл), и сушили в вакуумном сушильном шкафу, получая 5,30 г (75 %) (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5 -(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропионовой кислоты, соединение 192, в виде грязно-белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C35H41N3O4S: 599,3; m/z не наблюдали, tR = 11,10 минут (способ 10). Хиральная чистота составляла 98% e.e. (хиральный способ). 1H ЯМР (400 МГц, DMSO-d6) δ 12,87 (с, 1H), 9,17 (с, 2H), 8,68 (д, J=8,3 Гц, 1H), 8,47-8,17 (м, 2H), 7,96-7,71 (м, 2H), 7,64 (д, J=3,8 Гц, 1H), 7,55-7,29 (м, 2H), 7,26-7,02 (м, 2H), 6,93 (д, J=3,8 Гц, 1H), 4,79-4,48 (м, 1H), 4,03 (т, J=6,5 Гц, 2H), 3,27 (дд, J=13,9, 4,5 Гц, 1H), 3,12 (дд, J=13,9, 10,6 Гц, 1H), 1,90-1,58 (м, 2H), 1,58-1,01 (м, 17H), 1,01-0,69 (м, 3H).
Соединение 193 получали способом, аналогичным способу получения 192, исходя из (R)-трет-бутил 2-(((бензилокси)карбонил)амино)-3-(4-гидроксифенил)пропаноата.
Трет-бутил (4-(трет-бутил)бензоил)-L-тирозинат
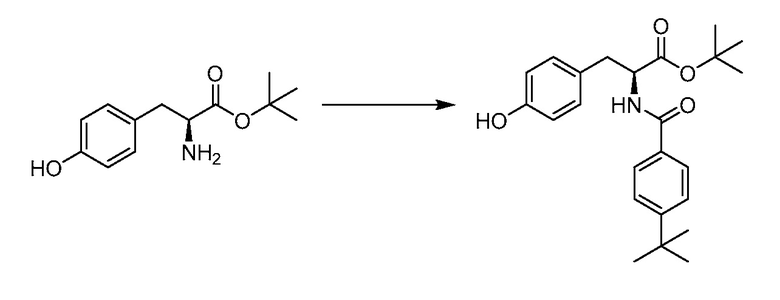
Получали, применяя общий способ 7. К раствору 4-(трет-бутил)бензойной кислоты (8,3 г, 46,4 ммоль) в DMF (100 мл) добавляли HATU (19,2 г, 50,6 ммоль), TEA (17,6 мл, 126,4 ммоль) и (S)-трет-бутил 2-амино-3-(4-гидроксифенил)пропаноат (10,0 г, 42,1 ммоль). Через 5 часов реакционную смесь разбавляли EA, промывали насыщенным водным NaHCO3 и соляным раствором, затем сушили (Na2SO4), концентрировали и очищали хроматографией (EA/гексан), получая 12,9 г (69%) трет-бутил (4-(трет-бутил)бензоил)-L-тирозината. LCMS-ESI (m/z) рассчитанная для C24H31NO4: 397,5; m/z не наблюдали, tR = 3,59 минут (способ 1). 1H ЯМР (400 МГц, CDCL3) δ 7,71-7,65 (м, 2H), 7,47-7,39 (м, 2H), 7,04 (т, J=5,7 Гц, 2H), 6,78-6,70 (м, 2H), 6,59 (д, J=7,5 Гц, 1H), 4,91 (дт, J=7,5, 5,6 Гц, 1H), 3,15 (кв.д, J=14,0, 5,6 Гц, 2H), 1,45 (с, 9H), 1,33 (с, 9H).
Трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(((трифторметил)сульфонил)окси)фенилпропаноат (INT-12)
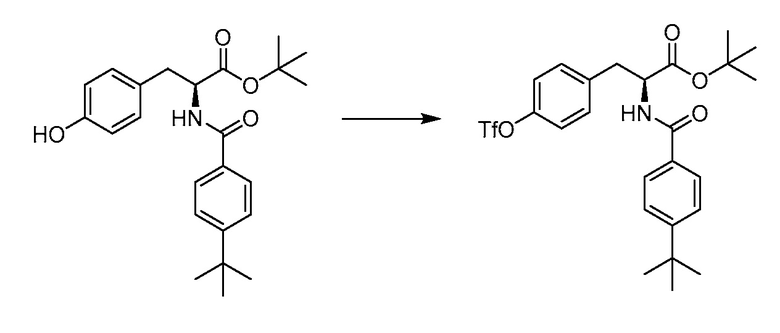
Получали, применяя общий способ 9. К раствору трет-бутил (4-(трет-бутил)бензоил)-1-тирозината (8,0 г, 17,9 ммоль) добавляли DIEA (3,7 мл, 1,2 ммоль) и N-фенилбис(трифторметансульфимид) (7,0 г, 19,7 ммоль). После перемешивания в течение 36 часов, реакционную смесь разбавляли DCM, затем промывали 10% водной лимонной кислотой и насыщенным водным NaHCO3. Органический слой сушили над Na2SO4 и концентрировали, получая 9,5 г (100%) трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(((трифторметил)сульфонил)окси)фенил)пропаноата INT-12, который применяли без дополнительной очистки. LCMS-ESI (m/z) рассчитанная для C25H30F3NO6S: 529,6; m/z не наблюдали, tR = 4,42 минут (способ 1). 1H ЯМР (400 МГц, CDCl3) δ 7,71-7,65 (м, 2H), 7,49-7,43 (м, 2H), 7,32-7,26 (м, 2H), 7,22-7,16 (м, 2H), 6,69 (д, J=7,0 Гц, 1H), 4,94 (дт, J=6,9, 5,9 Гц, 1H), 3,24 (т, J=7,1 Гц, 2H), 1,41 (с, 9H), 1,33 (с, 9H).
Трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(4, 4, 5, 5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноат (INT-13)

К дегазированному раствору (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(((трифторметил)сульфонил)окси)фенил)пропаноата INT-12 (9,5 г, 24 ммоль), KOAc (7,0 г, 72 ммоль) и биспинаколатоборана (9,1 г, 36 ммоль) в DMSO (20 мл) добавляли Pd(dppf)Cl2 (0,87 г, 1 ммоль). Реакционную смесь нагревали при 100°C в течение 12 часов в атмосфере N2. Реакционную смесь разбавляли EA, затем промывали насыщенным водным NaHCO3 и H2O. Органический слой сушили над Na2SO4, концентрировали и очищали хроматографией (EA/гексан), получая 7,2 г (60%) трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноата INT-13. LCMS-ESI (m/z) рассчитанная для C30H42BNO5: 507,5; m/z не наблюдали, tR = 4,53 минут (способ 1). 1H ЯМР (400 МГц, CDCL3) δ 7,74 (д, J=8,0 Гц, 2H), 7,72-7,67 (м, 2H), 7,48-7,43 (м, 2H), 7,21 (д, J=8,0 Гц, 2H), 6,59 (д, J=7,4 Гц, 1H), 5,05-4,92 (м, 1H), 3,27 (кв.д, J=13,7, 5,4 Гц, 2H), 1,47 (с, 9H), 1,36 (м, 21H).
Трет-бутил (S)-3-(4-(5-бромпиримидин-2-ил)фенил)-2-(4-(трет- бутил)бензамидо)пропаноат (INT-14)
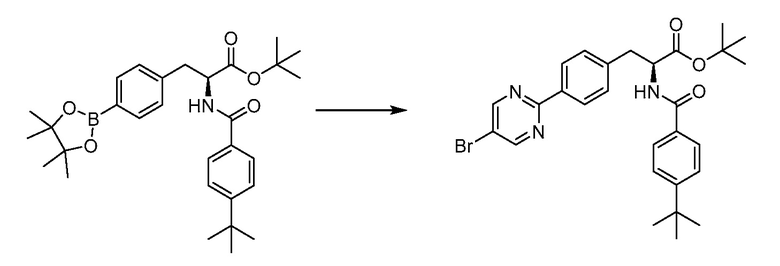
Получали, применяя общий способ 10. К дегазированному раствору (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноата INT-13 (1,0 г, 2,0 ммоль), Na2HCO3 (420 мг, 3,9 ммоль) и 5-бром-2-йодпиримидина (615 мг, 2,2 ммоль) в 2/2/1 MeCN/THF/H2O добавляли Pd(dppf)Cl2 (140 мг, 0,2 ммоль). Реакционную смесь нагревали при 110°C в течение 1 часа в микроволновом реакторе. Реакционную смесь концентрировали, растворяли в DCM и промывали H2O. Органический слой сушили над Na2SO4, концентрировали и очищали хроматографией (EA/гексан), получая 630 мг (58%) трет-бутил (S)-3-(4-(5-бромпиримидин-2-ил)фенил)-2-(4-(трет-бутил)бензамидо)пропаноат INT-14. LCMS-ESI (m/z) рассчитанная для C28H32BrN4O3: 538,5; m/z не наблюдали, tR = 4,66 минут (способ 1). 1H ЯМР 400 МГц, CDCL3) δ 8,84-8,78 (с, 2H), 8,31 (т, J=7,0 Гц, 2H), 7,75-7,64 (м, 2H), 7,46-7,38 (м, 2H), 7,30 (дд, J=12,9, 7,1 Гц, 2H), 6,65 (д, J=7,2 Гц, 1H), 5,10-4,94 (м, 1H), 3,43-3,20 (м, 2H), 1,45 (с, 9H), 1,32 (с, 9H).
Соединения 194-236 получали из трет-бутил (S)-3-(4-(5-бромпиримидин-2-ил)фенил)-2-(4-(трет-бутил)бензамидо)пропаноата INT-14, применяя последовательно общие способы 10 и 8.
Трет-бутил (5-(трет-бутил)тиофен-2-карбонил)-L-тирозинат
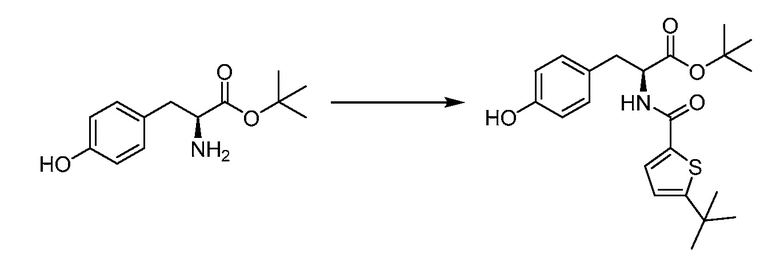
Получали, применяя общий способ 7. К раствору 5-(трет-бутил)тиофен-2-карбоновой кислоты (1,93 г, 10,0 ммоль) в DMF (20 мл) добавляли HATU (4,56 г, 12,0 ммоль) и TEA (4,18 мл, 30,0 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и добавляли (S)-трет-бутил 2-амино-3-(4-гидроксифенил)пропаноат (2,37 г, 10,0 ммоль). Через 1 час, реакционную смесь выливали в 400 мл ледяной воды, и твердый остаток фильтровали. Твердый остаток растворяли в DCM и EA, сушили над MgSO4, концентрировали и очищали хроматографией (EA/гексан), получая 3,6 г (89%) трет-бутил (5-(трет-бутил)тиофен-2-карбонил)-1-тирозината. LCMS-ESI (m/z) рассчитанная для C22H29NO4S: 403,2; найденная: 426,1 [M+Na]+, tR = 9,07 минут (способ 2).
Трет-бутил (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4- (((трифторметил)сульфонил)окси)фенил)пропаноат (INT-15)

Получали, применяя общий способ 9. К раствору трет-бутил (5-(трет-бутил)тиофен-2-карбонил)-L-тирозината (3,52 г, 8,72 ммоль) добавляли DIEA (4,56 мл, 26,17 ммоль) и N-фенилбис(трифторметансульфимид) (3,27 г, 9,16 ммоль). После перемешивания в течение 18 часов, реакционную смесь разбавляли DCM, затем промывали насыщенным водным NaHCO3. Органический слой сушили над MgSO4 и концентрировали. Неочищенный продукт очищали хроматографией, получая 4,10 г (87,6 %) трет-бутил (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо) -3-(4-(((трифторметил)сульфонил)окси)фенил)пропаноата INT-15. LCMS-ESI (m/z) рассчитанная для C23H28F3NO6S2: 535,1; m/z не наблюдали, tR = 4,22 минут (способ 3).
Трет-бутил (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноат (INT-16)
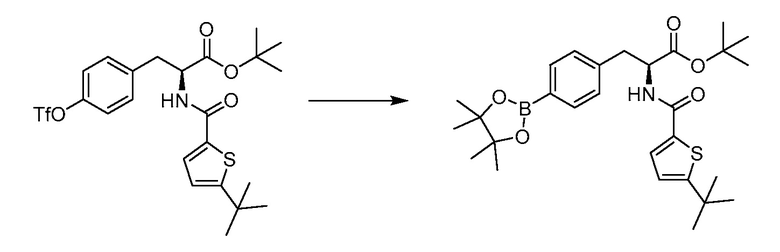
К дегазированному раствору трет-бутил (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(((трифторметил)сульфонил)окси)фенил)пропаноата INT-15 (3,89 г, 7,26 ммоль), KOAc (2,14 г, 21,79 ммоль) и биспинаколатоборана (2,40 г, 9,44 ммоль) в DMSO (50 мл) добавляли Pd(dppf)Cl2 (0,27 г, 0,36 ммоль). Реакционную смесь нагревали при 100°C в течение 18 часов в атмосфере N2. Реакционную смесь выливали в 600 мл ледяной воды, и твердый остаток фильтровали. Осадок разбавляли EA, сушили над MgSO4, концентрировали и очищали хроматографией (EA/гексан), получая 3,68 г (99%) трет-бутил (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноата INT-16. LCMS-ESI (m/z) рассчитанная для C28H40BNO5S: 513,3; m/z не наблюдали, tR = 4,51 минут (способ 3).
Трет-бутил (S)-3-(4-(5-бромпиримидин-2-ил)фенил)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)пропаноат (INT-17)

Получали, применяя общий способ 10. К дегазированному раствору трет-бутил (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноата INT-16 (510 мг, 1,0 ммоль) и 5-бром-2-йодпиримидина (570 мг, 2,0 ммоль) в 2/2/1 MeCN/THF/насыщенный водный NaHCO3 (10 мл) добавляли Pd(dppf)Cl2 (30 мг, 0,4 ммоль). Реакционную смесь нагревали при 120°C в течение 1 часа в микроволновом реакторе. Реакционную смесь разбавляли водой (100 мл) и EA (50 мл) и фильтровали через целит. Водный слой экстрагировали EA (3×30 мл), и объединенный органический слой сушили над MgSO4, концентрировали и очищали хроматографией (EA/гексан), получая 342 мг (63%) трет-бутил (S)-3-(4-(5-бромпиримидин-2-ил)фенил)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)пропаноата INT-17. LCMS-ESI (m/z) рассчитанная для C26H30BrN3O3S: 543,1; найденная: 488,0 [M-tBu+H]+, tR = 10,95 минут (способ 2).
Соединения 237-247 получали из трет-бутил (S)-3-(4-(5-бромпиримидин-2-ил)фенил)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)пропаноата INT-17, применяя последовательно общие способы 10 и 8.
Трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-цианопиримидин-2-ил)фенил)пропаноат (INT-18)
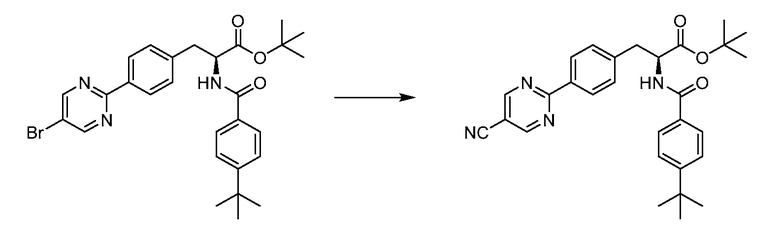
Получали, применяя общий способ 1. К дегазированному раствору (S)-3-(4-(5-бромпиримидин-2-ил)фенил)-2-(4-(трет-бутил)бензамидо)пропаноата INT-14 (100 мг, 0,190 ммоль) и Zn(CN)2 (44 мг, 0,370 ммоль) в NMP (5 мл) добавляли Pd(Ph3)4 (2 мг, 0,002 ммоль). Смесь нагревали в течение 45 минут при 80°C в микроволновом реакторе, затем распределяли между DCM и H2O. Органический слой сушили над Na2SO4, концентрировали и очищали хроматографией (EA/гексан), получая 75 мг (84%) трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-цианопиримидин-2-ил)фенил)пропаноата INT-18. LCMS-ESI (m/z) рассчитанная для C29H32N4O3: 484,60; m/z не наблюдали, tR = 4,17 минут (способ 1). 1H ЯМР 400 МГц, CDCL3) δ 8,97 (с, 2H), 8,38 (д, J=7,9 Гц, 2H), 7,67 (д, J=8,0 Гц, 2H), 7,46-7,35 (м, 2H), 7,33 (д, J=7,9 Гц, 2H), 6,77 (д, J=6,8 Гц, 1H), 4,96 (д, J=6,1 Гц, 1H), 3,27 (дд, J=13,1, 8,0 Гц, 2H), 1,37 (д, J=34,5 Гц, 9H), 1,26 (д, J=21,0 Гц, 9H).
Трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(N-гидроксикарбамимидоил)пиримидин-2-ил)фенил)пропаноат
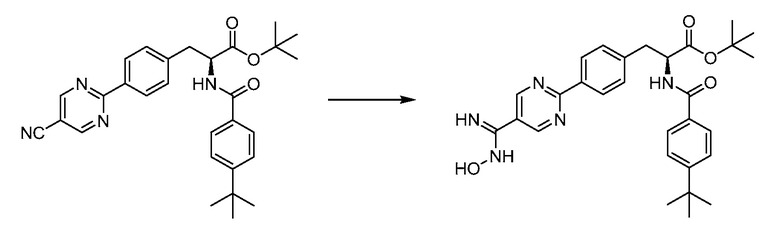
Получали, применяя общий способ 2. Раствор (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-цианопиримидин-2-ил)фенил)пропаноата INT-18 (35 мг, 0,07 ммоль), гидроксиламина (25 мкл, 0,36 ммоль, 50% раствор в H2O) и NEt3 (11 мкл, 0,08 ммоль) в EtOH (5 мл) нагревали при 80°C в течение 1,5 часов. Реакционную смесь концентрировали, растворяли в DCM и промывали H2O, получая 22 мг трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(N-гидроксикарбамимидоил)пиримидин-2-ил)фенил)пропаноата. LCMS-ESI (m/z) рассчитанная для C29H35N504: 517,6; найденная 462,2 [M-tBu+H]+, tR = 3,72 минут (способ 1). 1H ЯМР 400 МГц, CDCL3) δ 9,19 (с, 2H), 8,42 (д, J=8,2 Гц, 2H), 7,67 (дд, J=8,5, 2,1 Гц, 2H), 7,40 (дд, J=9,2, 8,0 Гц, 2H), 7,34 (дд, J=10,3, 8,4 Гц, 2H), 6,74 (дд, J=7,1, 4,7 Гц, 1H), 5,00 (кв., J=5,6 Гц, 1H), 2,83 (д, J=5,3 Гц, 2H), 1,44 (с, 9H), 1,28 (д, J=22,0 Гц, 9H).
Трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(5-гексил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)фенил)пропаноат (соединение 248)
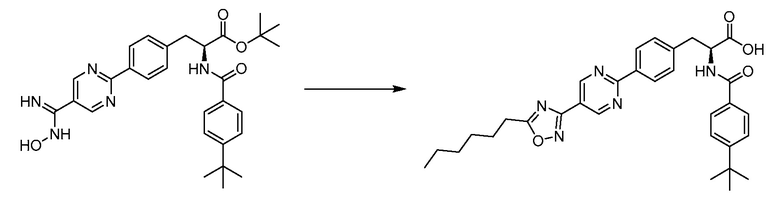
Получали, применяя общий способ 5. Раствор гептановой кислоты (7 мг, 0,05 ммоль), HOBt (12 мг, 0,09 ммоль) и EDC (13 мг, 0,09 ммоль) нагревали при 80°C в течение 2 часов. Реакционную смесь разбавляли EtOAc и промывали NaHCO3. Органический слой сушили над Na2SO4 и концентрировали. Полученную в результате смесь растворяли в EtOH (2 мл) и нагревали в течение 45 минут при 80°C в микроволновом реакторе. Смесь концентрировали и очищали препаративной ВЭЖХ, получая 1,5 мг трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(5-гексил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)фенил)пропаноата. LCMS-ESI (m/z) рассчитанная для C36H45N5O4: 611,8; m/z не наблюдали, tR = 5,5 минут (способ 1). 1H ЯМР 400 МГц, CDCL3) δ 9,45 (с, 2H), 8,44 (д, J=8,3 Гц, 2H), 7,71 (д, J=8,5 Гц, 2H), 7,48 (д, J=8,5 Гц, 2H), 7,38 (д, J=8,3 Гц, 2H), 6,80 (д, J=7,3 Гц, 1H), 5,04 (дд, J=12,7, 5,5 Гц, 1H), 3,37 (ддд, J=18,9, 13,8, 5,5 Гц, 2H), 3,02 (т, J=7,6 Гц, 2H), 1,92 (дт, J=15,3, 7,5 Гц, 2H), 1,49 (с, 9H), 1,44-1,28 (м, 15H), 0,93 (т, J=7,1 Гц, 3H).
(S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(5-гексил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)фенил)пропаноат деблокировали, применяя общий способ 8, получая 1,4 мг (6% суммарно) (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(5-гексил-1,2,4-оксадиазол-3-ил)пиримидин-2-ил)фенил)пропионовой кислоты, соединения 248. LCMS-ESI (m/z) рассчитанная для C32H37N5O4: 555,68; m/z не наблюдали, tR = 11,03 минут (способ 2). 1H ЯМР (400 МГц, CDCL3) δ 9,41 (с, 2H), 8,47 (д, J=8,2 Гц, 2H), 7,66 (д, J=8,4 Гц, 2H), 7,42 (дд, J=15,1, 8,4 Гц, 4H), 6,60 (д, J=6,8 Гц, 1H), 5,21-4,95 (м, 1H), 3,43 (ддд, J=20,0, 14,0, 5,6 Гц, 2H), 3,05-2,90 (м, 2H), 1,98-1,76 (м, 2H), 1,55-1,22 (м, 15H), 0,91 (т, J=7,0 Гц, 3H).
Трет-бутил (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4-гидроксифенил)пиримидин-2-ил)фенил)пропаноат
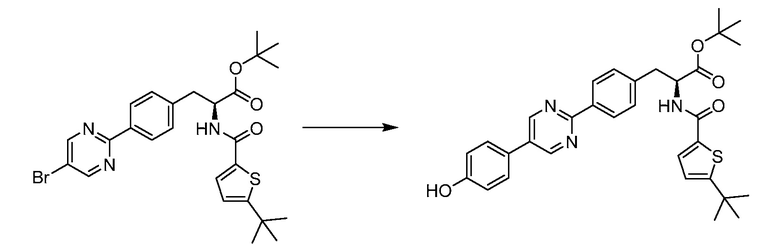
Получали, применяя общий способ 10. К дегазированному раствору трет-бутил (S)-3-(4-(5-бромпиримидин-2-ил)фенил)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)пропаноата INT-17 (180 мг, 0,3 ммоль), карбоната натрия (70 мг, 0,7 ммоль) и 4-гидроксифенилбороновой кислоты (55 мг, 0,4 ммоль) в 5 мл 2/2/1 MeCN/THF/H2O добавляли Pd(dppf)Cl2 (24 мг, 0,03 ммоль). Реакционную смесь нагревали при 110°C в течение 45 минут в микроволновом реакторе. Смесь фильтровали через целит, концентрировали, затем растворяли в DCM и промывали H2O. Органический слой концентрировали и очищали препаративной ВЭЖХ, получая 131 мг (78%) трет-бутил (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4-гидроксифенил)пиримидин-2-ил)фенил)пропаноата. LCMS-ESI (m/z) рассчитанная для C32H35N3O4S: 557,7; m/z не наблюдали, tR = 4,08 минут (способ 1). 1H ЯМР (400 МГц, CDCI3) δ 8,98 (с, 2H), 8,35 (д, J=8,1 Гц, 2H), 7,49 (д, J=8,6 Гц, 2H), 7,40-7,31 (м, 3H), 6,94 (д, J=8,5 Гц, 2H), 6,81 (д, J=3,8 Гц, 1H), 6,51 (д, J=7,5 Гц, 1H), 5,00 (дд, J=12,9, 5,8 Гц, 1H), 3,28 (кв.д, J=13,8, 5,6 Гц, 2H), 1,47 (с, 9H), 1,39 (с, 9H).
(S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4-(децилокси)фенил)пиримидин-2-ил)фенил)пропионовая кислота (соединение 249)
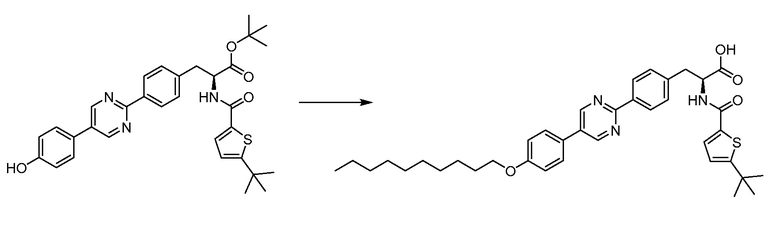
Получали, применяя общий способ 12. К раствору трет-бутил (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4-гидроксифенил)пиримидин-2-ил)фенил)пропаноата (20 мг, 0,04 ммоль) в DMF (0,5 мл) добавляли 1-бромдекан (8 мкл, 0,05 ммоль) и K2CO3 (8 мг, 0,05 ммоль). Реакционную смесь нагревали при 40°C в течение 18 часов, затем разбавляли DCM и промывали H2O. Органический слой сушили над Na2SO4 и концентрировали. Неочищенный материал деблокировали, применяя общий способ 8, затем очищали препаративной ВЭЖХ, получая 3,9 мг (17%) (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4-(децилокси)фенил)пиримидин-2-ил)фенил)пропионовой кислоты, соединения 249. LCMS-ESI (m/z) рассчитанная для C38H47N3O4S: 641,9; m/z не наблюдали, tR = 13,49 минут (способ 2). 1H ЯМР (400 МГц, CDCI3) δ 9,01 (с, 2H), 8,36 (д, J=8,1 Гц, 2H), 7,56 (д, J=8,7 Гц, 2H), 7,44 (д, J=8,2 Гц, 2H), 7,33 (д, J=3,8 Гц, 1H), 7,03 (д, J=8,8 Гц, 2H), 6,80 (д, J=3,8 Гц, 1H), 6,54 (д, J=6,8 Гц, 1H), 5,13 (д, J=6,8 Гц, 1H), 4,01 (т, J=6,6 Гц, 2H), 3,44 (д, J=4,9 Гц, 2H), 1,91-1,72 (м, 2H), 1,47 (дд, J=15,0, 7,3 Гц, 2H), 1,38 (с, 9H), 1,28 (с, 12H), 0,88 (т, J=6,8 Гц, 3H).
Соединения 250-252 получали из трет-бутил (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4-гидроксифенил)пиримидин-2-ил)фенил)пропаноата, применяя общий способ 12, с последующим общим способом 8.
(S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(трет-бутил)пиперидин-1-ил)пиримидин-2-ил)фенил)пропионовая кислота (соединение 253)

Получали, применяя общий способ 11. К дегазированному раствору INT-14 (50 мг, 0,09 ммоль), трет-бутоксида натрия (18 мг, 0,19 ммоль) и 4-трет-бутилпиперидина HCl (23 мг, 0,11 ммоль) в диоксане (2,5 мл) добавляли Pd2(dba)3 (9 мг, 0,01 ммоль) и 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил (6 мг, 0,015 ммоль). Реакционную смесь нагревали в течение 45 минут при 120°C в микроволновом реакторе. Смесь разбавляли EA и промывали NaHCO3. Органический слой сушили над Na2SO4, концентрировали и очищали препаративной ВЭЖХ. Выделенное промежуточное соединение деблокировали, применяя общий способ 8, получая 2,9 мг (6%) (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(трет-бутил)пиперидин-1-ил)пиримидин-2-ил)фенил)пропионовой кислоты, соединения 253. LCMS-ESI (m/z) рассчитанная для C33H42N4O3: 542,7; найденная 543,3 [M+H]+, tR = 10,79 минут (чистота). 1H ЯМР (400 МГц, CDCL3) δ 8,52 (с, 2H), 8,23 (д, J=8,0 Гц, 2H), 7,72 (д, J=8,4 Гц, 2H), 7,44 (дд, J=11,3, 8,4 Гц, 4H), 6,79 (д, J=6,8 Гц, 1H), 5,18 (д, J=6,5 Гц, 1H), 3,89 (д, J=11,9 Гц, 2H), 3,47 (д, J=5,2 Гц, 2H), 2,83 (т, J=11,5 Гц, 2H), 1,88 (д, J=12,0 Гц, 2H), 1,52-1,37 (м, 2H), 1,34 (с, 9H), 1,24 (дд, J=24,7, 12,8 Гц, 1H), 0,92 (с, 9H).
Соединение 254 получали из INT-14, применяя общий способ 11, затем общий способ 8.
Трет-бутил (S)-3-(4-(5-(2H-тетразол-5-ил)пиримидин-2-ил)фенил)-2-(4-(трет-бутил)бензамидо)пропаноат
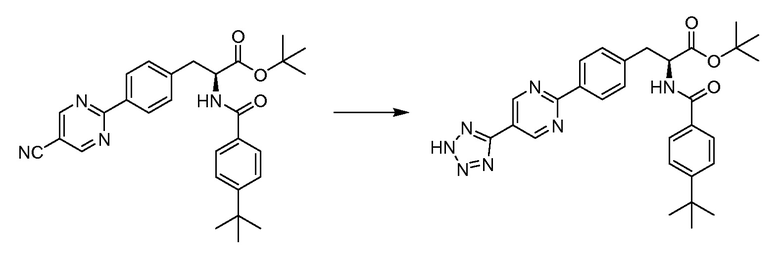
К раствору трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-цианопиримидин-2-ил)фенил)пропаноата INT-18 (34 мг, 0,07 ммоль) в DMF (2 мл) добавляли NH4Cl (7,5 мг, 1,4 ммоль) и NaN3 (7 мг, 0,1 ммоль). Реакционную смесь нагревали при 100°C в течение 3 часов, затем разбавляли EA и промывали NaHCO3. Органический слой сушили над Na2SO4, концентрировали и очищали препаративной ВЭЖХ, получая 4,6 мг (12%) трет-бутил (S)-3-(4-(5-(2H-тетразол-5-ил)пиримидин-2-ил)фенил)-2-(4-(трет-бутил)бензамидо)пропаноата. LCMS-ESI (m/z) рассчитанная для C29H33N7O3: 527,6; m/z не наблюдали, tR = 3,83 минут (способ 1). lH ЯМР (400 МГц, CDCL3) δ 9,35 (с, 2H), 8,42 (д, J=8,1 Гц, 2H), 7,75 (д, J=8,4 Гц, 2H), 7,47 (д, J=8,5 Гц, 2H), 7,43 (д, J=8,2 Гц, 2H), 7,11 (д, J=7,8 Гц, 1H), 5,13 (дд, J=14,4, 7,1 Гц, 1H), 3,28 (ддд, J=21,0, 13,6, 6,7 Гц, 2H), 1,47 (д, J=6,8 Гц, 9H), 1,33 (с, 9H).
Соединение 255 получали из трет-бутил (S)-3-(4-(5-(2H-тетразол-5-ил)пиримидин-2-ил)фенил)-2-(4-(трет-бутил)бензамидо)пропаноата, применяя общий способ 12, затем общий способ 8.
Соединение 256 получали из INT-14 и 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изоиндолин-1-она, применяя общие способы 10, 12 и 8.
Соединение 257 получали из INT-14 и пинаколового эфира 6-гидроксипиридин-3-бороновой кислоты, применяя общие способы 10, 12 и 8.
Соединение 258 получали из INT-13 и 5-(бензилокси)-2-хлорпиримидина, применяя общий способ 10, с последующим общим способом 8.
Соединения 259 и 260 получали из INT-14 и подходящей бороновой кислоты, применяя общие способы 10, затем 8.
Трет-бутил 4-(4-(гептилокси)фенил)-3-оксопиперазин-1-карбоксилат
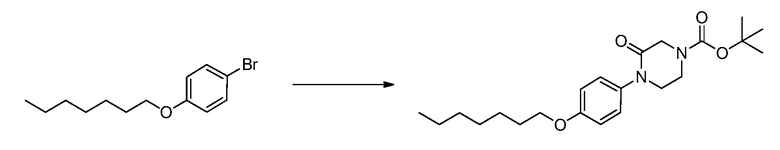
К перемешиваемому раствору 1-бром-4-(гептилокси)бензола (447 мг, 1,65 ммоль) в диоксане (5 мл) добавляли трет-бутил 3-оксопиперазин-1-карбоксилат (330 мг, 1,65 ммоль), йодид меди I (31,4 мг, 0,17 ммоль), (1R,2R)-N1,N2-диметилциклогексан-1,2-диамин (234 мг, 1,65 ммоль) и карбонат калия (456 мг, 3,30 ммоль). Реакционную смесь нагревали при 120°C в течение 16 часов. Реакционную смесь пропускали через слой целита, элюировали EA (50 мл). Органические слои промывали хлоридом аммония (25 мл), водой (25 мл) и соляным раствором (25 мл), затем сушили над MgSO4 и концентрировали, получая 602 мг (89%) трет-бутил 4-(4-(гептилокси)фенил)-3-оксопиперазин-1-карбоксилата. LCMS-ESI (m/z) рассчитанная для C22H34N2O4: 390,5; найденная 319,0 [M+H]+, tR = 2,90 минут (способ 4).
1-(4-(гептилокси)фенил)пиперазин-2-он

К трет-бутил 4-(4-(гептилокси)фенил)-3-оксопиперазин-1-карбоксилату (540 мг, 1,38 ммоль) добавляли 4M HCl в диоксане (2,07 мл, 8,30 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Осадок фильтровали, промывали гексаном (5 мл) и сушили. Неочищенный продукт очищали колоночной хроматографией (79/20/1 DCM/MeOH/NH4), получая 325 мг (80%) 1-(4-(гептилокси)фенил)пиперазин-2-она в виде бесцветного твердого остатка. LCMS-ESI (m/z) рассчитанная для C17H26N2O2: 290,4; найденная 291,0 [M+H]+, tR = 1,49 минут (способ 4).
Соединение 261 получали из INT-12 и 1-(4-(гептилокси)фенил)пиперазин-2-она, применяя общие способы 11 и 8.
Соединение 262 получали аналогичным способом из INT-12 и 1-(4-(гептилокси)фенил)имидазолидин-2-она, применяя общие способы 11 и 8.
Соединение 263 получали, применяя гидрохлорид (S)-метил 2-амино-3-(4-нитрофенил)пропаноата, 4-(трет-бутил)бензойную кислоту и 1-(4-(гептилокси)фенил)пиперидин-4-он, применяя общие способы 7, 14, 15, затем 4.
Трет-бутил 4-(4-(гептилокси)фенил)-4-гидроксипиперидин-1-карбоксилат
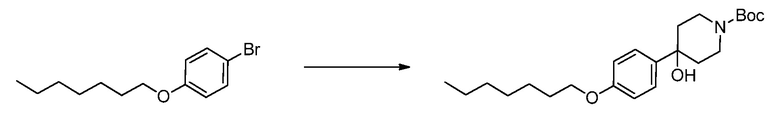
К перемешиваемому раствору 1-бром-4-(гептилокси)бензола (668 мг, 2,46 ммоль) в THF (5 мл) при -78°C добавляли бутиллитий (985 мкл, 2,46 ммоль). Через 30 минут добавляли раствор трет-бутил 4-оксопиперидин-1-карбоксилата (491 мг, 2,46 ммоль) в THF (2 мл). Через 10 минут, охлаждающую баню удаляли, и реакционную смесь перемешивали в течение 16 часов. Реакционную смесь выливали в NH4Cl (50 мл) и экстрагировали Et2O (3×20 мл). Объединенные органические слои промывали водой (20 мл), сушили над MgSO4 и упаривали. Неочищенный продукт очищали колоночной хроматографией (5-70% AcMe в изогексане), получая 0,4 г (33%) трет-бутил 4-(4-(гептилокси)фенил)-4-гидроксипиперидин-1-карбоксилата. LCMS-ESI (m/z) рассчитанная для C23H37NO4: 391,5; найденная 414,0 [M+Na]+, tR = 2,24 минут (способ 4).
4-(4-(гептилокси)фенил)пиперидин (INT-19)
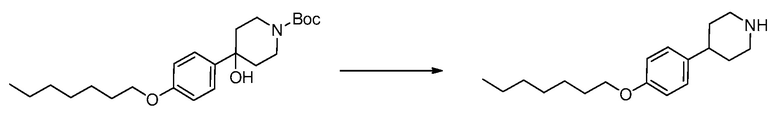
К перемешиваемому раствору трет-бутил 4-(4-(гептилокси)фенил)-4-гидроксипиперидин-1-карбоксилата (388 мг, 0,99 ммоль) и триэтилсилана (791 мкл, 4,95 ммоль) в DCM (2 мл), охлажденному до -30ºC, медленно добавляли по каплям 2,2,2-трифторуксусную кислоту (379 мкл, 4,95 ммоль). Реакционную смесь медленно нагревали и перемешивание продолжали в течение 16 часов. Реакционную смесь выливали в ледяную воду/NaOH (50 мл/5 мл, 2 M) и экстрагировали DCM (3×20 мл). Объединенные органические экстракты промывали последовательно водой (50 мл) и NaHCO3 (20 мл), сушили над MgSO4 и упаривали, получая 166 мг (58%) 4-(4-(гептилокси)фенил)пиперидина INT-19 в виде белого воскообразного остатка. LCMS-ESI (m/z) рассчитанная для C18H29NO: 275,4; найденная 276,0 [M+H]+, tR = 2,88 минут (способ 11).
Соединение 264 получали, применяя INT-12 и INT-19, применяя общие способы 11, затем 8.
Соединение 265 получали способом, аналогичным способу получения 264, применяя INT-12 и 3-(4-(гептилокси)фенил)пирролидин, применяя общий способ 11, затем 8.
Соединение 266 можно получить, применяя INT-12 и 1-([1,1'-бифенил]-4-ил)пиперазин, применяя общий способ 11, затем 8.
Соединение 267 получали, применяя INT-12, трет-бутил 4-(4-гидроксифенил)пиперазин-1-карбоксилат и 1-бромгептан, применяя общие способы 12, 8, 11, затем 8.
Соединение 268 получали, применяя INT-12, трет-бутил 1,4-диазепан-1-карбоксилат и 1-бром-4-(гептилокси)бензол, применяя общие способы 11, 8, 11, затем 8.
Соединение 269 получали, применяя 5-бром-2-йодпиридин, INT-13 и (4-(гептилокси)фенил)бороновую кислоту, применяя последовательно общие способы 10, 10 и 8.
Соединение 270 получали, применяя 5-бром-2-йодпиридин, (4-(гептилокси)фенил)бороновую кислоту и INT-13, применяя последовательно общие способы 10, 10 и 8.
Соединение 271 получали, применяя 5-бром-2-йодпиримидин, (4-(гептилокси)фенил)бороновую кислоту и INT-13, применяя последовательно общие способы 10, 10 и 8.
Соединение 272 получали, применяя 2-бром-5-йодпиразин, (4-(гептилокси)фенил)бороновую кислоту и INT-13, применяя последовательно общие способы 10, 10 и 8.
Соединение 273 получали, применяя 3-хлор-6-йодпиридазин, (4-(гептилокси)фенил)бороновую кислоту и INT-13, применяя последовательно общие способы 10, 10 и 8.
3-(4-бромфенил)-6-(4-(гептилокси)фенил)-1,2,4-триазин (INT-20)

К перемешиваемому раствору 4-бромбензогидразида (1,85 г, 8,62 ммоль) в этаноле (10 мл) добавляли уксусную кислоту (1 мл). Реакционную смесь перемешивали при 60°C в течение 30 минут, затем добавляли 2-бром-1-(4-(гептилокси)фенил)этанон (1,35 г, 4,31 ммоль) INT-4 и ацетат натрия (0,389 г, 4,74 ммоль), и смесь кипятили с обратным холодильником в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры, и полученный в результате осадок фильтровали и промывали изогексаном (20 мл), затем сушили. Твердый остаток растворяли в NMP и нагревали при 120ºC в течение 16 часов. Неочищенный материал охлаждали до комнатной температуры, разбавляли Et2O (4 мл), фильтровали, растирали с этанолом (3×2 мл), фильтровали и сушили, получая 241 мг (13%) 3-(4-бромфенил)-6-(4-(гептилокси)фенил)-1,2,4-триазина INT-20 в виде оранжевого твердого остатка. LCMS-ESI (m/z) рассчитанная для C22H24BrN3O: 425,1; найденная 426,3 [M+H]+, tR = 3,40 минут (способ 8).
Соединение 274 получали способом, аналогичным способу получения 79, применяя 3-(4-бромфенил)-6-(4-(гептилокси)фенил)-1,2,4-триазин INT-20 вместо 2-(4-бромфенил)-4-(4-(гептилокси)фенил)тиазола.
6-(4-бромфенил)-3-(4-(гептилокси)фенил)-1,2,4-триазин (INT-21)

