Область техники, к которой относится изобретение
Настоящее изобретение имеет отношение к новым соединениям, способам их получения, композициям, включающим эти соединения, и их применению при лечении или предупреждении заболеваний, которые поддаются регулированию посредством ингибирования клеточной адгезии. Более детально, настоящее изобретение имеет отношение к новым производным фенилаланина, которые ингибируют клеточную адгезию, опосредованную интегрином α4, и которые считаются полезными при лечении или предупреждении воспалительных заболеваний.
Уровень техники
Множественные адгезивные взаимодействия между лейкоцитами и эндотелиальными клетками или белками внеклеточного матрикса являются ключевым фактором в регулировании иммунитета и воспаления. Ранняя фаза миграции лейкоцитов из сосудистой сети в место воспаления включает сворачивание лейкоцитов с последующими изменениями авидности интегрина, которые приводят к последующей прочной адгезии (для анализа см. Butcher, Cell 67: 1033-1036 (1991); Harlan, Blood 3: 513-525 (1985); Hemler, Annu. Rev. Immunol. 8: 365-400 (1990); Osbom, Cell 62: 3-6 (1990); Shimizu et al., Immunol. Rev. 114: 109-143 (1990); Springer, Nature 346: 425-434 (1990), и Springer, Cell 76: 301-314 (1994)). В ответ на хемотаксические факторы лейкоциты мигрируют через две смежные эндотелиальные клетки и в ткани, которые частично состоят из белка внеклеточного матрикса фибронектина (FN) (см. Wayner et al., J. Cell Biol. 105: 1873-1884 (1987)) и коллагена (CN) (см. Bornstein et al., Ann. Rev. Biochem. 49: 957-1003 (1980), и Miller, Chemistry of the collagens and their distribution, in "Extracellular Matrix Biochemistry", K.A. Piez and A.H. Reddi, editors, Elsevier, Amsterdam, 41-78 (1983)). Молекулы, важные для узнавания, которые принимают участие в этих адгезивных реакциях, принадлежат к суперсемейству интегриновых генов (для обзора см. Hemler, Annu. Rev. Immunol. 8: 365-400 (1990); Hynes, Cell 48: 549-554 (1987); Shimizu et al., Immunol. Rev. 114: 109-143 (1990), и Springer, Nature 346: 425-434 (1990)).
Интегрины - гетеродимеры, состоящие из нековалентно связанных субъединиц, относящихся к альфа- (α) и бета- (β) субъединицам. До настоящего времени было идентифицировано 8 интегриновых β-субъединиц, которые ассоциированы с 16 различными субъединицами, участвующими в образовании, по меньшей мере, 23 различных интегринов.
Интегрин α4β1, также известный как VLA-4 (Very Late Antigen-4, очень поздний антиген-4) конститутивно экспрессируется на поверхности лейкоцитов, включая лимфоциты, моноциты, эозинофилы и базофилы (см. Hemler et al., J. Bio. Chem. 262: 11478-11485 (1987), и Bochner et al., J. Exp.Med. 173: 1553-1556 (1991)). Сообщалось, что VLA-4 присутствует на нейтрофилах у септических пациентов (см. Ibbotson et al., Nature Med. 7:465-470 (2001)). VLA-4 связывается с сосудисто-клеточной адгезивной молекулой-1 (VCAM-1) на активированных эндотелиальных клетках, что приводит к экстравазации лейкоцитов (Elices et al., Cell 60: 577-584 (1990)). Как только клетки достигают экстраваскулярного пространства, VLA-4 может соединяться со связывающим сегментом 1 (CS-1), областью альтернативного сплайсинга А-цепи FN (Wayner et al., J. Cell Biol. 109: 1321-1330 (1989)). Кроме того, известно, что VLA-4 связывается с остеопонтином, белком, осуществляющим апрегуляцию в артериосклеротических бляшках (см. Bayless et al.,J. Cell Science 111: 1165-1174(1998)).
Интегрин α4β7, также известный как LPAM-1 (Lymphocyte-Peyer's patch Adhesion Molecule-1, молекула адгезии лимфоцитов и пейеровых бляшек 1 типа), взаимодействует с тремя известными лигандами (VCAM-1, CS-1, MAdCAM-1). Одним из лигандов, который демонстрирует абсолютную специфичность для α4β7, является адрессин 1 типа, молекула клеточной адгезии слизистых оболочек (MAdCAM-1) (см. Andrew et al., J. Immunol. 153: 3847-3861 (1994); Briskin et al., Nature 363: 461-464 (1993), и Shyjan et al., J. Immunol 156: 2851-2857 (1996)). MAdCAM-1 в значительных количествах экспрессируется в высоких эндотелиальных венулах пейеровых бляшек, в лимфатических узлах брыжейки и на собственной пластинке кишечника и венулах молочных желез (Berg et al., Immunol. Rev. 108: 5-18 (1989)). Показано, что интегрин α4β7 и MAdCAM-1 важны для регулирования передвижения лимфоцитов к нормальному кишечнику (Holzmann et al. Cell 56: 37-46 (1989)).
Также было показано, что принимаемые внутрь биоаккумулируемые непептидные небольшие молекулы антагониста интегринов α4, α4β1 и α4β7 могли бы быть полезными при лечении или предупреждении состояний, таких как астма, воспаление кишечника, ревматоидный артрит, множественный склероз и другие заболевания (см. патентные заявки WO 99/36393 и WO 02/18320, содержание которых включено путем отсылки).
В настоящее время открыт новый класс соединений, который включен в основную цель патентной заявки WO 99/36393, но не раскрыт там специальным образом.
Раскрытие изобретения
Настоящее изобретение, таким образом, в первом аспекте обеспечивает соединение, имеющее формулу (I), или его фармацевтически приемлемое производное
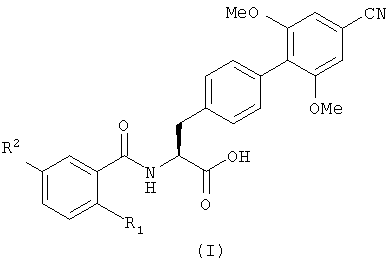
где R1 является бромом и
R2 является галогеном, C1-6 алкилом или С1-6 алкокси.
Предпочтительно R2 является галогеном или C1-6 алкокси.
Более предпочтительно R2 является фтором, метокси или этокси.
В следующем аспекте настоящее изобретение обеспечивает соединения Е1-Е7 (как описано ниже) или их фармацевтически приемлемое производное, например
(S)-2-{[1-(2-бром-5-этоксифенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовую кислоту;
(S)-2-{[1-(2-бром-5-фторфенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовую кислоту;
(S)-2-{[1-(2-бром-5-метоксифенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовую кислоту;
(S)-2-{[1-(2-бром-5-метилфенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовую кислоту;
(S)-2-{[(2-бром-5-хлорфенил)метаноил]амино}-3-[4'-циано-2',6'-диметоксибифенил-4-ил]пропионовую кислоту;
(S)-2-{[(2,5-дибромфенил)метаноил]амино}-3-[4'-циано-2',6'-диметоксибифенил-4-ил]пропионовую кислоту;
(S)-2-{[(5-(изопропокси)-2-бромфенил)метаноил]амино}-3-[4'-циано-2',6'-диметоксибифенил-4-ил]пропионовую кислоту
или их фармацевтически приемлемое производное.
По всему тексту настоящего подробного описания, если не оговорено специально:
термин «галоген» применяют для описания группы, выбранной из фтора, хлора, брома или иода;
термин «С1-6 алкил» применяют для описания группы или части группы, включающей линейную или разветвленную группу, содержащую от 1 до 6 атомов углерода; примеры таких групп включают метил, этил, пропил, изопропил, н-бутил, изобутил, трет-бутил, пентил или гексил;
термин «С1-6 алкокси» применяют для описания группы или части группы, в которой атом кислорода присоединен к указанной выше С1-6 алкильной группе; примеры таких групп включают метокси, этокси, пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси, пентокси или гексокси.
Характерной особенностью настоящих соединений является введение цианогруппы в 4'-положение бифенильного ядра вместе с заявленной 2,5-дизамещенной бензоильной группой.
Соединения, имеющие формулу (I), или их фармацевтически приемлемое производное обладают потенциальной ингибирующей активностью, направленной на опосредованную интегрином α4 клеточную адгезию. Кроме этого, было обнаружено, что некоторые примеры демонстрируют превосходное биоаккумулирование после перорального введения и/или хорошее системное воздействие.
Соединения E1, E2 и Е3 (как описано ниже) демонстрируют полезную комбинацию указанных выше свойств.
Понятно, что соединения с формулой (I) или их фармацевтически приемлемое производное могут иметь более одного асимметрического атома углерода и, таким образом могут возникать диастереоизомеры. Все эти изомерные формы включены в настоящее изобретение, включая их смеси.
Разделение диастереоизомеров можно достигнуть с помощью общепринятых методов, например с помощью дробной кристаллизации, хроматографии или ВЭЖХ. Также можно получить единственную стереоизомерную форму соединения из соответствующего оптически чистого интермедиата или с помощью разделения, например, с помощью ВЭЖХ соответствующего рацемата с применением подходящего хирального носителя, или с помощью дробной кристаллизации диастереоизомерных солей, образованных с помощью реакции соответствующего рацемата с подходящей оптически активной кислотой или основанием, выбираемых по обстановке. Альтернативно, смесь энантиомеров можно разделить с помощью химической реакции с подходящим хиральным соединением с образованием новых ковалентно связанных разновидностей, например с помощью связывания рацемической карбоновой кислоты с хиральным амином или спиртом для получения диастереоизомерной смеси (в случае амидов или сложных эфиров соответственно), которую можно разделить с помощью общепринятых методов, таких как колоночная хроматография, ВЭЖХ или дробная кристаллизация. Отдельные диастереоизомеры можно затем превратить в отдельные энантиомеры требуемого соединения с помощью подходящей реакции, такой как гидролитическое расщепление новой ковалентной связи.
