Область техники
Настоящее изобретение относится к соединениям феноксикарбоновой кислоты, предназначенным для доставки активных веществ, таких как биологически или химически активные вещества, к тканям-мишеням. Указанные соединения пригодны для образования нековалентных смесей с активными веществами, предназначенными для перорального, внутрикишечного, легочного и других способов введения лекарственных средств животным. Данное изобретение относится также к способам получения и введения таких композиций.
Предпосылки изобретения
Применение известных средств доставки активных веществ к тканям часто ограничено биологическими, химическими и физическими барьерами. Указанные барьеры обычно создаются средой, в которой производится доставка, средой, окружающей ткани-мишени, куда осуществляется доставка, и/или самими тканями-мишенями. Биологически и химически активные вещества особенно чувствительны к таким барьерам.
В организме животного существуют различные барьеры, препятствующие проникновению биологически и химически активных фармакологических и лекарственных средств. В качестве примеров физических барьеров можно привести кожу, двойные липидные слои и оболочки различных органов, которые являются относительно непроницаемыми для некоторых активных веществ, но должны быть преодолены на пути к мишеням, таким как сердечно-сосудистая система. Химические барьеры включают, но не ограничиваются ими, разные значения рН в желудочно-кишечном тракте (GI) и расщепляющие ферменты.
Преодоление указанных барьеров особенно важно при создании пероральных систем доставки лекарственных средств. Пероральное введение многих биологически или химически активных веществ является предпочтительным способом введения лекарственных средств животным, если ему не препятствуют биологические, химические и физические барьеры. К многочисленным средствам, которые обычно непригодны для перорального введения, относятся биологически или химически активные пептиды, такие как кальцитонин и инсулин; полисахариды и, в частности, мукополисахариды, включающие, но не ограничивающиеся ими, гепарин; гепариноподобные вещества; антибиотики и другие органические вещества. Указанные средства быстро становятся неэффективными или разрушаются в желудочно-кишечном тракте под действием кислотного гидролиза, ферментов и тому подобное. Кроме того, всасыванию могут препятствовать размер и структура макромолекулярных лекарственных средств.
Нестойкие фармацевтические средства ранее вводили вместе с адъювантами (например, резорцинами и неионогенными поверхностно-активными веществами, такими как полиоксиэтилен-олеиловый эфир и н-гексадецилполиэтиленовый эфир), позволяющими искусственно улучшить проницаемость стенок кишечника, а также с ингибиторами ферментов (например, ингибиторы трипсина поджелудочной железы, диизопропил-фторфосфат (DFF) и тразилол), позволяющими ингибировать ферментативное расщепление. В качестве систем доставки, инсулина и гепарина используют также липосомы. Однако спектр действия таких систем доставки лекарственных средств весьма ограничен по следующим причинам: (1) необходимость использовать адъюванты или ингибиторы в токсических количествах; (2) отсутствие приемлемых низкомолекулярных активных веществ; (3) плохая стабильность и недостаточный срок годности; (4) сложность изготовления; (5) неспособность таких систем защитить активное вещество; (6) неблагоприятное изменение активного вещества под действием системы; или (7) неспособность таких систем стимулировать всасывание активного вещества.
Для доставки фармацевтических средств недавно было предложено использовать белковые микросферы. См., например, патенты США №№ 5401516, 5443841 и заменяющий патент 35862. Кроме того, для доставки лекарственных средств используют некоторые модифицированные аминокислоты. См., например, патенты США №№ 5629020, 5643957, 5766633, 5776888 и 5866536.
Однако по-прежнему существует потребность в простых и дешевых системах доставки лекарственных средств, которые можно легко изготовить и которые обеспечивают доставку широкого спектра активных веществ при различных способах введения.
Краткое изложение существа изобретения
Настоящее изобретение относится к соединениям, облегчающим доставку активных веществ, и/или композициям для доставки активных веществ к тканям организма нуждающегося в этом животного, а также к применению вышеуказанных соединений для доставки активных веществ к тканям организма нуждающегося в этом животного. Соединения для доставки активных веществ к тканям организма нуждающегося в этом животного согласно настоящему изобретению включают соединения нижеследующей формулы:
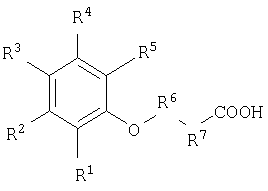
Где:
* Термин «napa-Ph» означает парафенилен.
Более предпочтительные соединения без ограничения включают соединения №№5, 7, 11, 12, 43 и 75.
Данное изобретение относится также к композиции, предназначенной для доставки активных веществ к тканям организма нуждающегося в этом животного, которая содержит по меньшей мере одно из ниже приведенных соединений, и по меньшей мере одно активное вещество. В частности, указанное композиция содержит по меньшей мере одно из соединений формулы
выбранное из группы, включающей:
и их фармацевтически приемлемые соли.
Указанные композиции доставляют активные вещества в выбранные биологические системы, обеспечивая более высокую или улучшенную биологическую доступность активного вещества по сравнению с введением активного вещества без соединения доставляющего агента. Предпочтительным соединением для применения в вышеуказанной композиции являются соединения №11, 12, 43, 47 и 75 и их фармацевтически приемлемые соли.
Настоящее изобретение относится также к стандартным (унифицированным) лекарственным формам, содержащим указанные композиции. Стандартная (унифицированная) лекарственная форма может быть жидкой или твердой, такой как таблетка, капсула или частицы, включая порошок или саше.
Другой вариант реализации изобретения относится к способу введения активного вещества нуждающемуся в этом животному в виде композиции, содержащей по меньшей мере одно соединение доставляющего агента вышеуказанной формулы, включая соединения, исключенные оговоренными условиями, и активное вещество. Предпочтительные способы введения включают пероральный, внутрикишечный и легочный способы.
Другой вариант реализации изобретения относится к способу лечения заболевания или достижения требуемого физиологического эффекта у животного в результате введения композиции по настоящему изобретению.
Другой вариант реализации изобретения относится к способу получения композиции по настоящему изобретению путем смешивания по меньшей мере одного соединения доставляющего агента вышеуказанной формулы, включая соединения, исключенные оговоренными условиями, и по меньшей мере одного активного вещества.
Еще один вариант реализации настоящего изобретения относится к применению соединения для доставки активных веществ к тканям организма нуждающегося в этом животного, где указанное соединение, имеет следующую формулу
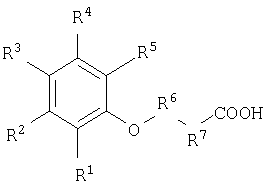
и выбрано из группы, включающей:
и их фармацевтически приемлемые соли.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения доставляющих агентов
Термины «алкил» и «алкенил» в используемом здесь значении означают соответственно алкильные и алкенильные заместители с линейной и разветвленной цепью.
Соединения доставляющих агентов могут представлять собой карбоновую кислоту или ее соли. Приемлемые соли включают, но не ограничиваются ими, органические и неорганические соли, например соли щелочных металлов, таких как натрий, калий и литий; соли щелочно-земельных металлов, таких как магний, кальций или барий; соли аммония; основные аминокислоты, такие как лизин или аргинин; и органические амины, такие как диметиламин или пиридин. Предпочтительными солями являются соли натрия. Указанные соли могут быть одно- или многовалентными солями, такими как мононатриевые и динатриевые соли. Предпочтительной динатриевой солью является данатриевая соль соединения 47. Данные соли могут быть также сольватами, включая сольваты этанола, и гидратами.
Соли соединений доставляющих агентов по настоящему изобретению можно получить способами, известными в данной области. Например, натриевые соли можно получить, растворяя соединение доставляющего агента в этаноле и добавляя водный раствор гидроксида натрия.
Соединение доставляющего агента можно очистить перекристаллизацией или фракционированием на одном или нескольких твердых хроматографических носителях, используемых отдельно или соединенных последовательно. Приемлемые системы растворителей для перекристаллизации включают, но не ограничиваются ими, ацетонитрил, метанол и тетрагидрофуран. Фракционирование может быть выполнено на приемлемом хроматографическом носителе, таком как оксид алюминия, используя смеси метанол/н-пропанол в качестве подвижной фазы; хроматографией с обращенной фазой, используя смеси трифторуксусная кислота/ацетонитрил в качестве подвижной фазы; и ионообменной хроматографией, используя воду или приемлемый буфер в качестве подвижной фазы. При выполнении анионообменной хроматографии предпочтительно используют градиент 0-500 мМ хлорида натрия.
Активные вещества
Активными веществами, пригодными для использования в настоящем изобретении, являются биологически и химически активные вещества, которые включают, но не ограничиваются ими, пестициды, фармакологические и лекарственные средства.
Например, биологически или химически активные вещества, пригодные для использования в настоящем изобретении, включают, но не ограничиваются ими, белки; полипептиды; пептиды; гормоны; полисахариды и, в частности, смеси мукополисахаридов; углеводы; липиды; небольшие полярные органические молекулы (то есть полярные органические молекулы с молекулярной массой 500 Дальтон или менее); другие органические соединения; и, в частности, соединения, которые сами по себе не способны (или способны лишь частично) проникать сквозь оболочку желудочно-кишечного тракта и/или подвержены химическому расщеплению кислотами и ферментами в желудочно-кишечном тракте; либо комбинации указанных веществ.
Другие примеры включают, но не ограничиваются ими, синтетические, природные или рекомбинантные источники нижеследующих соединений, таких как гормоны роста, включая гормоны роста человека (hGH), рекомбинантные гормоны роста человека (rhGH), бычьи гормоны роста и гормоны роста свиней; гормоны, стимулирующие выделение гормона роста; интерфероны, включая α, β и γ; интерлейкин-1; интерлейкин-2; инсулин, включая инсулин свиней, бычий инсулин, инсулин человека и рекомбинантный инсулин человека, необязательно имеющий противоионы цинка, натрия, кальция и аммония; инсулиноподобный фактор роста, включая IGF-1; гепарин, включая нефракционированный гепарин, гепариноподобные вещества, дерматаны, хондроитины, низкомолекулярный гепарин, очень низкомолекулярный гепарин и ультранизкомолекулярный гепарин; кальцитонин, включая кальцитонин лосося, угря, свиньи и человека; эритропоэтин; естественный фактор предсердия; антигены; моноклональные антитела; соматостатин; ингибиторы протеазы; адренокортикотропин; гормон, стимулирующий выделение гонадотропина; окситоцин; гормон, стимулирующий выделение лютеинизирующего гормона; фолликулостимулирующий гормон; глюкоцереброзидазу; тромбопоэтин; филграстим; простагландины; циклоспорин; вазопрессин; кромолиннатрий (натрий- или динатрийхромогликат); ванкомицин; десферриоксамин (DFO); бисфосфонаты, включая алендронат, тилудронат, этидронат, клодронат, памидронат, олпадронат и инкадронат; паращитовидный гормон (РТН), включая его фрагменты; противомикробные средства, включая антибиотики, антибактериальные средства и противогрибковые средства; витамины; аналоги, фрагменты, миметики или модифицированные полиэтиленгликолем (PEG) производные указанных соединений; или любые их комбинации. Неограничивающие примеры антибиотиков включают воздействующие на грамположительные бактерии, бактерицидные, липопептидные и циклические пептидные антибиотики, такие как даптомицин и его аналоги.
Предпочтительным активным веществом является даптомицин. Даптомицин описан в публикации Baltz, Biotechnology of Antibiotics, 2nd Ed., ed, W.R. Strohl (New York: Marcel Dekker, Inc.), 1997, pp. 415-435. Даптомицин представляет собой циклический липопептидный антибиотик, который может быть получен ферментацией Streptomyces roseosporus. Даптомицин является членом семейства антибиотиков фактора А-21978С0 S. roseosporus и имеет н-деканоильную боковую цепь, связанную цепью из трех аминокислот с N-концевым триптофаном циклического пептида, состоящего из 10 аминокислот. В настоящее время указанное соединение используется в различных составах для лечения серьезных инфекций, вызываемых бактериями, которые включают, но не ограничиваются ими, устойчивые к метициллину Staphylococcus aureus (MRSA) и устойчивые к ванкомицину энтерококки (VRE). Способы синтеза даптомицина описаны в патентах США №№ 32333, 32455, 5800157, 4885243, 32310, 32311, 4537717, 4482487 и 4524135.
Системы доставки активного вещества к тканям
Композиция по настоящему изобретению содержит одно или несколько соединений доставляющих агентов по данному изобретению, включая соединения, исключенные оговоренными условиями, и одно или несколько активных веществ. Соединение доставляющего агента и активное вещество обычно смешивают до введения, получая таким образом предназначенную для введения композицию.
Предпочтительные комбинации соединений доставляющих агентов и активных веществ включают, но не ограничиваются ими, соединение 12 и кальцитонин, в частности кальцитонин лосося; соединение 12 и гепарин; соединение 5 и кальцитонин, в частности кальцитонин лосося; любое из соединений 7, 11 и 43 и даптомицин; соединение 7 и кромолин, в частности кромолиннатрий; и соединение 47 и гормон роста человека.
Предназначенные для введения композиции могут быть жидкостями. В качестве растворяющей среды можно использовать воду (например, для кальцитонина лосося, паращитовидного гормона и эритропоэтина), 25% водный раствор пропиленгликоля (например, для гепарина) и фосфатный буфер (например, для rhGH). Еще одним растворителем является полиэтиленгликоль. Лекарственные растворы получают, смешивая раствор соединения доставляющего агента с раствором активного вещества непосредственно перед введением. Альтернативно, раствор соединения доставляющего агента (или активного вещества) можно смешивать с активным веществом (или соединением доставляющего агента) в твердой форме. Соединение доставляющего агента и активное вещество можно также смешивать в виде сухих порошков. Соединение доставляющего агента и активное вещество можно смешивать в процессе изготовления лекарственного средства.
Лекарственные растворы могут необязательно содержать добавки, такие как соли фосфатного буфера, лимонную кислоту, гликоли или другие диспергаторы. В указанный раствор можно вводить стабилизирующие добавки, предпочтительно в концентрации от 0,1 до 20% (мас./об.).
Предназначенные для введения композиции альтернативно могут быть получены в твердой форме, такой как таблетка, капсула или частицы, например порошок или саше. Твердые лекарственные формы можно получить, смешивая твердое соединение доставляющего агента с твердым активным веществом. Альтернативно, твердое вещество можно получить из раствора соединения доставляющего агента и активного вещества способами, известными в данной области, такими как сушка вымораживанием (лиофилизация), осаждение, кристаллизация и диспергирование в твердом состоянии.
Предназначенные для введения композиции по настоящему изобретению могут также включать один или несколько ингибиторов ферментов. Указанные ингибиторы ферментов включают, но не ограничиваются ими, такие соединения, как актинонин или эпиактинонин и их производные. Другие ингибиторы ферментов включают, но не ограничиваются ими, апротинин (тразилол) и ингибитор Боумана-Бирка.
Количество активного вещества в предназначенной для введения композиции по настоящему изобретению должно обеспечивать эффективное воздействие конкретного активного вещества на ткань-мишень. Количество активного вещества в указанных композициях обычно является фармакологически, биологически, терапевтически или химически эффективным количеством. Однако фактическое количество может быть меньше указанного количества при использовании данной композиции для изготовления стандартной лекарственной формы, так как такая лекарственная форма может содержать несколько композиций соединения доставляющего агента/активного вещества или разделенную дозу фармакологически, биологически, терапевтически или химически эффективного количества. В данном случае общее эффективное количество вводят в виде кумулятивных (суммарных) доз, содержащих эффективное количество активного вещества.
Общее количество активного вещества можно определить способами, известными специалистам в данной области. Однако благодаря тому что композиции по данному изобретению обеспечивают более эффективную доставку активных веществ, чем композиции, содержащие только активное вещество, нуждающемуся субъекту можно вводить более низкие дозы биологически или химически активных веществ по сравнению с ранее использовавшимися лекарственными формами или системами доставки лекарственного средства при достижении таких же уровней в крови и/или аналогичного лечебного действия.
Описываемые соединения доставляющих агентов облегчают доставку биологически и химически активных веществ, в частности, при пероральном, назальном, подъязычном, интрадуоденальном, подкожном, трансбуккальном, внутрикишечном, ректальном, вагинальном, слизистом, легочном, чрескожном, внутрикожном, парентеральном, внутривенном, внутримышечном и глазном введении, а также преодоление гематоэнцефалического барьера.
Стандартные (унифицированные) лекарственные формы могут также содержать используемые отдельно или в комбинации наполнители, разбавители, дезинтеграторы, смазывающие вещества, пластификаторы, красители, ароматизаторы, маскирующие вкус вещества, сахара, подслащивающие вещества, соли и растворители, включающие, но не ограничивающиеся ими, воду, 1,2-пропандиол, этанол, оливковое масло или любые их комбинации.
Соединения и композиции по настоящему изобретению пригодны для введения биологически или химически активных веществ любым животным, включая, но не ограничиваясь ими, птиц, таких как куры; млекопитающих, таких как грызуны, крупный рогатый скот, свиньи, собаки, кошки, приматы и, в частности, человек; а также насекомых.
Указанная система является особенно пригодной для введения таких химически или биологически активных веществ, которые в противном случае были бы разрушены или стали менее эффективными под действием условий, существующих в организме животного, прежде чем активное вещество достигнет участка действия (то есть участка, где должно происходить высвобождение активного вещества из композиции, используемой для его доставки). Соединения и композиции по настоящему изобретению пригодны для перорального введения активных веществ, которые обычно не предназначены для такого способа введения или требуют более эффективной доставки.
Композиции, содержащие указанные соединения и активные вещества, обеспечивают эффективную доставку активных веществ к выбранным биологическим системам и более высокую или лучшую биологическую доступность активного вещества по сравнению с введением данного активного вещества без доставляющего агента. Действие лекарственного средства можно улучшить, доставляя большее количество активного вещества в течение определенного периода времени или доставляя активное вещество в требуемый период времени (то есть осуществляя более быструю или медленную доставку) или через определенный период времени (то есть осуществляя пролонгированную доставку).
Другой вариант осуществления изобретения относится к способу лечения или профилактики заболевания, а также достижения требуемого физиологического действия, указанного в нижеследующей таблице, благодаря введению животному композиции по настоящему изобретению. Специальные показания для использования активных веществ представлены в справочнике Physicians′ Desk Reference (54th Ed., 2000, Medical Economics Company, Inc., Montvale, NJ), который включен в данное описание изобретения в качестве ссылки. Активные вещества, приведенные в нижеследующей таблице, включают их аналоги, фрагменты, миметики и производные, модифицированные полиэтиленгликолем.
Один вариант осуществления настоящего изобретения относится, например, к способу лечения субъекта, страдающего или подверженного диабету, путем введения инсулина и по меньшей мере одного из соединений доставляющих агентов по настоящему изобретению.
После введения лекарственного препарата активное вещество, присутствующее в композиции или стандартной унифицированной лекарственной форме, всасывается в систему кровообращения. Биологическую доступность активного вещества можно легко оценить, определяя известную фармакологическую активность в крови, например увеличение времени свертывания крови, вызываемое гепарином, или уменьшение уровней кальция в крови, вызываемое кальцитонином. Альтернативно, можно произвести прямое измерение уровней активного вещества в крови.