К перемешиваемому раствору 4-(гептилокси)бензогидразида (400 мг, 1,60 ммоль) в этаноле (15 мл) добавляли уксусную кислоту (1 мл). Реакционную смесь перемешивали при 60°C в течение 30 минут, затем добавляли 2-бром-1-(4-бромфенил)этанон (222 мг, 0,80 ммоль) и ацетат натрия (72,1 мг, 0,88 ммоль), и раствор кипятили с обратным холодильником в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, и полученные в результате кристаллы фильтровали, промывали изогексаном (20 мл), затем сушили, получая 108 мг (31%) 6-(4-бромфенил)-3-(4-(гептилокси)фенил)-1,2,4-триазина INT-21. LCMS-ESI (m/z) рассчитанная для C22H24BrN3O: 425,1; найденная 426,1 [M+H]+, tR = 3,38 минут (способ 8).
Соединение 275 получали способом, аналогичным способу получения 274, применяя 6-(4-бромфенил)-3-(4-(гептилокси)фенил)-1,2,4-триазин INT-21 вместо 3-(4-бромфенил)-6-(4-(гептилокси)фенил)-1,2,4-триазина.
Соединение 276 получали, применяя 274, применяя общие способы 7 и 8.
Соединения 277 и 278 получали, применяя INT-16 и 5-бром-2-йодпиридин, применяя последовательно общие способы 10, 10 и 8.
Соединения 279 и 280 получали, применяя INT-16 и 3-хлор-6-йодпиридазин, применяя последовательно общие способы 10, 10 и 8.
Соединения 281 и 282 получали, применяя INT-16 и 2-бром-5-йодпиразин, применяя последовательно общие способы 10, 10 и 8.
Соединение 283 получали из соединения 279 и трет-бутилглицината, применяя последовательно общие способы 7 и 8.
Соединение 284 получали из соединения 281 и трет-бутилглицината, применяя последовательно общие способы 7 и 8.
Соединение 285 получали из соединения 277 и трет-бутилглицината, применяя последовательно общие способы 7 и 8.
2-(4-(гептилокси)фенил)-2-оксоэтил 4-бромбензоат
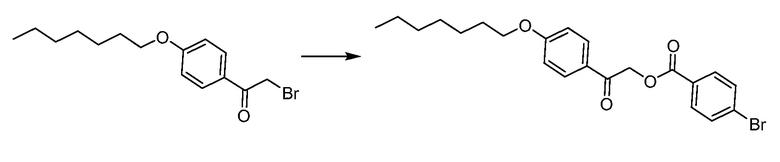
К раствору 2-бром-1-(4-(гептилокси)фенил)этанона INT-4 (1,3 г, 4,2 ммоль) и 4-бромбензойной кислоты (0,70 г, 3,5 ммоль) в ACN (30 мл) добавляли TEA (0,72 мл, 5,2 ммоль). После перемешивания в течение ночи смесь выливали в водную лимонную кислоту и EA, затем перемешивали в течение 10 минут перед тем, как собирать твердый остаток фильтрованием. Остаток на фильтре промывали водой и изогексаном, затем сушили, получая 905 мг (57%) 2-(4-(гептилокси)фенил)-2-оксоэтил 4-бромбензоата. LCMS-ESI (m/z) рассчитанная для C22H25BrO4: 432,1; найденная 433,2 [M+H]+, tR = 3,24 минут (способ 8).
2-(4-бромфенил)-5-(4-(гептилокси)фенил)-1H-имидазол
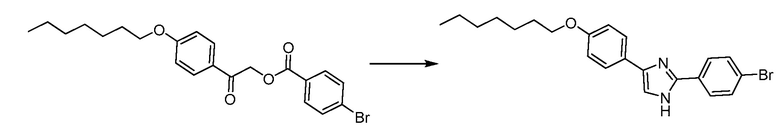
К раствору 2-(4-(гептилокси)фенил)-2-оксоэтил 4-бромбензоата (905 мг, 2,09 ммоль) в толуоле (6 мл) добавляли CH3COONH4 (1600 мг, 20,9 ммоль). После нагревания в течение ночи при 115°C, реакционную смесь разбавляли водным NaHCO3 и экстрагировали в DCM. Органические слои смешивали, сушили над MgSO4, фильтровали, и растворитель удаляли при пониженном давлении. Неочищенную реакционную смесь очищали хроматографией (EA/ гексан), получая 370 мг (33%) 2-(4-бромфенил)-5-(4-(гептилокси)фенил)-1H-имидазола. LCMS-ESI (m/z) рассчитанная для C22H25BrN2O: 412,1; найденная 413,2 [M+H]+, tR = 2,33 минут (способ 8).
2-(4-бромфенил)-5-(4-(гептилокси)фенил)-1-((2-(триметилсилил)этокси)метил)-1H-имидазол
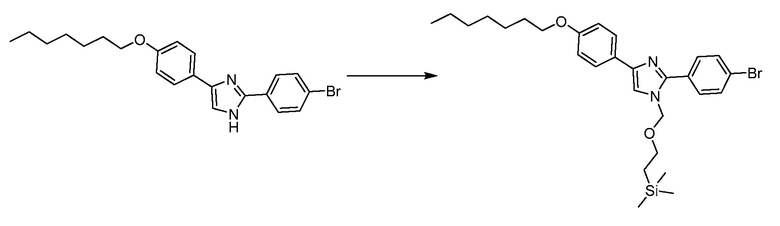
К раствору 2-(4-бромфенил)-5-(4-(гептилокси)фенил)-1H-имидазола (370 г, 900 ммоль) в DMF (4 мл) добавляли NaH (40 мг, 980 ммоль). Через 2 часа добавляли по каплям 2-(триметилсилил)этоксиметилхлорид (160 г, 990 ммоль) в THF (2 мл), и неочищенную реакционную смесь перемешивали в течение ночи. Реакционную смесь разбавляли EA и промывали водным NaHCO3. Органические слои сушили над MgSO4, фильтровали, и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали хроматографией (EA/гексан), получая 32 мг (65%) 2-(4-бромфенил)-5-(4-(гептилокси)фенил)-1-((2-(триметилсилил)этокси)метил)-1H-имидазола в виде коричневого твердого остатка. LCMS-ESI (m/z) рассчитанная для C28H39BrN2O2Si: 542,2; найденная 543,3 [M+H]+, tR = 3,35 минут (способ 8).
(S)-метил 2-((трет-бутоксикарбонил)амино)-3-(4-(4-(4-(гептилокси)фенил)-1-((2-триметилсилил)этокси)метил)-1H-имидазол-2-ил)фенил)пропаноат

Перемешиваемую суспензию цинка (68 мг, 1,03 ммоль) в DMF (2 мл) обрабатывали I2 (12 мг, 0,05 ммоль). После исчезновения цвета добавляли ((R)-метил 2-((трет-бутоксикарбонил)амино)-3-йодпропаноат (110 мг, 0,34 ммоль) и дополнительное количество I2 (12 мг, 0,05 ммоль). Через 30 минут, смесь дегазировали, затем добавляли 2-(4-бромфенил)-5-(4-(гептилокси)фенил)-1-((2-(триметилсилил)этокси)метил)-1H-имидазол (170 мг, 0,31 ммоль), дициклогексил(2',6'-диметокси-[1,1'-бифенил]-2-ил)фосфин (7 мг, 0,02 ммоль) и Pd2(dba)3 (8 мг, 7,8 мкмоль). После дополнительного дегазирования добавляли DMF (2 мл), и реакционную смесь нагревали при 50°C в течение ночи. Реакционную смесь очищали колоночной хроматографией (EA/гексан), получая 55 мг (25%) (S)-метил 2-((трет-бутоксикарбонил)амино)-3-(4-(4-(4-(гептилокси)фенил)-1-((2-(триметилсилил)этокси)метил)-1H-имидазол-2-ил)фенил)пропаноат в виде бесцветного масла. LCMS-ESI (m/z) рассчитанная для C37H55N3O6Si: 665,9; найденная 666,4 [M+H]+, tR = 3,10 минут (способ 8).
(S)-метил 2-амино-3-(4-(4-(4-(гептилокси)фенил)-1H-имидазол-2-ил)фенил)пропаноат

(S)-метил 2-амино-3-(4-(4-(4-(гептилокси)фенил)-1H-имидазол-2-ил)фенил)пропаноат получали из (S)-метил 2-((трет-бутоксикарбонил)амино)-3-(4-(4-(4-(гептилокси)фенил)-1-((2-(триметилсилил)этокси)метил)-1H-имидазол-2-ил)фенил)пропаноата, применяя общий способ 8. LCMS-ESI (m/z) рассчитанная для C26H33N3O3: 435,6; найденная 436,3 [M+H]+, tR = 1,43 минут (способ 8).
Гидрохлорид (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(4-(4-(гептилокси)фенил)-1H-имидазол-2-ил)фенил)пропионовой кислоты (соединение 286)
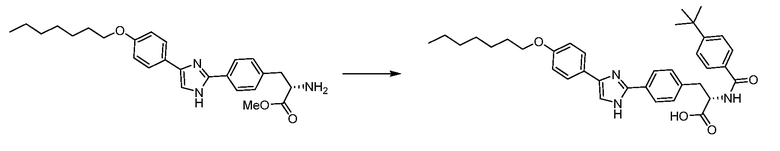
К раствору 4-(трет-бутил)бензойной кислоты (25 мг, 0,14 ммоль), (S)-метил 2-амино-3-(4-(4-(4-(гептилокси)фенил)-1H-имидазол-2-ил)фенил)пропаноата (55 мг, 0,13 ммоль) и TEA (53 мкл, 0,38 ммоль) в DMF (1 мл) добавляли HATU (53 мг, 0,14 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, разбавляли в DCM и промывали водным NaHCO3. Органический слой сушили, концентрировали и очищали хроматографией (EA/гексан), получая 14 мг (17%) метил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(4-(4-(гептилокси)фенил)-1H-имидазол-2-ил)фенил)пропаноата. LCMS-ESI (m/z) рассчитанная для C37H45N3O4: 595,8; найденная 596,4 [M+H]+, tR = 2,33 минут (способ 8).
Выделенное эфирное промежуточное соединение деблокировали, применяя общий способ 4, получая 14 мг (17,5%) гидрохлорида (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(4-(4-(гептилокси)фенил)-1H-имидазол-2-ил)фенил)пропионовой кислоты, соединения 286, в виде светло-коричневого твердого остатка. LCMS-ESI (m/z) рассчитанная для C36H43N3O4: 581,8; найденная 582,4 [M+H]+, tR = 6,56 минут (способ 9).
4-бром-1-4-(гептилокси)фенил)-1H-имидазол
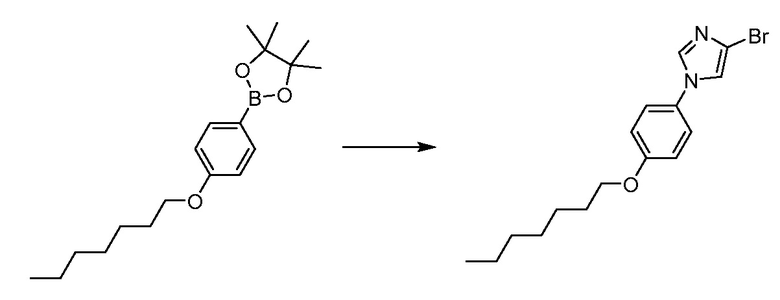
В пробирку помещали (4-(гептилокси)фенил)бороновую кислоту (1,00 г, 4,24 ммоль), 4-бром-1H-имидазол (0,31 г, 2,1 ммоль), Cu-(TMEDA)2(OH)2Cl2 (0,10 г, 0,21 ммоль) и DCM (12 мл). После перемешивания при комнатной температуре в течение 42 часов, смесь очищали хроматографией (EA/гексан), получая 80 мг продукт с примесями. Дополнительная очистка хроматографией (CAN/DCM) давала 42 мг (6%) 4-бром-1-(4-(гептилокси)фенил)-1H-имидазола в виде бесцветного масла. LCMS-ESI (m/z) рассчитанная для C16H21BrN2O: 336,1; найденная 337,1 [M+H]+, tR = 2,71 минут (способ 8).
(S)-2-(4-(трет-бутил)бензамидо)-3-(4-(1-(4-(гептилокси)фенил)-1H-имидазол-4-ил)фенил)пропионовая кислота (соединение 287)

Получали, применяя общий способ 10. В пробирку, содержащую INT-13 (96 мг, 0,19 ммоль) и 4-бром-1-(4-(гептилокси)фенил)-1H-имидазол (64 мг, 0,19 ммоль) в 2/2/1 THF/CAN/H2O (3 мл) добавляли Na2CO3 (40 мг, 0,38 ммоль). Реакционную смесь дегазировали и добавляли Pd(dppf)Cl2 (14 мг, 0,02 ммоль). После нагревания при 120°C в течение 30 минут в микроволновом реакторе, смесь разбавляли EA, промывали водным NaHCO3, сушили над MgSO4 и концентрировали. Очистка хроматографией (EA/гексан) давала 14 мг (12%) промежуточного соединения, трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(1-(4-(гептилокси)фенил)-1H-имидазол-4-ил)фенил)пропаноата, в виде белого твердого остатка.
Промежуточное соединение деблокировали согласно общему способу 8, получая 9 мг (8%) (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(1-(4-(гептилокси)фенил)-1H-имидазол-4-ил)фенил)пропионовой кислоты, соединения 287, в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C36H43N3O4: 581,3; найденная 582,2 [M+H]+, tR = 8,33 минут (способ 9).
(S)-2-(4-(трет-бутил)бензамидо)-3-(4-(1-(4'-метил-[1,1'-бифенил]-4-ил)-1H-пиразол-4-ил)фенил)пропионовая кислота (соединение 288)
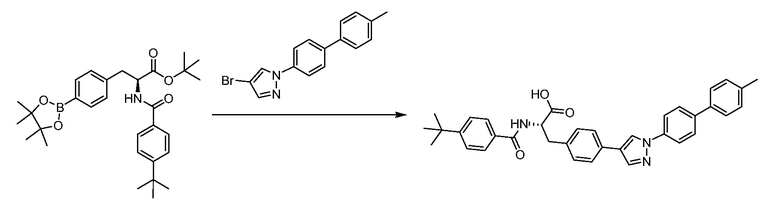
Получали, применяя общий способ 10. В пробирку, содержащую INT-13 (100 мг, 0,20 ммоль) и 4-бром-1-(4'-метил-[1,1'-бифенил]-4-ил)-1H-пиразол (63 мг, 0,201 ммоль) в 2/1 ACN/H2O (3 мл) добавляли насыщенный водный NaHCO3 (670 мкл, 0,60 ммоль). Реакционную смесь дегазировали и добавляли Pd(dppf)Cl2 (15 мг, 0,02 ммоль). После нагревания при 120°C в течение 60 минут в микроволновом реакторе, смесь разбавляли DCM, промывали водным NaHCO3, пропускали через картридж для разделения фаз и концентрировали. Очистка хроматографией (EA/гексан) давала 58 мг (47%) промежуточного соединения, трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(1-(4'-метил-[1,1'-бифенил]-4-ил)-1H-пиразол-4-ил)фенил)пропаноата в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C40H43N3O3: 613,8; найденная 614,0 [M+H]+, tR = 3,02 минут (способ 8). Промежуточное соединение перемешивали в 4M HCl/диоксан в течение 132 часов и фильтровали. Полученный в результате твердый остаток промывали гексаном, получая 13 мг твердого продукта. Фильтрат загружали на сильную анионообменную (SAX) колонку, промывали MeOH и элюировали 5% AcOH в MeOH. Элюированные растворы смешивали с растертым твердым остаток и концентрировали в вакууме, получая 18 мг (32%) (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(1-(4'-метил-[1,1'-бифенил]-4-ил)-1H-пиразол-4-ил)фенил)пропионовой кислоты 288 в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C36H35N3O3: 557,3; найденная 558,0 [M+H]+, tR = 9,37 минут (способ 9).
Метил 2-(4-бромфенил)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)ацетат
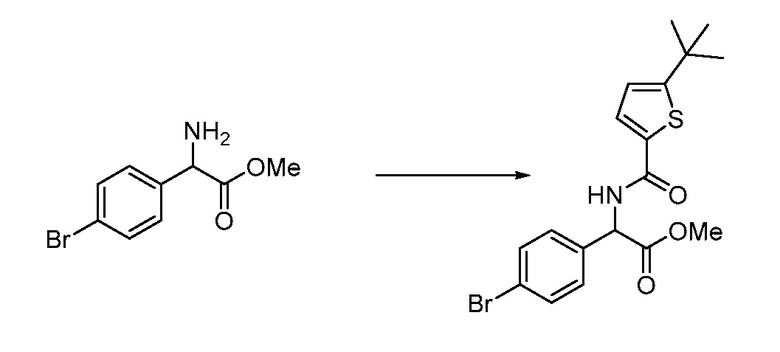
Получали, применяя общий способ 7. К раствору метил 2-амино-2-(4-бромфенил)ацетата, HCl (730 мг, 2,6 ммоль), 5-(трет-бутил)тиофен-2-карбоновой кислоты (480 мг, 2,6 ммоль) и TEA (1090 мкл, 7,8 ммоль) в DMF (10 мл) добавляли HATU (1090 мг, 2,9 ммоль). После перемешивания в течение ночи, реакционную смесь разбавляли EA (100 мл) и промывали 1M HCl (100 мл) и соляным раствором. Органический слой сушили над MgSO4, концентрировали и очищали хроматографией (EA /гексан), получая 900 мг (76%) метил 2-(4-бромфенил)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)ацетата в виде белого порошка. LCMS-ESI (m/z) рассчитанная для C18H20BrNO3S: 410,3; найденная 412,0 [M+2]+, tR = 2,71 минут (способ 8).
Метил 2-(5-(трет-бутил)тиофен-2-карбоксамидо)-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетат
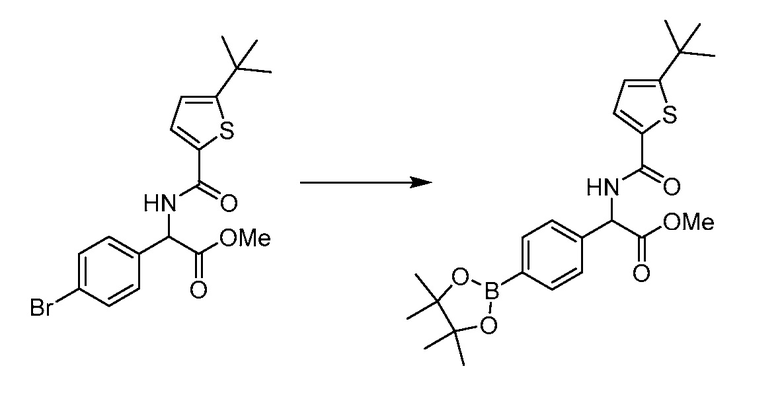
Получали, применяя общий способ 10. Дегазировали раствор 2-(4-бромфенил)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)ацетата (900 мг, 2,2 ммоль), KOAc (650 мг, 6,6 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2,-би(1,3,2-диоксаборолана) (670 мг, 2,6 ммоль) в DMSO (10 мл) при 40°C. Добавляли PdCl2dppf (80 мг, 0,11 ммоль), и смесь нагревали при 100°C в течение 3 часов. Реакционную смесь очищали хроматографией (EA/гексан с 1% TEA), получая 491 мг (41%) метил 2-(5-(трет-бутил)тиофен-2-карбоксамидо)-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетата. LCMS-ESI (m/z) рассчитанная для C24H32BNO5S: 457,4; найденная 458,0 [M+H]+, tR = 2,89 минут (способ 8).
2-(4-(5-бромпиримидин-2-ил)фенил)-2-(5-(трет-бутил)тиофен-2- карбоксамидо)уксусная кислота
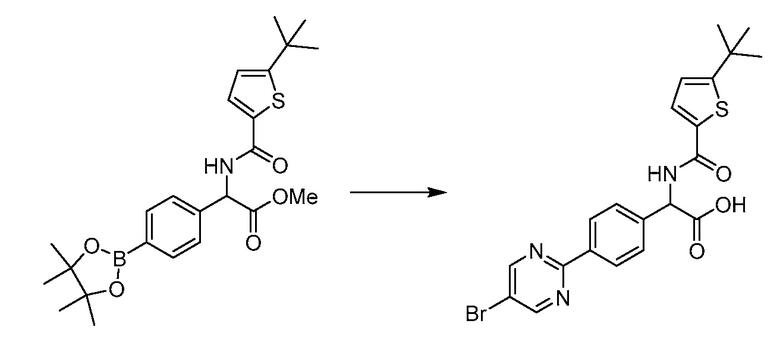
Получали, применяя общий способ 10. Смесь метил 2-(5-(трет-бутил)тиофен-2-карбоксамидо)-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)ацетата (320 мг, 0,71 ммоль) и 5-бром-2-йодпиримидина (220 мг, 0,78 ммоль) в THF (2 мл) и MeCN (2 мл) обрабатывали насыщенным водным NaHCO3 (1600 мкл, 1,40 ммоль) и дегазировали (N2 барботирование). Добавляли PdCl2dppf (26 мг, 0,04 ммоль), и смесь нагревали при 120°C в течение 30 минут в микроволновом реакторе. Смесь выливали в H2O (30 мл), подкисляли AcOH и экстрагировали EA (3×15 мл). Объединенные органические слои сушили над MgSO4, упаривали и очищали хроматографией (EA/гексан с 1% AcOH), получая 160 мг (46%) 2-(4-(5-бромпиримидин-2-ил)фенил)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)уксусной кислоты в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C21H20BrN3O3S: 473,0; найденная 474,0 [M+H]+, tR = 2,68 минут (способ 8).
(S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропионовая кислота (соединение 289)

Получали, применяя общий способ 10. Раствор 2-(4-(5-бромпиримидин-2-ил)фенил)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)уксусной кислоты (160 мг, 0,34 ммоль), (4-(гептилокси)фенил)бороновой кислоты (94 мг, 0,40 ммоль) и насыщенного водного NaHCO3 (930 мкл, 0,84 ммоль) в ACN (1,5 мл) и THF (1,5 мл) дегазировали (N2 барботирование). Добавляли PdCl2(dppf) (262 мг, 0,34 ммоль), и реакционную смесь нагревали при 110°C в микроволновом реакторе в течение 50 минут. Реакцию распределяли между EA и H2O. Органический слой сушили над MgSO4, фильтровали, концентрировали и очищали хроматографией (EA/гексан c 1% AcOH), получая 113 мг (55%) 2-(5-(трет-бутил)тиофен-2-карбоксамидо)-2-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)уксусной кислоты, соединения 289, в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C34H39N3O4S: 585,3; найденная 586,0 [M+H]+, tR = 3,37 минут (способ 9).
(S)-N-(1-амино-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)-1-оксопропан-2-ил)-4-(трет-бутил)бензамид
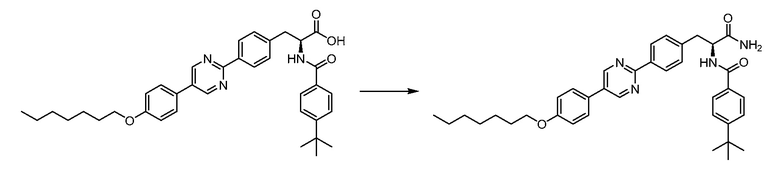
Раствор соединения 85 (245 мг, 0,413 ммоль) в DMF (5 мл) обрабатывали NH4Cl (180 мг, 3,3 ммоль), DIEA (760 мкл, 4,1 ммоль) и HATU (170 мг, 0,4 ммоль). После перемешивания в течение ночи, реакционную смесь разбавляли EA (50 мл), промывали водным 0,5 M HCl (100 мл) и соляным раствором (20 мл), затем сушили над MgSO4 и концентрировали. Остаток высаживали из ACN (4 мл), получая 204 мг (77%) (S)-N-(1-амино-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)-1-оксопропан-2-ил)-4-(трет-бутил)бензамида в виде мелкодисперсного белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C37H44N4O3: 592,3; найденная 593,0 [M+H]+, tR = 3,43 минут (способ 6).
(S)-метил 3-(4-(трет-бутил)бензамидо)-4-(4-гидроксифенил)бутаноат
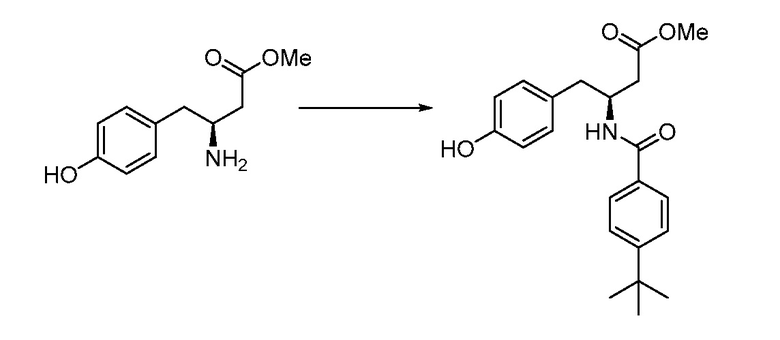
Получали, применяя общий способ 7. Раствор гидрохлорида (S)-метил 3-амино-4-(4-гидроксифенил)бутаноата (2,1 г, 8,7 ммоль), 4-(трет-бутил)бензойной кислоты (1,6 г, 9,0 ммоль) и DIEA (3,5 мл, 18,8 ммоль) в DMF (20 мл) и DCM (20 мл) обрабатывали HATU (3,3 г, 8,5 ммоль). Через 1 час, смесь выливали в 1M HCl (100 мл) и экстрагировали EA (3×50 мл). Объединенные органические экстракты промывали последовательно 1M HCl (50 мл), водой (50 мл) и соляным раствором (20 мл), затем сушили над MgSO4 и концентрировали. Полученный в результате остаток очищали хроматографией (EA/ гексан), получая 2,3 г (72%) (S)-метил 3-(4-(трет-бутил)бензамидо)-4-(4-гидроксифенил)бутаноата в виде белых игольчатых кристаллов. LCMS-ESI (m/z) рассчитанная для C22H27NO: 369,4, найденная 370,0 [M+H]+, tR = 2,52 минут (способ 6).
(S)-метил 3-(4-(трет-бутил)бензамидо)-4-(4-(((трифторметил)сульфонил)окси)фенил)бутаноат
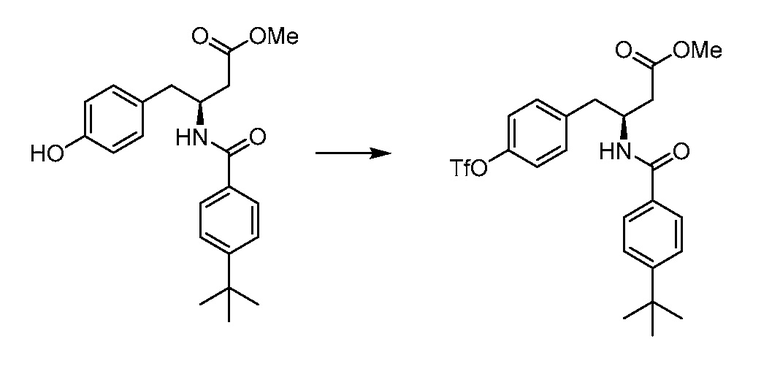
Получали, применяя общий способ 9. Перемешиваемый раствор (S)-метил 3-(4-(трет-бутил)бензамидо)-4-(4-гидроксифенил)бутаноата (2,30 г, 6,3 ммоль) в DCM (25 мл) обрабатывали DIEA (1,4 мл, 7,6 ммоль), затем 1,1,1-трифтор-N-фенил-N-((трифторметил)сульфонил)метансульфамидом (2,5 г, 6,9 ммоль). Через 18 часов реакционную смесь разбавляли DCM (100 мл), H2O (50 мл) и NaHCO3 (75 мл) и перемешивали в течение 1 часа. Органический слой выделяли, промывали NaHCO3 (100 мл), сушили над MgSO4, концентрировали и очищали хроматографией (EA/гексан), получая 2,5 г (75%) (S)-метил 3-(4-(трет-бутил)бензамидо)-4-(4-(((трифторметил)сульфонил)окси)фенил)бутаноата в виде густого масла. LCMS-ESI (m/z) рассчитанная для C23H26F3NO6S: 501,5, найденная 502 [M+H]+, tR = 3,20 минут (способ 6).
(S)-метил 3-(4-(трет-бутил)бензамидо)-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)бутаноат

В пробирку в атмосфере N2 добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (530 мг, 2,1 ммоль), (S)-метил 3-(4-(трет-бутил)бензамидо)-4-(4-(((трифторметил)сульфонил)окси)фенил)бутаноат (810 мг, 1,6 ммоль), KOAc (280 мг, 4,8 ммоль) и DMSO (14 мл). Раствор дегазировали. Добавляли Pd(dppf)Cl2 (59 мг, 0,08 ммоль), и раствор нагревали при 80°C в течение 6 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли EA (100 мл) и промывали насыщенным водным NaHCO3 (50 мл) и соляным раствором (50 мл). Органический слой сушили над MgSO4, концентрировали и очищали хроматографией (EA/гексан), получая 446 мг (57%) (S)-метил 3-(4-(трет-бутил)бензамидо)-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)бутаноата в виде бесцветного кристаллического остатка. LCMS-ESI (m/z) рассчитанная для C28H38BNO5: 479,4, найденная 480,3 [M+H]+, tR = 2,86 минут (способ 6).
(S)-метил 4-(4-(5-бромпиримидин-2-ил)фенил)-3-(4-(трет-бутил)бензамидо)бутаноат
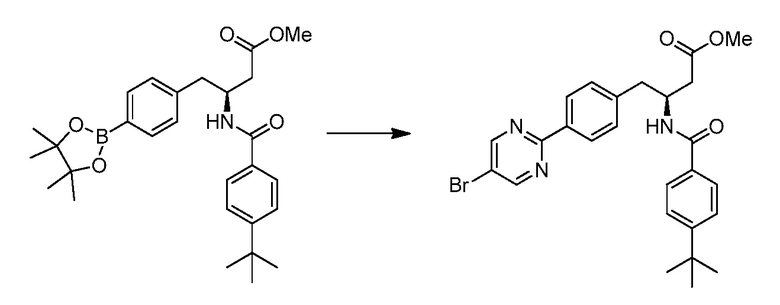
Получали, применяя общий способ 10. В пробирку добавляли (S)-метил 3-(4-(трет-бутил)бензамидо)-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)бутаноат (390 мг, 0,81 ммоль), 5-бром-2-йодпиримидин (240 мг, 0,85 ммоль), Na2CO3 (170 мг, 1,6 ммоль), THF (1,5 мл), ACN (1,5 мл) и H2O (0,75 мл). Раствор дегазировали и добавляли PdCl2(dppf) (60 мг, 0,08 ммоль). Реакционную смесь нагревали в микроволновом реакторе при 110°C в течение 60 минут. Образец охлаждали, разбавляли EA (50 мл) и промывали насыщенным водным NaHCO3 (30 мл). Органические слои сушили над MgSO4, фильтровали, концентрировали и очищали хроматографией (EA/гексан), получая 205 мг (49%) (S)-метил 4-(4-(5-бромпиримидин-2-ил)фенил)-3-(4-(трет-бутил)бензамидо)бутаноата в виде бесцветного твердого остатка. LCMS-ESI (m/z) рассчитанная для C26H28BrN3O3: 510,4, найденная 512,2 [M+H]+, tR = 2,77 минут (способ 6).
(S)-3-(4-(трет-бутил)бензамидо)-4-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)масляная кислота (соединение 291)
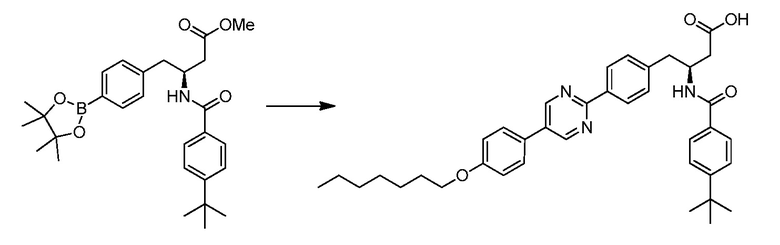
Получали, применяя общие способы 10 и 4. В пробирку добавляли (S)-метил 4-(4-(5-бромпиримидин-2-ил)фенил)-3-(4-(трет-бутил)бензамидо)бутаноат (180 мг, 0,35 ммоль), (4-(гептилокси)фенил)бороновую кислоту (98 мг, 0,41 ммоль), Na2CO3 (73 мг, 0,69 ммоль), ACN (1,2 мл), THF (1,2 мл) и H2O (0,7 мл). Раствор дегазировали, добавляли Pd(dppf)Cl2 (25 мг, 0,03 ммоль), и реакционную смесь нагревали в микроволновом реакторе при 110°C в течение 80 минут. Реакционную смесь разбавляли EA (50 мл) и промывали насыщенным водным NaHCO3 (30 мл). Органические слои сушили над MgSO4, концентрировали и очищали хроматографией (EA/гексан), получая 44 мг промежуточного соединения, метилового эфира. Твердый остаток растворяли в THF (1 мл) и 1M LiOH (1 мл). Раствор перемешивали при температуре окружающей среды в течение 1 часа, концентрировали и добавляли 1M HCl (1,5 мл). Твердый остаток собирали фильтрованием, промывали водой (2×5 мл) и гексаном (2×5 мл), получая 19 мг (9%) (S)-3-(4-(трет-бутил)бензамидо)-4-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)масляной кислоты, соединения 291, в виде бесцветного твердого остатка. LCMS-ESI (m/z) рассчитанная для C38H45N3O4: 607,8, найденная 608,4 [M+H]+, tR = 10,99 минут (способ 10).
5-бром-2-хлор-4-метоксипиримидин
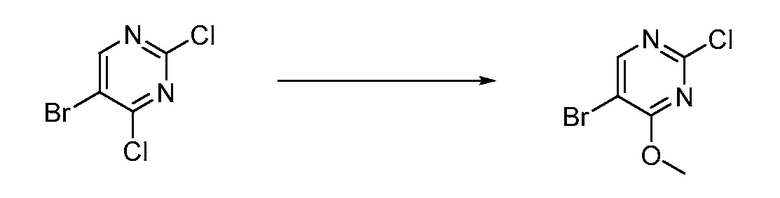
К перемешиваемому раствору 5-бром-2,4-дихлорпиримидина (500 мг, 2,19 ммоль) в MeOH (5 мл) добавляли 30% раствор метоксида натрия (0,40 мл, 2,26 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали. Остаток растворяли в воде (5 мл) и экстрагировали EA (3×5 мл). Объединенный органический слой промывали соляным раствором, сушили над MgSO4 и концентрировали, получая 432 мг (88%) 5-бром-2-хлор-4-метоксипиримидина в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C5H4BrClN2O: 223,4; найденная 224,2 [M+H]+, tR = 7,66 минут (способ 2).
5-бром-2-йод-4-метоксипиримидин
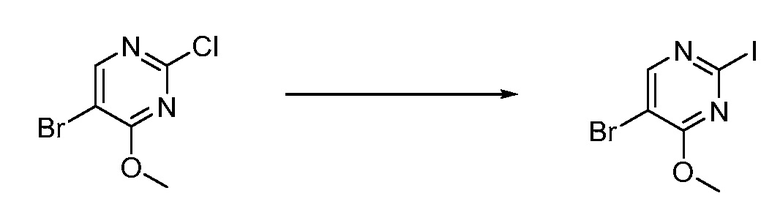
Получали, применяя общий способ 16: к перемешиваемому раствору 5-бром-2-хлор-4-метоксипиримидина (100 мг, 0,447 ммоль) в 57 % водном HI (1,0 мл) добавляли йодид натрия (125 мг, 0,838 ммоль). Реакционную смесь перемешивали при 40°C в течение 16 часов, охлаждали, затем гасили NaHCO3 (5 мл) и экстрагировали EA (3×5 мл). Объединенные органические слои промывали соляным раствором, сушили над MgSO4 и концентрировали, получая 22,0 мг (16%) 5-бром-2-йод-4-метоксипиримидина в виде грязно-белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C5H4BrIN2O: 314,9; найденная 315,9 [M+H]+, tR = 8,22 минут (способ 2). 1H ЯМР (400 МГц, DMSO) δ 8,25 (с, 1H), 4,07 (с, 3H).
Трет-бутил (S)-3-(4-(5-бром-4-метоксипиримидин-2-ил)фенил)-2- (4-(трет-бутил)бензамидо)пропаноат
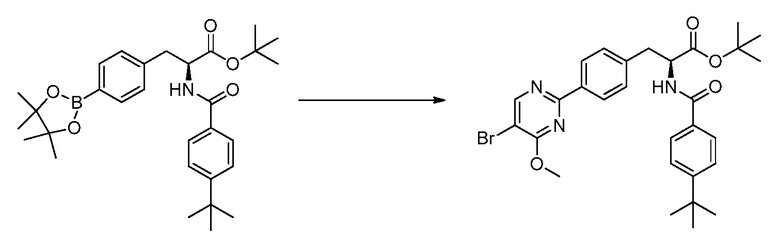
Получали, применяя общий способ 10: смесь трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноата INT-13 (30,0 мг, 0,06 ммоль), 5-бром-2-йод-4-метоксипиримидина (22,3 мг, 0,07 ммоль) и карбоната натрия (12,5 мг, 0,12 ммоль) в ацетонитриле (0,80 мл), THF (0,80 мл) и H2O (0,40 мл) дегазировали в течение 10 минут. Добавляли Pd(dppf)Cl2:CH2Cl2 (5 мг, 0,005 ммоль), и реакционную смесь грели при 110°C в микроволновой печи в течение 30 минут. После охлаждения реакцию разбавляли NaHCO3 (5 мл), экстрагировали EA (3×5 мл), и объединенные органические слои сушили над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией (EA/гексан), получая 20,0 мг (60%) трет-бутил (S)-3-(4-(5-бром-4-метоксипиримидин-2-ил)фенил)-2-(4-(трет-бутил)бензамидо)пропаноата в виде белого твердого остатка. LCMS-ESI (m/z) рассчитанная для C29H34BrN3O4: 568,5; найденная 514,2 [M-tBu+H]+, tR = 11,0 минут (способ 2).
Трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-4-метоксипиримидин-2-ил)фенил)пропаноат