Термин «фармацевтически приемлемое производное», употребляемый в тексте, означает любую фармацевтически приемлемую соль или пролекарственную форму, например сложный эфир соединения настоящего изобретения, которые после введения реципиенту способны обеспечить (прямо или косвенно) соединение настоящего изобретения или его активный метаболит, или их остатки. Такие производные признаются специалистами в этой области техники без дополнительных экспериментальных проверок. Тем не менее в текст включена отсылка к учебнику "Burger's Medicinal Chemistry and Drug Discovery, 5th Edition, Vol 1: Principles and Practice" для полного представления о таких производных. Предпочтительными фармацевтически приемлемыми производными являются соли и сложные эфиры.
Специалистам в области органической химии понятно, что множество органических соединений могут образовывать комплексы с растворителем, в котором они вступают в реакцию или из которого их осаждают или кристаллизуют. Эти комплексы известны как «сольваты». Например, комплекс с водой известен как «гидрат». Сольваты соединений настоящего изобретения входят в объем изобретения.
Термин «пролекарство», который используется в тексте, означает соединение, которое превращается в организме, например, с помощью гидролиза в крови в активную форму, которая обладает лечебными эффектами. Фармацевтически приемлемые пролекарства описаны в работе Т.Higuchi and V. Stella, Prodrugs as Novel Delivery Systems, Vol.14 of the A.C.S. Symposium Series, Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, и в работе D.Fleisher, S.Ramon and H.Barbra "Improved oral drug delivery: solubility limitations overcome by the use of prodrugs", Advanced Drug Delivery Reviews (1996) 19(2) 115-130, каждая из которых включена в текст путем отсылки.
Пролекарства являются ковалентно связанными носителями, которые высвобождают in vivo соединение с формулой (I) или его фармацевтически приемлемое производное, когда такое пролекарство вводят пациенту. Пролекарства обычно получают с помощью модификации функциональных групп таким образом, что такая модификация отщепляется или с помощью общепринятых способов, или in vivo, что приводит к появлению исходного соединения. В случае карбоновой кислоты (-СООН) можно использовать сложные эфиры, такие как метиловые эфиры, этиловые эфиры, двойные эфиры и т.п. Эфиры могут быть эффективными сами по себе и/или быть способными к гидролизу в условиях in vivo в организме человека. Подходящие фармацевтически приемлемые сложноэфирные группы, способные к гидролизу in vivo, включают те группы, которые без труда отщепляются в организме человека, чтобы отделиться от исходной кислоты или ее соли.
Соединения настоящего изобретения могут быть в форме и/или могут приниматься внутрь как фармацевтически приемлемая соль. Для обзора подходящих солей см. работу Berge et al., J. Pharm. Sci., 1977, 66, 1-19.
Обычно фармацевтически приемлемая соль может по обстановке быть без труда получена с применением желаемой кислоты или основания. Соль может осаждаться из раствора и ее можно собрать с помощью фильтрации или можно получить после выпаривания растворителя.
В случае соединения с формулой (I) подходящие фармацевтически приемлемые соли образуются из фармацевтически приемлемых оснований, которые включают соли аммония; соли щелочных металлов, такие как соли натрия и калия; соли щелочноземельных металлов, такие как соли кальция и магния, и соли органических оснований, включающие соли первичных, вторичных и третичных аминов, таких как изопропиламин, диэтиламин, этаноламин, триметиламин, дициклогексиламин, N-метил-D-глюкамин и трис(гидроксиметил)метиламин.
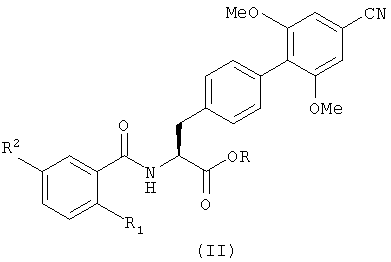
В следующем аспекте настоящее изобретение также обеспечивает способ получения соединения с формулой (I), который включает гидролиз производного с формулой (II), представляющего собой сложный эфир карбоновой кислоты:
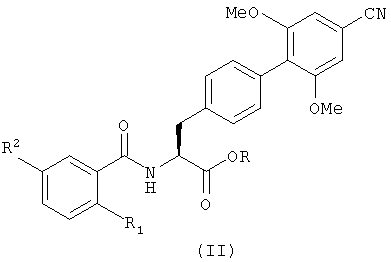
в котором R1 и R2 определены так же, как в формуле (I), и R является группой, способной к образованию сложного эфира с карбоновой кислотой, и не обязательно после этого образует из него фармацевтически приемлемое производное.
Примером подходящей группы R является C1-6 алкил, такой как метил или трет-бутил, предпочтительно метил. Гидролиз может происходить при использовании кислой или щелочной среды. Примером гидролиза в щелочной среде может быть обработка соединения с формулой (II), например, гидроокисью щелочного металла в подходящем растворителе, например обработка гидроокисью лития в водном тетрагидрофуране. Примером гидролиза в кислой среде может быть обработка соединения с формулой (II) минеральной кислотой в подходящем растворителе при повышенной температуре, например обработка с помощью 5 н. соляной кислоты в диоксане при 60°С в течение ночи. Такие методы известны специалистам в этой области техники.
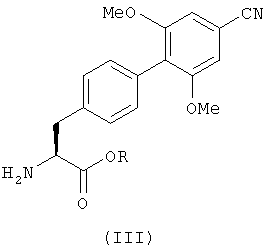
Соединения с формулой (II) можно получить с помощью реакции соединения, имеющего формулу (III), или его кислотно-аддитивной соли
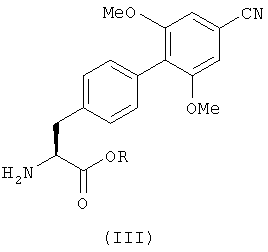
где R определен так же, как в формуле (II), с соединением с формулой (IV)
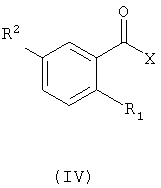
в котором R1 и R2 определены так же, как в формуле (I), и группа Х является гидроксильной группой или уходящей группой.
Соответствующий пример кислотно-аддитивной соли соединения с формулой (III) является гидрохлорид. Если Х является уходящей группой, соответствующим примером уходящей группы Х является галоген, в особенности хлор. Реакции между соединениями с формулами (III) и (IV) обычно проводят в инертном органическом растворителе, таком как тетрагидрофуран или дихлорметан, или в смешанной системе органический растворитель/вода при комнатной или повышенной температуре в присутствии подходящего основания, например органического основания (такого как триэтиламин), карбонатной соли щелочного металла (такой как карбонат калия) или гидрокарбонатной соли щелочного металла (например, гидрокарбоната натрия). Если Х является гидроксильной группой, реакцию между соединениями с формулой (III) и (IV) проводят с помощью стандартной методологии сочетания, например О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметиламмоний, гексафторфосфат, триэтиламин в безводном диметилформамиде перемешивали при комнатной температуре в течение ночи.
Осуществление изобретения
Соединения с формулой (III) можно получить с помощью сочетания соединения с формулой (V)
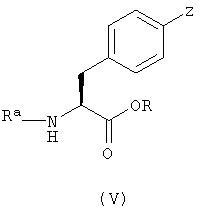
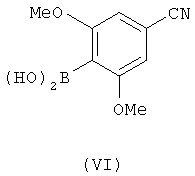
в котором Z является уходящей группой и Rа является защитной группой, с соединением, имеющим формулу (VI)
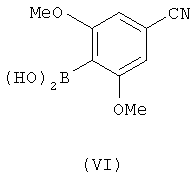
с последующим удалением защитной группы Rа.
Соответствующими примерами уходящей группы Z являются галоген, алкансульфонилоксигруппа (например, метансульфонильная группа), галоалкансульфонилоксигруппа (например, трифторметансульфонилоксигруппа) или арилсульфонилоксигруппа (например, н-толуолсульфонилоксигруппа).
Соответствующий пример защитной группы Rа включает примеры, перечисленные ниже, в особенности трет-бутилоксикарбонил (Вос).
Соединения с формулой (III) также можно получить с помощью сочетания соединения с формулой (V) с соединением с формулой (VII)
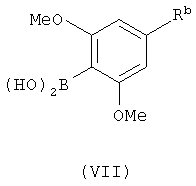
в котором Rb является гидроксиметильной группой, с последующим удалением защитной группы Ra и превращением Rb в цианогруппу. Реакцию между соединениями с формулой (V) и формулой (VII) проводили сходным путем, как в случае реакции между соединениями с формулой (V) и формулой (VI). Защитную группу в полученном продукте можно удалить с помощью способов, описанных выше. Группу Rb можно превратить в цианогруппу с помощью процедуры, известной специалистам в этой области техники и описанной здесь.
Реакцию между соединениями с формулой (V) и формулой (VI) или формулой (VII) можно, например, проводить в условиях реакции сочетания Сузуки, которые известны специалистам в этой области техники. В качестве иллюстрации реакцию можно проводить, используя палладий в качестве катализатора (например, тетракис(трифенилфосфин)палладий(0)) в подходящем растворителе (например, в 1-метил-2-пирролидоне) при повышенной температуре в присутствии подходящего основания (например, триэтиламина). Защитную группу полученного продукта можно удалить с помощью методов, известных специалистам в этой области техники. Если защитная группа является трет-бутилоксикарбонильной, депротекцию можно провести с помощью обработки кислотой (например, с помощью соляной кислоты) в подходящем растворителе (например, в спирте, таком как этанол или изопропанол).
Промежуточное соединение с формулой (VI) можно получить из имеющейся в продаже 4-бром-3,5-диметоксибензойной кислоты с применением процедур, описанных здесь. Очевидно, что промежуточные соединения с формулами (II), (III) и (VI) являются новыми, и они составляют еще один аспект этого изобретения.
Настоящее изобретение предоставляет соединение с формулой (II).
Настоящее изобретение в особенности обеспечивает соединение с формулой (II), в котором R2 является С1-6 алкокси или фтором.
