[1] Настоящая заявка испрашивает приоритет согласно предварительной заявке на патент США No. 60/576,088, поданной 1 июня 2004, предварительной заявке на патент США No. 60/576,397, поданной 1 июня 2004, предварительной заявке на патент США No. 60/576,105, поданной 1 июня 2004, предварительной заявке на патент США No. 60/571,090, поданной 14 мая 2004, предварительной заявке на патент США No. 60/571,092, поданной 14 мая 2004, предварительной заявке на патент США No. 60/571,195, поданной 14 мая 2004, предварительной заявке на патент США No. 60/571,194, поданной 14 мая 2004, предварительной заявке на патент США No. 60/571,093, поданной 14 мая 2004, предварительной заявке на патент США No. 60/571,055, поданной 14 мая 2004, предварительной заявке на патент США No. 60/571,151, поданной 14 мая 2004, предварительной заявке на патент США No. 60/571,315, поданной 14 мая 2004 г., предварительной заявке на патент США No. 60/571,144, поданной 14 мая 2004 г., и предварительной заявке на патент США 60/571,089, поданной 14 мая 2004 г., описание каждой из которых полностью включено в описание данной заявки посредством ссылок.
Область техники
[2] Настоящее изобретение относится к соединениям и составам для доставки активных веществ, таких как биологически или химически активные вещества, к мишени (органу). Эти соединения прекрасно подходят для формирования нековалентных смесей с активными веществами для перорального или другого способа введения в организм животных. Также описаны способы приготовления и введения таких композиций.
Предпосылки создания изобретения
[3] Обычные способы доставки активных веществ часто строго ограничены биологическими, химическими и физическими барьерами. Типично, эти барьеры налагает окружающая среда, через которую происходит доставка, окружающая среда непосредственно вокруг мишени доставки и/или сама мишень. Особенно уязвимы по отношению к таким барьерам биологически и химически активные вещества.
[4] При доставке в организм животного биологически и химически активных фармакологических и терапевтических агентов барьеры создает организм. Примерами физических барьеров являются кожа, эпителий, липидные бислои и мембраны различных органов, которые относительно непроницаемы для определенных активных веществ, но которые необходимо преодолеть для достижения мишени доставки, например, кровеносной системы. Химические барьеры без ограничения включают изменения рН в желудочно-кишечном (ЖКТ) тракте и разрушение ферментами.
[5] Эти барьеры имеют особое значение при разработке систем пероральной доставки. Пероральная доставка многих активных веществ была бы выбрана способом введения их в организм животных, если бы не биологические, химические и физические барьеры. К многочисленным веществам, которые традиционно не подлежат пероральному введению, относятся биологически или химически активные пептиды, например, кальцитонин или инсулин; полисахариды, такие как мукополисахариды, включая, без ограничений, гепарин; гепариноиды; антибиотики и другие органические субстанции. В желудочно-кишечном тракте под действием кислотного гидролиза, ферментов и т.п. эти вещества могут быстро инактивироваться или разрушаться. Кроме того, поглощению могут препятствовать размеры и структура макромолекулярных лекарств.
[6] Раннее известные способы перорального введения уязвимых фармакологических агентов основаны на совместном введении адъювантов (например, резорцинов и неионных поверхностно-активных веществ, таких как олеиловый эфир полиоксиэтилена и н-гексадецилполиэтиленовый эфир) с целью искусственного увеличения проницаемости кишечных стенок, а также на совместном введении ингибиторов ферментов (например, ингибиторах трипсина, диизопропилфторфосфата (ДФФ) и трасилола) для предотвращения разложения (агентов) ферментами. В качестве систем доставки инсулина и гепарина также были описаны липосомы. Однако широкое использование таких систем доставки лекарственных препаратов невозможно, поскольку (1) эти системы требуют токсичных количеств адъювантов или ингибиторов; (2) не найдено подходящих нагрузок (транспортируемых агентов) с низкомолекулярным весом, т.е. активных веществ; (3) эти системы обладают низкой стабильностью и недостаточным сроком годности при хранении; (5) системы не в состоянии защитить активное вещество (нагрузку); (6) системы изменяют активное вещество, делая его вредным; или (7) системы не позволяют или не стимулируют абсорбцию активного вещества.
[7] Для доставки лекарственных препаратов используют протеиновые микрогранулы. См., например, Патент США Nos. 5,401,516; 5,443,841; и Re. 35,862. Кроме того, для доставки лекарств используют специфически модифицированные аминокислоты. См., например, патент США Nos. 5,629,020; 5,643,957; 5,766,633; 5,776,888; и 5,866,536, и публикации международных заявок WO 98/49135; WO 00/06534; WO 00/07979; WO 00/40203; WO 00/47188; WO 00/50386; WO 00/59863; WO 01/32130, WO 01/32596, WO 01/44199, WO 01/51454, WO 02/02509, WO 02/15959, WO 02/16309, WO 02/20466, WO 02/19969, WO 02/69937, WO 03/45306.
[8] Еще недавно для получения полимерных веществ, обеспечивающих доставку, полимер конъюгировали с модифицированной аминокислотой или ее производным через связывающую группу. Модифицированным полимером может быть любой полимер, но предпочтительными полимерами являются, но не ограничиваются ими, полиэтиленгликоль (ПЭГ) и его производные. См., например, публикацию международной заявки WO 00/40203.
Однако все еще существует необходимость в простых дешевых системах доставки, которые легко приготовить и которые могут различными путями доставлять в организм широкий ряд активных веществ.
Краткое описание изобретения
[9] Настоящее изобретение обеспечивает соединения и композиции, которые способствуют доставке активных веществ. К соединениям, способствующим доставке (соединения-носители), предложенным в настоящем изобретении, относятся соединения, показанные ниже, и их фармацевтически допустимые соли:

в котором:
R1 представляет собой -(CH2)m-R8, где m=0 или 1;
R2-R6 независимо выбраны из группы, включающей водород, гидроксил, галоген, алкил C1-C4, алкенил С2-С4, алкинил С2-С4, алкокси C1-C4 и циано;
R7 выбран из группы, включающей алкил C1-С10, алкенил C2-С10 и алкинил С2-С10;
R8 выбран из группы, включающей циклопентил, циклогексил и фенил, при этом, когда R8 является фенилом, m=1; причем
R8 возможно замещен алкилом C1-С4, алкокси C1-С4, галогеном или гидроксилом или их комбинацией.
[10] В одном примере реализации R7 является С1 алкилом.
[11] В другом примере реализации R7 является С2 алкилом.
[12] В другом примере реализации R7 является С3 алкилом.
[13] В другом примере реализации R7 является C4 алкилом.
[14] В другом примере реализации R7 является C5 алкилом.
[15] В другом примере реализации R7 является С6 алкилом.
[16] В другом примере реализации R7 является С7 алкилом.
[17] В другом примере реализации R7 является С8 алкилом.
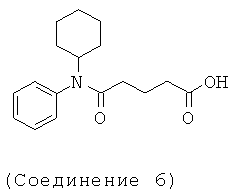
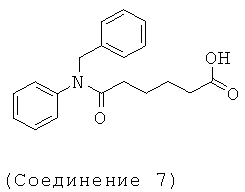
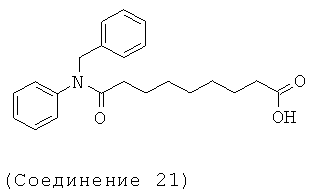
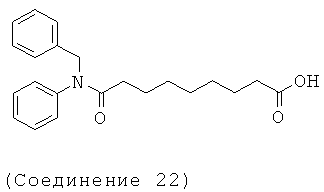
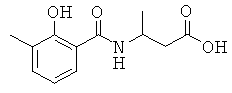
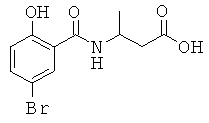
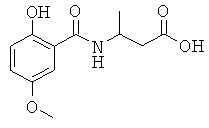
[18] Предпочтительными соединениями являются, помимо прочего, следующие соединения и их соли, пригодные для фармацевтического использования:
















[19] К другим соединениям-носителям, предложенным в

настоящем изобретении, относятся соединения, имеющие формулу: (Соединение В) и их фармацевтические допустимые соли, при этом:
R1 является алкилом C1-С6 или алкенилом С2-С6,
R2-R6 выбирают независимо друг от друга из группы, содержащей водород, гидроксил, галоген, алкил C1-С4, алкенил С2-C4, алкинил С2-C4, алкокси C1-С4, и циано, и
R7 выбирают из группы, содержащей алкил C1-С10, алкенил С2-С10, и алкинил С2-С10.
[20] В одном примере реализации R2-R6 представляют собой независимо друг от друга водород, метил, галоген, метокси.
[21] В другом примере реализации R2-R6 представляют собой независимо друг от друга водород, метил, хлор, метокси.
[22] В другом примере реализации R2-R6 представляют собой независимо друг от друга водород, метил, фтор, метокси.
[23] В другом примере реализации R2-R6 представляют собой независимо друг от друга водород, метил, йод, метокси.
[24] В другом примере реализации R2-R6 представляют собой независимо друг от друга водород, метил, бром, метокси.
[25] В другом примере реализации R1 является алкилом C1-С3.
[26] В другом примере реализации R1 является метилом.
[27] В другом примере реализации R1 является этилом.
[28] В другом примере реализации R1 является изопропилом.
[29] В другом примере реализации R2 является метилом.
[30] В другом примере реализации R2 является галогеном.
[31] В другом примере реализации R2 является хлором.
[32] В другом примере реализации R2 является фтором.
[33] В другом примере реализации R4 является метилом.
[34] В другом примере реализации R4 является метокси.
[35] В другом примере реализации R4 является галогеном.
[36] В другом примере реализации R4 является хлором.
[37] В другом примере реализации R4 является фтором.
[38] В другом примере реализации R4 является циано.
[39] В другом примере реализации R7 является C1 алкилом.
[40] В другом примере реализации R7 является C2 алкилом.
[41] В другом примере реализации R7 является С2 алкилом, разветвленным метилом.
[42] В другом примере реализации R7 является С3 алкилом.
[43] В другом примере реализации R7 является С3 алкилом, разветвленным метилом.
[44] В другом примере реализации R7 является C4 алкилом.
[45] В другом примере реализации R7 является C5 алкилом.
[46] В другом примере реализации R7 является С6 алкилом.
[47] В другом примере реализации R7 является С7 алкилом.
[48] В другом примере реализации R7 является С8 алкилом.
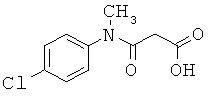
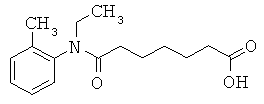
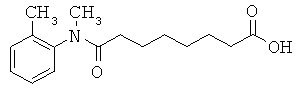
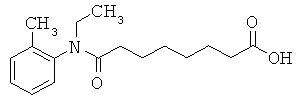
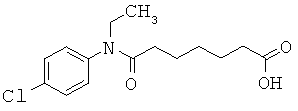
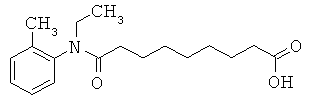
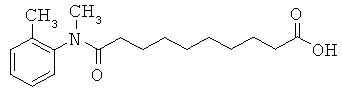
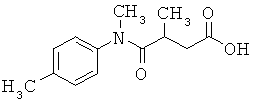
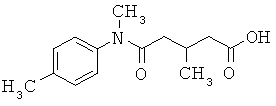
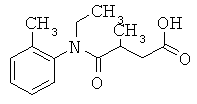
[49] К предпочтительным соединениям относятся, помимо прочего, следующие соединения и их фармацевтически допустимые соли:



























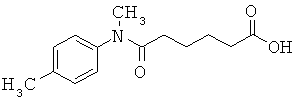



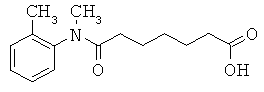

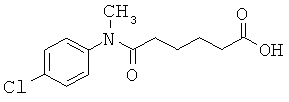






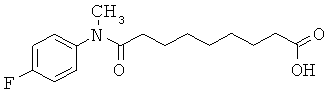























[50] К другим соединениям-носителям, предложенным в настоящем изобретении, относятся соединения, имеющие формулу:

и их фармацевтические допустимые соли, при этом
n = от 1 до 9, и
R1-R5 представляют собой, независимо друг от друга, водород, алкил C1-C4, алкокси C1-С4, алкенил C2-C4, галоген, гидроксил, -NH-С(O)-СН3, или -O-С6Н5.
[51] Предпочтительными соединениями веществ-носителей, являются, без ограничений, соединения, имеющие следующие формулы, и их соли:
[52] В одном примере реализации n=2-8.
[53] В другом примере реализации n=8.
[54] В другом примере реализации n=7.
[55] В другом примере реализации n=6.
[56] В другом примере реализации n=5.
[57] В другом примере реализации n=4.
[58] В другом примере реализации n=3.
[59] В другом примере реализации n=2 и остальные R группы являются водородами.
[60] В другом примере реализации n=8 и остальные R группы являются водородами.
[61] В другом примере реализации n=7 и остальные R группы являются водородами.
[62] В другом примере реализации n=6 и остальные R группы являются водородами.
[63] В другом примере реализации n=5 и остальные R группы являются водородами.
[64] В другом примере реализации n=4 и остальные R группы являются водородами.
[65] В другом примере реализации n=3 и остальные R группы являются водородами.
[66] В другом примере реализации n=2 и остальные R группы являются водородами.
[67] В другом примере реализации R1 и R5 являются водородами.
[68] В другом примере реализации R1 и R5 являются водородами и n=2.
[69] В другом примере реализации R3 является гидроксилом.
[70] В другом примере реализации R3 является гидроксилом и N=8.
[71] В другом примере реализации R1 является гидроксилом.
[72] В другом примере реализации R1 является гидроксилом и N=8.
[73] В другом примере реализации R3 является метокси.
[74] В другом примере реализации R3 является метокси и N=2.
[75] В другом примере реализации R3 является метокси и N=3.
[76] В другом примере реализации R2 и R4 являются галогенами и N=2.
[77] В другом примере реализации R2 и R4 являются фторами.
[78] В другом примере реализации R2 и R4 являются фторами и N=2.
[79] В другом примере реализации R1 и R3 являются метилами.
[80] В другом примере реализации R1 и R3 являются метилами и N=2.
[81] В другом примере реализации R2 и R4 являются метилами, R3 является метокси и N=4.
[82] В другом примере реализации R3 является изопропилом.
[83] В другом примере реализации R3 является изопропилом и N=3.
[84] В другом примере реализации R1 является метокси.
[85] В другом примере реализации R1 является метокси и N=2.
[86] В другом примере реализации R3 является галогеном.
[87] В другом примере реализации R3 является галогеном и N=2.
[88] В другом примере реализации R3 является фтором и N=2.
[89] В другом примере реализации R3 является метокси.
[90] В другом примере реализации R3 является метокси и N=4.
[91] В другом примере реализации R2 и R4 являются метилами.
[92] В другом примере реализации R2 и R4 являются метилами и N=2.
[93] В другом примере реализации R2 и R4 являются метилами и N=4.
[94] В другом примере реализации R2 и R4 являются метилами и N=6.
[95] В другом примере реализации R2 и R3 являются метилами и N=4.
[96] В другом примере реализации R2 и R3 являются метилами и N=2.
[97] В другом примере реализации R1 и R4 являются метилами и N=2.
[98] В другом примере реализации R1 и R4 являются галогенами.
[99] В другом примере реализации R1 и R4 являются галогенами и N=2.
[100] В другом примере реализации R1 и R4 являются галогенами и N=4.
[101] В другом примере реализации R1 и R4 являются хлорами.
[102] В другом примере реализации R1 и R4 являются хлорами и N=2.
[103] В другом примере реализации R1 и R4 являются хлорами и N=4.
[104] В другом примере реализации R1 и R4 являются гидроксилами.
[105] В другом примере реализации R1 и R4 являются гидроксилами и N=8.
[106] В одном примере реализации соединения 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 128, 129, 130, 132, 133, 134, 136 и/или 138 исключены из соединения С.
[107] К предпочтительным соединениям относятся, без ограничений, соединения, показанные ниже.




















[108] Другие соединения веществ-носителей, предложенные в настоящем изобретении, включают соединения, имеющие формулу:

и их фармацевтически допустимые соли, при этом
R1-R4 представляют собой, независимо друг от друга водород, алкил C1-C4, алкенил С2-С4, галоген, алкокси C1-C4 или гидроксил.
[109] В одном примере реализации R1-R4 представляют собой, независимо друг от друга, водород, метил, метокси, галоген, или изопропил.
[110] В одном примере реализации R1-R4 все являются водородами.
[111] В другом примере реализации R2 и R4 являются галогенами, предпочтительно бромом или предпочтительно хлором, или предпочтительно йодом, или предпочтительно фтором.
[112] В другом примере реализации R2 и R4 являются галогенами, предпочтительно бромом или предпочтительно хлором, или предпочтительно йодом, и R1 и R3 являются водородами.
[113] В другом предпочтительном примере реализации R2 и R4 являются изопропилами.
[114] В другом предпочтительном примере реализации R2 и R4 являются изопропилами, и R1 и R3 являются водородами.
[115] В другом предпочтительном примере реализации R4 является метилом.
[116] В другом предпочтительном примере реализации R4 является метилом и R1-R3 являются водородами.
[117] В другом предпочтительном примере реализации R3 является галогеном, предпочтительно хлором.
[118] В другом предпочтительном примере реализации R3 является галогеном, предпочтительно хлором и R1, R2 и R4 являются водородами.
[119] В другом предпочтительном примере реализации R3 является метокси.
[120] В другом предпочтительном примере реализации R3 является метокси, и R1, R2 и R4 являются водородами.
[121] В другом предпочтительном примере реализации R2 является галогеном, предпочтительно бромом.
[122] В другом предпочтительном примере реализации R2 является галогеном, предпочтительно бромом, и R1, R2 и R4 являются водородами.
[123] В другом предпочтительном примере реализации R2 является галогеном, предпочтительно хлором.
[124] В другом предпочтительном примере реализации R2 является галогеном, предпочтительно хлором, и R1, R3 и R4 являются водородами.
[125] В другом предпочтительном примере реализации R2 является метокси.
[126] В другом предпочтительном примере реализации R2 является метокси, и R1, R3 и R4 являются водородами.
[127] В другом предпочтительном примере реализации R2 является метилом.
[128] В другом предпочтительном примере реализации R2 является метилом, и R1, R3 и R4 являются водородами.
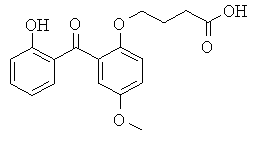
[129] Предпочтительные соединения-носители включают, без ограничений, соединения, имеющие следующие формулы, и их соли:







[130] Другие соединения-носители, предложенные в настоящем изобретении, включают соединения, имеющие формулу:

и их фармацевтически допустимые соли, при этом:
одна из групп R1-R5 имеет общую структуру -(СН2)n-СООН,
где n=0-6;
остальные четыре члена R1-R5 независимо друг от друга представляют собой водород, C1-C4 алкил, С2-С4 алкенил, галоген, C1-C4 алкокси, или гидроксил; и
R6-R10 независимо друг от друга представляют собой водород, C1-C4 алкил, С2-С4 алкенил, галоген, C1-C4 алкокси, или гидроксил.
[131] В одном примере реализации, n=0-4.
[132] В другом примере реализации, n=0.
[133] В другом примере реализации, n=1.
[134] В другом примере реализации, R1-R10 предпочтительно являются, независимо друг от друга, водородом, галогеном, метилом и метокси.
[135] В другом примере реализации, R1-R10 предпочтительно являются, независимо друг от друга, хлором, галогеном, метилом и метокси.
[136] В другом примере реализации, когда общая структура -(СН2)n-СООН присоединена к R1, остальные R группы являются водородами.
[137] В другом примере реализации, когда общая структура -(СН2)n-СООН присоединена к R1, остальные R группы являются водородами и n=0.
[138] В другом примере реализации, когда общая структура -(СН2)n-СООН присоединена к R1, остальные R группы являются водородами и n=1.
[139] В другом примере реализации, когда общая структура -(СН2)n-СООН присоединена к R3, остальные R группы являются водородами.
[140] В другом примере реализации, когда общая структура -(СН2)n-СООН присоединена к R3, остальные R группы являются водородами, и n=0.
[141] В другом примере реализации, когда общая структура -(СН2)n-СООН присоединена к R3, остальные R группы являются водородами.
[142] В другом примере реализации, когда общая структура -(СН2)n-СООН присоединена к R3, остальные R группы являются водородами и n=1.
[143] В другом примере реализации, R5 является метокси, когда общая структура -(СН2)n-СООН присоединена к R2.
[144] В другом примере реализации, R5 является метокси, когда общая структура -(СН2)n-СООН присоединена к R2, остальные R группы являются водородами.
[145] В другом примере реализации, R5 является метокси, когда общая структура -(СН2)n-СООН присоединена к R2, и n=0.
[146] В другом примере реализации, R5 является метокси, когда общая структура -(СН2)n-СООН присоединена к R2, и n=0, остальные R группы являются водородами.
[147] В другом примере реализации, R1 и R5 являются метилами, когда общая структура -(СН2)n-СООН присоединена к R3.
[148] В другом примере реализации, R1 и R5 являются метилами, когда общая структура -(СН2)n-СООН присоединена к R3, остальные R группы являются водородами.
[149] В другом примере реализации, R1 и R5 являются метилами, когда общая структура -(СН2)n-СООН присоединена к R3 и n=0.
[150] В другом примере реализации, R1 и R5 являются метилами, когда общая структура -(CH2)n-СООН присоединена к R3 и n=0, остальные R группы являются водородами.
[151] В другом примере реализации, R1 или R5 является метокси, когда общая структура -(СН2)n-СООН присоединена к R3 и n=0.
[152] В другом примере реализации, R1 или R5 является метокси, когда общая структура -(СН2)n-СООН присоединена к R3 и n=0, остальные R группы являются водородами.
[153] В другом примере реализации, R2 или R4 является галогеном, предпочтительно хлором, когда общая структура -(СН2)n-СООН присоединена к R3.
[154] В другом примере реализации, R2 или R4 является галогеном, предпочтительно хлором, когда общая структура -(СН2)n-СООН присоединена к R3, остальные R группы являются водородами.
[155] В другом примере реализации, R2 или R4 является галогеном, предпочтительно хлором, когда общая структура -(СН2)n-СООН присоединена к R3 и n=0.
[156] В другом примере реализации, R2 или R4 является галогеном, предпочтительно хлором, когда общая структура -(СН2)n-СООН присоединена к R3 и n=0, остальные R группы являются водородами.
[157] В одном примере реализации, соединения 152, 153, 154, 155, 156, 157, и/или 158 исключены из соединения Е.
[158] Другие соединения-носители для доставки веществ, предложенные в настоящем изобретении, включают соединения формулы:

и их фармацевтически допустимые соли, при этом:
n = от 1 до 9; и
R1-R9 представляют собой, независимо друг от друга, водород, C1-C4 алкил, С2-С4 алкенил, галоген, C1-С4 алкокси, или гидроксил.
[159] Согласно одному предпочтительному примеру реализации, n=3-7, предпочтительно, в одном предпочтительном примере реализации, n=3, предпочтительно, в другом предпочтительном примере реализации, n=4; предпочтительно, в другом предпочтительном примере реализации, n=5; предпочтительно, в другом предпочтительном примере реализации, n=6; предпочтительно, в другом предпочтительном примере реализации, n=7.
[160] Согласно другому предпочтительному примеру, R1-R8 является водородом.
[161] Согласно другому предпочтительному примеру, R3 является галогеном, предпочтительно, в одном примере реализации, R3 является хлором, предпочтительно, в другом примере реализации, R3 является бромом.
[162] Согласно другому предпочтительному примеру, R2 является метокси.
[163] Согласно другому предпочтительному примеру, R2 является метилом.
[164] Согласно другому предпочтительному примеру, R3 является метокси.
[165] Согласно другому предпочтительному примеру, R3 является метилом.
[166] Согласно другому предпочтительному примеру, R6 является метокси.
[167] Согласно другому предпочтительному примеру, R9 является водородом.
[168] Согласно другому предпочтительному примеру, R9 является гидроксилом.
[169] Согласно другому предпочтительному примеру, R9 является галогеном, предпочтительно, в одном примере реализации хлором.
[170] Согласно другому предпочтительному примеру, R3 и R6 оба являются метокси.
[171] Согласно другому предпочтительному примеру, R3 и R6 оба являются метокси и остальные R группы являются водород.
[172] Согласно другому предпочтительному примеру, R2 является метилом и R3 является хлором.
[173] Согласно другому предпочтительному примеру, R2 является метилом и R3 является хлором и остальные R группы являются водородами.
[174] Согласно другому предпочтительному примеру, R2 является метилом и R9 является хлором.
[175] Согласно другому предпочтительному примеру, R2 является метилом и R9 является хлором и остальные R группы являются водородами.
[176] Согласно другому предпочтительному примеру, R3 является метилом и R9 является хлором.
[177] Согласно другому предпочтительному примеру, R3 является метилом и R9 является хлором и остальные R группы являются водородами.
[178] Предпочтительные соединения-носители для доставки веществ включают, без ограничений, соединения, имеющие следующие формулы, и их соли:
[179] Другие соединения-носители для доставки, предложенные в настоящем изобретении, включают соединения, имеющие формулу:

и их фармацевтически допустимые соли, при этом:
R1-R5 представляют собой, независимо друг от друга, C1-С4 алкил, C2-С4 алкенил, галоген, C1-С4 алкокси, гидроксил, или -O-(СН2)n-СООН (где n=1-12);
по меньшей мере одна из R1-R5 групп имеет общую структуру
-O-(СН2)n-СООН
где n=1-12; и
R6-R10 представляют собой, независимо друг от друга, водород, C1-C4 алкил, С2-С4 алкенил, галоген, C1-С4 алкокси, или гидроксил.
[180] Предпочтительно, чтобы только одна из R1-R5 групп имела формулу -(СН2)n-СООН. Другими словами, четыре члена R1-R5 представляют собой, независимо друг от друга, водород, C1-C4 алкил, C2-C4 алкенил, галоген, C1-С4 алкокси, или гидроксил, и оставшийся член R1-R5 является -O-(СН2)n-СООН (где n=1-12).
[181] В одном предпочтительном примере реализации n=1-12.
[182] В другом предпочтительном примере реализации n=1-10.
[183] В другом предпочтительном примере реализации n=1-6.
[184] В другом предпочтительном примере реализации n=1-4.
[185] В другом предпочтительном примере реализации n=10.
[186] В другом предпочтительном примере реализации n=4.
[187] В другом предпочтительном примере реализации n=1.
[188] Когда общая структура -(СН2)n-СООН присоединена к R1, все другие R группы являются водородами.
[189] Когда общая структура -(СН2)n-СООН присоединена к R1, все другие R группы являются водородами и n=3.
[190] Когда общая структура -(СН2)n-СООН присоединена к R3, все другие R группы являются водородами.
[191] Когда общая структура -(СН2)n-СООН присоединена к R3, все другие R группы являются водородами и n=1.
[192] Когда общая структура -(СН2)n-СООН присоединена к R3, все другие R группы являются водородами и n=4.
[193] Когда общая структура -(СН2)n-СООН присоединена к R3, все другие R группы являются водородами и n=10.
[194] Предпочтительные соединения включают, но не ограничиваются ими, следующие соединения и их фармацевтически допустимые соли:



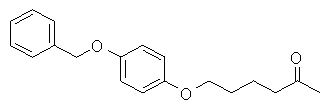
[195] Можно использовать также смеси этих соединений-носителей.
[196] Изобретение также обеспечивает состав, содержащий соединение-носитель, предложенный в настоящем изобретении, и, по меньшей мере одно активное вещество. Эти составы доставляют активные вещества в выбранные биологические системы, при этом биологическая доступность активных веществ увеличивается или улучшается по сравнению с введением активных веществ без соединений-носителей.
[197] Также обеспечиваются единичные дозируемые формы, содержащие эти составы. Единичные дозы могут быть в жидком или твердом виде, например таблетки, капсулы или частиц, в том числе порошка или саше.
[198] Другим вариантом реализации является способ введения активного вещества в организм животного путем введения состава, содержащего по меньшей мере одно из соединений-носителей, предложенных в настоящем изобретении, и активное вещество. Способы введения включают пероральное введение, введение в толстую кишку и введение через легкие.
[199] Еще одним вариантом реализации является способ лечения заболевания или достижения желаемого физиологического эффекта в организме животного путем введения состава, предложенного в настоящем изобретении.
[200] Еще одним примером реализации является введение состава, предложенного в настоящем изобретении, в организм животного, которому этот состав принесет благоприятный эффект, и/или животному, которое нуждается в активном веществе.
[201] Еще одним примером реализации является способ приготовления состава, предложенного в настоящем изобретении, путем смешивания по меньшей мере одного соединения-носителя, предложенного в настоящем изобретении, и, по меньшей мере одного активного вещества.
Подробное описание изобретения
Определения
[202] Используемые здесь и в прилагаемой формуле изобретения формы единственного числа включают многочисленные объекты, если содержание ясно не указывает обратное. Таким образом, например, термин «молекула» подразумевает одну или более таких молекул, термин «реактив» подразумевает один или более таких различных реактивов, термин «способ» включает равнозначные стадии или способы, известные типичному специалисту в данной области, которые могут быть модифицированы или заменены способами, описанными здесь.
[203] Термин «полиформ» относится к различным кристаллографическим формам вещества.
[204] Использующийся здесь термин «гидрат» включает, без ограничений, (i) вещество, содержащее воду и объединенное с ней в молекулярную форму и (ii) кристаллическое вещество, содержащее одну или более молекул воды, включенных в кристаллическую решетку, или кристаллическое вещество, содержащее свободную воду
[205] Используемый здесь термин «сольват» включает, без ограничений, молекулярный или ионный комплекс молекул или ионов растворителя с молекулами или ионами вещества-носителя.
[206] Термин «вещество-носитель» относится к любым соединениям-носителям, раскрытым или оформленным здесь ссылкой, включая их фармацевтически допустимые соли.
[207] «Эффективное количество фармацевтического состава» представляет собой количество описанного фармацевтического состава, которое является эффективным для лечения или предотвращения состояния субъекта, которому оно вводится в течение определенного периода времени, например, обеспечивает терапевтический эффект во время необходимого интервала дозирования.
[208] Термин «лечить», «лечение» или «подвергнутый лечению» относится к профилактическому предупреждению, выздоровлению, заживлению, облегчению, смягчению, изменению, излечению, улучшению, или влиянию на состояние (например, заболевание), симптомы состояния, или предрасположенность к состоянию.
[209] «Эффективное количество вещества для доставки» представляет собой количество вещества-носителя, которое способствует абсорбции необходимого количества активного вещества.
[210] Термин «субъект» включает млекопитающих, таких как грызуны, коровы, свиньи, собаки, кошки, приматы и, в частности, люди.
[211] Используемый здесь термин «ППК» означает площадь под кривой «концентрация плазмы - время», которую рассчитывают по формуле трапеции в течение всего интервала дозирования, например, 24-часового интервала.
[212] Термин «среднее», стоящий перед фармакокинетическим значением (например, средний пик) представляет собой среднее арифметическое значение фармакокинетического параметра, если не указано иное.
[213] Использующийся здесь термин «приблизительно» означает в пределах 10% отданной величины, предпочтительно в пределах 5%, и более предпочтительно в пределах 1% от данной величины. В качестве альтернативы, термин «приблизительно» означает, что значение может уменьшаться в пределах научно допустимого интервала погрешности для этого типа значения, который зависит от того, насколько качественно можно выполнить измерение, используя имеющиеся инструментальные средства.
[214] «Показание» означает использование, при котором вводят лекарство либо для предотвращения, либо лечения состояния, и может применяться попеременно с термином «лечение», «подвергнутый лечению» или «лечить».
[215] Использующийся здесь термин «замещенный» включает, без ограничений, замещение любым одним или любыми комбинациями следующих заместителей: галогены, гидроксид, алкил C1-C4 и алкокси C1-C4.
[216] Термины «алкил», «алкокси», «алкилен», «алкенилен», «алкил (арилен)», и «арил (алкилен)» включает, без ограничений, линейные и разветвленные алкил, алкокси, алкилен, алкенилен, алкил(арилен), и арил(алкилен)-группы, соответственно.
[217] Термином «пептид YY» или «ПYY» обозначают полипептид Пептид YY, полученный или извлеченный из любого биологического вида. Таким образом, термин «ПYY» включает как человеческий пептид полной длины, содержащий 36 аминокислот, как изложено в SEQ ID NO: в Международной публикации No. WO 02/47712 (которая является РСТ дубликатом Публикации патентов США No. 2002/0141985, оформленной здесь ссылкой) и Tatemoto, Proc Natl Acad Sci U.S.A. 79:2514-8, 1982, так и видовые вариации ПYY, в том числе, например, ПУУ мышей, хомяков, цыплят, коров, крыс и собак. Под термином «агонист ПYY» подразумевается любое соединение, которое вызывает воздействие, аналогичное действию ПYY, снижающее доступность питательных веществ, например соединение (1), обладающее активностью при приеме пищи, опорожнении желудка, панкреатической секреции, или пробе потери веса, как описано в Примерах 1, 2, 5, или 6 в WO 02/47712 и в публикации патентов США No. 2002/0141985, и (2) которое при анализе специфично связывается рецептор Y (Пример 10 в WO 02/47712 и Публикации патентов США No. 2002/0141985) или при анализе конкурентного связывания с меченным ПYY или ПYY [3-36] из определенных тканей, содержащих избыток Y рецепторов, в том числе, например, из области пострема (Пример 9 в WO 02/47712 и в Публикации патентов США No. 2002/0141985), при этом агонист ПYY не является панкреатическим полипептидом. Предпочтительно, чтобы агонисты ПYY связывались в таких способах анализа со сродством, большим чем приблизительно 1 мкМ, и более предпочтительно со сродством, большим чем от приблизительно 1 до приблизительно 5 мкМ.
[218] Такие агонисты могут содержать полипептид, имеющий функциональный домен ПYY, активный фрагмент ПYY, химический реагент или малую молекулу. Агонисты ПYY могут быть пептидными или непептидными соединениями и включать «аналоги агониста ПYY», к которым относится любое соединение, структурно похожее на ПYY и обладающее активностью ПYY, типично в силу связывания или иного способа прямого или косвенного взаимодействия с рецептором ПYY или другим рецептором или рецепторами, с которыми ПYY сам может взаимодействовать, вызывая биологический отклик. Такие соединения включают производные ПYY, фрагменты ПYY, удлиненные молекулы ПYY, содержащие более 36 аминокислот, укороченные молекулы ПYY, содержащие менее 36 аминокислот, и замещенные молекулы ПYY, содержащие одну или более разных аминокислот, или любые комбинации вышеуказанных соединений. Эти соединения также можно модифицировать, используя такие способы, как пегиляция (pegylation), амидирование, гликозилирование, ацилирование, сульфатирование, фосфорилирование, ацетилирование и циклизацию.
[219] Одним таким аналогом агониста ПYY является ПYY [3-36], обозначенный как SEQ ID NO: в WO 02/47712 и Публикации патентов США No. 2002/0141985; Eberlein, Eysselein et al., Peptides 10:797-803 (1989); и Grandy, Schimiczek et al., Regul Pept 51:151-9 (1994). Полипептиды с числами в скобках относятся к укороченным полипептидам, имеющим последовательность пептида с полной длиной во всех положениях аминокислот, приведенных в скобках. Таким образом, ПYY [3-36] имеет последовательность, идентичную ПYY по всем аминокислотам от 3 до 36. Иммунореактивность ПУУ [3-36] составляет приблизительно 40% от общей пептидной YY-подобной иммунореактивности в интестинальных экстрактах человека и собаки и от приблизительно 36% от общей иммунореактивности пептида YY в плазме при голодном состоянии субъекта до немного выше 50% в сытом состоянии. ПYY [3-36] по видимому представляет продукт расщепления пептида YY дипептидил-пептидазой-IV (ДПП4). Пептид YY[3-36] является, по имеющимся сообщениям, селективным лигандом у Y2 и Y5 рецепторов, которые представляются фармакологически уникальными в укороченных Y аналогах нейропептидов предпочтительно с атомами N на концах (например, С концевые фрагменты). Агонист ПYY может связываться с ПYY рецептором с более высоким или низким сродством, при этом он демонстрирует более длинный или более короткий период полувыведения in vivo или in vitro, или является более или менее эффективным, чем природный DYY.
[220] Другие подходящие агонисты ПYY включают агонисты, описанные в Международной публикации No. WO 98/20885, которая оформлена ссылкой.
[221] Используемый здесь термин «гепарин» относится ко всем формам гепарина, включая, без ограничений, нефракционированный гепарин, гепариноиды, дерматаны, хондроитины, гепарин с низкомолекулярным весом (например, тинзапарин (в том числе тинзапарин натрия)), гепарин с очень низкомолекулярный весом, и гепарин с ультранизкомолекулярный весом. Неограничивающие примеры включают, нефракционированный гепарин, например, гепарин натрия (например, гепарин натрия USP, имеющийся в Scientific Protein Labs of Waunakee, Висконсин). В общем случае, гепарин имеет молекулярный вес от приблизительно 1,000 или 5,000 до приблизительно 30,000 дальтонов. Термин «гепарин с низкомолекулярным весом» в общем случае относится к гепарину, в котором, по меньшей мере, приблизительно 80% (по весу) гепарина имеет молекулярный вес между приблизительно 3000 и приблизительно 9000 дальтонов. Неограничивающие примеры гепарина с низкомолекулярным весом включают, тинзапарин, эноксаприн и далтипарин. Тинзапарин был одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA US, США) для лечения острого симптоматического тромбоза глубоких вен с легочной эмболией или без нее при его введении совместно с варфарином натрия. Натриевую соль тиназапарина от Pharmion Corporation™ можно приобрести под торговой маркой Innohep of Boulder, Колорадо. Термин «гепарин с очень низкомолекулярным весом» в общем случае относят к гепарину, в котором по меньшей мере 80% (по весу) гепарина имеет молекулярный вес между приблизительно 1500 и приблизительно 5000 дальтонов. Неограничивающим примером гепарина с очень низкомолекулярным весом является бемипарин. Термин «гепарин с ультранизким молекулярным весом» в общем случае относится к гепарину, в котором по меньшей мере приблизительно 80% (по весу) гепарина имеет молекулярный вес между приблизительно 1000 и приблизительно 2000 дальтонов. Неограничивающим примером гепарина с ультранизким молекулярным весом является фондипаринакс.
Реагенты-носители
[222] Реагенты-носители, предложенные в настоящем изобретении, могут быть в форме свободной кислоты или фармацевтически допустимой соли. Подходящие фармацевтически допустимые соли включают, без ограничений, органические и неорганические соли, например, соли щелочных металлов, таких как натрий, калий и литий; соли щелочно-земельных металлов, таких как магний, кальций и барий; соли аммония; основные аминокислоты, такие как лизин и аргинин; и органические амины, такие как диметиламин или пиридин. В одном примере реализации соли представляют собой натриевые соли. Соли могут быть моно- или поливалентными солями, такими как мононатриевые соли и динатриевые соли. Соли могут также представлять собой сольваты, в том числе сольваты этанола и гидраты. Примеры фармацевтически допустимых солей включают, без ограничений, натриевые соли, соляную кислоту, серную кислоту, фосфорную кислоту, лимонную кислоту, уксусную кислоту, сульфаты, фосфаты, хлориды, бромиды, иодиды, ацетаты, пропионаты, бромистоводородную кислоту, гидроксид натрия, гидроксид калия, гидроксид аммония, и карбонат калия. Эти соли могут быть приготовлены способами, известными в данной области. Например, соли натрия можно приготовить путем растворения реагента-носителя в этаноле и добавления водного гидроксида натрия. Реагент-носитель можно очистить путем перекристаллизации или фракционирования на одном или более твердых хроматографических носителях, единичных или соединенных последовательно. Подходящие растворители для перекристаллизации включают, без ограничений, ацетонитрил, метанол и тетрагидрофуран. Фракционирование можно выполнять (i) на подходящем хроматографическом носителе, таком как оксид алюминия, с использованием смесей метанол/н-пропанол в качестве подвижной фазы, (ii) с помощью обратнофазной хроматографии, используя смеси трифторуксусная кислота/ацетонитрил в качестве подвижной фазы, или (iii) путем ионно-обменной хроматографии с использованием воды или подходящего буфера в качестве подвижной фазы. При использовании анионно-обменной хроматографии, предпочтительно, чтобы градиент содержания хлорида натрия составлял 0-500 мМ.
[224] Реагент-носитель может содержать полимер, конъюгированный с ним с помощью связывающей группы, выбранной из группы, состоящей из -NHC(O)NH-, -C(O)NH-,-NHC(O), -ООС-, -СОО-, -NHC(O)O-, -OC(O)NH-, -CH2NH -NHCH2-, -CH2NHC(O)O-, -OC(O)NHCH2-, -CH2NHCOCH2O-, -OCH2C(O)NHCH2-, -NHC(O)CH2O-, -OCH2C(O)NH-, -NH-, -O-, и связи углерод - углерод, при условии, что полимерный реагент-носитель не является полипептидом или полиаминокислотой. Полимер может являться любым полимером, включая, без ограничений, чередующиеся сополимеры, блоксополимеры и случайные сополимеры, которые безопасны для млекопитающих. Предпочтительные полимеры включают, без ограничений, полиэтилен; полиакрилаты; полиметакрилаты; поли(оксиэтилен); поли(пропилен); полипропиленгликоль; полиэтиленгликоль (ПЭГ); и их производные и комбинации. Молекулярный вес полимера в общем случае лежит в диапазоне от приблизительно 100 до приблизительно 200,000 дальтонов. Предпочтительно, чтобы молекулярный вес полимера находился в диапазоне от приблизительно 200 до приблизительно 10,000 дальтонов. В одном примере реализации молекулярный вес полимера лежит в интервале от приблизительно 200 до приблизительно 600 дальтонов и более предпочтительно лежит в интервале от приблизительно 300 до приблизительно 550 дальтонов. Патент США No. 6,627,228 оформлен здесь полной ссылкой.
[225] Количество реагента-носителя в твердом фармацевтическом составе является эффективным количеством и может быть определено для конкретного состава способами, известными специалисту в данной области. В общем случае, весовое соотношение реагента-носителя к активному веществу находится в интервале от примерно 0.1:1 до примерно 1000:1. Весовое соотношение будет варьировать в зависимости от активного вещества и конкретного показания, согласно которому активное вещество вводят в организм.
[226] Другие подходящие реагенты-носители, предложенные в настоящем изобретении, описаны в Патентах США Nos. 6,846,844, 6,699,467, 6,693,208, 6,693,208, 6,693,073, 6,663,898, 6,663,887, 6,646,162, 6,642,411, 6,627,228, 6,623,731, 6,610,329, 6,558,706, 6,525,020, 6,461,643, 6,461,545, 6,440,929, 6,428,780, 6,413,550, 6,399,798, 6,395,774, 6,391,303, 6,384,278, 6,375,983, 6,358,504, 6,346,242, 6,344,213, 6,331,318, 6,313,088, 6,245,359, 6,242,495, 6,221,367, 6,180,140, 5,541,155, 5,693,338, 5,976,569, 5,643,957, 5,955,503, 6,100,298, 5,650,386, 5,866,536, 5,965,121, 5,989,539, 6,001,347, 6,071,510, и 5,820,881. Реагенты-носители, предложенные в настоящем изобретении, также описаны в Публикации патентных заявок США Nos. 20050009748, 20040110839, 20040106825, 20040068013, 20040062773, 20040022856, 20030235612, 20030232085, 20030225300, 20030198658, 20030133953, 20030078302, 20030072740, 20030045579, 20030012817, 20030008900, 20020155993, 20020127202, 20020120009, 20020119910, 20020102286, 20020065255, 20020052422, 20020040061, 20020028250, 20020013497, 20020001591, 20010039258, и 20010003001. Реагенты-носители, предложенные в настоящем изобретении, также описаны в Международных публикациях Nos. WO 2005/020925, WO 2004/104018, WO 2004/080401, WO 2004/062587, WO 2003/057650, WO 2003/057170, WO 2003/045331, WO 2003/045306, WO 2003/026582, WO 2002/100338, WO 2002/070438, WO 2002/069937, WO 02/20466, WO 02/19969, WO 02/16309, WO 02/15959, WO 02/02509, WO 01/92206, WO 01/70219, WO 01/51454, WO 01/44199, WO 01/34114, WO 01/32596, WO 01/32130, WO 00/07979, WO 00/59863, WO 00/50386, WO 00/47188, WO 00/40203, и WO 96/30036. Каждый из вышеперечисленных патентов США, опубликованных заявок США и опубликованных международных заявок, оформлены здесь ссылкой. Указанные реагенты-носители можно приготовить способами, известными в данной области, описанными, например, в вышеупомянутых патентах и опубликованных патентных заявках. Например, SNAC можно приготовить способами, известными в данной области, которые, например, описаны патентах США Nos. 5,650,386 и 5,866,536, и Публикации патентных заявок США No. 2002/0065255.
Активные вещества
[227] Активные вещества, подходящие для использования в настоящем изобретении, включают биологически активные вещества и химически активные вещества, в том числе, без ограничений, пестициды, фармакологические препараты и терапевтические агенты. Подходящие активные вещества включают вещества, которые становятся менее эффективными, неэффективными или разрушаются в желудочно-кишечном тракте, в том числе под действием кислотного гидролиза, ферментов и т.п. К подходящим активным веществам относят также те макромолекулярные вещества, физиохимические характеристики которых, в том числе, размер, структура или заряд, препятствуют или задерживают абсорбцию активных веществ при их пероральном приеме.
[228] Например, вещество, которое должно попасть в организм или которое может благоприятно влиять благодаря улучшенной фармакокинетике, в том числе доставке, например, когда пероральная биодоступность ограничена или несущественна. К этим веществам, являющимся биологически или химически активными веществами, пригодными для использования в настоящем изобретении, относятся, без ограничений, макромолекулы, такие как пептиды, в том числе белки и полипептиды, включая дипептиды; гормоны; и сахариды, в том числе, моносахариды, полисахариды, включая дисахариды, смеси мукополисахаридов; карбогидраты; липиды; и малые полярные органические молекулы (то есть полярные органические молекулы, имеющие молекулярный вес 500 дальтонов или меньше); нуклеозиды, другие органические соединения; и, в частности, соединения, не обладающие пероральной биодоступностью или с ограниченной пероральной биодоступностью, в том числе те соединения, которые сами не проникают (или у которых проникает только часть принятой дозы) через слизистую оболочку желудочно-кишечного тракта и/или восприимчивы к химическому разложению под действием кислот и ферментов в желудочно-кишечном тракте; или любые их комбинации.
[229] Дополнительными примерами являются, без ограничений, следующие вещества, в том числе их синтетические, природные или рекомбинантные источники:
[230] в том числе стимуляторы секреции, аналоги, фрагменты, миметики или полиэтиленгликоль (ПЭГ)-модифицированные производные этих соединений; или любые их комбинации.
Системы доставки
[231] Состав, предлагаемый в настоящем изобретении, содержит одно или более соединений-носителей, предлагаемых в настоящем изобретении, и одно или более активных веществ. В одном примере реализации одно или более соединений-носителей или соли этих соединений, или полиаминокислоты или пептиды, из которых эти соединения или соли образуют один или более блоков, можно использовать в качестве реагентов-носителей путем смешивания с активным веществом перед их введением для получения состава введения.
[232] Составы для введения могут быть в виде жидкости. Растворяющей средой может быть вода (например, для кальцитонина лосося, паратиреоидного гормона и эритропоэтина), 25% водный пропиленгликоль (например, для гепарина) и фосфатный буфер (например, для рчГР роста). Другие дозируемые носители содержат полиэтиленгликоль. Дозируемые растворы можно приготовить путем смешивания раствора соединения-носителя с раствором активного вещества непосредственно до введения. В другом примере реализации раствор соединения-носителя (или активного вещества) можно смешать с твердой формой активного вещества (или соединения-носителя). Соединение-носитель и активное вещество можно также смешивать в виде сухих порошков. Соединение-носитель и активное вещество также можно подмешивать во время технологического процесса.
[233] Дозируемые растворы могут по выбору содержать добавки, такие как фосфатные буферные соли, лимонную кислоту, гликоли или другие диспергирующие агенты. В раствор могут быть введены стабилизирующие добавки, предпочтительно с концентрацией в интервале между приблизительно 0.1 и 20% (вес./объем).
[234] С другой стороны, составы введения могут быть в твердом виде, например, таблетки, капсулы или частицы, например, порошка или саше. Твердые дозируемые формы можно приготовить путем смешивания твердой формы соединения с твердой формой активного вещества. В другом примере реализации твердое вещество можно получить из раствора соединения и активного вещества способами, известными в данной области, такими как сублимационная сушка (лиофилизация), осаждение, кристаллизация и твердофазное диспергирование.
[235] Составы для введения, предложенные в настоящем изобретении, могут также содержать один или несколько ингибиторов ферментов. Такие ингибиторы ферментов включают, без ограничений, такие соединения, как актинонин или эпиактинонин и их производные. Другие ингибиторы ферментов включают, без ограничений, апротинин (Трасилол) и ингибитор Боумен-Бирка.
[236] Количество активного вещества, используемого в составе для введения, предложенного в настоящем изобретении, является эффективным количеством для достижения цели, для которой конкретное активное вещество было назначено пациенту. В общем, количество активного вещества в составе является фармакологически, биологически, терапевтически или химически эффективным количеством. Однако это количество может быть меньше, чем в случае состава, используемого в единичной дозируемой форме, так как такая форма может содержать много составов соединение-носитель/активные вещества или может содержать фармакологически, биологически, терапевтически или химически эффективное количество, разделенное на несколько доз. Тогда общее эффективное количество можно ввести в кумулятивных дозах, содержащих, в сумме, эффективное количество активного вещества.
[237] Общее количество активного вещества, которое необходимо использовать, можно определить способами, известными специалисту в данной области. Однако так как составы, предложенные в настоящем изобретении, могут переносить активные вещества более эффективно, чем составы, содержащие одно активное вещество, субъекту можно вводить более низкие количества биологически или химически активных веществ, чем количества, используемые в прежних единичных дозируемых формах или системах доставки, при этом достигаются те же самые уровни в крови и/или терапевтические эффекты.
[238] Описанные в настоящем изобретении соединения-носители способствуют доставке биологически и химически активных веществ, в частности, при пероральном, интраназальном, сублингвальном, желудочном, интестинальном (кишечном) введении, в том числе, при интрадуоденальном введении, введении через тонкую кишку и подвздошную кишку, при подкожном, ротовом введении, введении через толстую кишку, прямую кишку, вагинальной, внутримышечной и офтальмологической системах введения, введении через слизистую оболочку, а также при преодолении гематоэнцефалического барьера.
[239] Единичные дозируемые формы могут также содержать любой один или комбинацию наполнителей, разбавителей, дезинтегрантов, лубрикантов, пластификаторов, красителей, ароматизирующих веществ, вкусовых добавок, сахаров, заменителей сахара, солей и дозируемых наполнителей, в том числе, без ограничений, воду, 1,2-пропандиол, этанол, оливковое масло или их комбинацию.
[240] Соединения и составы, предложенные в содержании изобретения, применимы для введения биологически и химически активных веществ в организм любого животного, включая, без ограничений, птиц, например цыплят; млекопитающих, таких как грызуны, коровы, свиньи, собаки, кошки, приматы, и в частности, человек; и насекомых.
[241] Эта система особенно эффективна для доставки химически или биологически активных веществ, которые в противном случае разрушились бы или стали менее эффективными вследствие условий, в которые они попадают прежде чем достичь своей зоны мишени (т.е. участка, в котором активное вещество, содержащееся в транспортирующей композиции, должно высвободиться) внутри организма животного, в которое они введены. В частности, соединения и составы, предложенные в настоящем изобретении, являются полезными при пероральном введении активных веществ, особенно тех, которые обычно не транспортируют пероральным способом или для которых желательно использовать улучшенный способ доставки.
[242] Составы, содержащие соединения и активные вещества, полезны при доставке активных веществ в выбранные биологические системы, поскольку увеличивают или улучшают биодоступность активных веществ по сравнению с их введением без реагента-носителя. Доставку можно улучшить путем доставки более активного вещества в течение периода времени, или при доставке активного вещества за конкретный промежуток времени (например, осуществить более быструю или замедленную доставку), или при доставке активного вещества за определенное время, или в течение некоторого периода времени (например, длительная доставка).
[243] Другим примером реализации настоящего изобретения является способ лечения или предупреждения заболевания или достижения желаемого физиологического эффекта, как, например, способы, перечисленные в таблице ниже, у животных при введении состава, предложенного в настоящем изобретении. Предпочтительно для лечения или предупреждения заболевания или достижения желаемого физиологического эффекта вводить эффективное количество состава. Конкретные показания применения активных веществ можно найти в (1) «Physicians' Desk Reference» (58th Ed., 2004, Medical Economics Company, Inc., Montvale, NJ), и (2) Fauci, AS, et. al., Harrison's Principles of Internal Medicine (14th Ed., 1998, McGraw-Hill Health Professions Division, New York), оба из которых оформлены здесь ссылкой. К активным веществам, представленным в таблице ниже, относятся их аналоги, фрагменты, миметики и полиэтиленгликольмодифицированные производные.
[244] Например, один пример реализации настоящего изобретения представляет собой способ лечения пациентов, имеющих диабет или предрасположенных к нему, путем введении инсулина в фармацевтическом составе, предложенном в настоящем изобретении. Другие активные вещества, в том числе приведенные в таблице выше, также можно использовать в соединении с фармацевтическими составами, предложенными в настоящем изобретении.
[245] При последующем введении активное вещество, присутствующее в составе или единичной дозируемой форме вовлекается в кровообращение. Биодоступность вещества можно легко оценить, измеряя известную фармакологическую активность в крови, например, увеличение время свертывания крови, вызванное гепарином, или уменьшение уровней циркуляции кальция, вызываемое кальцитонином. Периодически можно непосредственно измерять уровни циркуляции самого активного вещества.
Добавки
