Область техники
Изобретение относится к диссеминированному внутрисосудистому свертыванию (ДВС-синдром, генерализованный тромбогеморрагический синдром). В частности, изобретение относится к терапевтическому воздействию на диссеминированное внутрисосудистое свертывание.
Известный уровень техники
Диссеминированное внутрисосудистое свертывание (ДВС, DIC) - вторичная болезнь и может быть (по)следствием любого из широкого ряда первичных (основных) заболеваний. (See Bick, Disseminated Intravascular Coagulation and Related Syndromes. CRC Press, Boca Raton, (1983)). Среди его характерных особенностей - бесконтрольная (повышенная) активация свертывания крови, что приводит к генерации и отложению фибрина, ведущему к закупорке капилляров в различных органах, что приводит к нарушению функции этих органов. Bick et al, Clin. Appl Thrombosis Hemostasis 1:3-23 (1995) указывает на другую характерную особенность ДВС, заключающуюся в системной циркуляции плазмина в русле крови, глобального протеолитического фермента, который может биодеградировать различные плазменные белки (факторы, гормоны и т.д.) и может расщеплять фибриноген/фибрин с получением продуктов деградации фибриногена/фибрина. Эти продукты ухудшают гемостаз и приводят к кровотечению.
Наиболее серьезные формы ДВС характеризуются интенсивным расходом свертывающих белков (факторов), существенным отложением фибрина и кровотечением.
Пациенты, получившие травму, имеют повышенный риск для развития ДВС, особенно в тех случаях, когда имеются широко распространенные области повреждения ткани (особенно головного мозга), сепсис и множественное повреждение органов. Травма головы представляет общую причину ДВС особенно у младенцев и детей вследствие высокого содержания тромбопластина головного мозга и пропорционально повышенного соотношения площади поверхности головы к суммарной площади поверхности тела.
Сепсис может иметь место у приблизительно 40% пациентов, получивших травму, и представляет важную первостепенную причину для всех пациентов. Клиническое состояние ухудшается вторичным фибринолизом, который приводит к образованию ПДФ (FDP's) (продукты деградации фибриногена/фибрина) или "D-димеров", которые препятствуют нормальному образованию фибрина и функции тромбоцитов.
Отложение фибрина при ДВС может привести к дальнейшей дисфункции органа. ДВС - основная причина острой почечной недостаточности и, кроме того, способствует нарушению функционирования этого органа. Правдивым является и обратное утверждение в отношении того, что поврежденные органы способствуют развитию ДВС.
В настоящее время единственное общепринятое лечение ДВС ограничивается попытками облегчить первичное расстройство. Без контроля ДВС будет возобновляться, несмотря на предпринимаемые формы лечения, направленные на корректирование кровотечения или проблему, имеющие отношение к тромбозу. В некоторых случаях, в которых имеет место существенное кровотечение, заместительная терапия свежезамороженной плазмой, криопреципитатом компонентов плазмы (например, антитромбин III) и/или концентратами тромбоцитов может быть полезной до тех пор, пока первичная проблема не будет взята под контроль, однако эти лечения недопустимо дороги. Использование гепарина при ДВС является крайне спорным и обычно гепарин не используют для пациентов, у которых основной проблемой является травма.
Поэтому существует потребность в новых и более подходящих соединениях и способах лечения ДВС [Смотри также, например, de Jonge et al, Drugs 55:767-777 (1998) and Levi et al., Thrombosis and Haemostasis 82: 695 (1999)].
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩЕСТВА ИЗОБРЕТЕНИЯ
Изобретение обеспечивает новые и улучшенные соединения и способ лечения ДВС. Неожиданно было обнаружено, что те противосвертывающие соединения, которые обладают ингибирующим действием как на свободные, так и на связанные со сгустком тромбин и фактор Ха и, кроме того, являются ингибирующими по отношению к плазмину и активаторам плазминогена, могут быть полезными для лечения ДВС.
В первом аспекте изобретение относится к композиции вещества, включающей пептидиларгиналь формулы (I)
где Xaa представляет остаток альфа-замещенной угольной кислоты формулы (II)
где Q представляет С1-3 алкилоксикарбониламиногруппу, метиламиногруппу или гидроксильную группу, и R представляет С7-9 циклоалкилметильную группу или С5-7 циклоалкильную группу или 1-адамантилметильную группу, и Xbb представляет остаток L-пролина или L-азетидин-2-карбоновой кислоты, и его аддитивные соли кислоты, образованные с органической или неорганической кислотой.
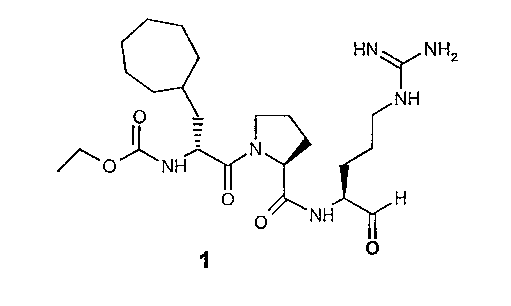
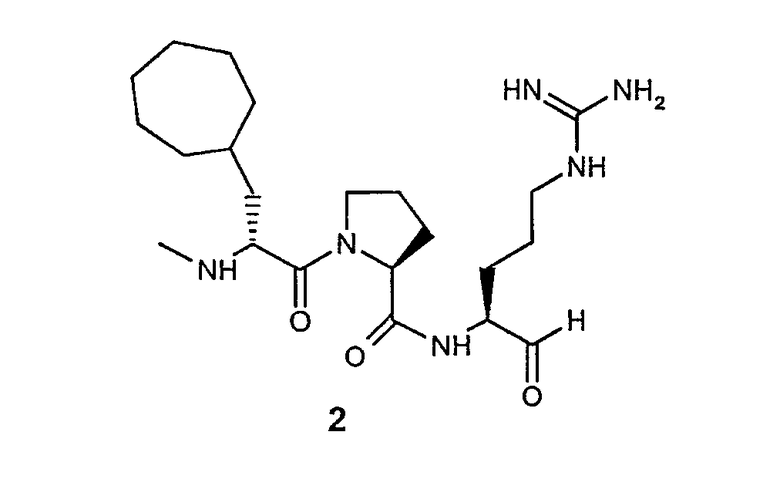
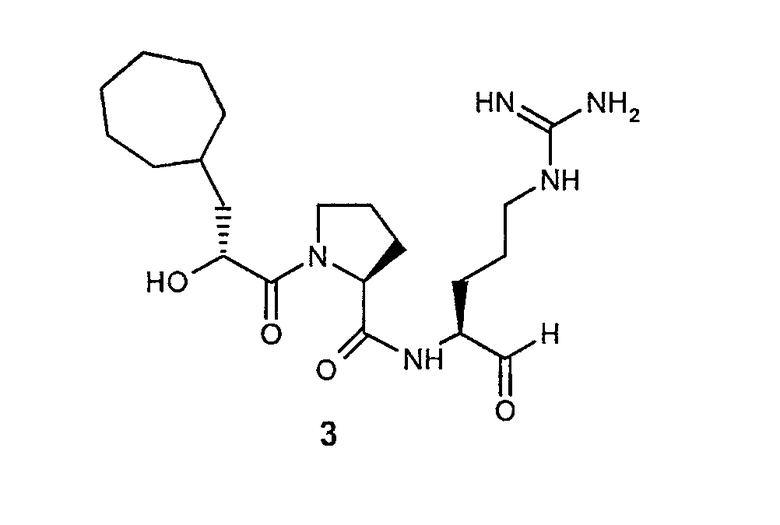
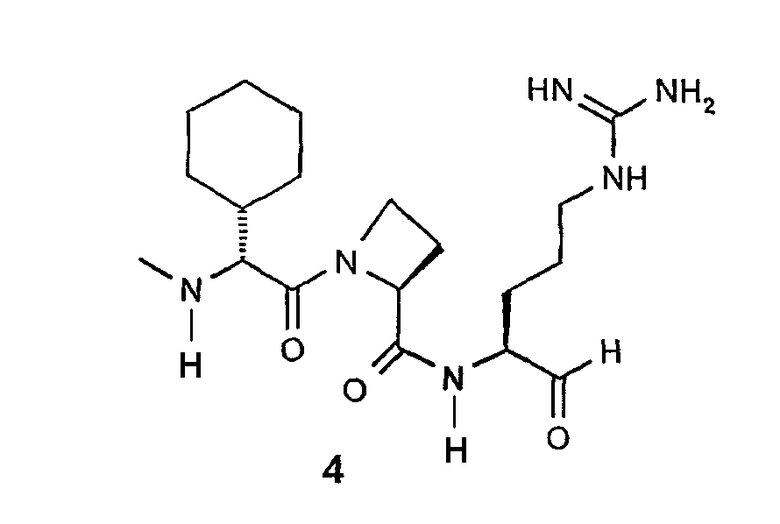
В особенно предпочтительных вариантах воплощения такие соединения могут иметь следующие структуры: 1 (этоксикарбонил-D-циклогептилаланил-L-пролил-L-аргининальдегид, Eoc-D-cHpa-Pro-Arg-H), или 2 (N-метил-D-циклогептилаланил-L-пролил-L-аргининальдегид, N-Me-D-cHpa-Pro-Arg-H), или 3 (D-циклогептиллактил-L-пролил-L-аргининальдегид, D-cHpl-Pro-Arg-H), или 4 (N-метил-D-циклогексилглицил-L-азетидин-2-карбонил-L-аргининальдегид, N-Me-D-Chg-Aze-Arg-H), которые соответствуют формуле (I), где Xaa представляет остатки альфа-замещенной алкилугольной кислоты формулы (II), где R представляет циклогептилметильную и циклогексильную группу, соответственно, Q представляет этоксикарбониламино-, метиламино- и гидроксильную группу, соответственно, и Xbb представляет остаток L-пролина и L-азетидинил-2-карбоновой кислоты соответственно.
Во втором аспекте изобретение относится к фармацевтической композиции, включающей препятствующий свертыванию крови пептидиларгиналь или его фармацевтически приемлемую соль согласно первому аспекту и фармацевтически приемлемый носитель, наполнитель или разбавитель.
В третьем аспекте, изобретение относится к способу лечения диссеминированного внутрисосудистого свертывания, и этот способ включает введение пациенту, страдающему диссеминированным внутрисосудистым свертыванием, препятствующего свертыванию крови пептидиларгиналя, соответствующего формуле (I)
где Xaa представляет остаток альфа-замещенной угольной кислоты формулы (II)
где Q представляет С1-3 алкилоксикарбониламиногруппу, метиламиногруппу или гидроксильную группу, и R представляет С7-9 циклоалкилметильную группу или 1-адамантилметильную группу, или С5-7 циклоалкильную группу и Xbb представляет остаток L-пролина или L-азетидин-2-карбоновой кислоты, и его фармацевтически приемлемой аддитивной соли кислоты. В особенно предпочтительных вариантах воплощения такие соединения могут иметь следующие структуры: 1 (этоксикарбонил-D-циклогептилаланил-L-пролил-L-аргининальдегид, Eoc-D-cHpa-Pro-Arg-H) или 2 (N-метил-D-циклогептилаланил-L-пролил-L-аргининальдегид, N-Me-D-cHpa-Pro-Arg-H), или 3 (D-циклогептиллактил-L-пролил-L-аргининальдегид, D-cHpl-Pro-Arg-H), или 4 (N-метил-D-циклогексилглицил-L-азетидин-2-карбонил-L-аргининальдегид, N-Me-D-Chg-Aze-Arg-H), которые соответствуют формуле (I), где Xaa представляет остатки альфа-замещенной алкилугольной кислоты формулы (II), где R представляет циклогептилметильную и циклогексильную группу соответственно, Q представляет этоксикарбониламино-, метиламино- и гидроксильную группу соответственно, и Xbb представляет остаток L-пролина и L-азетидинил-2-карбоновой кислоты соответственно.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Изобретение относится к диссеминированному внутрисосудистому свертыванию. В частности, изобретение относится к терапевтическому воздействию на диссеминированное внутрисосудистое свертывание. Изобретение обеспечивает новые и улучшенные (более подходящие) соединения и способ лечения ДВС. Соединения согласно изобретению обладают ингибирующим действием как на свободные, так и на связанные со сгустком тромбин и фактор Ха, а также на плазмин и активаторы плазминогена.