Настоящее изобретение также предоставляет соединение с формулой (III) или его кислотно-аддитивную соль.
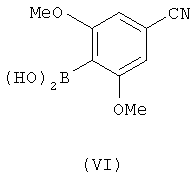
Настоящее изобретение также предоставляет соединение с формулой (VI).
Промежуточные соединения (IV) и (V) или имеются в продаже, или могут быть получены с помощью способов, описанных здесь, с применением методов, известных специалистам в этой области техники или также с помощью аналогичных методов.
Специалистам в этой области техники понятно, что при получении соединения с формулой (I) или его фармацевтически приемлемого производного может потребоваться и/или будет желательно защитить одну или несколько чувствительных групп в молекуле для предупреждения нежелательных побочных реакций.
Подходящие защитные группы для применения в настоящем изобретении хорошо известны специалистам в этой области техники и их можно применять общепринятым способом. См., например, "Protective groups in organic synthesis" by T.W.Greene and Р.Г.М.Wuts (John Wiley&Sons, 1991) или "Protecting Groups" by P.J.Kocienski (Georg Thieme Verlag, 1994). Примеры подходящих групп, защищающих аминогруппы, включают защитные группы ацильного типа (например, формил, трифторацетил, ацетил), ароматические защитные группы уретанового типа (например, бензилоксикарбонил (Cbz) и замещенный Cbz), алифатические защитные группы уретанового типа (например, 9-флуоренилметоксикарбонил (Fmoc), трет-бутилоксикарбонил (Boc), изопропилоксикарбонил, циклогексилоксикарбонил) и защитные группы алкильного типа (например, бензил, тритил, хлортритил). Примеры подходящих групп, защищающих кислород, могут включать, например, алкильные силильные группы, такие как триметилсилил или трет-бутилдиметилсилил; простые алкильные эфиры, такие как тетрагидрофуранил или трет-бутил, или сложные эфиры, такие как ацетат.
Соединения этого изобретения могут быть протестированы in vitro на биологическую активность в соответствии со следующим анализом.
Сцинтилляционный анализ близкого расстояния Jurkat J6 (SPA)
Для исследования взаимодействия VLA-4, экспрессированного на мембране клеток Jurkat J6, с анализируемыми соединениями использовали сцинтилляционный анализ близкого расстояния Jurkat J6. Клетки J6 (1 миллион клеток/лунку) оставляли для связывания с SPA-бусинками, покрытыми агглютинином зародышей пшеницы (Amersham, 1 мг/лунку), в буфере для измерения, содержащем 50 мМ HEPES, 100 мМ NaCl и 1 мМ MnCl2 (pH доводили до 7,5 с помощью 4 М NaOH). Тритированное 3H стандартное соединение А (конечная концентрация в среде измерения 1-3 нМ) и тестируемые соединения растворяли в подходящем растворителе и разводили в буфере для измерения (верхний предел измеряемой концентрации 2,5 мкМ; десять точек в дозозависимой кривой). Соединения анализировали в двойных повторах, применяли четырехпараметровую подгонку кривой. Равновесную константу диссоциации для каждого соединения рассчитывали с помощью метода Cheng & Prusoff (Biochem. Pharmacol., 22(23): 3099-3108 (1973)). Результаты представлены в виде среднего значения pKi.
Стандартное соединение А является калиевой солью (2S)-3-[4-({[4-(аминокарбонил)-1-пиперидинил]карбонил}окси)фенил]-2-[((2S)-4-метил-2-{[2-(2-метилфенокси)ацетил]амино}пентаноил)амино]пропионовой кислоты, которая описана в патентной заявке WO 00/37444 (Glaxo Group Ltd. et al.). Тритированные 3H-производные можно получить, используя общепринятые методы.
Все образцы, полученные в соответствии с этим изобретением, были проанализированы согласно этой процедуре, и было обнаружено, что они имеют значения pKi≥8,6.
Соединения с формулой (I) или их фармацевтически приемлемое производное ингибируют клеточную адгезию, опосредованную интегрином α4. Полагают, что клеточная адгезия, опосредованная интегрином α4, вовлечена в целый ряд состояний, таких как ревматоидный артрит (RA); астма; аллергические состояния, такие как ринит; респираторный дистресс-синдром у взрослых; СПИД-деменция; болезнь Альцгеймера; сердечно-сосудистые заболевания; тромбоз или повреждающая агрегация тромбоцитов; реокклюзия с последующим тромболизисом; реперфузионное повреждение; воспалительные заболевания кожи, такие как псориаз, экзема, контактный дерматит и атопический дерматит; диабет (например, инсулинзависимая сахарная болезнь, аутоиммунный диабет); множественный склероз; системная красная волчанка (SLE); воспалительные заболевания кишечника, такие как язвенный колит, болезнь Крона (региональный энтерит) и воспаление резервуара (паучит, например, возникший после проктоколектомии и илеоанального анастомоза); заболевания, ассоциированные с инфильтрацией лейкоцитов в желудочно-кишечный тракт, такие как болезнь Целиак, нетропическая спру, энтеропатия, ассоциированная с серонегативными артропатиями; лимфоцитарный или коллагенозный колит и эозинофильный гастроэнтерит; заболевания, ассоциированные с инфильтрацией лейкоцитов в другие ткани, покрытые эпителием, такие как кожа, мочевой тракт, дыхательные пути и суставный синовиум; панкреатит; мастит (молочные железы); гепатит; холецистит; холангит или перихолангит (желчные протоки и ткань, окружающая печень); бронхит; синусит; воспалительные заболевания легких, приводящие к интерстициальному фиброзу, такие как гиперчувствительная пневмония; коллагеновые заболевания (при SLE и RA); саркоидоз; остеопороз; остеоартрит; атеросклероз; неопластические заболевания, включающие метастазы при неопластических или злокачественных опухолях; раны (долго незаживающие раны); некоторые заболевания глаз, такие как отслоение сетчатки, аллергический конъюнктивит и аутоиммунный увеит; синдром Шегрена; отторжение (хроническое или острое) после трансплантации органов; заболевания «хозяин против трансплантата» и «трансплантат против хозяина»; интимальная гиперплазия; артериосклероз (включая артериосклероз трансплантанта после трансплантации); реинфаркт или рестеноз после хирургической операции, такой как чрезкожная транслюминальная коронарная ангиопластия (РТСА) и чрезкожная транслюминальная реканализация артерий; нефрит; опухолевый ангиогенез; злокачественные опухоли; множественная миелома и индуцированная миеломой ресорбция костей; сепсис; повреждения центральной нервной системы, такие как паралич, травматическое повреждение мозга и спинного мозга и болезнь Меньера.
Соединения настоящего изобретения могут предпочтительно применяться при лечении или предупреждении астмы, аллергических состояний, таких как ринит, воспаление кишечника, такое как язвенный колит и болезнь Крона, ревматоидный артрит, атопический дерматит, множественный склероз и отторжение органов после трансплантации. В особенности соединения настоящего изобретения могут применяться при лечении или предупреждении воспаления кишечника или множественного склероза.
Кроме того, настоящее изобретение обеспечивает способ лечения или предупреждения состояний, в которых полезен ингибитор клеточной адгезии, опосредованной интегрином α4, который при необходимости включает введение пациенту в безопасном и эффективном количестве соединения с формулой (I) или его фармацевтически приемлемого производного. Настоящее изобретение в особенности обеспечивает способ лечения или предупреждения указанных выше состояний.
Настоящее изобретение также обеспечивает соединение с формулой (I) или его фармацевтически приемлемое производное для применения в терапии, в особенности для лечения или предупреждения указанных выше заболеваний.
В другом аспекте настоящее изобретение обеспечивает применение соединения с формулой (I) или его фармацевтически приемлемого производного в производстве лекарственного средства для лечения или предупреждения состояний, в которых полезен ингибитор клеточной адгезии, опосредованной интегрином α4, в особенности для указанных выше заболеваний.
Несмотря на то, что соединения настоящего изобретения можно принимать внутрь в том виде, как они есть, предпочтительно составлять фармацевтические композиции в соответствии с общепринятой фармацевтической практикой. Поэтому изобретение также обеспечивает фармацевтическую композицию, которая включает терапевтически эффективное количество соединения с формулой (I) или его фармацевтически приемлемого производного вместе с фармацевтически приемлемым носителем или разбавителем.
Кроме того, изобретение обеспечивает фармацевтическую композицию, включающую соединение с формулой (I) или его фармацевтически приемлемое производное вместе с другим терапевтически активным веществом.
Кроме того, настоящим изобретением обеспечивается способ получения фармацевтической композиции, который включает смешивание, по меньшей мере, одного соединения изобретения или его фармацевтически приемлемого производного с фармацевтически приемлемым носителем или разбавителем.
Фармацевтические композиции могут предназначаться для применения людьми или животными в человеческой и ветеринарной медицине и обычно включают один или несколько фармацевтически приемлемых разбавителей, носителей или эксципиентов. Подходящие носители или разбавители для терапевтического применения хорошо известны в этой области техники, и они описаны, например, в работе Remington's Pharmaceutical Sciences, Mack Publishing Co. (A.R.Gennaro edit. 1985). Выбор фармацевтического носителя, эксципиента или разбавителя может быть сделан в соответствии с предполагаемым способом приема и обычной фармацевтической практикой. Носитель или разбавитель должны быть приемлемыми в смысле безвредности для реципиентов. Фармацевтически приемлемые носители или разбавители могут быть, например, связующими веществами (например, сиропом, гуммиарабиком, желатином, сорбитом, трагакантом, поливинилпирролидоном), эксципиентами (например, лактозой, сахарозой, кукурузным крахмалом, фосфатом калия, сорбитом, глицином), смазочными веществами (например, стеаратом магния, тальком, полиэтиленгликолем, кремнеземом), дезинтегрирующими веществами (например, картофельным крахмалом), увлажняющими веществами (например, лаурилсульфатом натрия) и т.п.