Получали, применяя общий способ 10: смесь трет-бутил (S)-3-(4-(5-бром-4-метоксипиримидин-2-ил)фенил)-2-(4-(трет-бутил)бензамидо)пропаноата (18,0 мг, 0,031 ммоль), (4-(гептилокси)фенил)бороновой кислоты (10,0 мг, 0,042 ммоль) и карбоната натрия (8,97 мг, 0,084 ммоль) в ацетонитриле (0,80 мл), THF (0,80 мл) и H2O (0,40 мл) дегазировали в течение 10 минут. Добавляли Pd(dppf)Cl2:CH2Cl2 (3,09 мг, 0,003 ммоль), и реакционную смесь грели при 110°C в микроволновой печи в течение 30 минут. После охлаждения, реакцию разбавляли NaHCO3 (5 мл) и экстрагировали EA (3×5 мл). Объединенные органические слои сушили над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией (EA:гексан), получая 20,0 мг (60%) трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-4-метоксипиримидин-2-ил)фенил)пропаноат в виде бледно-желтого твердого остатка. LCMS-ESI (m/z) рассчитанная для C42H53N3O5: 679,8; ион не наблюдали, tR = 13,83 минут (способ 2).
(S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-4-метоксипиримидин-2-ил)фенил)пропионовая кислота (соединение 292)
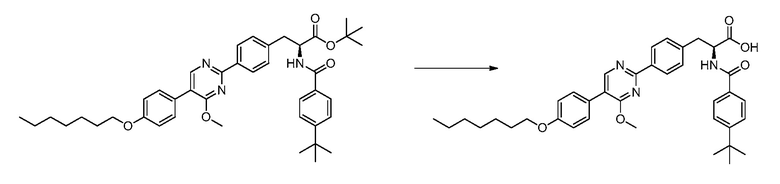
Получали, применяя общий способ 8: раствор трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил-4-метоксипиримидин-2-ил)фенил)пропаноата (20,0 мг, 0,029 ммоль) в DCM (1 мл) обрабатывали TFA (0,350 мл). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Растворитель концентрировали, и продукт очищали препаративной ВЭЖХ, получая 15,0 мг (82%) (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)-4-метоксипиримидин-2-ил)фенил)пропионовой кислоты, соединения 292, в виде бледно-желтого твердого остатка. LCMS-ESI (m/z) рассчитанная для C38H45N3O5: 623,8; ион не наблюдали, tR = 12,17 минут (способ 2).
Соединение 293 получали, применяя трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноат INT-13 и 5-бром-2-хлор-N,N-диметилпиримидин-4-амин, применяя последовательно общие способы 10, 10 и 8.
Соединение 294 получали, применяя трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноат INT-13 и 5-бром-2-йод-4-метилпиридин, применяя последовательно общие способы 10, 10 и 8.
5-бром-2-йод-4-(трифторметил)пиридин
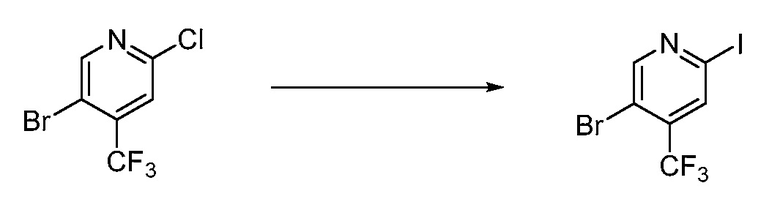
Получали, применяя общий способ 17: к перемешиваемому раствору 5-бром-2-хлор-4-(трифторметил)пиридина (150 мг, 0,576 ммоль) в ацетонитриле (2 мл) добавляли йодид натрия (518 мг, 3,45 ммоль). Реакционную смесь нагревали до 40°C и добавляли ацетилхлорид (26,0 мг, 0,345 ммоль). Реакционную смесь перемешивали при 40°C в течение 90 минут. После охлаждения реакцию гасили NaHCO3 (5 мл) и экстрагировали EA (3×5 мл). Объединенные органические слои промывали соляным раствором (10 мл), сушили над MgSO4 и концентрировали, получая 80,0 мг (40%) 5-бром-2-йод-4-(трифторметил)пиридина в виде белого кристаллического остатка, который применяли в следующей стадии без очистки. LCMS-ESI (m/z) рассчитанная для C6H2BrF3IN: 351,9; найденная 352,5 [M+H]+, tR = 3,91 минут (способ 1).
Соединение 295 получали применением трет-бутил (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноата INT-13 и 5-бром-2-йод-4-(трифторметил)пиридина, применяя последовательно общие способы 10, 10 и 8.
(S)-(2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропанoил)глицин (соединение 297)
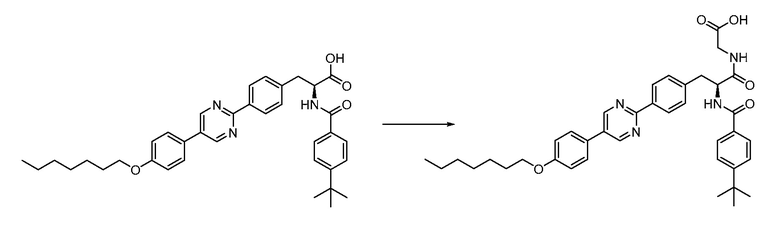
Получали, применяя общие способы 7 и 8: к раствору (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропионовой кислоты, соединения 85 (185 мг, 0,312 ммоль), гидрохлорида трет-бутил 2-аминоацетата (52,2 мг, 0,312 ммоль) и DIEA (163 мкл, 0,935 ммоль) в DMF (3 мл) добавляли HATU (124 мг, 0,327 ммоль). Смесь перемешивали в течение 1 часа при комнатной температуре. Неочищенный материал разбавляли в EA (50 мл), промывали насыщенным водным бикарбонатом натрия (20 мл) и соляным раствором (20 мл). Органический слой сушили над MgSO4, фильтровали, и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали хроматографией (EA/гексан), получая промежуточное соединение, трет-бутиловый эфир (110 мг).
Трет-бутиловый эфир растворяли в DCM (1 мл) и добавляли TFA (2 мл). Раствор перемешивали при комнатной температуре в течение 3 часов, и растворитель удаляли при пониженном давлении. Неочищенную смесь растворяли в DMSO (0,8 мл) и осаждали добавлением воды (3 мл). Осадок фильтровали, промывали водой (3 мл) и гексаном (2×2 мл), получая 58 мг (28%) (S)-(2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропанoил)глицина, соединения 297, в виде бесцветного твердого остатка. LCMS-ESI (m/z) рассчитанная для C39H46N4O5: 650,4; найденная 651,4 [M+H]+, tR = 10,43 минут (способ 10). Рассчитывали, что хиральная чистота составляла 92% e.e. (хиральный способ). 1H ЯМР (400 МГц, DMSO-d6) δ 12,62 (с, 1H), 9,15 (с, 2H), 8,60 (д, J=8,6 Гц, 1H), 8,49-8,40 (м, 1H), 8,35-8,25 (м, 2H), 7,84-7,70 (м, 4H), 7,58-7,49 (м, 2H), 7,48-7,41 (м, 2H), 7,16-7,02 (м, 2H), 4,90-4,75 (м, 1H), 4,03 (т, J=6,5 Гц, 2H), 3,93-3,75 (м, 2H), 3,25 (дд, J=13,8, 3,8 Гц, 1H), 3,09 (дд, J=13,7, 11,2 Гц, 1H), 1,79-1,68 (м, 2H), 1,51-1,21 (м, 17H), 0,94-0,80 (м, 3H).
(S)-3-(2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропанамидо)пропионовая кислота (соединение 298)
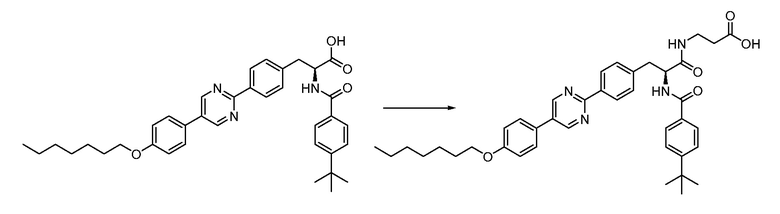
Получали, применяя общие способы 7 и 8: HATU (116 мг, 0,31 ммоль) добавляли к перемешиваемому раствору (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропионовой кислоты, соединения 85 (173 мг, 0,29 ммоль), гидрохлорида трет-бутил 3-аминопропаноата (53 мг, 0,29 ммоль) и DIEA (153 мкл, 0,87 ммоль) в DMF (3 мл). Неочищенный материал разбавляли в EA (50 мл), промывали насыщенным водным бикарбонатом натрия (20 мл) и соляным раствором (20 мл). Органический слой сушили над MgSO4, фильтровали, и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали хроматографией (EA/гексан), получая промежуточное соединение, трет-бутиловый эфир (122 мг).
трет-Бутиловый эфир растворяли в DCM (1 мл) и добавляли TFA (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, и растворитель удаляли при пониженном давлении. Неочищенную смесь растворяли в DMSO (0,8 мл) и осаждали добавлением воды (3 мл). Осадок фильтровали, промывали водой (3 мл) и гексаном (2×2 мл), получая 48 мг (25%) (S)-3-(2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропанамидо)пропионовой кислоты, соединения 298, в виде бесцветного твердого остатка. LCMS-ESI (m/z) рассчитанная для C40H48N4O5: 664,4; найденная 665,4 [M+H]+, tR = 10,36 минут (способ 10). 1H ЯМР (400 МГц, DMSO-d6) δ 12,26 (с, 1H), 9,15 (с, 2H), 8,51 (д, J=8,5 Гц, 1H), 8,40-8,25 (м, 2H), 8,25-8,14 (м, 1H), 7,96-7,65 (м, 4H), 7,65-7,36 (м, 4H), 7,28-6,99 (м, 2H), 4,84-4,64 (м, 1H), 4,03 (т, J=6,5 Гц, 2H), 3,32-3,24 (м, 2H), 3,17 (дд, J=13,7, 4,4 Гц, 1H), 3,06 (дд, J=13,7, 10,4 Гц, 1H), 2,41 (т, J=6,9 Гц, 2H), 1,81-1,68 (м, 2H), 1,50-1,20 (м, 17H), 0,88 (т, J=6,7 Гц, 3H).
(S)-4-(трет-бутил)-N-(3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)-1-(метилсульфамидо)-1-оксопропан-2-ил)бензамид (соединение 299)
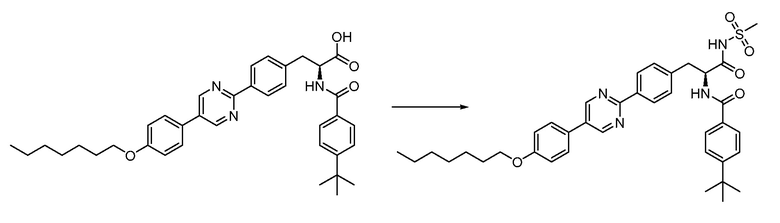
К раствору (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропионовой кислоты, соединения 85 (78,0 мг, 0,13 ммоль), метансульфамида (20,0 мг, 0,21 ммоль) и DMAP (16,1 мг, 0,13 ммоль) в DMF (1,5 мл) добавляли EDC (40,3 мг, 0,21 ммоль), и раствор перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли в EA (50 мл), промывали насыщенным водным бикарбонатом натрия (2×20 мл) и соляным раствором (20 мл). Органический слой сушили над MgSO4, фильтровали, и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали хроматографией (гексан/EA), получая 36 мг (40%) (S)-4-(трет-бутил)-N-(3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)-1-(метилсульфамидо)-1-оксопропан-2-ил)бензамида, соединения 299, в виде бесцветного твердого остатка. LCMS-ESI (m/z) рассчитанная для C38H46N4O5S; 670,3; найденная 671,3 [M+H]+, tR = 11,01 минут (способ 10).
Соединения 300-304 получали из (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропионовой кислоты, соединения 85, применяя общие способы 3 или 7, с последующим 4 или 8.
Соединения 305-317 получали из (S)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)-2-(4-изопропилбензамидо)пропионовой кислоты, соединения 94, применяя общие способы 3 или 7, с последующим 4 или 8.
Соединение 318 получали из (S)-2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гексилокси)фенил)пиримидин-2-ил)фенил)пропионовой кислоты, соединения 225, применяя общий способ 7, с последующим 8.
(S)-(2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропаноил)глицин (соединение 319)
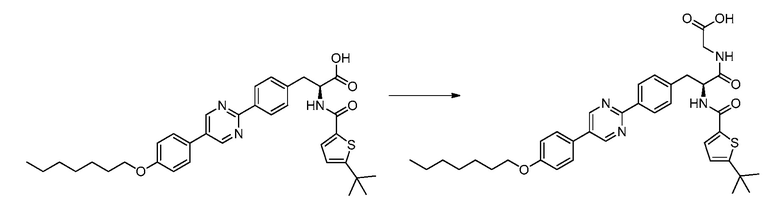
Получали, применяя общие способы 7 и 4: TEA (93 мкл, 0,67 ммоль) добавляли к раствору (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропионовой кислоты, соединения 192 (100 мг, 0,167 ммоль), гидрохлорида метил 2-аминоацетата (23,03 мг, 0,18 ммоль) и HATU (76 мг, 0,20 ммоль) в DMF (2 мл). Раствор перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли EA (25 мл) и промывали насыщенным водным NaHCO3 (2×25 мл) и 1 M HCl (2×25 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Твердый остаток очищали хроматографией (EA/гексан), получая, промежуточное соединение, метиловый эфир, в виде бесцветного твердого остатка. Твердый остаток растворяли в THF (3 мл) и добавляли 1 M LiOH (333 мкл, 0,33 ммоль). Полученный в результате желтый раствор перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь подкисляли до pH 1, применяя 1M HCl, и THF удаляли в вакууме. Остаток суспендировали в воде, и смесь фильтровали в вакууме. Твердый остаток азеотропно упаривали с MeOH и сушили в вакуумном сушильном шкафу, получая 48 мг (44 %) (S)-(2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропаноил)глицина, соединения 319, в виде желтого твердого остатка. LCMS-ESI (m/z) рассчитанная для C37H44N4O5S: 656,3; найденная 657,0 [M+H]+, tR = 10,34 минут (способ 10). Рассчитывали, что хиральная чистота составляла 95% e.e. (хиральный способ). 1H ЯМР (400 МГц, DMSO-d6) δ 12,61 (с, 1H), 9,16 (с, 2H), 8,62 (д, J=8,7 Гц, 1H), 8,51-8,41 (м, 1H), 8,36-8,26 (м, 2H), 7,84-7,75 (м, 2H), 7,68 (д, J=3,8 Гц, 1H), 7,55-7,43 (м, 2H), 7,14-7,05 (м, 2H), 6,92 (д, J=3,8 Гц, 1H), 4,84-4,72 (м, 1H), 4,03 (т, J=6,5 Гц, 2H), 3,89-3,73 (м, 2H), 3,22 (дд, J=13,9, 3,7 Гц, 1H), 3,10-2,96 (м, 1H), 1,78-1,66 (м, 2H), 1,31 (с, 17H), 0,94-0,81 (м, 3H).
((S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4- (гептилокси)фенил)пиримидин-2-ил)фенил)пропаноил)-L-глютамин (соединение 320)
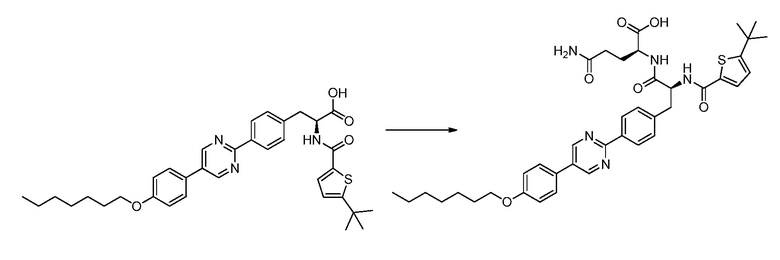
Получали, применяя общие способы 7 и 8: к перемешиваемому раствору (S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропионовой кислоты, соединения 192 (250 мг, 0,42 ммоль), гидрохлорида (S)-трет-бутил 2,5-диамино-5-оксопентаноата (109 мг, 0,46 ммоль) и TEA (145 мкл, 1,04 ммоль) в DMF (4 мл) добавляли HATU (190 мг, 0,50 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли EA (50 мл), промывали 1M HCl (50 мл) и соляным раствором (100 мл), сушили над сульфатом магния и концентрировали. Неочищенный продукт растворяли в DCM (5 мл) и добавляли TFA (3 мл). Через 3 часа добавляли толуол (10 мл), и растворитель упаривали. Соединение очищали препаративной ВЭЖХ, получая 78 мг (25%) ((S)-2-(5-(трет-бутил)тиофен-2-карбоксамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропаноил)-1-глютамина, соединение 320, в виде белого порошка. LCMS-ESI (m/z) рассчитанная для C40H49N5O6S: 727,3; найденная 728,0 [M+H]+, tR = 10,71 минут (способ 10). Хиральная чистота составляла 90% d.e. (хиральный способ). 1H ЯМР (400 МГц, DMSO-d6) δ 9,15 (с, 2H), 8,56 (д, J=8,6 Гц, 1H), 8,42-8,34 (м, 1H), 8,34-8,27 (м, 2H), 7,84-7,75 (м, 2H), 7,66 (д, J=3,9 Гц, 1H), 7,54-7,48 (м, 2H), 7,32 (с, 1H), 7,12-7,04 (м, 2H), 6,90 (д, J=3,8 Гц, 1H), 6,77 (с, 1H), 4,81-4,65 (м, 1H), 4,19-4,11 (м, 1H), 4,03 (т, J=6,5 Гц, 2H), 3,20 (дд, J=14,1, 3,5 Гц, 1H), 3,07-2,96 (м, 1H), 2,24-2,09 (м, 2H), 2,06-1,93 (м, 1H), 1,90-1,79 (м, 1H), 1,78-1,68 (м, 2H), 1,47-1,20 (м, 17H), 0,93-0,82 (м, 3H).
Соединения 321-350 получали из соединения 192, применяя общие способы 3 или 7, с последующим 4 или 8.
Соединения 351-368 получали из соединения 165, применяя общий способ 7, с последующим 4 или 8.
Соединение 369 получали из соединения 139, применяя общий способ 7, с последующим 8.
Соединение 370 получали из соединения 167, применяя общий способ 7, с последующим 8.
Соединение 371 получали из соединения 142, применяя общий способ 7, с последующим 8.
Соединение 372 получали из соединения 143, применяя общий способ 7, с последующим 8.
Соединение 373 получали из соединения 182, применяя общий способ 7, с последующим 8.
Соединения 374-379 получали из соединения 193, применяя общий способ 3 или 7, с последующим 4 или 8.
Соединение 380 получали из соединения 191, применяя общий способ 7, с последующим 8.
(S)-4-(трет-бутил-N-(3-(4-(5-(4-(гептилокси)фенилпиримидин-2-ил)фенил-1-((2-(метилсульфамидо)-2-оксоэтил)амино)-1-оксопропан-2-ил)бензамид (соединение 381)
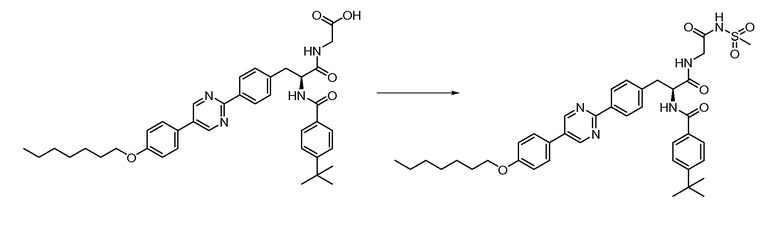
TEA (32,1 мкл, 0,23 ммоль) добавляли к суспензии (S)-(2-(4-(трет-бутил)бензамидо)-3-(4-(5-(4-(гептилокси)фенил)пиримидин-2-ил)фенил)пропаноил)глицина, соединения 297 (75,0 мг, 0,11 ммоль), метансульфамида (12,1 мг, 0,13 ммоль), HATU (52,6 мг, 0,14 ммоль) и DMAP (1,41 мг, 0,01 ммоль) в DCM (2 мл). Полученную в результате желтую суспензию перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь промывали насыщенным водным NaHCO3 (2 мл), и смесь пропускали через картридж для разделения фаз. Органическую фазу концентрировали в вакууме, получая желтый твердый остаток. Неочищенный продукт очищали хроматографией (EA/1% AcOH в гексане), получая 9 мг (11%) (S)-4-(трет-бутил)-N-(3-(4-(5-(4- (гептилокси)фенил)пиримидин-2-ил)фенил)-1-((2-(метилсульфамидо)-2-оксоэтил)амино)-1-оксопропан-2-ил)бензамида, соединения 381, в виде желтого твердого остатка. LCMS-ESI (m/z) рассчитанная для C40H49N5O6S: 727,3; найденная 728,0 [M+H]+, tR = 10,51 минут (способ 10).
Выборочные соединения и их соответствующие аналитические данные показаны в таблице 1, представленной в конце описания, где LCMS данные получали, применяя указанный способ.
БИОЛОГИЧЕСКИЕ АНАЛИЗЫ
Способы анализа
GLP-1 PAM цАМФ анализ на замедление подвижности в геле: дозозависимый эффект пептидного лиганда в присутствии фиксированной концентрации соединения.
GLP-1R экспрессирующую CRE-bla CHO-Kl клеточную линию приобретали у Invitrogen. Клетки высевали в 384-луночные белые планшеты с плоским дном при 5000 клеток/лунка/20 мкл ростовая среда (DMEM-High глюкоза, 10% диализированная FBS, 0,1 мМ NEAA, 25 мМ Hepes, 100 ед./мл пенициллина/100 мкг/мл стрептомицина, 5 мкг/мл бластицидина, 600 мкг/мл гигромицина) и выдерживали в течение 18 часов при 37°C в 5% CO2.
Ростовую среду заменяли 12 мкл буфера для анализа (сбалансированный солевой раствор Хенкса, 10 мМ Hepes, 0,1% BSA, pH 7,4). Кривую 5x зависимости эффекта от дозы пептида (12 точек) получали в буфере для анализа, содержащем 1,5 мМ IBMX, 12,5% DMSO и 50 мкм соединения. Пептидный лиганд представлял собой GLP- 1(9-36). Добавляли Смесь 5x дозозависимого эффекта пептида плюс соединения (3 мкл), и клетки выдерживали в течение 30 минут при 37°C. Прямое определение цАМФ осуществляли, применяя DiscoveRx HitHunter цАМФ набор согласно инструкции производителя, и люминесценцию регистрировали, применяя SpectraMax M5 ридер-планшет. Люминесценцию анализировали нелинейной регрессией, определяя EC50 и Emax. GLP-1(7-36) дозозависимый эффект включали для определения максимальной эффективности.
EC20 GLP-1(9-36) PAM цАМФ анализ: дозозависимый эффект соединения в присутствии фиксированной концентрации GLP-1 (9-36).
GLP-1R CRE-bla CHO-K1 клетки, выращенные в ростовой среде (DMEM-High глюкоза, 10% диализированная FBS, 0,1 мМ NEAA, 25 мМ Hepes, 100 ед./мл пенициллина/100 мкг/мл стрептомицина, 5 мкг/мл бластицидина, 600 мкг/мл гигромицина), подвергали трипсинолизу и помещали в суспензию в 384-луночные белые планшеты с плоским дном при 5000 клеток/лунка в 12 мкл буфера для анализа (сбалансированный солевой раствор Хенкса, 10 мМ Hepes, 0,1% BSA, pH 7,4). Кривую 5x зависимости эффекта от дозы пептида (12 точек) получали в буфере для анализа, содержащем 1,5 мМ IBMX, 12,5% DMSO. GLP-1(9-36) разбавляли до 4,2 мкМ в буфере для анализа, содержащем 1,5 мМ IBMX и 12,5% DMSO. Добавляли 5x дозозависимый эффект соединения (3 мкл), с последующим добавлением 0,5 мкл GLP-1(9-36), и клетки инкубировали в течение 30 минут при 37°C. Прямое определение цАМФ осуществляли, применяя DiscoveRx HitHunter цАМФ набор, согласно инструкции производителя, и люминесценцию регистрировали, применяя SpectraMax M5 ридер-планшет. Люминесценцию переводили в суммарный цАМФ, применяя цАМФ стандартную кривую, и данные анализировали нелинейной регрессией для определения EC50 и Emax.
Пептидные последовательности
GLP-1(7-36) (SEQ ID NO:2):
HAEGTFTSDVSSYLEGQAAKEFIAWLVKGR-NH2.
GLP-1(9-36) (SEQ ID NO:3): EGTFTSDVSSYLEGQAAKEFIAWLVKGR-NH2.
GLP-1 (7-36) приобретали у GenScript. GLP-1 (9-36) приобретали у Biopeptide Co., Inc.
Приведенные GLP-1 активности
Данные по активности для выборочных GLP-1 модуляторов показаны в таблице 2, представленной в конце описания. EC20 GLP-1 (9-36) PAM диапазон активностей показан следующим образом: + показывает активность < 0,8 мкМ, ++ показывает активность между 0,8 и 2,5 мкМ, +++ показывает активность между 2,5 и 5 мкМ, и ++++ показывает активность 5-10 мкМ.
IN VIVO АНАЛИЗЫ
In Vivo способы
Пероральное испытание на толерантность к глюкозе на C57BI/6 мышах.
Применение настоящих соединений для снижения глюкозы можно оценить на мышах, применяя пероральное испытание на толерантность к глюкозе (oGTT). Протокол описан Duez et. al (Endocrinology, January 2009, 150(l):56-62). C57BL/6 мужских особей мышей длительно канюлировали. После 5 дней восстановления, мышей держали без питания в течение 4 часов. Исходные образцы крови собирали перед внутривенным введением болюса или соляного раствора или эксендина-4, или перед введением соединения. Сразу после мышам вводили болюс глюкозы (1,5 г/кг) пероральным зондовым питанием (момент времени 0). Образцы крови собирали через частые интервалы времени из кончика хвоста для определения глюкозы (BD глюкометр; Becton-Dickinson, Lincoln Park, NJ). Для определения инсулина в плазме образец крови отбирали из хвостовой вены через 5 минут после введения глюкозы.
Пероральное испытание на толерантность к глюкозе на fa/fa крысах.
Применение настоящих соединений для снижения глюкозы можно оценить на крысах, применяя пероральное испытание на толерантность к глюкозе (oGTT). Протокол описан Pederson et. al. (Diabetes, Vol. 47, August 1998, 1253-1258). После выдерживания натощак в течение ночи, тощим или толстым животным вводили перорально глюкозу шприцом и питательной трубкой (1 г/кг) в виде 40% раствора (вес/объем). Соединение растворяли и вводили вместе с глюкозой. В контрольных экспериментах, пустышку вводили вместе с глюкозой. Образцы крови собирали из хвостовой вены крыс в сознательном состоянии без ограничения движения в гепаринизированные капиллярные трубки через 0 и 5, 10, 20, 30 и 60 минут после введения глюкозы. Образцы крови центрифугировали при 4°C, и плазму хранили при -20°C до анализа на измерение глюкозы и инсулина. Концентрации глюкозы измеряли, применяя способ глюкооксидазы (Beckman анализатор глюкозы; Fullerton, CA).
Различные варианты осуществления, описанные выше, можно комбинировать, получая дополнительные варианты осуществления. Все из США патентов, опубликованных патентных заявок США, патентных заявок США, иностранных патентов, иностранных патентных заявок и непатентных публикаций, на которые ссылаются в настоящем описании и/или которые перечислены в краткой инструкции по применению, включены в настоящее изобретение с помощью ссылки во всей своей полноте. Аспекты вариантов осуществления можно изменять, при необходимости, применяя концепции различных патентов, заявок и публикаций, получая еще дополнительные варианты осуществления. Данные и другие изменения можно осуществлять на вариантах осуществления в свете вышеуказанного описания. В общем, в следующей формуле изобретения применяемые термины не следует считать ограничивающими формулу изобретения конкретными вариантами осуществления, описанными в описании и формуле изобретения, но следует считать, что они включают все возможные варианты осуществления вместе с полным объемом эквивалентов, для которых формула изобретения является основанием. Соответственно, формула изобретения не ограничивается настоящим описанием.
| название | год | авторы | номер документа |
|---|---|---|---|
| АЗАБИЦИКЛИЧЕСКИЕ И ДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ОФТАЛЬМОЛОГИЧЕСКИХ НАРУШЕНИЙ | 2018 |
|
RU2795521C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
| МАКРОЦИКЛИЧЕСКИЕ АНТИБИОТИКИ ШИРОКОГО СПЕКТРА ДЕЙСТВИЯ | 2016 |
|
RU2766543C2 |
| ИНГИБИТОРЫ ИОННОГО КАНАЛА, ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ И ПРИМЕНЕНИЕ | 2016 |
|
RU2746188C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ИНГИБИТОРЫ TrkA КИНАЗЫ, ОСНОВАННЫЕ НА НИХ КОМПОЗИЦИИ И СПОСОБЫ | 2015 |
|
RU2672583C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2799820C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| УЛУЧШЕННЫЕ АГОНИСТЫ АПЕЛИНОВОГО РЕЦЕПТОРА (APJ) И ИХ ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2766148C1 |
| АГОНИСТЫ PPAR, СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2017 |
|
RU2746602C2 |
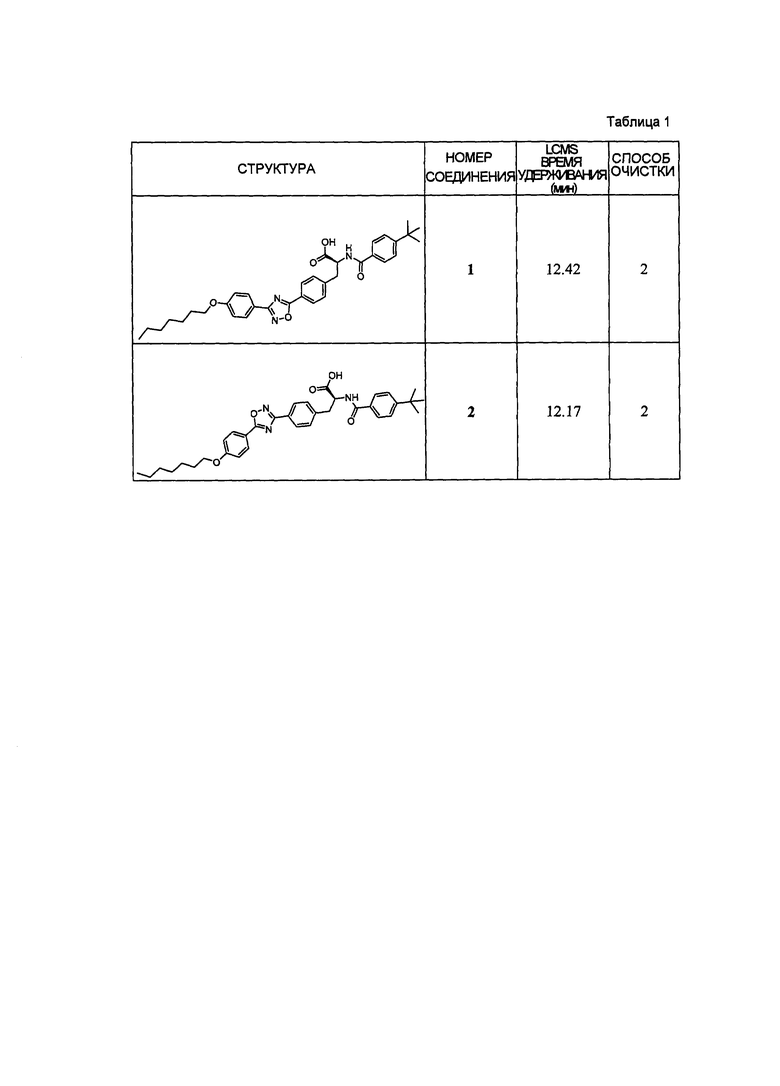
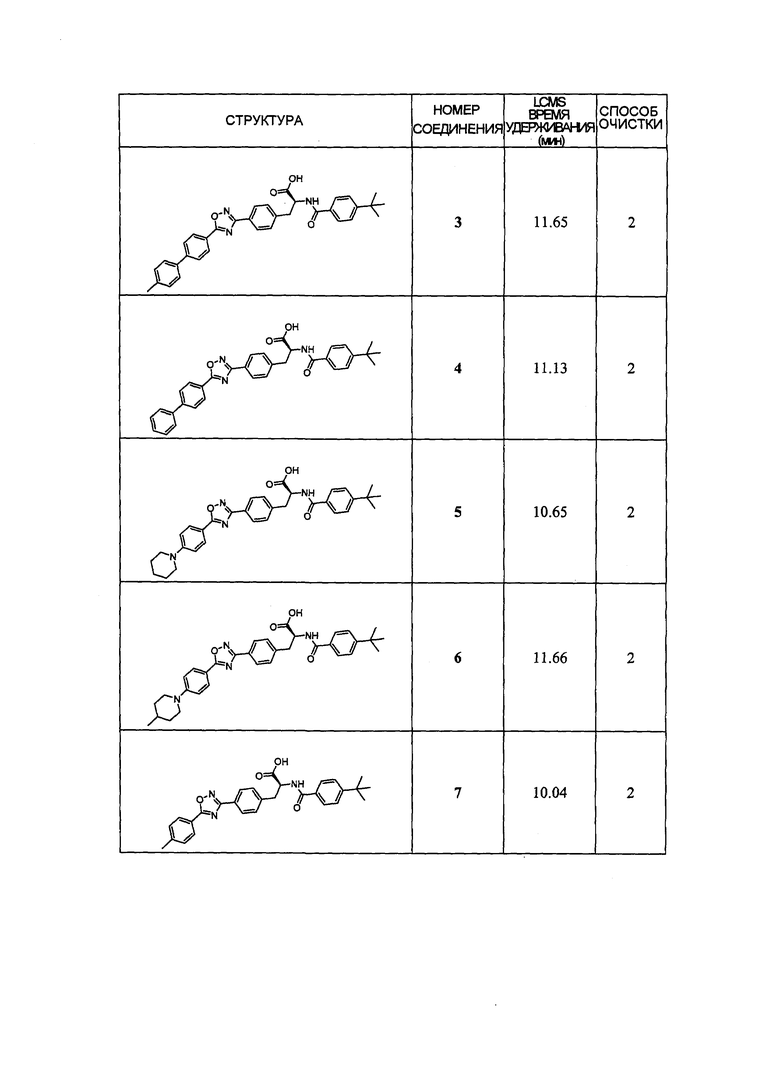
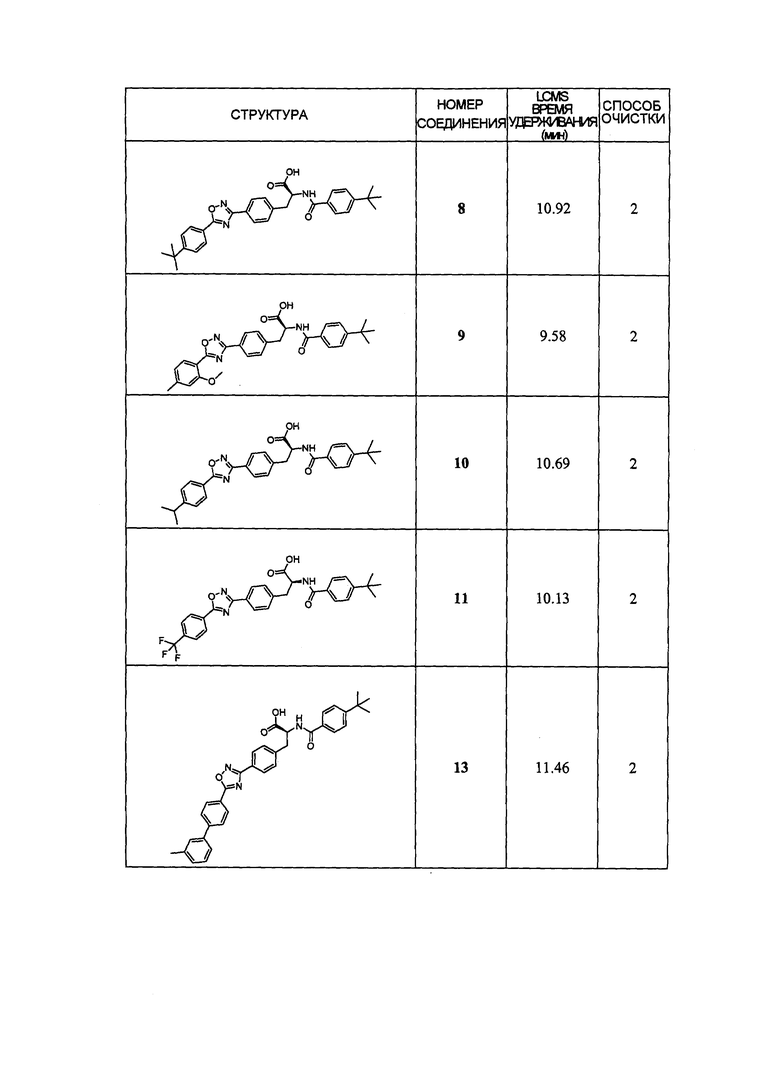