Патенты и публикации, цитируемые здесь, отражают уровень техники в данной области и введены в настоящее описание в качестве ссылки. Любая несогласованность между вышеуказанными патентами и публикациями и раскрытием существа настоящего изобретения должна быть разрешена в пользу существа настоящего изобретения.
В первом аспекте изобретение относится к пептидиларгиналю, имеющему формулу (I)
где Xaa представляет остаток альфа-замещенной угольной кислоты формулы (II)
где Q представляет С1-3 алкилоксикарбониламиногруппу, метиламиногруппу или гидроксильную группу, и R представляет С7-9 циклоалкилметильную группу, 1-адамантилметильную группу или С5-7 циклоалкильную группу и Xbb представляет остаток L-пролина или L-азетидинил-2-карбоновой кислоты, и его аддитивным солям кислоты, образованным с органической или неорганической кислотой.
Особенно предпочтительный вариант воплощения согласно вышеуказанному аспекту изобретения соответствует структуре 1 (этоксикарбонил-D-циклогептилаланил-L-пролил-L-аргининальдегид, Eoc-D-cHpa-Pro-Arg-H):
Другой особенно предпочтительный вариант осуществления согласно вышеуказанному аспекту изобретения соответствует структуре 2 (N-метил-D-циклогептилаланил-L-пролил-L-аргининальдегида, N-Me-D-cHpa-Pro-Arg-H):
Следующий особенно предпочтительный вариант осуществления согласно вышеуказанному аспекту изобретения соответствует структуре 3 (D-циклогептиллактил-L-пролил-L-аргининальдегида, D-Hpl-Pro-Arg-H):
Очередной особенно предпочтительный вариант осуществления согласно вышеуказанному аспекту изобретения соответствует структуре 4 (N-метил-D-циклогексилглицил-L-азетидин-2-карбонил-L-аргининальдегида, N-Me-D-Chg-Aze-Arg-H):
Соединения согласно формулам 1, 2, 3 и 4 получают, например, конденсацией кислотного компонента с двумя кислотными остатками, которые N- или O-защищены, с L-аргининлактамом, защищенным по гуанидиногруппе бензилоксикарбонильной группой, и восстановлением полученного трипептидлактама до защищенного трипептидальдегида, удалением защитной группы с гуанидиногруппы аргинина, а в случае Q = метиламино или гидроксил в формуле (II), также с концевой метиламино- или гидроксильной группы, и выделением пептидного производного формулы (I) в виде его аддитивной соли, образованной с органической или неорганической кислотой.
Соединения, представленные формулой (I), получают и используют в форме аддитивных солей кислоты вследствие более высокой стабильности солевых форм. В аддитивных солях кислоты соединения формулы (I) активность, присущая основанию и кислоте, имеет меньшее значение, хотя для терапевтических целей предпочтительно использование фармацевтически приемлемых аддитивных солей кислоты. Примеры вышеупомянутых подходящих кислот включают (a) неорганические кислоты: хлористоводородную, бромистоводородную, фосфорную, метафосфорную и серную кислоты, (b) органические кислоты: винную, уксусную, лимонную, яблочную, молочную, фумаровую, бензойную, гликолевую, глюконовую, янтарную, памоевую и арилсульфоновые кислоты, например п-толуолсульфоновую кислоту. Предпочтительной аддитивной солью кислоты является сульфат, особенно гемисульфатная соль.
Аддитивные соли кислоты получают обычным способом, например нейтрализацией формы свободного основания соединения формулы (I) кислотой.
Кислотный компонент с двумя остатками может быть представлен как D-Xaa-Xbb, где Xaa представляет остаток α-замещенной карбоновой кислоты формулы Q-CH(R)-CO, где Q означает С1-3 алкоксикарбониламино группу, R означает группу, определенную выше, и Xbb представляет остаток L-пролина или L-азетидин-2-карбоновой кислоты. Когда Xaa, α-замещенная алкилкислота, представляет α-метиламино или α-гидроксикислоту, т.е. Q представляет метиламино- или гидроксильную группу, кислотный компонент с двумя остатками может быть представлен как P-D-Xaa-Xbb, где P представляет N-защитную группу, такую как бензилоксикарбонил (Z) или трет-бутоксикарбонил (Boc) группу, или O-защитную группу, предпочтительно тетрагидропиранильную (THP) группу.
Ацилдипептид, используемый в качестве исходного вещества для соединений, содержащих остаток α-амино или α-метиламинокислоты, получают ацилированием α-аминокислоты соответствующим сложным эфиром хлормуравьиной кислоты, получая С1-3 алкоксикарбониламинокислоту и бензилоксикарбониламинокислоту, которые затем связывают с L-пролином или L-азетидин-2-карбоновой кислотой, получая D-Xaa-Xbb и Z-аминоацил Xbb, который N-метилируют с получением требуемого P-D-Xaa-Xbb.
D-Xaa, необходимый для связывания с Xbb, преимущественно можно получить ацилированием рацемического соединения DL-Xaa, превращением DL-ацетиламинокислоты в ее метиловый сложный эфир и ферментативным разделением рацемического сложного эфира ацетил-DL-Xaa-OMe. Затем полученный таким образом ацетил-D-Xaa-OMe омыляют и деацетилируют, затем превращают в необходимую N-защищенную D-аминокислоту.
Необходимую D-α-гидроксикислоту преимущественно можно получить из соответствующей D-α-аминокислоты. Затем ее превращают в соответствующую O-защищенную форму и связывают с Xbb, получая необходимую P-D-Xaa-Xbb.
Во втором аспекте изобретение относится к фармацевтической композиции, содержащей пептидиларгиналь согласно первому аспекту изобретения и фармацевтически приемлемый носитель, наполнитель или разбавитель.
Фармацевтические композиции включают эффективное количество соединения общей формулы (I) или его фармацевтически приемлемой соли и известные фармацевтически приемлемые носители, наполнители, разбавители и/или другие фармацевтические наполнители.
Вышеупомянутые носители, разбавители или наполнители могут представлять воду, спирты, желатин, лактозу, сахарозу, крахмал, пектин, стеарат магния, стеариновую кислоту, тальк, различные масла животного или растительного происхождения, кроме того, гликоли, например, пропиленгликоль или полиэтиленгликоль.
Фармацевтические наполнители могут представлять консерванты, различные натуральные или синтетические эмульгаторы, диспергирующие или увлажняющие средства, красители, ароматизирующие средства, буферы, вещества, промотирующие дезинтеграцию, и другие вещества, улучшающие биодоступность активного ингредиента.
Фармацевтические композиции по изобретению можно получить в виде обычных составов, таких как оральные композиции (вводимые через рот, такие как таблетки, капсулы, порошки, пилюли, драже или гранулы), а также в виде парентеральных композиций (лекарственные средства, вводимые в организм, но не через желудочно-кишечный тракт (систему), такие как инъекции, вливания, суппозитории, пластыри или мази).
В третьем аспекте изобретение обеспечивает способ лечения пациента, страдающего синдромом диссеминированного внутрисосудистого свертывания, и этот способ включает введение пациенту, страдающему синдромом диссеминированного внутрисосудистого свертывания, пептидиларгиналя, соответствующего формуле Xaa-Xbb-Arg-H (I), где Xaa и Xbb такие, как определены выше, или его фармацевтически приемлемой аддитивной соли кислоты. Пептидиларгинали также называют производными пептидиларгининальдегида.
Согласно вышеприведенному аспекту изобретения, изобретение обеспечивает способ лечения синдрома диссеминированного внутрисосудистого свертывания, и этот способ включает введение пациенту-животному, включая пациента-человека, пептидиларгиналей согласно изобретению. В способе согласно вышеуказанному аспекту изобретения, терапевтически эффективное количество предлагаемого пептидиларгиналя вводят на протяжении терапевтически эффективного периода времени животному, включая человека, у которого имеется диссеминированное внутрисосудистое свертывание. Предпочтительно, такое введение осуществляют при помощи внутривенной или подкожной инъекции, наиболее предпочтительно внутривенное введение. Введение терапевтических композиций можно осуществлять, используя известные способы, при дозах и в течение периодов времени, эффективных для ослабления (снижения) симптомов или маркеров, идентифицирующих заболевание ДВС. При системном введении, терапевтическую композицию предпочтительно вводят при дозе, достаточной для достижения уровня пептидиларгиналей в крови от около 6 мкМ до около 100 мкМ. Предпочтительно, суммарная дозировка может варьироваться от около 0,1 мг до около 50 мг пептидиларгиналя на кг массы тела в день. Возможно введение индивидууму одновременно или последовательно терапевтически эффективного количества одной или нескольких терапевтических композиций изобретения в виде разового эпизода лечения.
Нижеследующие примеры предназначены для более подробной иллюстрации некоторых особенно предпочтительных вариантов осуществления изобретения и, при этом подразумевается, что они никоим образом не ограничивают объема данного изобретения. Если не оговорено особо, для нижеследующих экспериментов человеческий тромбин (3000 NIH Е/мг), человеческий альбумин и человеческий фибриноген получали от Sigma Aldrich Kft. (Budapest, Hungary), а человеческий фактор Xa (8 мкг/Е) от Enzyme Research Laboratories (Swansea, UK). Реагент APTT доступен от REANAL (Budapest Hungary) и реагент PT, Симпластин D, приобретали у ORGANON, TEKNIKA (Eppelheim, Germany).
Аббревиатуры аминокислот, пептидов, заместителей и реагентов используют в соответствии с правилами, соответствующими номенклатуре IUPAC-IUB. Вышеупомянутые аббревиатуры, встречающиеся в данной заявке, выглядят следующим образом. Arg = L-аргинин, Boc = трет-бутоксикарбонил, Bzl = бензил, Chg = L-циклогексилглицин, DCHA = дициклогексиламин, DHP = дигидропиран, Eoc = этоксикарбонил, Gly = глицин, Me = метил, MePhe = N-метил-L-фенилаланин, Moc = метоксикарбонил, Pro = L-пролин, pNA = п-нитроанилино, ТФК (TFA)= трифторуксусная кислота, THP = тетрагидропиранил, Tos = п-толуолсульфонил, Z = бензилоксикарбонил, КТ (RT) = комнатная температура. Аббревиатуры необычных кислот, используемых в настоящей заявке, представляют Ada = адамантил-L-аланин, Aze = L-азетидин-2-карбоновая кислота, N-Me-D-cHpa = N-метил-D-циклогептилаланин, D-cHpa = D-циклогептилаланин или (R)-2-амино-3-циклогептилпропионовая кислота, D-Hla - D-циклогептилмолочная кислота или (R)-2-гидрокси-3-циклогептилпропионовая кислота.
Значения Rf, приводимые в примерах, определяли тонкослойной хроматографией, используя силикагель в качестве адсорбента (DC-Alufolien Kieselgel 60 F254, Merck, Darmstadt), в нижеследующих проявляющих системах. Номера используемых систем приводятся в скобках после аббревиатуры Rf.