Способы введения (доставки) композиции изобретения включают, но не ограничиваются только ими, один или несколько путей: пероральный (например, в виде таблеток, капсул или пищевого раствора), местный, мукозальный (например, в виде назального пульверизатора или аэрозоля для ингаляции), назальный, парентеральный (например, с помощью инъецируемой формы), желудочно-кишечный, интраспинальный, внутрибрюшинный, внутримышечный, внутривенный, внутриматочный, внутриглазной, внутрикожный, внутричерепной, внутривагинальный, внутрицеребровентрикулярный, внутримозговой, подкожный, глазной (включая интравитреальный или интракамеральный), чрезкожный, ректальный, буккальный, эпидуральный, сублингвальный.
Например, соединения можно вводить перорально в форме таблеток, капсул, овулей, эликсира, растворов или суспензий, которые могут содержать ароматизирующие или красящие вещества, для немедленного, замедленного, модифицированного, поддерживающего, импульсного способа применения или применения с контролируемым высвобождением. Таблетки могут содержать эксципиенты, такие как микрокристаллическая целлюлоза, лактоза, цитрат натрия, карбонат кальция, двухосновный фосфат кальция и глицин, дезинтеграторы, такие как крахмал (предпочтительно кукурузный, картофельный или тапиоковый крахмал), крахмальный гликолат натрия, кросскармелоза натрия и некоторые комплексные силикаты, и гранулированные связующие средства, такие как поливинилпирролидон, гидроксипропилметилцеллюлоза (НРМС), гидроксипропилцеллюлоза (НРС), сахароза, желатин и гуммиарабик. Дополнительно могут быть включены смазочные вещества, такие как стеарат магния, стеариновая кислота, глицерилбегенат и тальк. Также можно применять твердые композиции такого же типа в виде наполнителей в желатиновых капсулах. Предпочтительные эксципиенты в этом случае включают лактозу, крахмал, целлюлозу, молочный сахар или высокомолекулярные полиэтиленгликоли. Для водных суспензий и/или эликсиров вещество может быть смешано с другими подсластителями или ароматизаторами, красящими веществами и красками, с эмульгирующими и/или суспендирующими агентами и с разбавителями, такими как вода, этанол, пропиленгликоль и глицерин и их смеси.
Соединения настоящего изобретения могут быть измельчены с помощью известных процедур измельчения, таких как влажное измельчение для получения частиц с размерами, подходящими для формирования таблеток или для других типов композиций. Мелко измельченные (в виде наночастиц) препараты соединений изобретения можно получить с помощью способов, известных в этой области техники, например, см. патентную заявку International Patent Application No. WO 02/00196 (SmithKline Beecham).
Если соединение настоящего изобретения вводится парентерально, примеры такого введения включают один или несколько способов: внутривенное, внутриартериальное, внутрибрюшинное, интратекальное, интравентрикулярное, внутриуретальное, внутригрудинное, внутричерепное, внутримышечное или подкожное введение соединения; и/или ведение с помощью инфузионных методов. Для парентерального введения наилучшим образом соединения применяют в форме стерильного водного раствора, который может содержать другие вещества, например достаточное количество солей или глюкозы для получения раствора, изотоничного с кровью. Водные растворы при необходимости должны быть подходящим образом забуферены (предпочтительно, чтобы рН находился в диапазоне от 3 до 9). Приготовление подходящих парентеральных композиций в стерильных условиях легко осуществить с помощью обычных фармацевтических способов, хорошо известных специалистам в этой области техники.
Как было указано, соединение настоящего изобретения может быть введено интраназально или с помощью ингаляции, оно обычно выпускается в форме ингалятора с сухим порошком или аэрозольного пульверизатора, помещенного в контейнер с повышенным давлением, насоса, спрея или пульверизатора с подходящим газом-вытеснителем, например дихлордифторметаном, гидрофторэтаном, гидрофторалканом, таким как 1,1,1,2-тетрафторэтан (HFA 134AT'''') или 1,1,1,2,3,3,3-гептафторпропан (HFA 227ЕА) (например, от фирмы «Ineos Fluor»), двуокись углерода или подходящий газ. В случае аэрозолей, находящихся под давлением, дозировочную единицу можно определить с помощью клапана для подачи отмеряемого количества. Контейнер с повышенным давлением, насос, спрей или пульверизатор могут содержать раствор или суспензию активного соединения, например, в которых используется смесь этанола и газа-вытеснителя в качестве растворителя, которые могут дополнительно содержать смазочное вещество, например сорбитана триолеат. Капсулы и кассеты (сделанные, например, из желатина) для применения в ингаляторах или инжекторах могут быть разработаны таким образом, что содержат порошковую смесь соединения и подходящей порошкообразной основы, такой как лактоза или крахмал.
Альтернативно, соединение настоящего изобретения может быть введено в виде суппозитория или пессария или может применяться местно в форме геля, гидрогеля, лосьона, раствора, крема, мази или присыпки. Соединение настоящего изобретения может также введено дермальным или чрезкожным способом, например, с помощью кожного пластыря. Они также могут быть введены с помощью легочного или ректального способа. Они также могут быть введены глазным путем. При глазном применении соединения могут быть разработаны в виде тонкоизмельченных суспензий в изотонических стерильных солевых растворах с доведенным рН или предпочтительно в виде изотонических стерильных солевых растворах с доведенным рН, необязательно в комбинации с консервантами, такими как бензилалкония хлорид. Альтернативно, они могут быть разработаны в виде мази, такой как вазелин.
Для местного применения на коже соединение настоящего изобретения может быть разработано в виде подходящей мази, содержащей активное соединение, суспендированное или растворенное, например, в смеси с одним или несколькими из следующих компонентов: минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтиленгликоль, эмульгированный воск и вода. Альтернативно, оно может быть изготовлено в виде подходящего лосьона или крема, суспендированного или растворенного, например, в смеси с одним или несколькими из следующих компонентов: минеральное масло, сорбитана моностеарат, полиоксиэтиленгликоль, жидкий парафин, полисорбат 60, сложные цетиловые эфиры воска, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и вода.
Композиции настоящего изобретения могут быть введены с помощью прямой инъекции.
В предпочтительном воплощении соединения настоящего изобретения вводятся систематично (например, перорально, буккально, сублингвально), более предпочтительно перорально.
Таким образом, предпочтительно соединение находится в форме, которая подходит для перорального приема.
Обычно лечащий врач определяет действующую дозу, которая будет наилучшим образом подходить субъекту. Специфический уровень дозы и частота приема для любого человека может варьировать и будет зависеть от множества факторов, включающих активность специфических применяемых соединений, метаболическую стабильность и продолжительность действия соединений, возраст, вес тела, общее здоровье, пол, особенности питания, способ и время введения соединений, скорость экскреции, комбинация лекарств, тяжесть индивидуального состояния и вид индивидуальной терапии.
При пероральном способе введения пациентам суточная дозировка соединения может быть единственной дозой или может быть поделена на несколько доз.
Предполагаемая доза соединений в соответствии с настоящим изобретением для приема внутрь человеком (с приблизительным весом тела 70 кг) составляет от 0,1 мг до 2 г, более типично от 0,1 мг до 1 г, активного ингредиента на единичную дозу, выраженную в виде веса свободной кислоты. Единичная доза может приниматься, например, от 1 до 4 раз в сутки. Доза зависит от способа приема. Понятно, что обычно могут потребоваться изменения в дозировке, которые зависят от возраста и веса пациента, а также от тяжести состояния. Дозировка также зависит от способа приема. Точная дозировка и способ приема в конечном счете будут установлены по усмотрению лечащего врача или ветеринара.
Соединения настоящего изобретения могут также применяться вместе с другими терапевтическими средствами. Изобретение, таким образом, обеспечивает в следующем аспекте композицию, включающую соединение изобретения или его фармацевтически приемлемое производное вместе с другим терапевтическим средством.
Если соединение с формулой (I) или его фармацевтически приемлемое производное применяется вместе со вторым терапевтическим средством, активным против той же самой стадии заболевания, дозу каждого соединения можно изменить по сравнению с тем случаем, когда соединение применяют само по себе. Подходящие дозы будут понятны для специалистов в этой области техники. Понятно, что количество соединения настоящего изобретения, требуемое для лечения или предупреждения заболевания, будет различаться в зависимости от природы состояния и возраста и состояния пациента, и, в конце концов, оно будет установлено по усмотрению лечащего врача или ветеринара. Примеры других активных соединений, которые можно комбинировать с соединением, имеющим формулу (I), или его фармацевтически приемлемым производным включают, но не ограничиваются только ими: (а) другие антагонисты VLA-4; (б) антагонисты H1-гистаминовых рецепторов; (в) нестероидные противовоспалительные лекарственные средства (NSAID's); (г) антидиабетические вещества, например глитазоны; (д) антихолинергические вещества; (е) ингибиторы COX-2, например 2-(4-этоксифенил)-3-(4-метансульфонилфенил)пиразоло[1,5-b]пиридазин (как раскрыто в патентной заявке WO 99/12930); (ж) ингибиторы PDE-IV; (з) стероиды, например кортикостероиды; (и) бета-агонисты; (к) антагонисты хемокиновых рецепторов, например CCR-2, CCR-3, CCR-5 и CCR-8; (л) подходящие средства лечения или предупреждения множественного склероза, например интерферон; (м) антагонисты LFA-1; (н) ингибиторы TNF; (о) сульфасалазин и 5-аминосалицилаты и (п) иммунодепрессанты.
Указанные выше смеси могут применяться общепринятым способом в виде фармацевтической композиции, и, таким образом, фармацевтические композиции, включающие, как указано выше, фармацевтически приемлемый носитель или эксципиент, составляют следующий аспект изобретения. Отдельные компоненты таких составов могут быть введены или последовательно, или одновременно в отдельных или смешанных фармацевтических композициях общепринятым способом. Если прием средства последовательный, то или соединение изобретения, или второе терапевтическое средство могут приниматься первыми. Если прием средства совместный, смесь можно принимать или в одной и той же композиции или в различных фармацевтических композициях.