[246] Твердый фармакологический состав и единичная дозируемая форма, предлагаемые в настоящем изобретении, могут содержать другие активные вещества и фрамацевтически допустимые добавки, например, наполнители, носители, разбавители, стабилизаторы, пластификаторы, связующие вещества, глиданты, дезинтегранты, разрыхлители, лубриканты, красители, пленкообразующие вещества, ароматизаторы, вкусовые добавки, сахара, заменители сахара, консерванты, дозируемые наполнители, поверхностно-активные вещества и любую комбинацию из любых вышеуказанных веществ. Предпочтительно, чтобы эти добавки были пригодными для фармацевтического использования, как, например, описанные в монографии Remington's, The Science и Practice of Pharmacy, (Gennaro, A.R., ed., 20th edition, 2003, Mack Pub. Co.), оформленной здесь ссылкой.
[247] Подходящие связующие вещества включают, без ограничений, крахмал, желатин, сахара (такие как, сахарозу, мелассу и лактозу), дигидрат гидроортофосфата кальция, природную и синтетическую камеди (такие как, аравийскую камедь), альгинат натрия, карбоксиметилцеллюлозу, метилцеллюлозу, поливинилпирролидон, полиэтиленгликоль, этилцеллюлозу и воска.
[248] Подходящие глиданты включают, без ограничений, тальк, и диоксид кремния (кварц) (например, плавленые кварц и коллоидный диоксид кремния).
[249] Подходящие дезинтегранты включают, без ограничений, крахмалы, крахмалгликолат натрия, кроскармелоза натрия, кросповидон, глины, целлюлозы (например, очищенная целлюлоза, метилцеллюлоза и натрийкарбоксисиметилцеллюлоза), альгинаты, прежелатизированный кукурузный крахмал, и камеди (такие как, агар, гуар, плоды рожкового дерева, караю, пектиновые и трагакантовые смолы). Предпочтительным дезинтегрантом является крахмалгликолат натрия.
[250] Подходящие наполнители включают, без ограничений, крахмалы (такие как рисовый крахмал), микрокристаллическая целлюлоза, лактоза (например, моногидрат лактозы), сахароза, декстроза, маннитол, сульфат кальция, сульфат кальция и сульфат трикальция.
[251] Подходящие лубриканты включают, без ограничений, стеариновую кислоту, стеараты (такие как стеарат кальция и стеарат магния), тальк, борная кислота, бензоат натрия, ацетат натрия, фумарат натрия, хлорид натрия, полиэтиленгликоль, гидрированное хлопковое масло и касторовые масла.
[252] Подходящие поверхностно-активные вещества всключают, без ограничений, лаурилсульфат натрия, гидроксилированный соевый лецитин, полисорбаты, и блок-сополимеры пропиленоксида и этиленоксида.
Дозируемые формы
[253] Твердый фармацевтический состав, предлагаемый в настоящем изобретении, который содержит активное вещество и реагент-носитель, можно определить как твердую единичную дозируемую форму. Дозируемая форма может быть, например, в виде таблетки, саше или капсулы, в частности, твердой или мягкой капсулы из желатина. Дозируемая форма может обеспечить мгновенное, длительное или контролируемое выделение реагента-носителя, гепарина и по выбору дополнительных активных веществ.
[254] Твердый фармацевтический состав и твердую единичную дозируемую форму можно приготовить следующим образом. Реагент-носитель в твердом виде перерабатывают (например, измельчают через сито 35 меш (число отверстий сита на линейный дюйм)) для получения порошка, имеющего относительно малые и предпочтительно одинаковые размеры частиц. Затем реагент-носитель смешивают с активным веществом и, по выбору, наполнителем и/или увлажняющим веществом используя, например, V-смеситель или схожий аппарат для получения порошковой смеси.
[255] Отдельно готовят смесь увлажняющего вещества путем смешивания увлажняющего вещества, гепарина и реагента-носителя. Смесь также может содержать, например, воду. Рецептуру увлажняющей смеси выбирают таким образом, чтобы смочить гепарин при его смешивании с вышеуказанной порошковой смесью. Согласно одному предпочтительному примеру, увлажняющую смесь составляют так, чтобы частично растворить реагент-носитель при его смешивании с порошковой смесью.
[256] Порошковую смесь малыми количествами добавляют к увлажняющей смеси при постоянном перемешивании. Для получения однородного состава перемешивание продолжают в течение достаточного времени (например, 15 минут) после того, как добавят всю порошковую смесь. Конечный состав обычно является полутвердым, гелеобразным или жидким.
[257] Затем состав можно перевести в дозируемую форму, такую как капсула, способами, известными в данной области. Согласно одному предпочтительному примеру, конечную композицию упаковывают в мягкие или твердые желатиновые капсулы (например, в твердые желатиновые капсулы размера 0 Licap Capsugel). Другие подходящие способы описаны в Патентах США Nos. 6,605,298, 6,458,383, 6,261,601, 5,714,477, и 3,510,561; в Публикации патентных заявок США Nos. 2003/0077303 и 2001/0024658; и Международной публикации No. WO 88/10117, которые все оформлены здесь ссылками.
ПРИМЕРЫ
[258] Следующие примеры демонстрируют настоящее изобретение без ограничения. Все процессы приведены по весу, если не указано иное.
[259] Соединения, перечисленные ниже, были исследованы способом протонного ядерного магнитного резонанса (1Н ЯМР) на спектрометре Bruker® при 300 МГц (Bruker-Physik AG, Silberstreifen, GERMANY) или на спектрометре JEOL при 400 МГц (JEOL USA, Inc., Peabody, MA), с использованием диметилсульфоксида (ДМСО-d6) в качестве растворителя, если не указано иное.
[260] Были выполнены анализы на жидкостном хроматографе/масс-спектрометре (ЖХ-МС) с использование Agilent Technologies (Palo Alto, Калифорния), ЖХ/МС 1100 (единичный квадруплет), имеющих следующие параметры:
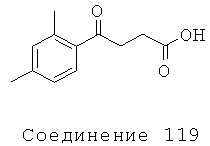
[261] Подвижная фаза А: 50:950:5 ацетонитрил: вода: уксусная кислота (об./об./об.).
[262] Подвижная фаза В: 950:50:5 ацетонитрил: вода: уксусная кислота (об./об./об.).
[263] Градиентное элюирование: 4-минутный линейный градиент 0-100% В; общее время одного ввода - 11 минут.
[264] Объем вводимой пробы: 5 мкл.
[265] Колонка: ZORBAX Rapid Resolution Cartridge, SB-C18, 2.1×30 мм, 3.5 мкм.
[267] Размер частиц, каталог # 873700-902.
[268] Температура колонки: 40°С.
[269] УФ детектирование при 244 нм.
[270] МСД параметры:
[271] Источник: API-ES, положительная полярность.
[272] Параметры сканирования:
Диапазон массовых чисел: 125.00-600.00
Фрагментор: 60 В
Прирост: 1.0 ЕМВ
Порог: 150
Распылительная камера:
Температура газа 350 гр.
Осушающий газ: 12.0 л/мин
Давление; 275.79 кПа
Допустимая нагрузка 4000 В
положительный/отрицательный.
Пример 1 - Приготовление соединений 1-22
[273] Соединения 1-22 были получены по способу, описанному в патенте США No. 6,384,278, который оформлен здесь полной ссылкой.
[274] Соответствующий N-замещенный анилин смешивают с соответствующим моноэфиром дикарбоновой кислоты и нагревают в ксилоле в присутствии борной кислоты в качестве катализатора.
Пример 2 - Приготовление соединений 23-34 и 59
[275] Просушенную, 200-мл круглодонную колбу с 3-мя горловинами оборудовали магнитной мешалкой с тефлоновым покрытием и ловушкой Дина-Старка с вакуумной рубашкой, на которую сверху установили дефлегматор с входным отверстием, приспособленным для подачи азота. В реакционный сосуд помещали N-изопропил-N-фениламин (8,11 г, 60 ммоль), борную кислоту (0,93 г, 15 ммоль) и ксилол (88 мл). К реакционной смеси при перемешивании добавляли одной порцией 7-этокси-7-оксогептановую кислоту (11,29 г, 60 ммоль). Для возврата флегмы реакционную смесь нагревали, используя нагревательный кожух. Воду, которая начала выделяться при азеотропной перегонке, улавливали в ловушку Дина-Старка. После 16 часов орошения вода была собрана, и реакционную смесь оставляли охлаждаться до комнатной температуры. Реакционную смесь разбавляли этилацетатом (100 мл), и промывали 2 н. водным раствором HCI (50 мл), и затем насыщенным раствором бикарбоната натрия (60 мл). Большую часть органического растворителя удаляли в вакууме. К остатку добавляли 2 н. водный раствор гидроксида натрия (60 мл). Смесь нагревали при 60°С в течение 4 часов. После охлаждения до комнатной температуры смесь промывали 60 мл этилацетата. После того, как водную фазу осторожно отделили от органического слоя, ее выпаривали с целью удаления любых остатков этилацетата. К водному раствору добавляли лед, а затем водный раствор HCl (2 н., 60 мл), что приводило к выпадению в осадок твердого белого вещества. Прежде чем собрать осадок с помощью спеченной воронки, в течение дополнительных 30 минут продолжали перемешивание. Собранное твердое белое вещество эффективно промывали водой и гексаном, прежде чем поместить в вакуум при комнатной температуре на 12 часов для получения 7,49 г (45%) 7-[изопропил(фенил)амино]-7-оксогептановой кислоты в виде белого твердого вещества. ВЭЖХ: одинарный пик на 4.83 мин.; Мр: 62-63°С. 1H ЯМР (ДМСО-d6,) δ: 0.95-0.97 (d, 6H), 1.08-1.10 (m, 2Н), 1,34-1.40 (m, 4H), 1.76-1,79 (m, 2Н), 2,09-2,13 (m, 2Н), 4.81-4.85 (m, 1H), 7.18-7.20 (m, 2Н), 7.44-7.46 (m, 3Н). Масса (М+1): 278. Анал. расчет для C16H23NO3: С, 69.29; Н, 8.36; N 5.05. Найдено: С, 69.06; Н, 8.45; N, 4.99.
[276] Соединения 24-34 и 59 были приготовлены из соответствующих исходных материалов с использованием вышеупомянутого способа.
Соединение (24)
[277] ВЭЖХ: одинарный пик на 4,43 мин. Масса (М+1): 264. 1H ЯМР (400 МГц, ДМСО-d6) δ: 0.95(d, 6H), 1.30(m, 2Н), 1.40 (m, 2Н), 1.80(m, 2Н), 2.00(m, 2Н), 4.80(m, 1H), 7.15 (m, 2Н), 7.40 (m, 3Н). 13С ЯМР (100МГц, ДМСО-d6) δ: 21.0, 24.0, 24.5, 33.0, 34.0, 45.0, 128.0, 129.0, 130.0, 138.5, 170.5, 174.0.
Соединение (25)
[278] ВЭЖХ: одинарный пик на 4,62 мин. Масса (М+1): 264. 1H ЯМР (400 МГц, ДМСО-d6) δ: 0.78 (d, 3Н), 0.94-0.95(d, 6H), 1.70-1.72(m, 1H), 1.80-1.92 (m, 2Н), 2.08-2.15(m, 1H), 2.20-2.30(m, 1H), 4.75-4.90(m, 1H), 7.10-7.20(m, 2H), 7.35-7.50(m, 3H). 13C ЯМР (100МГц, ДМСО-d6) δ: 19.5, 21.0, 27.0, 40.5, 41.0, 45.0, 128.0, 129.0, 130.5, 138.5, 170.0, 174.0.
Соединение (26)
[279] ВЭЖХ: одинарный пик на 4,19 мин. Масса (М+1): 250. 1Н ЯМР (400 МГц, ДМСО-d6) δ: 0.65 (d, 3H), 0.84-0.86(1, 3H), 1.80-1.90(m, 3H), 2.01-2.12(m, 2H), 3.49-3.53(q, 2H), 7.09-7.11(d, 2H), 7.20-7.25(m, 1H), 7.30-7.32 (m, 2H). 13C ЯМР (100МГц, ДМСО-d6) δ: 9.18, 15.87, 17.30, 23.35, 39.50, 123.98, 124.72, 125.92, 138.39, 166.17, 168.27, 169.80.
Соединение (27)
[280] ВЭЖХ: одинарный пик на 3.92 мин. Масса (М+1): 250. 1H ЯМР (400 МГц, ДМСО-d6) δ: 1.13 (m, 2H), 1.37-1.46 (m, 4H), 1.99 (m, 2H), 2.10-2.15 (t, 2H), 3.15 (s, 3H), 7.29-7.37 (m, 3H), 7.42-7.47 (m, 2H).
Соединение (28)
[281] ВЭЖХ: одинарный пик на 3.72 мин. Масса (М+1): 236. 1H ЯМР (400 МГц, ДМСО-d6) δ: 0.79-0.81 (d, 3H), 1.93-2.02 (m, 3H), 2.16-2.30 (m, 2H), 3.15 (s, 3H), 7.27-7.37 (m, 3H), 7.43-7.48 (m, 2H).
Соединение (29)
[282] ВЭЖХ: одинарный пик на 3.88 мин. Масса (М+1): 242. 1H ЯМР (400 МГц, ДМСО-d6) δ: 2.21(m, 2H), 2.49 (m, 2H), 3.13 (s, 3H), 7.37 (m, 2H), 7.58 (m, 2H), 12.10 (br., 1H). 13C ЯМР (100 МГц, ДМСО-d6) δ: 28.81, 29.0, 36.5, 129.32, 129.58, 132.0, 142.66, 170.58, 173.63.
Соединение (30)
[282] ВЭЖХ: одинарный пик на 4,82 мин. Масса (М+1): 278. 1H ЯМР (400 МГц, ДМСО-d6) δ: 1.02 (m, 4H), 1.32 (m, 4H), 1.86 (m, 2H), 2.05(m, 2H), 2.21 (s, 3H), 3.00 (s, 3H), 7.00 (m, 2H), 7.12 (m, 2H), 11.85 (br., 1H).
Соединение (31)
[283] ВЭЖХ: одинарный пик на 4,44 мин. Масса (М+1): 294. 1H ЯМР (400 МГц, ДМСО-d6) δ: 1.10 (m, 4H), 1.39 (m, 4H), 1.93 (m, 2H), 2.11 (m, 2H), 3.07 (s, 3Н), 3.75 (s 3Н), 6.96 (m, 2H), 7.20 (m, 2H), 11.93 (br., 1H).
Соединение (32)
[284] ВЭЖХ: одинарный пик на 4.81 мин. Масса (М+1): 278. 1H ЯМР (400 МГц, ДМСО-d6) δ: 0.97 (t, 3Н), 1.10 (m, 4H), 1.39 (m, 4H), 1.90 (m, 2H), 2.13 (m, 2H), 3.58-3.63 (q, 2H), 7.09-7.24 (d, 2H), 7.34 (m, 1H), 7.41-7.45 (m, 2H).
Соединение (33)
[285] ВЭЖХ: одинарный пик на 5.48 мин. Масса (М+1): 312. 1H ЯМР (400 МГц, ДМСО-d6) δ: 0.96 (t, 3Н), 1.10 (m, 4H), 1.40 (m, 4H), 1.91 (m, 2H), 2.12 (m, 2H), 3.60 (q, 2H), 7.27 (d, 2H), 7.46 (m, 2H), 11.93 (br., 1H).
Соединение (34)
[286] ВЭЖХ: одинарный пик на 4,52 мин. Масса (М+1): 282. 1H ЯМР (400 МГц, ДМСО-с1б) 5: 1.09(т, 4H), 1.39(т, 4H), 1.93(т, 2H), 2.10-2.14(т, 2H), 3.09(s. ЗН), 3.75(8 ЗН), 7.19(т, 2H), 7.30(т, 2H), 11.91(br., 1H).
Соединение (59)
[287] ВЭЖХ: одинарный пик на 4,71 мин. Масса (М+1): 284. 1Н ЯМР (400 МГц, ДМСО-d6) δ: 0.90(t, 3Н), 1.35-1.37 (m, 4H), 1.87 (t, 2H), 2.04 (t, 2H), 3.52-3.57 (q, 2H), 7.25 (m, 2H), 7.43 (m, 2H), 11.94 (8, 1H).
Пример 3 - приготовление соединений 111-139
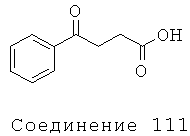
Соединение 111 - 4-Оксо-4-фенилмасляная кислота:
[289] 10 г (56 ммоль) 3-бензоилпропионовой кислоты (доступна в продаже в Sigma-Aldrich Co., Сент-Луис, МО) добавляли к 10 мл воды. Смесь перемешивали и добавляли 28 мл 2 н. гидроксида натрия (водного). Конечный раствор перемешивали в течение 2 часов и после высушивания сублимационным способом, собирали твердый продукт. 1H ЯМР (d6-ДМСО): δ 7.9, d, 2H, (арильные Н); δ 7.6, t, 1H, (арильные Н); δ 7.5, t, 2H, (арильные Н); δ 3.1, t, 2H (СН2 α к карбонилу); δ 2.2, t, 2H (СН2 α к СООН); пик СООН не наблюдали из-за присутствия воды в пробе.
Соединение 113 - 10-(4-Гидрокси-фенил)-10-оксодекановая кислота:
[290] В 500-мл колбу, оборудованную дефлегматором, в инертной атмосфере помещали декандионовую кислоту (20 г, 296 ммоль) и уксусный ангидрид (280 мл, 2,96 моль). С целью орошения смесь нагревали в течение 5 часов. Уксусную кислоту и избыток уксусного ангидрида удаляли при пониженном давлении. Продукт использовали без дальнейшей очистки.
[292] В 500-мл колбу, оборудованную механической мешалкой, в инертной атмосфере добавляли оксациклоундекан-2,11-дион (20 г, 108,5 ммоль), фенол (10,22 г, 108,5 ммоль) и 200 мл дисульфида углерода. Добавляли трихлорид алюминия (III) (72,34 г, 542 ммоль) и реакционную смесь перемешивали в течение 72 часов. Дисульфид углерода декантировали, и осторожно добавляли лед до тех пор, пока большая часть смеси не растворялась. Нерастворимое вещество собирали вакуум-фильтром и промывали 2×100 мл водой. Затем твердое вещество растворяли в 100 мл 1 М водного гидроксида натрия и осторожно подкисляли 1 М соляной кислотой (водной) до рН=7,5 Образовавшийся сухой остаток удаляли путем фильтрования и исходный раствор продолжали подкислять до рН 2,5. Осадок неочищенного продукта отфильтровывали и промывали 1×100 мл водой. Неочищенный продукт растворяли в 100 мл 1 М водного раствора гидроксида натрия, осторожно подкисляли 1 М водным раствором соляной кислоты до рН=7,5 и отфильтровывали осажденные примеси. Исходный раствор подкисляли затем до рН 2. Неочищенный продукт отфильтровывали и промывали 2×50 мл водой. Затем продукт перекристаллизовывали из ацетона. Выделенный продукт (1,2 г, 4%) собирали путем фильтрации. Найдено: С 69.00, Н 7.81%; C16H22O4 требует С: 69.04, Н: 7.97% 1Н ЯМР (d6-ДМСО): δ 12.0, bs, 1H (СООН); δ 10.3, bs, 1H (арил-гидроксил); δ 7.8 d, 2H (арильные Н); δ 6.8, d, 2H, (арильные Н); δ 2.9, t, 2H (СН2 α к карбонилу); δ 2.2, t, 2H (СН2 α к СООН); δ 1.5, мультиплет, 4Н (СН2-группы β к карбонилу и β к СООН), δ 1.3, мультиплет, 8Н (остальные СН2-группы).
Соединение 114 - 10-(2-Гидрокси-фенил)-10-оксодекановая кислота:
[293] В 100-мл колбу помещали метиленхлорид (50 мл), 9-бромнонанол (7,63 г, 34,2 ммоль) и триметилсилилхлорид (4,5 мл, 35,5 ммоль) и перемешивали в атмосфере азота в течение 20 минут. Затем добавляли триэтиламин (5,0 мл, 35,9 ммоль) и конечную реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Затем реакционную смесь разбавляли 80 мл гексана, отфильтровывали, и затем концентрировали при пониженном давлении. Конечный остаток снова разбавляли 80 мл гексана, отфильтровывали, и затем концентрировали при пониженном давлении, получая 9,7 г (96%) желтой жидкости, которую использовали без дальнейшей очистки.
[294] 5,69 г (19,3 ммоль) (9-бром-нонилокси)-триметилсилана добавляли по каплям в инертной атмосфере в 50-мл колбу, содержащую металлический магний (0,59 г, 24,3 ммоль), 20 мл тетрагидрофурана и использовали маленький кристалл йода для инициирования реакции Гриньяра. В 100-мл колбе в инертной атмосфере охлаждали раствор салицилового альдегида (2,1 мл, 19,7 ммоль) в 20 мл тетрагидрофурана, используя наружную ледяную баню. Затем охлажденный раствор альдегида обрабатывали 1,0 М литий-бис(триметилсилил)амидом (20,0 мл, 20 ммоль). После перемешивания в течение 1 часа реакционную смесь Гриньяра охлаждали с помощью наружной ледяной бани. Затем при постоянном перемешивании охлажденную реакционную смесь Гриньяра добавляли по каплям через канюлю к раствору альдегида в течение 5-минутного периода. Конечную реакционную смесь оставляли нагреваться до комнатной температуры и продолжали перемешивать всю ночь. В реакционную смесь вливали 40 мл этилацетата и гасили 15 мл насыщенного водного раствора бикарбоната натрия. Органический слой отделяли и промывали 2×25 мл порциями 4% раствора водной соляной кислоты, а затем 1×20 мл порцией соляного раствора. Органический слой высушивали над сульфатом натрия, отфильтровывали, и растворитель удаляли при пониженном давлении. Остаточный салицилальдегид удаляли методом перегонки Кюгельроха и конечный осадок использовали без дальнейшей очистки.
[295] В 100-мл колбу помещали 1-(2-гидрокси-фенил)-ундекан-1,11-диол (5,0 г, 18,9 ммоль) и 50 мл диметилформамида. Туда же добавляли бихромат пиридина (32,9 г, 87,5 ммоль). (Добавка являлась умеренно экзотермической.) Реакционную смесь перемешивали при комнатной температуре всю ночь. Реакционную смесь наливали в 50 мл этилацетата и промывали 200 мл воды, 30 мл 4% водного раствора соляной кислоты, 30 мл воды, и, наконец, 30 мл соляного раствора. Затем органический слой в течение 15 минут перемешивали с 10 г силикагеля, высушивали сульфатом натрия, отфильтровывали, и удаляли растворитель при пониженном давлении. Неочищенный продукт грязно-белого цвета перекристаллизовывали из смеси этанол/вода. Продукт (0,5 г, 10%) выделяли в виде грязно-белого твердого вещества с температурой плавления 85-87°С. Органический элементарный анализ: найдено: С 69.01, Н 8.36%; C16H22O4 требует С: 69.54, Н: 8.02% 1Н ЯМР (d6-ДМСО): δ 12.0, s, 1H (СООН); δ 7.9 dd, 1H (арильный Н); δ 7.5, dt, 1H, (арильный Н); δ 6.9, сложный мультиплет, 2Н (арильные Н), 3.1, t, 2H (СН2 α к карбонилу); δ 2.2, t, 2H (СН2 α к СООН); δ 1.6, мультиплет, 2H (СН2 β к карбонилу), δ 1.5, мультиплет, 2H (СН2 β к СООН), δ 1.3, мультиплет, 8Н (остаток СН2-группы).
Соединение 115 - 4-(4-Метокси-фенил)-4-оксомасляная кислота:
[296] В 500-мл круглодонную колбу, оборудованную магнитной мешалкой, помещали в инертной атмосфере (газообразный азот) 5,25 мл (48,3 ммоль) анизола, 4,83 г (48,3 ммоль) янтарного ангидрида, 125 мл 1,1,2,2-тетрахлопентана и 125 мл нитробензола. Реакционный сосуд охлаждали при помощи наружной водяной бани и перемешивали реакционную смесь в течение 30 минут. Трихлорид алюминия (14,2 г, 106,4 ммоль) добавляли к бледно-желтому раствору, который затем окрашивался в темный красновато-коричневый цвет. Ледяную баню убирали и реакционную смесь продолжали перемешивать при комнатной температуре в течение 36 часов. Затем реакционную смесь снова охлаждали наружной водяной баней. Приготовленый кислый раствор путем наливания раствора 1 н. хлористого водорода в 100 мл химический стакан заполнили льдом. Этот раствор осторожно добавляли в реакционную смесь, вначале по каплям до тех пор, пока реакционная смесь с появлением белого осадка не стала прозрачной. После этого момента порцию 10 мл осторожно добавляли к пробе на реактивность и затем добавляли оставшуюся смесь лед/кислота. Добавляли вторые 100 мл смеси лед/кислота, убирали наружную ледяную баню и осветленную эмульсию перемешивали в течение 2 часов. Белый осадок извлекали из эмульсии способом вакуум-фильтрации. Это твердое вещество растворяли в 300 мл 0,3 М раствора гидроксида натрия, промывали 100 мл этилацетата, и подкисляли до ~рН 11 М соляной кислотой. Собранный способом вакуумной фильтрации белый осадок промывали 3×100 мл деионизированной водой и высушивали. Продукт (4,7 г, 47%) выделяли в виде белого твердого вещества с температурой плавления 149-150°С. Органический элементарный анализ: найдено: С 63.52, Н 5.78%; C11H12O4 требует С: 63.45, Н: 5.81% 1Н ЯМР (d6-ДМСО): δ 12.2, s, 1H (СООН); δ 7.9 d, 2H (арильные Н); δ 7.0, d, 2H, (арильные Н); δ 3.8, s, 3Н (ОМе H's); δ 3.2, t, 2H (СН2 α к карбонилу); δ 2.5, t, 2H (CH2 α к СООН).
Соединение 116 - 4-Метокси-фенил)-5-оксопентановая кислота:
[297] Соединение 116 было получено аналогично соединению 15, за исключением использования глутаминового ангидрида вместо янтарного ангидрида, температура плавления 141-142°С. Найдено: С 64.65, Н 6.34%; C12H14O4 требует С: 64.85, Н: 6.35% 1H ЯМР (d6-ДМСО): δ 12.2, s, 1H (СООН); δ 7.9 d, 2H (арильные Н); δ 7.0, d, 2H, (арильные Н); δ 3.8, s, 3Н (ОМе H's); δ 3.0, t, 2H (CH2 α к карбонилу); δ 2.3, t, 2H (CH2 α к СООН)); δ 1.8 квинтуплет, 2H (CH2 между другими двумя).
[298] Соединение 117 было приобретено у Aldrich (Сент-Луис, МО), номер в каталоге 514683.
[299] Соединение 118 было приобретено у Aldrich (Сент-Луис, МО), номер в каталоге В12687.
[300] Соединение 119 было приобретено у Aldrich (Сент-Луис, МО), номер в каталоге S346810.
[301] Соединение 120 было приобретено у Reike, Aldrich (Сент-Луис, МО), номер в каталоге 7013D.
[302] Соединение 121 было приобретено у Reike, Aldrich (Сент-Луис, МО), номер в каталоге 7148С и
Соединение 121 - натриевая соль 5-(4-изопропил-фенил)-5-оксопентановой кислоты:
[303] 5-(4-изопропил-фенил)-5-оксо-пентановую кислоту (5 г, 21,3 ммоль) растворяли в 75 мл этанола в 250-мл колбе. Добавляли гидроксид натрия (0,85 г, 21,3 ммоль) и реакционную смесь на роторном испарителе перемешивали всю ночь при пониженном давлении. Твердое вещество высушивали в вакууме и использовали без дальнейшей очистки. Найдено: С 60.24, Н 6.66, Na 9.21%; C14H17O3Na требует С: 61.28, Н: 6.98, Na 8.38% 1H ЯМР (D2O): 5 7.7, d, 2H (арильные Н); δ 7.2 d, 2H (арильные Н); δ 2.9, t, 2H (СН2 α к карбонилу); δ 2.8, мультиплет, 1H, (СН изопропил-группы); δ 2.1, t, 2H (СН2 α к СООН); δ 1.8, q, 2H (СН2 β к обоим карбонилу и СООН), δ 1.1, d, 6H (CH3's изопропил-группы).
[304] Соединение 122 было приобретено у Aldrich (Сент-Луис, МО), номер в каталоге В13802.
[305] Соединение 123 было приобретено у Reike, Aldrich (Сент-Луис, МО), номер в каталоге 7060В.
[306] Соединение 124 было приобретено у Fischer-Scientific (Хамптон, Нью-Хэмпшир), Acros, номер в каталоге 17.522.62
[300] Соединение 125 было приобретено у Reike, Aldrich (Сент-Луис, МО), номер в каталоге 7011D.
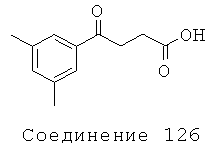
[301] Соединение 126 было приобретено у Reike, Aldrich (Сент-Луис, МО),номер в каталоге 7036В.
[302] Соединение 128 было приобретено у Reike, Aldrich (Сент-Луис, МО), номер в каталоге 7012D.
[303] Соединение 129 было приобретено у Reike, Aldrich (Сент-Луис, МО), номер в каталоге 7012В.
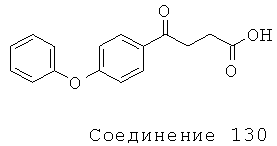
[304] Соединение 130 было приобретено у Reike, Aldrich (Сент-Луис, МО), номер в каталоге 7055В
[305] Соединение 132 было приобретено у Reike, Aldrich (Сент-Луис, МО), номер в каталоге 7005b.
[306] Соединение 133 было приобретено у Reike, Aldrich (Сент-Луис, МО), номер в каталоге 7036F.
[307] Соединение 134 было приобретено у Reike, Aldrich (Сент-Луис, МО), номер в каталоге 7144D
[308] Соединение 136 было приобретено у Reike, Aldrich (Сент-Луис, МО), номер в каталоге 7144В.
[309] Соединение 138 было приобретено у Reike, Aldrich (Сент-Луис, МО), номер в каталоге 7036D.
Соединение 139 - 10-(2,5-дигидрокси-фенил)-10-оксодекановая кислота:
[310] В 500-мл колбу, оборудованную дефлегматором, в инертной атмосфере помещали декандионовую кислоту (20 г, 296 ммоль) и уксусный ангидрид (280 мл, 2,96 моль). С целью орошения смесь нагревали в течение 5 часов. Уксусную кислоту и избыток уксусного ангидрида удаляли при пониженном давлении. Продукт использовали без дальнейшей очистки.
[311] В 500-мл колбу, оборудованную механической мешалкой, в инертной атмосфере помещали предварительно синтезированный оксациклоундекан-2,11-дион (37,95 г, 206 ммоль), 1,4-диацетоксибензол (20 г, 103 ммоль) и 200 мл дисульфида углерода. Добавляли трихлорид алюминия (III) (68,7 г, 515 ммоль) и реакционную смесь перемешивали в течение 72 часов. Дисульфид углерода декантировали, и осторожно добавляли лед до тех пор, пока большая часть смеси не растворилась. Нерастворимое вещество собирали способом вакуум-фильтрации и промывали 2×100 мл водой. Затем твердое вещество растворяли в 50 мл 1 М водного раствора гидроксида натрия и перемешивали в течение 1 часа. Раствор подкисляли 1 М водным раствором соляной кислоты до рН=2. Осадок неочищенного продукта собирали фильтрованием, повторно растворяли в ацетонитриле (50 мл) и метиленхлориде (15 мл) и оставляли медленно осаждаться в течение недели. Образовавшийся коричневый продукт отфильтровывали и перекристаллизовывали из смеси уксусная кислота:вода 10:3. Продукт выделяли (0,8 г, 3%) фильтрованием. Найдено: С 65.55, Н 7.69%; C16H22O5 требует С: 65.29, Н: 7.53% 1Н ЯМР (d6-ДМСО): δ 12.0, s, 1Н (СООН); δ 11.4, s, 1H (арил-гидроксил); δ 9.2, s, 1Н (арил-гидроксил); δ 7.2 d, 1Н (арильный Н); δ 7.0, dd, 1Н, (арильный Н); δ 6.8, d, 1Н (арильные Н), 3.0, t, 2H (СН2 α к карбонилу); δ 2.2, t, 2H (CH2 α к СООН); δ 1.6, мультиплет, 2H (СН2 β к карбонилу), δ 1.5, мультиплет, 2H (СН2 β к СООН), δ 1.3, мультиплет, 8Н (остаток СН2-группы).
Пример 4 - Приготовление соединений 140-151
[312] В общем случае, эти соединения готовили в четыре стадии. Первое, смешивали салициловую кислоту, замещенную соответствующим образом, и этиловый эфир 3-аминомасляной кислоты с этилендихлоридом (ЭДХ)/гидроксибензолтриазолом (ГБТ)/дихлорметаном (ДХМ). Второе, добавляли основную ионно-обменную смолу А-15/А-21 (доступен в продаже в Rohm и Haas, Филадельфия, Пенсильвания). Третье, после частичной обработки, продукт реагировал с триметилсиланолатом калия (ТМСК)/тетрагидрофураном (ТГФ). Четвертое, добавляли смолу IRC-50 (Rhohm & Haas, Филадельфия, Пенсильвания).
[313] В каждую сцинтилляционную пробирку помещали салициловую кислоту (4,57 ммоль), ДХМ (10 мл), ЭДХ (1,05 г, 5,48 ммоль), ГБТ (838 мг, 5,48 ммоль), ДМФ (2 мл), и этил-3-аминобутират (600 мг, 4,57 ммоль). Все пробирки плотно закрывали пробками, помещали на реакционный блок J-Kem (J-Kem Scientific Inc., Сент-Луис, МО), и встряхивали при нагревании (150 об/мин, 35°С) всю ночь. Контролируемые с помощью ТСХ, все реакции имеют одну преобладающую точку. В каждую виалу добавляли смолы Amberlyst-21 и Amberlyst-15 (приблизительно 2,5 г, 11 ммоль) и продолжали встряхивать при температуре окружающей среды всю ночь. Реакционную смесь отфильтровывали, смолы промывали ДХМ (2×5 мл), и объединенные фильтраты каждой реакционной смеси собирали в чистую сцинтилляционную пробирку. Фильтраты продували в токе азота до объема приблизительно 2 мл.
[314] В каждую пробирку помещали 1,2 М раствор триметилсилонолата калия (ТМСК) в ТГФ (10 мл, 12 ммоль). В некоторые реакционные смеси добавляли больше ТГФ, когда необходимо было получить суспензии, пригодные к встряхиванию. Все пробирки плотно закрывали пробками, помещали на реакционный блок J-Kem, и встряхивали при нагревании (150 об/мин, 60°С 6 ч). Реакционный блок охлаждали и в каждую пробирку добавляли смолу IRC-50 (3 г, 30 ммоль) для гашения соли калия. При необходимости суспендировать смолу и облегчить встряхивание добавляли ДХМ. Реакционные смеси трясли всю ночь. Реакционные смеси отфильтровывали, смолы промывали ДХМ (2×5 мл), при необходимости, чтобы растворить твердые вещества, промывали ДМФ и объединенные фильтраты каждой реакционной смеси собирали в чистую сцинтилляционную пробирку. На этой стадии отбирали маленькие аликвоты фильтрата и разбавляли смесью 1:1 АЦН/Н2О для ЖХ-ЯМ. Фильтраты продували в потоке азота. Для удаления следовых количеств ДМФ, пробирки помещали в вакуумную печь при 50°С.
[315] Согласно данным ЖХ-МС анализа, некоторые реакционные смеси все еще содержали значительные количества эфира. Эти смеси повторно обрабатывали ТМСК. В каждую пробирку добавляли 1,2 М раствор ТМСК в ТГФ (8 мл, 9,6 ммоль). Все пробирки плотно закрывали крышкой, помещали в реакционный блок Пирса, перемешивали и нагревали (60°С, 5 час). Реакционный блок охлаждали, и в каждую пробирку добавляли смолу IRC-50 (2 г, 20 ммоль) для гашения соли натрия. При необходимости суспендировать смолу и облегчить встряхивание добавляли ДХМ. Реакционные смеси перемешивали все выходные дни. Реакционные смеси отфильтровывали через слой из кварца, смолы и кварц промывали ДХМ (2×5 мл), затем смесью 2:5 МеОН/ДХМ (3×7 мл), и объединенные фильтраты каждой реакционной смеси собирали в чистую сцинтилляционную пробирку. На этой стадии отбирали маленькие аликвоты фильтрата и разбавляли смесью 1:1 АЦН/H2O для ЖХ-МС. Фильтраты продували в токе азота.
[316] Все другие реакционные смеси, остальные от первой обработки ТМСК, помещали в смесь 10:1 ДХМ/МеОН и отфильтровывали через слой из кварца, элюируя большим количеством 10:1 ДХМ/МеОН. На этой стадии отбирали маленькие аликвоты фильтрата и разбавляли смесью 1:1 АЦН/H2O для ЖХ-ЯМ. Фильтраты продували в токе азота.
Альтернативное приготовление соединения 140-151:

[317] В 1-л колбу с круглым дном помещали 3,5-диизопропилсалициловую кислоту (25,0 г, 112,5 ммоль), ГБТ (20,6 г, 135,0 ммоль), этил-3-аминобутират (18,0 г, 123,7 ммоль) и диоксан (400 мл). Полученную смесь перемешивали при температуре окружающей среды. ЭДХ (25,9 г, 135,0 ммоль) добавляли порциями и продолжали перемешивать всю ночь. ВЭЖХ-анализ реакционной смеси на этой стадии показал наличие ГБТ, возможно следовых количеств исходной салициловой кислоты и один новый продукт в преобладающих количествах. Добавляли еще порцию ЭДХ (5 г, 26,0 ммоль) и оставляли перемешиваться на всю ночь. Еще один ВЭЖХ-анализ показал отсутствие существенных изменений. Реакционную смесь гасили водой (400 мл) и, используя роторный испаритель, отгоняли диоксан. Образующуюся смесь масло/вода наливали в 1-л делительную воронку и добавляли ДХМ (400 мл). Образовалось большое количество белого твердого вещества. Пытаясь разделить слои, добавляли EtOAc, но безуспешно. Смесь сливали из разделительной колонки и отгоняли из нее легкие органические фракции на роторном испарителе. Смесь вода/масло экстрагировали EtOAc (500 мл, затем 200 мл). Объединенные слои EtOAc промывали водным раствором HCl (10%, 2×200 мл), водным раствором NaOH (10%, 2×200 мл) и соляным раствором (50 мл, затем 200 мл). Органику высушивали над Na2SO4 и отгоняли легкие фракции на роторном испарителе, получая коричневое масло, которое содержало небольшие количества твердого белого вещества. ВЭЖХ анализ показал, что белое твердое вещество представляет собой остаточные количества ГБТ, а требуемым продуктом является коричневое масло. Коричневое масло отбирали пипеткой из колбы, стараясь, чтобы в пипетку не попало твердое белое вещество. Коричневое масло помещали в EtOAc (500 мл), промывали NaOH (10%, 2×200 мл) и высушивали над Na2SO4. EtOAc отгоняли на роторном испарителе, получая коричневое масло. На этой стадии результаты ВЭЖХ анализа показывали один преобладающий пик и отсутствие ГБТ.
[318] Вязкое масло растворяли в ТГФ (200 мл) и добавляли ТМСК (31,7 г, 247,4 ммоль). Получившуюся вязкую смесь перемешивали всю ночь. ВЭЖХ анализ свидетельствовал об окончании реакции наличием одного пика. Добавляли смолу IRC-50 (37 г, 370 ммоль, 1,5 экв.) и 100 мл ДХМ для суспендирования смолы, затем перемешивали несколько часов. Отфильтровывали, промывали смолу ДХМ (3×50 мл) и концентрировали на роторном испарителе до коричневого масла. Попытка перекристаллизовать из смеси АЦН/ацетон была неуспешной. Основываясь на растворимости, на этой стадии определили, что преобладающим веществом являлась соль калия. Масло помещали в смесь Н2О/АЦН, нагревали до тех пор, пока оно не становилось прозрачным, отфильтровывали в горячем состоянии, и охлаждали до комнатной температуры. Фильтрат обрабатывали водным раствором HCl, выделяли образовавшийся твердый осадок и измельчали в порошок для получения Е1528: 9,13 грамм, ВЭЖХ: время удерживания 6.7 мин 100%, KF 0.47, ЯМР согласующийся со структурой, элементарный анализ теоретически С:66.11, Н:8.21, N:4.54, найдено С:65.62, Н:8.19, М:4.46.
3-(2-гидрокси-бензоиламино)масляная кислота
Пример 5 - Получение соединений 152-160
[319] Соединение 152 - было приобретено у Transworld Chemical (Южный Мельбурн, Австралия).
[320] Соединение 153 - было приобретено у Lancaster (Windham, NH).
[321] Соединение 154 - было приобретено у Avocado (Heysham, Lancashire, Англия).
[322] Соединение 155 - было приобретено у Aldrich, номер в каталоге 42919 (Сент-Луис, МО).
[323] Соединение 156 - было приобретено у Sigma-Aldrich (Сент-Луис, МО).
[324] Соединение 157 - было приобретено у Sigma (Сент-Луис, МО).
[325] Соединение 158 - было приобретено у Matrix Scientific (Колумбия, SC).
Время
удерживания
Прото
кол
Ра
плавления,
интервал
значений
С
Соединение 160:
[326] Гидроксид калия (10,37 г, 184,8 ммоль) измельчали в ступке пестиком до порошка и помещали в 250-мл колбу, содержащую 75 мл диметилсульфоксида и метиловый эфир 2-гидрокси-бензойной кислоты (7,03 г, 46,2 ммоль). К этой смеси добавляли бензилбромид (7,91 г, 46,2 ммоль) и приготавливали смесь в течение 4 часов при перемешивании. Добавляли воду (100 мл) и перемешивали реакционную смесь еще в течение 30 минут. Затем реакционную смесь охлаждали с помощью наружной ледяной бани до 0°С и подкисляли концентрированной соляной кислотой до рН 1. Смесь экстрагировали 3×230 мл этилацетатом. Органические слои объединяли и растворитель удаляли при пониженном давлении. Образовавшуюся желтую жидкость растворяли в этилацетате (50 мл) и промывали 2×30 мл водой, а затем 2×30 мл соляным раствором. Органический слой высушивали над сульфатом натрия, отфильтровывали, и растворитель удаляли при пониженном давлении. Образовавшуюся желтую жидкость сушили над вакуумом в течение нескольких дней, при этом образовалось белое кристаллическое твердое вещество. Твердый продукт собирали и затем высушивали над вакуумом. Продукт (8,04 г, 76%) выделяли в виде белого кристаллического твердого вещества с температурой плавления 67-70°С. Органический элементарный анализ: найдено: С 73.08, Н 5.37%; С14Н12О3 требует С 73.45, Н 5.32%;
Пример 6 - Приготовление соединений 160-167
[326] Соединение F приготовили в соответствии с общей схемой, при этом 2-гидроксибензофенон алкилировали бромалкилэфиром в присутствии основания, что сопровождалось разложением эфирной части при использовании триметилсиланоата калия.
[327] Соединение 160 - 6-(2-(2-гидроксибензоил) фенокси) гексановая кислота:
В 250-мл круглодонную колбу, оборудованную магнитной мешалкой и дефлегматором, помещали 10,32 г (48,2 ммоль) 2,2'-дигидроксибензофенона и 100 мл диметилсульфоксида (ДМСО). К прозрачному раствору добавляли гидроксид калия (2,91 г, 51,9 ммоль), предварительно размельченный до порошка. Реакционную смесь нагревали до 45°С, до тех пор, пока большая часть твердого вещества не растворилась. Образовавшуюся красную суспензию обрабатывали 8,80 мл (11,04 г, 49,5 ммоль) этил-6-бромгексаноатом. После перемешивания в течение 20 часов при 25°С, прозрачную реакционную смесь разбавляли водным 1% раствором соляной кислоты и метил-трет-бутиловым эфиром (МТБЭ). Слои разделялись. Органическую фазу промывали водой (2×50 мл) и соляным раствором (1×40 мл), высушивали над сульфатом натрия и концентрировали. Смолу помещали в 100 мл тетрагидрофурана (ТГФ) и обрабатывали триметилсиланоатом калия (15,09 г, 118 ммоль). Оранжевый раствор перемешивали в течение 20 часов при 25°С, разбавляли 4% водным раствором соляной кислоты до рН 7.5 и промывали МТБЭ. Органическую фазу экстрагировали водным 3% раствором бикарбоната натрия. Объединенные водные фазы подкисляли до рН 2 4% водным раствором соляной кислоты и экстрагировали 60 мл МТБЭ. Эту органическую фазу промывали соляным раствором (1×40 мл), высушивали над сульфатом натрия и концентрировали. Остаток очищали способом флеш-хроматографии, используя 80% смесь гексан/этилацетат (введенную с 0,5% уксусной кислотой). Продукт (4,2 г, 27%) выделяли в виде грязно-белого твердого вещества с температурой плавления 89-91°С. Органический элементарный анализ: найдено: С 69.50, Н 6.04%; C19H20O5 требует С: 69.50, Н: 6.14% 1Н ЯМР (d6-ДМСО): δ 12.0, bs, 1Н (СООН); δ 11.5, bs, 1Н (ОН); δ 7.5, t, 2H, (арильные Н); δ 7.4, dd, 1Н (арильный Н); δ 7.3, dd, 1Н (арильный Н); δ 7.15, d, 1Н (арильный Н); δ 7.1, t, 1Н (арильный Н); δ 7.0, d, 1Н (арильный Н); δ 6.9, t, 1Н (арильный Н); δ 3.9, t, 2H, (СН2 α к О); δ 2.05, t, 2H (СН2 α к СООН); δ 1.4, m, 4H (другие две CH2's); δ 1.0, р, 2H (СН2 в середине цепи).
[328] Следующие соединения были приготовлены из соответствующих исходных веществ с использованием того же способа: Соединение 161, Соединение 162, Соединение 163, Соединение 164, Соединение 165, Соединение 166 и Соединение 167.
[329] Соединение 161 - Натрий 8-(2-(2-гидроксибензоил) фенокси) октаноат:
При использовании 2,2'-дигидроксибензофенона и этил-8-бромоктаноата как исходных веществ, было приготовлено названное соединение и затем переведено в натриевую соль следующим образом: свободную кислоту (3,56 г, 9,99 ммоль) растворяли в 40 мл изопропанола и обрабатывали раствором гидроксида натрия (1,7 мл), приготовленным из гидроксида натрия (0,90 г, 22,5 ммоль) и воды (3,7 мл). Добавляли изопропанол и МТБЭ, что привело к осаждению твердого вещества. При нагревании этой смеси большая часть твердого вещества растворилась. Оставшееся твердое вещество было удалено фильтрацией. Грязно-белое твердое вещество, образовавшееся при охлаждении сухим льдом, выделяли фильтрацией и высушивали при пониженном давлении. Органический элементарный анализ: найдено: С 65.02%, Н 6.22%; C21H23O5Na требует С 66.00, Н 6.65%, 1H ЯМР (d6-ДМСО): δ 12.6, bs, 1H (ОН); δ 7.41, t, 1H, (арильный Н); δ 7.31, t, 1H (арильный Н); δ 7.27, dd, 1H (арильный Н); δ 7.15, dd, 1H (арильный Н); δ 7.03, d, 1H (арильный Н); δ 6.97, t, 1H (арильный Н); δ 6.91, d, 1H (арильный Н); δ 6.65, t, 1H (арильный Н); δ 3.83, t, 2H, (СН2 α к О); δ 1.82, t, 2H (CH2 α к COONa); δ 1.3, m, 4H (еще два CH2's); δ 1.0, m, 6H (CH2's в середине цепи). 13С ЯМР (d6-ДМСО): 198.59, 177.35, 161.35, 156.10, 134.56, 131.98, 131.78, 129.55, 128.57, 123.57, 120.18, 118.00, 117.09, 112.51, 67.74, 37.87, 28.83, 28.35, 28.27, 25.84, 24.87.
[330] Соединение 162 - 5-(2-(2-гидроксибензоил)-4-метоксифенокси) валериановая кислота (приведены данные основных изомеров): ЖХ-МС анализ: m+1 установленный пик (345). Результаты 1H ЯМР-анализа: (d6-ДМСО): δ 12.4, ds, 1H (СООН); δ 11.9, bs, 1H (ОН); δ 7.47, t, 1H, (арильный Н); δ 7.26, dd, 1H (арильный Н); δ 7.14, d, 1H (арильный Н); δ 7.13, d, 1H (арильный Н); δ 7.03, t, 1H (арильный Н); δ 6.49, d, 1H (арильный Н); δ 6.42, dd, 1H (арильный Н); δ 3.95, t, 2H, (CH2 α к О); δ 3.79, s, 3H, (СН3О); δ 2.07, t, 2H (СН2 β к СООН); δ 1.48, р, 2H (CH2 в цепи); δ 1.34, р, 2H (CH2 в цепи). 13С ЯМР (d6-ДМСО): 199.60, 174.18, 165.97, 163.34, 155.14, 135.14, 131.77, 128.29, 127.83, 120.46, 114.06, 112.69, 107.41, 100.70, 67.51, 55.76, 33.05, 27.80, 20.77.
[331] Соединение 163 - 5-(2-(2-гидроксибензоил) фенокси) валериановая кислота: ЖХ-МС анализ: m+1 установленный пик (315). Результаты 1Н ЯМР-анализа: (d6-ДМСО): δ 11.9, bs, 1H (СООН); δ 11.5, bs, 1H (ОН); δ 7.50, dt, 1H, (арильный Н); δ 7.48, dt, 1H, (арильный Н); δ 7.35, dd, 1H (арильный Н); δ 7.25, dd, 1H (арильный Н); δ 7.14, d, 1H (арильный Н); δ 7.06, t, 1H (арильный Н); δ 6.96, d, 1H (арильный Н); δ 6.85, t, 1H (арильный Н); δ 3.93, t, 2H, (СН2 α к О); δ 2.06, t, 2Н (СН2 α к СООН); δ 1.42, р, 2Н (СН2 в цепи); δ 1.29, р, 2Н (СН2 в цепи). 13С ЯМР (d6-ДМСО): 200.59, 174.15, 160.43, 155.71, 135.94, 132.69, 132.22, 128.58, 128.02, 121.50, 120.46, 119.06, 117.30, 112.67, 67.50, 33.05, 27.75, 20.70.
[332] Соединение 164 - 5-(2-(2-гидрокси-5-метоксибензоил)-4-метоксифенокси) валериановая кислота: ЖХ-МС анализ: m+1 установленный пик (375). Результаты 1H ЯМР-анализа: (d6-ДМСО): δ 12.4, bs, 1H (СООН); δ 12.0, bs, 1H (ОН); δ 7.25, d, 1H, (арильный Н); δ 7.21, d, 1H, (арильный Н); δ 6.66, d, 1H (арильный Н); δ 6.62, dd, 1H (арильный Н); δ 6.48, d, 1H (арильный Н); δ 6.42, dd, 1H (арильный Н); δ 3.96, t, 2Н, (СН2 α к 0); δ 3.81, s, 3Н, (СН3О); δ 3.80, s, 3Н, (СН3О); δ 2.08, t, 2Н (СН2 α к СООН); δ 1.48, р, 2Н (СН2 в цепи); δ 1.34, р, 2Н (СН2 в цепи). 13С ЯМР (d6-ДМСО): 198.85, 174.20, 165.62, 164.14, 162.54, 157.11, 135.18, 130.22, 120.60, 114.44, 107.04, 105.51, 100.63, 99.24, 67.55, 55.69, 55.48, 33.06, 27.75, 20.77.
[333] (Соединение 166) 4-(2-(2-гидроксибензоил) фенокси) масляная кислота: ЖХ-МС анализ: m+1 установленный пик (333). Результаты 1H ЯМР-анализа: (d6-ДМСО): δ 12.0, bs, 1H (СООН); δ 7.46, m, 2Н (арильные Н); δ 7.33, dt, 1H (арильный Н); δ 7.29, d, 1H (арильный Н); δ 6.82, t, 1H (арильный Н); δ 3.77, t, 2Н, (СН2 α к О); δ 1.85, t, 2Н (СН2 α к СООН); δ 1.35, р, 2Н (срединный СН2 в цепи). ^С ЯМР (d6-ДМСО): 200.47, 173.92, 160.40, 155.57, 135.97, 132.64, 132.27, 128.64, 128.00, 121.52, 120.56, 119.10, 117.34, 112.62, 66.99, 29.55, 23.92.
[334] Соединение 167 - 4-(2-хлорбензоил-4-метилфенокси)масляная кислота: ЖХ-МС анализ: m+1 установленный пик (333). Результаты 1H ЯМР-анализа: (d6-ДМСО): δ 12.4, bs, 1H (СООН); δ 12.0, bs, 1H (ОН); δ 7.23, d, 1H (арильный Н, о к О); δ 3.74, t, 2Н, (СН2 α к О); δ 2.25, s, 3Н, СН3); δ 1.84, t, 2Н (СН2 α к СООН); δ 1.33, р, 2Н (срединный СН2 в цепи). 13С ЯМР (d6-ДМСО): 198.76, 173.97, 165.63, 164.10, 162.58, 156.99, 135.11, 130.29, 120.55, 114.45, 107.14, 105.67, 100.67, 99.16, 67.03, 55.69, 55.50, 29.56, 23.85.
Пример приготовления соединений 168-173
[335] (Соединение 168) 4-(2-бензоил-5-метоксифенокси)масляная кислота:
В 100-мл мини-блочную трубку, оборудованную магнитной мешалкой, помещали 4,56 г (20,0 ммоль) 2-гидрокси-4-метоксибензофенона, 2,70 мл (3,68 г, 18,9 ммоль) этил 4-бромбутирата и 40 мл диметилформамида (ДМФ). К прозрачному раствору добавляли карбонат калия (2,96 г, 21,4 ммоль). Реакционную смесь нагревали до 80°С. После перемешивания в течение 20 часов при 25°С, прозрачную реакционную смесь разбавляли водой. Образовавшееся твердое вещество выделяли фильтрацией. Это твердое вещество помещали в 30 мл тетрагидрофурана (ТГФ) и обрабатывали 3,10 г (24,0 ммоль) триметилсиланоата калия. Оранжевый раствор перемешивали в течение 20 часов при 25°С, разбавляли 4% водным раствором соляной кислоты до рН 7.5 и промывали МТБЭ. Органическую фазу экстрагировали 3% водным раствором бикарбоната натрия. Объединенные водные фазы подкисляли до рН=2 4% водным раствором соляной кислоты и экстрагировали 60 мл МТБЭ. Эту органическую фазу промывали соляным раствором (1×40 мл), высушивали над сульфатом натрия и концентрировали. Образовавшееся твердое вещество очищали измельчением в порошок, используя смеси МТБЭ/гексаны. Большую часть продукта выделяли из маточного раствора. ЖХ-МС анализ: m+1 установленный пик (315). Результаты 1Н ЯМР-анализа: (d6-ДМСО): δ 12.0, bs, 1Н (СООН); δ 7.6, d, 2Н, (фенильные Н, о к СО); δ 7.56, t, 1H (фенильный Н, p к СО); δ 7.44, t, 2Н (фенильные Н, m к СО); δ 7.35, d, 1Н (арильный Н, о к СО); δ 6.64, m, 2Н (арильные Н, m к СО); δ 3.88, t, 2Н, (СН2 α к О); δ 3.82, s, 3Н, (СН3О); δ 1.84, t, 2Н (СН2 α к СООН); δ 1.53, р, 2Н (срединный СН2 в цепи). 13С ЯМР (d6-ДМСО): 195.08, 173.91, 163.17, 158.33, 138.84, 132.37, 131.37, 128.67, 128.24, 120.87, 105.87, 99.02, 66.89, 55.53, 29.45, 23.79.
[336] Другие реагенты-носители синтезированы тем же способом: Соединение 169, Соединение 170, Соединение 171, Соединение 172 и Соединение 173.
[337] (Соединение 169) 4-(2-бензоил-4-хлорфенокси)масляная кислота: ЖХ-МС анализ: m+1 установленный пик (319). Результаты 1Н ЯМР-анализа: (d6-ДМСО): δ 11.9, bs, 1Н (СООН); δ 7.64, d, 2H, (фенильные Н, о к СО); δ 7.59, t, 1Н (фенильный Н, р к СО); δ 7.51, dd, 1Н (арильный Н, р к СО); δ 7.45, t, 2H (фенильные Н, m к СО); δ 7.36, d, 1Н (арильный Н, о к СО); δ 7.14, d, 1Н (арильный Н, m к СО); δ 3.87, t, 2H, (CH2 α к О); δ 1.84, t, 2H (CH2 α к СООН); 51.53, р, 2H (серединный СН2 в цепи). 13С ЯМР (d6-ДМСО): 194.37, 173.82, 154.74, 136.96, 133.42, 131.56, 130.05, 128.97, 128.62, 128.29, 124.48, 114.61, 67.38, 29.37, 23.79.
[338] (Соединение 170) 4-(2-бензоил-4-бромфенокси)масляная кислота: ЖХ-МС анализ: m+1 установленный пик (363). Результаты 1Н ЯМР-анализа: (d6-ДМСО): δ 11.9, bs, 1Н (СООН); δ 7.6, m, 3H, (арильные Н); δ 7.60, t, 1Н (фенильный Н, р к СО); δ 7.49, dd, 1Н (арильный Н, о к СО); δ 7.46, t, 2H (фенильные H, m к СО); δ 7.09, d, 1Н (арильный Н, m к СО); δ 3.89, t, 2H, (CH2 α к О); δ 1.82, t, 2H (СН2 α к СООН); δ 1.53, р, 2H (срединный СН2 в цепи). 13С ЯМР (d6-ДМСО): 194.28, 173.81, 155.19, 136.97 134.48, 133.42, 131.06, 130.48, 128.97, 128.62, 115.08, 112.02, 67.33, 29.35, 23.77.
[339] (Соединение 171) 4-(2-(2-хлорбензоил-5-метилфенокси) масляная кислота: ЖХ-МС анализ: m+1 установленный пик (333). Результаты 1Н ЯМР-анализа: (d6-ДМСО): δ 12.0, bs, 1Н (СООН); δ 7.54, d, 1Н, (арильный Н); δ 7.4, m, 2H (арильные Н); δ 7.33, dt, 1Н (арильный Н); δ 7.29, d, 1H (арильный Н); δ 6.86, m, 2H (арильные Н, о к О); δ 3.77, t, 2H, (CH2 α к О); δ 2.31, s, 3H, СН3); δ 1.85, t, 2H (СН2 α к СООН); δ 1.35, р, 2H (срединный CH2 в цепи). 13С ЯМР (d6-ДМСО): 193.31, 173.81, 158.34, 145.98, 141.38, 130.99, 130.56, 129.48, 129.43, 128.38, 127.00, 123.95, 121.46, 113.43, 66.95, 29.65, 23.70, 21.48.
[340] (Соединение 172) 4-(2-(2-хлорбензоил-4-метилфенокси) масляная кислота: ЖХ-МС анализ: m+1 установленный пик (333). Результаты 1Н ЯМР-анализа: (d6-ДМСО): δ 11.95, bs, 1Н (СООН); δ 7.43, m, 3H, (арильные Н); δ 7.34, m, 3H (арильные Н); δ 6.92, d, 1H (арильный Н, о к О); δ 3.74, t, 2H, (СН2 α к О); δ 2.25, s, 3H, СН3); δ 1.84, t, 2H (CH2 α к СООН); δ 1.33, р, 2H (срединный СН2 в цепи). 13С ЯМР (в6-ДМСО): 193.92, 173.81, 156.15, 140.95, 135.37, 131.24, 130.40, 129.65, 129.56, 129.49, 128.62, 127.02, 126.45, 112.95, 67.07,29.65,23.75, 19.80.
[341] (Соединение 173) 4-(2-бензоил-4-хлор-5-метилфенокси) масляная кислота: ЖХ-МС анализ: m+1 установленный пик (333). Результаты 1H ЯМР-анализа: (d6-ДМСО): δ 11.9, bs, 1H (СООН); δ 7.61, d, 2H, (фенильные Н, о to СО); δ 7.57, t, 1 Н (фенильный Н, р to СО); δ 7.44, t, 2H (фенильные Н, m к СО); δ 7.33, s, 1H (арильный Н, о к СО); δ 7.14, s, 1H (арильный Н, m к СО); δ 3.87, t, 2H, (СН2 α к О); δ 2.33, s, 3H, СН3); δ 1.81, t, 2H (СН2 α к СООН); δ 1.49, р, 2H (срединный СН2 в цепи). 13С ЯМР (d6-ДМСО): 194.31, 173.83, 154.78, 139.80, 137.39, 133.17, 128.91, 128.84, 128.51, 127.55, 124.69, 115.57, 67.32, 29.37, 23.80, 20.03.
Возможный способ приготовления соединения F
[342] Соединение F можно приготовить иначе, используя метод ацилирования ароматических соединений Фриделя-Крафтса:
[343] Используют фенол, замещенный подходящим образом, смешивают его с соответствующим бромэфиром, используют К2СО3 в качестве основания, проводят реакцию продукта с соответствующим хлоридом ароматической кислоты в присутствии AlCl3; или берут салициловую кислоту, замещенную подходящим образом, и смешивают ее с соответствующим бромэфиром, используя K2CO3 в качестве основания. Продукт переводят в кислый хлорид SOCl2, который затем реагирует с бензолом, замещенным подходящим образом, в присутствии AlCl3.
Пример 8 - приготовление соединения 174
[344] Соединение 174 было приготовлено в три стадии:
А. О-ацетил-5-хлорсалициловая кислота