1 Этилацетат
2 Этилацетат - н-гексан (1:4)
3 Этилацетат - н-гексан (1:1)
4 Этилацетат - циклогексан (15:85)
5 Хлороформ - ацетон (95:5)
6 Этилацетат-пиридин-уксусная кислота-вода (960:20:6:11)
7 Этилацетат-пиридин-уксусная кислота-вода (480:20:6:11)
8 Этилацетат-пиридин-уксусная кислота-вода (240:20:6:11)
9 Этилацетат-пиридин-уксусная кислота-вода (120:20:6:11)
10 Этилацетат-пиридин-уксусная кислота-вода (90:20:6:11)
11 Этилацетат-пиридин-уксусная кислота-вода (60:20:6:11)
12 Этилацетат-пиридин-уксусная кислота-вода (45:20:6:11)
13 Этилацетат-пиридин-уксусная кислота-вода (30:20:6:11)
Коэффициенты емкости (k'), указанные в примерах, определяли на установке "Pharmacia LKB Analytical HPLC System Two" следущим образом:
Колонка: LiChrospher ОФ-18: 12 мкм 240x4 мм
Температура колонки: температура окружающей среды
Элюенты: Растворитель A, 0,1% ТФК/вода, Растворитель B, 0,1% ТФК/ацетонитрил
Профиль градиента: 0 → 15 мин. 30 → 60% B, затем изократический режим (с постоянным составом элюента) 60% B.
Скорость потока растворителя: 1 мл/мин.
Детектор: УФ-детектор LKB 2141, длина волны: 214 нм.
Инжектор: Rheodyne 7125. Петля для пробы: 100 мкл.
Насосы: тип 2 LKB 2148. Система контроля: LKB HPLC Manager.
Концентрация пробы: 1 мг/мл в Растворителе A, вводимый объем 25 мкл.
Время анализа 40 мин.
Ациларгининальдегиды присутствуют в равновесных структурах, т.е. в формах альдегида, гидрата альдегида и двух формах аминоциклола. Во время анализа ВЭЖХ (HPLC) гидрат альдегида и одна или две формы аминоциклола проявляются в виде двух или трех отдельных пиков. Ациларгининальдегиды, описанные в примерах, определяются двумя или тремя значениями k'.
Масс-спектрометрия. Измерения с использованием ионизации ББА (FAB) с регистрацией положительных ионов осуществляют на приборе Finnigan MAT 8430. Образцы растворяли в матрице - м-нитробензиловом спирте (NBA) и пробы вводили непосредственно в ионный источник. В спектре пептидил-аргининальдегидов был детектирован дополнительный молекулярный ион, который представлял аддукт соединения, образованный сNBA: [M+H]+ и [M+H+NBA]+. В примерах приведены соответственно данные спектров ББА (FAB). Измерения с использованием электрораспылительной ионизации с регистрацией положительных ионов осуществляют на установке VG Quattro (Fisons). Образцы растворяли в смеси ацетонитрил-вода (1:1), содержащей 1% (об/об) муравьиной кислоты, и пробы вводили при помощи 10-мл петли для пробы в ионный источник при скорости потока 15-25 мл/мин.
Пример 1
Синтез этоксикарбонил-D-циклогептилаланил-L-пролил-L-аргининальдегида гемисульфата
Стадия 1: Этоксикарбонил-D-циклогептилаланил-L-пролил-NG-бензилоксикарбонил-L-аргининлактам
7,85 г (20,1 мМ) трет-бутилоксикарбонил-NG-бензилоксикарбонил-L-аргининлактама [(Bajusz et al, J. Med. Chem. 33, 1729 (1990)] суспендируют в 20 мл хлороформа, затем при перемешивании и охлаждении льдом добавляют 20 мл этилацетата, насыщенного газом-HCl (0,11-0,15 г/мл). Отщепление Boc-группы (бензилоксикарбонильной) контролируют тонкослойной хроматографией [Rf(11)=0,5 (свободное соединение); 1,0 (Boc-соединение)]. По окончании реакции суспензию разбавляют 40 мл простого диэтилового эфира, образовавшуюся массу кристаллов отфильтровывают, промывают 10 мл ацетона и 10 мл диэтилового эфира и сушат при пониженном давлении над KOH. Полученный NG-бензилоксикарбонил-L-аргининлактам гидрохлорид растворяют в 20 мл диметилформамида, охлаждают до -20°C и добавляют к следующему смешанному ангидриду.
7,12 г (20,1 мМ) этоксикарбонил-D-циклогептилаланил-L-пролина (Пример 1, Стадия J) растворяют в 20 мл диметилформамида, охлаждают до -15°С, затем при перемешивании добавляют 2,23 мл (20,1 мМ) N-метилморфолина и 2,65 мл (20,1 мМ) изобутилхлорформиата. После перемешивания в течение 10 минут, добавляют вышеупомянутый раствор NG-бензилоксикарбонил-L-аргининлактама в диметилформамиде, затем триэтиламин в количестве, достаточном для доведения рН реакционной смеси до 8 (требуется около 2,8 мл). Реакционную смесь перемешивают при -10°C в течение 30 минут, затем при 0°C в течение одного часа. После этого соли отфильтровывают и фильтрат разбавляют 100 мл этилацетата. Полученный раствор промывают 3x25 мл воды, 10 мл 1M KHSO4 и 3x10 мл воды, сушат над безводным Na2SO4 и упаривают при 2,0-2,5 кПа (kPa). Полученный продукт подвергают колоночной хроматографии на силикагеле, используя 200 г Kieselgel 60 (0,040-0,063 мм) в качестве адсорбента и этилацетат в качестве элюента. Фракции, содержащие исключительно чистый продукт [(Rf(1)=0,60], объединяют и выпаривают при 2,0-2,5 кПа. Остаток после выпаривания перекристаллизовывают из диизопропилового эфира.
Выход 10,84 г (86,1%), Rf(1)=0,55-0,65.
Масс-спектр (МС-ББА) (627 [M+H]+) подтвердила предполагаемую структуру.
Стадия 2: Этоксикарбонил-D-циклогептилаланил-L-пролил-NG-бензилоксикарбонил-L-аргининальдегид
8,02 г (12,8 мМ) этоксикарбонил-D-циклогептилаланил-L-пролил-NG-бензилоксикарбонил-L-аргининлактама (Пример 1, Стадия 1) растворяют в 15 мл тетрагидрофурана и затем при перемешивании и при температуре, не превышающей -50°C, добавляют раствор 3,6 мМ LiAlH4, растворенного в тетрагидрофуране. Протекание (реакции) восстановления контролируют тонкослойной хроматографией, используя (растворитель 7) в качестве проявляющего растворителя, и, если требуется, добавляют дополнительную порцию LiAlH4. К этой реакционной смеси добавляют по каплям 0,5M KHSO4 при постоянном перемешивании и охлаждении до тех пор, пока pH реакционной смеси не достигнет 3, затем добавляют 35 мл воды. Полученный раствор экстрагируют 2x15 мл гексана, затем 3x20 мл дихлорметана. Экстракты дихлорметана объединяют, промывают 3x15 мл воды, 15 мл холодного 5% раствора гидрокарбоната натрия и снова 15 мл воды, сушат над безводным Na2SO4 и выпаривают при 2,0-2,5 кПа. Остаток после выпаривания обрабатывают диизопропиловым эфиром, фильтруют и сушат при пониженном давлении.
Выход 7,08 г (88%), Rf(8)=0,40-0,50.
Масс-спектр (МС-ББА) (629 [M+H]+, 782 [M+H+NBA]+) подтверждает предполагаемую структуру.
Стадия 3: Этоксикарбонил-D-циклогептилаланил-L-пролил-L-аргининальдегид гемисульфат
6,91 г (11,0 мМ) этоксикарбонил-D-циклогептилаланил-L-пролил-NG-бензилоксикарбонил-L-аргининальдегида (Пример 1, Стадия 2) растворяют в 85 мл этанола и 11,25 мл 0,5M серной кислоты, затем добавляют 0,7 г катализатора Pd-C, суспендированного в 14 мл воды, и смесь гидрируют при около 10°C. Протекание реакции контролируют тонкослойной хроматографией. После завершения реакции (около 15 минут), катализатор отфильтровывают и фильтрат концентрируют до приблизительно 7-9 мл при 2,0-2,5 кПа. Остаток разбавляют 80 мл воды, экстрагируют 4x15 мл дихлорметана и водный раствор выдерживают при 20-22°C в течение 24 часов. Раствор снова экстрагируют 3x15 мл дихлорметана и доводят рН до 3,5 при помощи ионообменной смолы (ионита) Dowex AG 1-X8 (HO), затем раствор подвергают лиофильной сушке (сушке вымораживанием).
Выход 4,90 г (82%). Rf(11)=0,35-0,45. [α]D 20=-77,6° (c=1,018; вода).
ВЭЖХ: k'=1,695 и 2,328.
Масс-спектр (МС-ББА) (495 [M+H]+, 648 [M+H+NBA]+) подтверждает предполагаемую структуру.
Синтез исходных веществ:
Этоксикарбонил-D-циклогептилаланил-L-пролин
Стадия A: 1-циклогептилацетил-2,5-диметилпиразол
58,6 г (375 мМ) циклогептилуксусной кислоты [Protiva et al, Collect. Czech., Chem., Commun., 55: 1278-1289 (1990)] растворяют в 375 мл тетрагидрофурана, охлаждают до -15°С, затем при перемешивании добавляют 41,3 мл (375 мМ) N-метилморфолина, 49,50 мл (375 мМ) изобутилхлорформиата, и, после перемешивания в течение 10 минут при -10°С, добавляют раствор 37,85 г (393,75 мМ) 3,5-диметилпиразола в 300 мл тетрагидрофурана при -10°C. Перемешивание продолжают при -10°C в течение 30 мин, при 0°C в течение часа и при комнатной температуре в течение 3 часов. После чего соли отфильтровывают, фильтрат выпаривают при пониженном давлении и остаток растворяют в 900 мл этилацетата. Полученный раствор последовательно промывают 3x80 мл 1M NaOH, 89 мл воды, 3x80 мл 1M HCl и водой до нейтральности. (Основные промывные воды объединяют и подкисляют, регенерируя ˜5,8 г, 37,1 мМ циклогептилуксусной кислоты.) Этилацетатный раствор сушат над безводным Na2SO4, затем упаривают при 2,0-2,5 кПа. Получают маслянистый продукт в виде 340 мМ 1-циклогептилацетил-2,5-диметилпиразола, и его используют на следующей стадии без дополнительной очистки. Rf(2)=0,8-0,9 (кислота: 0,3-0,4).
Стадия B: 1-циклогептилацетальдегид
К 350 мл тетрагидрофурана, охлажденного до -30°C, добавляют 12,83 г (338 мМ) LiAlH4. После этого раствор маслянистого продукта (пример 1, Стадия A) в 250 мл холодного тетрагидрофурана вводят по каплям при перемешивании при -25°C. Ход реакции контролируют ТСХ. Если требуется, добавляют 30-40 мл раствора LiAlH4 в тетрагидрофуране (3 г/100 мл). По завершении восстановления холодную смесь подкисляют 6M HCl и разбавляют этилацетатом (500 мл). Водную фазу экстрагируют этилацетатом и органические растворы объединяют, промывают водой до нейтральности, сушат над безводным Na2SO4 и затем выпаривают при 2,0-2,5 кПа. Полученный маслянистый продукт представляет неочищенный 1-циклогептилацетальдегид, загрязненный 2,5-диметилпиразолом (ДМР, DMP). Rf(2)=0,7-0,8 (ДМП: 0,25-0,35).