Описание предпочтительных вариантов осуществления изобретения
Приведенные ниже примеры иллюстрируют данное изобретение, не ограничивая его объем. Все части являются массовыми частями, за исключением особо оговоренных случаев.
Нижеследующие соединения анализируют спектроскопией протонного ядерного магнитного резонанса (1Н ЯМР), которую выполняют при помощи спектрометра Брукера 300 МГц, используя в качестве растворителя диметилсульфоксид (ДМСО-d6), за исключением особо оговоренных случаев.
Пример 1. Получение соединений
Получение соединения 1
Гидроксид калия (8,82 г, 157,2 ммоль) измельчают в ступке до порошкообразного состояния и переносят в 125 мл колбу Эрленмейера, содержащую 60 мл диметилсульфоксида. Полученную смесь перемешивают в течение 5 минут, после чего добавляют 5,35 г (39,3 ммоль) 2′-гидроксиацетофенона. Смесь перемешивают еще 15 минут и добавляют 5,39 г (25,1 ммоль) 4-(бромметил)бензойной кислоты. Реакционную смесь перемешивают при комнатной температуре в течение примерно четырех часов. К коричневой реакционной смеси добавляют дистиллированную воду (200 мл) и полученный раствор охлаждают до 0°С. Затем добавляют концентрированный водный раствор соляной кислоты до достижения рН раствора около 5. Образовавшееся твердое вещество собирают фильтрованием и перекристаллизовывают из смеси этанол:вода (50:50), получая 3,59 г (52,9%) светло-коричневого порошка. Температура плавления: 170,5-172,0°С. Анализ сжиганием: %С: 71,10 (вычислено), 70,81 (найдено); %Н: 5,22 (вычислено), 5,25 (найдено). 1H ЯМР (ДМСО-d6): δ 13,0, с, 1Н; 8,00-7,97, д, 2Н; 7,64-7,59, м, 3Н; 7,55-7,49, дт, 1Н; 7,25-7,22, д, 1Н; 7,07-7,01, дт, 1Н; 5,33, с, 2Н; 2,54, с, ЗН.
Соединения 63, 62 и 64 получают вышеописанным способом, используя соответствующие исходные вещества.
Соединение 63. Температура плавления: 91-94°С. Анализ сжиганием: %С: 69,62 (вычислено), 69,91 (найдено); %Н: 8,53 (вычислено), 8,28 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, шир.с, 1Н; 7,9, д, 2Н; 7,0, д, 2Н; 4,0, т, 2Н; 3,0, kb, 2H; 2,2, т, 2Н; δ 1,7, п, 2H; 1,5, п, 2H; 1,35, м, 6Н; 1,05, т, 3Н.
Соединение 62. Температура плавления: 125-129°С. Анализ сжиганием: %С: 69,04 (вычислено), 68,91 (найдено); %Н: 7,97 (вычислено), 8,04 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, шир.с, 1Н; 7,9, д, 2H; 7,02, д, 2H; 4,01, т, 2H; 2,52, с, 3Н; 2,23, т, 2Н; 1,7, п, 2Н; 1,5, п, 2Н; 1,38, м, 6Н.
Соединение 64. Температура плавления: 62-65°С. Анализ сжиганием: %С: 69,06 (вычислено), 69,32 (найдено); %Н: 7,91 (вычислено), 7,97 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,5, д, 1Н; 7,4, м, 2Н; 7,19, дд, 1Н; 4,02, т, 2Н; 2,55, с, 3Н; 2,2, т, 2Н; 1,7, п, 2Н; 1,5, п, 2Н; 1,3, м, 6Н.
Соединения 66 и 52 также получают аналогично способу получения соединения 1, заменяя 2′-гидроксиацетофенон указанным в скобках соединением: 66 (фенол) и 52 (салициламид).
Соединение 66. Температура плавления: 219-221°С. Анализ сжиганием: %С: 73,67 (вычислено), 73,70 (найдено); %Н: 5,30 (вычислено), 5,22 (найдено). 1H ЯМР (ДМСО-d6): δ 13,0, с, 1Н; 7,97, д, 2Н; 7,57, д, 2Н; 7,30, м, 2Н; 7,01, м, 2Н; 6,95, м, 1Н; 5,19, с, 2Н.
Соединение 52. Температура плавления: 242-243°С. Анализ сжиганием: %С: 66,08 (вычислено), 65,74 (найдено); %Н: 4,86 (вычислено), 4,79 (найдено); %N: 5,14 (вычислено), 4,78 (найдено) 1H ЯМР (ДМСО-d6): δ 13,0, с, 1Н; 7,97, д, 2Н; 7,75, дд, 1Н; 7,64, шир.с, 1Н; 7,62, д, 2Н; 7,56, шир.с, 1Н; 7,44, дт, 1Н; 7,17, д, 1Н; 7,03, т, 1Н; 5,35, с, 2Н.
Получение соединения 2
Гидроксид калия (9,88 г, 176 ммоль) измельчают в ступке до порошкообразного состояния и переносят в 125 мл колбу Эрленмейера, содержащую 80 мл диметилсульфоксида и 5,54 г (50,3 ммоль) катехина. Полученную смесь перемешивают в течение 45 минут, слегка нагревая до 35°С. Темную смесь обрабатывают раствором 6,94 г (40,7 ммоль) 4-(хлорметил)бензойной кислоты и 30 мл диметилсульфоксида. Реакционную смесь перемешивают при комнатной температуре в течение примерно 17 часов. Смесь подкисляют 4% водным раствором соляной кислоты, что вызывает образование твердого вещества. Твердое вещество собирают фильтрованием. Полученный продукт перекристаллизовывают из смеси этилацетат/метил трет-бутиловый эфир/гексаны и подвергают флэш-хроматографии с использованием в качестве элюента смеси 70% гексанов/этилацетат/1% уксусной кислоты, получая соединение 2 в виде белого твердого вещества (1,10 г) (выход 11%). Температура плавления: 196-198°С. Анализ сжиганием: %С: 68,85 (вычислено), 68,60 (найдено); %Н: 4,95 (вычислено), 4,82 (найдено). 1H ЯМР (ДМСО-d6): δ 12,96, с, 1H; 9,03, с, 1H; 7,97, д, 2Н; 7,61, д, 2Н; 6,95, дд, 1Н; 6,83, дд, 1Н; 6,78, тд, 1H; 6,70, дт, 1H; 5,18, с, 2Н.
Соединения 79 и 59 получают аналогично способу получения соединения 2.
Соединение 79. Температура плавления: 176-178°С. Анализ сжиганием: %С: 65,69 (вычислено), 65,53 (найдено); %Н: 5,15 (вычислено), 5,00 (найдено). 1H ЯМР (ДМСО-d6): δ 13,0, шир.с, 1H; 8,7, шир.с, 1H; 7,9, д, 2Н; 7,6, д, 2Н; 6,9, д, 1H; 6,75, м, 2Н; 6,7, м, 1H; 5,9, шир.с, 1H; 5,0, м, 1H; 4,1, дд, 1H; 3,85, дд, 1H.
Соединение 59. Температура плавления: 164-167°С. Анализ сжиганием: %С: 70,31 (вычислено), 70,18 (найдено); %Н: 4,72 (вычислено), 4,83 (найдено). 1H ЯМР (ДМСО-d6): δ 13,0, шир.с, 1Н; 10,5, с, 1Н; 8,7, шир.с, 1Н; 7,9, д, 2Н; 7,6, д, 2Н; 6,9, д, 1Н; 6,75, м, 2Н; 6,7, м, 1Н; 5,9, шир.с, 1Н; 5,0, м, 1Н; 4,1, дд, 1Н; 3,85, дд, 1Н.
Получение соединения 3
Соединение 3 предоставлено фирмой Lancaster Synthesis Inc. (Windham, NH).
Получение соединения 6
В 200 мл круглодонную колбу помещают 11,2 г (4 экв.) порошкообразного гидроксида калия и 100 мл диметилсульфоксида. Полученную смесь перемешивают при комнатной температуре в течение 5 минут. Добавляют 2-бензилоксифенол (10 г, 1 экв.), после чего сразу же добавляют этил 7-бромгептаноат (14,6 мл, 1,5 экв.). Полученный раствор перемешивают при комнатной температуре в течение 1 часа.
Реакционную смесь выливают в 200 мл дистиллированной воды и экстрагируют 5×100 мл метиленхлорида. Объединенные органические слои промывают водой и насыщенным раствором соли (по 20 мл) и концентрируют. Полученную жидкость растворяют в 125 мл водного метанола. Добавляют твердый гидроксид натрия (3 экв., 3,7 г) и полученный раствор нагревают до 80°С в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры и выпаривают метанол. Водный слой экстрагируют 150 мл простого эфира и подкисляют до рН ˜ 2 концентрированным водным раствором соляной кислоты. Водный слой экстрагируют этилацетатом (2×300 мл), фильтруют и сушат, получая 19 г (2-бензилоксифенил)- 7-оксигептановой кислоты.
Получают суспензию (2-бензилоксифенил)-7-оксигептановой кислоты (19 г, 58 ммоль), 150 мл этилового спирта и 150 мг палладиевой черни и помещают в автоклав Парра. В реакторе повышают давление до 7 атм (100 фунтов/кв.дюйм), подавая водород. Смесь перемешивают при 50°С в течение 17 часов. Палладий отфильтровывают и фильтрат концентрируют, получая продукт в виде бледно-желтого твердого вещества. Сырое вещество очищают хроматографией на силикагеле, используя в качестве элюента смесь 30-60% этилацетат/гексаны, и получают 5 г (42%) (2-гидроксифенил)-7-оксигептановой кислоты в виде не совсем белого твердого вещества. Температура плавления: 47-50°С. Анализ сжиганием: %С: 65,53 (вычислено), 65,12 (найдено); %Н: 7,61 (вычислено), 7,82 (найдено). МС (EI): 238 (вычислено), 238 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 8,8, с, 1Н; 6,89-6,86, м, 1Н; 6,80-6,87, м, 3Н; 3,94, т, 2Н; 2,21, т, 2Н; 1,72-1,67, м, 2Н; 1,55-1,25, м, 6Н.
Получение соединения 7
В 200 мл круглодонную колбу помещают 22,9 г (3 экв.) только что измельченного гидроксида калия и 100 мл диметилсульфоксида. Полученную смесь перемешивают при 25°С в течение 5 минут. Добавляют катехин (15 г, 1 экв.), после чего сразу же добавляют этил 8-бромоктаноат (34,2 г, 1 экв). Темно-коричневый раствор перемешивают при 25°С в течение 2 часов.
Добавляют дистиллированную воду (100 мл) и полученный раствор нагревают до 85°С в течение 2 часов. Смесь охлаждают, подкисляют до рН ˜2 концентрированным водным раствором соляной кислоты и экстрагируют этилацетатом (2×300 мл). Объединенные органические слои сушат над сульфатом магния, фильтруют и выпаривают растворитель. Сырое вещество очищают хроматографией на силикагеле, используя в качестве элюента смесь 30-60% этилацетат/гексаны. Требуемый продукт собирают и сушат, получая 6,6 г (19%) 8-(2-гидроксифенокси)октановой кислоты в виде не совсем белого твердого вещества. Температура плавления: 60-64°С. Анализ сжиганием: %С; 66,65 (вычислено), 66,65 (найдено); %Н: 7,99 (вычислено), 8,10 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 8,8, с, 1Н; 6,90-6,86, м, 1Н; 6,80-6,76, м, 3Н; 3,92, т, 2Н; 2,21, т, 2Н; 1,75-1,66, м, 2Н; 1,56-1,29, м, 8Н.
Соединения 4, 35, 38, 92 и 98 также получают вышеописанным способом, используя соответствующие исходные вещества.
Соединение 4. Температура плавления: 64-66°С. Анализ сжиганием: %С: 61,22 (вычислено), 61,32 (найдено); %Н: 6,16 (вычислено), 6,27 (найдено). 1H ЯМР (ДМСО-d6): δ 12,1, с, 1Н; 8,75, с, 1Н; 6,90-6,87, м, 1Н; 6,81-6,68, м, 3Н; 3,98, т, 2Н; 2,51, т, 2Н; 1,98-1,89, м, 2Н.
Соединение 35. Температура плавления: 77-80°С. Анализ сжиганием: %С: 67,65 (вычислено), 67,40 (найдено); %Н: 8,33 (вычислено), 8,37 (найдено). 1H ЯМР (ДМСО-d6): δ 11,9, с, 1Н; 6,96-6,85, м, 4Н; 3,94, т, 2Н; 3,74, с, 3Н; 2,23, т, 2Н; 1,72-1,65, м, 2Н; 1,53-1,48, м, 2Н; 1,39-1,29, м, 6Н.
Соединение 38. Температура плавления: 75-76°С. Анализ сжиганием: %С: 65,29 (вычислено), 65,42 (найдено); %Н: 7,53 (вычислено), 7,47 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 11,9, с, 1Н; 7,35, т, 1Н; 6,56, дд, 2Н; 4,04, т, 2Н; 2,55, с, 3Н; 2,27, т, 2Н; 1,79-1,70, м, 2Н; 1,55-1,48, м, 2Н; 1,45-1,37, м, 2Н; 1,32-1,14, м, 4Н.
Соединение 92. Температура плавления: 107-108°С. Анализ сжиганием: %С: 63,89 (вычислено), 63,98 (найдено); %Н: 7,74 (вычислено), 7,72 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, шир.с, 2Н; 6,95, м, 2Н; 6,85, м, 2Н; 3,9, т, 4Н; 3,0, кв, 2H; 2,2, т, 4Н; 6 1,7, п, 4Н; 1,55, п, 4Н; 5 1,4, п, 4Н.
Соединение 98. Температура плавления: 75-77°С. Анализ сжиганием: %С: 63,16 (вычислено), 62,81 (найдено); %Н: 8,01 (вычислено), 8,17 (найдено); %N: 3,2 (вычислено), 3,05 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 2H; 7,60, с, 1Н; 7,45, с, 1Н; 7,03-7,21, м, ЗН; 3,9, м, 4Н; 2,14, т, 4Н; 1,61, м, 4Н; 1,22-1,55, м, 16Н.
Альтернативное получение соединения 7
В 500 мл колбу Эрленмейера помещают 28 г (4 экв.) порошкообразного гидроксида калия и 400 мл диметилсульфоксида. Полученную смесь перемешивают при комнатной температуре в течение 5 минут. Добавляют 2-бензилоксифенол (25 г, 1 экв.), после чего сразу же добавляют этил 8-бромоктаноат (37,6 г, 1,2 экв.). Полученный раствор перемешивают при комнатной температуре в течение 2 часов.
Реакционную смесь выливают в 200 мл дистиллированной воды и нагревают до 80°С в течение 3 часов. Затем смесь подкисляют концентрированным водным раствором соляной кислоты до рН около 2. Образуется осадок не совсем белого твердого вещества. Образовавшееся твердое вещество выделяют вакуумным фильтрованием и сушат в течение ночи при комнатной температуре в вакууме. Полученное вещество этерифицируют, подвергая сырую кислоту взаимодействию с 1 л метанола и 5 мл серной кислоты, и нагревают до 80°С в течение ночи. Смесь охлаждают и трижды экстрагируют 400 мл этилацетата, сушат над сульфатом магния, фильтруют и упаривают, получая сложный метиловый эфир с количественным выходом.
Сырой сложный эфир растворяют в 150 мл этанола и смешивают с 1 г 10% палладия на активированном угле. Полученную смесь помещают в автоклав Парра. В реакторе повышают давление до 14 атм (200 фунтов/кв.дюйм), подавая водород. Гетерогенную смесь перемешивают при 50°С в течение 18 часов. Палладий отфильтровывают и фильтрат концентрируют, получая дебензилированный продукт.
Сложный метиловый эфир омыляют, используя 10 г гидроксида натрия, 400 мл метанола и 50 мл воды. Раствор нагревают до 80°С в течение одного часа и перемешивают при комнатной температуре в течение ночи. Метанол выпаривают. Добавляют еще 100 мл воды и водный слой подкисляют концентрированным водным раствором соляной кислоты до рН 2. Водную фазу экстрагируют этилацетатом (3×300 мл), сушат и упаривают, получая целевое вещество. Сырое вещество очищают хроматографией на силикагеле, используя в качестве элюента смесь 30-60% этилацетат/гексаны, и получают 22,24 г (71%) 8-(2-гидроксифенокси)октановой кислоты в виде не совсем белого твердого вещества. Температура плавления: 65-68°С. Анализ сжиганием: %С: 66,65 (вычислено), 66,98 (найдено); %Н: 7,99 (вычислено), 8,22 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 8,8, с, 1Н; 6,90-6,87, м, 1Н; 6,80-6,67, м, 3Н; 3,94, т, 2Н; 2,23, т, 2Н; 1,73, п, 2Н; 1,53-1,29, м, 8Н.
Соединения 5, 8 и 72 также получают вышеописанным способом, используя соответствующие исходные вещества.
Соединение 5. Температура плавления: 51-53°С. Анализ сжиганием: %С: 64,27 (вычислено), 64,26 (найдено); %Н: 7,19 (вычислено), 7,00 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, шир.с, 1Н; 8,80, шир.с, 1Н; 6,90-6,85, м, 1Н; 6,80-6,68, м, 3Н; 3,94, т, 2Н; 2,26, т, 2Н; 1,76-1,67, м, 2Н; 1,61-1,52, м, 2Н; 1,48-1,40, м, 2Н.
Соединение 8. Температура плавления: 54-57°С. Анализ сжиганием: %С: 68,55 (вычислено), 68,78 (найдено); %Н: 8,63 (вычислено), 8,43 (найдено). 1H ЯМР (ДМСО-d6): δ 8,8, шир.с, 1Н; 6,92-6,89, м, 1Н; 6,82-6,71, м, 3Н; 3,96, т, 2Н; 2,24, т, 2Н; 1,75-1,68, м, 2Н; 1,54-1,39, м, 4Н; 1,30, шир.с, 8Н.
Соединение 72. Температура плавления: 58-60°С. Анализ сжиганием: %С: 69,36 (вычислено), 69,12 (найдено); %Н: 8,90 (вычислено), 8,89 (найдено). 1H ЯМР (ДМСО-d6): δ 6,88-6,85, м, 1H; 6,80-6,66, м, 3Н; 3,93, т, 2Н; 2,20, т, 2Н; 1,74-1,65, м, 2Н; 1,50-1,35, м, 4Н; 1,25, шир.с, 10Н.
Получение соединения 12
Гидроксид калия (10,72 г, 191,1 ммоль) измельчают в ступке до порошкообразного состояния и помещают в 250 мл круглодонную колбу, содержащую 80 мл диметилсульфоксида. Полученную смесь перемешивают в течение 5 минут, после чего добавляют 6,47 г (47,5 ммоль) 2-гидроксиацетофенона и 24,04 г (95,7 ммоль) этил 8-бромоктаноата. Реакционную смесь перемешивают при комнатной температуре в течение одного часа. Оранжевую реакционную смесь выливают в 200 мл дистиллированной воды, затем пять раз экстрагируют 300 мл (всего) метиленхлорида. Органические слои промывают двумя 50 мл порциями воды, затем концентрируют, получая ярко-желтую жидкость.