[345] 10 г (57,9 ммоль) 5-хлорсалициловой кислоты взвешивали в 100-мл круглодонной колбе, затем добавляли уксусный ангидрид (12,8 мл, 115,9 ммоль). Прежде чем добавить концентрированную серную кислоту (2 капли), смесь перемешивали в течение 5 минут. Реакционную смесь орошали в течение 3 часов. Протекание реакции контролировали с помощью ВЭЖХ. Реакционную смесь охлаждали до комнатной температуры и наливали в химический стакан, содержащий 2 н, HCl (200 мл) для осаждения продукта. Продукт собирали способом вакуумной фильтрации. Чистоту проверяли с помощью ВЭЖХ, что позволило обнаружить присутствие примесей. В 200-мл круглодонной колбе осадок всю ночь перемешивали в воде (150 мл). Нерастворимое твердое вещество собирали способом вакуумной фильтрации. Наличие примеси проверяли с помощью ВЭЖХ, показавшей, что в неочищенном продукте отсутствуют примеси. Продукт высушивали всю ночь в вакууме, получая 12 г о-ацетил-5-хлорсалициловой кислоты (56 ммоль, выход 97%).
В. О-ацетил-5-хлорсалицоилхлорид
[346] Тионилхлорид (~100 мл) помещали в 250-мл круглодонную колбу и перемешивали в ледяной бане в течение 15 минут. О-ацетил-5-хлорсалициловую кислоту (6,0 г, 27,9 ммоль) медленно добавляли к охлажденному тионилхлориду. Для растворения кислоты в реакционную смесь добавляли ДМФ (2 капли). Реакционную смесь перемешивали всю ночь, получив гомогенный раствор. Избыток тионилхлорида отгоняли в вакууме. Оставшийся остаток высушивали в вакууме всю ночь.
С. 3-(N-2-гидрокси-5-хлорбензоил)аминопропионовая кислота
[347] (3-Аланин (2,5 г, 28,0 ммоль) взвешивали в 250-мл круглодонной колбе. В колбу добавляли метиленхлорид (100 мл) и смесь перемешивали в течение 5 минут. Затем в колбу добавляли по капле хлортриметилсилан (6,06 г, 55,7 ммоль). При орошении реакционную смесь нагревали в течение 15 минут. Смесь оставляли охлаждаться до комнатной температуры и помещали в ледяную баню на 15 минут. Триэтиламин (8,5 г, 84,0 ммоль) медленно добавляли в охлажденную колбу. О-Ацетил-5-хлорсалицоилхлорид (7,6 г, 27,9 ммоль) растворяли в метиленхлориде (30 мл) и добавляли к реакционной смеси в течение 0,5 часов. Реакционную смесь перемешивали всю ночь, и оставляли нагреваться до комнатной температуры. Протекание реакции подтверждали с помощью ВЭЖХ. Растворитель выпаривали под вакуумом. Оставшийся остаток перемешивали в 2 н. NaOH (~100 мл) в течение 2 часов и медленно нагревали до 60°С. Затем раствор охлаждали до комнатной температуры и отфильтровывали способом безнапорной фильтрации. Фильтрат медленно подкисляли концентрированной HCl до образования осадков. Неочищенный продукт собирали при рН смеси, равной 6. Продукт перекристаллизовывали, используя смесь МеОН-Н2О. Чистоту проверяли с помощью ВЭЖХ, показавшей наличие примесей. Продукт несколько раз подвергали очистке и перекристаллизации, пока не получали чистое соединение. Конечный продукт перемешивали всю ночь в метиленхлориде, собирали способом фильтрации, и высушивали под вакуумом всю ночь, получая розоватый порошок (3,98 г, 16,3 ммоль, выход 58,5%); температура плавления 181-183°С; 1Н-ЯМР (ДМСО-d6) δ 2.47-2.58 (t, 2Н), 3.44-3.54 (q, 2Н), 6.93-6.98 (d, 1H), 7.39-7.44 (dd, 1H), 7.91-7.96 (d, 1H), 8.93-9.01 (t, 1H), 12.1-12.3 (s, 1H). Величина KF=1.615%. Результаты расчета C10H10NO4Cl*0.2220H2O: С, 48.50, Н, 4.25, N, 5.66. Найдено: С, 48.20; Н, 4.03; N, 5.43.
Пример 9 - Приготовление соединения 175-178
Соединение 175 -4-(2-бензилокси-фенокси)-масляная кислота:
[346] В 250-мл колбу, оборудованную дефлегматором и магнитной мешалкой, в инертной атмосфере добавляли 2-бензилоксифенол (8,0 г, 40 ммоль), этиловый эфир 4-бромбутановой кислоты (5,7 мл, 40 ммоль), карбонат калия (7,2 г, 52 ммоль), и этанол (100 мл). При орошении реакционную смесь нагревали при перемешивании в течение 8 часов. Затем реакционную смесь охлаждали до комнатной температуры и удаляли нерастворимый побочный продукт способом вакуум-фильтрации. К фильтрату добавляли 2 н. водный раствор гидроксида натрия (30 мл). Этот раствор нагревали до 50°С в течение 2 часов. Затем раствор охлаждали до комнатной температуры, этанол удаляли при пониженном давлении, и устанавливали рН образовавшегося раствора равным 9. Водный раствор промывали этилацетатом (2×30 мл), а оставшийся этилацетат удаляли при пониженном давлении. Раствор охлаждали до 0°С, используя наружную ледяную баню, и затем подкисляли до рН=2 6 н. водным раствором соляной кислоты. Осажденный продукт собирали способом вакуум-фильтрации и высушивали под вакуумом. Продукт (7,2 г, 63%) выделяли в виде белого порошка. 1Н-ЯМР (400МГц, ДМСО-d6): δ 12.0, s, 1H (СООН); δ 7.4, мультиплет, 5Н (бензиловые арильные Н); δ 7.0, мультиплет, 2Н (арильные Н); δ 6.9, мультиплет, 2Н (арильные Н); δ 5.0, s, 2Н (бензиловый СН3); δ 4.0, t, 2Н (CH2 α к фенокси); δ 2.4, t, 2Н (CH2 α к СООН); δ 1.9, мультиплет, 2Н (оставшаяся CH2 группа).
[347] Соединение 176 - (4-Бензилокси-фенокси)уксусная кислота была приобретена у Lancaster.
[348] Соединение 177 - 11-(2-Бензилоксифенокси)ундекановая кислота:
[349] В 250-мл колбу Эрленмейера поместили только что размолотый гидроксид калия (4,2 г, 74,91 ммоль) и 100 мл диметилсульфоксида. Добавили 2-бензилоксифенол (5 г, 24,97 ммоль) и метиловый эфир 11-бромундекановой кислоты (7 г, 25,07 ммоль) и перемешивали смесь при комнатной температуре всю ночь. Затем добавили воду (75 мл) и нагревали раствор до 85°С при перемешивании в течение трех часов. Реакционную смесь охлаждали до комнатной температуры и подкисляли концентрированной соляной кислотой до рН=2. Подкисленный раствор охлаждали до 4°С в течение 2 часов, и затем собирали осадок способом вакуум-фильтрации. Продукт перекристаллизовывали из смеси этанол/вода. Продукт (8,88 г, 93%) выделяли в виде светло-коричневого твердого вещества с температурой плавления 62-63°С. Органический элементарный анализ: найдено: С 74.71 Н 8.08%; C24H32O4 требует С 74.97, Н 8.39%;
[350] Соединение 178 - 5-(4-бензилокси-фенокси)пентановая кислота:
[351] В 500-мл колбу с тремя горловинами, оборудованную дефлегматором, в инертной атмосфере помещали 4-бензилоксифенол (30,64 г, 150 ммоль), этиловый эфир 5-бромпентановой кислоты (31,99 г, 150 ммоль), карбонат калия (22,80 г, 165 ммоль), и 270 мл 2-бутанона. При орошении реакционную смесь нагревали в течение 23 часов, охлаждали и затем разбавляли этилацетатом (150 мл) и экстрагировали на фоне воды (500 мл). Органический слой промывали водой (1×250 мл) и соляным раствором (1×250 мл) и растворитель удаляли при пониженном давлении. Образовавшееся масло высушивали под вакуумом в течение 4 дней, на протяжении которых образовались белые кристаллы. Белые кристаллы растворяли в этилацетате (100 мл), промывали водным 1 н. раствором гидроксида натрия (3×50 мл), и удаляли растворитель при пониженном давлении. Образовавшееся масло высушивали под вакуумом всю ночь, получая белые кристаллы. Продукт перекристаллизовывали из смеси 1:1 этанол/вода, собирали способом вакуум-фильтрации и высушивали под вакуумом. Этот продукт использовали без дальнейшей очистки.
[352] В 1-л круглодонную колбу, оборудованную дефлегматором, помещали этиловый эфир 5-(4-бензилокси-фенокси) пентановой кислоты (15,13 г, 46 ммоль) и 2 н. водный раствор гидроксида натрия (47 мл). Смесь перемешивали в течение 30 минут. Добавляли воду (200 мл). Смесь перемешивали в течение 20 минут, затем нагревали при орошении в течение 2 часов, образуя коричневый раствор. Добавляя лед, раствор быстро охлаждали до комнатной температуры. Охлажденный раствор подкисляли 2н водным раствором соляной кислоты (50 мл) и собирали образовавшийся белый осадок способом вакуум-фильтрации, промывали водой (2×100 мл), гексаном (2×100 мл), и сушили над вакуумом всю ночь. Порошок мелко размалывали и промывали гексаном (1×250 мл) и диэтиловым эфиром (1×250 мл), получая белый порошок. В 1-л химическом стакане полученный порошок растворяли в смеси этилацетата (300 мл) и диэтилэфира (200 мл). Раствор нагревали в течение 10 минут, добавляли метанол (5 мл), нагревали еще 10 минут, и затем отфильтровывали через целитный слой, получая прозрачный желтый раствор. Продукт кристаллизовали, медленно добавляя гексан. Собирали первую партию кристаллов способом фильтрации и к маточному раствору добавляли гексан (200 мл). Затем раствор концентрировали при пониженном давлении до объема 400 мл и оставляли на некоторое время. Вторую порцию кристаллов собирали фильтрованием и соединяли с первой. Продукт (8,92 г, 65%) выделяли в виде белого кристаллического вещества с температурой плавления 127-128°С. Органический элементарный анализ: найдено: С 71.01 Н 6.98%; C18H20O4 требует С 71.98, Н 6.71%;
Пример 10 - Пероральная доставка ПYY [3-361 в твердой форме в организм крыс
[353] Использовали исходный раствор ПYY [3-36] (80 мг/мл), приготовленный с использованием деионизированной воды (ПYY можно приобрести в Bachem California Inc., Торранс, Калифорния).
[354] Приблизительно 0,08 мг/таблетка (приблизительно 0,3 мг/кг) ПYY (приблизительно 1 мкл) добавляли и смешивали с примерно 13,5 или 27 мг/таблетка (50 или 100 мг/кг) свободной кислоты или натриевой соли соединения-носителя, как указано ниже. Верхний перфоратор, нижний перфоратор и пресс-форму ручного пресса для производства таблеток Carver 4350 со стандартным образцом в виде капсулы, производства Natoli Engineering Company, Inc. (Сент-Чарльз, Миссури) обрабатывали стеаратом магния (0,1%). Приблизительно 13,58 мг или приблизительно 27,08 мг смешанного порошка загружали в пресс-форму и при давлении приблизительно 6894.74 кПа производили мини-таблетку в виде гранулы. Получившаяся твердая дозируемая форма имеет приблизительно размеры стандартной капсулы 9 (диаметр приблизительно 2,65 мм и длина приблизительно 8,40 мм) для таблетки 27,08 мг, а для таблетки 13,58 мг диаметр составляет приблизительно 2,65 мм и длина приблизительно 4,20 мм.
[355] Самцов крыс Sprague Dawley (приблизительно от 260 до 280 г) не кормили всю ночь и затем подвергали анестезии используя стандартный способ CO2-ингаляции в течение приблизительно 10-30 секунд, что приводило к состоянию анестезии на время, меньшее чем приблизительно 1 минута, предпочтительно приблизительно от 10 до 30 секунд.
[356] Использовали дозирующую трубку для перорального введения. Дозирующую трубку вставляли крысам в рот и осторожно проникали в глотку и пищевод крыс примерно на 8-15 см в зависимости от веса крысы (в общем случае на приблизительно 11 см). Твердую дозируемую форму вводили в дистальный пищевод и/или желудок, надавливая на поршень дозирующей трубки для перорального ввода.
[357] Пробы крови брали ретро-орбитальным способом обычно через 0, 15, 30, 60 и 90 минут. Концентрации DYY в сыворотке количественно определяли с помощью радиоиммунологического обследования на содержание ПYY [3-36] (Каталог #RK-059-02 от Phoenix Pharmaceuticals, Inc., Белмонт, Калифорния). Результаты, полученные у животных в каждой группе, усредняли для каждого момента времени. Максимальное значение этих средних величин (т.е., средняя максимальная концентрация ПYY [3-36] в сыворотке ± стандартное отклонение (СО)) приведено ниже.
Пример 11 - Пероральная доставка ПУУГ3-361 в жидкой форме в организм крыс
[358] Дозируемые растворы для перорального ввода соединения-носителя и пептидных YY остатков 3-36 (ПYY [3-36]) (можно приобрести в Bachem California Inc. of Torrance, Калифорния) в деионизированной воде были приготовлены следующим образом.
[359] Исходный раствор ПYY [3-36] (80 мг/мл) был приготовлен с использование деионизированной воды. Были приготовлены композиции для перорального ввода, содержащие 200 мг/кг соединения-носителя и 0,3 мг/кг ПYY в водном растворе. Раствор соединения 23 приготавливали, добавляя один эквивалент гидроксида натрия для перевода агента-носителя в виде свободной кислоты в ее натриевую соль.
[360] Самцов крыс Sprague-Dawley с весом между 240-320 г перед экспериментами не кормили максимум 24 часа и перед введением тестируемого изделия вводили кетамин (44 мг/кг) и торазин (1,5 мг/кг) путем внутримышечной инъекции. Затем, анестезированным животным перорально вводили тестируемый состав. Группе из пяти животных вводили один из дозируемых растворов. Для перорального ввода, 11 см французский катетер Rusch 8 прикрепляли к 1 мл шприцу с кончиком пипетки. Шприц наполняли дозируемым раствором путем протягивания раствора через катетер, который затем вытирали досуха. Катетер помещали в пищевод, оставляя 1 см трубки за резцами. Дозируемый раствор вводили, нажимая на поршень шприца.
[361] Пробы крови отбирали серийно из хвостовой артерии или путем пункции сердца, обычно через 0, 15, 30, 45, 60 и 90 минут. Концентрацию ПYY в сыворотке количественно определяли с помощью радиоиммунологического обследования на содержание ПYY [3-36] (Каталог #RK-059-02 от Phoenix Pharmaceuticals, Inc., Белмонт, Калифорния). Результаты, полученные у животных в каждой группе, усредняли для каждой временной точки.
Максимальное значение этих средних величин (т.е. средняя максимальная концентрация ПYY [3-36] в сыворотке ± стандартное отклонение (СО)) приведено ниже в таблице 2. Когда животным перорально вводили только ПYY [3-36], существенного содержания ПYY [3-36] в крови не обнаружили.
доза)
Пример 12 - Пероральная доставка человеческого рекомбинантного инсулина в организм крыс
[362] Инсулин (человеческий рекомбинантный) был приобретен в ICN Biomedicals (Аврора, Огайо) в виде нерасфасованного порошка. Для приготовления исходных растворов инсулин растворяли в деионизированной воде (рН~6,5), получая концентрацию 15 мг/мл. До использования исходные растворы хранили замороженными при -20°С в 1,0-мл аликвотах. Для того чтобы получить конечную концентрацию соединения-носителя в дозируемых растворах 200 мг/мл, соединение-носитель растворяли и деионизированной воде. Реагент-носитель в форме свободной кислоты переводили в натриевую соль, добавляя один эквивалент гидроксида натрия. Растворы встряхивали, обрабатывали ультразвуком и нагревали и, при необходимости, еще раз добавляли гидроксид натрия в мкл-количествах для достижения равномерной растворимости. рН растворов устанавливали в интервале 3,5-8,5 путем добавления или хлорной кислоты, или гидроксида натрия. Затем для получения конечной концентрации 0,5 мг/мл исходный раствор инсулина (обычно 66,7 мкл) добавляли к раствору реагента-носителя. После растворения и добавления лекарственного препарата растворы доводили до конечного объема, добавляя деионизированную воду.
[363] Пероральным способом в самцов крыс вводили или только инсулин, или инсулин в комбинации с реагентом-носителем Emisphere. Обычно перед введением крыс не кормили в течение 18-24 часов. Для введения французский катетер Rusch 8 укорачивали до 11 см в длину и адаптировали для использования со шприцем на 1 мл. Шприц наполняли дозируемым раствором, и катетер досуха вытирали, убирая избыток раствора. Катетер вставляли в рот крысы и направляли в пищевод (10,0 см). Дозируемый раствор вводили путем надавливания на поршень шприца, держа крысу в положении стоя.
Отбор и обработка проб: инсулин
[364] При отборе проб крови непосредственно до каждого отбора крыс быстро (~10 сек) подвергали воздействию диоксида углерода, до тех пор, пока они не впадали в состояние прострации. Для отбора крови 77-мм капиллярную трубку вставляли в ретро-орбитальную полость. Обычно пробы крови отбирали до введения (время 0) и через 15, 30, 45, и 60 минут после введения. Пробы собирали в трубки CAPIJECT® (Terumo Corporation, Токио, Япония), содержащие активатор свертывания крови (красный колпачок, разделительные трубки для сыворотки). Пробы оставляли свертываться в течение ~20 минут при 4°С. После свертывания, для того, чтобы отделить сыворотку, пробы центрифугировали при 10,000 оборот/мин в течение 4 минут при 6°С. Сыворотку собирали в трубки Эппендорфа и охлаждали при -20°С до начала анализа.
Отбор и обработка проб: глюкоза в цельной крови
[365] Для того чтобы определить фармакодинамический отклик, для измерения содержания глюкозы в цельной крови после введения инсулина или инсулина с реагентом-носителем использовали ручной глюкометр (OneTouch Ultra, LifeScan® (Johnson & Johnson, Нью-Брансуик, Нью-Джерси)). Пробы отбирали или из ретро-орбитальной полости (см. Отбор и обработка проб: инсулин) или из хвостовой артерии (т.е. путем обрезания хвоста). При обрезании хвоста кончик хвоста отрезали скальпелем приблизительно на 5 мм. После отбрасывания первых капель крови, маленькой пробой (~5-10 мкл) прикасались к тестовой полоске глюкометра (OneTouch Ultra, LifeScan) и на приборе появлялось показание содержания глюкозы в крови. При каждом последующем отборе, сгусток крови, образующийся на кончике хвоста, разрушали и отбирали свежую пробу. В общем случае, пробы отбирали до введения (время 0) и через 15, 30, 45, и 60 минут после введения.
Пример 13 - Человеческий цинковый инсулин - пероральная доставка
[366] Составы соединения-носителя и человеческого цинкового инсулина для перорального введения (минимум 26 МЕ/мг, можно приобрести в Calbiochem - Novabiochem Corp, Ла Джолла, Калифорния) были приготовлены в деионизированной воде. 500 мг соединения-носителя добавляли к 1,5 мл воды. Свободную кислоту, входящую в состав соединения-носителя, переводили в натриевую соль путем перемешивания полученного раствора и добавления одного эквивалента гидроксида натрия. Раствор встряхивали, затем нагревали (приблизительно 37°С) и обрабатывали ультразвуком. рН устанавливали в диапазоне примерно 7-8,5 с помощью NaOH или HCl. Для достижения равномерной растворимости, при необходимости, добавляли дополнительные количества NaOH, и рН снова устанавливали в интервале примерно от 7 до 8,5. Затем водой доводили общий объем до примерно 2,4 мл и встряхивали. Приблизительно 1,25 мг инсулина из исходного раствора инсулина (15 мг/мл, приготовленных путем смешивания 0,5409 г инсулина и 18 мл деионизированной воды, установки рН=8,15 с помощью HCl и NaOH и, для получение прозрачного раствора, использования 40 мл концентрированной HCl, 25 мл 10н NaOH и 50 мл 1н NaOH) добавляли к раствору и перемешивали путем переворачивания. Приведены конечные дозы соединения-носителя, инсулина и объемы доз.
[367] Самцов крыс Sprague-Dawley, весивших приблизительно 200-250 г, не кормили в течение 24 часов и вводили кетамин (44 мг/кг) и хлорпромазин (1,5 мг/кг) за 15 минут до введения и еще раз, при необходимости сохранить анестезию. Группе из пяти животных вводили один из дозируемых растворов. Для перорального ввода, 11 см французский катетер Rusch 8 прикрепляли к 1 мл шприцу с кончиком пипетки. Шприц наполняли дозируемым раствором путем протягивания раствора через катетер, который затем вытирали досуха. Катетер помещали в пищевод, оставляя 1 см трубки за резцами. Дозируемый раствор вводили, нажимая на поршень шприца.