К полученному маслянистому продукту (62,6 г), растворенному в 350 мл метанола, добавляют при перемешивании раствор 36,4 г NaHSO3 в 70 мл воды. Реакционную смесь выдерживают в холодильнике на протяжении ночи. Выпавшее в осадок вещество отфильтровывают, промывают холодной смесью 35 мл метанола и 7 мл воды, затем диэтиловым эфиром и сушат. Твердое вещество представляет аддукт бисульфита натрия (73,87 г, 302,38 мМ), Rf(14)=0,55-0,65), который добавляют к смеси в 450 мл метиленхлорида и 450 мл воды, содержащей 47,7 г (450 мМ) Na2CO3. Реакционную смесь перемешивают на протяжении ночи. Две фазы разделяют, органическую фазу промывают метиленхлоридом (2x100 мл). Объединенные органические слои промывают водой, сушат над безводным Na2SO4, затем выпаривают при 2,0-2,5 кПа. Полученный маслянистый продукт (36,97 г, 263,64 мМ) представлял неочищенный 1-циклогептилацетальдегид, который непосредственно используют в следующей реакции. Rf(2)=0,7-0,8.
Стадия С: 5-циклогептилметилгидантоин
К раствору альдегида (Пример 1, Стадия B) в 50% водном этаноле (857 мл; 3,25 мл/мМ) 15,5 г (290 мМ; 1,1 экв.) хлорида аммония, 27,87 г (659 мМ; 2,5 экв.) карбоната аммония и 18,9 г (290 мМ; 1,1 экв.) цианида калия добавляют при 50-55°C при перемешивании, и перемешивание продолжают при 50°C в течение 48 часов. Выпавшее в осадок вещество отфильтровывают, промывают 50% этанолом (100 мл) и сушат в вакуумном эксикаторе. В виде первой порции получают 42,26 г (206,97 мМ) вещества. Из маточного раствора отгоняют этанол, остаток экстрагируют этилацетатом (1x250 мл и 2x100 мл), органические растворы объединяют, промывают водой (3x50 мл), сушат над безводным Na2SO4, затем выпаривают при 2,0-2,5 кПа. Остаток растирают с диизопропиловым эфиром, фильтруют и сушат. В виде второй порции, получают 3,6 г (17,12 мМ) вещества; получают суммарный выход 5-циклогептилметилгидантоина - 45,86 г (218 мМ, 82,5%). Т.пл: 240,9°C. Rf(6)=0,73-0,78.
Анализ для C11H18N2O2 (210,28). Рассчитано: C%=62,83; H%=8,63; N%=13,32. Найдено: C%=63,09; H%=8,67; N%=13,25.
Стадия D: DL-циклогептилаланин гидрохлорид
87 г (1,55 M) KOH растворяют в 435 мл н-бутанола при перемешивании и подогревании и добавляют 45,71 г (217,4 мМ) 5-циклогептилметилгидантоина (Пример 1, Стадия C). Полученный таким образом раствор кипятят с обратным холодильником в течение 72 часов, затем разбавляют водой и упаривают при пониженном давлении. Остаток растворяют в 220 мл воды, подкисляют до pH 2 при помощи концентрированнойHCl (˜130 мл) и выдерживают в холодильнике на протяжении ночи. Выпавшее в осадок вещество отфильтровывают, промывают 50 мл воды и сушат в вакуумном эксикаторе. Полученный таким образом продукт (106,5 г) выделяют в виде 217 мМ DL-циклогептилаланина и его используют для дальнейших реакций. Rf(11)=0,2-0,3.
Стадия E: Гидрохлорид метилового сложного эфира DL-циклогептилаланина
23,65 мл (325,25 мМ) SOCl2 по каплям добавляют в 217 мл метанола при температуре между 0°C и -5°C при перемешивании. После этого добавляют DL-циклогептилаланин (217 мМ из Примера 1, Стадия D) и перемешивают в течение 24 часов. Превращение контролируют ТСХ Rf(11)=0,75-0,85 (сложный эфир), (0,3-0,4 (кислота)). В случае обнаружения непрореагировавшей аминокислоты, реакционную смесь охлаждают до -5°C, в нее по каплям добавляют 11,8 мл SOCl2 и реакционную смесь перемешивают в течение еще 24 часов. После этого нерастворенные соли отфильтровывают, промывают метанолом (2x50 мл) и объединенные метанольные растворы выпаривают. Остаток вновь растворяют в метаноле и снова выпаривают. Наконец, остаток растирают с диизопропиловым эфиром, фильтруют, промывают диизопропиловым эфиром и сушат в вакуумном эксикаторе над KOH и P2O5. Получают 33,68 г (142,85 мМ) гидрохлорида метилового сложного эфира DL-циклогептилаланина. Rf(11)=0,5-0,6. Т.пл. 95,4-96,5°С.
Стадия F: Метиловый эфир ацетил-DL-циклогептилаланина
К раствору 33,68 г (142,85 мМ) гидрохлорида метилового эфира DL-циклогептилаланина (Пример 1, Стадия E) в 140 мл пиридина, по каплям добавляют уксусный ангидрид (16,2 мл, 171,43 мМ) в течение часа при перемешивании и охлаждении на бане со смесью лед-вода. Реакционную смесь перемешивают в течение 24 часов при температуре окружающей среды, затем выпаривают при пониженном давлении. Остаток растворяют в 300 мл этилацетата, промывают 1M KHSO4 (3x50 мл) и водой (3x50 мл), сушат над безводным Na2SO4 и затем выпаривают при 2,0-2,5 кПа. Полученный маслянистый продукт растирают с диизопропиловым эфиром до (получения) твердого вещества, которое отфильтровывают, промывают диизопропиловым эфиром, затем водой и сушат в эксикаторе. Получают 24,36 г (100,94 мМ, 70,4%) метил ацетил-DL-циклогептилаланината (ацетил-DL-сложный эфир). Rf(1)=0,55-0,65. Т.пл.: 69-71°С
Анализ для C13H23NO3 (241,332). Рассчитано: C%=64,70; H%=9,61; N%=5,80. Найдено: C%=64,75; H%=9,76; N%=5,85.
Стадия G: Метиловый эфир ацетил-D-циклогептилаланина (ферментативное разделение метилового эфира ацетил-DL-циклогептилаланина)
К раствору 8,69 г (36 мМ) метилового эфира ацетил-DL-циклогептилаланина, ацетил-DL-сложного эфира, (Пример 1, Стадия F) в 36 мл толуола, добавляют 72 мл воды и 36 мг Subtilisin Carlsberg (Протеаза Типа VIII, Sigma). Ферментативный гидролиз L-энантиомера, ацетил-L-сложного эфира, протекает при pH 7,0, который поддерживают посредством аутотитратора, заполненного 3M NaOH. После прекращения потребления NaOH (до 5,8 мл, 17,4 мМ), реакционную смесь разбавляют 36 мл толуола, и два слоя разделяют. Водную фазу промывают 2x30 мл толуола. Объединенные толуольные растворы содержали ацетил-D-сложный эфир, а объединенные водные растворы содержали натриевую соль ацетил-L-кислоты.
После сушки над безводным Na2SO4 объединенные толуольные растворы выпаривают при пониженном давлении, получая 3,8 г (15,75 мМ) ацетил-D-сложного эфира, Rf(1)=0,55-0,65, который непосредственно используют на следующей стадии.
Объединенные водные растворы подкисляют и экстрагируют 3x30 мл этилацетата. Объединенные этилацетатные растворы промывают водой, сушат над безводным Na2SO4 и выпаривают при пониженном давлении, получая 4,0 г (17,6 мМ) ацетил-L-кислоты. Rf(7)=0,38-0,42.
Подобное получение, исходя из 9,65 г (40 мМ) ацетил-DL-сложного эфира (Пример 1, Стадия F) дает 4,6 г (19,06 мМ) ацетил-D-сложного эфира и 4,44 г (19,55 мМ) ацетил-L-кислоты.
Стадия H: D-циклогептилаланин гидрохлорид
7,24 г (30 мМ) ацетил-D-сложного эфира (Пример 1, Стадия G) суспендируют в 120 мл 6M HCl и кипятят с обратным холодильником в течение 3 часов. Свободную аминокислоту выделяют в виде кристаллов. Реакционную смесь охлаждают, выдерживают в холодильнике на протяжении ночи, фильтруют, промывают холодной водой и простым эфиром, затем сушат в вакуумном эксикаторе. Получают 5,9 г (26,69 мМ, 89%) гидрохлорида D-циклогептилаланина. Rf(12)=0,10-0,15. [α]D 20=-11° (c=0,4; 1M HCl).
Анализ для C10H19NO2·HCl (221,728). Рассчитано: C%=54,17; H%=9,09; N%=6,32; Cl%=15,99. Найдено: C%=54,27; H%=9,27; N%=6,30; Cl%=16,2.
Стадия I: Этоксикарбонил-D-циклогептилаланин
К раствору 4,43 г (20 мМ) гидрохлорида D-циклогептилаланина (Пример 1, Стадия H) в 20 мл диметилформамида добавляют 5,6 мл (40 мМ) триэтиламина и 3,95 г (21 мМ) (N-гидроксисукцинимидил)этилкарбоната.* После перемешивания при комнатной температуре в течение 3 часов, реакционную смесь выпаривают, остаток растворяют в 40 мл этилацетата и промывают 2x30 мл 1M KHSO4 и водой до нейтральности. После этого органический слой сушат над безводным Na2SO4, затем выпаривают при 2,0-2,5 кПа. Полученный маслянистый продукт (4,47 г, ˜17 мМ) представлял этоксикарбонил-D-циклогептилаланин [Rf(7)=0,85-0,90], который непосредственно используют на следующей стадии. [α]D 20=+6,3° (c=1, метанол).
*Получение (N-гидроксисукцинимидил)этилкарбоната
11,5 г (100 мМ) N-гидроксисукцинимида растворяют в 100 мл тетрагидрофурана, охлаждают до -10°С, затем при перемешивании добавляют 15,4 мл (110 мМ) триэтиламина и 10,45 мл (110 мМ) этилхлоркарбоната. После перемешивания в течение 2 часов при комнатной температуре смесь фильтруют и фильтрат выпаривают при пониженном давлении. Маслянистый остаток кристаллизуется при охлаждении. Кристаллическое вещество суспендируют в низкокипящем петролейном эфире, фильтруют и сушат в вакуумном эксикаторе. Выход 12,78 г (68,3%). Т.пл.: 39,4-39,7°С.
Анализ для C7H9NO5 (187,15). Рассчитано: С, 44,92; H, 4,85; N, 7,48. Найдено: C%=44,67; H%=4,81; N%=7,27.
Стадия J: Этоксикарбонил-D-циклогептилаланил-L-пролин
Раствор этоксикарбонил-D-циклогептилаланина (˜17 мМ, Пример 1, Стадия I) в 17 мл ТГФ (THF) объединяют с 374 г (20 мМ) N-гидроксисукцинимида, охлаждают до 10°С и объединяют с раствором 4,12 г (20 моль) 1,3-дициклогексилкарбодиимида в приблизительно 20 мл ТГФ. Смесь перемешивают в течение приблизительно 5 ч при 22°C, после чего полноту образования активного сложного эфира оценивали по данным ТСХ.