Если средства объединены в одну и ту же композицию, то понятно, что два соединения должны быть стабильны и совместимы друг с другом и с другими компонентами композиции. Если средства рецептированы по отдельности, то они могут поставляться в любой удобной для применения композиции таким образом, как известно для таких соединений в этой области техники.
Все публикации, включающие, но не ограничивающиеся патентами и патентными заявками, процитированные в этом описании, включены сюда путем отсылки, как если бы для каждой такой публикации было конкретно и в индивидуальном порядке указано, что они включены сюда путем отсылки даже при полном их объяснении.
Приведенные ниже разделы Получение препаратов и Примеры иллюстрируют получение соединений настоящего изобретения.
Основной способ
В экспериментальном разделе, там, где указано, термин MDAP означает Mass Directed Auto Preparation, автоматическую систему очистки соединений с помощью препаративной ВЭЖХ с детекцией и сбором соединений с желаемьми массами с применением масс-спектрометра и препаративной системы ВЭЖХ. Далее применяли систему Waters FractionLynx MDAP с подходящей обратно-фазовой колонкой при использовании градиента смеси вода/ацетонитрил, оба растворителя содержали 0,1%-ную муравьиную кислоту.
Получение препарата 1
Метиловый эфир (S)-2-трет-бутоксикарбониламино-3-(4-гидроксифенил)пропионовой кислоты (Р1)
Ди-трет-бутилбикарбонат (200 г, 0,92 моля) добавляли порциями к смеси метилового эфира и гидрохлорида L-тирозина (200 г, 0,86 моля, фирма «Aldrich») и гидрокарбонат натрия (100 г, 1,19 моля) в дихлорметане (1 л) и воде (1 л). Смесь перемешивали в течение 2 час при комнатной температуре. Органический слой отделяли и водную фазу вновь экстрагировали с помощью дихлорметана (500 мл). Объединенный органический слой высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении для получения неочищенного указанного в заголовке соединения в виде бесцветного стеклообразного вещества, которое использовали в следующей стадии без очистки. LC/MS (ES-ve): [M-H]- при m/z 294 (C15H21NO5 требует [М-Н]- при m/z 294).
Получение препарата 2
Метиловый эфир (S)-2-трет-бутоксикарбониламино-3-(4-трифторметансульфонилоксифенил)пропионовой кислоты (Р2)
Пиридин (58 мл, 0,72 моля) добавляли к раствору неочищенного метилового эфира (S)-2-трет-бутоксикарбониламино-3-(4-гидроксифенил)пропионовой кислоты (Р1, 70,5 г, 0,24 моля) в дихлорметане (1 л) под аргоном. Раствор охлаждали на льду и затем при перемешивании по каплям добавляли трифторметансульфоновый ангидрид (52 мл, 0,31 моля). После того как добавление было завершено, смесь перемешивали на льду в течение 2 час, промывали 2 М соляной кислотой (500 мл), высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на силикагеле (Biotage 75L, 800 г силикагеля), элюируя с помощью смеси этилацетат : гексан (15:85) для получения указанного в заголовке соединения в виде бесцветного масла, которое медленно затвердевало с получением белого твердого вещества. LC/MS (ES+ve): [М-ВОС+Н]+ при m/z 328 (C16H20NO7S требует [М+Н]+ при m/z 428).
Получение препарата 3
4-Гидроксиметил-2,6-диметоксифенилбороновая кислота (Р3)
Раствор (3,5-диметоксифенил)метанола (53 г, 0,31 моля, фирма «Aldrich») в безводном тетрагидрофуране (1 л) охлаждали до температуры от -50 до -70°С в токе аргона и обрабатывали н-бутиллитием (450 мл, 1,6 М в гексане, 0,72 моля) в течение 30 мин. После добавления реакционную смесь оставляли нагреваться до 0°С в течение 45 мин и затем оставляли стоять при комнатной температуре в течение 2 час. Реакционную смесь вновь последовательно охлаждали до -60°С и обрабатывали, добавляя порциями триметилборат (135 мл, 1,15 моля). После добавления смесь оставляли нагреваться до комнатной температуры и перемешивали в течение последующих 18 час. Реакцию останавливали при 0°С, добавляя порциями водный раствор лимонной кислоты (75 г лимонной кислоты в 300 мл воды). Водный слой насыщали хлористым натрием и продукт экстрагировали этилацетатом (2×1 л).
Объединенные органические слои высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении. К остатку добавляли этилацетат (100 мл) и полученный бесцветный осадок собирали фильтрованием и высушивали при 40°С при пониженном давлении для получения указанного в заголовке соединения в виде бесцветного твердого вещества, которое использовали в следующей реакции без очистки.
Получение препарата 4
Метиловый эфир (S)-2-трет-бутоксикарбониламино-3-(4'-гидроксиметил-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (Р4)
Триэтиламин (28 мл, 0,20 моля) добавляли к раствору метилового эфира (S)-2-трет-бутоксикарбониламино-3-(4-трифторметансульфонилоксифенил)пропионовой кислоты (Р2, 42,73 г, 0,10 моля) и 4-гидроксиметил-2,6-диметоксифенилбороновой кислоты (Р3, 29,7 г, 0,14 моля) в безводном диметилформамиде (250 мл). После дегазирования раствора с помощью аргона добавляли тетракис(трифенилфосфин)палладий(0) (5,8 г, 5 ммоля) и смесь нагревали до 90°С в течение 1 час в токе аргона [примечание: наблюдалась небольшая экзотермия]. После охлаждения до комнатной температуры смесь разводили этилацетатом (1 л) и водой (700 мл) и разделяли. Органический слой промывали водой (2×300 мл), высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении.
Неочищенный продукт очищали с помощью хроматографии на силикагеле (Biotage 75L, 800 г силикагеля), элюируя смесью этилацетат : гексан (50:50) для получения указанного в заголовке соединения в виде бесцветного твердого вещества. LC/MS (ES+ve): [М-BOC+H]+ при m/z 346 (C24H31NO7 требует [M+H]+ при m/z 446).
Получение препарата 5
Метиловый эфир (S)-2-трет-бутоксикарбониламино-3-(4'-формил-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (Р5)
Двуокись марганца (230 г, 2,64 моля) добавляли порциями к раствору метилового эфира (S)-2-трет-бутоксикарбониламино-3-(4'-гидроксиметил-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (Р4, 28,98 г, 65,1 ммоля) в дихлорметане (1 л). Полученную суспензию перемешивали при комнатной температуре в течение 1 час и затем фильтровали через целит, промывая затем целитовую прокладку с помощью дихлорметана (1 л). Фильтрат концентрировали при пониженном давлении для получения продукта в виде бесцветного вспененного вещества, которое использовали без дальнейшей очистки. LC/MS (ES+ve): [M-BOC+H]+ при m/z 344 (C24H29NO7 требует [М+Н]+ при m/z 444).
Получение препарата 6
Метиловый эфир (S)-2-трет-бутоксикарбониламино-3-[4'-(гидроксииминометил)-2',6'-диметоксибифенил-4-ил]пропионовой кислоты (Р6)
Гидроксиламина гидрохлорид (7,4 г, 106 ммолей) и диизопропилэтиламин (18 мл, 106 ммолей) добавляли к раствору метилового эфира (S)-2-трет-бутоксикарбониламино-3-(4'-формил-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (Р5, 23,6 г, 53 ммоля) в тетрагидрофуране (300 мл). Реакционную смесь нагревали с обратным холодильником в течение 2 час и затем оставляли охлаждаться до комнатной температуры. Раствор концентрировали при пониженном давлении и затем вновь растворяли в этилацетате (500 мл), промывали 10%-ным водным раствором лимонной кислоты, водой и солевым раствором (каждого по 500 мл), высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении для получения бесцветного вспененного вещества, которое использовали на следующей стадии без дополнительной очистки. LC/MS (ES+ve): [М-ВОС+Н]+ при m/z 359 (С24Н30N2O7 требует [M+H]+ при m/z 459).
Получение препарата 7
Гидрохлорид метилового эфира (S)-2-амино-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (Р7)
Тионилхлорид (30 мл, 411 ммолей) по каплям добавляли к раствору метилового эфира (S)-2-трет-бутоксикарбониламино-3-[4'-(гидроксииминометил)-2',6'-диметоксибифенил-4-ил]пропионовой кислоты (Р6, 46,5 г, 101 ммолей) в дихлорметане (500 мл) при 0°С в токе аргона. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 3 дней. Выпавшее в осадок твердое вещество собирали с помощью фильтрования, промывали дихлорметаном (200 мл) и сушили при 45°С при пониженном давлении. Указанное в заголовке соединение получали в виде белого твердого вещества. LC/MS (ES+ve): [М+Н]+ при m/z 341 (C19H20N2O4 требует [М+Н]+ при m/z 341).
Получение препарата 8
Метиловый эфир 2-бром-5-гидроксибензойной кислоты (Р8)
2-Бром-5-метоксибензойную кислоту (30,0 г, 130 ммолей, фирма «Aldrich») перемешивали в токе аргона в безводном дихлорметане (600 мл), охлажденном в смеси сухой лед/ацетон, по мере того как в нее в течение 15 мин добавляли трехбромистый бор (1 М в дихлорметане, 280 мл, 280 ммолей). Смесь затем перемешивали при комнатной температуре в течение 2,5 час, получая осадок. Затем осторожно по каплям добавляли метанол (300 мл) (внимание: в начале реакция чрезвычайно экзотермична и происходит бурное вспенивание) в течение 30 мин, что приводило в конце реакции к получению темного гомогенного раствора, к которому добавляли концентрированную серную кислоту (15 мл). Раствор перемешивали с обратным холодильником в течение 1 час, охлаждали и концентрировали при пониженном давлении. Осадок растворяли в дихлорметане (500 мл), промывали солевым раствором (500 мл), высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении для получения указанного в заголовке соединения в виде твердого вещества. LC/MS (ES+ve): [М+Н]+ при m/z 231, 233 (C8H7BrO3 требует [М+Н]+ при m/z 231, 233).