Полученную жидкость растворяют в 25 мл диоксана. Добавляют водный раствор гидроксида натрия (1 н. раствор, 20 мл) и полученную жидкость перемешивают и нагревают (65°С) в течение двух часов. Реакционную смесь охлаждают до 0°С, подкисляют до рН 1 концентрированным водным раствором соляной кислоты, затем экстрагируют двумя 100 мл порциями этилацетата. Органический слой концентрируют, получая ярко-желтое масло. Масло кристаллизуют смесью метанол:вода (1:1), затем перекристаллизовывают один раз смесью метанол:вода (1:1) и еще один раз смесью метиленхлорид:гексаны (1:4), получая 5,70 г (43,1%) твердого вещества от бледно-желтого до не совсем белого цвета. Температура плавления: 71,5-73,5°С. Анализ сжиганием: %С: 69,04 (вычислено), 68,77 (найдено); %Н: 7,97 (вычислено), 8,04 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,57, дд, 1Н; 7,52, дт, 1Н; 7,15, д, 1Н; 7,00, дт, 1Н; 4,09, т, 2Н; 2,52, с, 3Н; 2,20, т, 2Н; 1,78, п, 2Н; 1,46, м, 4Н; 1,32, м, 4Н.
Соединения 9, 10, 11 и 71 также получают вышеописанным способом, используя соответствующие исходные вещества.
Соединение 9. Температура плавления: 94,5-9°С. Анализ сжиганием: %С: 64,85 (вычислено), 64,81 (найдено); %Н: 6,35 (вычислено), 6,30 (найдено). 1H ЯМР (300 МГц, ДМСО-d6): δ 12,0 (с, 1Н), 7,58, дд, 1Н; 7,5, дт, 1Н; 7,15, дд, 1Н; 7,0, дт, 1Н; 4,15, т, 2Н; 2,55, с, 3Н; 2,45, т, 2Н; 2,0, п, 2Н.
Соединение 10. Температура плавления: 76-77°С. Анализ сжиганием: %С: 66,09 (вычислено), 65,83 (найдено); %Н: 6,83 (вычислено), 6,76 (найдено). 1H ЯМР (300 МГц, ДМСО-d6): δ 7,58, дд, 1Н; 7,5, дт, 1Н; 7,15, дд, 1Н; 7,0, дт, 1Н; 4,1, т, 2Н; 2,55, с, 3Н; 2,3, т, 2Н; 1,8, дп, 2Н; 1,6, дп, 2Н.
Соединение 11. Температура плавления: 44-4°С. Анализ сжиганием: %С: 67,18 (вычислено), 67,32 (найдено); %Н: 7,25 (вычислено), 7,26 (найдено). 1H ЯМР (300 МГц, ДМСО-d6): δ 12,0, с, 1Н; 7,58, дд, 1Н; 7,5, дт, 1Н; 7,15, д, 1Н; 7,0, т, 1Н; 4,1, т, 2Н; 2,55, с, 3Н; 2,25, т, 2Н; 1,8, п, 2Н; 1,6, п, 2Н; 1,45, п, 2Н.
Соединение 71. Температура плавления: 62-63°С. Анализ сжиганием: %С: 61,76 (вычислено), 61,69 (найдено); %Н; 6,66 (вычислено), 6,59 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, шир.с, 1Н; 7,13-7,30, м, 2Н; 6,94-7,02, м, 1Н; 3,98-4,02, т, 2Н; 2,17-2,22, т, 2Н; 1,65-1,72, м, 2Н; 1,28-1,52, м, 8Н.
Приведенные ниже соединения также получают вышеописанным способом, заменяя 2′-гидроксиацетофенон указанным в скобках соединением: 18 (2-пропенилфенол), 20 (2-нитрофенол), 24 (2-ацетамидофенол), 26-29 (2-гидроксипропиофенон), 32 (метилсалицилат) и 39 (6-метокси-2-гидроксиацетофенон). Соединения 18 и 20 далее очищают колоночной хроматографией, используя в качестве элюента 50% этилацетат в гексанах.
Соединение 18. Температура плавления: 79-81°С. Анализ сжиганием: %С: 73,88 (вычислено), 73,85 (найдено); %Н: 8,75 (вычислено), 8,77 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,38-7,41, дд, 1Н; 7,13-7,18, м, 1Н; 6,93-6,95, д, 1Н; 6,84-6,89, т, 1Н; 6,59-6,65, дд, 1Н; 6,21-6,28, м, 1Н; 3,94-3,98, т, 2Н; 2,18-2,23, т, 2Н; 1,83-1,86, дд, 2Н; 1,69-1,78, м, 2Н; 1,31-1,53, м, 9Н.
Соединение 20. Температура плавления: 81-88°С. Анализ сжиганием: %С: 59,78 (вычислено), 59,66 (найдено); %Н; 6,81 (вычислено), 6,96 (найдено); %N: 4,98 (вычислено), 4,69 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,82-7,85, дд, 1Н; 7,60-7,65, м, 1Н; 7,33-7,36, дд, 1Н; 7,06-7,11, м, 1Н; 4,12-4,16, т, 2Н; 2,15-2,27, т, 2Н; 1,66-1,75, м, 2Н; 1,28-1,54, м, 8Н.
Соединение 24. Температура плавления: 110-111°С. Анализ сжиганием: %С: 65,51 (вычислено), 65,47 (найдено); %Н: 7,90 (вычислено), 7,73 (найдено); %N: 4,77 (вычислено), 4,65 (найдено). 1H ЯМР (300 МГц, ДМСО-d6): δ 12,0, с, 1Н; 8,9, с, 1Н; 7,8, д, 1Н; 7,08-6,99, м, 2Н; 6,89-6,84, м, 1Н; 3,99, т, 2Н; 2,20, т, 2Н; 2,07, с, 3Н; 1,75, п, 2Н; 1,56-1,30, м, 8Н.
Соединение 26. Температура плавления: 70-71,5°С. Анализ сжиганием: %С: 66,09 (вычислено), 65,92 (найдено); %Н: 6,83 (вычислено), 6,67 (найдено). 1H ЯМР (ДМСО-d6): δ 12,15, с, 1Н; 7,56-7,45, м, 2Н; 7,12, д, 1Н; 7,00, т, 1Н; 4,10, т, 2Н; 2,92, кв, 2Н; 2,42, т, 2Н; 2,00, п, 2Н; 1,05, т, 3Н.
Соединение 27. Температура плавления: 68-69,5°С. Анализ сжиганием: %С: 68,16 (вычислено), 68,40 (найдено); %Н: 7,63 (вычислено), 7,60 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,54-7,46, м, 2Н; 7,13, д, 1Н; 6,99, т, 1Н; 4,08, т, 2Н; 2,93, кв, 2Н; 2,24, т, 2Н; 1,77, п, 2Н; 1,47, м, 2Н; 1,05, т, 3Н.
Соединение 28. Температура плавления: 85-86°С. Анализ сжиганием: %С: 69,84 (вычислено), 69,59 (найдено); %Н: 8,27 (вычислено), 7,98 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,54-7,46, м, 2Н; 7,13, д, 1Н; 6,99, т, 1Н; 4,08, т, 2Н; 2,93, кв, 2Н; 2,20, т, 2Н; 1,74, п, 2Н; 1,52-1,30, м, 8Н; 1,05, т, 3Н.
Соединение 29. Температура плавления: 67-69°С. Анализ сжиганием: %С: 71,22 (вычислено), 71,06 (найдено); %Н: 8,81 (вычислено), 9,02 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,54-7,45, м, 2Н; 7,12, д, 1Н; 6,99, т, 1Н; 4,06, т, 2Н; 2,93, кв, 2H; 2,18, т, 2Н; 1,76, п, 2Н; 1,51-1,36, м, 12Н; 1,05, т, 3Н.
Соединение 32. Температура плавления: 89-92°С. Анализ сжиганием: %С: 64,27 (вычислено), 63,96 (найдено); %Н: 7,19 (вычислено), 7,40 (найдено). 1H ЯМР (ДМСО-d6): δ 12,2, шир.с, 2H; 7,59, дд, 1Н; 7,45, дт, 1Н; 7,09, д, 1Н; 6,97, т, 1Н; 4,00, т, 2H; 2,20, т, 2H; 1,70, п, 2H; 1,54-1,27, м, 8Н.
Соединение 39. Температура плавления: 69-70,5°С. Анализ сжиганием: %С: 65,35 (вычислено), 65,39 (найдено); %Н: 7,89 (вычислено), 7,80 (найдено). 1H ЯМР (ДМСО-d6): δ 7,27, т, 1Н; 6,67, д, 2H; 3,95, т, 2H; 3,73, с, 3Н; 2,34, с, 3Н; 2,18, т, 2H; 1,63, п, 2H; 1,49, п, 2H; 1,40-1,27, м, 6Н.
Соединения 19, 21, 22 и 23 также получают вышеописанным способом, за исключением того, что используют один эквивалент соответствующего алкилирующего агента и два эквивалента гидроксида калия и промежуточные сложные эфиры очищают жидкостной хроматографией среднего давления (MPLC), используя в качестве подвижной фазы этилацетат и гексаны. Используют следующие составы растворителей: 19 и 21 (20% этилацетат ) и 22 и 23 (10% этилацетат).
Соединение 19. Температура плавления: 58-59°С. Анализ сжиганием: %С: 66,91 (вычислено), 66,73 (найдено); %Н: 8,42 (вычислено), 8,01 (найдено); %N: 5,57 (вычислено), 5,27 (найдено). 1H ЯМР (ДМСО-d6): δ 6,74-6,78, д, 1Н; 6,60-6,68, м, 2H; 6,46-6,52, м, 1Н; 3,88-3,93, т, 2H; 2,17-2,22, т, 2H; 1,66-1,76, м, 2H; 1,30-1,56, м, 8Н.
Соединение 21. Температура плавления: 115-117°С. Анализ сжиганием: %С: 63,14 (вычислено), 62,05 (найдено); %Н: 7,23 (вычислено), 7,11 (найдено); %N: 6,69 (вычислено), 6,37 (найдено). 1H ЯМР (ДМСО-d6): δ 6,74-6,77, дд, 1Н; 6,60-6,68, м, 2Н; 6,46-6,52, м, 1Н; 3,90-3,94, т, 2Н; 2,26-2,31, т, 2Н; 1,63-1,78, м, 4Н.
Соединение 22. Температура плавления: 69-71°С. Анализ сжиганием: %С: 58,84 (вычислено), 58,84 (найдено); %Н: 7,05 (вычислено), 7,08 (найдено); %N: 4,90 (вычислено), 4,83 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 6,72-6,74, д, 1Н; 6,62-6,63, д, 1Н; 6,44-6,48, дд, 1Н; 5,0, с, 2Н; 3,87-3,91, т, 2Н; 2,17-2,22, т, 2Н; 1,65-1,72, м, 2Н; 1,28-1,52, м, 8Н.
Соединение 23. Температура плавления: 80-81°С. Анализ сжиганием: %С: 54,22 (вычислено), 54,15 (найдено); %Н: 5,79 (вычислено), 5,74 (найдено); %N: 5,75 (вычислено), 5,66 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 6,72-6,75, д, 1Н; 6,62-6,63, д, 1Н; 6,45-6,49, дд, 1Н; 5,0, шир.с, 2Н; 3,89-3,39, т, 2Н; 2,25-2,30, т, 2Н; 1,63-1,75, м, 4Н.
Получение соединения 77
Соединение 77 в форме свободной кислоты получают аналогично общему способу получения соединения 12, используя соответствующие исходные вещества. Свободную кислоту соединения 77 (10,4 г, 38,43 ммоль) растворяют в этаноле (83,0 мл). Добавляют 10,0 н. водный раствор гидроксида натрия (3,80 мл) и смесь перемешивают при комнатной температуре в течение примерно 2 часов. Этанол выпаривают, получая гелеобразный мокрый остаток. Полученный остаток растворяют в деионизированной воде (200 мл) и экстрагируют этилацетатом (2×100 мл). Остаточный этилацетат удаляют, продувая реактор азотом. Водный раствор лиофилизуют, получая белый порошок (6,50 г, 22,1 ммоль, выход 58%). Температура плавления: >230°С с разложением. МС (FAB) (положительная), m/z 295,2 (М + Н)+ 317,2 (М + Na)+. 1H ЯМР (ДМСО-d6): δ 7,09-7,15, м, 3Н; 4,05-4,09, т, 2Н; 1,81-1,86, т, 2Н; 1,58-1,68, м, 2Н; 1,22-1,44, м, 8Н.
Альтернативное получение соединения 12
Гидроксид калия (43,28 г, 771,3 ммоль) измельчают в ступке до порошкообразного состояния, затем помещают в 500 мл колбу Эрленмейера, содержащую 250 мл диметилсульфоксида. Полученную смесь перемешивают в течение 15 минут, после чего добавляют 27,47 г (201,8 ммоль) 2-гидроксиацетофенона с последующим немедленным добавлением 50,7 г (201,9 ммоль) этил 8-бромоктаноата. Реакционную смесь перемешивают при комнатной температуре в течение трех часов. Мутную густую оранжевую реакционную смесь выливают в 150 мл дистиллированной воды и перемешивают, пока раствор не становится прозрачным (примерно 15 минут).
Прозрачный оранжевый раствор охлаждают до 0°С на ледяной бане, затем подкисляют концентрированным водным раствором соляной кислоты до образования твердого вещества (рН=7). Твердое вещество собирают фильтрованием и перекристаллизовывают из смеси этанол:вода (50:50), получая 38,08 г (67,8%) желтого твердого вещества. Температура плавления: 72-73°С. Анализ сжиганием: %С: 69,04 (вычислено), 69,10 (найдено); %Н: 7,97 (вычислено), 7,99 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,57, дд, 1Н; 7,52, дт, 1Н; 7,15, д, 1Н; 7,00, дт, 1Н; 4,09, т, 2Н; 2,52, с, 3Н; 2,20, т, 2Н; 1,78, п, 2Н; 1,46, м, 4Н; 1,32, м, 4Н.
Соединение 54 получают вышеописанным способом, используя соответствующие исходные вещества. Нижеследующие соединения также получают указанным способом, заменяя 2′-гидроксиацетофенон указанным в скобках соединением: 55 (2-гидрокси-5-метоксиацетофенон), 56 (2-гидрокси-4-метокси-ацетофенон) и 58 (2-гидрокси-5-метилацетофенон).
Соединение 54. Температура плавления: 71-73,5°С. Анализ сжиганием для C18H26О4·0,068Н2О: %С: 70,28 (вычислено), 69,98 (найдено); %Н: 8,56 (вычислено), 8,16 (найдено). 1Н ЯМР (300 МГц, ДМСО-d6): δ 11,8, с, 1Н; 7,55, дд, 1Н; 7,5, дт, 1Н; 7,15, д, 1Н; 7,0, дт, 1Н; 4,1, т, 2Н; 2,55, с, 3Н; 2,2, т, 2Н; 1,8, п, 2Н; 1,5, м, 2Н; 1,3, м, 10Н.
Соединение 55. Температура плавления: 120,5-121,5°С. Анализ сжиганием: %С: 66,21 (вычислено), 66,00 (найдено); %Н: 7,84 (вычислено), 7,54 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,1, м, 3Н; 4,03, т, 2Н; 3,72, с, 3Н; 2,54, с, 3Н; 2,20, кв, 2Н; 1,76, п, 2Н; 1,53-1,30, м, 8Н.
Соединение 56. Температура плавления: 106-107,5°С. Анализ сжиганием: %С: 65,87 (вычислено), 65,76 (найдено); %Н: 7,86 (вычислено), 7,57 (найдено). 1H ЯМР (ДМСО-d6): δ 7,65, д, 1Н; 6,61-6,55, м, 2Н; 4,08, т, 2Н; 3,82, с, 3Н; 2,49, с, 3Н; 2,19, кв, 2Н; 1,78, п, 2Н; 1,54-1,29, м, 8Н.
Соединение 58. Температура плавления: 121-123°С. Анализ сжиганием: %С: 68,16 (вычислено), 67,88 (найдено); %Н: 7,63 (вычислено), 7,65 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,37, м, 1Н; 7,30, м, 1Н; 7,04, д, 1Н; 4,04, т, 2Н; 2,52, с, 3Н; 2,24, м, 5Н; 1,76, п, 2Н; 1,59-1,41, м, 4Н.
Получение соединения 13
Соединение 13 предоставлено фирмой Aldrich Chemical Co. (Milwaukee, WI).
Получение соединения 15
Гидроксид калия (28,60 г, 0,511 моль) измельчают в ступке и помещают в 500 мл кругло донную колбу, содержащую диметилсульфоксид (215 мл). Полученную смесь перемешивают в течение 5 минут. К смеси добавляют фенол (12,00 г, 0,1277 моль). Затем сразу же добавляют этил 6-бромгексаноат (22,70 мл, 0,1277 моль). Смесь перемешивают в течение примерно 3 часов и выливают в 500 мл воды. Реакционную смесь нагревают при 90°С в течение 1,5 часов, после чего нагревание прекращают. Затем смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь подкисляют 2 н. водным раствором соляной кислоты, в результате чего образуется осадок белого твердого вещества. Белое твердое вещество выделяют вакуумным фильтрованием и сушат в течение ночи при комнатной температуре в вакууме. Получают 25,09 г (выход 94,5%) продукта. Температура плавления: 64-67°С. Анализ сжиганием: %С: 69,23 (вычислено), 68,84 (найдено); %Н: 7,69 (вычислено), 7,78 (найдено); %N: 0,00 (вычислено), <0,02 (найдено). 1H ЯМР (300 МГц, AMCO-d6): δ 11,95, с, 1Н; δ 7,27, м, 2Н; δ 6,90, м, 3Н; 3,93, т, 2Н; 2,20, т, 2Н; 1,70, п, 2Н; 1,50, п, 2Н; 1,30, м, 6Н.
Соединения 14, 16, 76, 75 и 68 также получают вышеописанным способом, используя соответствующие исходные вещества.
Соединение 14. Температура плавления: 57-60°С. Анализ сжиганием: %С: 66,67 (вычислено), 66,49 (найдено); %Н: 6,67 (вычислено), 6,56 (найдено). 1H ЯМР (300 МГц, ДМСО-d6): δ 12,2, (с, 1Н), 7,25 (м, 2Н), 6,90 (м, 3Н), 3,95 (т, 2Н), 2,35 (т, 2Н), 1,90 (п, 2Н).
Соединение 16. Температура плавления: 72-75°С. Анализ сжиганием: %С: 72,73 (вычислено), 72,45 (найдено); %Н: 9,09 (вычислено), 8,92 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,24, т, 2Н; 6,88, м, 3Н; 3,89, т, 2Н; 2,15, т, 2Н; 1,35, м, 4Н; 1,21, м, 8Н.