L-Пролин (1,95 г, 17 мМ) добавляют к перемешиваемой реакционной смеси с последующим добавлением 2,3 мл (17 мМ) триэтиламина. Реакционную смесь перемешивают при 22°C в течение приблизительно 15 ч, после чего определяют полноту расхода активного сложного эфира методом ТСХ. Реакционную смесь фильтруют, отфильтрованный осадок промывают 10 мг ТГФ и фильтрат выпаривают. Остаток растворяют в 30 мл этилацетата и 30 мл воды. Водную фазу промывают 2x20 мл этилацетата, подкисляют 20 мл 1M KHSO4 и экстрагируют 3x20 мл этилацетата. Объединенные этилацетатные растворы промывают водой до нейтральности, сушат над безводным Na2SO4, затем выпаривают при 2,0-2,5 кПа. После растирания диизопропиловым эфиром, остаток кристаллизуется. Эту суспензию кристаллов охлаждают в холодильнике, фильтруют петролейным эфиром (пределы выкипания которого 40-60°С) и сушат в вакуумном эксикаторе. Получают 4,2 г (11,85 мМ, 70%) этоксикарбонил-D-циклогептилаланил-L-пролина. Rf(7)=0,45-0,55.
Анализ для C18H30N2O4 (354,45). Рассчитано: С, 60,99; H, 8,53; N, 7,90. Найдено: C%=60,14; H%=8,55; N%=7,38.
Масс-спектр (МС-ББА) (355 [M+H]+) подтверждает предполагаемую структуру.
Пример 2
Синтез N-метил-D-циклогептилаланил-L-пролил-L-аргининальдегид сульфата
Стадия 1: Бензилоксикарбонил-N-метил-D-циклогептилаланил-L-пролил-NG-бензилоксикарбонил-L-аргининлактам
3,93 г (10 мМ) трет-бутилоксикарбонил-NG-бензилоксикарбонил-L-аргининлактама [(Bajusz et al, J. Med. Chem. 33, 1729 (1990)] суспендируют в 10 мл хлороформа, затем добавляют при перемешивании и охлаждении льдом 10 мл этилацетата, насыщенного газом-HCl (0,11-0,15 г/мл). Отщепление Boc-группы контролируют тонкослойной хроматографией [Rf(11)=0,5 (свободное соединение); 1,0 (Boc-соединение)]. К концу реакции суспензию разбавляют 20 мл диэтилового эфира, образовавшуюся массу кристаллов отфильтровывают, промывают 5 мл ацетона и 5 мл диэтилового эфира и сушат при пониженном давлении над KOH. Полученный NG-бензилоксикарбонил-L-аргининлактам гидрохлорид растворяют в 10 мл диметилформамида, охлаждают до -20°C и добавляют к следующему смешанному ангидриду.
4,32 г (10 мМ) бензилоксикарбонил-N-метил-D-циклогептилаланил-L-пролина (Пример 2, Стадия С) растворяют в 10 мл диметилформамида, охлаждают до -15°С, затем при перемешивании добавляют 1,12 мл (10,1 мМ) N-метилморфолина и 1,33 мл (10,1 мМ) изобутилхлорформиата. После перемешивания в течение 10 минут добавляют вышеупомянутый раствор NG-бензилоксикарбонил-L-аргининлактама в диметилформамиде, затем триэтиламин в количестве, достаточном для доведения рН реакционной смеси до 8 (требуется около 1,4 мл). Реакционную смесь перемешивают при -10°C в течение 30 минут, затем при 0°C в течение одного часа. После этого соли отфильтровывают и фильтрат разбавляют 100 мл этилацетата. Полученный раствор промывают 3x15 мл воды, 6 мл 1M KHSO4 и 3x6 мл воды, сушат над безводным Na2SO4 и выпаривают при 2,0-2,5 кПа. Полученный продукт подвергают колоночной хроматографии на силикагеле, используя 100 г Kieselgel 60 (0,040-0,063 мм) в качестве адсорбента и этилацетат в качестве элюента. Фракции, содержащие исключительно чистый продукт [(Rf(1)=0,70], объединяют и выпаривают при 2,0-2,5 кПа. Остаток после выпаривания перекристаллизовывают из диизопропилового эфира.
Выход 6,0 г (85%), Rf(1)=0,65-0,75.
Масс-спектр (МС-ББА) (703 [M+H]*) подтверждает предполагаемую структуру.
Стадия 2: Бензилоксикарбонил-N-метил-D-циклогептилаланил-L-пролил-NG-бензилоксикарбонил-L-аргининальдегид
5,62 г (8 мМ) бензилоксикарбонил-N-метил-D-циклогептилаланил-L-пролил-NG-бензилоксикарбонил-L-аргининлактама (Пример 2, Стадия 1) растворяют в 10 мл тетрагидрофурана и затем при перемешивании и при температуре, не превышающей -50°C, добавляют раствор 2,25 мМ LiAlH4 в тетрагидрофуране. Ход реакции восстановления контролируют тонкослойной хроматографией, используя (растворитель 7) в качестве проявляющего растворителя и, если требуется, добавляют дополнительную порцию LiAlH4. К этой реакционной смеси по каплям добавляют 0,5M KHSO4 при постоянном перемешивании и охлаждении до тех пор, пока рН не достигнет 3, затем добавляют 25 мл воды. Полученный раствор экстрагируют 2x10 мл гексана, затем 3x15 мл дихлорметана. Дихлорметановые экстракты объединяют, промывают 3x15 мл воды, 15 мл холодного 5% раствора NaHCO3 и снова 15 мл воды, сушат над безводным Na2SO4 и выпаривают при 2,0-2,5 кПа. Остаток после выпаривания обрабатывают диизопропиловым эфиром, фильтруют и сушат при пониженном давлении.
Выход 4,95 г (88%), Rf(1)=0,20-0,25.
Масс-спектр (МС-ББА) (705 [M+H]+, 858 [M+H+NBA]+) подтверждает предполагаемую структуру.
Стадия 3: N-метил-D-циклогептилаланил-L-пролил-L-аргининальдегид сульфат
4,6 г (6,5 мМ) бензилоксикарбонил-N-метил-D-циклогептилаланил-L-пролил-NG-бензилоксикарбонил-L-аргининальдегида (Пример 2, Стадия 2) растворяют в 65 мл этанола и 13,5 мл 0,5M серной кислоты, затем добавляют 0,4 г катализатора Pd-C, суспендированного в 10 мл воды, и смесь гидрируют при приблизительно 10°С. Ход реакции контролируют тонкослойной хроматографией. После завершения реакции (около 15 минут), катализатор отфильтровывают и фильтрат концентрируют до приблизительно 5-7 мл при 2,0-2,5 кПа. Остаток разбавляют 50 мл воды, экстрагируют 4x10 мл дихлорметана и водный раствор оставляют стоять при 20-22°C в течение 24 часов. Раствор снова экстрагируют 3x10 мл дихлорметана и доводят рН до 3,5 при помощи ионообменной смолы Dowex AG 1-X8 (HO), затем раствор сушат вымораживанием (лиофильная сушка).
Выход 2,85 г (82%). Rf(9)=0,35-0,45. [α]D 20=-79,6° (c=1; вода).
Масс-спектр (МС-ББА) (437 [M+Н]+, 590 [M+H+NBA]+) подтверждает предполагаемую структуру.
Синтез исходных веществ:
Бензилоксикарбонил-N-метил-D-циклогептилаланил-L-пролин
Стадия A: Синтез N-бензилоксикарбонил-D-циклогептилаланин
D-циклогептилаланин гидрохлорид (Пример 1, Стадия H) (11,09 г, 50 мМ) объединяют с 40 мл ТГФ и 40 мл воды при 0°С. Перемешиваемую смесь доводят до рН около 10, добавляя 5M раствор NaOH. К реакционной смеси добавляют бензилхлорформиат (8,22 мл, 55,4 мМ), поддерживая температуру около 3°C и приблизительно рН 10 путем добавления 5M NaOH, если требуется. По завершении добавления бензилхлорформиата, реакционную смесь перемешивают в течение 1 ч при 0°C и поддерживают рН около 10. Перемешивание продолжают на протяжении ночи при КТ и завершение реакции контролируют ТСХ (7). Если требуется, к реакционной смеси добавляют дополнительное количество бензилхлорформиата (1-2x0,75 мл, 5 мМ), поддерживая температуру около 3°C и приблизительно pH 10 путем добавления 5M NaOH. Добавляют т-бутилметиловый эфир (25 мл) и перемешиваемую смесь нагревают до 22°С. Водную фазу отделяют и промывают второй порцией т-бутилметилового эфира (25 мл). Содержание органической фазы контролируют ТСХ и, если это требуется, органическую фазу обратно экстрагируют 25 мл воды. Водные фазы и 25 мл этилацетата объединяют и доводят до рН 2 концентрированной HCl. Фазы разделяют и водную фазу экстрагируют второй порцией этилацетата (25 мл). Объединенные органические фазы промывают 20 мл 1M KHSO4 и 2x30 мл воды, сушат над безводным Na2SO4 и выпаривают при 2,0-2,5 кПа. Остаток после выпаривания растворяют в 45 мл ТГФ и этот раствор бензилоксикарбонил-D-циклогептилаланина сохраняют для использования на следующей стадии без дополнительной очистки.
Стадия B: Бензилоксикарбонил-D-циклогептилаланил-L-пролин
ТФК раствор бензилоксикарбонил-D-циклогептилаланина, полученного в Примере 2, Стадия A, объединяют с 5,9 г (51,24 мМ) N-гидроксисукцинимида, охлаждают до 10°С и объединяют с раствором 11 г (53,3 мМ) 1,3-дициклогексилкарбодиимида в приблизительно 25 мл ТГФ. Смесь перемешивают в течение приблизительно 4,5 ч при 22°C, после чего завершение образования активного сложного эфира оценивают ТСХ.
L-Пролин (5,9 г, 51,24 мМ) добавляют к перемешиваемой реакционной смеси с последующим добавлением 7,2 мл (51,24 мМ) триэтиламина. Реакционную смесь перемешивают при 22°C в течение приблизительно 15 ч, после чего полный расход активного сложного эфира определяют методом ТСХ. Реакционную смесь фильтруют, отфильтрованный осадок промывают 25 мл ТГФ и фильтрат выпаривают. Остаток растворяют в 50 мл этилацетата и 50 мл воды. Водную фазу промывают 2x20 мл этилацетата, подкисляют 20 мл 1M KHSO4 и экстрагируют 3x40 мл этилацетата. Объединенные этилацетатные растворы промывают до нейтральности, сушат над безводным Na2SO4, затем выпаривают при 2,0-2,5 кПа. Остаток, бензилоксикарбонил-D-циклогептилаланил-L-пролин, растворяют в 60 мл ТГФ и используют на следующей стадии без дополнительной очистки.