Получение препарата 9
Метиловый эфир 2-бром-5-этоксибензойной кислоты (Р9)
Гидрид натрия (60% в минеральном масле, 4,24 г, 106 ммолей) перемешивали в токе аргона в безводном диметилформамиде (150 мл), охлаждая на льду, по мере того как в него в течение 15 мин добавляли метиловый эфир 2-бром-5-гидроксибензойной кислоты (Р8, 20,41 г, 88 ммолей) в безводном диметилфомамиде (150 мл). Смесь перемешивали при комнатной температуре в течение 45 мин, вновь охлаждали на льду и обрабатывали иодэтаном (8,5 мл, 106 ммолей). Эту смесь перемешивали при комнатной температуре в течение 3 час, концентрировали при пониженном давлении, разводили этилацетатом (400 мл), промывали водой (3×400 мл) и солевым раствором (400 мл), высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении для получения указанного в заголовке соединения, загрязненного следовыми количествами соответствующего этилового эфира и остатками минерального масла. Этот материал использовали на следующей стадии без дополнительной очистки. LC/MS (ES+ve): [M+H]+ при m/z 259, 261 (С10H11BrO3 требует [М+Н]+ при m/z 259, 261).
Получение препарата 10
2-Бром-5-этоксибензойная кислота (Р10)
Метиловый эфир 2-бром-5-этоксибензойной кислоты (Р9, 20,53 г, 79 ммолей) перемешивали в тетрагидрофуране (600 мл) и обрабатывали гидроокисью лития (18,2 г, 791 ммоля) в воде (200 мл). Смесь перемешивали в течение 4 дней, концентрировали при пониженном давлении, разводили водой (500 мл), подкисляли 5 н. соляной кислотой и экстрагировали этилацетатом (2×300 мл). Объединенные экстракты промывали солевым раствором (250 мл), высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении для получения не совсем белого твердого вещества. После растирания в порошок с помощью гексана, фильтрования и сушки было получено указанное в заголовке соединение в виде белого порошка. LC/MS (ES+ve): [M+H]+ при m/z 245, 247 (C9H9BrO3 требует [М+H]+ при m/z 245, 247).
Получение препарата 11
Метиловый эфир (S)-2-{[1-(2-бром-5-этоксифенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (Р11)
Гидрохлорид метилового эфира (S)-2-амино-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (Р7, 37,1 г, 98,5 ммоля) растворяли в дихлорметане (500 мл) и воде (400 мл) и охлаждали до 0°С в токе аргона. Гидрокарбонат натрия (20,1 г, 239,3 ммоля) добавляли в реакционную смесь и затем по каплям добавляли 2-бром-5-этоксибензоилхлорид (27,2 г, 103,7 ммоля) (полученный из 2-бром-5-этоксибензойной кислоты (Р10) с помощью стандартной процедуры с применением оксалилхлорида (4 экв.) в дихлорметане и капли диметилформамида). Реакционную смесь перемешивали при 0°С в течение 1 час и затем разводили насыщенным водным раствором гидрокарбоната натрия (200 мл). После отделения органического слоя водный слой повторно экстрагировали дихлорметаном (2×400 мл). Объединенные органические слои высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Продукт очищали с помощью хроматографии на силикагеле (Biotage 75L, 800 г силикагеля), элюируя смесью этилацетат : дихлорметан (3:97) для получения указанного в заголовке соединения в виде бесцветного твердого вещества. MS (ES+ve): [М+Н]+ при m/z 567, 569 (С28Н27BrN2O6 требует [М+Н]+ при m/z 567, 569).
Получение препарата 12
Этиловый эфир (S)-2-{[1-(2-бром-5-метоксифенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (Р12)
2-Бром-5-метоксибензойную кислоту (0,355 г, 1,54 ммоля, фирма «Aldrich»), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (1,168 г, 2 экв.) и триэтиламин (1,069 мл, 5 экв.) добавляли к диметиформамиду (25 мл) в токе аргона при перемешивании при комнатной температуре. Через 0,5 час добавляли этиловый эфир, соответствующий Р7 (Р13), гидрохлорид этилового эфира (S)-2-амино-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (0,6 г, 1 экв.) и перемешивали в течение ночи. Затем растворитель выпаривали при пониженном давлении и остаток распределяли между этилацетатом и водой. Органический слой промывали водой (х2) и насыщали водным раствором бикарбоната натрия перед выпариванием для высушивания. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле градиентом 0-10% метанола в дихлорметане с последующей препаративной ВЭЖХ для получения указанного в заголовке соединения. MS (AP+ve): [M+H]+ при m/z 567, 569 (C29H27BrN2O6 требует [М+Н]+ при m/z 567, 569).
Получение препарата 13
Гидрохлорид этилового эфира (S)-2-амино-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (Р7 - соответствующий этиловый эфир) (Р13)
Тионилхлорид (7,71 мл, 105,7 ммоля) добавляли по каплям в течение 15 мин к раствору этилового эфира (S)-2-трет-бутоксикарбониламино-3-[4'-(гидроксииминометил)-2',6'-диметоксибифенил-4-ил]пропионовой кислоты (12,5 г, 26,5 ммоля) [полученного в аналогичном синтезе со стадиями Р1-Р6, за исключением начальной стадии получения препарата 1, где вместо гидрохлорида метилового эфира L-тирозина использовали гидрохлорид этилового эфира L-тирозина (от фирмы «Bachem»)] в дихлорметане (250 мл) при 0°С в токе аргона. Реакционную смесь перемешивали в течение 15 мин при температуре порции, затем оставляли нагреваться до комнатной температуры и продолжали перемешивать в течение ночи. Выпавшее в осадок соединение, указанное в заголовке, собирали фильтрованием, промывали дихлорметаном (2×15 мл) и сушили при пониженном давлении. LC/MS (ES+ve): [М+Н]+ при m/z 355 (C20H22N2O4 требует [М+Н]+ при m/z 355).
Пример 1
(S)-2-{[1-(2-Бром-5-этоксифенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовая кислота (Е1)
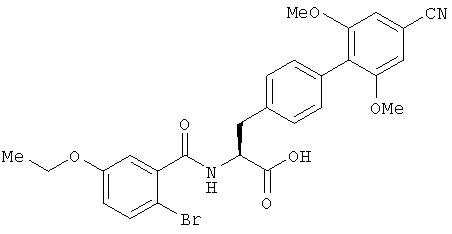
Раствор метилового эфира (S)-2-{[1-(2-бром-5-этоксифенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (Р11, 51,5 г, 90,8 ммоля) в тетрагидрофуране (950 мл) охлаждали до 0°С и обрабатывали 0,5 М водным раствором гидроокиси лития (700 мл). Реакционную смесь перемешивали при 0°С в течение 1 час и затем подкисляли 5 М соляной кислотой. Тетрагидрофуран выпаривали при пониженном давлении и остаток разводили этилацетатом. После отделения органического слоя водную фазу вновь экстрагировали этилацетатом. Объединенные органические слои промывали водой (х2), затем высушивали над сульфатом магния и выпаривали растворитель при пониженном давлении для получения соединения, указанного в заголовке, в виде бесцветного твердого вещества.
1Н NMR δ (DMSO-d6): 1,29 (3Н, t, J=7, 0 Гц), 2,96 (1H, dd, J=13,9, 10,91 Гц), 3,22 (1H, dd, J=14,1, 4,2 Гц), 3,71 (6H, s), 3,99 (2H, q, J=7, 0 Гц), 4,65 (1H, ddd, J=10,9, 8,4, 4,3 Гц), 6,69 (1H, d, J=3, 0 Гц), 6,91 (1H, dd, J=8,8, 3,1 Гц), 7,14 (2H, d, J=8,1 Гц), 7,24 (2H, s), 7,32 (2H, d, J=8,1 Гц), 7,47 (1H, d, J=8,8 Гц), 8,77 (1H, br. d, J=8,4 Гц), 12,83 (1H, br. s). MS (ES+ve) [M+H]+ при m/z 553, 555 (С27Н25BrN2O6 требует [М+Н]+ при m/z 553, 555).
Пример 1 (альтернативная процедура синтеза)
Соединение, указанное в заголовке, также получали с помощью процедуры, приведенной ниже.
4-Бром-3,5-диметоксибензамид
Тионилхлорид (60 мл, 0,82 моля) добавляли к перемешиваемой суспензии 4-бром-3,5-диметоксибензойной кислоты (от фирмы «Jintan Baocheng», 100 г, 0,38 моля) в толуоле (700 мл) и диметиформамиде (1 мл). Реакционную смесь перемешивали с обратным холодильником в токе азота. ВЭЖХ через 2 час показала полное исчезновение кислоты после образования метилового эфира (тушение метанолом кислотного хлорида). Быстроиспаряющееся вещество удаляли перегонкой и полученное твердое соединение добавляли к амиаку (0,88, 350 мл). Смесь перемешивали при комнатной температуре в течение 45 мин. Продукт фильтровали, промывали водой (2×200 мл) и сушили при пониженном давлении в течение ночи для получения соединения, указанного в заголовке, в виде не совсем белого твердого вещества.
1Н NMR δ (DMSO-d6): 3,89 (6Н, s), 7,23 (2H, s), 7,52 (1H, s), 8,2 (1H, s).
MS (ES+ve) [M+H]+ при m/z 260, 262 (C9Н10BrNO3 требует [М+Н]+ при m/z 260, 262).