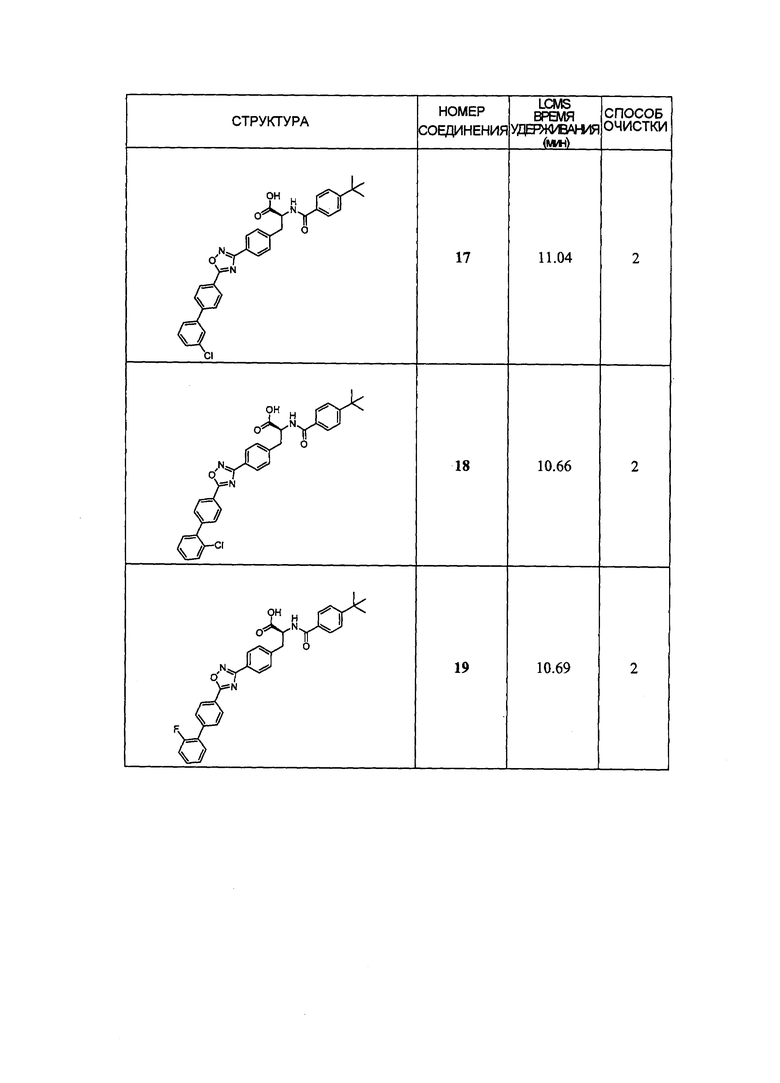
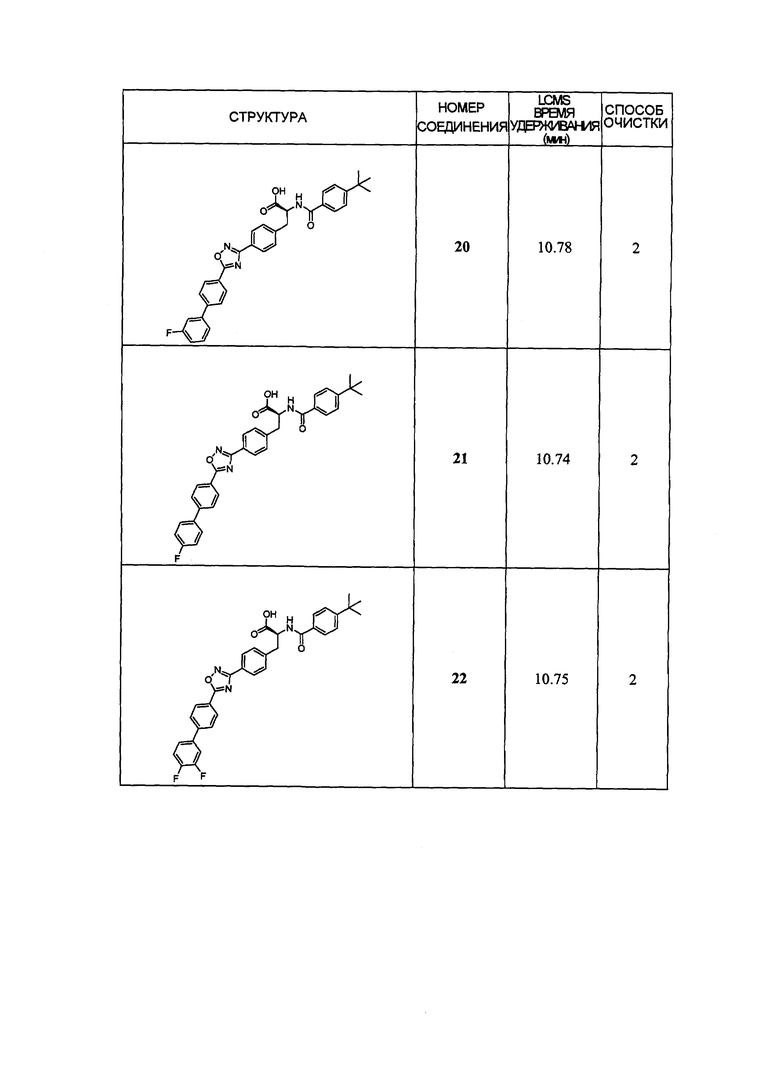
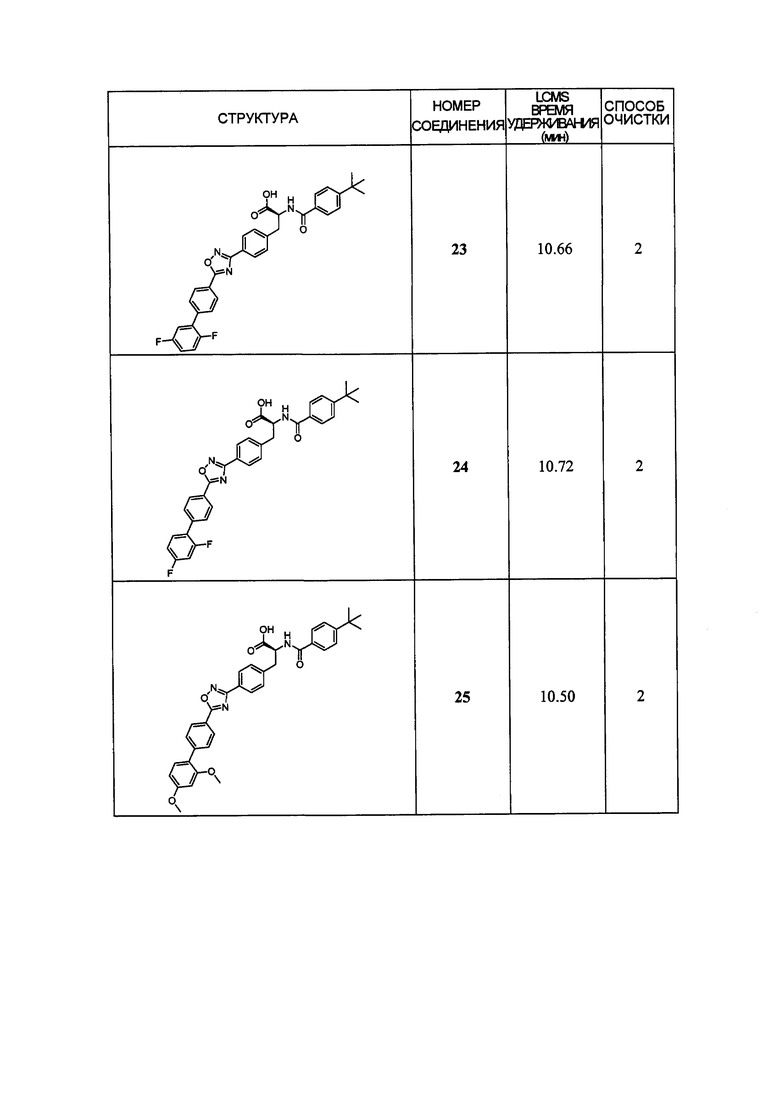
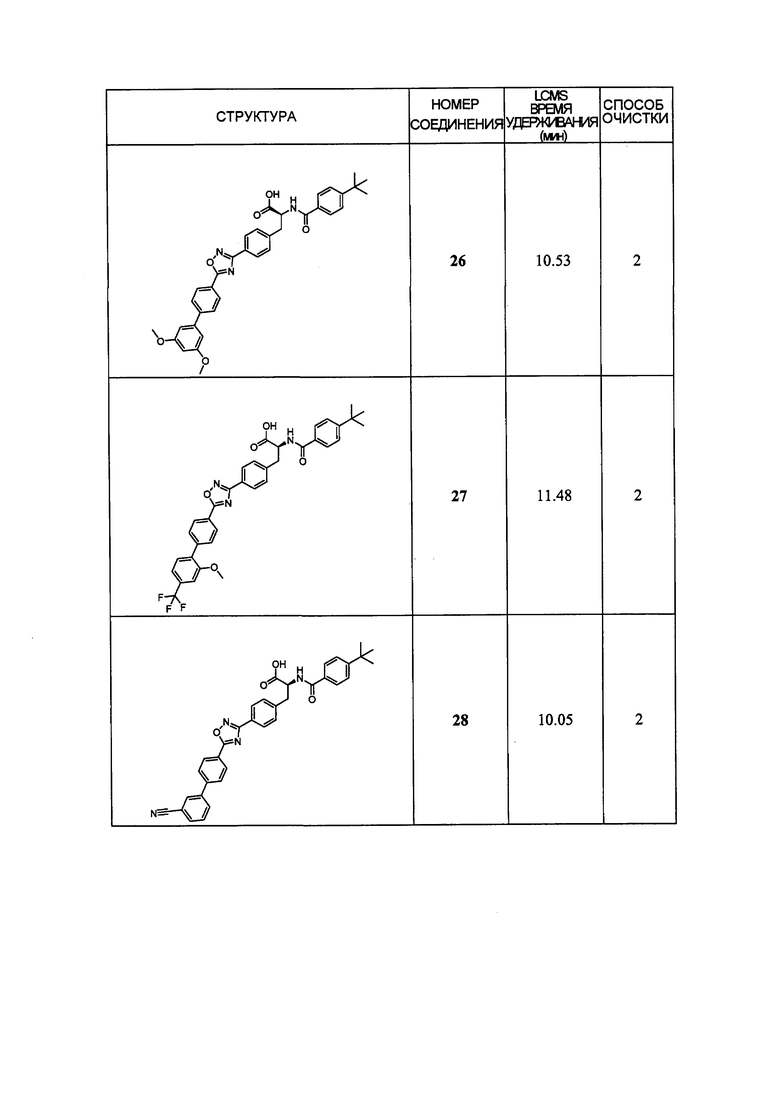
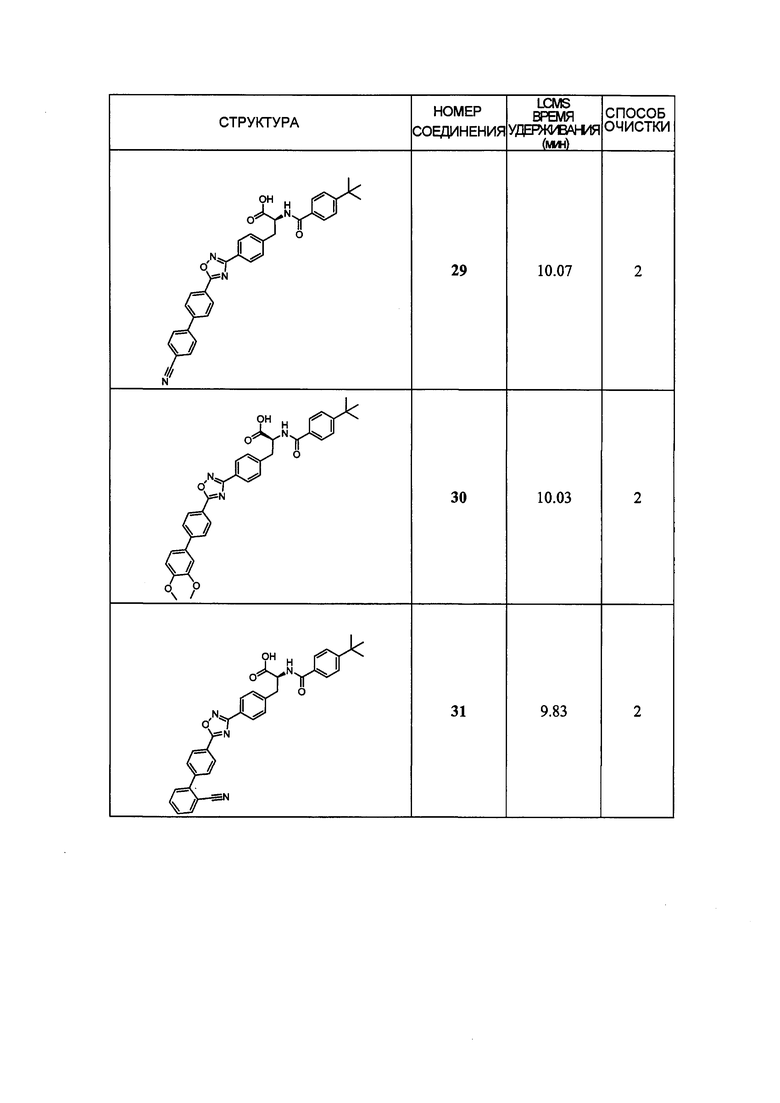
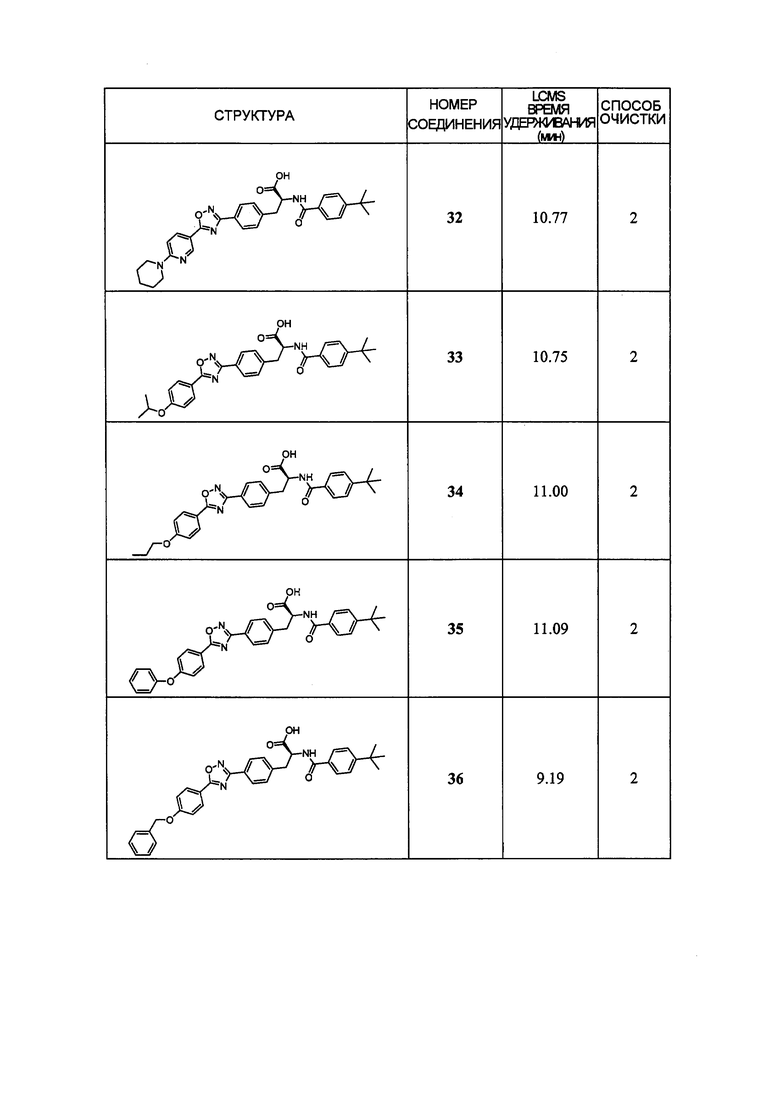
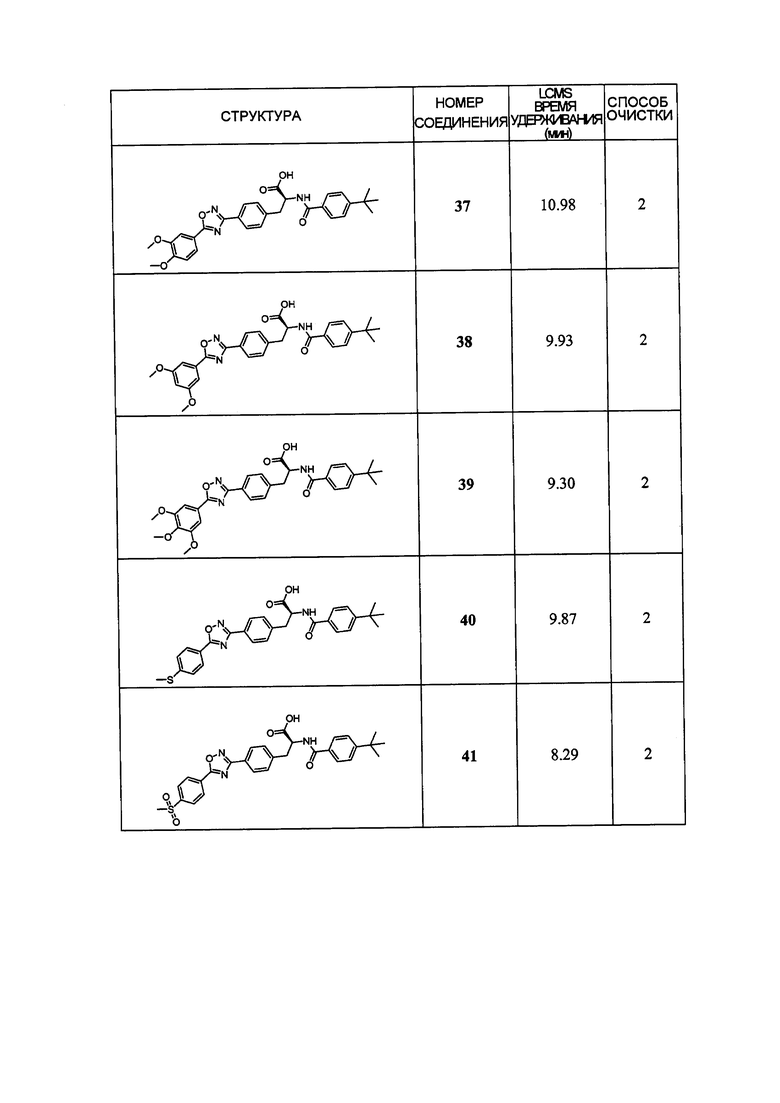
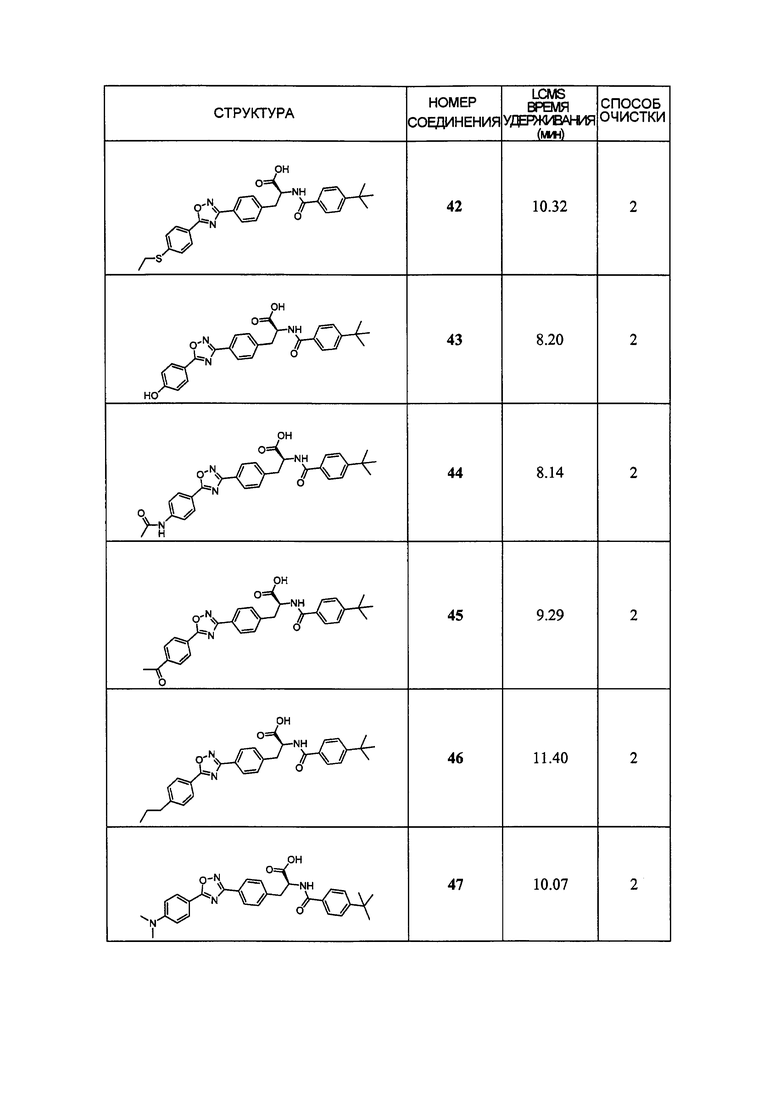
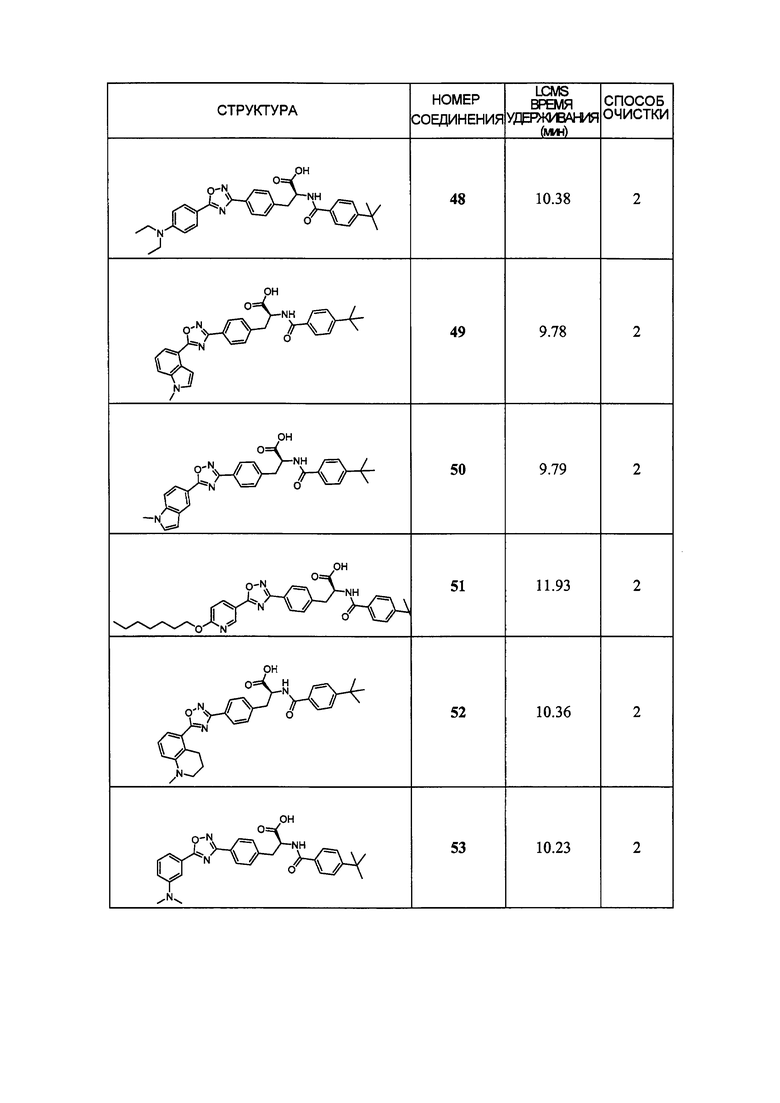
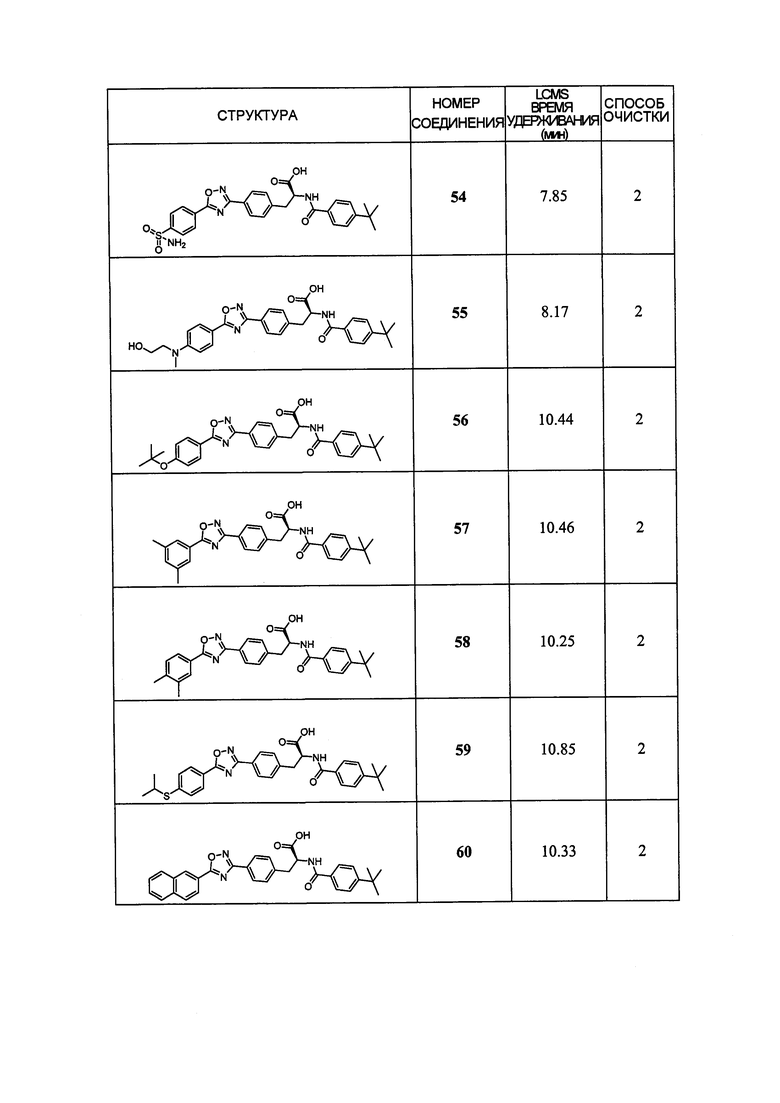
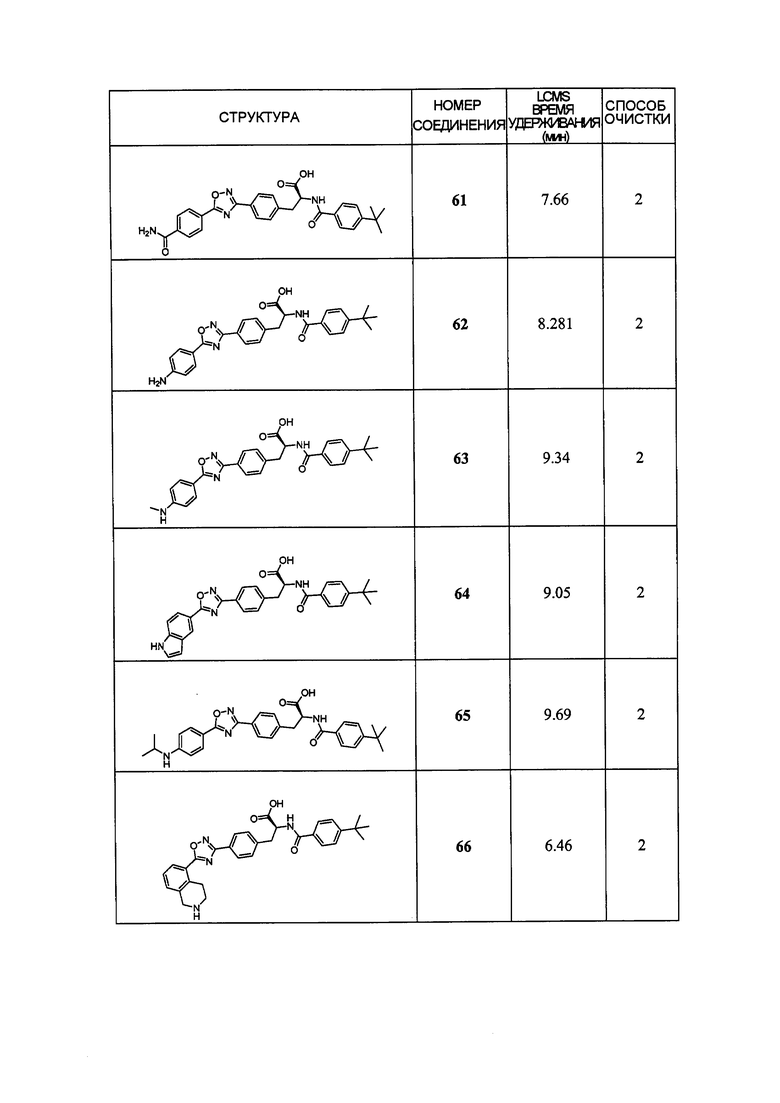
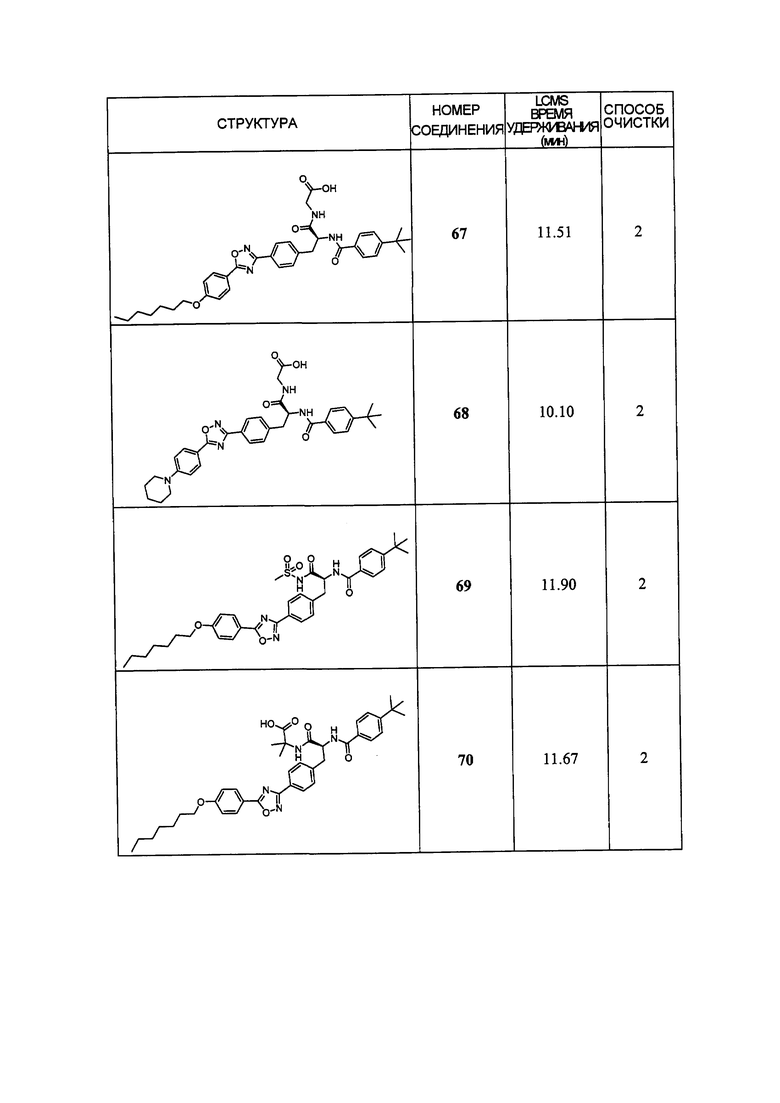
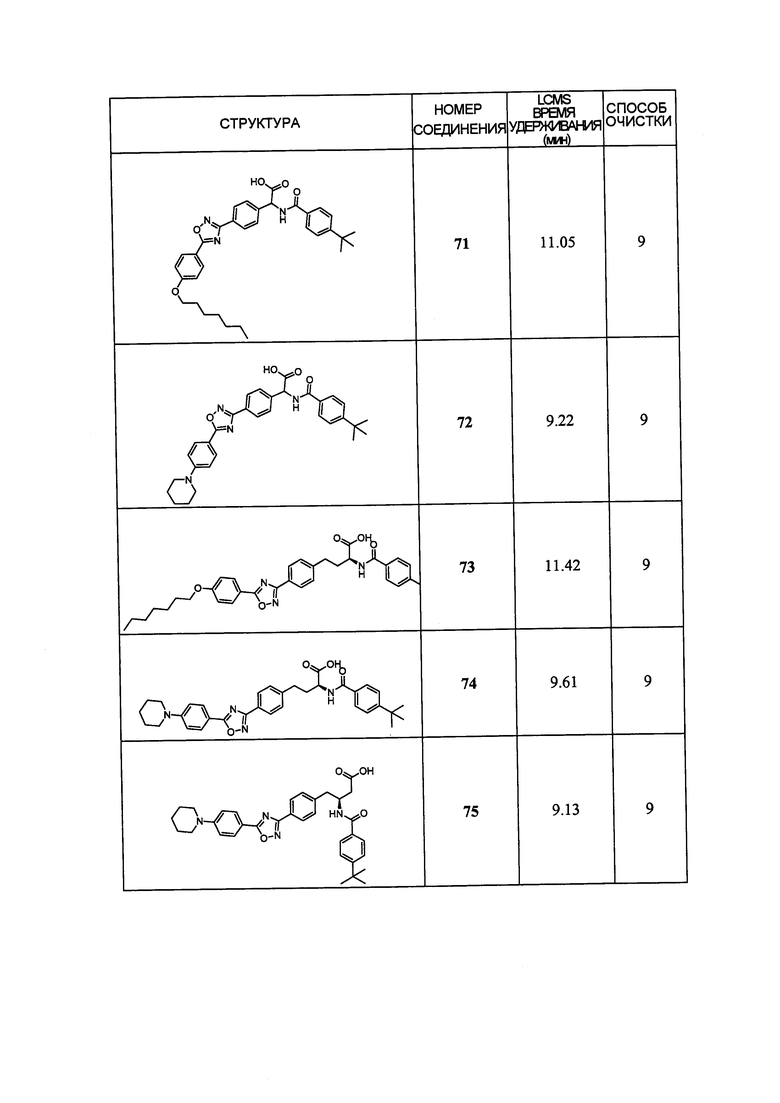
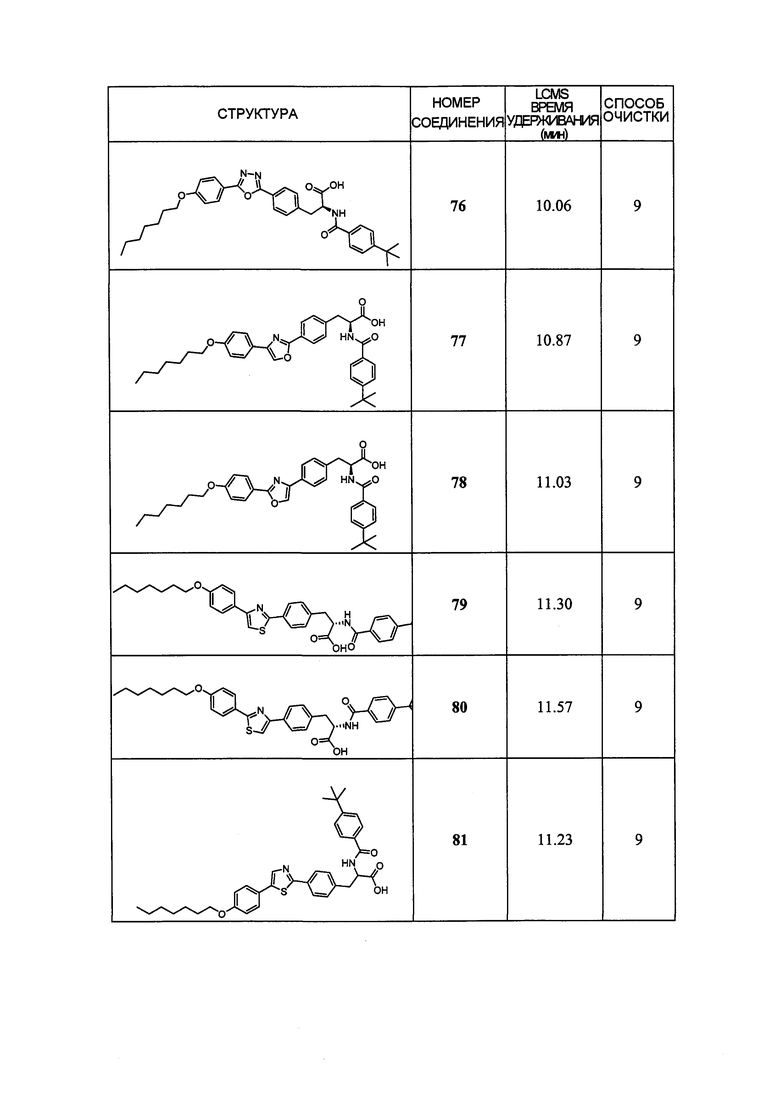
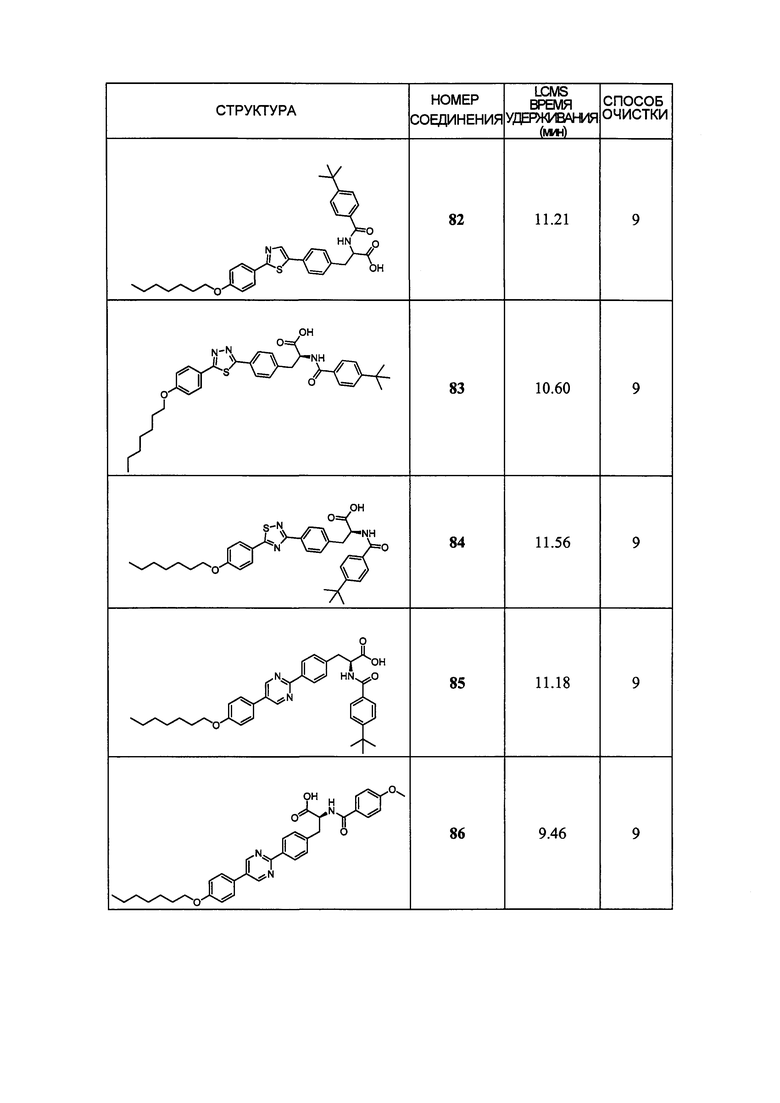

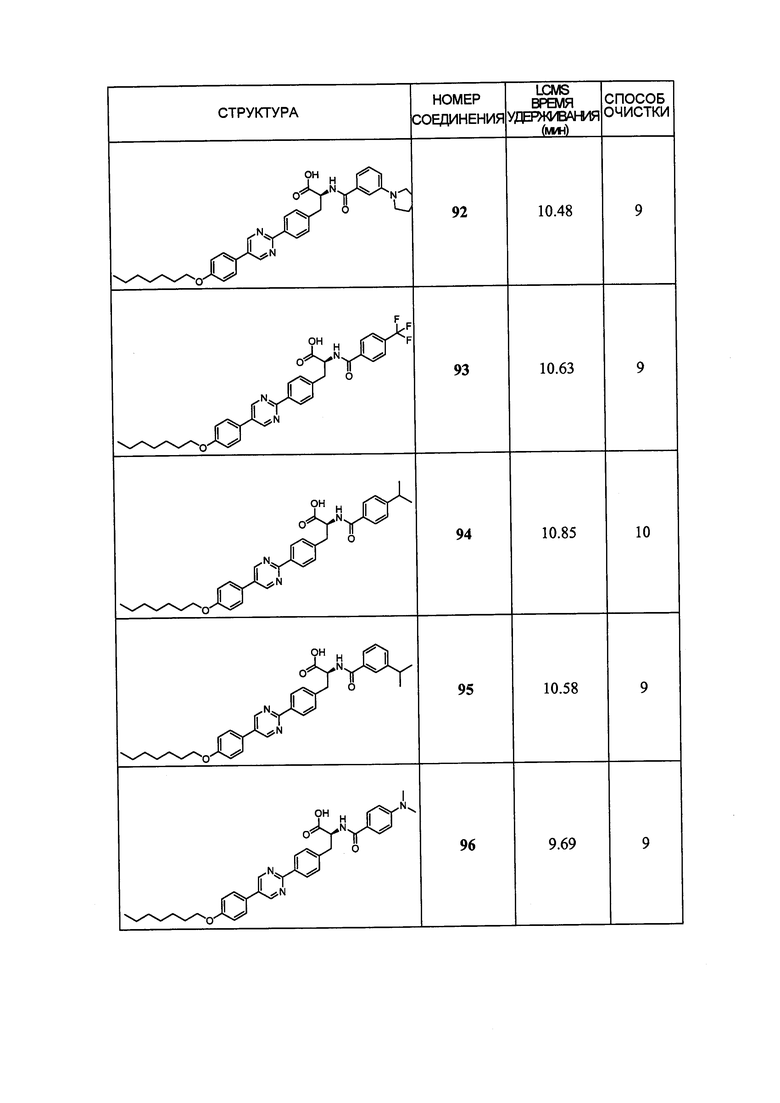


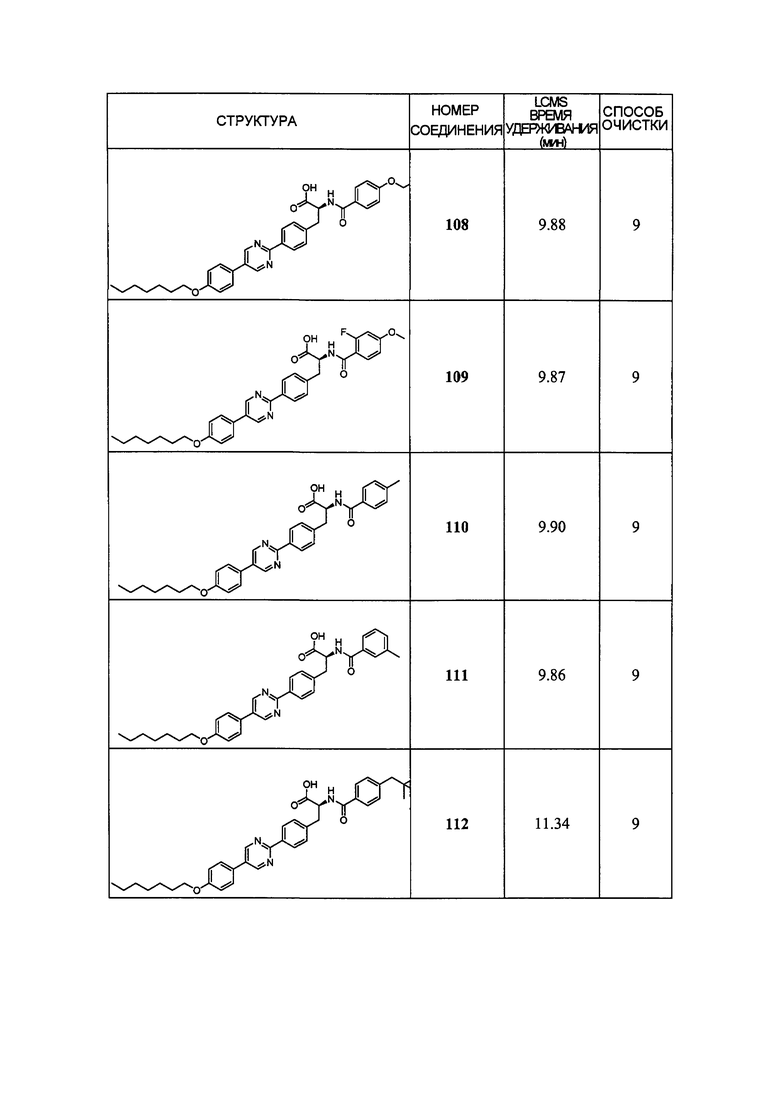
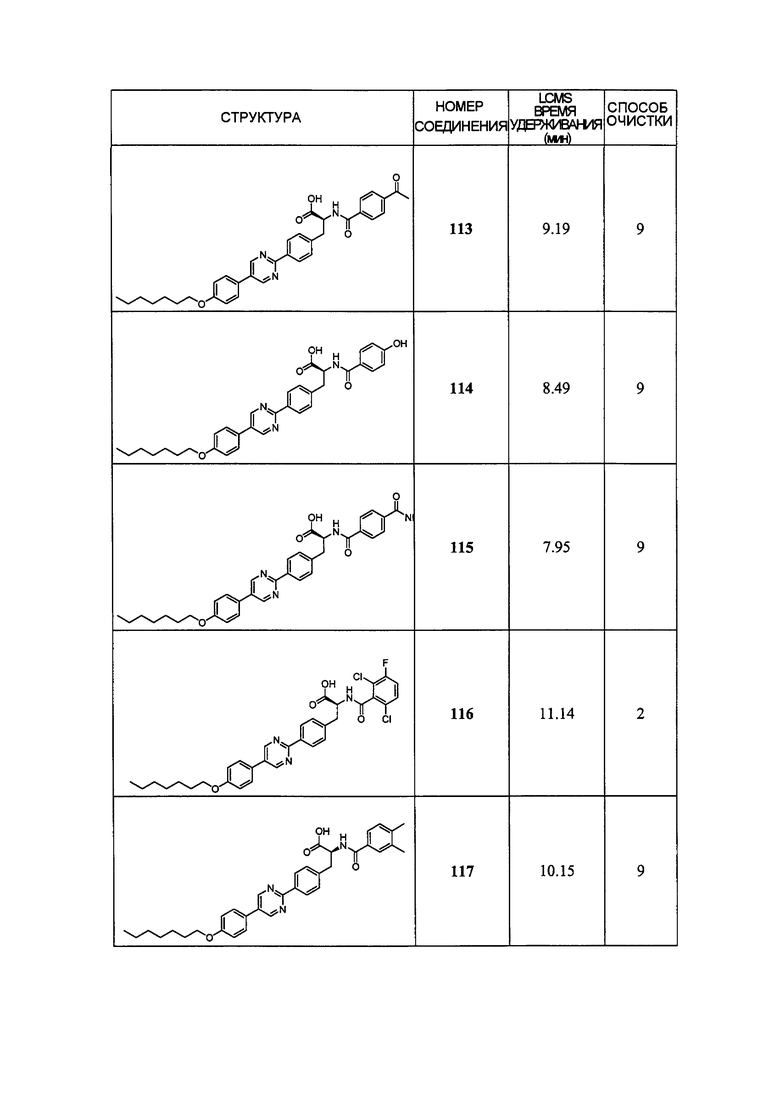
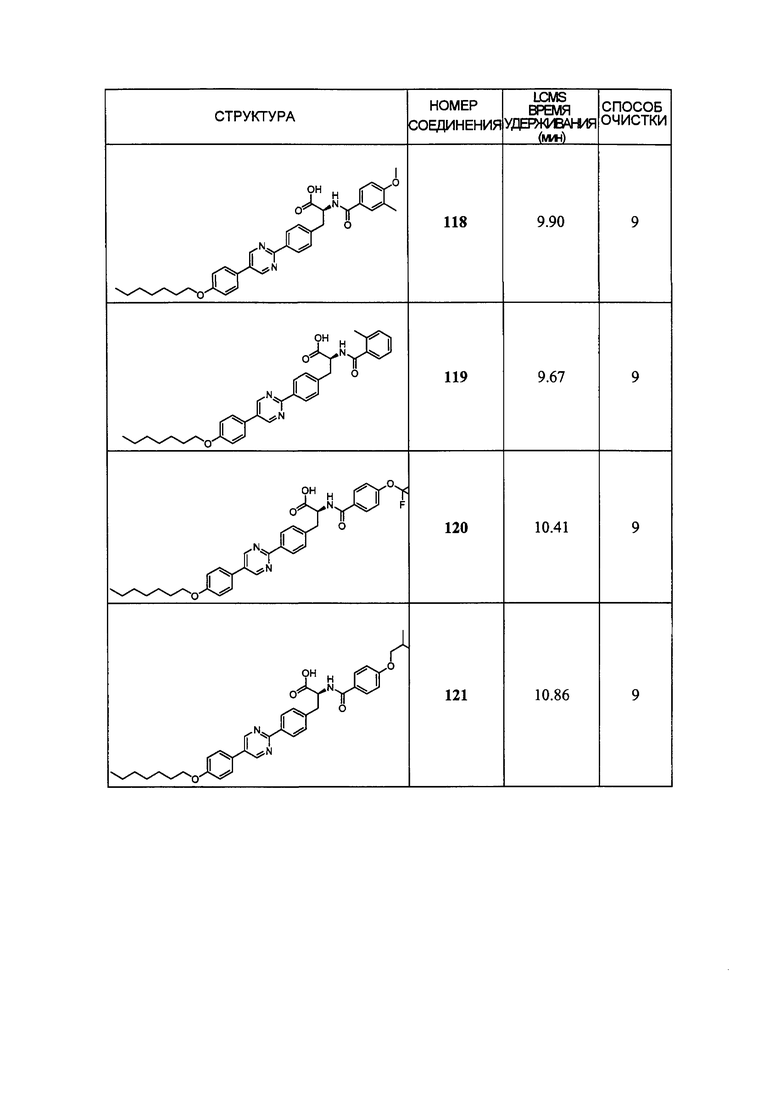
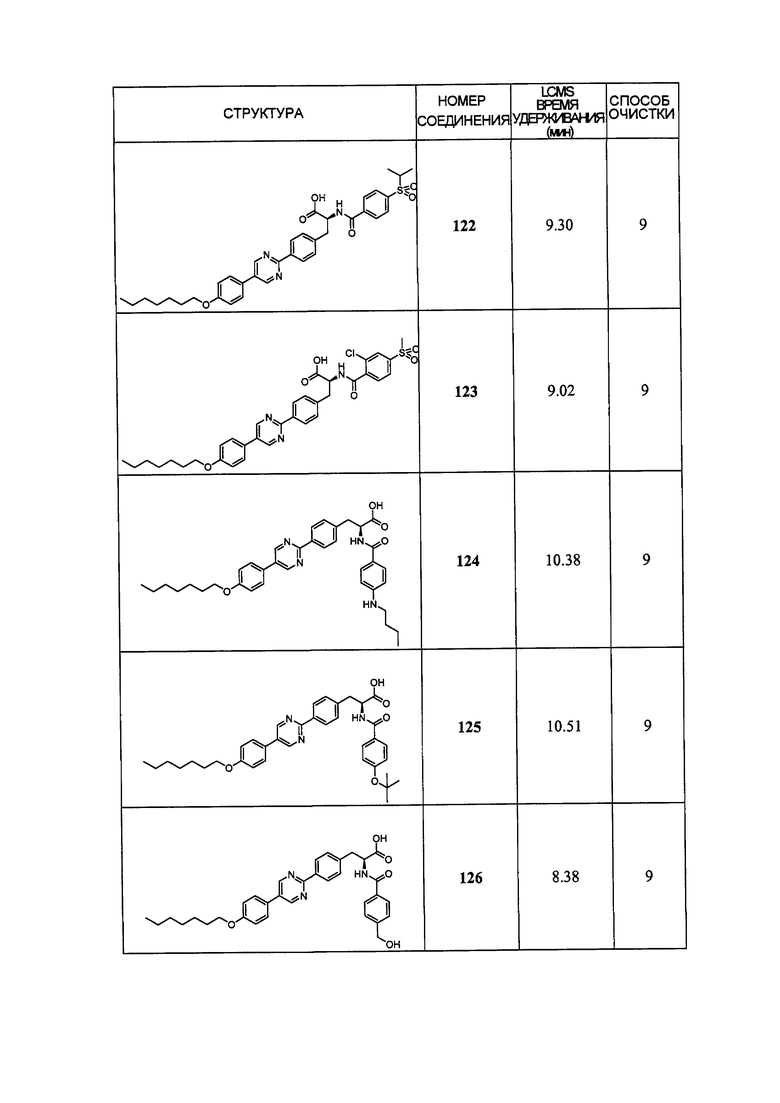
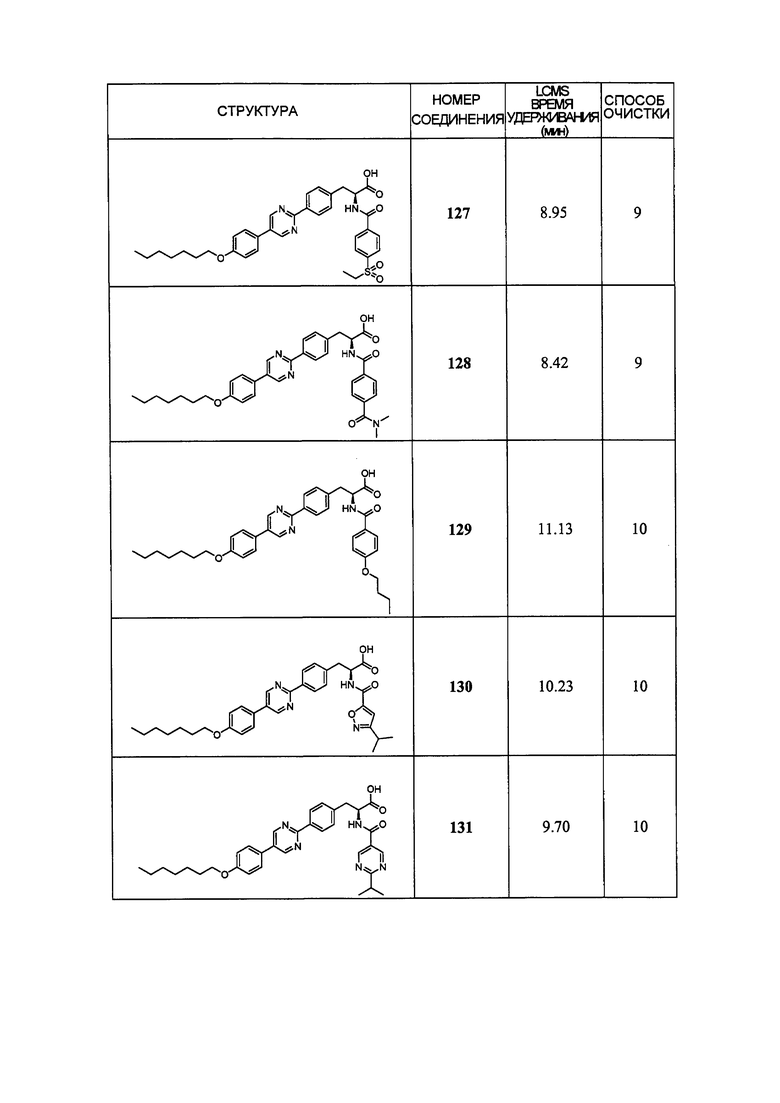