Соединение 75. Температура плавления: 55-57°С. Анализ сжиганием: %С: 62,26 (вычислено), 61,93 (найдено); %Н: 6,17 (вычислено), 5,89 (найдено); %F: 8,95 (вычислено), 9,11 (найдено). 1H ЯМР (ДМСО-d6): δ 7,25-7,10, м, 3Н; 6,95-6,83, м, 1Н; 4,05, т, 2Н; 2,31, т, 2Н; 1,77-1,62, м, 4Н.
Соединение 76. Температура плавления: 65-67°С. Анализ сжиганием: %С: 54,96 (вычислено), 54,62 (найдено); %Н: 5,0 (вычислено), 4,97 (найдено); %F: 21,73 (вычислено), 21,73 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,61, д, 2Н; 7,26, шир.д, 1Н; 7,10, шир.т, 1Н; 4,12, т, 2Н; 2,31, т, 2Н; 1,80-1,61, м, 4Н.
Соединение 68. Температура плавления: 67-68°С. Анализ сжиганием: %С: 62,11 (вычислено), 61,77 (найдено); %Н: 7,07 (вычислено), 6,94 (найдено); %Cl: 13,09 (вычислено), 13,05 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,32, т, 1Н; 7,00-6,95, м, 2Н; 6,91-6,88, м, 1Н; 3,99, т, 2Н; 2,23, т, 2Н; 1,78, п, 2Н; 1,62, п, 2Н; 1,45-1,30, м, 6Н.
Получение соединения 17
Соединение 17 предоставлено фирмой Aldrich Chemical Co. (Milwaukee, WI).
Получение соединения 25
В 250 мл круглодонную колбу поочередно помещают 5,57 г (33,9 ммоль) 2-гидроксикоричной кислоты, 80 мл метанола и 6 капель концентрированной серной кислоты. Полученный прозрачный раствор нагревают при температуре кипения с обратным холодильником в течение 6 часов и оставляют охлаждаться до комнатной температуры. Растворитель удаляют в вакууме и получают липкое белое твердое вещество. Полученное твердое вещество растворяют в 80 мл этилацетата и промывают тремя 40 мл порциями 10% водного раствора бикарбоната натрия, одной 40 мл порцией воды и двумя 25 мл порциями насыщенного раствора соли. Органический слой концентрируют в вакууме, получая 5,51 г (91,4%) метил 2-гидроксициннамата в виде белого твердого вещества.
Гидроксид калия (7,63 г, 136,0 ммоль) измельчают в ступке до порошкообразного состояния, затем помещают в 125 мл колбу Эрленмейера, содержащую 75 мл диметилсульфоксида. Полученную смесь перемешивают в течение 10 минут, после чего добавляют 5,49 г (30,8 ммоль) метил 2-гидроксициннамата и 7,81 г (31,1 ммоль) этил 8-бромоктаноата. Реакционную смесь перемешивают при комнатной температуре в течение примерно пяти часов, после чего добавляют 50 мл дистиллированной воды. Желтый раствор перемешивают при комнатной температуре в течение ночи, затем промывают двумя 80 мл порциями этилацетата. Водный слой охлаждают до 0°С. Добавляют концентрированный водный раствор соляной кислоты до достижения рН раствора, равного примерно 5. Полученное твердое вещество собирают фильтрованием и перекристаллизовывают из смеси этанол:вода (50:50), получая 4,31 г (45,7%) белого порошка. Температура плавления: 148-150°С. Анализ сжиганием: %С: 66,65 (вычислено), 66,59 (найдено); %Н: 7,24 (вычислено), 7,24 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, шир.с, 2Н; 6 7,86, с; 7,81, с, 1Н; 7,67-7,63, дд, 1Н; 7,39-7,33, дт, 1Н; 7,07-7,04, д, 1Н; 6,98-6,93, т, 1Н; 6,55, с; 6,50, с, 1Н; 4,04, т, 2Н; 2,19, т, 2Н; 1,76, п, 2Н; 1,50, м, 2Н; 1,43-1,28, м, 6Н.
Получение соединения 30
Салициламид (5,3 г, 0,03875 моль) помещают в одногорлую круглодонную колбу, содержащую (15,0 г, 0,03875 моль) этил 8-бромоктаноат. К полученной смеси одной порцией добавляют карбонат калия (6,43 г, 0,0465 моль), используя 35 мл ацетона в качестве растворителя. Реакционную смесь нагревают в течение примерно 4 часов. Затем нагревание прекращают, реакционную смесь охлаждают до комнатной температуры и перемешивают в течение выходных дней в конце недели. Реакцию прекращают, когда данные ВЭЖХ указывают на наличие одного пика, соответствующего времени удерживания, равному 6,44 минуты. Реакционную смесь подвергают вакуумному фильтрованию и лепешку на фильтре промывают ацетоном. Фильтрат концентрируют в вакууме, чтобы удалить избыток растворителя (ацетона).
Твердые вещества перемешивают в гексанах в течение нескольких часов, фильтруют, выделяют и сушат под вакуумом в течение ночи. Твердые вещества (10,93 г, 0,0439 моль) перемешивают в 1,5 экв. 2 н. раствора гидроксида натрия (32 мл, 0,0658 моль). Реакционную смесь нагревают и перемешивают до окончания реакции в соответствии с данными ВЭЖХ. Реакционную смесь охлаждают до комнатной температуры. Реактор помещают на водно-ледяную баню и подкисляют суспензию 2 н. водным раствором соляной кислоты. Твердые вещества выделяют вакуумным фильтрованием и лепешку на фильтре промывают водой. Твердые вещества сушат в вакууме в течение ночи, затем переносят в колбу Эрленмейера и перекристаллизовывают, используя смесь этанол/вода. Твердые вещества выпадают в осадок в течение ночи, после чего их отделяют и сушат, получая 8,08 г 8-(2-карбоксамидофенокси)каприловой кислоты. Температура плавления: 114-116°С. Анализ сжиганием: %С: 64,51 (вычислено), 64,50 (найдено); %Н: 7,52 (вычислено), 7,55 (найдено); %N: 5,02 (вычислено), 4,86 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,82, дд, 1Н; 7,55, шир.с, 2Н; 7,45, дт, 1Н; 7,12, д, 1Н; 7,01, т, 1Н; 4,10, т, 2Н; 2,20, т, 2Н; 1,77, п, 2Н; 1,54-1,29, м, 8Н.
Получение соединения 33
Получение N-этилсалициламида. Диметилацетамид (50 мл) и карзалам (10,00 г, 0,0613 моль) помещают в круглодонную колбу, оснащенную устройством для продувки азотом, конденсатором с водяным охлаждением и магнитной мешалкой. Добавляют карбонат натрия (6,50 г, 0,0613 моль) и иодэтан (4,38 мл, 0,0548 моль) и начинают нагревать реакционную смесь. Смесь нагревают при 80°С в течение 16 часов, после чего нагревание прекращают и реакционную смесь оставляют охлаждаться до комнатной температуры. Затем реакционную смесь фильтруют через воронку из спекшегося стекла и собирают фильтрат. К фильтрату добавляют воду до осаждения белого твердого вещества. Твердое вещество выделяют фильтрованием и переносят в колбу Эрленмейера, содержащую 2 н. водный раствор гидроксида натрия (200 мл). Полученную смесь нагревают при температуре кипения с обратным холодильником в течение примерно 1 часа и перемешивают в течение ночи при комнатной температуре. Смесь подкисляют 2 н. водным раствором соляной кислоты, в результате выделяется желтое масло. Реакционную смесь дважды экстрагируют 200 мл порциями этилацетата. Объединенные этилацетатные слои дважды промывают 200 мл порциями деионизированной воды, сушат сульфатом натрия и концентрируют в вакууме. N-этилсалициламид выделяют в виде желтого масла, которое сушат в течение ночи в вакууме и получают в количестве 7,93 г.
Получение О-ацетил-N-этилсалициламида. Полученный выше N-этилсалициламид (7,93 г, 0,0481 моль) и метиленхлорид (100 мл) помещают в круглодонную колбу, оснащенную устройством для продувки азотом, капельной воронкой и магнитной мешалкой. Полученный раствор охлаждают на водно-ледяной бане и добавляют триэтиламин (14,71 мл, 0,1057 моль). Ацетилхлорид (3,76 г, 0,0529 моль) помещают в капельную воронку и по каплям медленно добавляют к реакционной смеси в течение примерно 10 минут. Через 1 час водно-ледяную баню удаляют и реакционную смесь оставляют нагреваться до комнатной температуры в течение ночи. Реакционную смесь разбавляют дихлорметаном (100 мл) и экстрагируют сначала 100 мл 2 н. водного раствора соляной кислоты и затем двумя 100 мл порциями деионизированной воды. Метиленхлоридный слой сушат сульфатом натрия и концентрируют в вакууме. Полученное масло очищают элюированием через колонку с силикагелем. В качестве элюента используют смесь гексан:этилацетат (60:40) и собирают 75 мл фракции. Фракции, содержащие требуемый О-ацетил-N-этилсалициламид, объединяют и концентрируют в вакууме, получая 4,28 г продукта в виде желтого масла.
Получение 8-(2-(N-этилбензамид)окси)октановой кислоты. Вышеуказанный O-ацетил-N-этилсалициламид (4,28 г, 0,0207 моль) и диметилформамид (75 мл) помещают в 250 мл круглодонную колбу, оснащенную устройством для продувки азотом, капельной воронкой и магнитной мешалкой. Полученную смесь охлаждают на водно-ледяной бане. После перемешивания смеси в течение примерно 10 минут добавляют гидрид натрия (0,76 г, 0,0316 моль), после чего по каплям добавляют раствор этил 8-бромоктаноата (7,78 г, 0,0310 моль) в диметилформамиде (25 мл) в течение 25 минут. Затем водно-ледяную баню удаляют и реакционную смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют деионизированную воду (75 мл) и экстрагируют тремя 75 мл порциями дихлорметана. Объединенные дихлорметановые слои промывают тремя 75 мл порциями деионизированной воды, сушат сульфатом натрия и концентрируют в вакууме. Полученное коричневое масло поглощают водным раствором гидроксида натрия (2 н. раствор, 200 мл), нагревают при температуре кипения с обратным холодильником в течение примерно 2 часов и оставляют охлаждаться до комнатной температуры в течение ночи. Смесь подкисляют 2 н. водным раствором соляной кислоты и экстрагируют тремя 100 мл порциями этилацетата. Объединенные этилацетатные слои промывают тремя 100 мл порциями деионизированной воды и затем тремя 100 мл порциями насыщенного раствора соли. Этилацетатный слой сушат сульфатом натрия и концентрируют в вакууме. Полученное масло кристаллизуют из смеси этилацетат:гексан (30:70), получая 3,24 г требуемого продукта 8-(2-(N-этилбензамид)окси)октановой кислоты. Температура плавления: 94-95°С. Анализ сжиганием: %С: 67,29 (вычислено), 67,18 (найдено); %Н: 8,41 (вычислено), 8,55 (найдено); %N: 4,36 (вычислено), 4,26 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 7,93, д, 1H; 7,75, дд, 1Н; 7,40, тд, 1H; 7,10, д, 1H; 6,98, тд, 1H; 4,00, м, 3Н; 2,15, т, 2Н; 1,71, п, 2Н; 1,25, м, 8Н; 1,10, д, 6Н.
Соединения 31 и 34 также получают вышеописанным способом, используя соответствующее исходное вещество.
Соединение 31. Температура плавления: 91,5-94°С. Анализ сжиганием: %С: 65,51 (вычислено), 65,35 (найдено); %Н: 7,90 (вычислено), 8,03 (найдено); %N: 4,77 (вычислено), 4,46 (найдено). 1Н ЯМР (300 МГц, ДМСО-d6): δ 12,0, с, 1H; 8,02, шир.д, 1H; 7,72, дд, 1H; 7,42, дт, 1H; 7,11, д, 1H; 7,00, т, 1H; 4,08, т, 2Н; 2,80, д, 3Н; 2,20, т, 2Н; 1,77, п, 2Н; 1,53-1,25, м, 8Н.
Соединение 34. Температура плавления: 94-95°С. Анализ сжиганием: %С: 67,29 (вычислено), 67,18 (найдено); %Н: 8,41 (вычислено), 8,55 (найдено); %N: 4,36 (вычислено), 4,26 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0 (с, 1H), 7,93 (д, 1H), 7,75 (дд, 1H), 7,40 (тд, 1H), 7,10 (д, 1H), 6,98 (тд, 1H), 4,00 (м, 3Н), 2,15 (т, 2Н), 1,71 (п, 2Н), 1,25 (м, 8Н), 1,10 (д, 6Н).
Получение соединения 36
В 250 мл круглодонную колбу, оснащенную конденсатором, помещают 5,00 г (17,4 ммоль) соединения 12 и 170 мл этанола. Колбу продувают азотом. Борогидрид натрия (1,15 г, 30,4 ммоль) добавляют тремя порциями к прозрачному желтому раствору соединения 12. Реакционную смесь перемешивают в течение двух часов и проверяют окончание реакции по ВЭЖХ. Добавляют еще 0,38 г (10,0 ммоль) борогидрида натрия и реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь гасят добавлением 30 мл 10% водного раствора бикарбоната натрия, затем фильтруют через подушку из целита. Фильтрат концентрируют в вакууме, получая бледно-желтый гель. Полученный гель перемешивают в 60 мл 1 н. водного раствора гидроксида натрия в течение двух часов, охлаждают до 0°С, затем подкисляют до рН=1 концентрированным водным раствором соляной кислоты. Водный слой экстрагируют четырьмя 30 мл порциями этилацетата. Объединенные органические слои сушат над сульфатом натрия и концентрируют в вакууме, получая 2,46 г (48,8%) продукта в виде прозрачного вязкого желтого масла. Анализ сжиганием: %С: 67,51 (вычислено), 67,16 (найдено); %Н: 8,67 (вычислено), 8,56 (найдено). (Следует отметить, что при выполнении анализа сжиганием использовано 0,176 моль Н2О (исходя из значения KF) и 0,068 моль этилацетата (как показано в ЯМР). 1H ЯМР (300 МГц, ДМСО-d6): δ 7,45-7,42, дд, 1Н; 7,18-7,12, дт, 1Н; 6,93-6,88, т, 2Н; 5,03-4,97, 1Н; 3,99-3,91, м, 2Н; 2,20, т, 2Н; 1,72, п, 2Н; 1,51, м, 2Н; 1,39-1,30, м, 6Н; 1,27-1,25, д, 3Н.
Получение соединения 37
Раствор 10,0 мл (11,31 г, 83,1 ммоль) 2′-гидрокси-ацетофенона и 50 мл тетрагидрофурана помещают на ледяную баню и обрабатывают 120,0 мл (168,0 ммоль) 1,4 М раствора метиллития в тетрагидрофуране, который добавляют по каплям в течение 30 минут. Реакционная смесь сначала мутнеет и затем становится прозрачной. После перемешивания в течение 18 часов раствор подкисляют 4% водным раствором соляной кислоты. Слои разделяют. Органическую фазу промывают 30 мл насыщенного раствора соли, сушат над сульфатом натрия и концентрируют. Получают в общей сложности 12,05 г 2-(диметилгидрокси-метил)фенола.
Получают раствор 6,77 г (44,5 ммоль) 2-(диметилгидрокси-метил) фенола и 50 мл диметилсульфоксида и обрабатывают его 9,90 г (176 ммоль) только что измельченного гидроксида калия. Светло-зеленый раствор перемешивают в течение 20 минут и добавляют 9,85 г (45,8 ммоль) 4-(бромметил)бензойной кислоты и 0,40 г (2,67 ммоль) иодида натрия. Густую суспензию перемешивают в течение 4 часов, после чего добавляют еще 1,66 г (7,72 ммоль) 4-(бромметил)бензойной кислоты. Реакционную смесь перемешивают еще 2 часа и обрабатывают 50 мл воды. После перемешивания в течение 20 часов раствор подкисляют 4% водным раствором соляной кислоты, в результате чего образуется твердое вещество, которое выделяют фильтрованием. Твердое вещество перекристаллизовывают из смеси этанол/вода, получая 5,8 г продукта. Температура плавления: 171-172°С. Анализ сжиганием: %С: 71,31 (вычислено), 71,28 (найдено); %Н: 6,34 (вычислено), 6,14 (найдено). 1H ЯМР (300 МГц, ДМСО-d6): δ 13,0, с, 1Н; 8,0, д, 2Н; 7,7, дд, 1Н; 7,6, д, 2Н; 7,2, дт, 1Н; 7,1, д, 1Н; 7,0, т, 1Н; 5,25, с, 2Н; 5,0, с, 1Н; 1,55, с, 6Н.
Получение соединения 67
Раствор 50,1 г (455 ммоль) гидрохинона, 15,52 г (91,0 ммоль) α-хлор-п-толуиловой кислоты, 1 г (6,7 ммоль) иодида натрия, 75 мл (750 ммоль) 10 н. водного раствора гидроксида натрия и 300 мл воды нагревают до 70°С в течение 24 часов в атмосфере азота. Охлажденную реакционную смесь подкисляют 20% водным раствором соляной кислоты, в результате чего образуется коричневое твердое вещество. Полученное твердое вещество выделяют фильтрованием. Затем твердое вещество поглощают этилацетатом. Нерастворившееся твердое вещество отфильтровывают. Фильтрат промывают насыщенным раствором соли, сушат над сульфатом натрия и концентрируют. Остаток кристаллизуют из смеси этанол/вода, получая 8,1 г соединения 67, температура плавления >230°С. Анализ сжиганием: %С; 68,85 (вычислено), 68,44 (найдено); %Н: 4,95 (вычислено), 4,93 (найдено). 1H ЯМР (ДМСО-d6): δ 9,0, с, 1Н; 8,0, д, 2Н; 7,5, д, 2Н; 6,8, д, 2Н; 6,7, д, 2Н; 5,1, с, 2Н.
Соединения 78 и 73 получают аналогично способу получения соединения 67, используя соответствующие исходные вещества.
Соединение 78. Температура плавления: 178-181°С. Анализ сжиганием: %С: 64,01 (вычислено), 63,95 (найдено); %Н: 4,22 (вычислено), 4,25 (найдено). 1H ЯМР (ДМСО-d6): δ 8,0, д, 2Н; 7,6, д, 2Н; 7,45, дд, 1Н; 7,3, дт, 1Н; 7,2, дд, 1Н; 7,0, дт, 1Н; 5,3, с, 2Н.
Соединение 73. Температура плавления: 63-65°С. Анализ сжиганием: %С: 62,11 (вычислено), 62,02 (найдено); %Н: 7,07 (вычислено), 7,04 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, шир.с, 1H; 7,4, дд, 1Н; 7,3, дт, 1H; 7,1, дд, 1Н; 6,95, дт, 1Н; 4,0, т, 2Н; 2,2, т, 2Н; 1,75, п, 2Н; 1,5, м, 4Н; δ 1,35, м, 4Н.