4-Бром-3,5-диметоксибензонитрил
Трифторуксусный ангидрид (129 мл, 0,91 моля) добавляли к суспензии 4-бром-3,5-диметоксибензамида (104 г, 0,38 моля) в пиридине (127 мл, 1,57 моля) и тетрагидрофуране (950 мл). Температуру поддерживали при 20-25°С с помощью ледяной бани. Оранжево-коричневый раствор перемешивали при 20-25°С в токе азота в течение 1 час. ВЭЖХ показала полное исчезновение 4-бром-3,5-диметоксибензамида после получения продукта. Реакционную смесь концентрировали примерно в 50% от исходного объема. Добавляли воду (900 мл) и продукт отфильтровывали, промывали водой (2×250 мл) и сушили при 40°С при пониженном давлении для получения соединения, указанного в заголовке, в виде пушистого не совсем белого вещества.
1Н NMR δ (CDCl3): 3,96 (6Н, s), 6,8 (2H, s).
4-Циано-2,6-диметоксифенилбороновая кислота
Изопропилмагнийхлорид (2 М раствор в тетрагидрофуране, 206 мл, 0,412 моля) добавляли к жидкой кашице 4-бром-3,5-диметоксибензонитрила (50 г, 0,207 моля) в тетрагидрофуране (400 мл) при 5-10°С в токе азота. Полученный раствор оставляли нагреваться до комнатной температуры. Анализ ВЭЖХ показал полное поглощение исходного материала. Реакционную смесь охлаждали до 5-10°С и добавляли к раствору триметилбората (103 мл, 0,92 моля) в тетрагидрофуране (100 мл), поддерживая температуру во время добавления ниже 10°С. Реакционную смесь оставляли нагреваться до комнатной температуры. ВЭЖХ показала завершение реакции образования продукта. Реакционную смесь охлаждали до 5-10°С и добавляли 1 М соляную кислоту (500 мл), поддерживая температуру во время добавления ниже 10°С. Смесь перемешивали при комнатной температуре в течение 30 мин и затем быстро испаряющиеся вещества удаляли перегонкой. Продукт фильтровали, промывали водой и сушили при 40°C при пониженном давлении для получения соединения, указанного в заголовке, в виде твердого не совсем белого вещества.
1Н NMR δ (DMSO-d6): 3,78 (6Н, s), 7,0 (2H, s), 9,25 (2H, s).
Гидрохлорид метилового эфира (S)-2-амино-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты
Раствор ди-трет-бутилдикарбоната (17,9 г, 81,8 ммоля) в толуоле (60 мл) добавляли по каплям к раствору (S)-метилтирозина (от фирмы «Flamma», 15,2 г, 77,9 ммоля) в толуоле (120 мл) при 90°С. Реакционную смесь перемешивали при 90°С, по меньшей мере, в течение 30 мин. После того как ВЭЖХ подтвердила окончание реакции, реакционную смесь охлаждали до 0°С и добавляли пиридин (19 мл, 0,23 моля), поддерживая температуру 0°С. Трифторметансульфоновый ангидрид (16,7 мл) добавляли по каплям, поддерживая температуру 0°С. Оранжевую кашицу перемешивали при 0°С в течение, по меньшей мере, 2 час. После того как ВЭЖХ подтвердила окончание реакции, добавляли 2 М соляную кислоту (133 мл), поддерживая температуру при 0°С. Слои разделяли и верхний органический слой промывали 10%-ным (мас./мас.) водным раствором карбоната натрия (138 мл) и 36%-ным (мас./мас.) солевым раствором (138 мл). Органический слой концентрировали приблизительно в 60 мл и добавляли твердый хлорид натрия (30 г), а затем н-гептан (300 мл). Смесь фильтровали и фильтрат экстрагировали 1-метил-2-пирролидоном (2×150 мл). Остатки быстроиспаряющихся органических растворителей отгоняли при пониженном давлении. Добавляли 4-циано-2,6-диметоксифенилбороновую кислоту (17,5 г, 84,5 ммоля) и раствор дегазировали.
Добавляли тетракис(трифенилфосфин)палладий(0) (3 г, 2,7 ммоля) и триэтиламин (19,8 мл) и реакционную смесь нагревали до 70°С. Реакционную смесь перемешивали при 70°С, по меньшей мере, в течение 2 час. После того как ВЭЖХ подтвердила окончание реакции, реакционную смесь переносили в другой сосуд и разводили толуолом (300 мл). Добавляли 2 М соляную кислоту (300 мл) и смесь фильтровали. Отделяли органический слой и промывали водой (300 мл). Органический слой фильтровали и промывали толуолом (90 мл). Толуольный раствор смешивали с Silicycle silica, поддерживаемым N-функционализированной тиомочевиной (11,7 г), по меньшей мере, в течение 15 час. Силикагель фильтровали и промывали толуолом (30 мл). Раствор сушили азеотропно, затем медленно добавляли к раствору хлористый водород в изопропаноле (155 мл при концентрации 5 М) при 50°С. Реакционную смесь перемешивали при 50°С в течение 30 мин до завершения реакции, подтвержденного ВЭЖХ. Смесь охлаждали до 20°С, продукт отфильтровывали при пониженном давлении и промывали толуолом (60 мл). Твердое вещество сушили при пониженном давлении при 40°С, чтобы получить соединение, указанное в заголовке, в виде твердого не совсем белого вещества.
Метиловый эфир 2-бром-5-гидроксибензойной кислоты
Раствор трехбромистого бора (18 мл, 190,5 ммоля) в дихлорметане (18 мл) добавляли к перемешиваемой суспензии 2-бром-5-метоксибензойной кислоты (от фирмы «Wychem», 20 г, 86,6 ммоля) в дихлорметане (200 мл) при температуре от -15 до -10°С. Желтую суспензию оставляли нагреваться до 0°С. Анализ ВЭЖХ показал полное исчезновение кислоты. Реакцию останавливали добавлением метанола (100 мл). Темный прозрачный раствор перемешивали с обратным холодильником (приблизительно при 45°С) в течение 12 час, выпаривали почти досуха, распределяли между водой (400 мл) и этилацетатом (500 мл) и разделяли. Органический раствор промывали солевым раствором (250 мл) и выпаривали для получения соединения, указанного в заголовке, в виде твердого вещества кремового цвета.
Метиловый эфир 2-бром-5-этоксибензойной кислоты
Смесь метилового эфира 2-бром-5-гидроксибензойной кислоты (18,9 г, 81,8 ммоля), карбоната калия (34,4 г, 249,5 ммоля) и иодистого этила (13 мл, 16,7 ммоля) в метилизобутилкетоне (190 мл) перемешивали при 70-75°С в токе азота в течение 15 час. Анализ ВЭЖХ показал полное превращение в продукт. Добавляли воду (100 мл). Водную фазу удаляли и метилизобутилкетоновый раствор промывали солевым раствором (100 мл), затем концентрировали при пониженном давлении для получения соединения, указанного в заголовке, в виде оранжевого масла.
2-Бром-5-этоксибензойная кислота
Смесь метилового эфира 2-бром-5-этоксибензойной кислоты (20,7 г, 79,9 ммоля) и 10 М гидроокиси натрия (40 мл, 0,4 моля) в тетрагидрофуране (100 мл) перемешивали при комнатной температуре в течение 18 час. Анализ ВЭЖХ показал полное исчезновение исходного материала. Реакционную смесь охлаждали до 0-5°С и подкисляли до рН 1 5 М соляной кислотой. Органическую фазу отбирали и водную фазу экстрагировали третичным бутилметиловым эфиром (40 мл). Органические фазы объединяли, промывали солевым раствором (50 мл) и концентрировали при пониженном давлении для получения соединения, указанного в заголовке, в виде твердого вещества кремового цвета.
Метиловый эфир (S)-2-{[1-(2-бром-5-этоксифенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты
К суспензии гидрохлорида метилового эфира (S)-2-амино-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (5 г, 13,3 ммоля) в тетрагидрофуране (100 мл) добавляли триэтиламин (5,5 мл, 39,8 ммоля). Суспензию перемешивали в течение 60 мин при 20°С. Раствор 2-бром-5-этоксибензоилхлорида (приготовленного из 2-бром-5-этоксибензойной кислоты (3,2 г, 13 ммолей) с помощью стандартной процедуры с использованием тионилхлорида (3,3 экв.) в толуоле (16 мл)) (3,4 г, 13 ммолей) в толуоле (10 мл) добавляли по каплям в течение 10 мин, поддерживая температуру 0-5°С. Смесь перемешивали при 0-5°С, по меньшей мере, в течение 60 мин. После того как ВЭЖХ подтвердила окончание реакции, реакционную смесь последовательно промывали 2 М соляной кислотой (25 мл), 10%-ным (мас./мас.) водным раствором карбоната натрия (25 мл) и 36%-ным (мас./мас.) солевым раствором (25 мл). Растворитель удаляли перегонкой для получения твердого вещества кремового цвета. Его растворяли в тетрагидрофуране (75 мл) и перемешивали в течение ночи с Silicycle silica, поддерживаемым N-функционализированной тиомочевиной (0,75 г). Силикагель фильтровали и тетрагидрофурановый раствор концентрировали приблизительно в 30 мл. В течение 45 мин добавляли воду (75 мл). Кашицу перемешивали при комнатной температуре в течение 1 час. Продукт фильтровали, промывали водой (30 мл) и сушили при пониженном давлении при 40°С для получения соединения, указанного в заголовке, в виде твердого вещества не совсем белого цвета.
Тетрагидрофурановый сольват (S)-2-{[1-(2-бром-5-этоксифенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты
Раствор метилового эфира (S)-2-{[1-(2-бром-5-этоксифенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (5 г, 8,8 ммоля) в тетрагидрофуране (25 мл) охлаждали до 0-5°С. Добавляли по каплям 2 М гидроокись натрия (4,5 мл, 9 ммолей), поддерживая температуру ниже 5°С. Реакционную смесь перемешивали при 0-5°С, по меньшей мере, в течение 18 час. После того как ВЭЖХ подтвердила окончание реакции, смесь разводили водой (50 мл) перед тем, как довести рН до 1 с помощью 2 М соляной кислоты (приблизительно 10 мл).