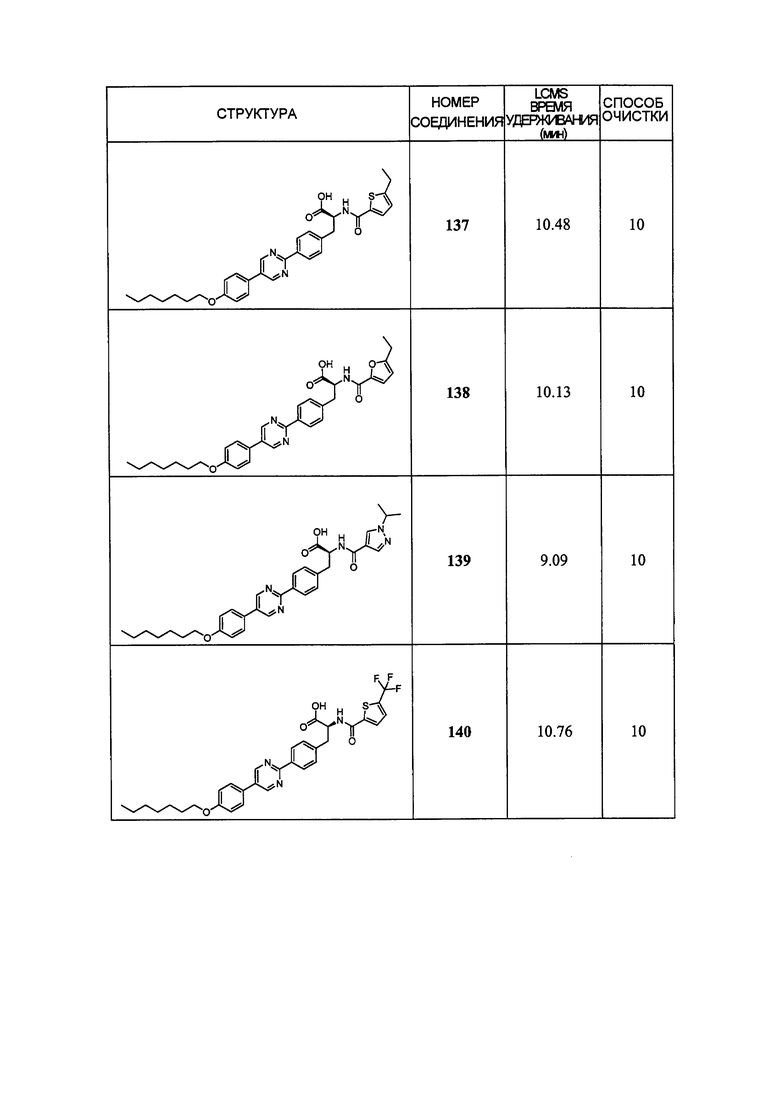
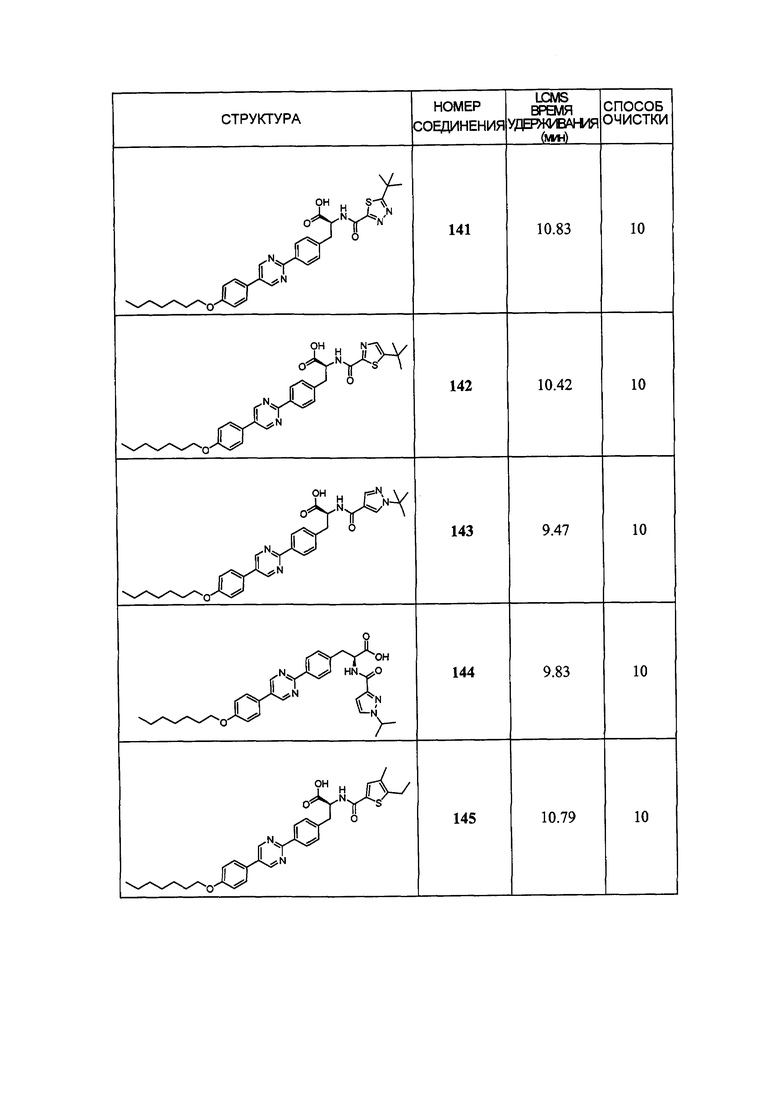
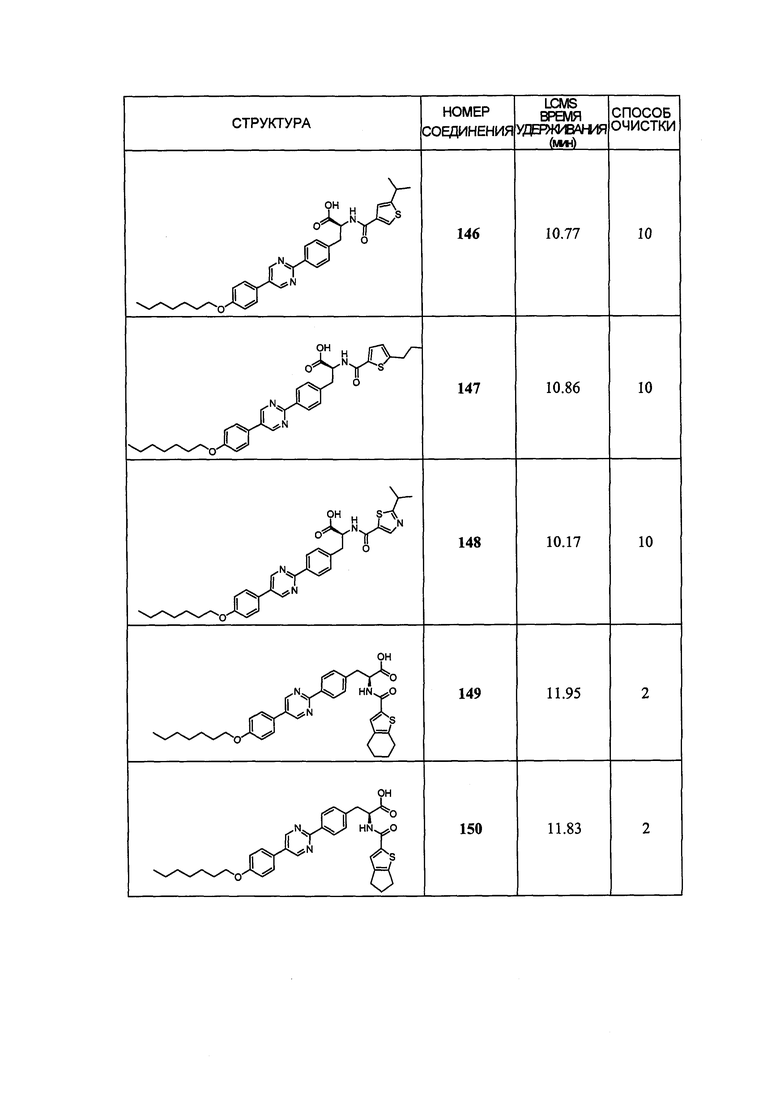
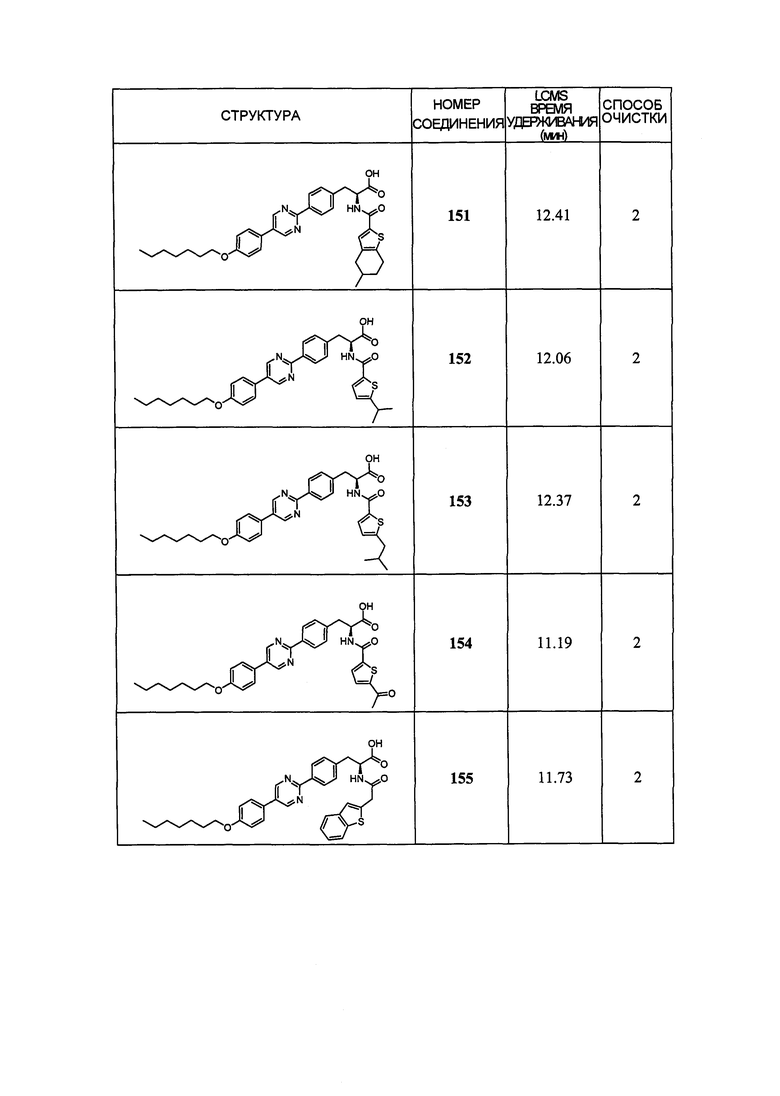
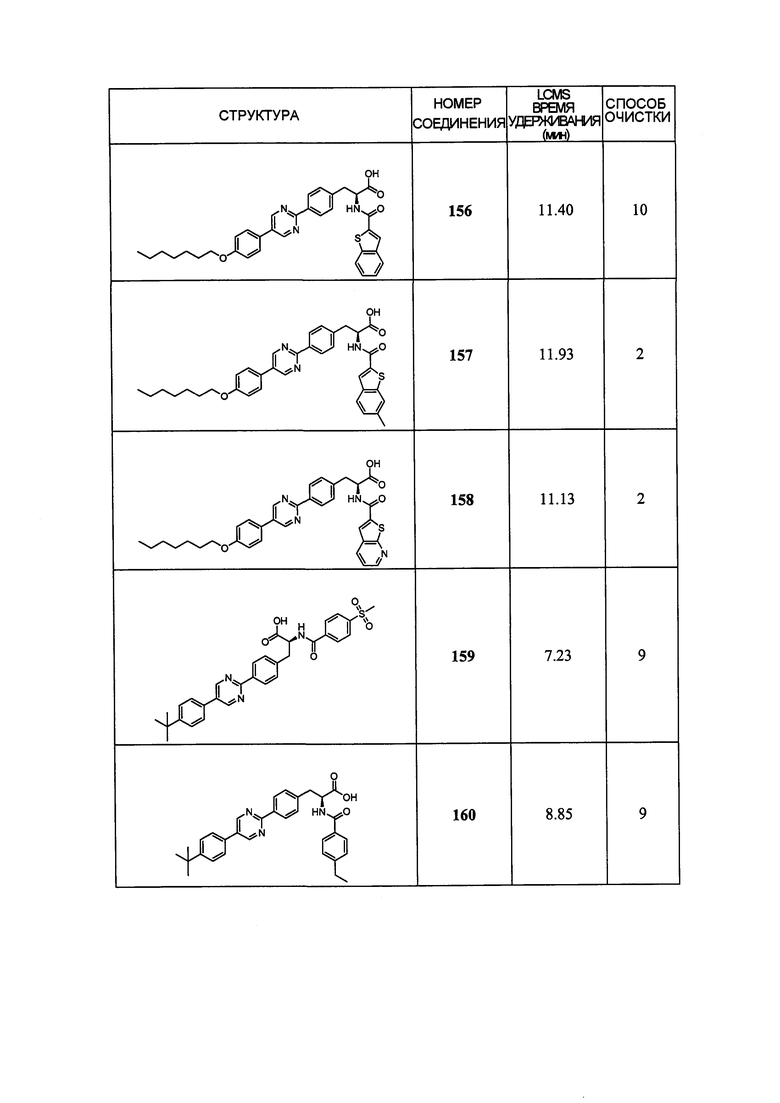
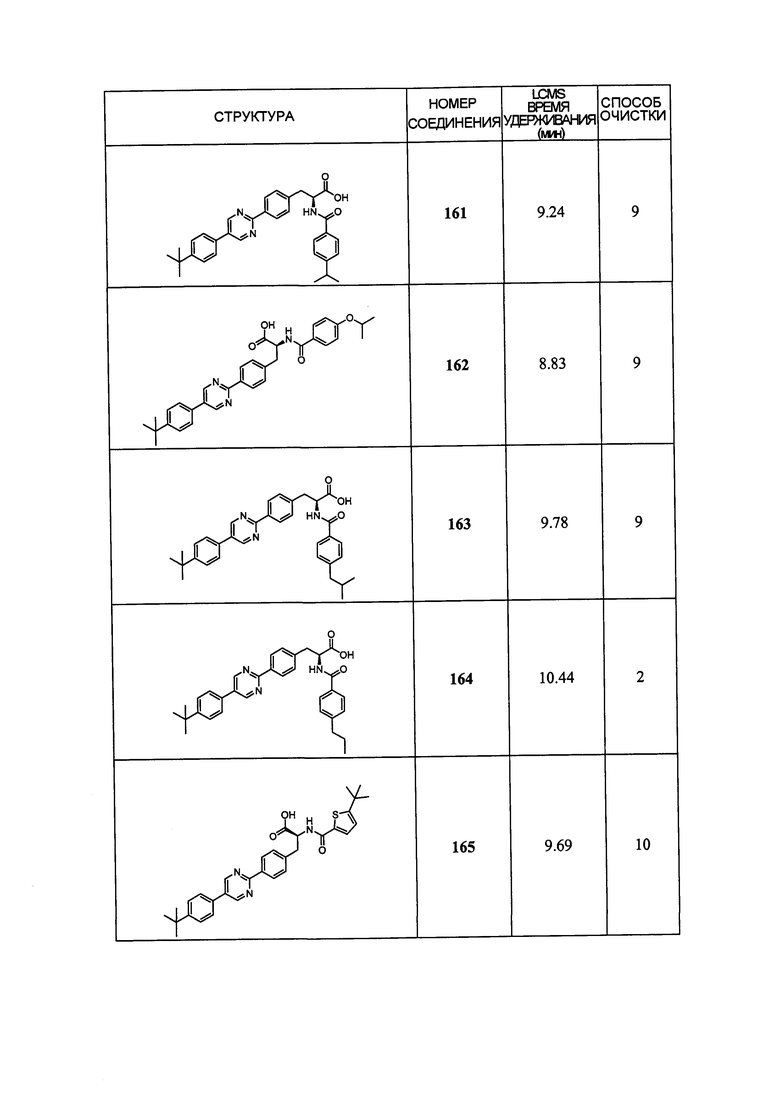


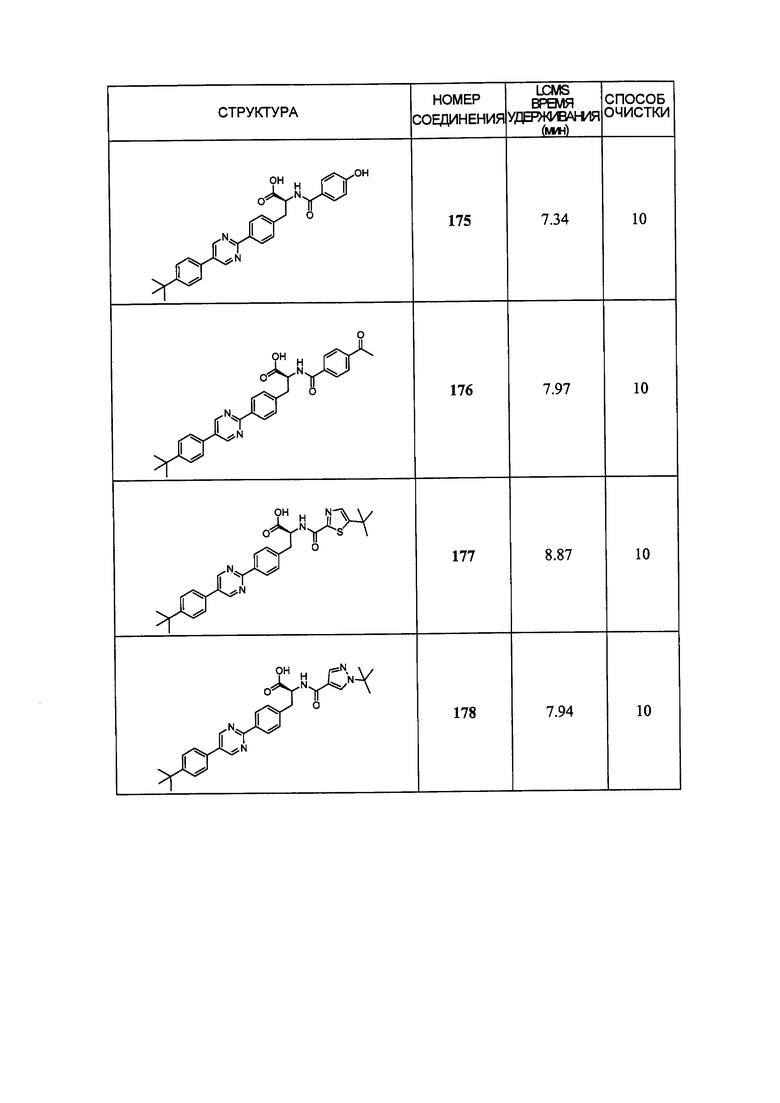
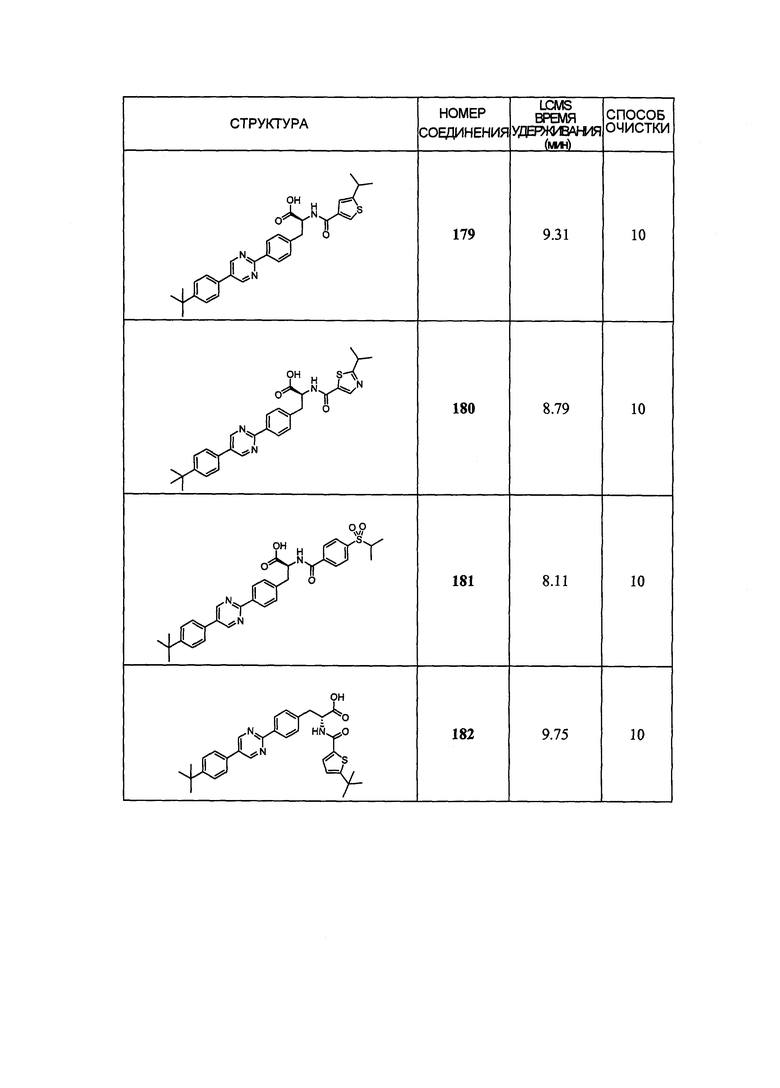
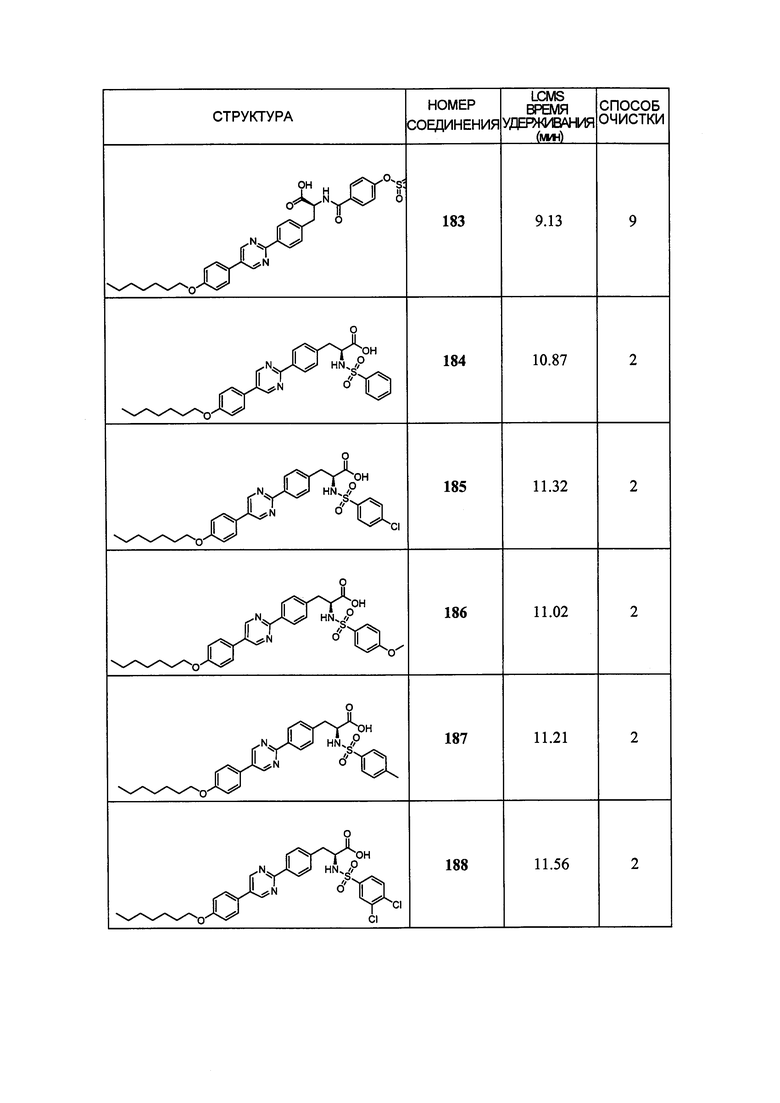

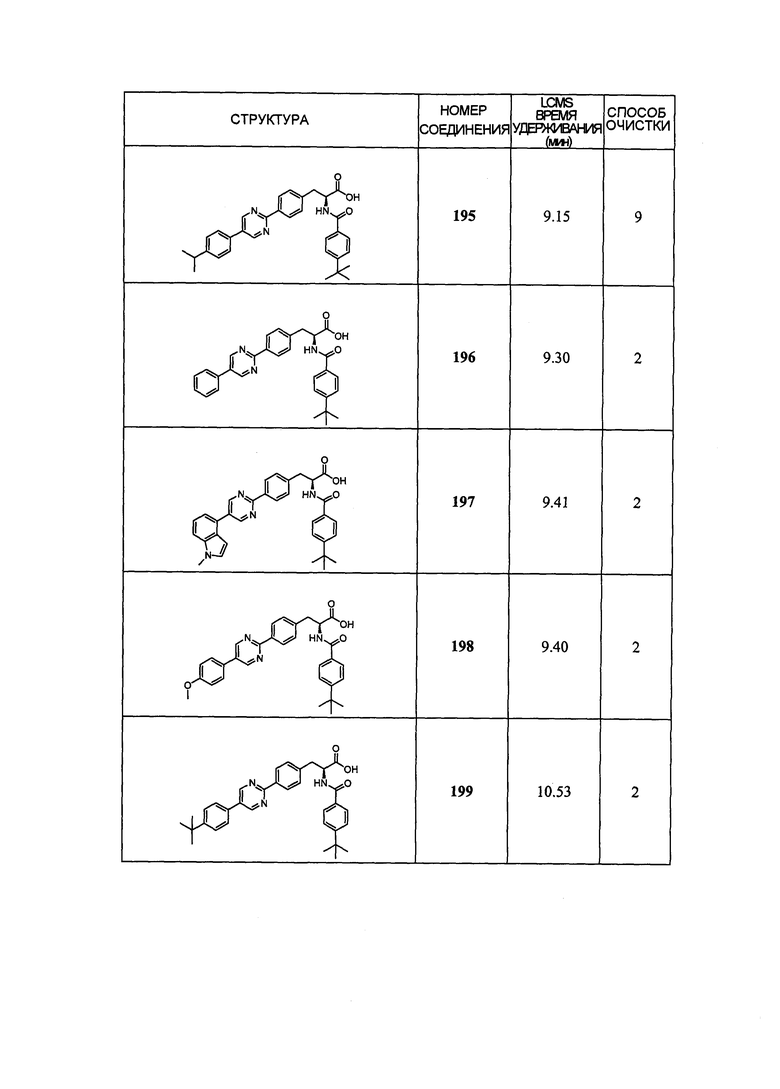
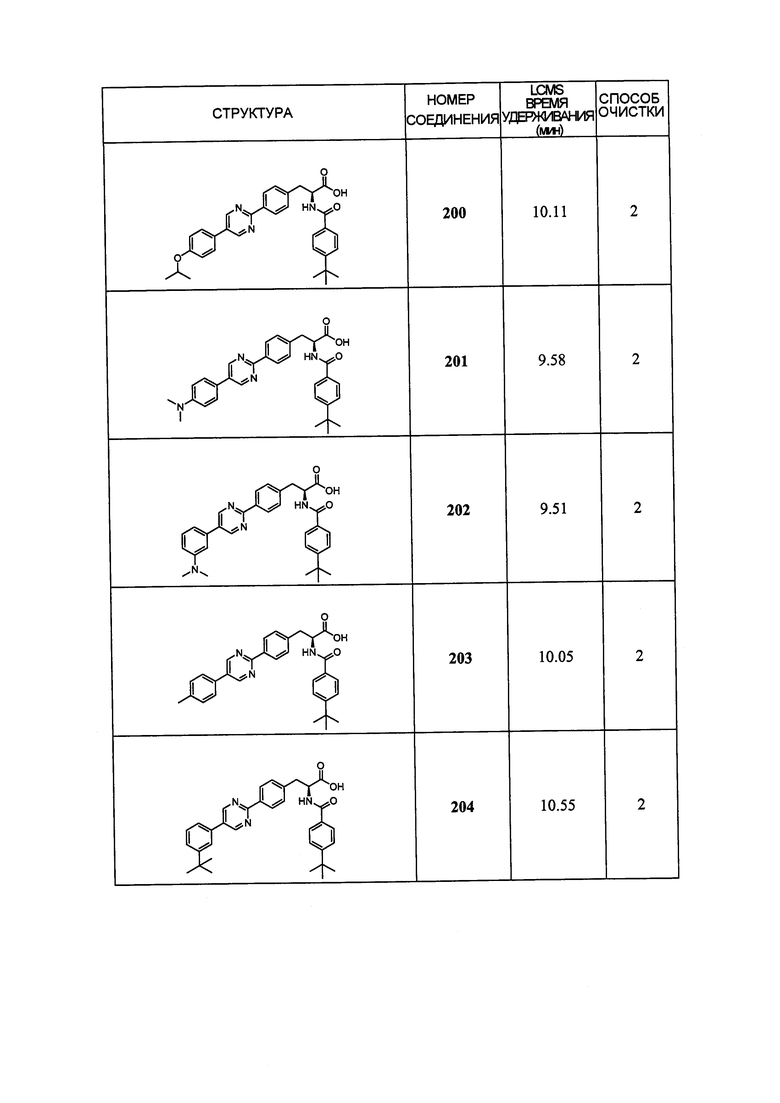
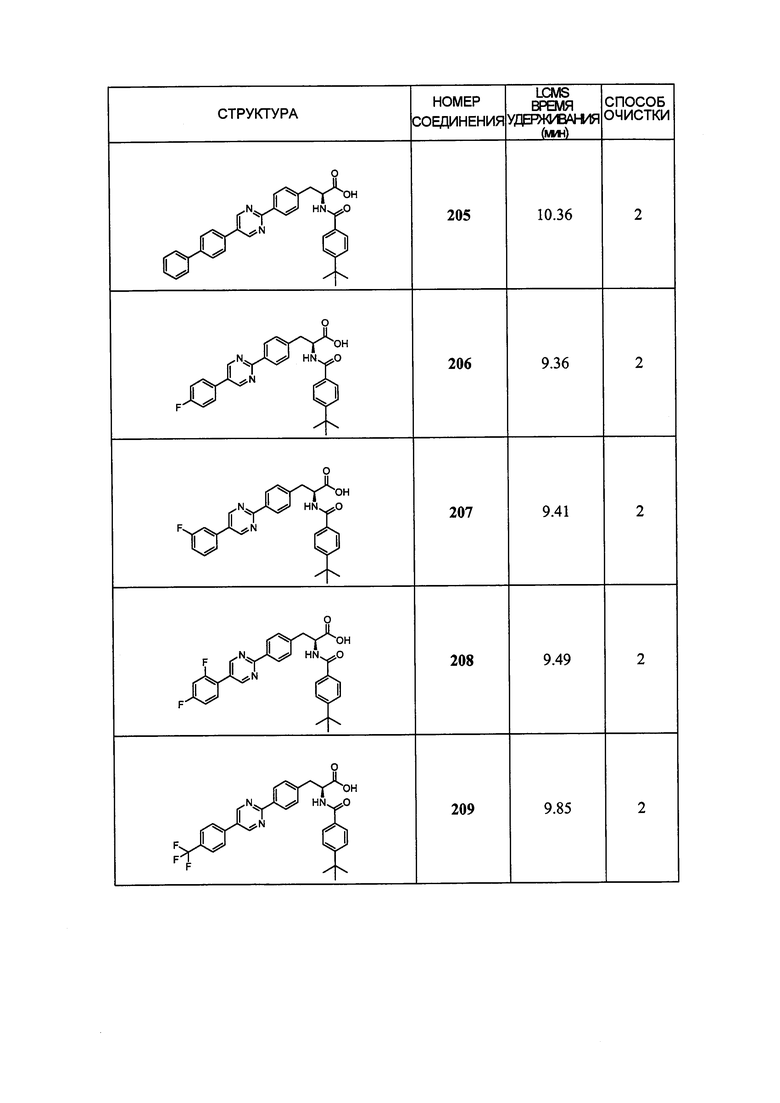