[368] Пробы крови отбирали серийно из хвостовой артерии, в общем случае через 15, 30, 60, 120 и 180 минут. Уровень инсулина в сыворотке определяли с помощью тестового комплекта Insulin ELISA (комплект # DSL-10-1600 из Diagnostic Systems Laboratories, Inc., Уэбстер, Техас), корректируя стандартный протокол для оптимизации чувствительности и линейной области калибровочной кривой для объемов и концентраций проб, используемых в настоящем протоколе. Концентрации человеческого инсулина в сыворотке (мкЕд/мл) измеряли для каждого момента времени для каждого из пяти животных в каждой группе. Пять значений для каждого момента времени усредняли и результаты наносили на график: концентрация инсулина - время.
(Предшествующие эксперименты не выявили измеримых уровней человеческого инсулина после его введения без реагента-носителя). Максимум (пик) и площадь под кривой (ППК) приведены ниже в таблице 4. Для % изменения относительно базовой линии для Blood Glucose the ONE TOUCH® (Life Scan, Johnson & Johnson, Нью-Брансуик, Нью-Джерси).
Инсулин - пероральная доставка
Пример 14 - Инсулин - доставка через легкие
[368] Были приготовлены дозируемые композиции соединения-носителя и человеческого инсулина в воде. В общем случае к 1,5 мг соединения-носителя добавляли деионизированную воду, доводя объем до 1,0 мл, и встряхивали раствор. Использовали или натриевую соль соединения-носителя, или свободную кислоту переводили в натриевую соль путем перемешивания полученного раствора, добавления одного эквивалента гидроксида натрия (10 н) и разбавления водой. Раствор встряхивали, затем нагревали (приблизительно 37°С) и обрабатывали ультразвуком. рН устанавливали от приблизительно 7,0 до 8,5 с помощью NaOH или HCl. К раствору добавляли 75 мкл исходного раствора человеческого инсулина (2 мг/мл). (Исходный раствор готовили следующим образом. К 0,02 г инсулина добавляли 3 мл раствора HCl в деионизированной воде с рН 3,0. рН образовавшегося раствора доводили с помощью HCl и NaOH до значений ниже 3,0 (приблизительно 2,6) до тех пор, пока раствор не становился прозрачным. Затем рН доводили до 7,6, используя NaOH и HCl. Конечный объем доводили до 10 мл деионизированной водой с рН 7,5. Конечное рН=7,59). Затем добавляли воду, доводя общий объем до 2,0 мл, и раствор осторожно переворачивали несколько раз. Согласно протоколу введения, раствор можно использовать сразу после приготовления, или в другом примере реализации, перед введением его можно поместить в водяную баню с температурой 37°С на один час. Конечная доза соединения-носителя, доза инсулина и объемы доз приведены ниже в таблице 4.
[369] Типичными протоколами введения и отбора проб являются следующие. Самцов крыс Sprague-Dawley, весивших между 200-250 г, не кормили в течение 24 часов и за 15 минут до введения вводили кетамин (44 мг/кг) и хлорпромазин (3,0 мг/кг) и, при необходимости сохранить анестезию, вводили их снова (используя те же количества кетамина и 1,5 мг/кг хлорпромазина). Обычно группе из пяти животных вводили один из дозируемых растворов. Контрольной группе из пяти животных вводили инсулин отдельно. Трахеальная капельница для грызунов, оборудованная источником света (можно приобрести в Perm Century, Inc., Питсбург, Пенсильвания), была заполнена дозируемым раствором и вставлена в глотку таким образом, что игла входила в трахею (подтверждено визуально). Дозируемый раствор вводился путем надавливания поршня.
[370] Пробы крови у каждого животного отбирали серийно из хвостовой артерии, в общем случае через 5, 15, 30, 60 и 120 минут после введения. Уровень инсулина в сыворотке определяли с помощью тестового комплекта Insulin ELISA (комплект # DSL-10-1600 из Diagnostic Systems Laboratories, Inc., Уэбстер, Техас), корректируя стандартный протокол для оптимизации чувствительности и линейной области калибровочной кривой для объемов и концентраций проб, используемых в настоящем протоколе. Концентрации инсулина в сыворотке (мкЕд/мл) измеряли для каждого момента времени для каждого из пяти животных в каждой группе. Пять значений для каждого момента времени усредняли и результаты наносили на график: концентрация инсулина - время. Отношение площади под кривой (ППК) для тестируемой группы относительно площади контрольной группы приведено ниже. Отношение максимальной концентрации инсулина в сыворотке (Смах) у тестируемой группы относительно максимальной концентрации у контрольной группы также приведено ниже.
Пример 15 - Доставка гепарина перорально и через толстую кишку
[371] Были приготовлены дозируемые растворы для перорального введения и введения через толстую кишку (ВТК), содержащие соединение-носитель и гепарин натрия USP в 25% водном растворе пропиленгликоля. Использовали натриевую соль этого соединения. В общем случае соединение-носитель и гепарин в виде сухих порошков (примерно 166-182 МЕ/мг) смешивали при встряхивании. Эту сухую смесь растворяли в 25% (по объему) водном растворе пропиленгликоля, встряхивали и помещали в прибор для обработки ультразвуком (приблизительно 37°С). рН устанавливали приблизительно 7 (от 6,5 до 8,5), используя водный раствор NaOH (2 н.). Дозируемый раствор обрабатывали ультразвуком, получая прозрачный раствор. Доводили конечный объем до 3,0 мл. Конечная доза соединения-носителя, доза гепарина и объемы доз приведены в таблице 6.
[372] Использовали следующие типичные протоколы введения и отбора проб. Самцов крыс Sprague-Dawley, весивших 275-350 г, не кормили в течение 24 часов и обезболивали гидрохлоридом кетамина (88 мг/кг) путем внутримышечной инъекции непосредственно до введения. Группе из пяти животных вводили один их дозируемых растворов. Для перорального ввода 11 см французский катетер Rusch 8 прикрепляли к шприцу на 1 мл с кончиком пипетки. Шприц наполняли дозируемым раствором путем протягивания раствора через катетер, который затем вытирали досуха. Катетер помещали в пищевод, оставляя 1 см трубки за резцами. Раствор вводили, нажимая на поршень шприца. При вводе через толстую кишку (ВТК) 7,5 см французский катетер Rusch 8 прикрепляли к шприцу на 1 мл с кончиком пипетки. Дозирующий катетер вставляли в толстую кишку через анальное отверстие до тех пор, пока трубка не переставала быть видимой. Дозируемый раствор медленно вводили в толстую кишку.
[373] Цитратные пробы крови отбирали способом пункции сердца, предварительно вводя кетамин (88 мг/кг), обычно через 0,25, 0,5, 1,0 и 1,5 часов. Активность гепарина определяли, используя время частичного активирования тромбопластина (ВЧАТ) в соответствии с способом Henry, J.B., Clinical Diagnosis и Management by Laboratory Methods, Philadelphia, PA, W.B. Saunders (1979). Согласно предварительным исследованиям, значения базовой линии составляют примерно 20 сек. Результаты, полученные при исследовании пяти крыс в каждой группе, усредняли для каждого момента времени. Полученный максимум приведен ниже в таблице 6.
Ха)
Пример 16 - Доставка паратироидного гормона (ПТГ 1-34) - Пероральная/внутритолстокишечная доставка
[374] Были приготовлены растворы соединения-носителя и остатков человеческого паратиреоидного гормона 1-34 (ПТГ) в воде для орального введения и/или введения через толстую кишку. Использовали натриевую соль соединения носителя. Обычно раствор этого соединения готовили в воде и перемешивали, добавляя один эквивалент гидроксида натрия (1,0 н), получая натриевую соль. Готовили конечные дозируемые растворы путем смешивания соединения с исходным раствором ПТГ (ПТГ получали из Eli Lilly и Co., Индианаполис, Индиана) (обычно имеющего концентрацию 5 мг ПТГ/мл) и разбавления до требуемого объема (обычно 3,0 мл). Конечное соединение, ПТГ и объемные количества доз приведены ниже в таблице 7.
[375] Использовали следующие типичные протоколы введения и отбора проб. Самцов крыс Sprague-Dawley, весивших 200-250 г, не кормили в течение 24 часов и вводили кетамин (44 мг/кг) и хлорпромазин (1,5 мг/кг) за 15 минут до введения. Группе из пяти крыс вводили один их дозируемых растворов. Для перорального ввода 11 см французский катетер Rusch 8 прикрепляли к шприцу на 1 мл с кончиком пипетки. Шприц наполняли дозируемым раствором путем протягивания раствора через катетер, который затем вытирали досуха. Катетер помещали в пищевод, оставляя 1 см трубки за резцами крысы. Раствор вводили, нажимая на поршень шприца. При вводе через толстую кишку (ВТК) 7,5 см французский катетер Rusch 8 прикрепляли к шприцу на 1 мл с кончиком пипетки Эппендорфа. Шприц наполняли дозируемым раствором путем протягивания раствора через катетерную трубку. Катетерную трубку вытирали досуха. Кончик пипетки смазывали гелем K-Y jelly, избегая соприкосновения с ушком трубки, и трубку вставляли в толстую кишку до тех пор, пока она была видна. Раствор вводили, нажимая на поршень шприца, и трубку удаляли.
[376] Пробы крови отбирали серийно из хвостовой артерии, в общем случае через 0, 15, 30, 45, 60 и 90 минут при пероральном введении и 0, 10, 20, 30, 60 и 90 минут при введении через толстую кишку. Концентрацию ПТГ в сыворотке количественно определяли с помощью комплекта радиоиммунологического анализа на содержание ПТГ (Комплект # RIK 6101 от Peninsula Laboratories, Inc. Сан-Карлос, Калифорния). Согласно предварительным исследованиям значения базовой линии равны примерно нулю. Результаты анализа крови пяти крыс в каждой группе усредняли для каждого момента времени. Максимальное значение приведено ниже в таблице 7.
Пример 17 - Интерферон - Пероральная доставка
[377] В деионизированной воде были приготовлены дозируемые растворы соединения-носители и интерферона альфакон-1 (ИФН) (можно приобрести в виде Infergen® в InterMune, Inc. Брисбен, Калифорния). Свободную кислоту, входящую в состав соединения-носителя, переводили в натриевую соль путем добавления одного эквивалента гидроксида натрия. Обычно готовили раствор соединения-носителя в воде и перемешивали, добавляя один эквивалент гидроксида натрия (1,0 н) для получения натриевой соли. Эту смесь встряхивали и помещали в прибор для обработки ультразвуком (приблизительно 37°С). рН устанавливали от приблизительно 7,0 до 8.5, добавляя водный раствор NaOH. Для получения однородной суспензии или раствора смесь встряхивали, а также, при необходимости, использовали прибор для обработки ультразвуком и нагревание. Для достижения однородной растворимости, при необходимости, добавляли дополнительное количество NaOH, и рН вновь устанавливали в районе от 7,0 до 8,5. Раствор соединения-носителя смешивали с исходным раствором ИНФ (приблизительно 22,0-27,5 мг/мл в фосфатном буферизованном солевом растворе) и разбавляли до требуемого объема (обычно 3,0 мл). Конечные дозы соединения-носителя и ИНФ и дозируемые объемы приведены в таблице 8.
[378] При введении и отборе проб обычно использовали следующие протоколы. Самцов крыс Sprague-Dawley, весивших приблизительно 200-250 г, не кормили в течение 24 часов и вводили кетамин (44 мг/кг) и хлорпромазин (1,5 мг/кг) за 15 минут до ввода и, при необходимости сохранить анестезию, вводили их снова. Группе из пяти животных вводили один из дозируемых растворов. 11 см французский катетер Rusch 8 прикрепляли к шприцу на 1 мл с кончиком пипетки. Шприц наполняли дозируемым раствором путем протягивания раствора через катетер, который затем вытирали досуха. Катетер помещали в пищевод, оставляя 1 см трубки за резцами. Дозируемый раствор вводили, нажимая на поршень шприца.
[379] Пробы крови отбирали серийно из хвостовой артерии, обычно через 0, 15, 30, 45, 60 и 90 минут. Концентрацию ИНФ количественно определяли, используя иммунологический комплект с цитоэкраном для определения человеческого альфа-ИНФ (каталог # КНС4012 от Biosource International, Камарилльо, Калифорния). Согласно предварительным исследованиям значения базовых линий равнялись приблизительно нулю. Результаты, полученные при анализе крови животных из каждой группы, усреднялись для каждого момента времени. Максимум этих значений (например, средняя максимальная концентрация ИНФ в сыворотке) приведен ниже в таблице 8.
Пример 18 - Пероральная доставка кальцитонина лосося (лКТ)
[380] Были приготовлены композиции для перорального ввода соединения-носителя и кальцитонина лосося (лКТ) в воде. Обычно 450 мг соединения-носителя добавляли к 2,0 мл воды. Использовали или натриевую соль указанного соединения, или свободную кислоту переводили в натриевую соль путем перемешивания образовавшегося раствора, добавления одного эквивалента гидроксида натрия (1,0 н) и разбавления водой. Раствор встряхивали, затем нагревали (приблизительно 37°С) и обрабатывали ультразвуком. Устанавливали рН приблизительно 7 (от 6,5 до 8,5), добавляя NaOH или HCl. 90 г лКТ из исходного раствора добавляли к раствору. Затем водой доводили общий объем до примерно 3,0 мл (варьирует в зависимости от растворимости соединения-носителя). Конечная доза соединения-носителя, доза лКТ и объемы доз приведены ниже в таблице 9.
[381] Самцов крыс Sprague-Dawley, весивших приблизительно 200-250 г, не кормили в течение 24 часов и вводили кетамин (44 мг/кг) и хлорпромазин (1,5 мг/кг) за 15 минут до введения. Группе из пяти животных вводили один из дозируемых растворов. Для перорального введения 11 см французский катетер Rusch 8 прикрепляли к шприцу на 1 мл с кончиком пипетки. Шприц наполняли дозируемым раствором путем протягивания раствора через катетр, который затем вытирали досуха. Катетер помещали в пищевод, оставляя 1 см трубки за резцами крысы. Дозируемый раствор вводили, нажимая на поршень шприца.
[382] Пробы крови отбирали серийно из хвостовой артерии, обычно через 0, 10, 20, 30, 60 и 90 минут. Концентрации лКТ в сыворотке определяли тестированием с использованием комплекта EIA (Комплект # EIAS-6003 от Peninsula Laboratories, Inc., Сан Карлос, Калифорния), корректируя стандартный протокол из комплекта следующим образом: выдерживали со смесью 50:1 пептид: антитело в течение 2 часов при встряхивании в темноте, промывали чашку, добавляли сыворотку и биотинилированный пептид и разбавляли 4 мл буфера, и в темноте стряхивали всю ночь. Показатели устанавливали в соответствии со значениями базовой линии, полученными при времени=0. Результаты анализа крови пяти крыс в каждой группе усредняли для каждого момента времени. Максимум приведен ниже в таблице 9.
Пример 19 - Доставка рекомбинантного человеческого гормона роста (рчГР) пероральным способом или через толстую кишку
[383] Были приготовлены растворы соединения-носителя и рчГР в фосфатном буфере для перорального ввода и/или ввода через толстую кишку (рчГР можно приобрести в Novartis, Базель, Швейцария). Натриевую соль соединения-носителя получали с помощью реакции свободной кислоты с одним эквивалентом гидроксида натрия. Конечные дозируемые растворы готовили путем смешивания соединения с исходным раствором рчГР (15 мг рчГР/мл) и разбавления до требуемого объема (обычно 3,0 мл). Дозируемые количества соединений и рчГР приведены ниже в таблице 10.
[384] Использовали следующие типичные протоколы введения и отбора проб. Самцов крыс Sprague-Dawley, весивших 200-250 г, не кормили в течение 24 часов и вводили кетамин (44 мг/кг) и хлорпромазин (1,5 мг/кг) за 15 минут до введения. Группе из пяти крыс вводили один их дозируемых растворов. Для перорального ввода 11 см французский катетер Rusch 8 прикрепляли к шприцу на 1 мл с кончиком пипетки. Шприц наполняли дозируемым раствором путем протягивания раствора через катетер, который затем вытирали досуха. Катетер помещали в пищевод, оставляя 1 см трубки за резцами крысы. Раствор вводили, нажимая на поршень шприца. При вводе через толстую кишку (ВТК) 7,5 см трубки катетера Rusch 8 (French 8 или 6) прикрепляли к шприцу на 1 мл с кончиком пипетки Эппендорфа. Шприц наполняли дозируемым раствором путем протягивания раствора через катетерную трубку. Катетерную трубку вытирали досуха. Кончик пипетки смазывали гелем K-Y jelly, избегая соприкосновения с ушком трубки, и трубку вставляли в толстую кишку через анальное отверстие до тех пор, пока она не станет невидна. Раствор вводили, нажимая на поршень шприца, и трубку удаляли.
[385] Пробы крови отбирали серийно из хвостовой артерии или ретро-орбитальной полости, обычно через 0, 15, 30, 45, 60 и 90 минут при пероральном введении и 0, 10, 20, 30, 60 и 90 минут при внутритолстокишечном введении. Пробы собирали в трубки CAPIJECT® (Terumo Corporation, Токио, Япония), содержащие активатор свертываемости (красный колпачок, разделительные трубки для сыворотки). Пробы оставляли свертываться в течение ~20 мин при 4°С. Концентрации рчГР в сыворотке количественно определяли с помощью тестового комплекта для иммунологического анализа рчГР (Kit#K1F4015 от Genzyme Corporation Inc., Кеймбридж, Массачусетс). Пять проб для каждого периода времени объединяли. Согласно предварительным исследованиям, величины базовых линий примерно были равны нулю.
[386] Максимальные концентрации для каждой группы приведены ниже в таблице 10.
Все публикации, ссылки, патенты, опубликованные патентные заявки, которые здесь упоминаются, оформлены ссылкой.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОСТАВЫ И СМЕСИ ДЛЯ ДОСТАВКИ АКТИВНЫХ АГЕНТОВ | 2005 |
|
RU2403237C2 |
| ПРОИЗВОДНОЕ ИМИДАЗОПИРИДИНА, ИСПОЛЬЗУЕМОЕ ДЛЯ ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА | 2013 |
|
RU2648242C2 |
| АНТИДИАБЕТИЧЕСКИЕ ОКСАЗОЛИДИНДИОНЫ | 2005 |
|
RU2355682C2 |
| ЗАМЕЩЕННЫЕ АМИНО ШЕСТИЧЛЕННЫЕ НАСЫЩЕННЫЕ ГЕТЕРОАЛИЦИКЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ DPP-IV ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2016 |
|
RU2720488C2 |
| МОДУЛЯТОРЫ НАТРИЕВОГО КАНАЛА ДЛЯ ЛЕЧЕНИЯ БОЛИ | 2014 |
|
RU2669367C2 |
| АМИНОИЗОХИНОЛИНОВЫЙ ИНГИБИТОР ТРОМБИНА С УЛУЧШЕННОЙ БИОДОСТУПНОСТЬЮ | 2007 |
|
RU2437879C2 |
| ПРОИЗВОДНЫЕ КОНДЕНСИРОВАННОГО 1,2,4-ТИАДИАЗИНА И КОНДЕНСИРОВАННОГО 1,4-ТИАЗИНА, ИХ ПОЛУЧЕНИЕ И ИСПОЛЬЗОВАНИЕ | 1997 |
|
RU2199542C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2011 |
|
RU2609018C2 |
| СПОСОБЫ, СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ СВЯЗАННЫХ С АНГИОТЕНЗИНОМ ЗАБОЛЕВАНИЙ | 2014 |
|
RU2688163C2 |
| КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ ДИФТОРМЕТАНФОСФОНАТЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНТИРОЗИНФОСФАТАЗЫ IB (PTP-1B) | 2008 |
|
RU2462469C2 |
Изобретение относится к соединениям или их фармацевтически приемлемым солям, которые могут быть использованы для доставки биологически активных веществ. Изобретение также относится к фармацевтическим составам, содержащим указанные соединения, и к способу введения биологически активных веществ. 9 н. и 8 з.п. ф-лы, 10 табл., 19 пр.
1. Соединение, выбранное из:


и их фармацевтически приемлемых солей.
2. Фармацевтический состав для доставки биологически активных веществ, содержащий:
(A) по меньшей мере одно биологически активное вещество, выбранное из пептида YY [PYY], инсулина и паратиреоидного гормона; и
(B) эффективное количество соединения, выбранного из




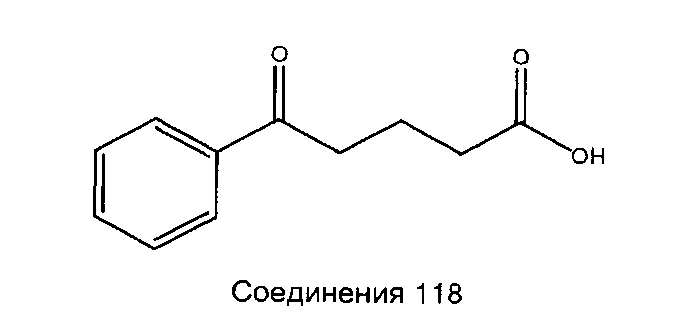





и их фармацевтически приемлемых солей.
3. Фармацевтический состав по п.2, отличающийся тем, что указанное биологически активное вещество представляет собой человеческий инсулин-цинк.
4. Фармацевтический состав по п.2, отличающийся тем, что указанное биологически активное вещество представляет собой PYY [3-36].
5. Фармацевтический состав по п.2, отличающийся тем, что указанное биологически активное вещество представляет собой человеческий паратиреоидный гормон (ПТГ-1-34).
6. Способ введения биологически активного вещества, выбранного из пептида YY [PYY], инсулина и паратиреоидного гормона, животному, которое нуждается в указанном веществе, включающий пероральное введение указанному животному фармацевтического состава по п.2.
7. Соединение, выбранное из следующих:





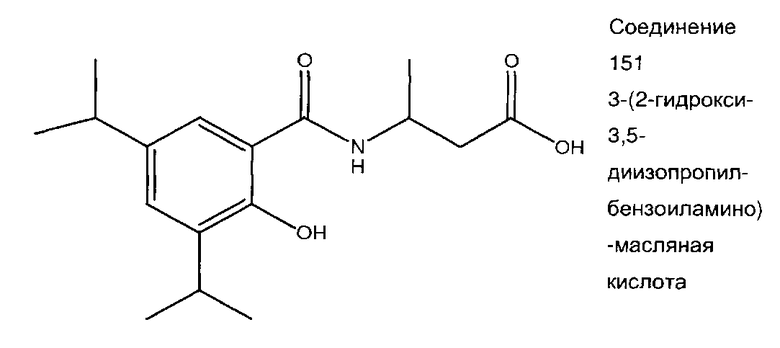
и их фармацевтически приемлемых солей.
8. Фармацевтический состав для доставки биологически активных веществ, содержащий:
(A) по меньшей мере одно биологически активное вещество, выбранное из пептида YY [PYY], инсулина, гепарина и интерферона; и
(B) эффективное количество соединения по п.7.
9. Фармацевтический состав по п.8, отличающийся тем, что указанное биологически активное вещество представляет собой интерферон альфакон-1.
10. Фармацевтический состав по п.8, отличающийся тем, что указанное биологически активное вещество представляет собой человеческий рекомбинантный инсулин.
11. Фармацевтический состав по п.8, отличающийся тем, что указанное биологически активное вещество представляет собой гепарин.
12. Фармацевтический состав по п.8, отличающийся тем, что указанное биологически активное вещество представляет собой PYY [3-36].
13. Способ введения биологически активного вещества, выбранного из пептида YY [PYY], инсулина, гепарина и интерферона, животному, которое нуждается в указанном веществе, включающий пероральное введение указанному животному фармацевтического состава по п.8.
14. Соединение, представляющее собой:
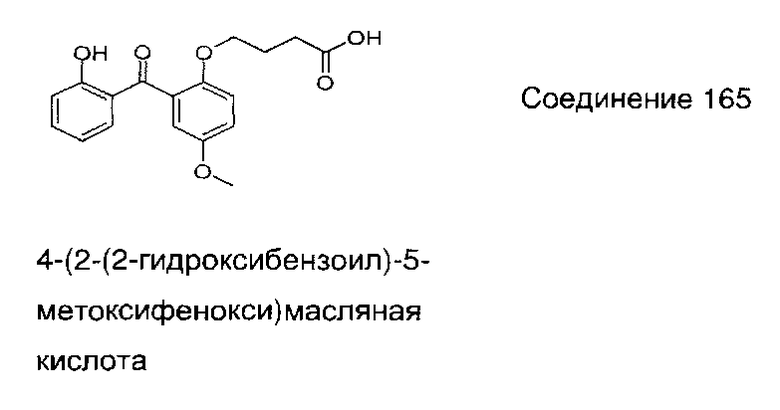
и его фармацевтические приемлемые соли.
15. Фармацевтический состав для доставки биологически активных веществ, содержащий:
(A) инсулин и
(B) эффективное количество соединения по п.14.
16. Фармацевтический состав по п.15, отличающийся тем, что указанный инсулин представляет собой человеческий рекомбинантный инсулин.
17. Способ введения инсулина животному, которое нуждается в указанном инсулине, включающий пероральное введение указанному животному фармацевтического состава по п.15.
| US3072473A, 08.01.1963 | |||
| Способ получения замещенных 4-пирролинонов-2 | 1983 |
|
SU1109389A1 |
| WO9929640 A2, 17.06.1999 | |||
| US5504244 A, 02.04.1996 | |||
| WO2002067304 A1, 29.08.2002 | |||
| EP757081 A2, 05.02.1997 | |||
| US5354496 A, 11.10.1994 | |||
| ФОРМА ДЛЯ ИЗГОТОВЛЕНИЯ СТРОИТЕЛЬНЫХ ИЗДЕЛИЙ | 0 |
|
SU300325A1 |
| US5643957 А, 01.07.1997 | |||
| WO2004026848 A1, 01.04.2004 | |||
| WO2003007923 A2, 30.01.2003 | |||
| СПОСОБ ПОЛУЧЕНИЯ БИТУМНОЙ ЭМУЛЬСИИ | 0 |
|
SU191724A1 |
| WO9601267 A1, 18.01.1998 . | |||