Получение соединения 60
Раствор 3,0 мл (3,44 г, 28,2 ммоль) салицилальдегида, 5,05 мл (6,33 г, 28,4 ммоль) этил 6-бромгексаноата и 50 мл этанола обрабатывают 5,07 г (36,7 ммоль) карбоната калия. Суспензию нагревают при температуре кипения с обратным холодильником. Через 20 часов реакционную смесь охлаждают до 25°С, фильтруют через подушку из целита и концентрируют. Остаток промывают гексанами и поглощают этанолом и 10 мл 2 н. водного раствора гидроксида натрия. Через 6 часов этанол отгоняют. Смесь подкисляют 4% водным раствором соляной кислоты и экстрагируют этилацетатом. Органическую фазу промывают 30 мл насыщенного раствора соли, сушат над сульфатом натрия и концентрируют. Полученный продукт перекристаллизовывают из смеси этанол/вода, получая 3,0 г соединения 60 в виде коричневого твердого вещества. Температура плавления: 58-60°С. Анализ сжиганием: %С: 66,09 (вычислено), 61,39 (найдено); %Н: 6,83 (вычислено), 6,98 (найдено). МС 236 (М+ пик). 1H ЯМР (ДМСО-d6): δ 12,0, шир.с, 1H; 10,4, с, 1H; 7,7, дд, 1H; 7,65, дт, 1H; 7,2, д, 1H; 7,05, 1,5, м, 2Н.
Соединение 61 получают аналогично способу получения соединения 60, используя соответствующие исходные вещества. Температура плавления: 59-62°С. Анализ сжиганием: %С: 68,18 (вычислено), 67,59 (найдено); %Н: 7,57 (вычислено), 7,63 (найдено). МС 264 (M+ пик). 1H ЯМР (ДМСО-d6): δ 12,0, шир.с, 1H; 10,4, с, 1Н; 8,0, д, 2Н; 7,75, дд, 1Н; 7,65, дт, 1Н; 7,65, д, 2Н; 7,3, д, 1H; 7,1, т, 1H; 5,4, с, 2Н.
Получение соединения 51
Карзалам (30,00 г, 0,1840 моль), иодметан (10,23 мл, 0,1643 моль), карбонат натрия (19,51 г, 0,1840 моль) и диметилформамид (150 мл) помещают в 500 мл круглодонную колбу. Реакционную смесь нагревают в течение ночи при 80°С. Затем реакционную смесь охлаждают до комнатной температуры, фильтруют и собирают белое твердое вещество. Полученное вещество промывают водой, после чего оставшееся твердое вещество переносят в 250 мл круглодонную колбу. К фильтрату, полученному первоначальным фильтрованием, добавляют воду, в результате чего происходит осаждение белого твердого вещества. Полученное вещество объединяют с твердым веществом, находящимся в 250 мл колбе, и добавляют 2 н. водный раствор гидроксида натрия (150 мл). Смесь нагревают в течение одного часа, после чего нагревание прекращают и реакционную смесь оставляют охлаждаться в течение ночи. Белое твердое вещество, выпавшее в осадок в течение ночи, выделяют фильтрованием и сушат в вакууме. Получают 21,52 г N-метилсалициламида.
N-метилсалициламид (21,52 г, 0,1425 моль) и метиленхлорид (300 мл) помещают в литровую круглодонную колбу. Колбу охлаждают на водно-ледяной бане и добавляют триэтиламин (43,62 мл, 0,3135 моль). Затем по каплям добавляют ацетилхлорид в течение пяти минут. Баню удаляют и реакционную смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют метиленхлорид (300 мл). Смесь промывают двумя 300 мл порциями 1 н. водного раствора соляной кислоты и затем тремя 300 мл порциями деионизированной воды. Раствор в метиленхлориде сушат сульфатом натрия и концентрируют в вакууме, получая оранжевое твердое вещество, которое перекристаллизовывают из смеси этилацетат:гексан (70:30). Получают 12,15 г О-ацетил, N-метилсалициламида.
О-ацетил, N-метилсалициламид (17,74 г, 0,919 моль), полученный вышеописанным способом, переносят в литровую круглодонную колбу, содержащую диметилформамид (300 мл). Колбу охлаждают на водно-ледяной бане и добавляют гидрид натрия (3,38 г, 0,1406 моль). Метил 8-бромдеканоат (36,54 г, 0,1379 моль) растворяют в дополнительной порции диметилформамида (100 мл) и полученный раствор по каплям добавляют к реакционной смеси в течение 25 минут. После перемешивания в течение примерно получаса ледяную баню удаляют и реакционную смесь продолжают перемешивать еще три дня при комнатной температуре. Добавляют воду (300 мл) и смесь экстрагируют двумя 250 мл порциями метиленхлорида. Объединенные метиленхлоридные слои трижды промывают 150 мл порциями воды, сушат сульфатом натрия и концентрируют в вакууме, получая коричневое масло. Полученное масло поглощают 2 н. водным раствором гидроксида натрия (200 мл) и нагревают в течение 45 минут. После перемешивания в течение ночи при комнатной температуре добавляют еще 200 мл 2 н. водного раствора гидроксида натрия и реакционную смесь нагревают, пока она не становится прозрачной. После охлаждения реакционную смесь подкисляют 2 н. водным раствором соляной кислоты и экстрагируют тремя 250 мл порциями этилацетата. Объединенные этилацетатные слои промывают тремя 250 мл порциями воды и затем тремя 250 мл порциями насыщенного раствора соли. Этилацетатный слой сушат сульфатом натрия и концентрируют в вакууме, получая желтовато-коричневое твердое вещество, которое перекристаллизовывают из смеси этилацетат:гексан (30:70). Продукт выделяют в виде белого твердого вещества с выходом, равным 26,30 г. Данные анализа соединения 51: Температура плавления: 81-84°С. Анализ сжиганием: %С: 67,29 (вычислено), 67,17 (найдено); %Н: 8,41 (вычислено), 8,70 (найдено); %N: 4,36 (вычислено), 4,36 (найдено). 1H ЯМР (ДМСО-d6): δ 12,00, с, 1Н; 7,98, д, 1Н; 7,70-7,75, дд, 1Н; 7,39-7,48, дт, 1Н; 7,09-7,15, д, 1Н; 6,95-7,05, тд, 1Н; 4,05, т, 2Н; 2,75, д, 3Н; 2,15, т, 2Н; 1,70, п, 2Н; 1,20-1,55, м, 12Н.
Получение соединения 65
Гидроксид калия (28,60 г, 0,511 моль) помещают в 500 мл круглодонную колбу. Добавляют диметилсульфоксид (215 мл) и начинают перемешивать смесь. После перемешивания в течение примерно 35 минут, добавляют фенол (12,00 г, 0,1277 моль) и этил 8-бромоктаноат (32,04 г, 0,1277 моль). Полученную смесь перемешивают при комнатной температуре в течение 3 часов и добавляют 500 мл деионизированной воды. Смесь нагревают при температуре кипения с обратным холодильником. Реакционную смесь охлаждают до комнатной температуры и подкисляют 2 н. водным раствором соляной кислоты. Полученное белое твердое вещество выделяют фильтрованием и сушат в вакууме в течение ночи. Получают 27,74 г 8-феноксиоктановой кислоты. Данные анализа соединения 65: Температура плавления: 65-68°С. Анализ сжиганием: %С: 71,19 (вычислено), 70,98 (найдено); %Н: 8,47 (вычислено), 8,70 (найдено). 1H ЯМР (ДМСО-d6): δ 11,95, с, 1Н; 7,23-7,31, м, 2Н; 6,87-6,95, м, 3Н; 3,90, т, 2Н; 2,15, т, 2Н; 1,62, п, 2Н; 1,45, п, 2Н; 1,22-1,45, м, 6Н.
Получение соединения 43
Гидроксид калия (2,62 г, 0,0467 моль) и диметилсульфоксид (90 мл) помещают в 500 мл круглодонную колбу в атмосфере азота. После перемешивания в течение 5 минут добавляют монобензоат резорцина (10,0 г, 0,0467 моль) и этил 8-бромоктаноат (11,73 г, 0,0467 моль). После перемешивания в течение ночи при комнатной температуре к полученной смеси добавляют дополнительную порцию гидроксида калия (2,62 г, 0,0467 моль) для завершения реакции. После перемешивания в течение дополнительных 5,5 часов к смеси добавляют воду (200 мл) и экстрагируют тремя порциями дихлорметана (100 мл порции). Объединенные дихлорметановые порции сушат сульфатом натрия и концентрируют в вакууме. Полученное коричневое масло с запахом диметилсульфоксида поглощают водой. Затем смесь экстрагируют тремя порциями этилацетата (100 мл порции). Объединенные этилацетатные слои промывают тремя порциями воды (100 мл порции). Этилацетатный слой сушат сульфатом натрия и концентрируют в вакууме. Полученное коричневое масло поглощают водным раствором гидроксида натрия (2 н. раствор, 100 мл). Затем добавляют тетрагидрофуран (50 мл) и смесь нагревают при температуре кипения с обратным холодильником в течение 2 часов, после чего нагревание прекращают. Тетрагидрофуран удаляют в вакууме и реакционную смесь подкисляют 2 н. водным раствором соляной кислоты. Полученное желтовато-коричневое твердое вещество несколько раз промывают водой с температурой 40-50°С и затем перекристаллизовывают из смеси вода:этанол (80:20). Полученное желтовато-коричневое твердое вещество сначала перекристаллизовывают из смеси гексан:этилацетат (90:10) и затем добавляют в кипящую воду. Этиловый спирт добавляют до тех пор, пока смесь не становится прозрачной. Во время охлаждения осаждается желтовато-коричневое твердое вещество, которое выделяют фильтрованием. Полученный продукт сушат в вакууме и выделяют с выходом 5,96 г. Данные анализа соединения 43: Температура плавления = 89-91°С. Анализ сжиганием: %С: 66,67 (вычислено), 66,68 (найдено); %Н: 7,94 (вычислено), 7,92 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 9,3, с, 1Н; 7,00, т, 1Н; 6,29, м, 3Н; 3,84, т, 2Н; 2,15, т, 2Н; 1,62, п, 2Н; 1,45, п, 2Н; 1,23, м, 6Н.
Соединения 44, 45, 74 и 46 получают вышеописанным способом, используя соответствующее исходное вещество.
Соединение 44. Температура плавления: 89-92°С. Анализ сжиганием: %С: 68,57 (вычислено), 68,71 (найдено); %Н: 8,57 (вычислено), 8,58 (найдено). 1H ЯМР (ДМСО-d6): δ 11,9, с, 1Н; 9,2, с, 1Н; 7,00, т, 1Н; 6,29, м, 3Н; 3,84, т, 2Н; 2,15, т, 2Н; 1,62, п, 2Н; 1,30, п, 2Н; 1,23, м, 8Н.
Соединение 45. Температура плавления: 98-99,5°С. Анализ сжиганием: %С: 64,29 (вычислено), 64,06 (найдено); %Н: 7,14 (вычислено), 7,12 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 9,3, с, 1Н; 7,00, т, 1Н; 6,29, м, 3Н; 3,84, т, 2Н; 2,17, т, 2Н; 1,62, п, 2Н; 1,49, п, 2Н; 1,35, м, 2Н.
Соединение 74. Температура плавления: 126-128°С. Анализ сжиганием: %С: 56,57 (вычислено), 56,72 (найдено); %Н: 6,39 (вычислено), 6,66 (найдено); %N: 4,71 (вычислено), 4,32 (найдено). 1H ЯМР (ДМСО-d6): δ 11,7, с, 1Н; 10,4, с, 1Н; 7,75-7,8, дд, 1Н; 7,68-7,73, д, 1Н; 6,92-6,99, д, 1Н; 4,00, т, 2Н; 2,15, т, 2Н; 1,67, п, 2Н; 1,22-1,55, м, 8Н.
Соединение 46. Температура плавления: 93-95°С. Анализ сжиганием: %С: 61,22 (вычислено), 61,20 (найдено); %Н: 6,12 (вычислено), 6,02 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 9,3, с, 1Н; 7,01, т, 1Н; 6,30, м, 3Н; 3,86, т, 2Н; 2,35, т, 2Н; 1,85, п, 2Н.
Получение соединения 47
Гидроксид калия (11,20 г, 200,0 ммоль) измельчают в ступке до порошкообразного состояния и помещают в 0,5 л круглодонную колбу, содержащую 90 мл диметилсульфоксида. Полученную смесь перемешивают в течение 5 минут, после чего добавляют 10,00 г (50,0 ммоль) 4-бензилоксифенола с последующим немедленным добавлением 12,55 г (50,0 ммоль) этил 8-бромоктаноата. Реакционную смесь перемешивают при комнатной температуре в течение двух с половиной часов. Затем реакционную смесь выливают в 200 мл дистиллированной воды. Смесь нагревают при температуре кипения с обратным холодильником. После окончания реакции реакционную смесь оставляют охлаждаться до комнатной температуры. Смесь подкисляют 2 н. водным раствором соляной кислоты и образовавшееся твердое вещество выделяют фильтрованием. Твердое вещество сушат в вакууме в течение ночи. Получают 17,96 г (4-бензилоксифенил)-8-оксиоктановой кислоты. Полученное вещество используют на следующей стадии без очистки.
(4-Бензилоксифенил)-8-оксиоктановую кислоту помещают в 0,5 л круглодонную колбу, содержащую 120 мл этилового спирта. Реакционную смесь барботируют азотом в течение 15 минут и добавляют 10% палладий на активированном угле. Затем в колбе создают вакуум и поверх колбы помещают баллон с водородом так, чтобы содержимое колбы находилось в атмосфере водорода. Смесь перемешивают в течение ночи при комнатной температуре и затем фильтруют через целит. Этиловый спирт удаляют в вакууме, получая белое твердое вещество, которое сначала перекристаллизовывают из смеси этиловый спирт:вода (90:10) и затем растворяют в 2 н. водном растворе гидроксида натрия. Смесь фильтруют и подкисляют 2 н. водным раствором соляной кислоты. Образовавшееся белое твердое вещество выделяют фильтрованием и сушат в вакууме. Получают 2,12 г (4-гидроксифенил)-8-оксиоктановой кислоты. Данные анализа соединения 47. Температура плавления: 97-100°С. Анализ сжиганием: %С: 66,67 (вычислено), 66,43 (найдено); %Н: 7,94 (вычислено), 7,80 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1Н; 9,00, с, 1H; 6,63, м, 4Н; 3,75, т, 2Н; 2,15, т, 2Н; 1,60, п, 2Н; 1,45, п, 2Н; 1,20, м, 6Н.
Соединения 48, 49 и 50 получают вышеописанным способом, используя соответствующее исходное вещество.
Соединение 48. Температура плавления: 99-100°С. Анализ сжиганием: %С: 68,57 (вычислено), 68,47 (найдено); %Н: 8,57 (вычислено), 8,67 (найдено). 1H ЯМР (ДМСО-d6): δ 6,63, м, 4Н; 3,75, т, 2Н; 2,15, т, 2Н; 1,60, п, 2Н; 1,45, п, 2Н; 1,20, м, 10Н.
Соединение 49. Температура плавления: 102-104°С. Анализ сжиганием: %С: 64,29 (вычислено), 64,53 (найдено); %Н: 7,14 (вычислено), 7,32 (найдено). 1H ЯМР (ДМСО-d6): δ 11,5, с, 1H; 8,5, с, 1H; 6,63, м, 4Н; 3,75, т, 2Н; 2,15, т, 2Н; 1,60, п, 2Н; 1,45, п, 2Н; 1,30, м, 2Н.
Соединение 50. Температура плавления: 117-120°С. Анализ сжиганием: %С: 58,43 (вычислено), 58,63 (найдено); %Н: 6,35 (вычислено), 6,40 (найдено). 1H ЯМР (ДМСО-d6): δ 12,0, с, 1H; 8,6, с, 1H; 6,62, м, 4Н; 3,80, т, 2Н; 2,50, т, 2Н; 1,80, п, 2Н.
Получение соединения 57
2,5-Дигидироксиацетофенон (5,00 г, 0,0329 моль), бензил-бромид (3,72 мл, 0,031 моль), карбонат калия (4,31 г, 0,031 моль) и ацетон (150 мл) помещают в 500 мл круглодонную колбу. Реакционную смесь нагревают в течение ночи при температуре кипения с обратным холодильником и охлаждают до комнатной температуры. К холодной реакционной смеси добавляют деионизированную воду (150 мл) и трижды экстрагируют 100 мл порциями диэтилового эфира. Объединенные эфирные слои сушат сульфатом натрия и концентрируют в вакууме, получая темное твердое вещество. Темное твердое вещество перекристаллизовывают из смеси этанол:вода (50:50), получая 3,09 г 2-гидрокси-5-бензилоксиацетофенона в виде желтых игл.
Гидроксид калия (11,11 г, 0,1983 моль) и диметил-сульфоксид (90 мл) помещают в 250 мл круглодонную колбу. Через 10 минут добавляют полученный выше 2-гидрокси-5-бензилоксиацетофенон (12,00 г, 0,0496 моль), затем этил 8-бромоктаноат (12,45 г, 0,496 моль). Реакционную смесь перемешивают в течение ночи при комнатной температуре. Добавляют деионизированную воду и реакционную смесь нагревают при температуре кипения с обратным холодильником в течение пяти часов. Затем реакционную смесь оставляют охлаждаться до комнатной температуры и подкисляют 2 н. водным раствором соляной кислоты. Полученное желтовато-коричневое твердое вещество выделяют фильтрованием и дважды промывают порциями деионизированной воды. После сушки в течение ночи в вакууме получают 16,75 г (4-бензилокси-2-ацетилфенил)-8-оксиоктановой кислоты.
(4-Бензилокси-2-ацетилфенил)-8-оксиактановую кислоту (16,75 г, 0,0435 моль) и этилацетат (85 мл) помещают в 300 мл реактор Парра. Добавляют 10% палладий на активированном угле (0,75 г), после чего реактор герметично закрывают, откачивают воздух и подают водород. Реактор нагревают при 50°С в течение ночи, открывают и добавляют еще 0,5 г 10% палладия на активированном угле. Реактор снова герметично закрывают, откачивают воздух и подают водород. Если реакционная смесь не изменяется, находясь в течение двух дней при комнатной температуре, реактор снова открывают и реакционную смесь фильтруют. Фильтрат концентрируют в вакууме и остаток снова помещают в реактор Парра. Затем остаток поглощают этилацетатом и добавляют 10% палладий на активированном угле. Реактор герметично закрывают, откачивают воздух, подают водород и нагревают в течение ночи при 50°С. После охлаждения до комнатной температуры реактор открывают, палладий на активированном угле отфильтровывают и реакционную смесь концентрируют в вакууме. Образовавшееся желтое твердое вещество перекристаллизовывают из смеси вода:этиловый спирт (80:20). Желтое твердое вещество, полученное в результате перекристаллизации, поглощают кипящим гексаном. Затем добавляют этилацетат, пока не образуется прозрачный раствор, и смесь оставляют охлаждаться до комнатной температуры. Осажденное желтовато-коричневое твердое вещество выделяют фильтрованием и сушат в вакууме. Получают 6,23 г (4-гидрокси-2-ацетилфенил)-8-оксиактановой кислоты. Данные анализа соединения 57. Температура плавления: 112-115°С. Анализ сжиганием: %С: 65,31 (вычислено), 65,32 (найдено); %Н: 7,48 (вычислено), 7,39 (найдено). 1H ЯМР (ДМСО-d6): δ 6,88-7/02, м, 3Н; 3,92, т, 2Н; 2,49, с, 3Н; 2,15, т, 2Н; 1,69, п, 2Н; 1,20-1,59, м, 8Н.