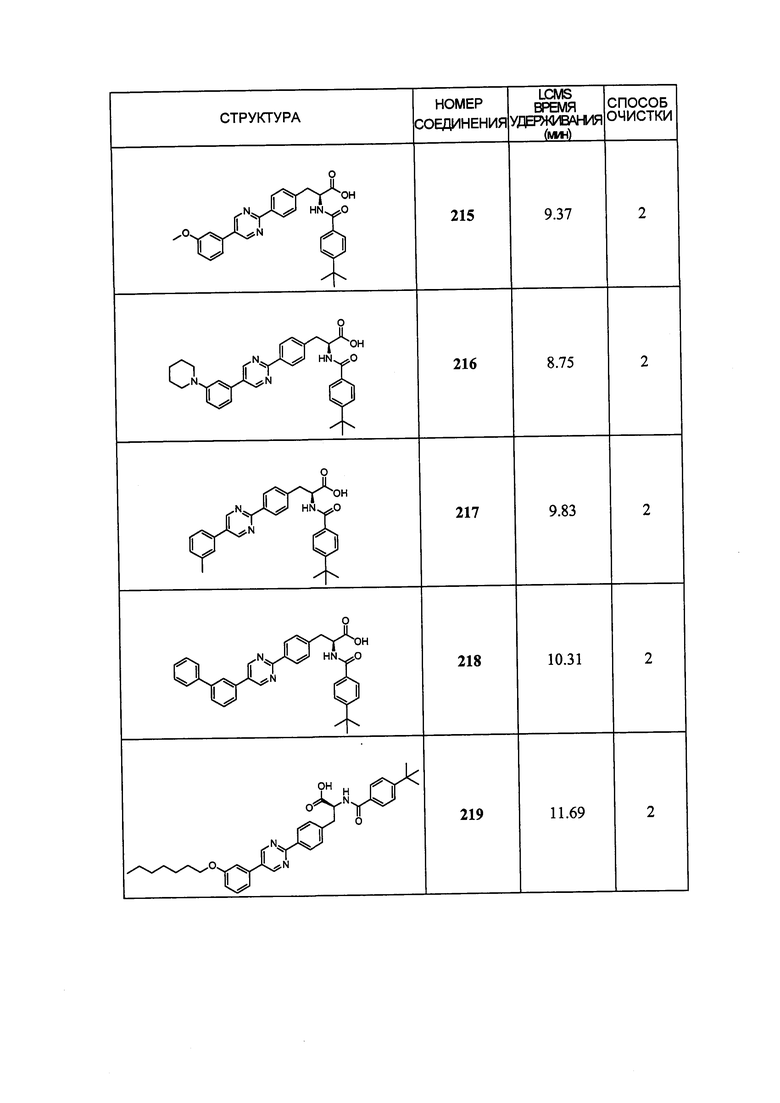
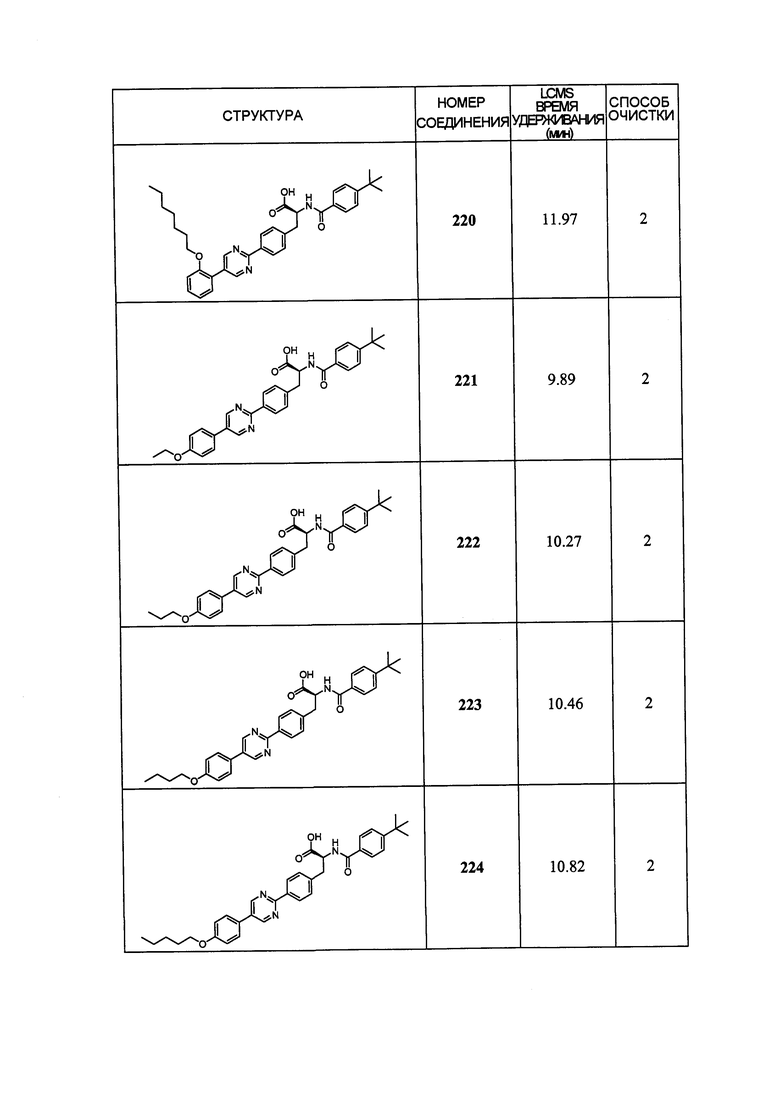
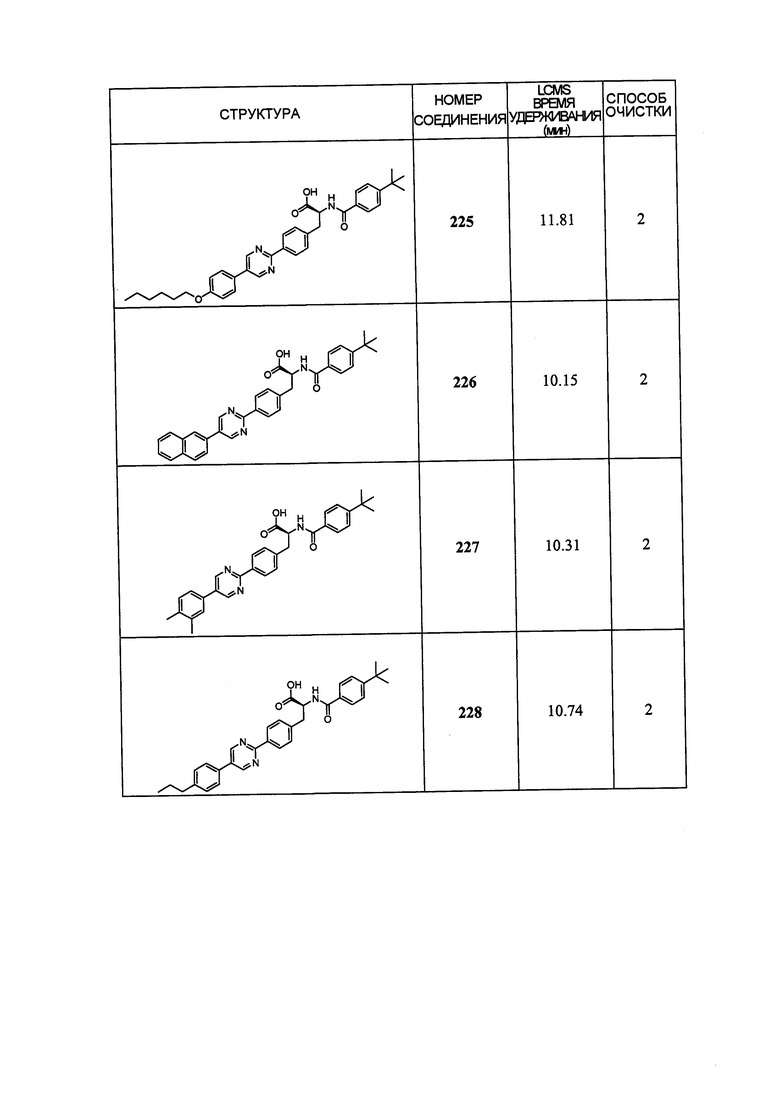
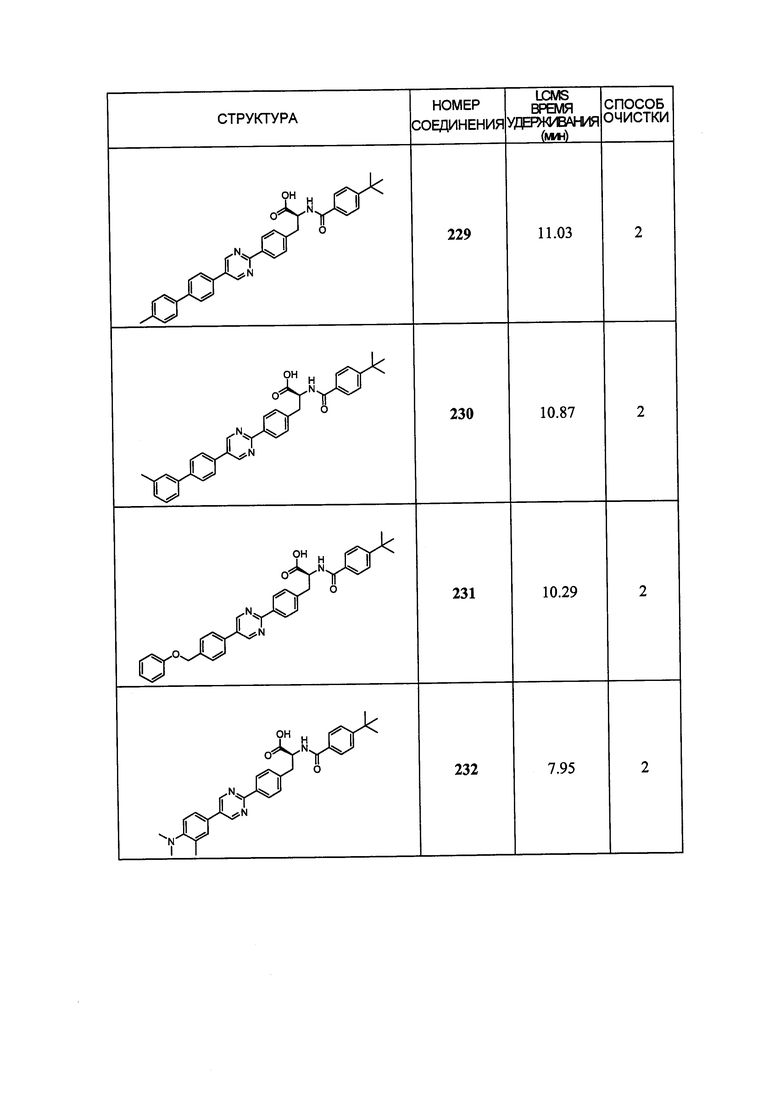
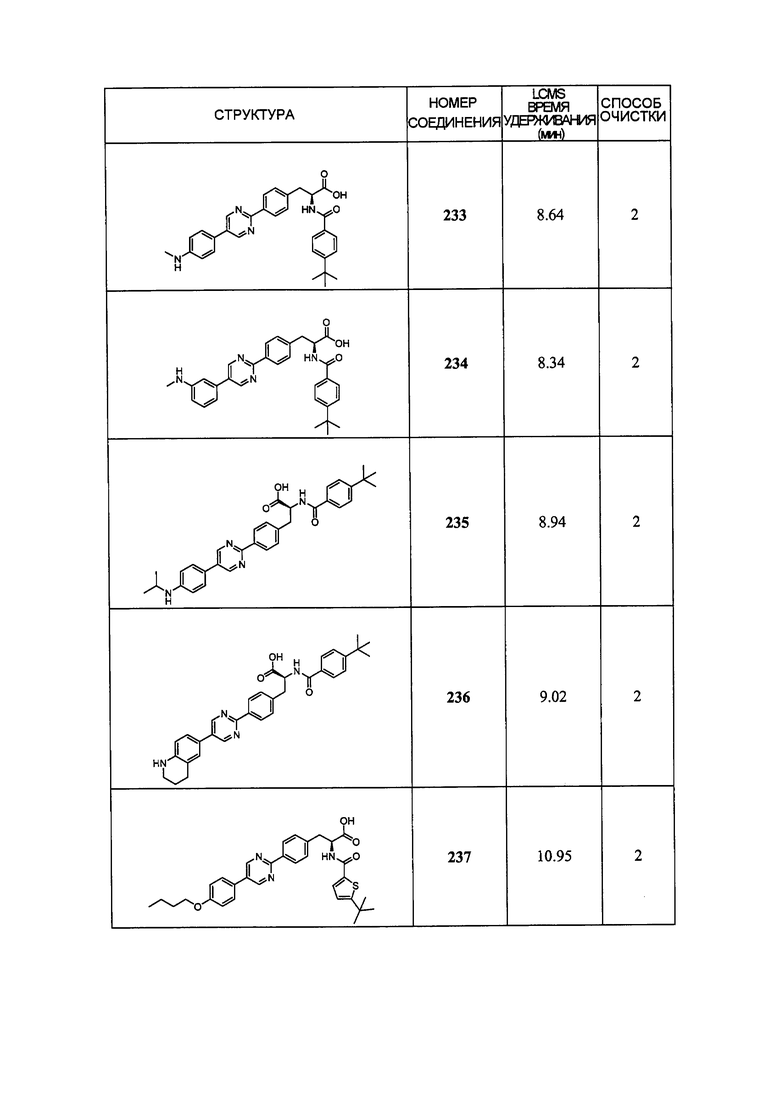
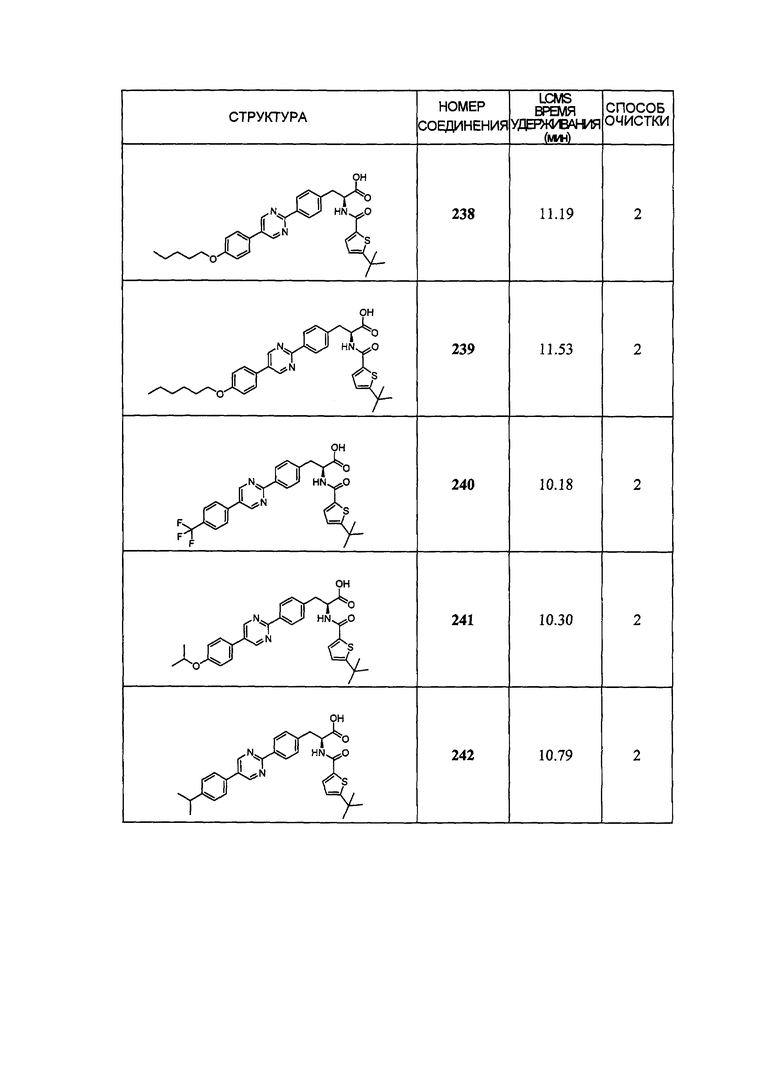
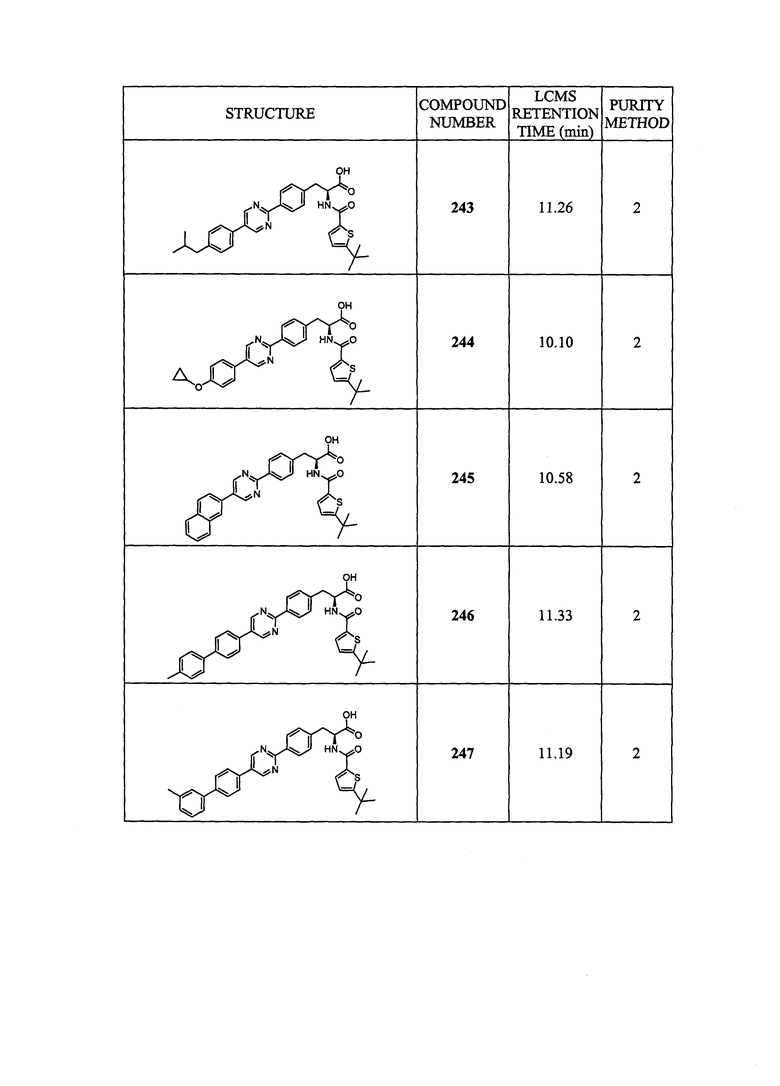
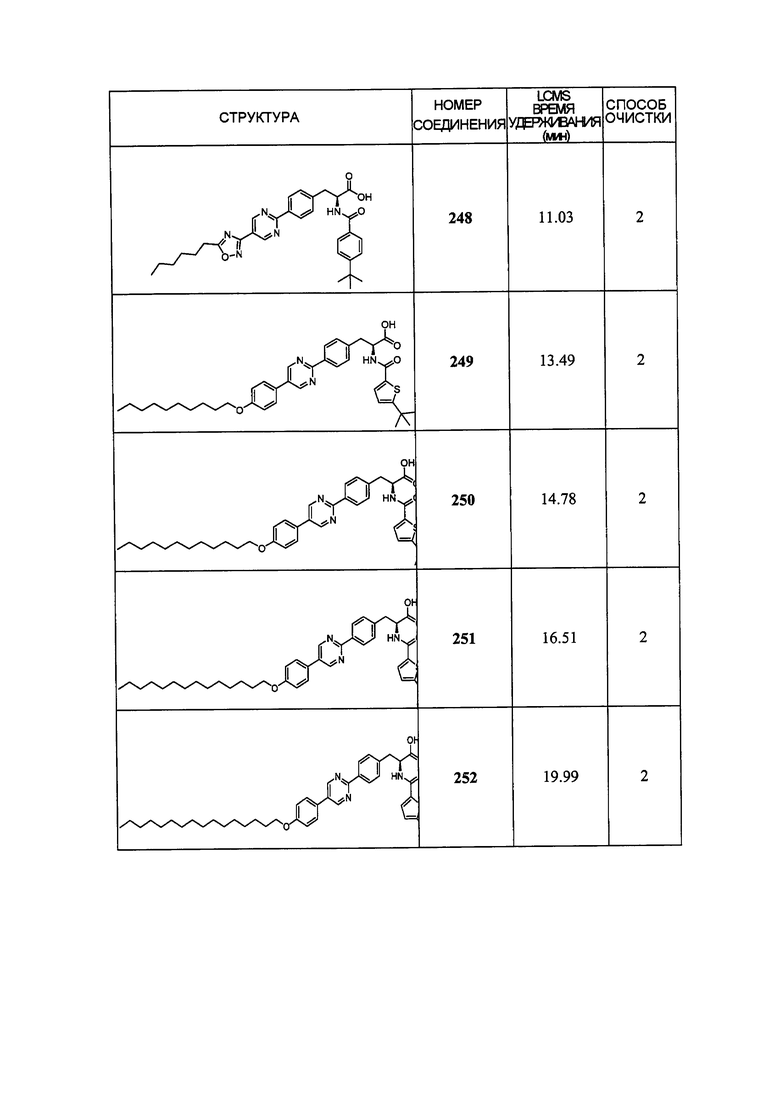
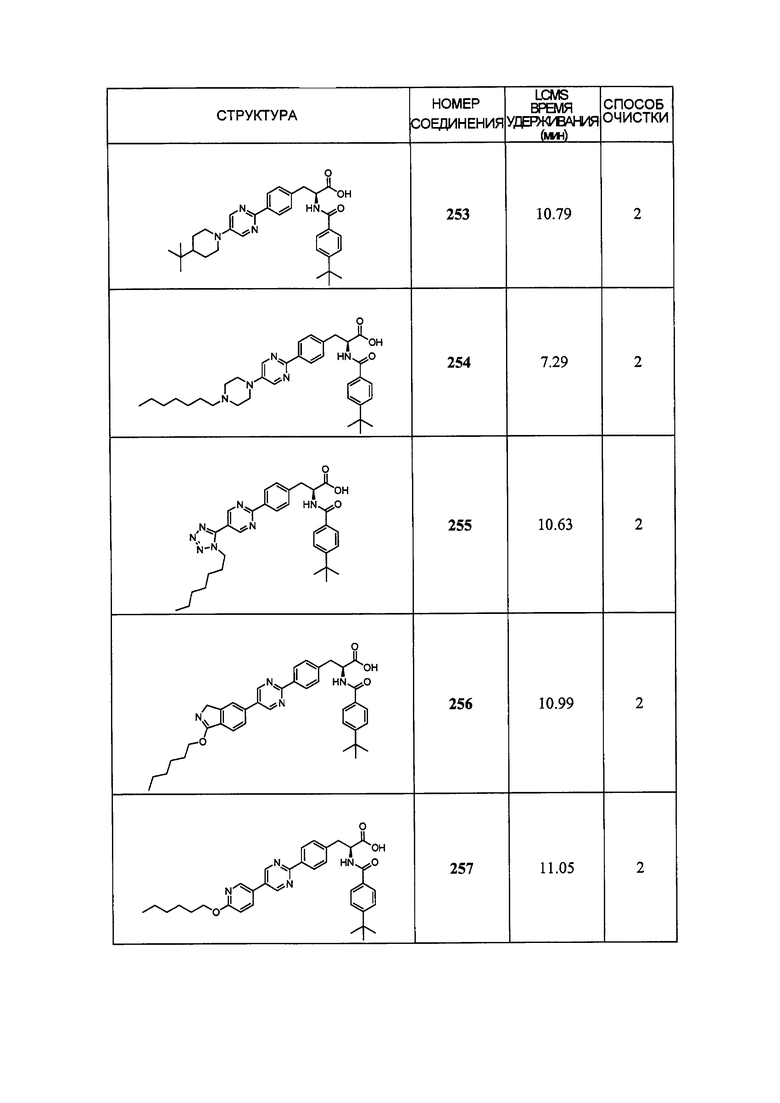
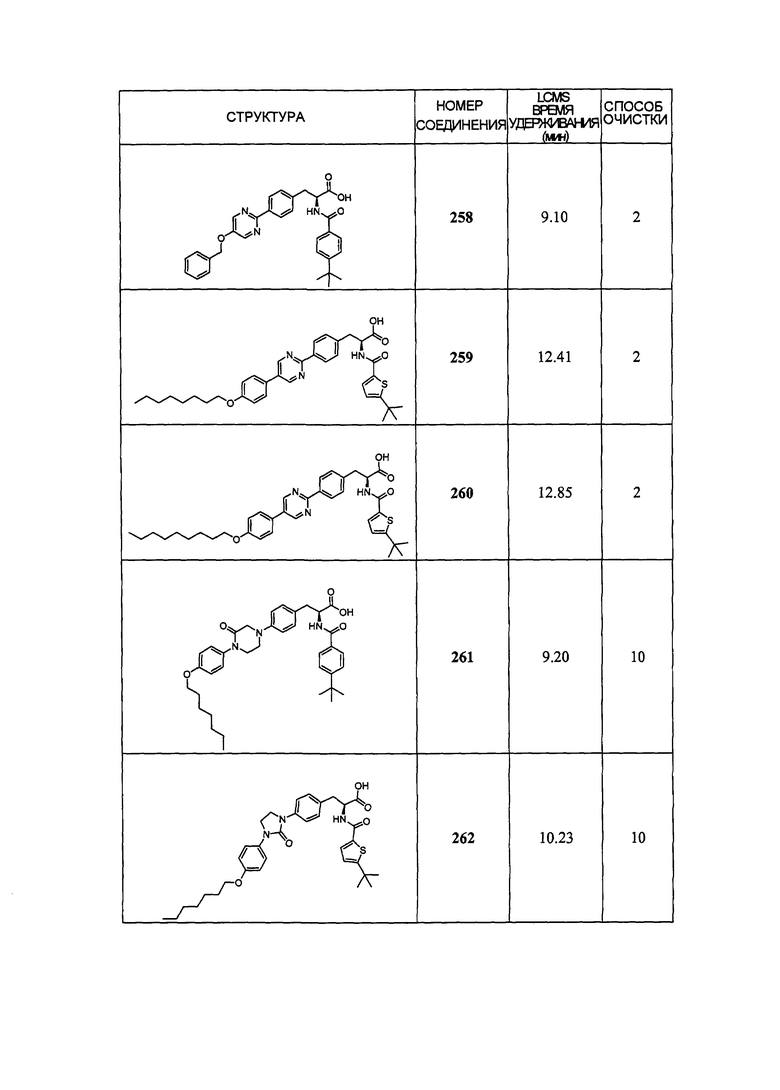

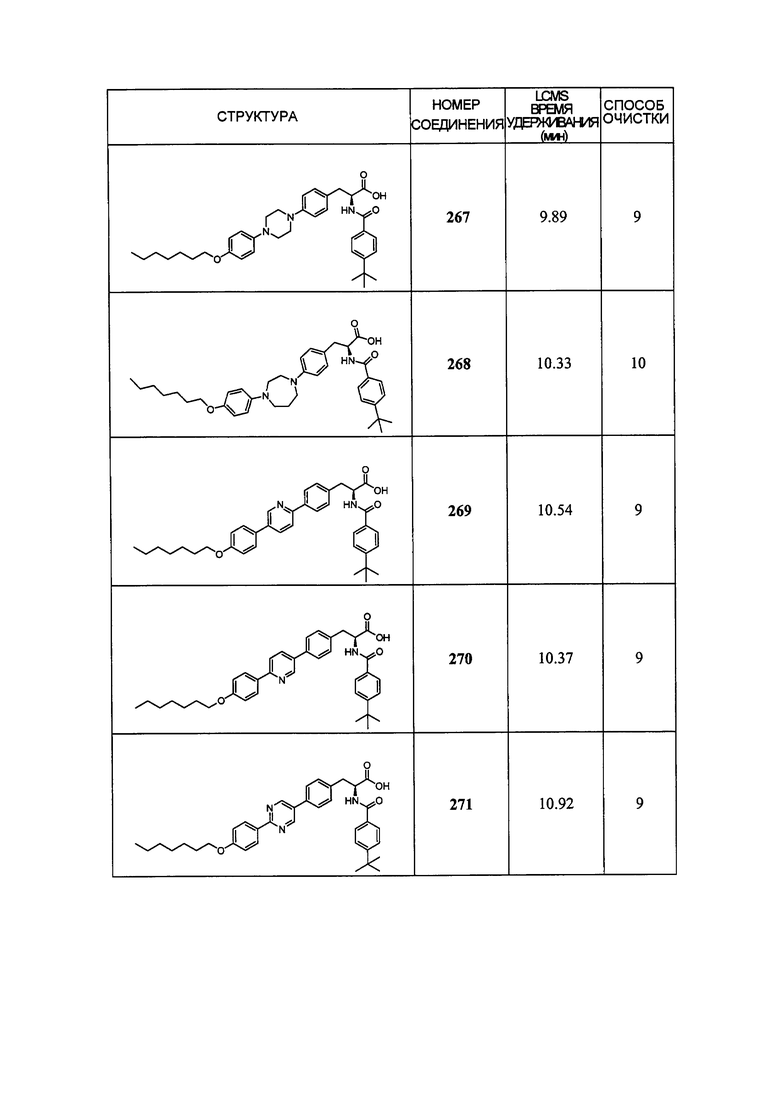

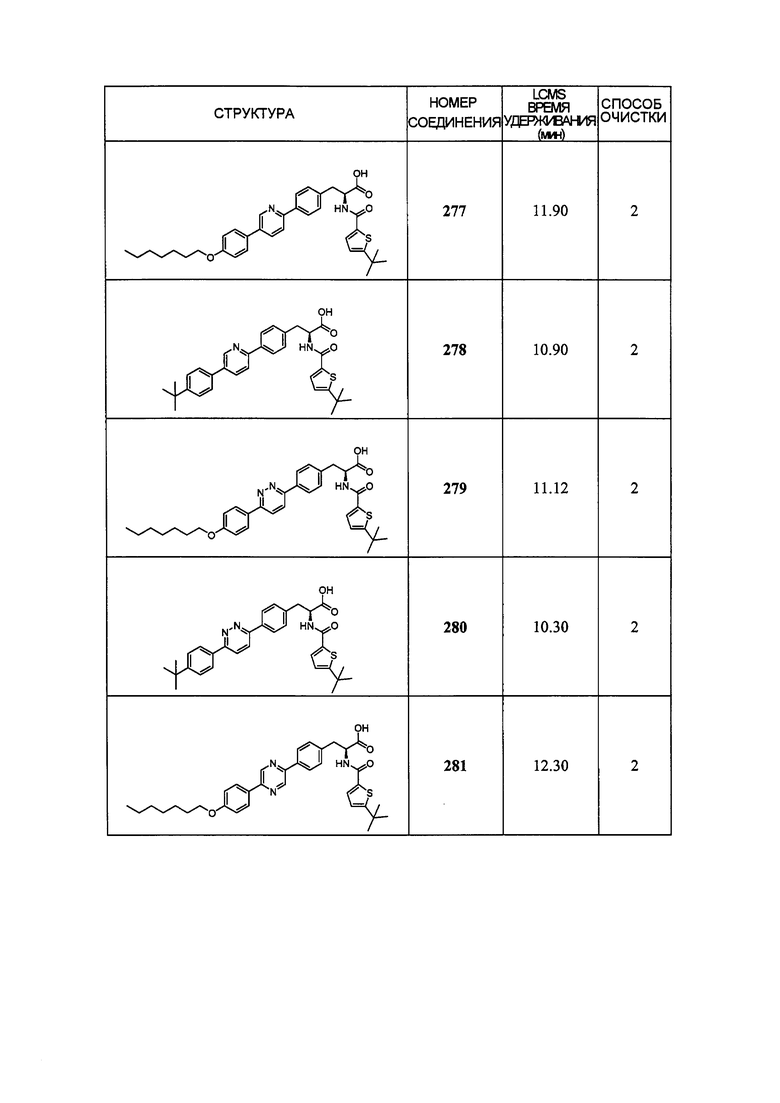
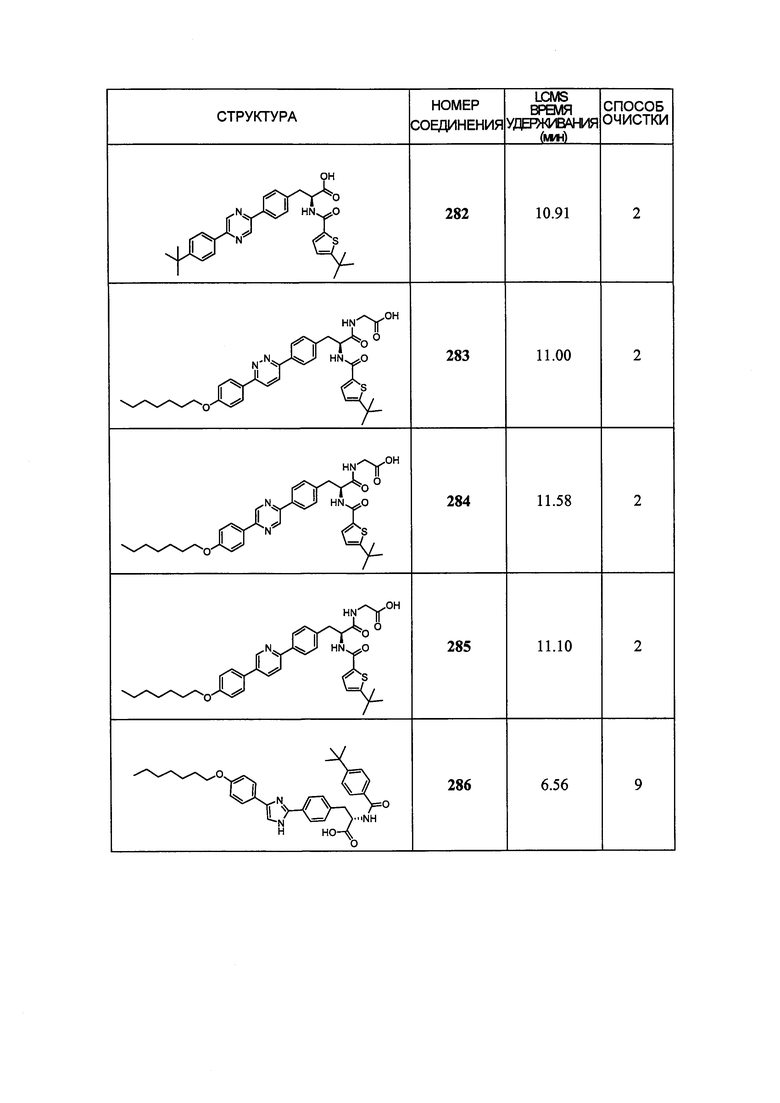
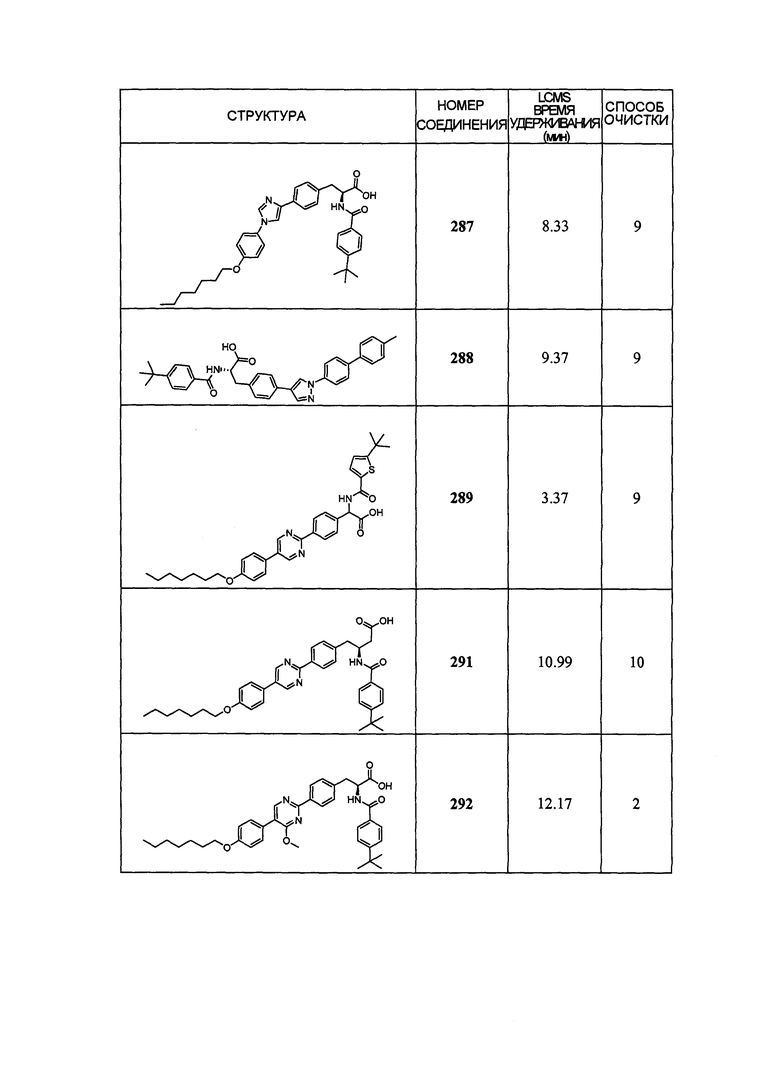
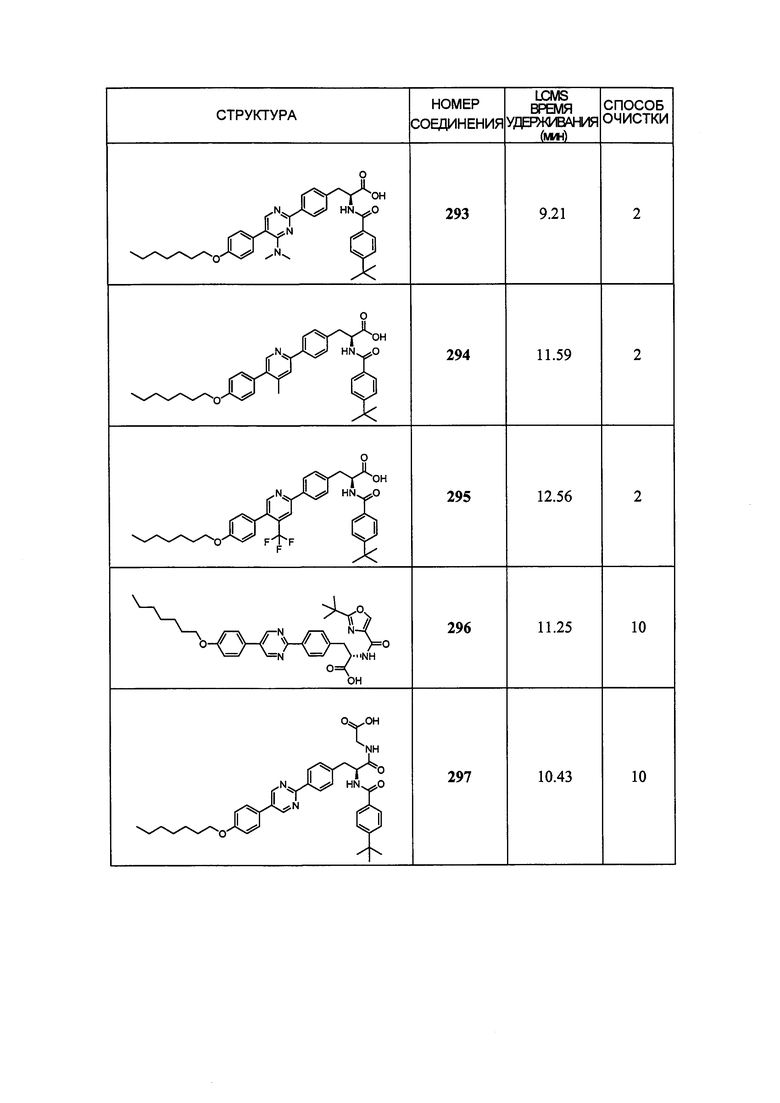
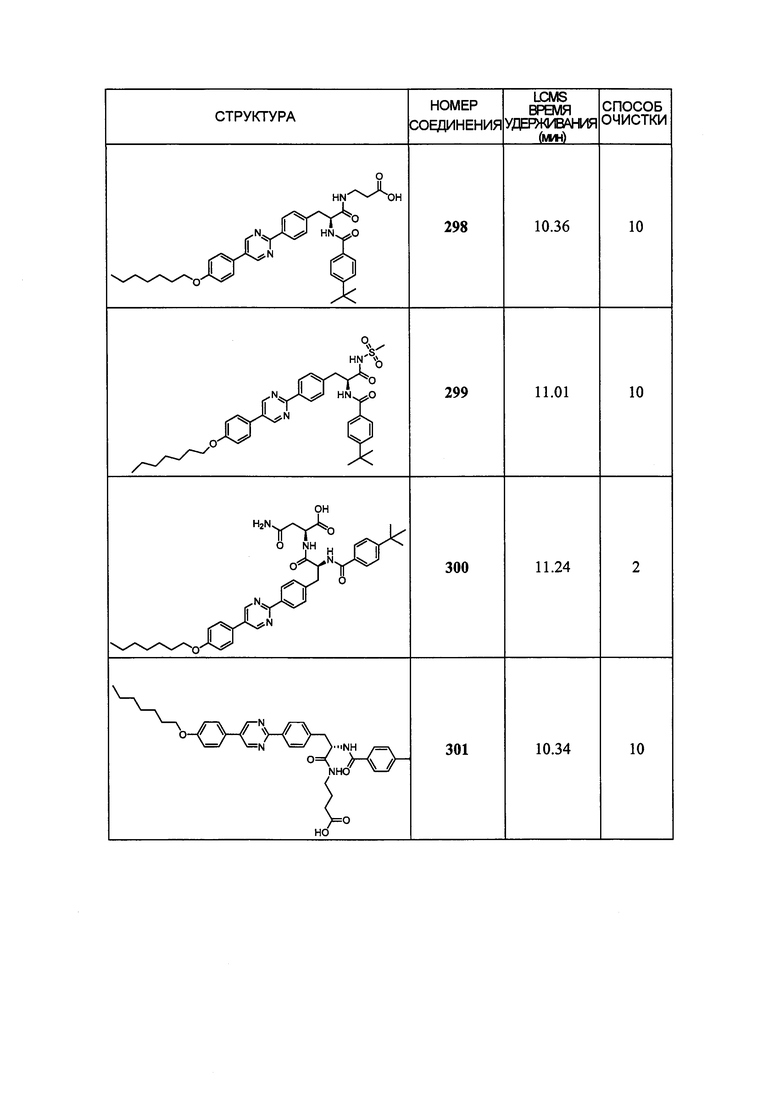
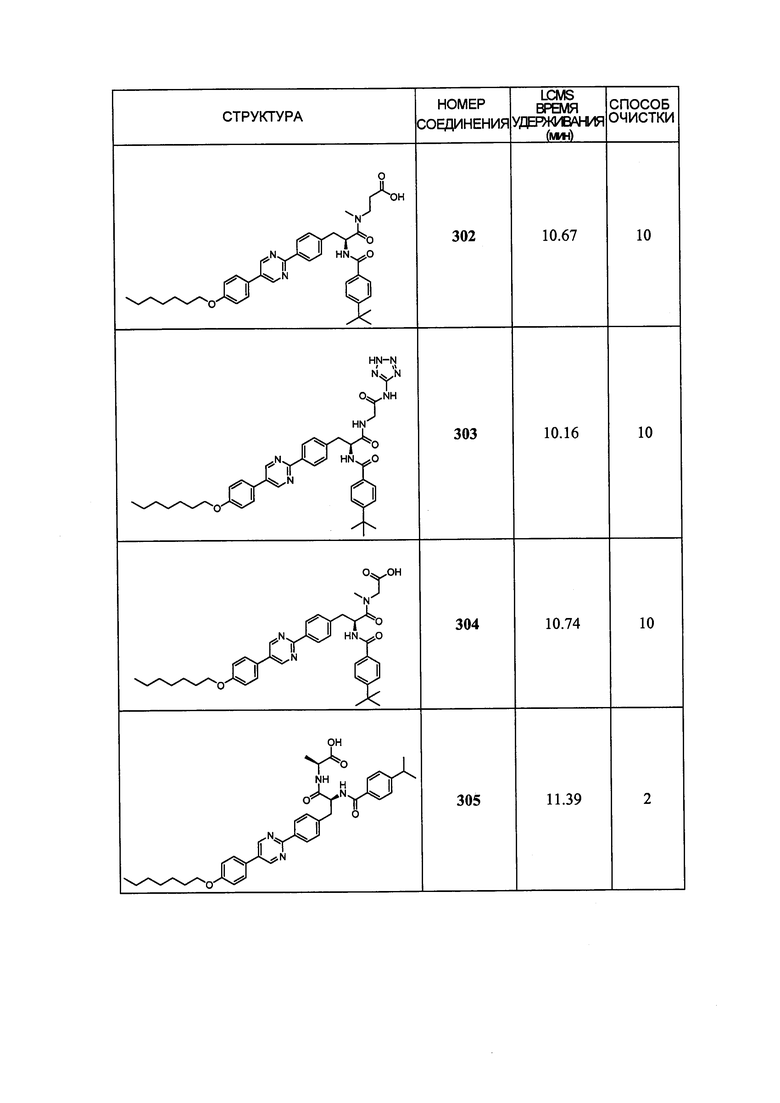