Полученную суспензию выдерживали, по меньшей мере, 30 мин при 0-5°С перед тем, как отфильтровать при пониженном давлении твердое вещество, и промывали водой (20 мл). Продукт затем сушили при пониженном давлении при 40°С для получения соединения, указанного в заголовке, в виде твердого вещества не совсем белого цвета.
(S)-2-{[1-(2-Бром-5-этоксифенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовая кислота (Е1)
Суспензию тетрагидрофуранового сольвата (S)-2-{[1-(2-бром-5-этоксифенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (5 г) в метаноле (50 мл) нагревали с обратным холодильником и перемешивали для образования раствора. Раствор охлаждали до 60°С и фильтровали. Затем раствор охлаждали до 0°С в течение 1,5 час. Полученную суспензию выдерживали при 0°С в течение 2 час. Твердое вещество фильтровали при пониженном давлении и промывали холодным метанолом (5 мл). Продукт сушили при пониженном давлении при 40°С для получения соединения, указанного в заголовке, в виде кристаллического твердого вещества белого цвета.
Пример 2
(S)-2-{[1-(2-Бром-5-фторфенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовая кислота (Е2)
Соединение, указанное в заголовке, получали способом, аналогичным тому, который был описан в разделе Получение препарата 11 и в Примере 1, используя Р13 и 2-бром-5-фторбензоилхлорид (от фирмы «Apollo»). [M+H]+ при m/z 527, 529.
Пример 3
(S)-2-{[1-(2-Бром-5-метоксифенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовая кислота (Е3)
Пример 3а
Соединение, указанное в заголовке, получали способом, аналогичным тому, который был описан в разделе Получение препарата 11 и в Примере 1, используя Р7 и 2-бром-5-метоксибензоилхлорид (от фирмы «Avocado»),
Пример 3б
Водную гидроокись лития (0,5 М, 25 мл) добавляли при перемешивании к раствору этилового эфира (S)-2-{[1-(2-бром-5-метоксифенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовой кислоты (P12, 0,539 г, 0,95 ммоля) в тетрагидрофуране (30 мл) при комнатной температуре. Перемешивание продолжалось 2 час, и затем реакцию останавливали избытком водного 10%-ного раствора лимонной кислоты. Смесь затем экстрагировали этилацетатом и органический слой промывали еще одной порцией лимонной кислоты и затем водой (х2). Органический слой выпаривали досуха и полученный неочищенный продукт очищали с помощью MDAP для получения соединения, указанного в заголовке, в виде твердого вещества белого цвета. [М+Н]+ при m/z 539, 541.
Пример 4
(S)-2-{[1-(2-Бром-5-метилфенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовая кислота (Е4)
Соединение, указанное в заголовке, получали способом, аналогичным тому, который был описан в разделе Получение препарата 12, используя Р13 и 2-бром-5-метилбензойную кислоту (от фирмы «Apin»), и полученный этиловый эфир гидролизовали по методу, описанному в Примере 3б. [М+Н]+ при m/z 523, 525.
Пример 5
(S)-2-{[1-(2-Бром-5-хлорфенил)метаноил]амино}-3-[4'-циано-2',6'-диметоксибифенил-4-ил]пропионовая кислота (Е5)
Соединение, указанное в заголовке, получали из соответствующего этилового эфира способом, аналогичным тому, который был описан в Примере 36. Этиловый эфир получали из Р13 и 2-бром-5-хлорбензойной кислоты (от фирмы «Lancaster»), используя метод, описанный в разделе Получение препарата 12. [М-Н]- при m/z 541, 543, 545.
Пример 6
(S)-2-{[1-(2,5-Дибромфенил)метаноил]амино}-3-[4'-циано-2',6'-диметоксибифенил-4-ил]пропионовая кислота (Е6)
Соединение, указанное в заголовке, получали из соответствующего этилового эфира способом, аналогичным тому, который был описан в Примере 36. Этиловый эфир получали из Р13 и 2,5-дибромбензойной кислоты (от фирмы «Lancaster»), используя метод, описанный в разделе Получение препарата 12. [М-Н]- при m/z 585, 587, 589.
Пример 7
(S)-2-{[(5-(Изопропокси)-2-бромфенил)метаноил]амино}-3-[4'-циано-2',6'-диметоксибифенил-4-ил]пропионовая кислота (Е7)
Соединение, указанное в заголовке, получали из соответствующего этилового эфира способом, аналогичным тому, который был описан в Примере 3б. Этиловый эфир получали из Р13 и 5-изопропокси-2-бромбензойной кислоты (полученной способом, аналогичным стадиям, описанным в разделе Получение препарата 8 - Получение препарата 10, за исключением использования изопропилбромида на стадии алкилирования, описанной в разделе Получение препарата 9) с помощью метода, описанного в разделе Получение препарата 12. [М-Н]- при m/z 565, 567.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ МОЧЕВИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2296120C2 |
| СОЕДИНЕНИЯ С МЕДИЦИНСКИМИ ЭФФЕКТАМИ, ОБУСЛОВЛЕННЫМИ ВЗАИМОДЕЙСТВИЕМ С РЕЦЕПТОРОМ ГЛЮКОКОРТИКОИДОВ | 2006 |
|
RU2430086C2 |
| АЛКИНИЛ-ЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2017 |
|
RU2729069C1 |
| СЕРУСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ НА ИХ ОСНОВЕ | 2000 |
|
RU2244708C2 |
| 2,2,2-ТРИФТОРЭТИЛ-ТИАДИАЗИНЫ | 2015 |
|
RU2692102C2 |
| ИНГИБИТОРЫ С-FMS КИНАЗЫ | 2007 |
|
RU2475483C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ У МЛЕКОПИТАЮЩИХ | 2004 |
|
RU2344121C2 |
| (3-ЦИКЛОАЛКИЛ-2,3,4,5-ТЕТРАГИДРО-1Н-БЕНЗО[d]АЗЕПИН-7-ИЛОКСИ)ПРОИЗВОДНЫЕ, ИХ ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ Н3 РЕЦЕПТОРОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОЛУЧЕНИЯ | 2003 |
|
RU2388752C2 |
| ПРОИЗВОДНОЕ ГЕТЕРОАРИЛ[4,3-с]ПИРИМИДИН-5-АМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2018 |
|
RU2764655C2 |
| ИМИДАЗО[1,2-А]ПИРИДИНОВЫЕ И ПИРАЗОЛ[2,3-А]ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2000 |
|
RU2248976C2 |
Изобретение относится к новым производным фенилаланина, которые ингибируют клеточную адгезию, опосредованную интегрином α4, и считаются полезными при лечении или предупреждении воспалительных заболеваний, а именно к соединениям, имеющим формулу (I), или к их фармацевтически приемлемой соли или эфиру, в котором R1 является бромом; R2 является галогеном, C1-6 алкилом или C1-6 алкокси. Изобретение также относится к способу получения соединения, имеющего формулу (I), к фармацевтической композиции, применению соединения, имеющего формулу (I), в производстве лекарственного средства, а также к промежуточным соединениям, имеющим формулу (II), (III) или (IV). 10 н. и 7 з.п. ф-лы.
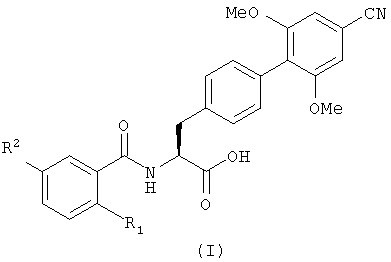
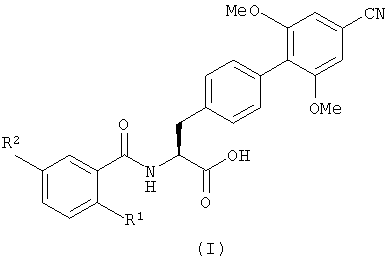
в котором R1 является бромом; и
R2 является галогеном, C1-6 алкил или C1-6 алкокси.
(S)-2-{[1-(2-бром-5-метилфенил)метаноил]амино}-3-(4'-циано-2',6'-диметоксибифенил-4-ил)пропионовую кислоту;
(S)-2-{[(2-бром-5-хлорфенил)метаноил]амино}-3-[4'-циано-2',6'-диметокси)бифенил-4-ил]пропионовую кислоту;
(S)-2-{[(2,5-дибромфенил)метаноил]амино)-3-[4'-циано-2',6'-диметокси)бифенил-4-ил]пропионовую кислоту;
(S)-2-{[(5-изо-пропокси)-2-бромфенил)метаноил]амино}-3-(4'-циано-2',6'-диметокси)бифенил-4-ил]пропионовую кислоту или их фармацевтически приемлемая соль или эфир.
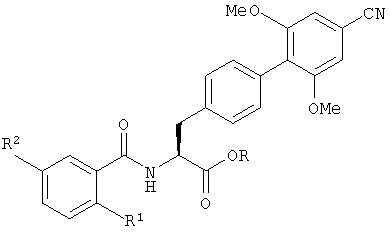
в которой R1 и R2 определены так же, как в формуле (I), и R представляет собой C1-6алкил, и при необходимости, затем, образование фармацевтически приемлемой соли или эфира.
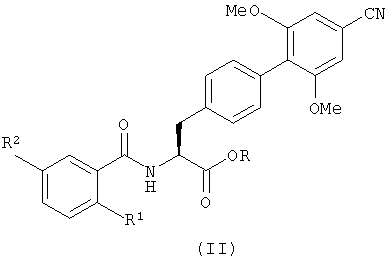
в котором R1 и R2 определены так же, как в формуле (I), и R представляет собой C1-6алкил.
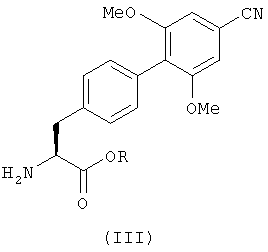
в котором R представляет собой C1-6алкил.
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| WO 9936393 A, 22.07.1999 | |||
| Этиловый эфир N @ ,О-дифенилацетил-трео-DL-фенилсерина, проявляющий противовоспалительное действие | 1981 |
|
SU961297A1 |