Получение соединения 81
Получение этилового эфира 8-(4-бензилоксифенокси)-2-метилоктановой кислоты
Перенос жидкостей осуществляют в безвоздушных условиях. 14,0 г этилового эфира 8-(4-бензилоксифенокси)октановой кислоты (0,03778 моль, 1 экв.) помещают в высушенную пламенем 250 мл трехгорлую круглодонную колбу, оснащенную мешалкой. Затем добавляют 80 мл безводного ТГФ. Смесь перемешивают в течение 10 минут или до полного растворения твердого вещества. Смесь охлаждают до -78°С на бане со смесью сухого льда с ацетоном. К полученной смеси добавляют 19,84 мл 2 М раствора диизопропиламида лития (0,03967 моль, 1,05 экв.). Добавление производят медленно, поддерживая температуру ниже -60°С. Закончив добавление, смесь перемешивают в течение 2,0 часов при -78°С, после чего суспензию медленно гасят 4,70 мл иодметана (0,07556 моль, 2,0 экв). Во время добавления не допускают повышения температуры выше -50°С. Реакционную смесь оставляют медленно нагреваться до комнатной температуры и перемешивают в течение 3 дней. Из раствора отфильтровывают осадок и супернатант концентрируют в вакууме до образования остатка. Полученный остаток поглощают 60 мл этилацетата и обрабатывают насыщенным раствором бикарбоната натрия (50 мл) и насыщенным раствором NaCl (50 мл). Этилацетатный слой сушат 4,5 г безводного сульфата натрия и фильтруют. Органический слой концентрируют в вакууме до образования остатка. Конечный продукт получают в виде золотистого масла с выходом сырого продукта, равным 9,10 г (выход 62,6%). Данные ВЭЖХ показывают, что полученный продукт содержит небольшое количество исходного вещества вместе с некоторым количеством диметилированного побочного продукта. Промежуточный продукт не исследуют и используют без очистки на следующей стадии.
Указанное соединение дебензилируют при помощи Pd/C и H2, как описано в способе получения соединения 47. Полученный продукт гидролизуют способами, описанными для соединения 47, получая соединение 81.
Выход равен 67,85%. Продукт представляет собой белое твердое вещество. Т.пл. равна 67-70°С. Элементный анализ: теоретические значения С=67,65%, Н=8,33%; найдено С=67,56%, Н=8,56%. Количественный анализ 13С ЯМР (ДМСО-d6): С=0 (1С, 177,477); CAr-O (2С, 151,467 и 151,015 м.д.); СAr-Н (4С, 115,617 и 115,209 м.д.); CH2-CH2-O (1C, 67,939 м.д.); СН-СН3 (1С, 38,689 м.д.); -CH2- (5C, 33,211, 28,769, 26,645, 25,451 м.д.); СН-СН3 (1C, 16,944 м.д.).
Получение соединения 82
Получение этилового эфира 8-(4-бензилоксифенокси)-2-(пропен-2-ил)октановой кислоты
Перенос жидкостей осуществляют в безвоздушных условиях. 10,0 г этилового эфира 8-(4-бензилоксифенокси)октановой кислоты (0,02699 моль, 1 экв.) помещают в высушенную пламенем 250 мл трехгорлую круглодонную колбу, оснащенную мешалкой. Затем добавляют 100 мл безводного ТГФ. Смесь перемешивают течение 10 минут или до полного растворения твердого вещества.
Смесь охлаждают до -78°С на бане со смесью сухого льда с ацетоном. К смеси добавляют 24,0 мл 2 М раствора диизопропиламида лития (0,0480 моль, 1,7 экв.). Добавление производят медленно, поддерживая температуру ниже -60°С. Закончив добавление, смесь перемешивают в течение 2,0 часов при -78°С, после чего суспензию медленно гасят 5,0 мл аллилбромида (0,0577 моль, 2,13 экв.). Во время добавления не допускают повышения температуры выше -50°С. Реакционную смесь оставляют медленно нагреваться до комнатной температуры и перемешивают в течение 16 часов. Из раствора отфильтровывают осадок и супернатант концентрируют в вакууме до образования остатка. Полученный остаток поглощают 60 мл этилацетата и обрабатывают насыщенным раствором бикарбоната натрия (50 мл) и насыщенным раствором NaCl (50 мл). Затем этилацетатный слой сушат 4,5 г безводного сульфата натрия и фильтруют. Органический слой концентрируют до образования масла и хроматографируют на силикагеле, используя смесь гексанов с этилацетатом (9:1). Конечный продукт получают в виде золотистого масла с выходом, равным 7,0 г (выход 64,1%). Количественный анализ 13С ЯМР (CDCl3-d): С=O (1С, 175,462 М.д.); СBn (6С, 137,188, 128,36, 127,679, 127,298 м.д.); СAr-O (2С, 153,303 и 152,683 м.д.); СAr-Н (4С, 115,607 и 115,176 м.д.); =СН2 (1С, 116,484 м.д.); (СН2)Bn (1С, 70,439 м.д.); (CH2-CH2-O (1С, 68,242 м.д.); СН3-СН2-O (1С, 59,946 м.д.); СН-СН3 (1С, 45,147 м.д.); -СН2-СН= (1С, 36,404 м.д.); -СН2- (5С, 31,609, 29,121, 27,054, 25,745 м.д.); CH2-СН3 (1С, 14,221 м.д.).
Указанное соединение дебензилируют при помощи Pd/C и Н2, как описано в способе получения соединения 47. Полученный продукт гидролизуют способами, описанными для соединения 47, получая соединение 82. Выход равен 67,66%. Продукт является белым твердым веществом. Т.пл. равна 98-100°С. Элементный анализ: теоретические значения С=69,36%, Н=8,90%, найдено С=69,33%, Н=8,96%. Количественный анализ 13С ЯМР (AMCO-d6): С=O (1С, 177,226); CAr-O (2С, 151,660 и 151,216 м.д.); СAr-Н (4С, 115/786 и 115,368 м.д.); CH2-CH2-O (1C, 67,908 м.д.); СН-СН2 (1С, 44,887 м.д.); -СН2- (5С, 34,341, 28,968, 27,071, 25,614 м.д.); CH-CH2 (1C, 32,060 м.д.); CH2-СН3 (1C, 20,312 м.д.); СН2-СН3 (1C, 14,007 м.д.).
Получение соединения 80
Получение этилового эфира 8-(4-метоксифенокси)октановой кислоты
В 500 мл трехгорлую круглодонную колбу помещают 14,90 г 4-метоксифенола (0,12 моль), 30,0 г 8-бромэтилоктаноата (0,1265 моль), 10,36 г карбоната калия (0,075 моль), 150 мл сухого ацетона и 2,5 мол.% иодида калия. Реакционную смесь помещают в атмосферу азота и нагревают с обратным холодильником в течение 2 дней. Гетерогенную смесь упаривают в вакууме до образования твердого остатка и смешивают с 600 мл используемых в равных частях воды и этилацетата. Две фазы разделяют и органический слой экстрагируют 3 н. раствором NaOH (3×150 мл). Органический слой еще раз экстрагируют насыщенным раствором NaCl. Затем органический слой сушат над безводным сульфатом магния и фильтруют. Затем органический раствор концентрируют до половины объема (˜180 мл) и дополняют равным количеством гексана. Полученную смесь помещают на ночь в холодильник. Образовавшиеся кристаллы подвергают вакуумному фильтрованию и затем воздушной сушке. Продукт не анализируют и используют без очистки на следующих стадиях.
Получение этилового эфира 8-(4-метоксифенокси)-2-метил-октановой кислоты
Перенос жидкостей осуществляют в безвоздушных условиях. 14,0 г полученного выше соединения (0,03778 моль, 1 экв.) переносят в высушенную пламенем 250 мл трехгорлую круглодонную колбу, оснащенную мешалкой. Добавляют 80 мл безводного ТГФ. Смесь перемешивают в течение 10 минут или до полного растворения твердого вещества. Затем смесь охлаждают до -78°С на бане со смесью сухого льда с ацетоном. К смеси добавляют 19,84 мл 2 М раствора диизопропиламида лития (0,03967 моль, 1,05 экв.). Добавление производят медленно, поддерживая температуру ниже -60°С. Закончив добавление, смесь перемешивают в течение 2 часов при -78°С, после чего суспензию медленно гасят 4,70 мл иодметана (0,07556 моль, 2,0 экв.). Во время добавления не допускают повышения температуры выше -50°С. Реакционную смесь оставляют медленно нагреваться до комнатной температуры и перемешивают в течение 16 часов. Из раствора отфильтровывают осадок и супернатант концентрируют в вакууме до образования остатка. Образовавшийся остаток поглощают 60 мл этилацетата и обрабатывают насыщенным раствором бикарбоната натрия (50 мл) и насыщенным раствором NaCl (50 мл). Этилацетатный слой сушат 4,5 г безводного сульфата натрия и фильтруют. Органический слой концентрируют в вакууме до образования остатка. Конечный продукт получают в виде золотистого масла с выходом сырого продукта, равным 9,10 г (выход 62,6 %). Данные ВЭЖХ показывают, что полученный продукт содержит небольшое количество исходного вещества вместе с некоторым количеством диметилированного побочного продукта. Промежуточный продукт не исследуют и используют без очистки на следующей стадии.
Продукт получают в виде прозрачной жидкости, которую подвергают вакуумной перегонке при 140°С и давлении 1 мм Hg. Конечный выход после перегонки равен 55,38%.
Количественный анализ 13С ЯМР (COCl3-d): C=O (1С, 176,508 м.д.); CAr-O (2С, 153,411 и 152,935 м.д.); СAr-Н (4С, 115,09 и 114,299 м.д.); СН2-СН2-O (1С, 68,186 м.д.); СН3-СН2-О (1С, 59,782); СН3О (1С, 55,339 м.д.); СН-СН3 (1С, 39,239 м.д.); -СН2- (5С, 33,465, 29,030, 26,888, 25,666 м.д.); СН-СН3 (1С, 16,835); СН2-СН3 (1С, 14,012 м.д.).
Полученный продукт гидролизуют способами, описанными для соединения 47, получая соединение 80. Выход равен 82,3%. Продукт представляет собой не совсем белое твердое вещество. Т.пл. равна 71-73°С. Элементный анализ: теоретические значения С=68,55%, Н=8,63%; найдено С=68,04%, Н=8,65%. Количественный анализ 13С ЯМР (ДМСО-d6): С=O (1С, 177,668); CAr-O (2С, 153,243 и 152,773 м.д.); CAr-H (4С, 115,265 и 114,554 м.д.); СН2-СН2-O (1С, 67,818 м.д.); ОСН3 (1С, 55,305 м.д.); СН-СН3 (1С, 33,340 м.д.); -CH2- (5C, 33,340, 28,805, 26,742, 25,487 м.д.); СН-СН3 (1С, 17,075 м.д.)
Получение соединения 69
Этиловый эфир 8-(4-метоксифенокси)-2-метилоктановой кислоты получают вышеописанным способом. Полученный продукт гидролизуют способами, описанными для соединения 47, получая соединение 69.
Выход равен 74,6%. Продукт представляет собой белое твердое вещество. Т.пл. равна 96-97°С. Элементный анализ: теоретические значения С=67,65%, Н=8,33%, найдено С=67,74%, Н=8,44%. Количественный анализ 13С ЯМР (ДМСО-d6): С=O (1С, 174,628); CAr-O (2С, 153,291 и 152,798 м.д.); CAr-H (4С, 115,251 и 114,562 м.д.); СН2-СН2-О (1С, 67,844 м.д.); ОСН3 (1С, 55,301 м.д.); СН2-С=O (1C, 39,343 м.д.); -CH2- (5C, 33,747, 28,700, 25,545, 24,575 м.д.).
Получение соединения 88
Смесь 6,525 г (40 ммоль) карзалама, 10,26 г (44 ммоль) 9-бром-1-нонанола и 5,30 г (50 ммоль) карбоната натрия в 30 мл N,N-диметилацетамида (ДМА) нагревают при 75-80°С в течение 3 часов. Данные ТСХ (элюент: этилацетат/гептан) свидетельствуют об окончании реакции. Реакционную смесь осторожно выливают в смесь воды со льдом. Образовавшееся белое твердое вещество перемешивают в течение 1 часа. Указанное вещество собирают на фильтре из спекшегося стекла, промывают водой, гексаном и сушат в вакууме, получая 9,80 г 3-(9-гидроксинонил)-2Н-1,3-бензоксазин-2,4(3Н)-диона (80%). К суспензии 6,11 г (20 ммоль) 3-(9-гидроксинонил)-2Н-1,3-бензоксазин-2,4(3Н)-диона в 10 мл N,N-диметилацетамида при комнатной температуре добавляют 20,4 мл (20,4 ммоль) раствора трет-бутоксида калия в ТГФ. Прозрачный коричневый раствор становится очень густым. Добавляют дополнительное количество ДМА (10 мл) и нагревают смесь при температуре кипения с обратным холодильником в течение 5 минут. Затем добавляют 0,664 г (4 ммоль) иодида калия, после чего по каплям добавляют 3,34 г (20 ммоль) этилбромацетата. Реакционную смесь кипятят с обратным холодильником в течение 1 часа, охлаждают примерно до 35°С и выливают в смесь воды со льдом. Образуется камедь. Супернатант сливают и добавляют свежую воду. Указанную процедуру повторяют дважды. Камедь растворяют в ТГФ. Раствор в ТГФ осторожно выливают в гексан. Образовавшееся твердое вещество собирают, промывают гексаном и сушат в вакууме. Масса требуемого продукта равна 2,36 г (35%). ВЭЖХ: 4,39 мин; Т.пл. 125-128°С. 1H ЯМР (ДМСО-d6): δ 1,25 (12Н, м), 1,37 (2Н, м), 1,53 (2Н, м), 3,28 (2Н, м), 3,36 (2Н, т), 4,85 (2Н, с), 7,07 (2Н, м), 7,45 (1Н, т), 7,88 (1Н, д), 8,70 (1Н, т). Элементный анализ: вычислено для C18H27NO5: С, 64,07; Н, 8,07; N, 4,15; найдено: С, 63,71; Н, 8,29; N, 4,31.
Получение соединения 93
Температура плавления: 57-59°С. Анализ сжиганием: %С: 72,69 (вычислено), 72,75 (найдено); %Н: 9,15 (вычислено), 9,44 (найдено).
Получение соединения 94
Температура плавления: 59-61°С. Анализ сжиганием: %С: 71,16 (вычислено), 71,08 (найдено); %Н: 8,53 (вычислено), 8,99 (найдено).
Получение соединения 95
Данное соединение предоставлено фирмой Contact Service Company, Москва, Россия.
Получение соединения 96
Данное соединение предоставлено фирмой Contact Service Company, Москва, Россия.
Получение соединения 97
Данное соединение предоставлено фирмой Sigma Company of Milwaukee, WI.
Пример 2. Кальцитонин лосося (sCT) - пероральное введение
Лекарственные композиции для перорального введения (РО), содержащие соединение доставляющего агента и кальцитонин лосося (sCT) в деионизированной воде, получают в соответствии с приведенной ниже таблицей 2. В частности, 450 мг соединения доставляющего агента добавляют к 2,0 мл воды. Обычно используют натриевую соль указанного соединения, либо свободную кислоту превращают в натриевую соль, для чего к перемешиваемому раствору добавляют один эквивалент гидроксида натрия (1,0 н. раствор) и разбавляют водой. Раствор подвергают вихревому перемешиванию, нагревают (около 37°С) и обрабатывают ультразвуком. Показатель рН доводят примерно до 7 (около 6,5- 8,5), добавляя NaOH или HCl. К полученному раствору добавляют 90 мкг sCT из исходного раствора sCT (раствор с концентрацией 2 мг/мл получают, добавляя к sCT 1000% фосфатного буфера с рН 4, после чего полученную смесь оставляют в покое примерно на 10-20 минут и периодически осторожно переворачивают). Затем добавляют воду, доводя общий объем до 3,0 мл (указанное количество может изменяться в зависимости от растворимости соединения доставляющего агента). Лекарственные растворы, содержащие соединения доставляющих агентов 3 и 15, необходимо дополнительно развести водой, причем для достижения требуемого количества соединения доставляющего агента и sCT вводимые конечные дозы соответственно должны быть равны 3 и 2 мл/кг. Конечные дозы соединения доставляющего агента, sCT и объемы доз приведены в нижеследующей таблице 2.
Обычно используют описанные ниже методики определения требуемой дозы и отбора проб. Самцов крыс Sprague-Dawley с массой тела 200-250 г не кормят в течение 24 часов, после чего им вводят кетамин (44 мг/кг) и хлорпромазин (1,5 мг/кг) за 15 минут до введения лекарственного средства и при необходимости производят повторное введение анестезирующих средств для обеспечения необходимой степени наркоза. Экспериментальной группе из пяти животных вводят один из лекарственных растворов. Для перорального введения используют французский катетер Rusch 8 длиной 11 см, соответствующий 1 мл шприцу с концом в виде пипетки. Шприц заполняют лекарственным раствором, пропуская раствор через катетер, который затем вытирают досуха. Катетер вводят в пищевод, оставляя перед резцами конец трубки длиной 1 см. Подачу раствора осуществляют нажатием на поршень шприца.
Пробы крови отбирают из хвостовой артерии через промежутки времени, равные обычно 0, 10, 20, 30, 60 и 90 минутам. Содержание sCT в сыворотке определяют при помощи тест-набора EIA (набор № EIAS-6003 фирмы Peninsula Laboratories, Inc., San Carlos, CA). Результаты сравнивают с базовыми значениями, полученными в промежуток времени = 0. Результаты, полученные у животных в каждой экспериментальной группе, усредняют для каждого промежутка времени. Усредненные максимальные значения приведены в нижеследующей таблице 2.
Пример 3. Рекомбинантный гормон роста человека (rhGH) - пероральное введение
Лекарственные растворы для перорального введения (РО) через желудочный зонд, содержащие соединение доставляющего агента и rhGH, получают в фосфатном буфере, смешивая компоненты. Для получения раствора соединения доставляющего агента обычно используют натриевую соль указанного соединения либо свободную кислоту превращают в натриевую соль. Раствор соединения доставляющего агента обычно получают в фосфатном буфере, причем для получения натриевой соли к перемешиваемому раствору добавляют один эквивалент гидроксида натрия (1,0 н. раствор). Конечные лекарственные растворы получают, смешивая раствор соединения доставляющего агента с исходным раствором rhGH (раствор с концентрацией 15 мг rhGH/мл получают, смешивая в виде порошков 15 мг rhGH, 75 мг D-маннита, 15 мг глицина и 3,39 мг двухосновного фосфата натрия и разбавляя 2% глицерином), после чего полученный раствор разводят до требуемого объема (обычно 3,0 мл). Показатель рН при необходимости доводят до 7-8,5. Конечные дозы соединений доставляющих агентов и rhGH приведены в нижеследующей таблице 3.