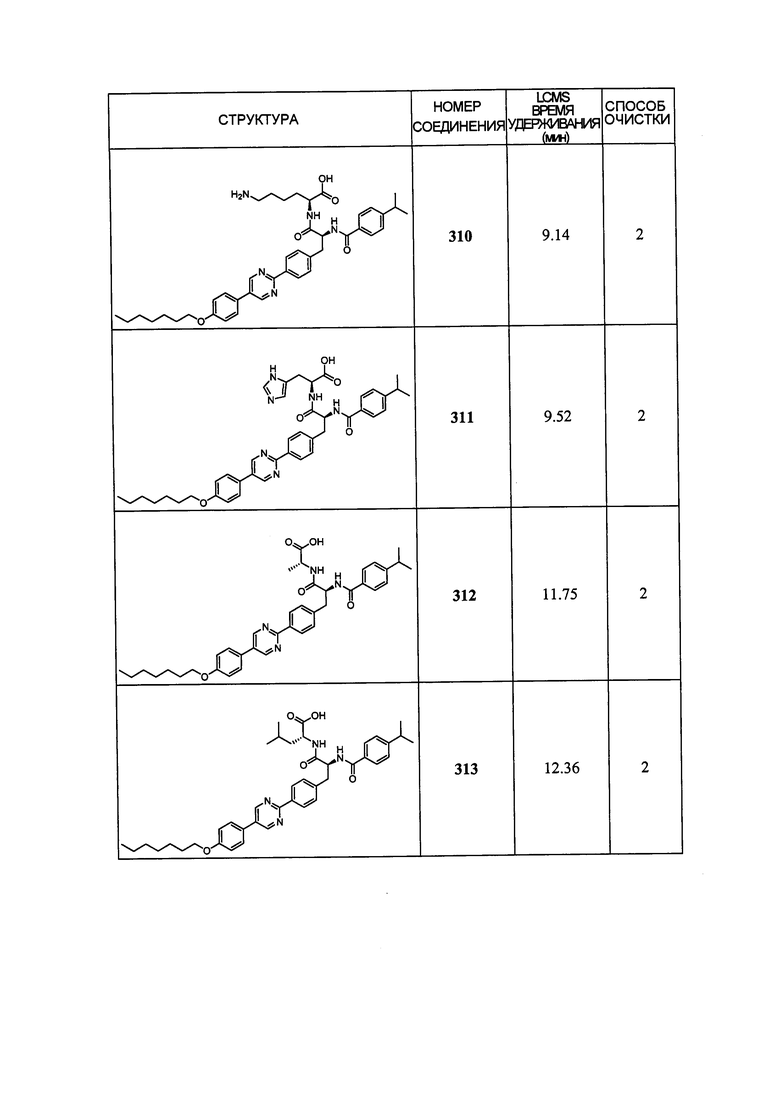
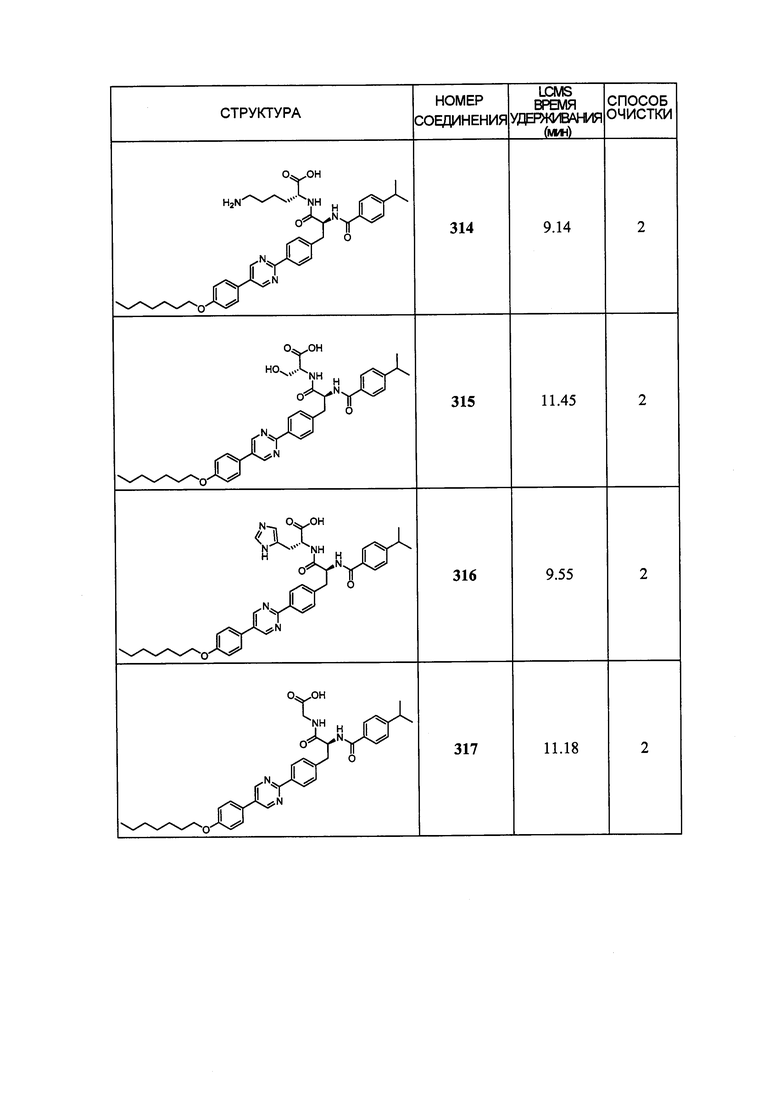
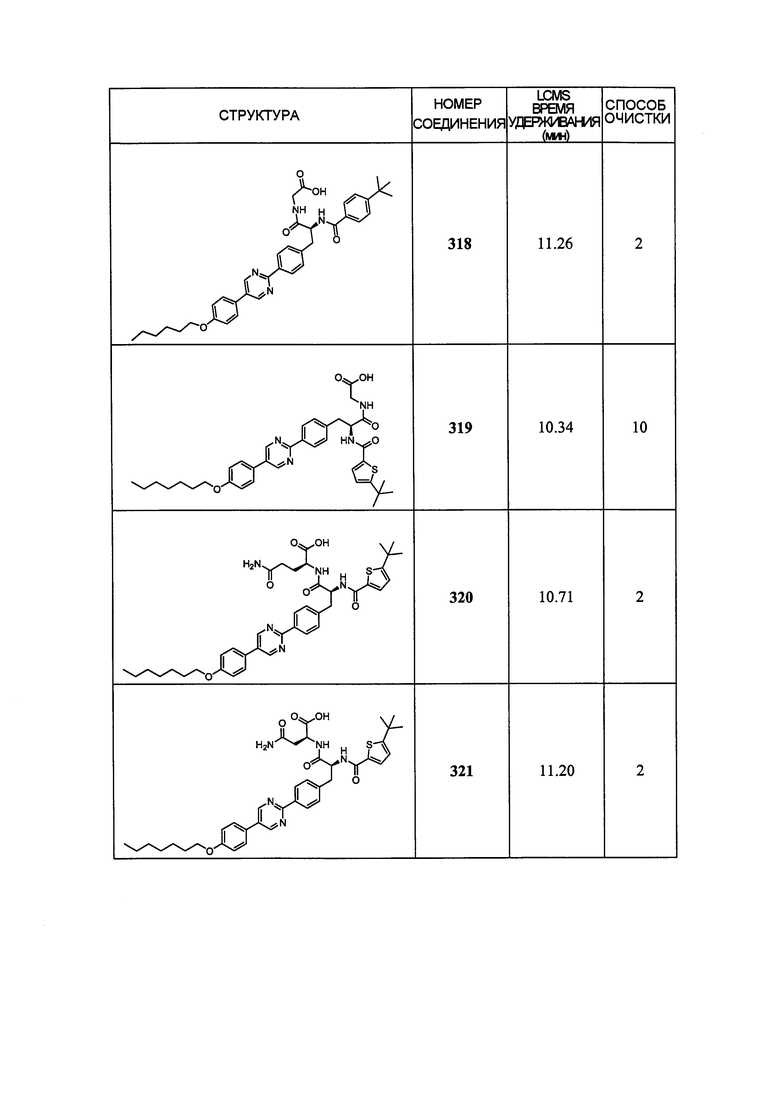
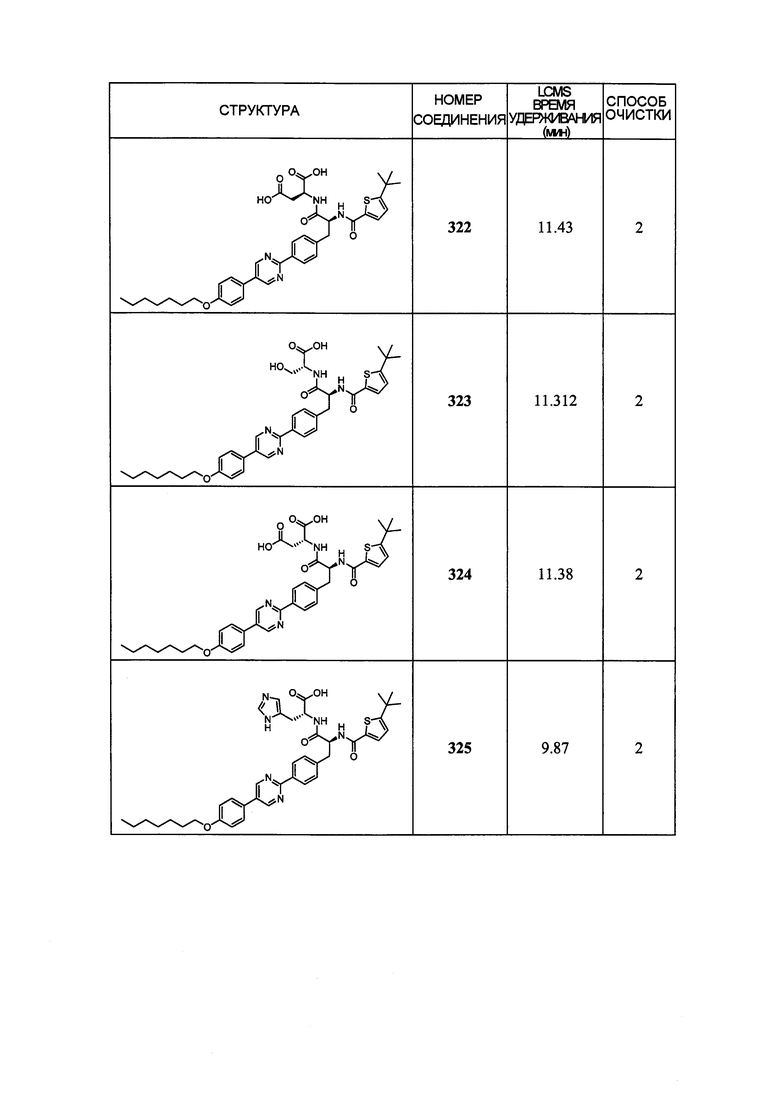
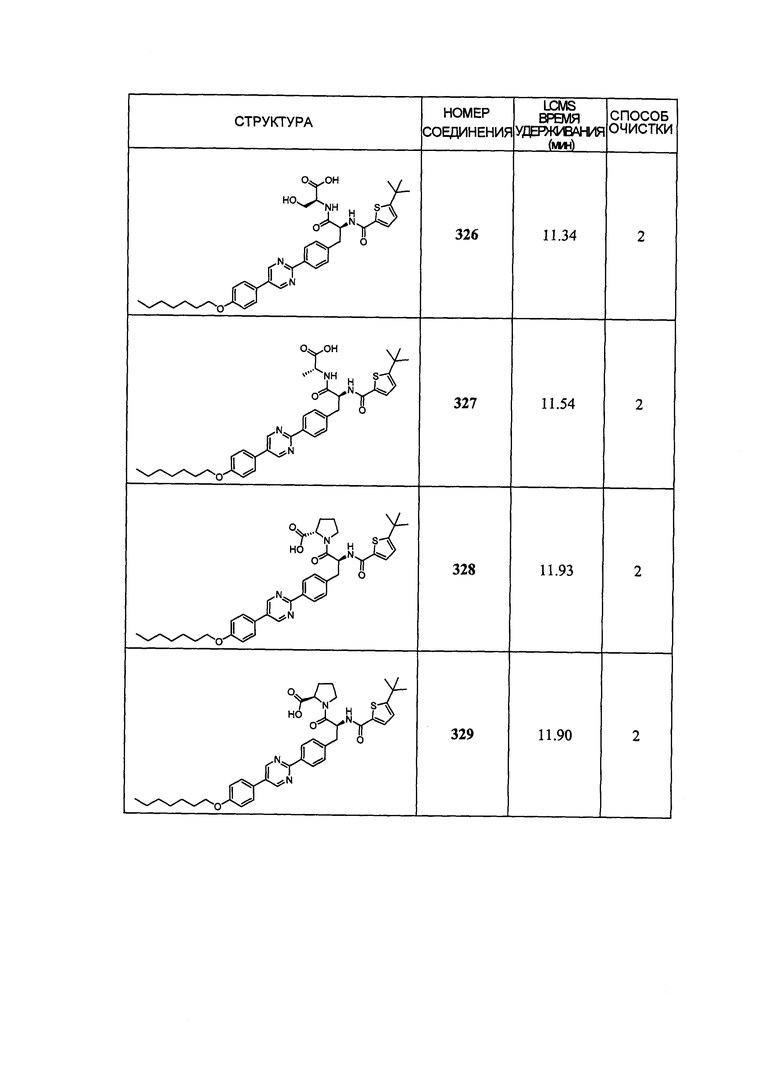

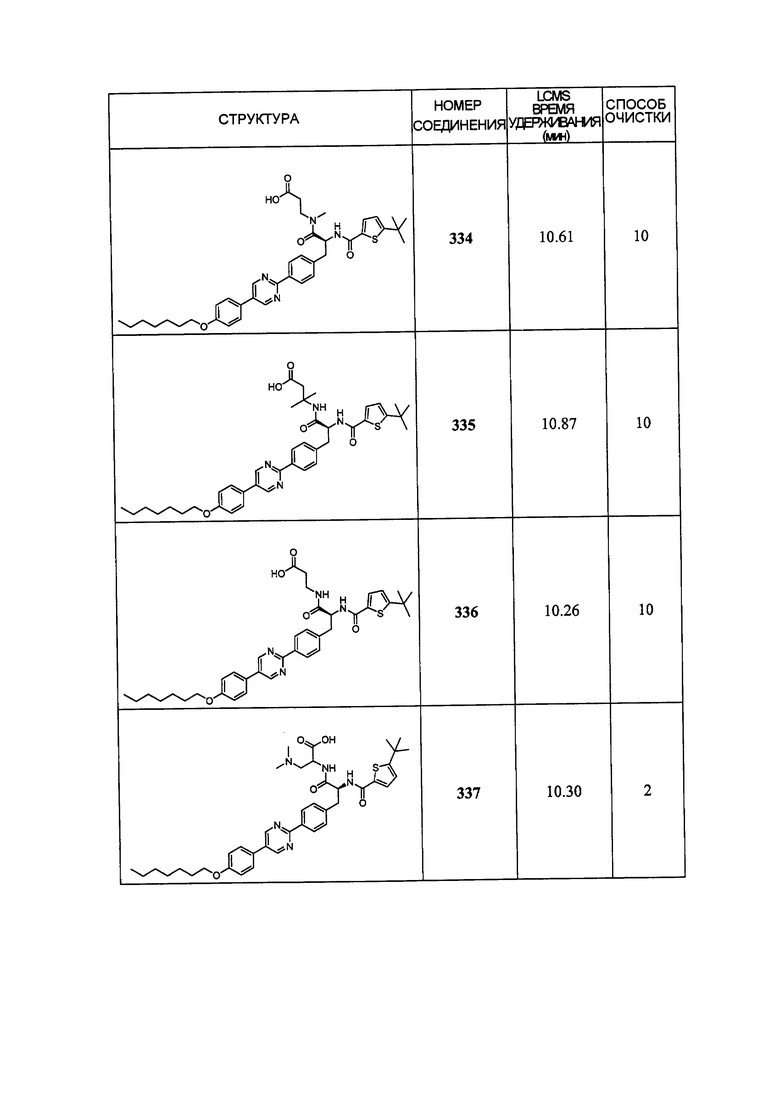

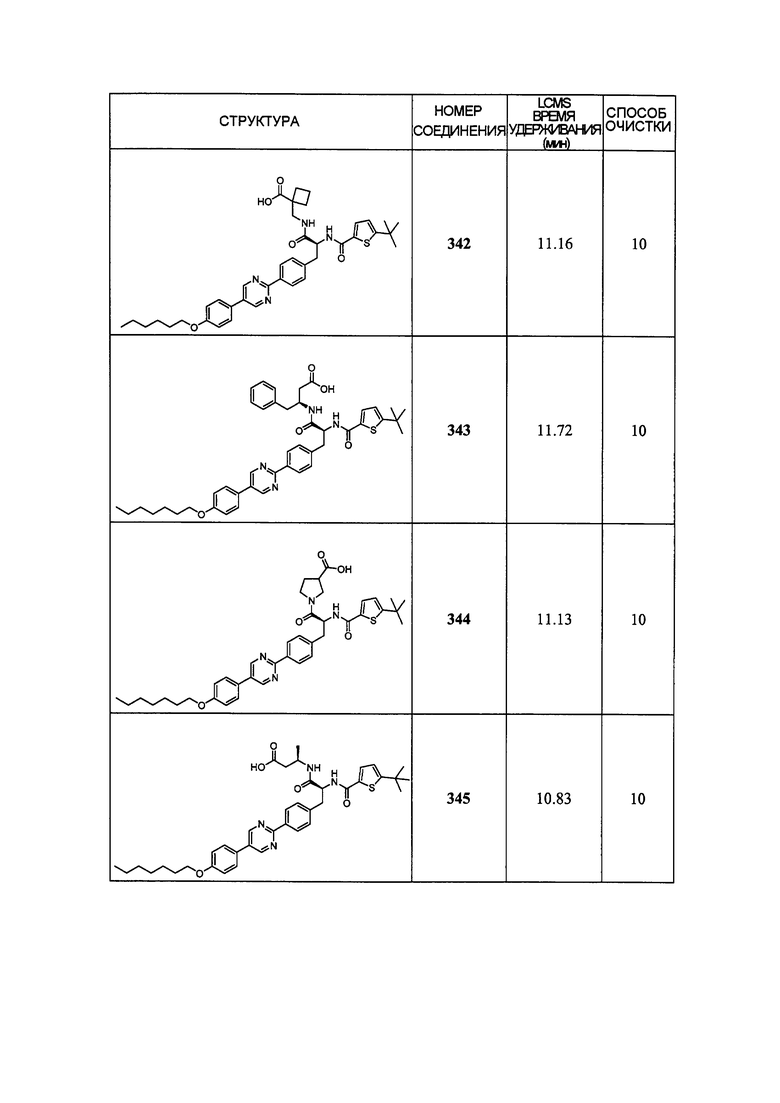
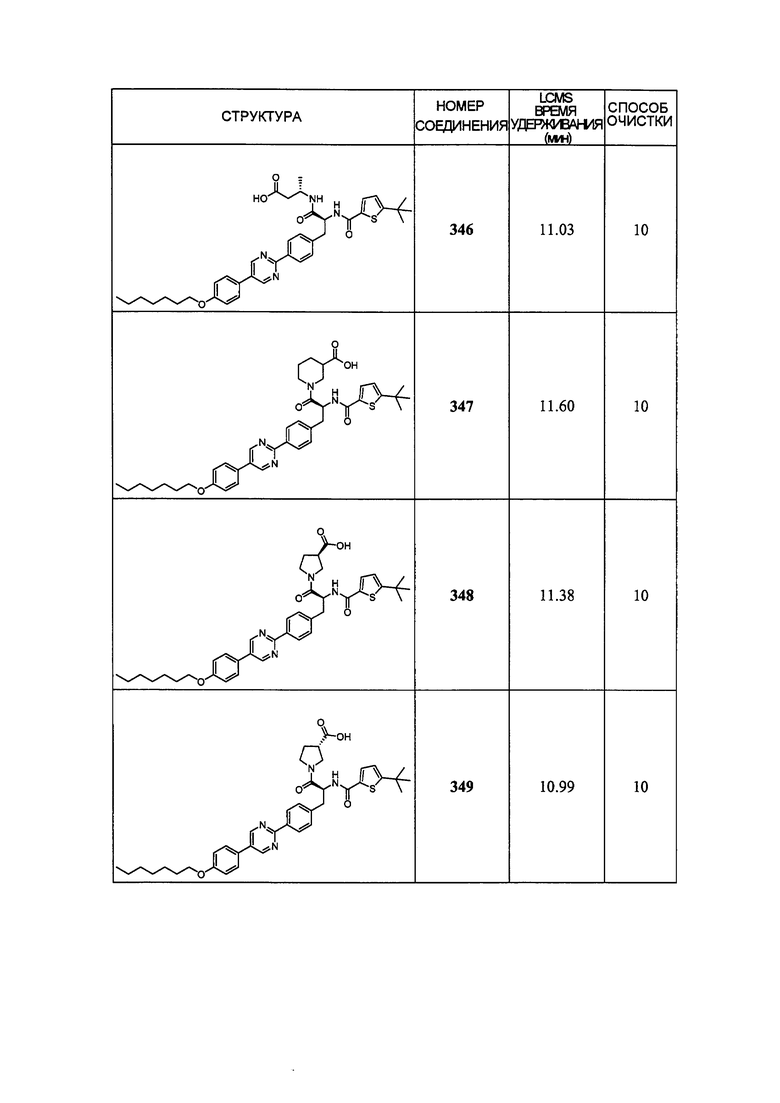
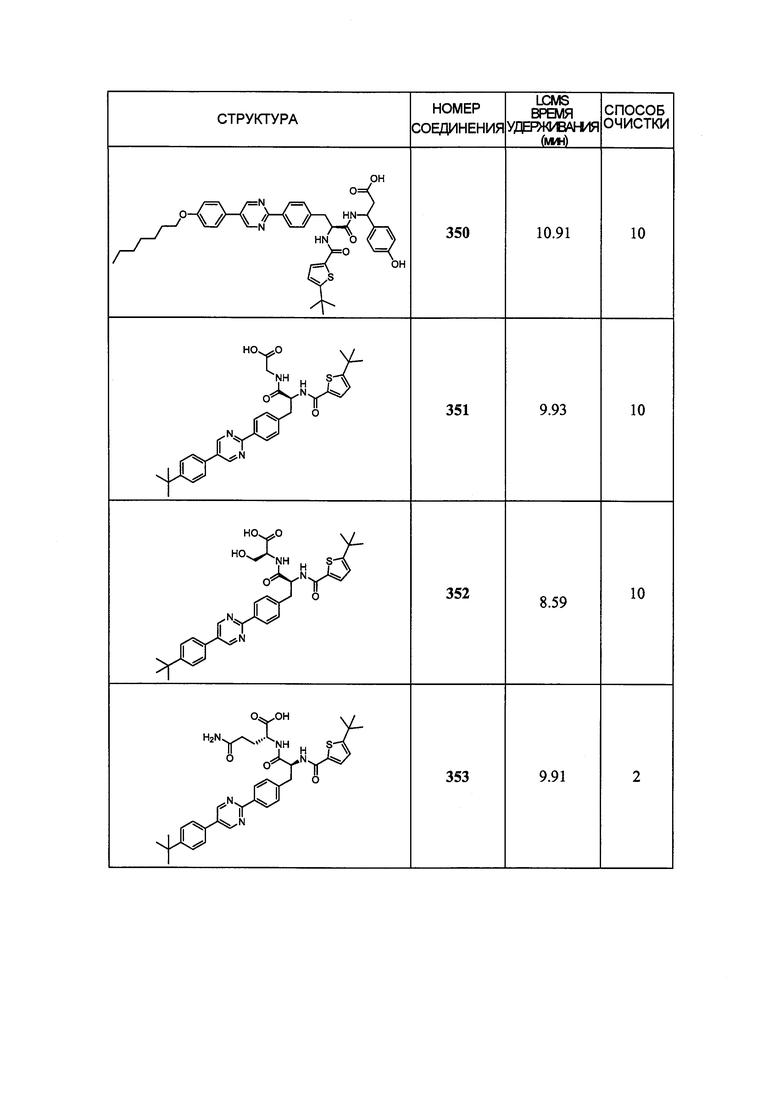
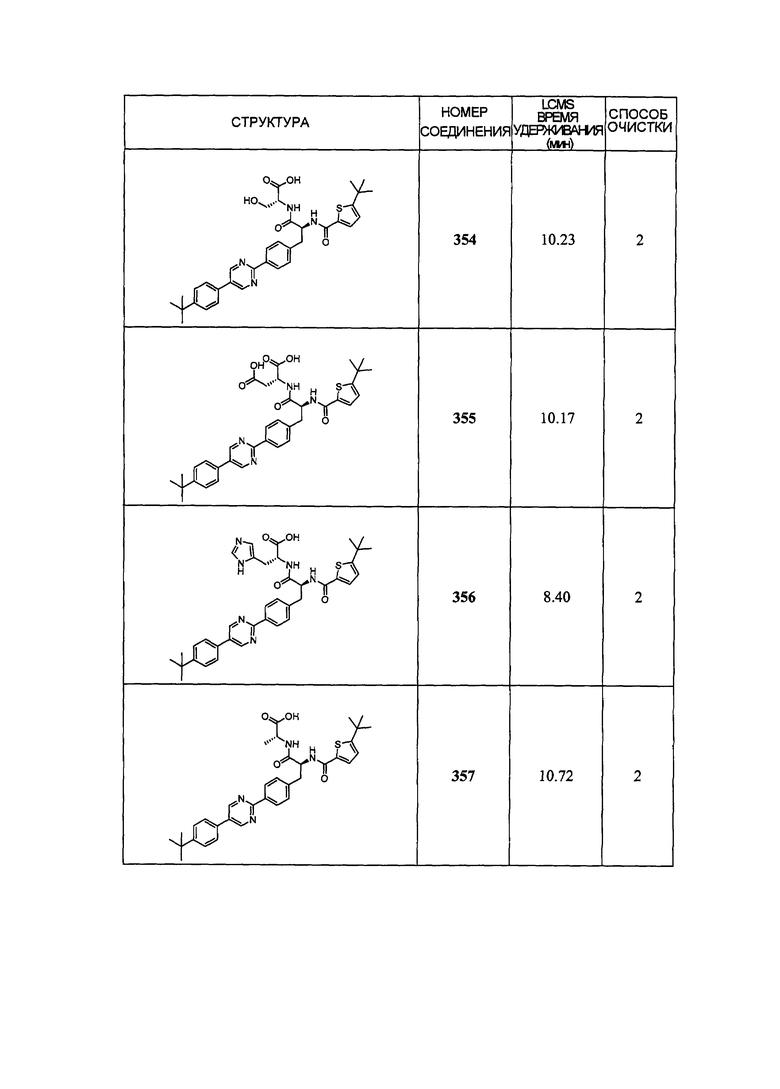
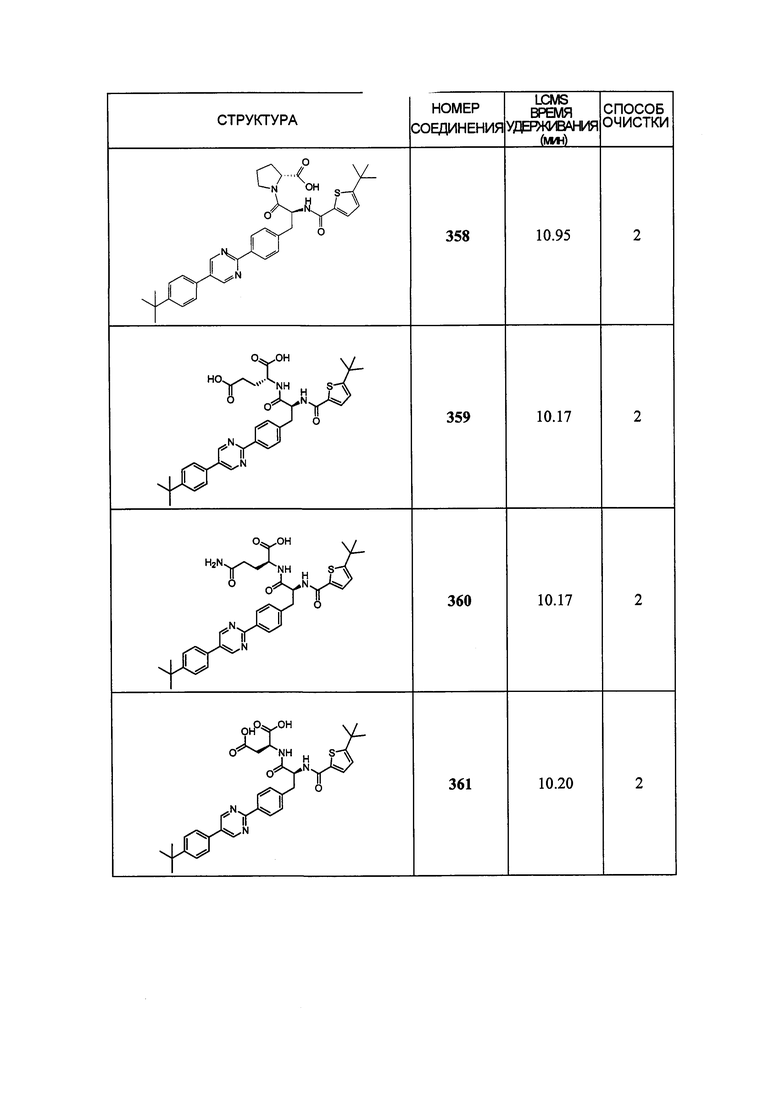

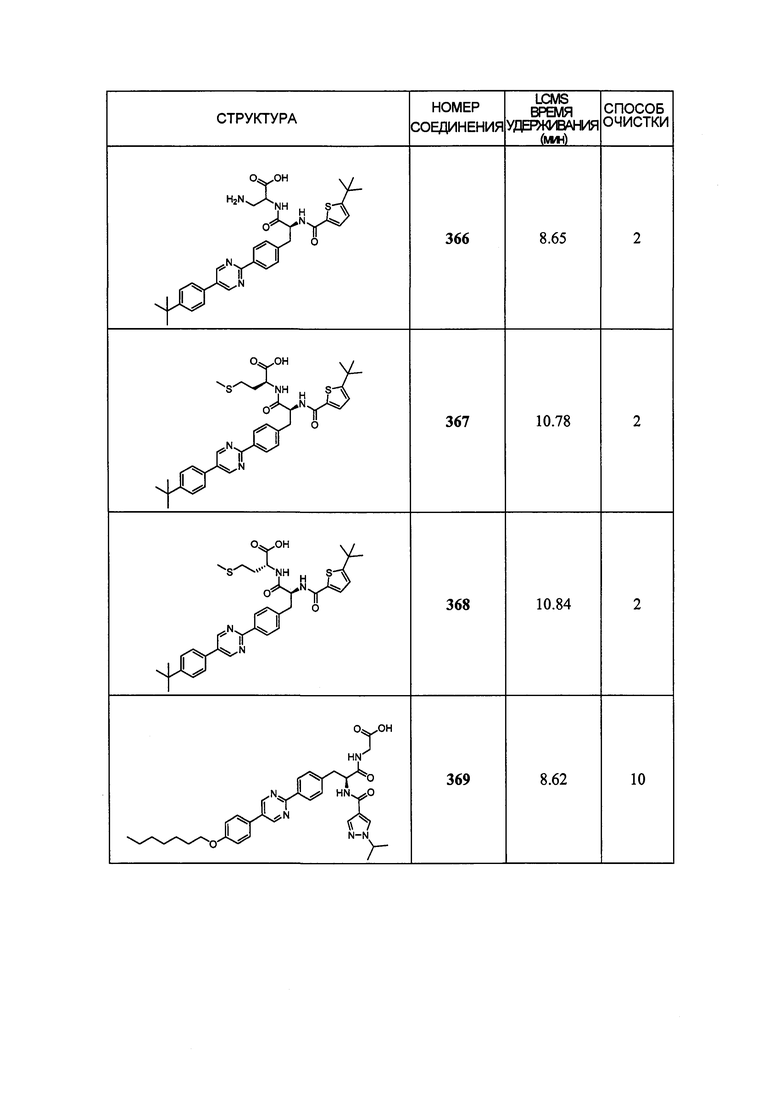
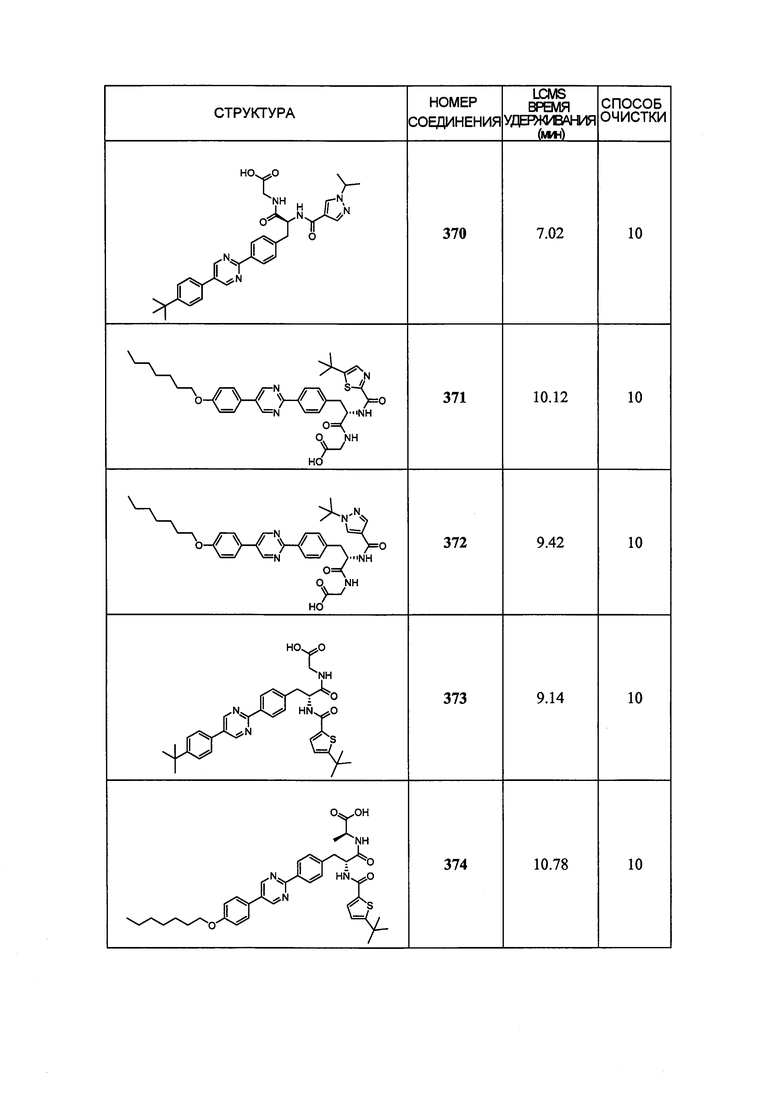
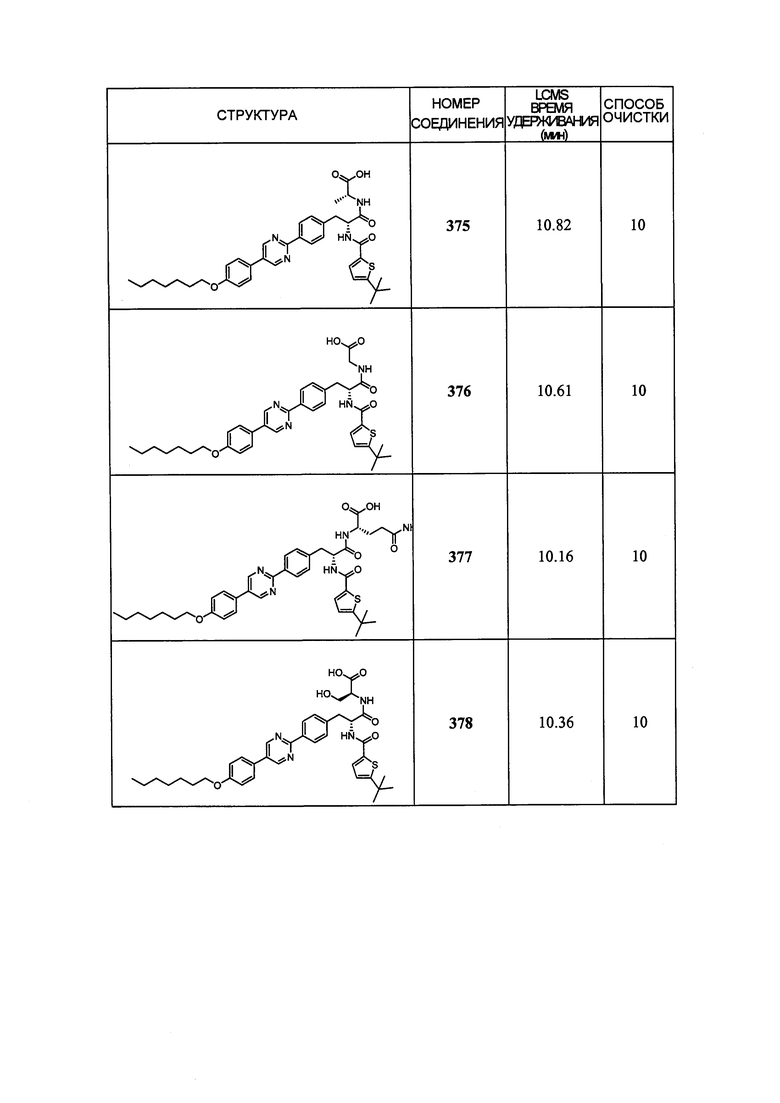
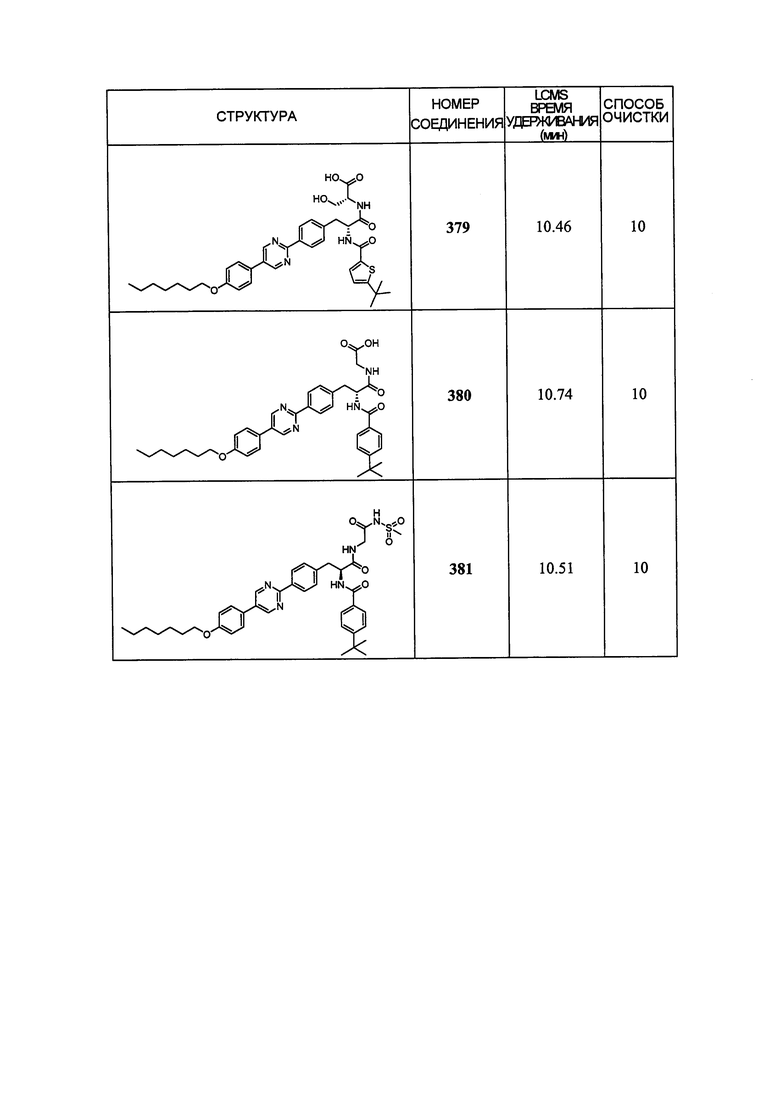
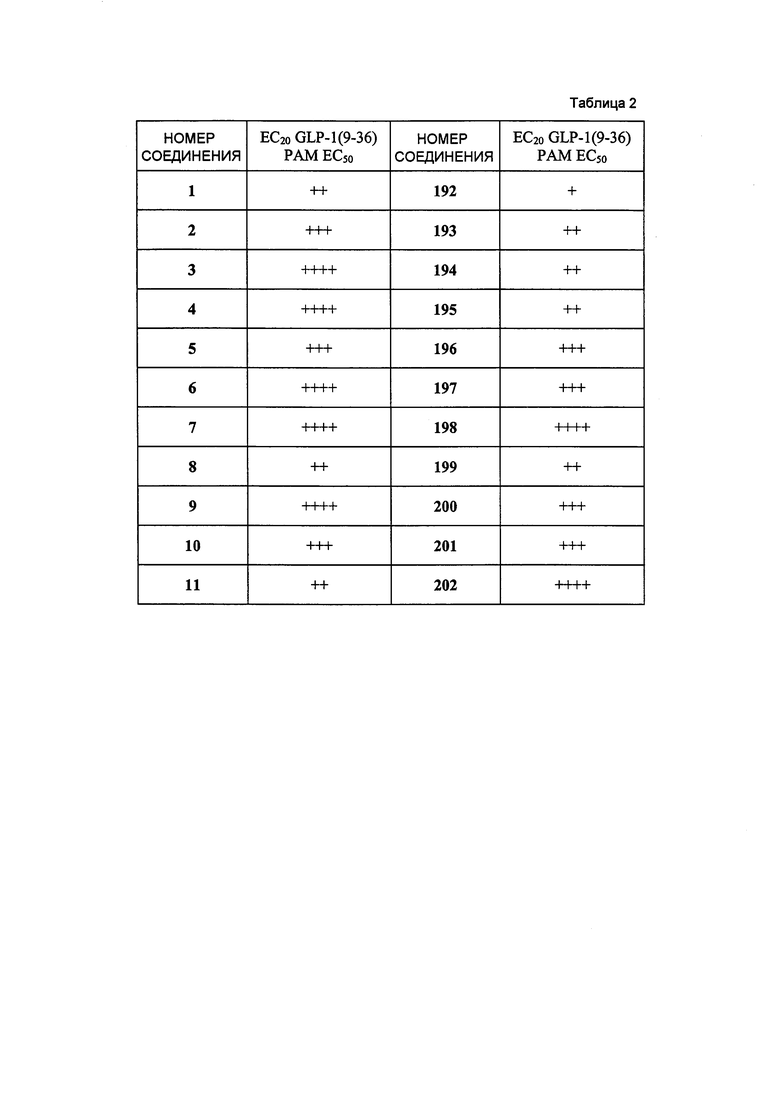