Стадия C: Бензилоксикарбонил-N-метил-D-циклогептилаланил-L-пролин
Иодометан (17,95 мл, 288 мМ) добавляют к раствору бензилоксикарбонил-D-циклогептилаланил-L-пролина в ТГФ из Примера 2, Стадия B. Этот раствор охлаждают до 8°C и переносят вместе с 20-мл ТГФ промывкой в перемешиваемую суспензию 4,75 г (119 мМ) гидрида натрия, 60%, в 35 мл THF, поддерживая температуру ниже 13°С. Реакционную смесь перемешивают при 11°C в течение 24 ч. Избыточный гидрид натрия разлагают осторожным добавлением в реакционную смесь 1,6 мл воды, поддерживая температуру ниже 13°C и регулируя пенообразование. Погашенную реакционную смесь перемешивают в течение приблизительно 20 мин при 22°C и затем концентрируют до приблизительно 40 мл при пониженном давлении при температуре ниже 30°С. К остатку добавляют воду (70 мл) с последующим добавлением 30 мл т-бутилметилового эфира. Фазы разделяют и водную фазу снова промывают 30 мл т-бутилметилового эфира. Водную фазу продукта объединяют с 40 мл этилацетата и доводят до рН 2,2 3M раствором серной кислоты. Фазы разделяют и водную фазу снова экстрагируют 40 мл этилацетата. Объединенную органическую фазу промывают 70 мл 5% раствора тиосульфата натрия. Фазы разделяют и органическую фазу концентрируют до небольшого объема при пониженном давлении (33-45 кПа), поддерживая температуру ниже 50°С. Остаток объединяют с 18,6 мл ТГФ и 100 мл воды и доводят до рН 8,5 циклогексиламином. Полученную суспензию концентрируют до 100 мл при пониженном давлении (9-33 кПа) при температуре от 25 до 55°С, доводят до 25°С, разбавляют 71,5 мл воды и перемешивают в течение приблизительно 10,5 ч. Суспензию фильтруют, промывают водой и подвергают воздушной сушке при 45°C, получая 16,72 г циклогексиламинную соль бензилоксикарбонил-N-метил-D-циклогептилаланил-L-пролина (выход 63%, исходя из D-циклогептилаланина). Rf(8)=0,55-0,65.
Пример 3
Синтез D-циклогептиллактил-L-пролил-L-аргининальдегид гемисульфата
Стадия 1: Тетрагидропиранил-D-циклогептиллактил-L-пролил-NG-бензилоксикарбонил-L-аргининлактам
5,08 г (13 мМ) трет-бутилоксикарбонил-NG-бензилоксикарбонил-L-аргининлактама [(Bajusz et al, J. Med. Chem. 33, 1729 (1990)] суспендируют в 13 мл хлороформа, затем добавляют при перемешивании и охлаждении льдом 13 мл этилацетата, насыщенного газом-HCl (0,11-0,15 г/мл). Отщепление Boc-группы контролируют тонкослойной хроматографией [Rf(11)=0,5 (свободное соединение); 1,0 (Boc-соединение)]. К концу реакции суспензию разбавляют 25 мл диэтилового эфира, образовавшуюся массу кристаллов отфильтровывают, промывают 7 мл ацетона и 7 мл диэтилового эфира и сушат при пониженном давлении над гидроксидом калия. Полученный NG-бензилоксикарбонил-L-аргининлактам гидрохлорид растворяют в 13 мл диметилформамида, охлаждают до -20°C и добавляют к нижеследующему смешанному ангидриду.
Раствор соли триэтиламмония тетрагидропиранил-D-циклогептил-лактил-L-пролина, полученной в Стадии I Примера 3 (12 мМ), охлаждают до -20°C, затем при перемешивании добавляют 1,6 мл (12 мМ) изобутилхлорформиата. После перемешивания в течение 10 минут добавляют вышеупомянутый раствор NG-бензилоксикарбонил-L-аргининлактама в диметилформамиде, затем триэтиламин в количестве, достаточном для доведения рН реакционной смеси до 8 (около 1,8 мл требуется). Реакционную смесь перемешивают при -10°C в течение 30 минут, затем при 0°C в течение одного часа. После этого соли отфильтровывают и фильтрат разбавляют 65 мл этилацетата. Полученный раствор промывают 3x25 мл воды, 7 мл 1M (раствора) гидросульфата калия и 3x7 мл воды, сушат над безводным Na2SO4 и выпаривают при 2,0-2,5 кПа. Полученный продукт подвергают колоночной хроматографии на силикагеле, используя 130 г Kieselgel 60 (0,040-0,063 мм) в качестве адсорбента и этилацетат в качестве элюента. Фракции, содержащие исключительно чистый продукт [(Rf(1)=0,60], объединяют и выпаривают при 2,0-2,5 кПа. Остаток после выпаривания перекристаллизовывают из диизопропилового эфира.
Выход 5,0 г (7,8 мМ, 64%), Rf(1)=0,6.
Масс-спектр (МС-ББА) (640 [M+H+]) подтверждает предполагаемую структуру.
Стадия 2: Тетрагидропиранил-D-циклогептиллактил-L-пролил-NG-бензилоксикарбонил-L-аргининальдегид
4,8 г (7,51 мМ) тетрагидропиранил-D-циклогептиллактил-L-пролил-NG-бензилоксикарбонил-L-аргининлактама (Пример 3, Стадия 1) растворяют в 15 мл тетрагидрофурана, затем при перемешивании при температуре, не превышающей -50°C, добавляют раствор 3,6 мМ литийалюминийгидрида, растворенного в тетрагидрофуране. Протекание восстановления контролируют тонкослойной хроматографией с проявляющим растворителем №8 и, если требуется, добавляют дополнительную порцию литийалюминийгидрида. К этой реакционной смеси добавляют по каплям 0,5M серной кислоты при постоянном перемешивании и охлаждении до тех пор, пока рН не достигнет 3, затем добавляют 35 мл воды. Полученный раствор экстрагируют 2x15 мл гексана, затем 3x20 мл дихлорметана. Дихлорметановые экстракты объединяют, промывают 3x15 мл воды, 15 мл холодного 5% раствора гидрокарбоната натрия и снова 15 мл воды, сушат над безводным сульфатом натрия и выпаривают при 2,0-2,5 кПа. Остаток после выпаривания обрабатывают диизопропиловым эфиром, фильтруют и сушат при пониженном давлении.
Выход 3,9 г (6,07 мМ, 81%). Rf(8)=0,40. [α]D 20=+16,0° (c=1, тетрагидрофуран).
Масс-спектр (МС-ББА) (642 [M+H+], 795 [M+H+NBA]+) подтверждает предполагаемую структуру.
Стадия 3: D-Циклогептиллактил-L-пролил-L-аргининальдегид гемисульфат
3,21 г (5 мМ) тетрагидропиранил-D-циклогептиллактил-L-пролил-NG-бензилоксикарбонил-L-аргининальдегида (Пример 3, Стадия 2) растворяют в 40 мл этанола и 5 мл 0,5M серной кислоты, затем добавляют 0,3 г Pd-C катализатора, суспендированного в 6 мл воды, и смесь гидрируют при приблизительно 10°С. Ход реакции контролируют тонкослойной хроматографией. После завершения реакции (около 15 минут), катализатор отфильтровывают, и фильтрат концентрируют до приблизительно 4-6 мл при 2,0-2,5 кПа. Остаток разбавляют 40 мл воды, экстрагируют 4x7 мл дихлорметана и водный раствор оставляют стоять при 20-22°C в течение 24 часов. Раствор экстрагируют 3x15 мл дихлорметана снова и доводят рН до 3,5 при помощи ионообменной смолы Dowex AG 1-X8 (НО-), затем раствор сушат вымораживанием (лиофильная сушка).
Выход 1,65 г (3,5 мМ, 70%) [α]D 20=-94,7° (c=1, вода)
ВЭЖХ: k'=2,702 и 3,010.
Масс-спектр (МС-ББА) (424 [M+H]+, 577 [M+H+NBA]+) подтверждает предполагаемую структуру.
Синтез исходных веществ:
Триэтиламмонийная соль тетрагидропиранил-D-циклогептиллактил-L-пролина
Стадия A: Метиловый эфир хлорацетил-DL-циклогептилаланина
К раствору 15,2 г (65 мМ) гидрохлорида метилового сложного эфира DL-циклогептилаланина (Пример 1, Стадия E) в 65 мл дихлорметана добавляют 9,1 мл (65 мМ) триэтиламина и 14,9 г (78 мМ) (N-гидроксисукцинимидил)хлорацетата*. После перемешивания при комнатной температуре в течение 3 часов, реакционную смесь разбавляют 65 мл дихлорметана и последовательно промывают 3x30 мл воды, 1M KHSO4, водой, 5% NaHCO3, и, наконец, водой до нейтральности. После этого органический слой сушат над безводным Na2SO4, затем выпаривают при 2,0-2,5 кПа. Полученный маслянистый продукт растирают с петролейным эфиром (пределы выкипания 40-60°С). Твердое вещество отфильтровывают, промывают петролейным эфиром (пределы выкипания 40-60°С) и сушат в вакуумном эксикаторе. Получают 17,54 г (63,6 мМ, ˜98%) метилового сложного эфира хлорацетил-DL-циклогептилаланина, который используют непосредственно на следующей стадии. Rf(7)=0,73-0,83. Т.пл.: 78-80°С.
Анализ для С13H22NO3Cl (275,777). Рассчитано: C%=56,62; H%=8,04; N%=5,08; Cl%=12,86. Найдено: C%=55,65; H%=7,93; N%=5,06; Cl%=12,72.
* Получение (N-гидроксисукцинимидил)хлорацетата
32 мл (450 мМ) хлорацетилхлорида добавляют к 23 г (200 мМ) N-гидроксисукцинимида и смесь кипятят с обратным холодильником в течение 10 минут, затем выливают на расколотый на кусочки лед, фильтруют, промывают холодной водой и сушат в вакуумном эксикаторе. Выход 20,23 г (105,9 мМ, 53%). Т.пл.: 113,3-113,7°C.
Анализ для C6H6NO4Cl (C7H9NO5) (191,55). Рассчитано: C%=37,62; H%=3,16; N%=7,31; Cl%=18,51. Найдено: C%=37,37; H%=3,16; N%=7,23; Cl%=18,35.
Стадия B: Метиловый сложный эфир хлорацетил-D-циклогептилаланина (ферментативное разделение метилового сложного эфира хлорацетил-DL-циклогептилаланина)
К раствору 8,71 г (31,6 моль) метилового сложного эфира хлорацетил-DL-циклогептилаланина, DL-сложного эфира, (Пример 3, Стадия A) в 30 мл толуола добавляют 70 мл воды и 50 мг Subtilisin Carlsberg (Протеаза Типа VIII, Sigma). Ферментативный гидролиз L-энантиомера, L-сложного эфира, протекает при pH 7,0, который поддерживают посредством аутотитратора, заполненного 3M NaOH. После прекращения потребления NaOH (до 5,522 мл, 16,57 мМ), реакционную смесь разбавляют 30 мл толуола, и два слоя разделяют. Водную фазу промывают 2x20 мл толуола. Объединенные толуольные растворы содержали D-сложный эфир, а объединенные водные растворы содержали натриевую соль L-кислоты.
После сушки над безводным Na2SO4, объединенные толуольные растворы выпаривают при пониженном давлении, получая 4,18 г (15,16 мМ) D-сложного эфира. Rf(7)=0,73-0,83, который непосредственно используют для получения D-циклогептилаланина.
Объединенные водные растворы подкисляют и экстрагируют 3x30 мл этилацетата. Объединенные этилацетатные растворы промывают водой, сушат над безводным Na2SO4 и выпаривают при пониженном давлении, получая 3,46 г (13,22 мМ) L-кислоты [Rf(7)=0,45-0,50], которую непосредственно используют для получения L-циклогептилаланина.
Подобное получение, исходя из 8,25 г (30 мМ) DL-сложного эфира (Пример 2, Стадия A), дает 4,08 г (14,79 мМ) D-сложного эфира и3,23 г (12,34 мМ) L-кислоты.
Стадия С: D-циклогептилаланин гидрохлорид
8,27 г (30 мМ) D-сложного эфира (Пример 3, Стадия B) суспендируют в 120 мл 6M HCl и кипятят с обратным холодильником в течение 3 часов. Свободную аминокислоту выделяют в виде кристаллов. Реакционную смесь охлаждают, выдерживают в холодильнике на протяжении ночи, фильтруют, промывают холодной водой и простым эфиром, затем сушат в вакуумном эксикаторе. Получают 5,9 г (26,69 мМ, 89%) гидрохлорида D-циклогептилаланина. Rf(12)=0,10-0,15. [α]D 20=-11° (c=0,4; 1M HCl).