Обычно используют описанные ниже методики определения требуемой дозы и отбора проб. Самцов крыс Sprague-Dawley с массой тела 200-250 г не кормят в течение 24 часов, после чего им вводят кетамин (44 мг/кг) и хлорпромазин (1,5 мг/кг) за 15 минут до введения лекарственного средства и при необходимости производят повторное введение анестезирующих средств для обеспечения необходимой степени наркоза. Экспериментальной группе из пяти животных вводят один из лекарственных растворов. Для перорального введения используют французский катетер Rusch 8 длиной 11 см, соответствующий 1 мл шприцу с концом в виде пипетки. Шприц заполняют лекарственным раствором, пропуская раствор через катетер, который затем вытирают досуха. Катетер вводят в пищевод, оставляя перед резцами конец трубки длиной 1 см. Подачу раствора осуществляют нажатием на поршень шприца.
Пробы крови отбирают из хвостовой артерии через промежутки времени, равные обычно 15, 30, 45, 60 и 90 минутам. Объединяют пять проб, взятых в каждый промежуток времени (за исключением проб, для которых указано стандартное отклонение (SD) и стандартная ошибка (SE)). Количественное определение концентрации rhGH в сыворотке производят при помощи набора для иммуноанализа rhGH (набор № K1F4015 фирмы Genzyme Corporation Inc., Cambridge, MA). Базовые значения, полученные в предшествующих исследованиях, практически равны нулю. Максимальные концентрации для каждой группы приведены в нижеследующей таблице 3.
Пример 4. Гепарин - Пероральное/внутрикишечное введение
Лекарственные растворы для перорального введения (РО) через желудочный зонд и/или внутрикишечного введения (IC), содержащие соединение доставляющего агента и гепариннатрий фармакопеи США (USP), получают в 25% водном растворе пропиленгликоля. Используют натриевую соль соединения доставляющего агента или свободную кислоту превращают в натриевую соль, добавляя один эквивалент гидроксида натрия. Соединение доставляющего агента и гепарин (примерно 166-182 ед./мг (обычно 166,9 ед./мг)) смешивают в виде сухих порошков, подвергая их вихревому перемешиванию. Сухую смесь растворяют в 25% (об./об.) водном растворе пропиленгликоля, подвергают вихревому перемешиванию и помещают в устройство для ультразвуковой обработки (около 37°С). Показатель рН доводят примерно до 7 (6,5-8,5) водным раствором NaOH (2 н. раствор). Лекарственный раствор обрабатывают ультразвуком, получая прозрачный раствор. Конечный объем доводят примерно до 3,0 мл. Конечные дозы соединения доставляющего агента, гепарина и объемы доз приведены в нижеследующей таблице 4.
Обычно используют описанные ниже методики определения требуемой дозы и отбора проб. Самцов крыс Sprague-Dawley с массой тела 275-350 г не кормят в течение 24 часов, после чего животных анестезируют, вводя им внутримышечно гидрохлорид кетамина (88 мг/кг) непосредственно перед введением лекарственного средства и при необходимости производят повторное введение анестезирующего средства для обеспечения необходимой степени наркоза. Экспериментальной группе из пяти животных вводят один из лекарственных растворов. Для перорального введения (РО) через желудочный зонд используют французский катетер Rusch 8 длиной 11 см, соответствующий 1 мл шприцу с концом в виде пипетки. Шприц заполняют лекарственным раствором, пропуская раствор через катетер, который затем вытирают досуха. Катетер вводят в пищевод, оставляя перед резцами конец трубки длиной 1 см. Подачу лекарственного раствора осуществляют нажатием на поршень шприца. Для внутрикишечного введения (IC) используют французский катетер Rusch 8 длиной 7,5 см, соответствующий 1 мл шприцу с концом в виде пипетки. Катетер вводят в ободочную кишку через анальное отверстие так, чтобы трубка была совершенно не видна. Лекарственный раствор медленно подают в ободочную кишку, нажимая на поршень шприца.
Пробы цитратной крови отбирают пункцией в сердце, которую делают после введения кетамина (88 мг/кг), обычно через 0,25, 0,5, 1,0 и 1,5 часа после введения лекарственного раствора. Всасывание гепарина определяют по увеличению времени свертывания крови, измеряемого с учетом времени появления частично активированного тромбопластина (АРТТ) в соответствии со способом, описанным Henry, J.B., Clinical Diagnosis and Management by Laboratory Methods, Philadelphia, PA, W.B. Saunders (1979). Базовые значения, полученные в предшествующих исследованиях, равны примерно 20 секундам. Результаты, полученные у животных в каждой группе, усредняют для каждого промежутка времени, при этом наибольшие из указанных средних значений (т.е. усредненный максимум АРТТ) приведены в нижеследующей таблице 4.
Таблица 4. Гепарин пероральное/внутрикишечное введение
Пример 5. Низкомолекулярный гепарин (LMWH) - пероральное/внутрикишечное введение
Лекарственные композиции для перорального введения (РО) и/или внутрикишечного введения (IC), содержащие соединение доставляющего агента и низкомолекулярный гепарин (LMWH), получают в 25% водном растворе пропиленгликоля. Используют натриевую соль соединения доставляющего агента или свободную кислоту превращают в натриевую соль, добавляя один эквивалент гидроксида натрия. Соединение доставляющего агента и LMWH (парнапарин, 91 ед./мг, средняя молекулярная масса около 5000, фирмы Opocrin, Modena, Italy) (обычно 90-105 ед./мг, средняя молекулярная масса около 5000) смешивают в виде сухих порошков, подвергая их вихревому перемешиванию. Сухую смесь растворяют в 25% (об./об.) водном растворе пропиленгликоля, подвергают вихревому перемешиванию и помещают в устройство для ультразвуковой обработки (около 37°С) для получения прозрачного раствора. Показатель рН доводят примерно до 7 (6,5-8,5) 2 н. водным раствором NaOH. Лекарственный раствор обрабатывают ультразвуком, получая прозрачный раствор. Конечный объем доводят примерно до 3,0 мл. Конечные дозы соединения доставляющего агента, LMWH и объемы доз приведены в нижеследующей таблице 5.
Обычно используют описанные ниже методики определения требуемой дозы и отбора проб. Самцов крыс Sprague-Dawley с массой тела 275-350 г не кормят в течение 24 часов, после чего животных анестезируют, вводя им внутримышечно гидрохлорид кетамина (88 мг/кг) непосредственно перед введением лекарственного средства и при необходимости производят повторное введение анестезирующего средства для обеспечения необходимой степени наркоза. Экспериментальной группе из пяти животных вводят один из лекарственных растворов. Для перорального введения (РО) через желудочный зонд используют французский катетер Rusch 8 длиной 11 см, соответствующий 1 мл шприцу с концом в виде пипетки. Шприц заполняют лекарственным раствором, пропуская раствор через катетер, который затем вытирают досуха. Катетер вводят в пищевод, оставляя перед резцами конец трубки длиной 1 см. Подачу лекарственного раствора осуществляют нажатием на поршень шприца. Для внутрикишечного введения (IC) используют французский катетер Rusch 8 длиной 7,5 см, соответствующий 1 мл шприцу с концом в виде пипетки. Катетер вводят в ободочную кишку через анальное отверстие так, чтобы трубка была совершенно не видна. Лекарственный раствор медленно подают в ободочную кишку, нажимая на поршень шприца.
Пробы нитратной крови отбирают пункцией в сердце, которую делают после введения кетамина (88 мг/кг), обычно через 0,5, 1,0, 2,0, 3,0 и 4,0 часа после введения лекарственного раствора. Всасывание LMWH определяют по увеличению концентрации LMWH в плазме, измеряемой при помощи анти-Ха-анализа гепарина с использованием набора для тестирования антител к фактору Ха CHROMOSTRATETM (фирмы Organon Teknika Corporation, Durham, NC). Величины концентрации LMWH в плазме, полученные у животных в каждой группе, усредняют для каждого промежутка времени и строят график средних значений концентрации LMWH в плазме в зависимости от времени. Усредненные максимальные значения концентрации LMWH в плазме приведены в нижеследующей таблице 5.
Пример 6. Параддитовидный гормон (РТН 1-34) - пероральное/внутрикишечное введение
Лекарственные растворы для перорального введения (РО) через желудочный зонд и/или внутрикишечного введения (IC), содержащие соединение доставляющего агента и остатки 1-34 паращитовидного гормона человека (РТН), получают в деионизированной воде. Для получения раствора соединения доставляющего агента используют натриевую соль указанного соединения, либо свободную кислоту превращают в натриевую соль. Раствор соединения доставляющего агента обычно получают в воде, для чего при получении натриевой соли к перемешиваемому раствору добавляют один эквивалент гидроксида натрия (1,0 н. раствор). Конечные лекарственные растворы получают, смешивая раствор соединения доставляющего агента с исходным раствором РТН (обычно в воде в концентрации 5 мг РТН/мл) и разбавляя его до требуемого объема (обычно 3,0 мл). Показатель рН при необходимости доводят примерно до 7-8,5. Конечные дозы соединения доставляющего агента, РТН и объемы доз приведены в нижеследующей таблице 6.
Обычно используют описанные ниже методики определения требуемой дозы и отбора проб. Самцов крыс Sprague-Dawley с массой тела 200-250 г не кормят в течение 24 часов, после чего им вводят кетамин (44 мг/кг) и хлорпромазин (1,5 мг/кг) за 15 минут до введения лекарственного средства и при необходимости производят повторное введение анестезирующих средств для обеспечения необходимой степени наркоза. Экспериментальной группе из пяти животных вводят один из лекарственных растворов. Для перорального введения через желудочный зонд (РО) используют французский катетер Rusch 8 длиной 11 см, соответствующий 1 мл шприцу с концом в виде пипетки. Шприц заполняют лекарственным раствором, пропуская раствор через катетер, который затем вытирают досуха. Катетер вводят в пищевод, оставляя перед резцами конец трубки длиной 1 см. Подачу раствора осуществляют нажатием на поршень шприца. Для внутрикишечного введения (IC) используют французский катетер Rusch 8 или 6 длиной 7,5 см, соответствующий шприцу с концом в виде пипетки Эппендорфа. Шприц заполняют лекарственным раствором, подавая раствор через трубку катетера. Катетер вытирают досуха. На конец трубки наносят гель K-J, избегая соприкосновения с отверстием трубки, после чего трубку вводят в ободочную кишку через анальное отверстие так, чтобы трубка была совершенно не видна. Лекарственный раствор медленно подают в ободочную кишку, нажимая на поршень шприца, после чего катетер удаляют.
Пробы крови отбирают из хвостовой артерии через промежутки времени, равные обычно 0, 15, 30, 45, 60 и 90 минутам при пероральном введении и 0, 10, 20, 30, 60 и 90 минутам при внутрикишечном введении. Количественное определение концентрации РТН в сыворотке производят при помощи набора для радиоиммуноанализа РТН (набор № RIK 6101 фирмы Peninsula Laboratories, Inc., San Carlos, CA). Базовые значения, полученные в предшествующих исследованиях, практически равны нулю. Результаты, полученные у животных в каждой группе, усредняют для каждого промежутка времени. Максимальные значения средних концентраций (то есть усредненные максимальные концентрации РТН в сыворотке) приведены в нижеследующей таблице 6.
Пример 7. Интерферон - пероральное введение
Лекарственные растворы, содержащие соединение доставляющего агента и интерферон человека (IFN), получают в деионизированной воде. Свободную кислоту соединения доставляющего агента превращают в натриевую соль, добавляя один эквивалент гидроксида натрия. Раствор соединения доставляющего агента обычно получают в воде, для чего при получении натриевой соли к перемешиваемому раствору добавляют один эквивалент гидроксида натрия (1,0 н. раствор). Затем смесь подвергают вихревому перемешиванию и помещают в устройство для ультразвуковой обработки (около 37°С). Показатель рН доводят примерно до 7,0-8,5 водным раствором NaOH. Смесь подвергают вихревому перемешиванию, получая однородную суспензию или раствор, при необходимости обрабатывают ультразвуком и нагревают. В случае необходимости добавляют дополнительное количество NaOH для достижения однородной растворимости и рН вновь доводят примерно до 7,0-8,5. Раствор соединения доставляющего агента смешивают с исходным раствором IFN (в концентрации примерно от 22,0 до 27,5 мг/мл в физиологическом растворе с фосфатным буфером) и разводят до требуемого объема (обычно 3,0 мл). Конечные дозы соединения доставляющего агента, IFN и объемы доз приведены в нижеследующей таблице 7.
Обычно используют описанные ниже методики определения требуемой дозы и отбора проб. Самцов крыс Sprague-Dawley с массой тела 200-250 г не кормят в течение 24 часов, после чего им вводят кетамин (44 мг/кг) и хлорпромазин (1,5 мг/кг) за 15 минут до введения лекарственного средства и при необходимости производят повторное введение анестезирующих средств для обеспечения необходимой степени наркоза. Экспериментальной группе из пяти животных вводят один из лекарственных растворов. Для этой цели используют французский катетер Rusch 8 длиной 11 см, соответствующий 1 мл шприцу с концом в виде пипетки. Шприц заполняют лекарственным раствором, пропуская раствор через катетер, который затем вытирают досуха. Катетер вводят в пищевод, оставляя перед резцами конец трубки длиной 1 см. Подачу раствора осуществляют нажатием на поршень шприца.
Пробы крови отбирают из хвостовой артерии через промежутки времени, равные обычно 0, 15, 30, 45, 60 и 90 минутам. Количественное определение концентрации IFN в сыворотке производят при помощи набора для иммуноанализа альфа-IFN человека Cytoscreen (№ в каталоге КНС4012 фирмы Biosource International, Camarilio, CA). Базовые значения, полученные в предшествующих исследованиях, практически равны нулю. Результаты, полученные у животных в каждой группе, усредняют для каждого промежутка времени. Максимальные значения средних концентраций (то есть усредненные максимальные концентрации IFN в сыворотке) приведены в нижеследующей таблице 7.
Пример 8. Инсулин - пероральное введение
Композиции для перорального введения (РО), содержащие соединение доставляющего агента и инсулин человека с противоионами цинка (минимум 26 ед./мг, фирмы Calbiochem - Novabiochem Corp., La Jolla, CA), получают в деионизированной воде. Обычно, 500 мг соединения доставляющего агента добавляют к 1,5 мл воды. Свободную кислоту соединения доставляющего агента превращают в натриевую соль, для чего к полученному раствору при перемешивании добавляют один эквивалент гидроксида натрия. Раствор подвергают вихревому перемешиванию, затем нагревают (около 37°С) и обрабатывают ультразвуком. Показатель рН доводят примерно до 7-8,5, добавляя NaOH или HCl. При необходимости добавляют дополнительное количество NaOH для достижения однородной растворимости и показатель рН повторно доводят примерно до 7-8,5. Затем добавляют воду до общего объема около 2,4 мл и подвергают раствор вихревому перемешиванию. К полученному раствору добавляют примерно 1,25 мг инсулина из исходного раствора инсулина (раствор с концентрацией 15 мг/мл получают из 0,5409 г инсулина и 18 мл деионизированной воды, доводя рН до 8,15 при помощи HCl и NaOH, причем для получения прозрачного раствора используют 40 мл концентрированной HCl, 25 мл 10 н. раствора NaOH и 50 мл 1 н. раствора NaOH) и подвергают вихревому перемешиванию. Конечные дозы соединения доставляющего агента, инсулина и объемы доз приведены в нижеследующей таблице 8.
Обычно используют описанные ниже методики определения требуемой дозы и отбора проб. Самцов крыс Sprague-Dawley с массой тела 200-250 г не кормят в течение 24 часов, после чего им вводят кетамин (44 мг/кг) и хлорпромазин (1,5 мг/кг) за 15 минут до введения лекарственного средства и при необходимости производят повторное введение анестезирующих средств для обеспечения необходимой степени наркоза. Экспериментальной группе из пяти животных вводят один из лекарственных растворов. Для перорального введения лекарственного средства используют французский катетер Rusch 8 длиной 11 см, соответствующий 1 мл шприцу с концом в виде пипетки. Шприц заполняют лекарственным раствором, пропуская раствор через катетер, который затем вытирают досуха. Катетер вводят в пищевод, оставляя перед резцами конец трубки длиной 1 см. Подачу раствора осуществляют нажатием на поршень шприца.
Пробы крови отбирают из хвостовой артерии через промежутки времени, равные обычно 15, 30, 60, 120 и 180 минутам. Уровни инсулина в сыворотке определяют при помощи тест-набора для анализа инсулина методом ELISA (набор № DSL-10-1600 фирмы Diagnostic Systems Laboratories, Inc., Webster, TX), причем в стандартную процедуру анализа вносят изменения для оптимизации чувствительности и линейного диапазона стандартной кривой, отображающей объемы и концентрации проб, используемых в данном анализе. Концентрации инсулина человека в сыворотке (мкед./мл) измеряют для каждого промежутка времени у каждого из пяти животных в каждой экспериментальной группе. Пять значений для каждого промежутка времени усредняют и строят график концентрации инсулина в сыворотке в зависимости от времени. (В предшествующих экспериментах не были обнаружены измеряемые концентрации инсулина человека после перорального введения одного инсулина человека). Максимальные значения (пики) и величины площади под кривой (AUC) приведены в нижеследующей таблице 8.
Пример 9. Инсулин - легочное введение
Лекарственные композиции, содержащие соединение доставляющего агента и инсулин человека, получают в воде. В частности, к 1,5 мг соединения доставляющего агента добавляют деионизированную воду, доводя объем до 1,0 мл, и подвергают раствор вихревому перемешиванию. Используют натриевую соль соединения доставляющего агента, либо свободную кислоту превращают в натриевую соль, для чего к полученному раствору при перемешивании добавляют один эквивалент гидроксида натрия (10 н. раствор) и разбавляют водой. Раствор подвергают вихревому перемешиванию, затем нагревают (около 37°С) и обрабатывают ультразвуком. Показатель рН доводят примерно до 7,0-8,5, добавляя NaOH или HCl. К полученному раствору добавляют 75 мкл исходного раствора инсулина человека (2 мг/мл). (Исходный раствор получают следующим образом. К 0,02 г инсулина добавляют 3 мл раствора HCl с рН 3,0 в деионизированной воде. Показатель рН полученного раствора понижают до 3,0 (примерно 2,6), добавляя HCl и NaOH, пока раствор не становится прозрачным. Затем показатель рН повышают до 7,6, используя NaOH и HCl. Конечный объем доводят до 10 мл деионизированной водой с рН 7,5. Конечный рН равен 7,59). Затем добавляют воду до достижения общего объема, равного 2,0 мл, после чего сосуд с раствором несколько раз осторожно переворачивают. Конечные дозы соединения доставляющего агента, инсулина и объемы дозы приведены в нижеследующей таблице 9.