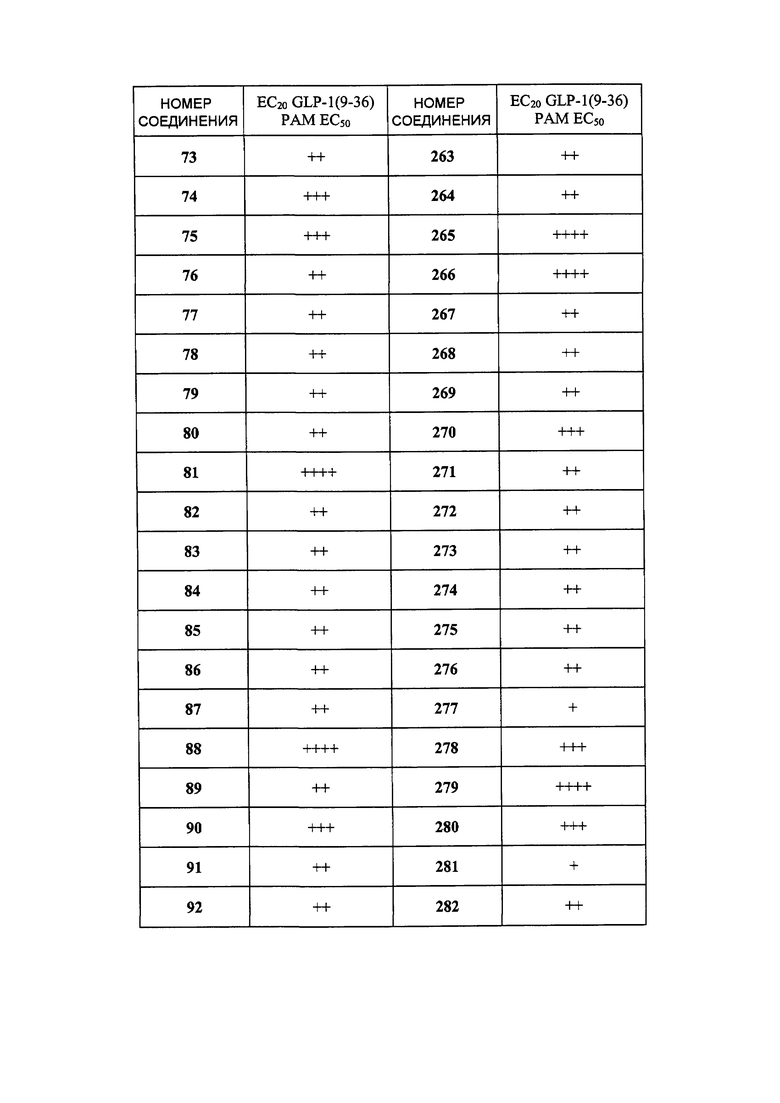

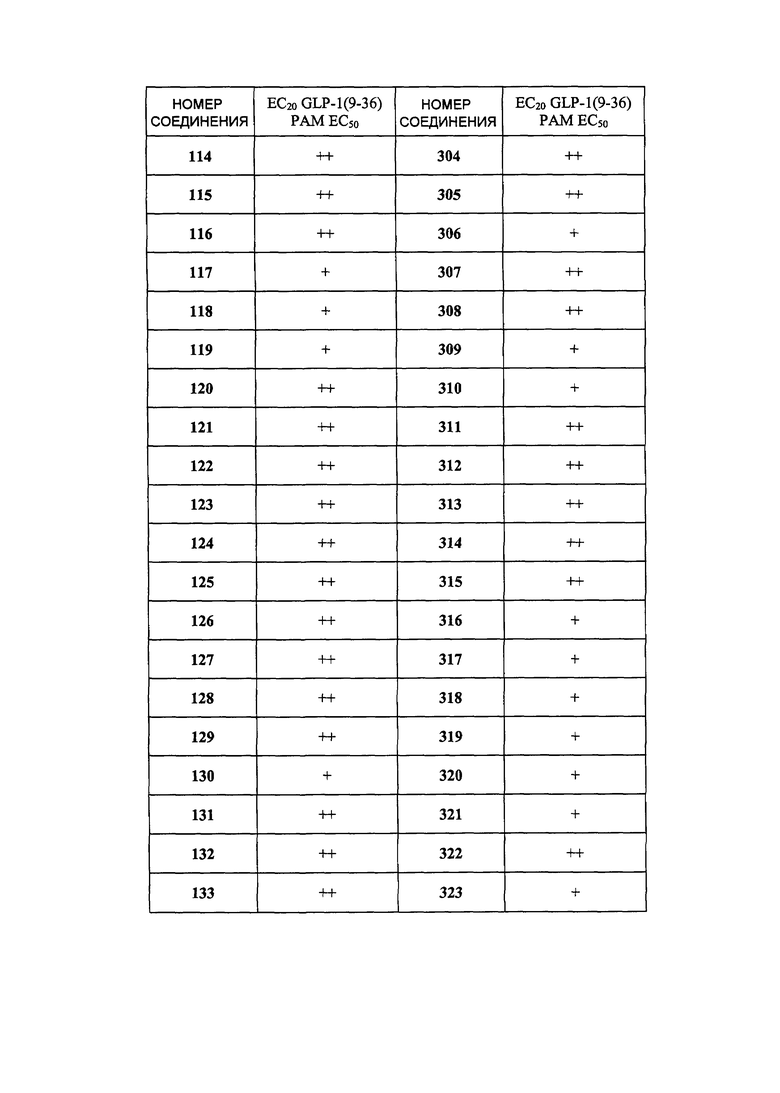
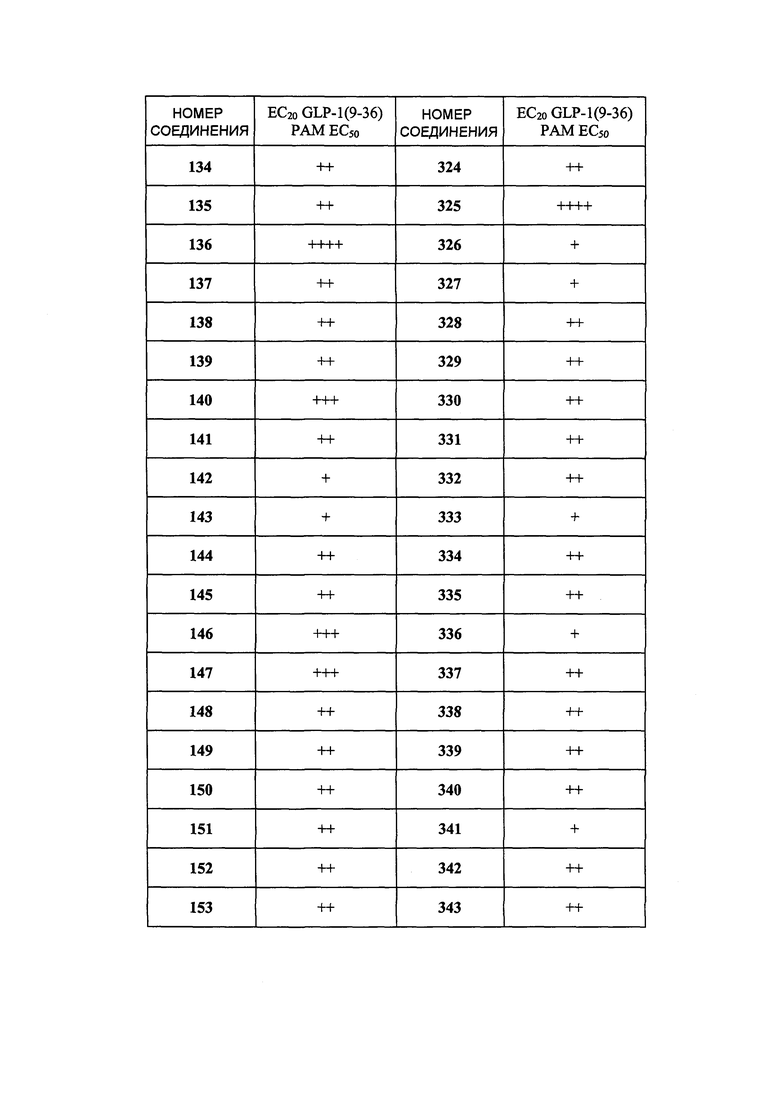

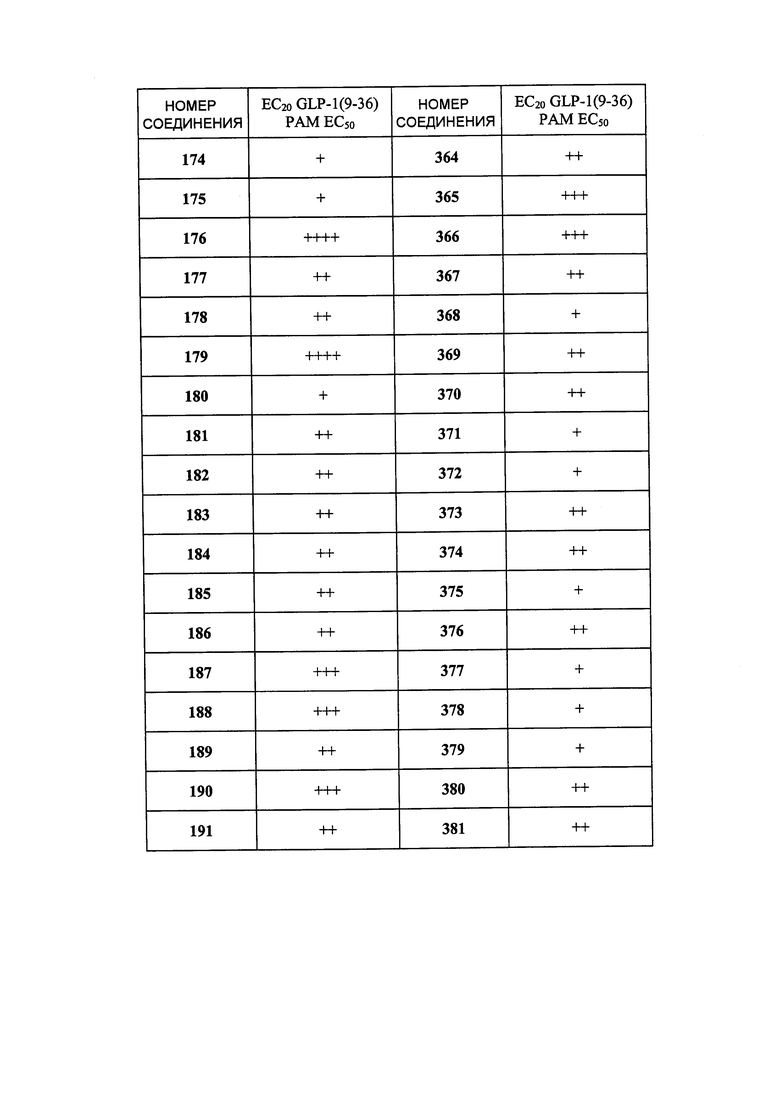

Изобретение относится к новым соединениям формулы I-R или I-S, его фармацевтически приемлемому изомеру, энантиомеру, рацемату или фармацевтически приемлемой соли. Соединения обладают активностью в отношении глюкагонподобного пептида 1 (GLP-1R) и могут найти применение для лечения заболеваний, для которых показано модулирование или потенцирование GLP-1R, в частности для лечения диабета. Соединения могут быть использованы в комбинации для снижения глюкозы с эксенатидом. В формуле I-R или I-S
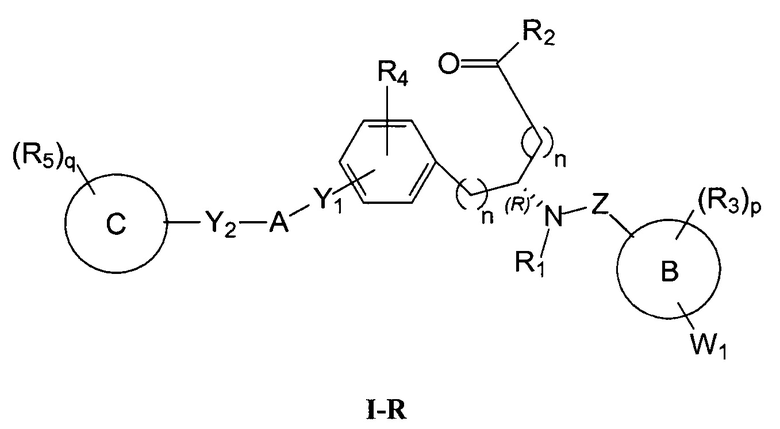

А представляет собой гетероарил, выбранный из имидазола, оксадиазола, тиадиазола, тиазола, оксазола, пиридина, пиримидина, пиридазина, триазина; В представляет собой гетероциклил, выбранный из изоксазола, оксазола, пиримидина, пиразола, тиазола, тиофенила, тиадиазола, азепина, необязательно конденсированного с другим азотсодержащим 6-членным гетероциклическим кольцом; С представляет собой арил, выбранный из фенила и нафтила, бензил; Y1 и Y2 отсутствуют; Z представляет собой -С(О)-; каждый R1 независимо представляет собой Н или С1-4 алкил; R2 представляет собой -O-R8, -N(R1)-SO2-R8, -NR41R42, -N(R1)-(CRaRb)m-COOH, -N(R1)-(CRaRb)m-CO-N(R1)-гетероциклил, -N(R1)-(CRaRb)m-CO-N(R1)(R7) или -N(R1)-гетероциклил, где R2 не является -ОН или -NH2, и гетероциклил представляет собой 5-6-членный гетероциклил, содержащий 1-3 гетероатома, выбранных из N и S; каждый R3 и R4 независимо представляет собой галоген, C1-С4алкил, С1-С4алкил, замещенный R31, С1-С4алкокси, галогенС1-С4алкил, пергалогенС1-С4алкил, галогенС1-С4алкокси, -ОН, -NR1R8, -C(O)R8, -C(O)NR1R8, -S(O)2R8, -OS(O)2R8, -(CRaRb)mNR1R8, -(CRaRb)mO(CRaRb)mR8; или любые две R3 или R4 группы на одном и том же атоме углерода, взятые вместе, образуют оксо, значения R31, R40, R41, R42 указаны в формуле изобретения; W1 отсутствует; каждый Ra и Rb имеют значения, указанные в формуле изобретения; R5 представляет собой R7, -(СН2)m-L2-(СН2)m-R7 или -(-L3-(CRaRb)r-)s-L3-R7; R7, R8 имеют значения, указанные в формуле изобретения; L2 независимо является, от ближнего к дальнему концу структуры формулы I-R или I-S, отсутствующей или -О-; каждая L3 независимо является отсутствующей, -О- или -N(R1)-, каждый m независимо равен 0, 1, 2, 3, 4, 5 или 6; каждый n независимо равен 0, или 1, или 2; p равен 0, 1, 2 или 3; q равен 0, 1, 2 или 3. 3 н. и 5 з.п. ф-лы, 2 табл., 381 пр.
1. Соединение, имеющее структуру формулы I-R или I-S, или его фармацевтически приемлемый изомер, энантиомер, рацемат или фармацевтически приемлемая соль
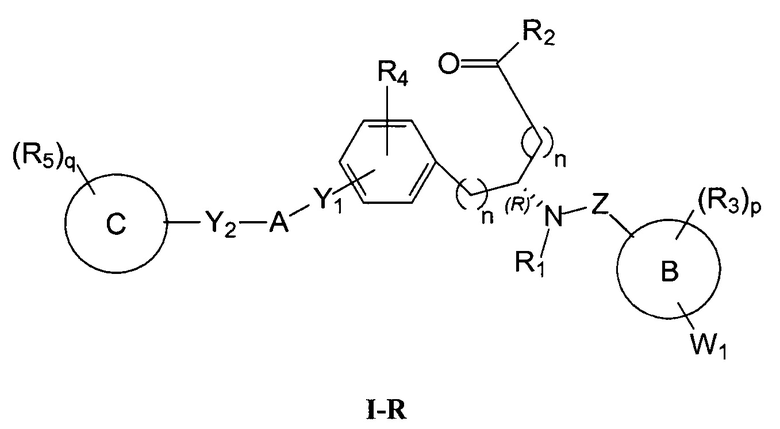
где
А представляет собой 5-, 6- или 7-членный гетероарил, содержащий один, два или три гетероатома, где каждый такой гетероатом независимо выбран из О, N и S, выбранный из имидазола, оксадиазола, тиадиазола, тиазола, оксазола, пиридина, пиримидина, пиридазина, триазина;
В представляет собой гетероциклил, представляющий собой 5-, 6- или 7-членный гетероциклил, содержащий один, два или три гетероатома, выбранных из О, N и S, и выбранный из изоксазола, оксазола, пиримидина, пиразола, тиазола, тиофенила, тиадиазола, азепина, необязательно конденсированного с другим азотсодержащим 6-членным гетероциклическим кольцом;
С представляет собой арил, выбранный из фенила и нафтила, бензил;
обе Y1 и Y2 являются отсутствующими;
Z представляет собой -С(О)-;
каждый R1 независимо представляет собой Н или С1-4алкил;
R2 представляет собой -O-R8, -N(R1)-SO2-R8, -NR41R42, -N(R1)-(CRaRb)m-COOH, -N(R1)-(CRaRb)m-CO-N(R1)-гетероциклил, -N(R1)-(CRaRb)m-CO-N(R1)(R7) или -N(R1)-гетероциклил, где R2 не является -ОН или -NH2, и гетероциклил представляет собой 5-6-членный гетероциклил, содержащий 1-3 гетероатома, выбранных из N и S;
каждый R3 и R4 независимо представляет собой галоген, C1-С4алкил, С1-С4алкил, замещенный R31, С1-С4алкокси, галогенС1-С4алкил, пергалогенС1-С4алкил, галогенС1-С4алкокси, -ОН, -NR1R8, -C(O)R8, -C(O)NR1R8, -S(O)2R8, -OS(O)2R8, -(CRaRb)mNR1R8, -(CRaRb)mO(CRaRb)mR8; или любые две R3 или R4 группы на одном и том же атоме углерода, взятые вместе, образуют оксо,
каждый R31 независимо представляет собой Н, галоген, гидроксил;
каждый R40 независимо представляет собой Н или С1-С4алкил;
каждый R41 и R42 независимо представляет собой R40 или -(СН2)n-COO-R40, или два, взятые вместе с N атомом, с которым они соединены, могут образовывать 5-6-членный гетероциклил;
W1 является отсутствующей;
каждый Ra и Rb независимо представляет собой Н, С1-С4алкил, С1-С4алкокси, фенил, 5-6-членный гетероциклил или 5-6-членный гетероциклилалкил, содержащий 1-2 гетероатома, выбранные из N, О и S, причем любой из данных алкилов, алкокси, фенила, гетероциклилов или гетероциклилалкилов может быть необязательно (одним-тремя заместителями) замещен R7, или -(СН2)mC(О)OR40, -(CH2)mOR40, -(СН2)mNR41R42, -(СН2)mC(О)NR41R42; или любые два Ra и Rb, взятые вместе с углеродом, с которым они соединены, образуют С3-С6циклоалкил или 5-6-членный насыщенный гетероциклил, содержащий N и О в качестве гетероатомов;
R5 представляет собой R7, -(СН2)m-L2-(СН2)m-R7 или -(-L3-(CRaRb)r-)s-L3-R7;
R7 представляет собой Н, галоген, С1-С6алкил, галогенС1-С6алкил, пергалогенС1-С6алкил, С1-С4алкокси, -ОН, -OR8, -NR1R8, -(CRaRb)mO(CRaRb)mR8, -NR1(CRaRb)mR8, -C(O)R8, -NR1(CRaRb)mCOOH, -C(O)NR1R8, -SR8, -S(O)2R8, -S(O)2NR1R8; или кольцевую группу, выбранную из С3-С6циклоалкила, фенила, где каждая кольцевая группа необязательно однократно или несколько раз замещена галогеном, -ОН, -CN, С1-С4алкилом, С1-С4алкокси;
каждая R8 независимо представляет собой Н, С1-С4алкил, С3-С6циклоалкил или фенил;
L2 независимо является, от ближнего к дальнему концу структуры формулы I-R или I-S, отсутствующей или -О-;
каждая L3 независимо является отсутствующей, -О- или -N(R1)-,
каждый m независимо равен 0, 1, 2, 3, 4, 5 или 6;
каждый n независимо равен 0, или 1, или 2;
p равен 0, 1, 2 или 3;
q равен 0, 1, 2 или 3.
2. Соединение по п. 1, где А представляет собой 5- или 6-членную гетероарильную группу.
3. Соединение по п. 2, где соединение имеет следующую структуру
 ,
,
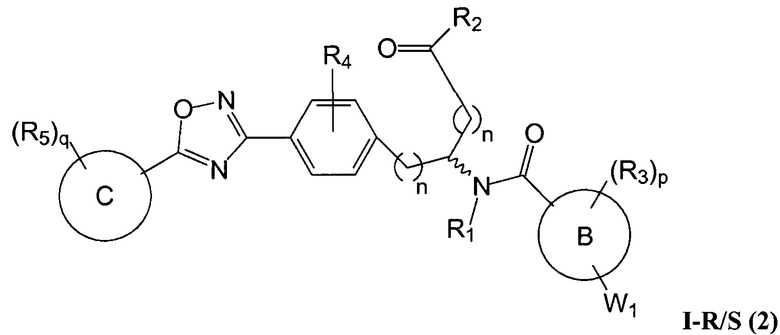 ,
,
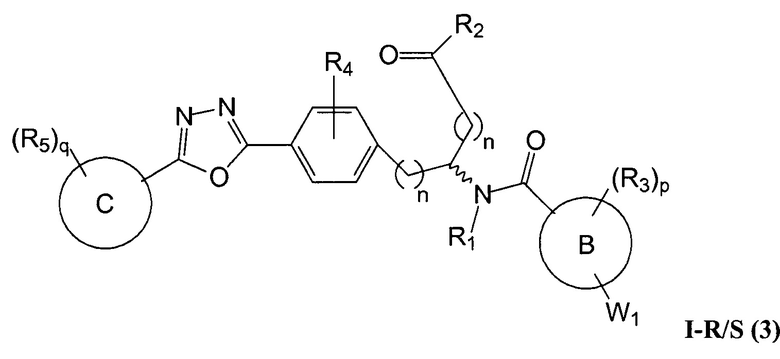 ,
,
 ,
,
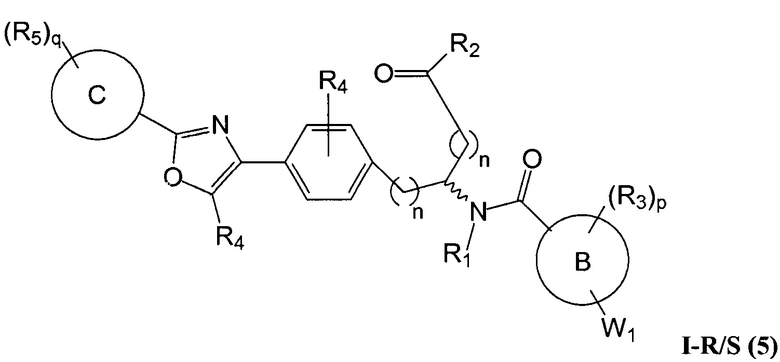 ,
,
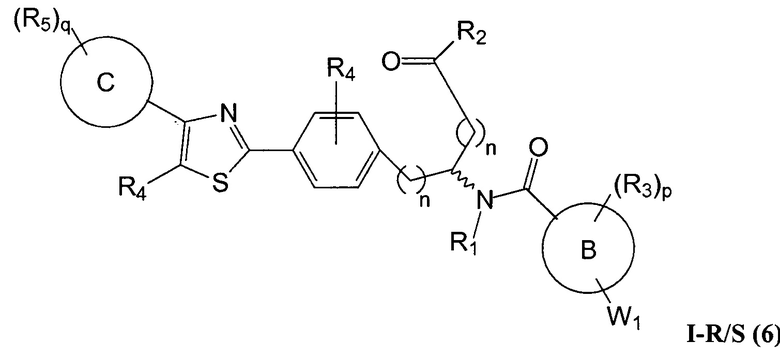 ,
,
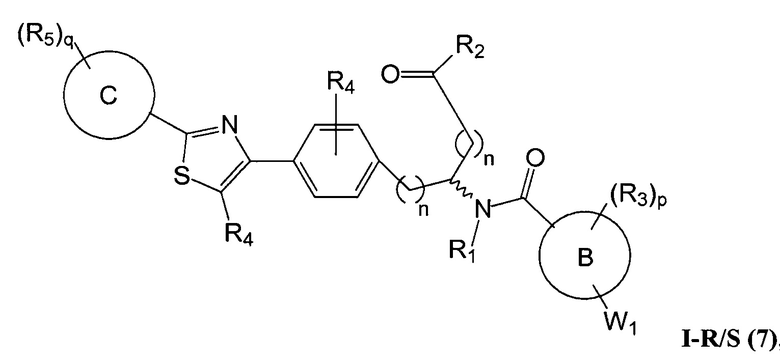 ,
,
 ,
,
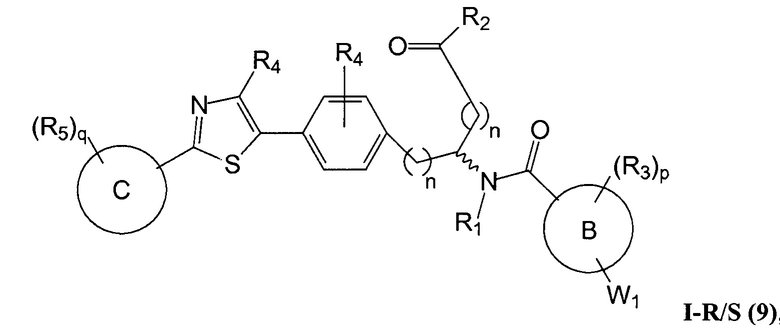 ,
,
 ,
,
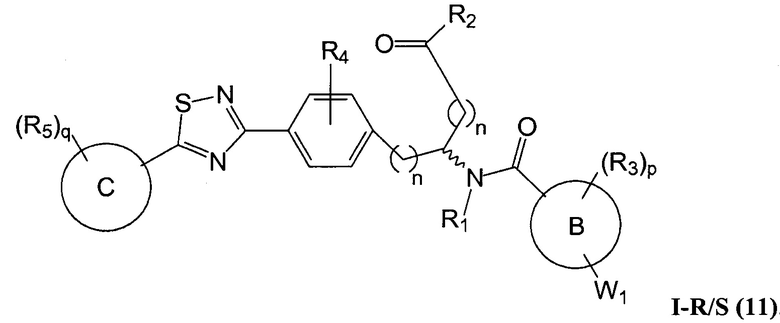 ,
,
 ,
,
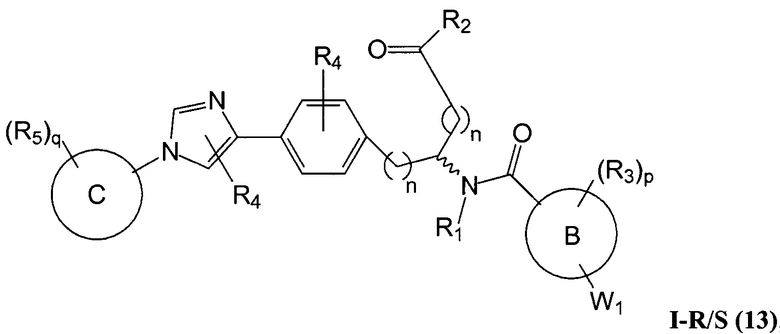 ,
,
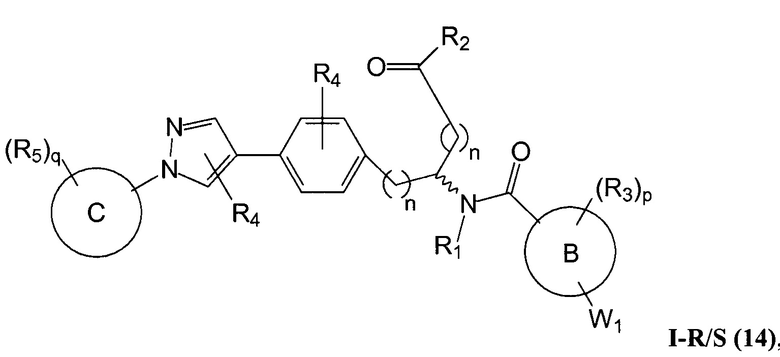 ,
,
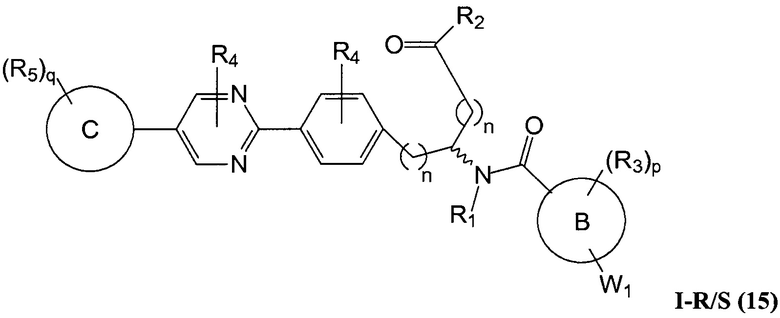 ,
,
 ,
,
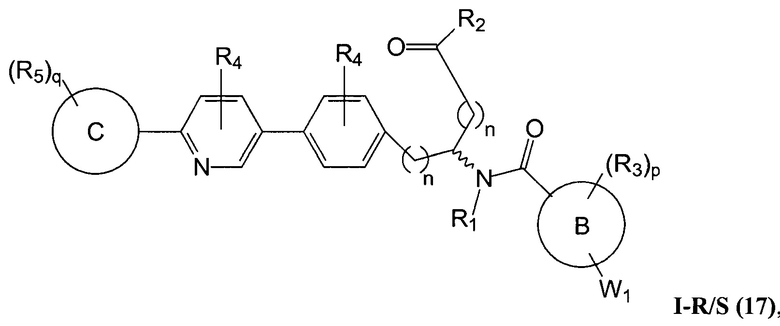 ,
,
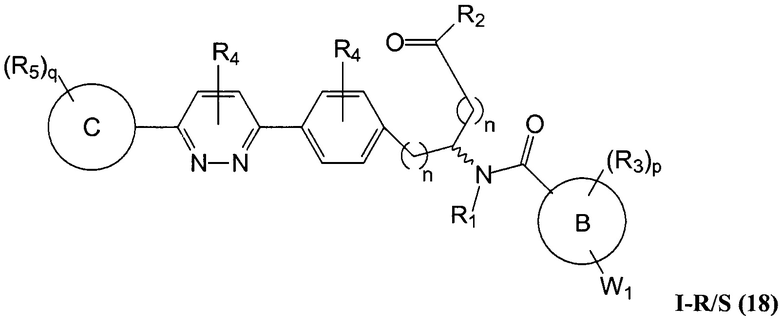 ,
,
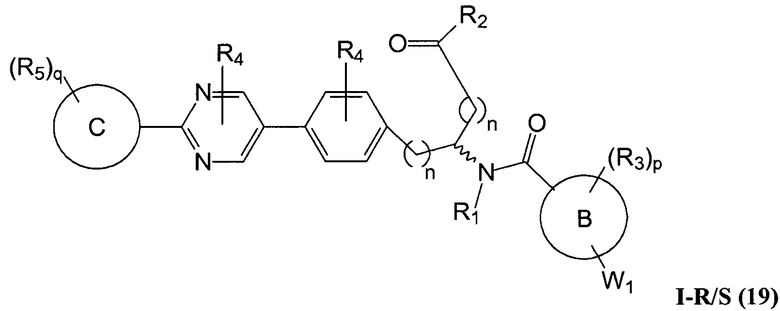 ,
,
 ,
,
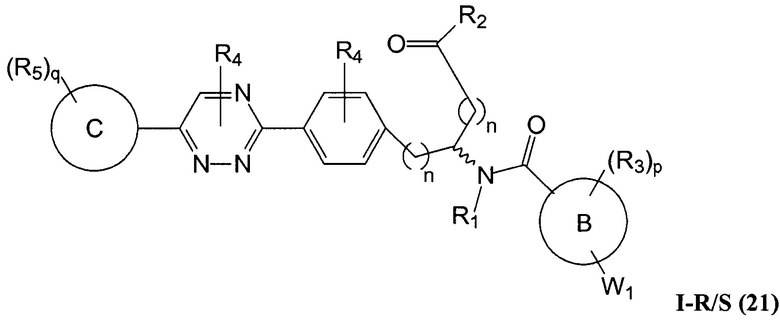 ,
,
или
 .
.
4. Соединение по п. 1, где С представляет собой арил.
5. Соединение по п. 4, где соединение имеет следующую структуру
 ,
,
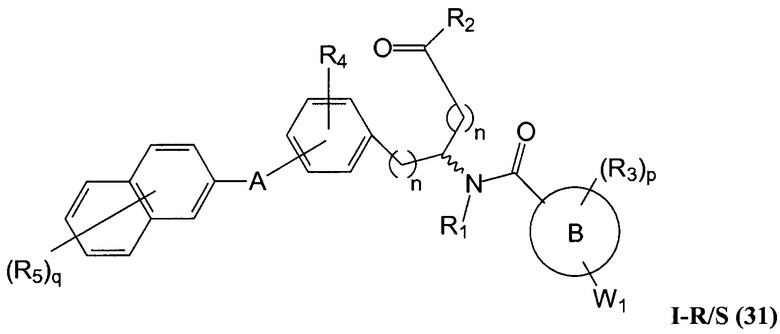 или
или
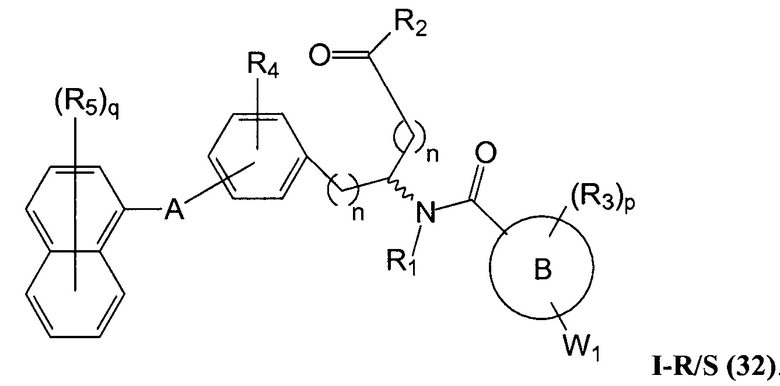 .
.
6. Соединение по п. 1, где соединение имеет следующую структуру
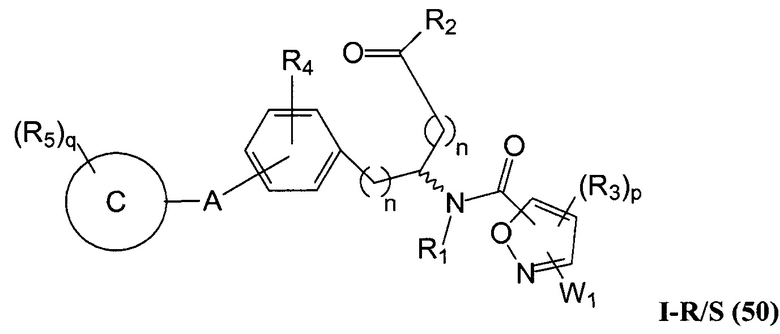 ,
,
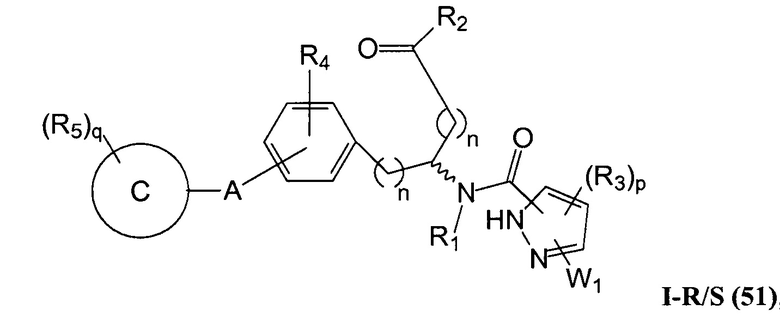 ,
,
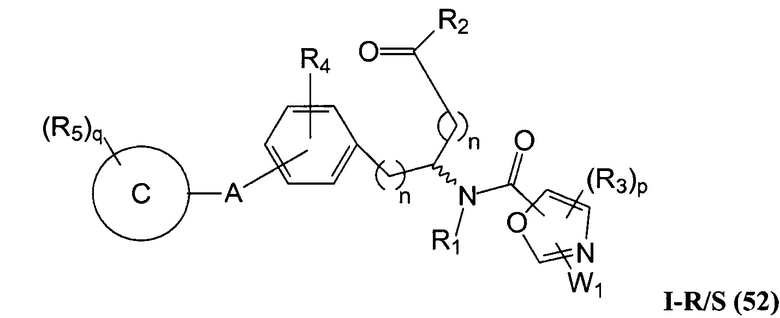 ,
,
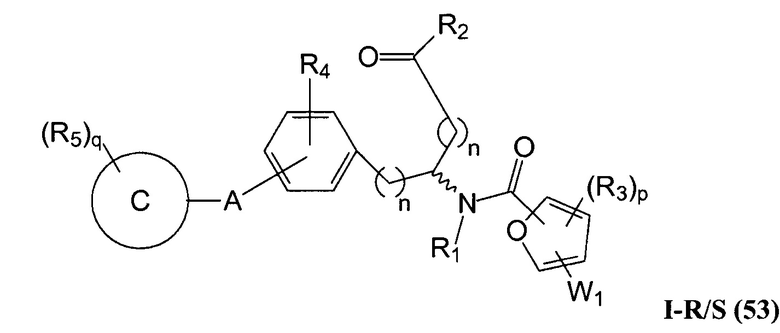 ,
,
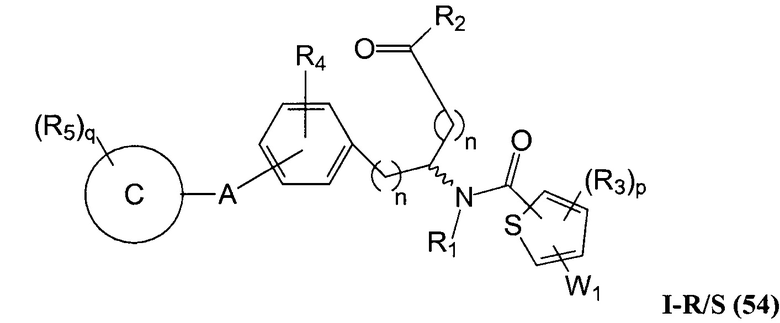 ,
,
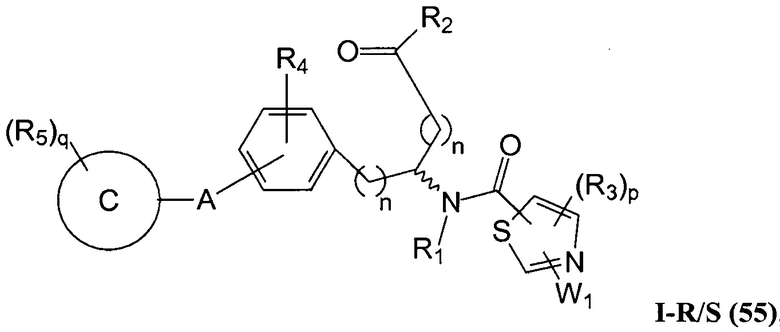 ,
,
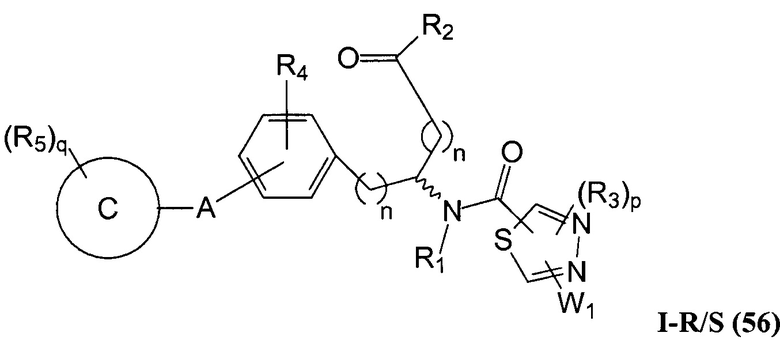 ,
,
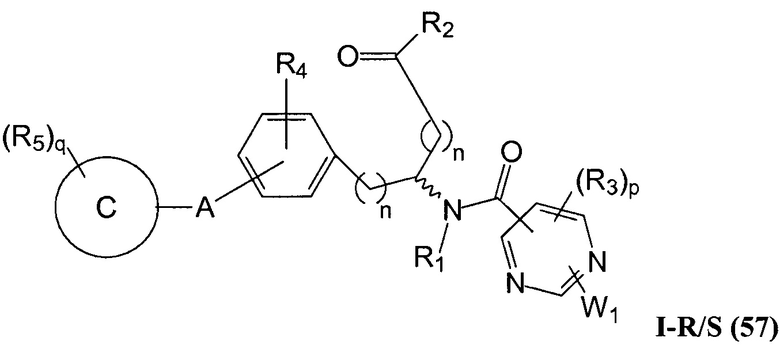 ,
,
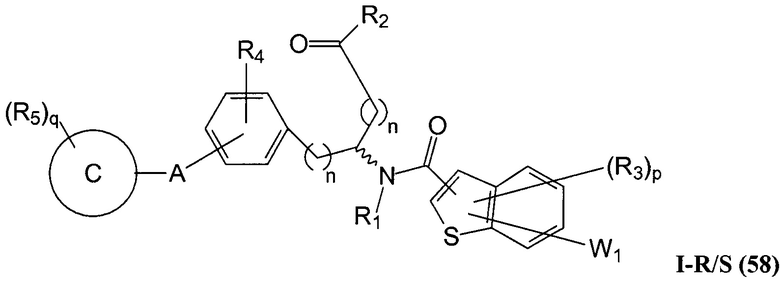 ,
,
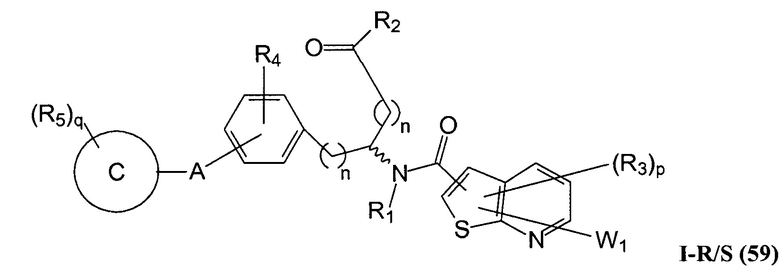 ,
,
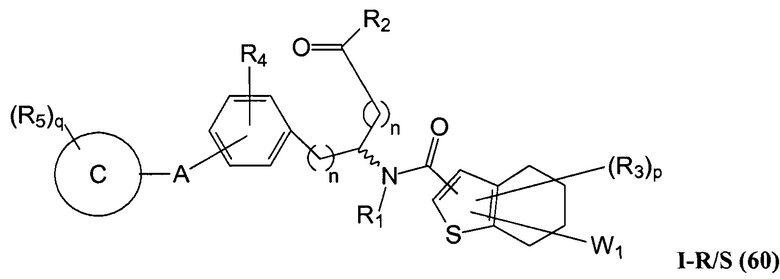 ,
,
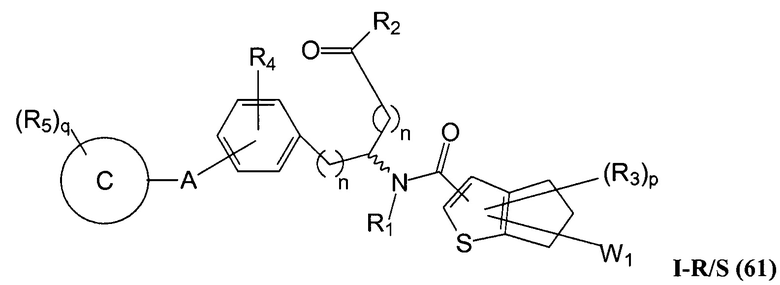 или
или
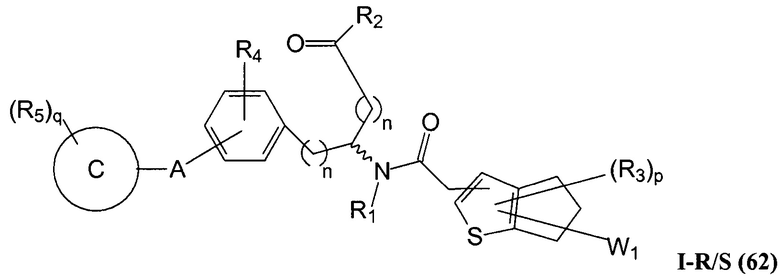 .
.
7. Фармацевтическая композиция, обладающая активностью в отношении глюкагонподобного пептида 1 (GLP-1R), содержащая соединение по п. 1 вместе, по меньшей мере, с одним фармацевтически приемлемым носителем, разбавителем или эксципиентом.
8. Фармацевтическая комбинация для снижения глюкоз, содержащая соединение по п. 1 и второе лекарственное средство, представляющее собой эксенатид.
| WO 2011094890 A1, 11.08.2011 | |||
| Установка для мойки деталей | 1987 |
|
SU1477482A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| WO 9910312 A1.04.03.1999 & RU 2220950 C2, 10.01.2004 | |||
| МОЛОТ ДЛЯ ДИНАМИЧЕСКОЙ ШТАМПОВКИ | 1989 |
|
SU1700850A1 |
| WO 2005077915 A1, 25.08.2005 | |||
| DATABASE CA [Online] Chemical Abstracts Service, COLUMBUS,OHIO,US;THORSETT, EUGENE D | |||
| et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| US 2005222141 A1, 06.10.2005 & RU 2390520 C2, 27.05.2010 | |||
| WO 9906437 A1, 11.02.1999. | |||