Анализ для C10H19NO2·HCl (221,728). Рассчитано: C%=54,17; H%=9,09; N%=6,32; Cl%=15,99. Найдено: C%=54,27; H%=9,27; N%=6,30; Cl%=16,2.
Стадия D: Дициклогексиламмонийная соль D-циклогептилмолочной кислоты
5,78 г (26,15 мМ) гидрохлорида D-циклогептилаланина (Пример 3, Стадия С) растворяют в 26 мл воды, разбавляют 105 мл воды и 52,5 мл ледяной уксусной кислоты и охлаждают до 5°C. К этой смеси по каплям добавляют раствор 18,0 г (261 мМ) NaNO2 в 30 мл воды при перемешивании и охлаждении. Перемешивание продолжают при 5°C в течение часа и при комнатной температуре на протяжении ночи. На следующий день реакционную смесь подкисляют 25 мл концентрированной HCl при перемешивании. После этого смесь выпаривают досуха при 50°C при пониженном давлении. Остаток растворяют в 100 мл воды и выпаривают подобным образом, растирают с толуолом и снова выпаривают. Конечный остаток растворяют в 50 мл этилацетата и 50 мл воды. Водную фазу промывают этилацетатом и объединенные этилацетатные растворы промывают водой до нейтральности, сушат над безводным Na2SO4 и выпаривают при пониженном давлении. Полученное твердое вещество растворяют в 20 мл диизопропилового эфира. К этому раствору добавляют 5 мл (25 мМ) дициклогексиламина, после чего выделяется кристаллическая соль. После охлаждения кристаллы отфильтровывают, промывают холодным простым эфиром и сушат в вакуумном эксикаторе, получая 5,5 г (14,96 мМ, 57,2%) соли дициклогексиламина D-циклогептилмолочной кислоты, D-cHla.DCHA. Rf(7)=0,53-0,60. [α]D 20=+19,5° (c=1; метанол.). Т.пл.: 147-150°С.
Анализ для C10H18O3(C22H41NO3). Рассчитано: C%=71,89; H%=11,24; N%=3,81. Найдено: C%=71,57; H%=11,34; N%=3,83.
Превращение 0,35 г (1 мМ) D-cHla.DCHA в свободную α-гидроксикислоту дало 0,15 г (0,8 мМ) D-cHla, [α]D 20=+10,1° (c=1; метанол); Т.пл.: 125-127°С.
Анализ для C10H18O3 (186,252). Рассчитано: C%=64,48; H%=9,74. Найдено: C%=64,54; H%=9,86.
Стадия E: Бензиловый сложный эфир D-циклогептилмолочной кислоты
К раствору 11,21 г (30,5 мМ) дициклогексиламмонийной соли D-циклогептилмолочной кислоты (Пример 3, Стадия D) в 30 мл диметилформамида добавляют 3,57 мл (30 мМ) бензилбромида. Смесь перемешивают при комнатной температуре в течение 24 часов, затем фильтруют и выпаривают при 2,0-2,5 кПа. Остаток растворяют в 20 мл 0,5M гидрокарбоната калия и 60 мл диэтилового эфира. Органическую фазу последовательно промывают 20 мл воды, 0,5M KHSO4 и водой, затем сушат над безводным сульфатом натрия и выпаривают при пониженном давлении. Маслянистый остаток представляет 8,3 г (˜30 мМ) бензилового сложного эфира D-циклогептилмолочной кислоты [Rf(3)=0,2-0,3], который непосредственно используют на стадии F.
Стадия F: Бензиловый сложный эфир тетрагидропиранил-D-циклогептилмолочной кислоты
К раствору 8,29 г (30 мМ) бензилового сложного эфира D-циклогептилмолочной кислоты (Пример 3, Стадия E) в 30 мл дихлорметана добавляют 3,01 мл (33 мМ) 3,4-дигидро-2H-пирана и 0,3 мл ˜3M HCl в этилацетате, и смесь оставляют стоять при комнатной температуре в течение 16 часов. После чего реакционную смесь разбавляют 40 мл дихлорметана, промывают 3x20 мл воды и сушат над безводным сульфатом натрия и выпаривают при 2,0-2,5 кПа. Остаток подвергают колоночной хроматографии на силикагеле, используя 200 г Kieselgel 60 (0,040-0,063 мм) в качестве адсорбента и смесь 85:15 циклогексана и этилацетата в качестве элюента. Фракции, содержащие исключительно чистый продукт [Rf(4)=0,60-0,70], объединяют и выпаривают при 2,0-2,5 кПа. Маслянистый остаток представлял 7,9 г (21,9 мМ, 73%) бензилового сложного эфира тетрагидропиранил-D-циклогептилмолочной кислоты, который непосредственно используют на следующей стадии.
Стадия G: Триэтиламмонийная соль тетрагидропиранил-D-циклогептилмолочной кислоты
7,21 г (20 мМ) бензилового сложного эфира тетрагидропиранил-D-циклогептилмолочной кислоты (Пример 3, Стадия F) растворяют в 20 мл диметилформамида, добавляют 2,8 мл (20 мМ) триэтиламина и гидрируют в присутствии 0,1 г катализатора Pd/C. Ход реакции контролируют тонкослойной хроматографией [Rf(1)=0,30 (сложный эфир), 0,00 (кислота)]. После завершения реакции катализатор отфильтровывают и промывают 2x5 мл диметилформамида. Фильтрат и промывки объединяют и используют на следующей стадии в виде раствора 20 мМ триэтиламмонийной соли тетрагидропиранил-D-циклогептилмолочной кислоты.
Стадия H: Бензиловый сложный эфир тетрагидропиранил-D-циклогептиллактил-L-пролина
Раствор триэтиламмонийной соли тетрагидропиранил-D-циклогептилмолочной кислоты, полученной на стадии G Примера 3 (20 мМ), охлаждают до +5°С и при перемешивании добавляют 2,7 г (20 мМ) 1-гидроксибензотриазола, 4,83 г (20 мМ) гидрохлорида бензилового сложного эфира L-пролина и 4,12 г (20 мМ) дициклогексилкарбодиимида. Реакционную смесь выдерживают при комнатной температуре на протяжении ночи, затем фильтруют и выпаривают при 2,0-2,5 кПа. Остаток растворяют в 50 мл этилацетата и промывают 20 мл воды и 5% гидрокарбонатом натрия, сушат над безводным Na2SO4 и выпаривают при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, используя 200 г Kieselgel 60 (0,040-0,063 мм) в качестве адсорбента и смесь 1:1 н-гексана и этилацетата в качестве элюента. Фракции, содержащие исключительно чистый продукт [Rf(3)=0,3-0,4], объединяют и выпаривают при 2,0-2,5 кПа. Маслянистый остаток представлял 5,95 г (13 мМ, 65%) бензилового сложного эфира тетрагидропиранил-D-циклогептиллактил-L-пролина, который непосредственно используют на следующей стадии.
Стадия I: Триэтиламмонийная соль тетрагидропиранил-D-циклогептиллактил-L-пролина
5,5 г (12 мМ) бензилового сложного эфира тетрагидропиранил-D-циклогептиллактил-L-пролина (Пример 3, Стадия H) растворяют в 12 мл диметилформамида, добавляют 1,68 мл (12 мМ) триэтиламина и гидрируют в присутствии 0,1 г катализатора Pd/C. Ход реакции контролируют тонкослойной хроматографией [Rf(1)=0,30 (сложный эфир), 0,00 (кислота)]. После завершения реакции катализатор отфильтровывают и промывают 2x2 мл диметилформамида. Фильтрат и промывки объединяют и используют в виде раствора, содержащего 12 мМ триэтиламмонийной соли тетрагидропиранил-D-циклогептиллактил-L-пролина.
Пример 4
Синтез гемисульфата N-метил-D-циклогексилглицил-L-азетидин-2-карбонил-L-аргининальдегида
Стадия 1: Бензилоксикарбонил-N-метил-D-циклогексилглицил-L-азетидин-2-карбонил-NG-бензилоксикарбонил-L-аргининлактам
1,97 г (5 мМ) трет-бутилоксикарбонил-NG-бензилоксикарбонил-L-аргининлактама [(Bajusz et al, J. Med. Chem. 33, 1729 (1990)] суспендируют в 5 мл хлороформа, затем добавляют 5 мл этилацетата, насыщенного газом-HCl (0,11-0,15 г/мл), при перемешивании и охлаждении льдом. Отщепление Boc-группы контролируют тонкослойной хроматографией [Rf(11)=0,5 (свободное соединение); 1,0 (Boc-соединение)]. К концу реакции суспензию разбавляют 10 мл диэтилового эфира, образовавшуюся массу кристаллов отфильтровывают, промывают 3 мл ацетона и 3 мл диэтилового эфира и сушат при пониженном давлении над KOH. Полученный NG-бензилоксикарбонил-L-аргининлактам гидрохлорид растворяют в 5 мл диметилформамида, охлаждают до -20°C и добавляют к следующему смешанному ангидриду.
1,95 г (5 мМ) бензилоксикарбонил-N-метил-D-циклогексилглицил-L-азетидин-2-карбоновой кислоты (Пример 4, Стадия B) растворяют в 5 мл диметилформамида, охлаждают до -15°С, затем при перемешивании добавляют 0,56 мл (5,05 мМ) N-метилморфолина и 0,665 мл (5,05 мМ) изобутилхлорформиата. После перемешивания в течение 10 минут, добавляют вышеупомянутый раствор NG-бензилоксикарбонил-L-аргининлактама в диметилформамиде, затем триэтиламин в количестве, достаточном для доведения pH реакционной смеси до 8 (около 0,7 мл требуется). Реакционную смесь перемешивают при -10°C в течение 30 минут, затем при 0°C в течение одного часа. После этого соли отфильтровывают и фильтрат разбавляют 50 мл этилацетата. Полученный раствор промывают 3x7 мл воды, 3 мл 1M KHSO4 и 3x3 мл воды, сушат над безводным Na2SO4 и упаривают при 2,0-2,5 кПа. Полученный продукт подвергают колоночной хроматографии на силикагеле, используя 50 г Kieselgel 60 (0,040-0,063 мм) в качестве адсорбента и этилацетат в качестве элюента. Фракции, содержащие исключительно чистый продукт [(Rf(1)=0,70], объединяют и выпаривают при 2,0-2,5 кПа. Остаток после выпаривания перекристаллизовывают из диизопропилового эфира.
Выход 2,35 г (71%), Rf(1)=0,45-0,55.
Масс-спектр (МС-ББА) (661 [M+H]+) подтверждает предполагаемую структуру.