Обычно используют описанные ниже методики определения требуемой дозы и отбора проб. Самцов крыс Sprague-Dawley с массой тела 200-250 г не кормят в течение 24 часов, после чего им вводят кетамин (44 мг/кг) и хлорпромазин (3,0 мг/кг) за 15 минут до введения лекарственного средства и при необходимости производят повторное введение анестезирующих средств для обеспечения необходимой степени наркоза (используют такое же количество кетамина и 1,5 мг/кг хлорпромазина). Экспериментальной группе из пяти животных вводят один из лекарственных растворов. Контрольной группе из пяти животных вводят только инсулин. Трахеальный инстиллятор для грызунов, оснащенный лампой (фирмы Perm Century, Inc., Pittsburgh, PA), заполняют лекарственным раствором и вводят в горло так, чтобы игла вошла в трахею (с визуальной проверкой). Подачу лекарственного раствора осуществляют нажатием на поршень.
Пробы крови отбирают у всех животных из хвостовой артерии через промежутки времени, равные обычно 5, 15, 30, 60 и 120 минутам. Уровни инсулина в сыворотке определяют при помощи тест-набора для анализа инсулина методом ELISA (набор № DSL-10-1600 фирмы Diagnostic Systems Laboratories, Inc., Webster, TX), причем в стандартную процедуру анализа вносят изменения для оптимизации чувствительности и линейного диапазона стандартной кривой, отображающей объемы и концентрации проб, используемых в данном анализе. Концентрации инсулина в сыворотке (мкед./мл) измеряют для каждого промежутка времени у каждого из пяти животных в каждой экспериментальной группе. Пять значений для каждого промежутка времени усредняют и строят график концентрации инсулина в сыворотке в зависимости от времени. Ниже приведены соотношения площадей под кривой (AUC) для экспериментальной группы и контрольной группы. В нижеследующей таблице приведены также соотношения максимальных концентраций инсулина в сыворотке (Cmax) для экспериментальной группы и контрольной группы.
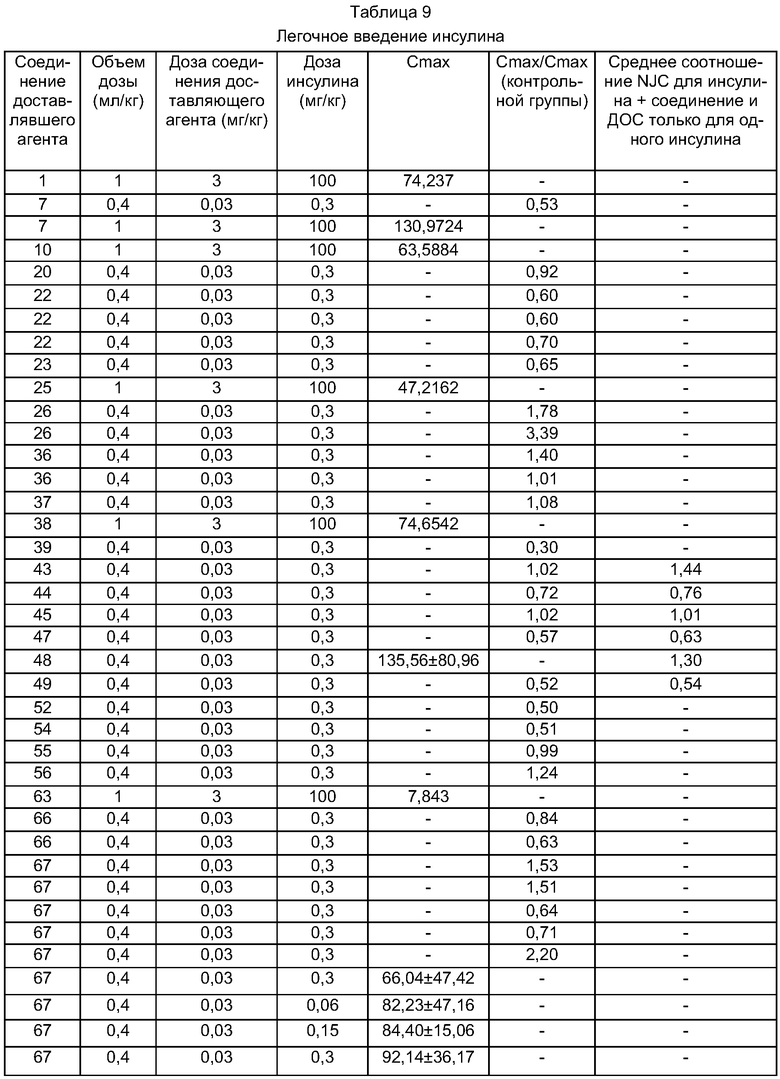
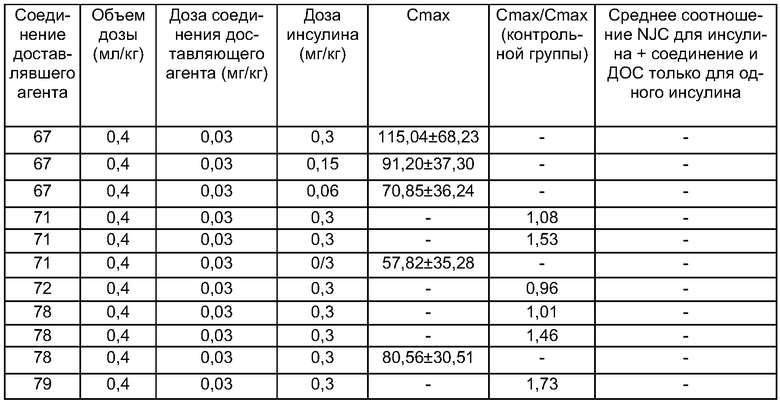
Пример 10. Кромолин - пероральное введение
Лекарственные растворы, содержащие соединение доставляющего агента (полученного по примеру 1) и динатриевую соль кромолина (кромолин) (фирмы Sigma Chemical Co., St. Louis, МО), получают в деионизированной воде. Свободную кислоту соединения доставляющего агента превращают в натриевую соль, добавляя один эквивалент гидроксида натрия. Полученную смесь подвергают вихревому перемешиванию и помещают в устройство для ультразвуковой обработки (около 37°С). Показатель рН доводят примерно до 7-7,5 водным раствором NaOH. При необходимости добавляют дополнительное количество NaOH для достижения однородной растворимости и повторно регулируют показатель рН. Смесь подвергают вихревому перемешиванию для получения однородного раствора, при необходимости производя обработку ультразвуком и нагревание. Раствор соединения доставляющего агента смешивают с кромолином из исходного раствора (175 мг кромолина/мл в деионизированной воде, показатель рН при необходимости доводят примерно до 7,0, добавляя NaOH или HCl, после чего исходный раствор хранят в замороженном виде завернутым в фольгу, перед использованием размораживают и нагревают примерно до 30°С). Смесь подвергают вихревому перемешиванию для получения однородного раствора, при необходимости производя обработку ультразвуком и нагревание. Показатель рН доводят примерно до 7-8 водным раствором NaOH. Затем раствор разбавляют водой до требуемого объема (обычно 2,0 мл) и концентрации и до использования хранят завернутым в фольгу. Конечные дозы соединения доставляющего агента и кромолина и объемы доз приведены в нижеследующей таблице 10.
Обычно используют описанные ниже методики определения требуемой дозы и отбора проб. Самцов крыс Sprague-Dawley с массой тела 200-250 г не кормят в течение 24 часов, после чего их анестезируют кетамином (44 мг/кг) и хлорпромазином (1,5 мг/кг) за 15 минут до введения лекарственного средства и при необходимости производят повторное введение анестезирующих средств для обеспечения необходимой степени наркоза. Экспериментальной группе из пяти животных вводят один из лекарственных растворов. Для этой цели используют французский катетер Rusch 8 длиной 11 см, соответствующий 1 мл шприцу с концом в виде пипетки. Шприц заполняют лекарственным раствором, пропуская раствор через катетер, который затем вытирают досуха. Катетер вводят в пищевод, оставляя перед резцами конец трубки длиной 1 см. Подачу раствора осуществляют нажатием на поршень шприца.
Пробы крови отбирают из хвостовой артерии через промежутки времени, равные обычно 0,25, 0,5, 1,0 и 1,5 часам. Концентрации кромолина в сыворотке измеряют при помощи ВЭЖХ. Образцы получают следующим образом: 100 мкл сыворотки помещают вместе с 100 мкл 3 н. раствора HCl и 300 мкл этилацетата в пробирку Эппендорфа. Содержимое пробирки подвергают вихревому перемешиванию в течение 10 минут и затем центрифугируют в течение 10 минут со скоростью 10000 оборотов/мин. 200 мкл этилацетатного слоя переносят в пробирку Эппендорфа, содержащую 67 мкл 0,1 М раствора фосфатного буфера. Содержимое пробирки подвергают вихревому перемешиванию в течение 10 минут и затем центрифугируют в течение 10 минут со скоростью 10000 оборотов/мин. Слой фосфатного буфера переносят в пробирку для ВЭЖХ и вводят в колонку для ВЭЖХ (колонка длиной 150 мм и внутренним диаметром 2 мм Keystone Exsil Amino, 5 мкм, 100 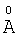 ; подвижная фаза = 35% буфера (68 мМ KH2PO4. доведенного до рН 3,0 при помощи 85% Н3PO4)/65% ацетонитрила; инъецируемый объем = 10 мкл; скорость потока= 0,30 мл/мин; время удерживания кромолина = 5,5 мин; оптическую плотность определяют при 240 нм). Базовые значения, полученные в предшествующих исследованиях, практически равны нулю.
; подвижная фаза = 35% буфера (68 мМ KH2PO4. доведенного до рН 3,0 при помощи 85% Н3PO4)/65% ацетонитрила; инъецируемый объем = 10 мкл; скорость потока= 0,30 мл/мин; время удерживания кромолина = 5,5 мин; оптическую плотность определяют при 240 нм). Базовые значения, полученные в предшествующих исследованиях, практически равны нулю.
Результаты, полученные для животных в каждой группе, усредняют для каждого промежутка времени, при этом наибольшие средние значения (т.е. усредненный максимум концентрации кромолина в сыворотке) приведены в нижеследующей таблице 10.
Таблица 10. Кромолин - пероральное введение
Пример 11. Даптомицин - пероральное введение
Лекарственные растворы, содержащие соединение доставляющего агента и даптомицин (Cubist Pharmaceuticals, Cambridge, МА.), получают в 0,9% изотоническом растворе. Для получения раствора соединения доставляющего агента или используют натриевую соль указанного соединения, или свободную кислоту превращают в его натриевую соль. Свободную кислоту соединения доставляющего агента превращают в натриевую соль, добавляя один эквивалент гидроксида натрия. Полученную смесь подвергают вихревому перемешиванию и помещают в устройство для ультразвуковой обработки (около 37°С). Показатель рН доводят примерно до 7,0-7,5 водным раствором HCl или NaOH. При необходимости добавляют дополнительное количество NaOH для достижения однородной растворимости и повторно регулируют показатель рН. Смесь подвергают вихревому перемешиванию для получения однородного раствора, при необходимости производя обработку ультразвуком. Раствор соединения доставляющего агента смешивают с даптомицином из исходного раствора (200 мг даптомицина/мл в 0,9% изотоническом растворе, и при необходимости показатель рН доводят до 6,0-7,0 с помощью NaOH или HCl). Исходный раствор хранят в замороженном виде (-20°С) завернутым в фольгу, перед использованием размораживают и постепенно нагревают примерно до 25°С. Смесь доставляющего агента и даптомицина подвергают вихревому перемешиванию при низкой скорости, получая однородный раствор. Показатель рН доводят примерно до 7,0-7,5 водным раствором NaOH. Раствор разводят 0,9% изотоническим раствором до требуемого объема (обычно 2,0 мл) и концентрации и до использования хранят завернутым в фольгу. Конечные дозы соединения доставляющего агента, даптомицина и объемы доз приведены в нижеследующей таблице 11.
Обычно используют описанные ниже методики определения требуемой дозы и отбора проб. Самцов крыс Sprague-Dawley с массой тела 200-250 г не кормят в течение 24 часов, после чего их анестезируют кетамином (44 мг/кг) и хлорпромазином (1,5 мг/кг) за 15 минут до введения лекарственного средства и при необходимости производят повторное введение анестезирующих средств для обеспечения необходимой степени наркоза. Экспериментальной группе из пяти животных вводят один из лекарственных растворов. Для этой цели используют французский катетер Rusch 8 длиной 11 см, соответствующий 1 мл шприцу с концом в виде пипетки. Шприц заполняют лекарственным раствором, пропуская раствор через катетер, который затем вытирают досуха. Катетер вводят в пищевод, оставляя перед резцами конец трубки длиной 1 см. Подачу раствора осуществляют нажатием на поршень шприца.
Пробы гепаринизированной крови крыс отбирают из вентральной хвостовой артерии через промежутки времени, равные обычно 0,25, 0,5, 0,75, 1,0, 2,0 и 4,0 часам, после введения лекарственного раствора и хранят на льду. Пробы крови центрифугируют со скоростью 11500 оборотов/мин в течение 4 минут при 4°С, получая плазму (супернатант), которую хранят при -70°С. Концентрации даптомицина в плазме измеряют при помощи изократической ВЭЖХ с обращенной фазой, выдерживая образцы во время анализа при температуре 4°С. Базовые значения, полученные при исследовании чистой плазмы, равны нулю.
Значения концентрации даптомицина в крови, полученные у животных в каждой экспериментальной группе, усредняют для каждого промежутка времени. Усредненные максимальные значения концентрации даптомицина (Сmax) и площади, соответствующие уровню даптомицина, под кривой (AUC) приведены в нижеследующей таблице 11.
Таблица 11. Даптомицин - пероральное введение
* AUC = Общая AUC (0 → ∞)
Приведенные выше патенты, заявки на патент, методы испытания и публикации полностью включены в данное описание изобретения в качестве ссылки.
Из приведенного выше подробного описания изобретения специалистам в данной области должны быть очевидны многие модификации настоящего изобретения. Все подобные модификации входят в объем прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ВИЧ-ИНТЕГРАЗЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2002 |
|
RU2284315C2 |
| СОЕДИНЕНИЯ И СОСТАВЫ ДЛЯ ДОСТАВКИ АКТИВНЫХ ВЕЩЕСТВ | 2005 |
|
RU2530889C2 |
| СОСТАВЫ И СМЕСИ ДЛЯ ДОСТАВКИ АКТИВНЫХ АГЕНТОВ | 2005 |
|
RU2403237C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛЗАМЕЩЕННЫХ 2-ОКСО-1,2-ДИГИДРОПИРИДИН-3,4-ДИКАРБОНИТРИЛОВ | 2015 |
|
RU2577526C1 |
| Четвертичные аммониевые соли на основе производных витамина В6 | 2015 |
|
RU2607522C1 |
| ПРОИЗВОДНЫЕ АРИЛМЕТОКСИ ИЗОИНДОЛИНА И КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ ИХ, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2011 |
|
RU2567753C2 |
| Азопроизводные аминофенолов, обладающие способностью ингибировать образование конечных продуктов гликирования | 2024 |
|
RU2839138C1 |
| Четвертичные аммониевые соединения на основе производных пентаэритрита и пиридоксина, обладающие антибактериальной активностью | 2023 |
|
RU2811203C1 |
| ГИДРОКСАМОВЫЕ КИСЛОТЫ, СОДЕРЖАЩИЕ ЯДРА 1,2,4- И 1,3,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ПОТЕНЦИАТОРОВ ДЕЙСТВИЯ АНТИБИОТИКОВ | 2023 |
|
RU2830361C1 |
| ЛИГАНДЫ, ТРОПНЫЕ К ПРОСТАТИЧЕСКОМУ СПЕЦИФИЧЕСКОМУ МЕМБРАННОМУ АНТИГЕНУ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ ДВОЙНЫХ КОНЪЮГАТОВ С ТЕРАПЕВТИЧЕСКИМИ АГЕНТАМИ НА ИХ ОСНОВЕ ДЛЯ КОМБИНИРОВАННОЙ ТЕРАПИИ ПСМА ЭКСПРЕССИРУЮЩИХ ОПУХОЛЕЙ | 2023 |
|
RU2841078C1 |
Изобретение относится к новым соединениям, предназначенным для доставки активных веществ к тканям, следующей формулы
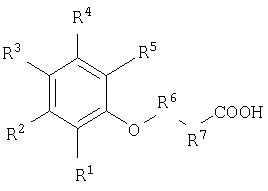
где значения для радикалов R1-R7 определены в п.1 формулы, и их фармацевтически приемлемым солям. Изобретение относится также к композициям, предназначенным для доставки активных веществ к тканям, содержащим: (А) активное вещество и (В) по меньшей мере, одно соединение, предназначенное для доставки активного вещества к тканям животного, формулы
где значения для радикалов R1-R7 определены в пп.3-5 формулы. Данное изобретение относится также к стандартной лекарственной форме, предназначенной для доставки активных веществ к тканям организма, способу получения указанных композиций и введения активных веществ для их доставки к тканям организма. 9 н. и 14 з.п. ф-лы, 11 табл.
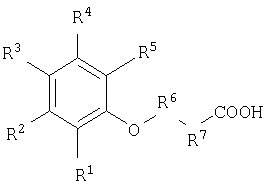
и выбрано из группы, включающей
и их фармацевтически приемлемые соли.
и выбрано из группы, включающей
и их фармацевтически приемлемые соли.
(A) активное вещество и
(B) по меньшей мере одно соединение, предназначенное для доставки активного вещества к тканям животного, формулы
которое выбрано из группы
и их фармацевтически приемлемые соли.
(A) активное вещество и
(B) по меньшей мере одно соединение, предназначенное для доставки активного вещества к тканям животного, формулы
и его фармацевтически приемлемые соли.
(A) активное вещество и
(B) по меньшей мере одно соединение, предназначенное для доставки активного вещества к тканям животного, формулы
которое выбрано из группы, включающей
и их фармацевтически приемлемые соли.
(A) композицию по пп.3-5 и
(B) (а) наполнитель,
(b) разбавитель,
(c) дезинтегратор,
(d) смазывающее вещество,
(e) пластификатор,
(f) краситель,
(g) носитель или
(h) любую их комбинацию.
(A) по меньшей мере одно активное вещество;
(B) по меньшей мере одно соединение по п.1 и,
(C) необязательно, носитель, приемлемый для введения.
и выбрано из группы, включающей
и их фармацевтически приемлемые соли.
Приоритет по пп.1-23 формулы установлен от 06.11.2000.
| US 4412041 A, 25.10.1983 | |||
| Способ бурения скважин и устройство для его осуществления | 1974 |
|
SU548711A1 |
| US 4410537 A, 18.10.1983 | |||
| US 4499299 A, 12.02.1985 | |||
| US 5783593 A, 21.07.1998 | |||
| DE 3206030 A1, 09.09.1982 | |||
| CASSEBAUM H | |||
| «Radiopaque media based on iodinated phenoxy fatty acids» CHEMICAL ABSTRACTS, 1964, vol 56, p.7191 | |||
| US 5747537 A, 05.05.1998 | |||
| Гербицидная композиция | 1977 |
|
SU668569A3 |
| Гербицидная композиция | 1977 |
|
SU730270A3 |
| SU | |||