Стадия 2: Бензилоксикарбонил-N-метил-D-циклогексилглицил-L-азетидин-2-карбонил-NG-бензилоксикарбонил-L-аргининальдегид
2,15 г (3,25 мМ) бензилоксикарбонил-N-метил-D-циклогексилглицил-L-азетидин-2-карбонил-NG-бензилоксикарбонил-L-аргининлактама (Пример 4, Стадия 1) растворяют в 5 мл тетрагидрофурана, и затем при перемешивании и при температуре, не превышающей -50°C, добавляют раствор 2,25 мМ LiAlH4, растворенного в тетрагидрофуране. Ход реакции контролируют тонкослойной хроматографией, используя (растворитель 7) в качестве проявляющего растворителя и, если требуется, добавляют дополнительную порцию LiAlH4. К этой реакционной смеси добавляют по каплям 0,5M KHSO4 при постоянном перемешивании и охлаждении до тех пор, пока не будет достигнут рН 3, затем добавляют 13 мл воды. Полученный раствор экстрагируют 2x5 мл гексана, затем 3x7 мл дихлорметана. Дихлорметановые экстракты объединяют, промывают 3x7 мл воды, 7 мл холодного 5% раствора NaHCO3 и снова 7 мл воды, сушат над безводным Na2SO4 и выпаривают при 2,0-2,5 кПа. Остаток после выпаривания обрабатывают диизопропиловым эфиром, фильтруют и сушат при пониженном давлении.
Выход 1,6 г (74%), Rf(7)=0,33-0,43.
Масс-спектр (МС-ББА) (663 [M+H]+) подтверждает предполагаемую структуру.
Стадия 3: N-метил-D-циклогексилглицил-L-азетидин-2-карбонил-L-аргининальдегид сульфат
1,53 г (2,3 мМ) бензилоксикарбонил-N-метил-D-циклогексилглицил-азетидин-2-карбонил-NG-бензилоксикарбонил-L-аргининальдегида (Пример 4, Стадия 2) растворяют в 23 мл этанола и 4,8 мл 0,5M серной кислоты, затем добавляют 0,15 г катализатора Pd-C, суспендированного в 3,5 мл воды, и смесь гидрируют при около 10°С. Ход реакции контролируют тонкослойной хроматографией. После завершения реакции (около 15 минут), катализатор отфильтровывают и фильтрат концентрируют до приблизительно 2-3 мл при 2,0-2,5 кПа. Остаток разбавляют 20 мл воды, экстрагируют 4x4 мл дихлорметана, и водный раствор оставляют стоять при 20-22°C в течение 24 часов. Раствор снова экстрагируют 3x4 мл дихлорметана, и pH доводят до 3,5 при помощи ионообменной смолы Dowex AG 1-X8 (HO), затем раствор сушат вымораживанием (лиофильная сушка).
Выход 1,02 г (90%). Rf(12)=0,40.
Масс-спектр (МС-ББА) (395 [M+H]+, 548 [M+H+NBA]+) подтверждает предполагаемую структуру.
Синтез исходных веществ:
Бензилоксикарбонил-N-метил-D-циклогексилглицил-L-азетидин-2-карбоновая кислота
Стадия A: Синтез 2,4,5-трихлорфенилового сложного эфира бензилоксикарбонил-D-циклогексилглицина
К перемешиваемой суспензии 5,42 г (20 мМ) трифторацетатной соли D-циклогексилглицина и 5,6 мл (40 мМ) триэтиламина в 50 мл диметилформамида, добавляют 8,8 г (22 мМ) бензил-пентахлорфенилкарбоната [Anteunis et al.: Bul. Soc. Chim. Belg. 96, 775 (1987)] и 6,16 мл (44 мМ) триэтиламина. После перемешивания в течение 3 часов реакционную смесь выпаривают при пониженном давлении, остаток растворяют в 60 мл диэтилового эфира и 60 мл воды. Фазы разделяют, органическую фазу промывают водой и объединенные водные фазы промывают диэтиловым эфиром, подкисляют 1M KHSO4 до pH 3, затем экстрагируют 3x30 мл этилацетата. Органическую фазу промывают водой до нейтральности, сушат над безводным Na2SO4 и выпаривают при 2,0-2,5 кПа.
Остаток после выпаривания представляет бензилоксикарбонил-D-циклогексилглицин, который растворяют в 20 мл тетрагидрофурана и объединяют с 4,54 г (22 мМ) 2,4,5-трихлорфенола и 4,54 г (22 мМ) дициклогексилкарбодиимида. Спустя три часа реакционную смесь фильтруют, фильтрат и промывки объединяют и выпаривают при пониженном давлении. Твердый остаток очищают колоночной хроматографией на силикагеле, используя 140 г Kieselgel 60 (0,040-0,063 мм) в качестве адсорбента и смесь 95:5 хлороформа и ацетона в качестве элюента. Фракции, содержащие исключительно чистый продукт [Rf(5)=0,7-0,8], объединяют и выпаривают при 2,0-2,5 кПа. Маслянистый остаток растирают с диэтиловым эфиром, фильтруют, промывают диэтиловым эфиром и сушат. Выход, 8,34 г (88,5%) чистого 2,4,5-трихлорфенилового сложного эфира бензилоксикарбонил-D-циклогексилглицина.
Стадия B: Синтез бензилоксикарбонил-N-метил-D-циклогексилглицил-L-азетидин-2-карбоновой кислоты
L-Азетидин-2-карбоновую кислоту (1,01 г, 10 мМ) и триэтиламин (1,4 мл, 10 мМ) добавляют к раствору 2,4,5-трихлорфенилового сложного эфира бензилоксикарбонил-D-циклогексилглицина (5,18 г, 11 мМ, из Примера 4, Стадия A) в 10 мл пиридина. После перемешивания на протяжении ночи реакционную смесь выпаривают при пониженном давлении, и остаток растворяют в 50 мл 5% NaHCO3 и 50 мл диэтилового эфира. Органическую фазу промывают водой и объединенные водные фазы промывают диэтиловым эфиром, подкисляют 1M KHSO4 до pH 3, затем экстрагируют 3x50 мл этилацетата. Этилацетатные экстракты объединяют, промывают водой до нейтральности, сушат над безводным Na2SO4 и выпаривают при 2,0-2,5 кПа.
Остаток после выпаривания представляет бензилоксикарбонил-D-циклогексилглицил-L-азетидин-2-карбоновую кислоту, которую растворяют в 10 мл тетрагидрофурана и объединяют с 5,0 мл (80 мМ) иодметана и охлаждают до 0°С. К этому раствору добавляют 1,2 г (30 мМ) гидрида натрия, 60%, и реакционную смесь перемешивают при КТ на протяжении ночи. Избыточный гидрид натрия разлагают осторожным добавлением 0,4 мл воды. Погашенную реакционную смесь концентрируют до приблизительно 10 мл при пониженном давлении при температуре ниже 30°С. Остаток разбавляют 15 мл воды и 10 мл т-бутилметилового эфира. Фазы разделяют и водную фазу промывают снова 10 мл т-бутилметилового эфира. Водную фазу продукта объединяют с 15 мл этилацетата и доводят до рН 2,23M раствором серной кислоты. Фазы разделяют и водную фазу экстрагируют обратно 10 мл этилацетата. Объединенную органическую фазу промывают 15 мл 5% раствора тиосульфата натрия. Фазы разделяют и органическую фазу выпаривают при (пониженном) давлении при температуре ниже 40°С. Маслянистый остаток представляет 3,75 г бензилоксикарбонил-N-метил-D-циклогексилглицил-L-азетидин-2-карбоновую кислоту (9,65 мМ, Rf(7)=0,85-0,95), которую растворяют в 9,65 мл тетрагидрофурана, и раствор выдерживают для использования на стадии 1 Примера 4.
Масс-спектр (МС-ББА) (403 [M+H]+) подтверждает предполагаемую структуру.
Пример 5
Ингибирование свертывания пептидиларгиналями
Ингибирующее действие на свертывание плазмы оценивали в тестах определения тромбинового времени(TT), активированного частичного тромбопластинового времени (APTT)и протромбинового времени (PT), используя цитратную человеческую плазму в качестве субстрата [Bagdy, D. et al: Thromb. Haemostas. 67, 325 (1992)]. Тест определения ТТ измеряет ингибирование одной стадии, коагуляцию (свертывание) фибриногена на действие экзогенного тромбина (конечная концентрация 2,5 NIH Е/мл). Свертывание плазмы в тестах определения APTT и PT индуцировали рекальцификацией (восполнение солей кальция в тканях). Эндогенный генерированный тромбин мог теоретически присутствовать при конечной концентрации вплоть до 50 NIH Е/мл (APTT, PT). Эти тесты обнаруживают сумму ингибирующих действий в отношении образования фибрина и генерации тромбина, и эта сумма ингибирующих действий включает несколько протеолитических реакций, опосредованных либо тромбином, либо другими ферментами, например фактором Xa (fXa). Противосвертывающую активность выражают в CT2, которая представляет концентрацию (нМ), необходимую для удваивания времени свертывания.
CT2(нМ)b
IC50(нМ)
LA50, мкМc
Xa
bCT2 = концентрация, необходимая для удваивания времен свертывания в тестах TT (тромбиновое время), APTT (активированное частичное тромбопластиновое время) и PT (протромбиновое время).
cLA50 = концентрация, необходимая для восстановления лизированной зоны на 50% от контроля, в тесте определения гемолиза в чашках Петри, PL = плазмин, tPA = тканевый активатор плазминогена, UK = урокиназа.
Некоторые из вышеуказанных соединений превосходят Эфегатран (C, D-MePhe-Pro-Arg-H, родственный пептидаргиналь) по своей способности пролонгировать время свертывания (Смотри Таблицу 1). В колонке A Таблицы 1 представлены противосвертывающие активности соединений формулы (I) изобретения (1-4) в сравнении с противосвертывающими активностями эфегатрана (C), известным антикоагулянтом [Bajusz, S. et al.: J. Med. Chem. 33, 1729 (1990); Патент США № 4703036 (1982); Bagdy, D. et al.: Thromb. Haemostas. 67, 357 and 68, 125 (1992); Jackson, C.V. et al.: Clin. Appl. Thrombosis/Hemostasis 2, 258 (1996)], и другими родственными пептидиларгиналями (например, C1 и C2) [Bajusz, S. et al.: Bioorg. & Med. Chem. 3, 1079 (1995); Патент США № 6121241 (2000); PCT Pub. No. WO97/46576]. Тест TT демонстрирует, что новые аналоги почти также эффективны, как и эфегатран в ингибировании реакции тромбин-фибриноген. APTT, с другой стороны, свидетельствует о том, что новые пептиды более эффективны, чем эфегатран в ингибировании стадий свертывания, предшествующих генерации тромбина.
Пример 6
Ингибирование тромбина и фактора Xa
Ингибирование фермента исследовали в обогащенных тромбоцитами сгустках плазмы, используя хромогенные субстраты, т.е. Tos-Gly-Pro-Arg-pNA (S1) для тромбина и Moc-D-Chg-Gly-Arg-pNA (S2) для фактора Ха, опубликованные в общих чертах (Bajusz, S. et al.: PCT Pub. No. WO97/46576). Испытания проводили при комнатной температуре в стеклянных пробирках и 96-луночных титрационных микропланшетах.
Растворы, (i) Буфер A: 0,1M фосфат натрия/0,05M NaCl (pH 8,5). (ii) Ингибиторы: 0,1, 1,0 и 10 мг/мл растворы в буфере A, содержащие 0,02% человеческого альбумина, (iii) Субстраты: 1 мМ S1 и 2 мМ S2 в дистиллированной воде.
(b) Получение сгустков плазмы. Обогащенные тромбоцитами образцы плазмы (200 мкл), помещенные в стеклянные пробирки, инкубировали при комнатной температуре с 80 мкл 40мМ CaCl2 в течение 60 мин. Сгустки промывали 2-мл аликвотами физиологического раствора (0,9% NaCl) при легком взбалтывании, чтобы удалить несвязанные ферменты. В случае осуществления успешного промывания, оптическая плотность реакционной смеси промывки и S1 составляет меньше, чем 5% от контроля. Полученный таким образом сгусток плазмы хранили под физиологическим раствором в пробирке до использования.
(c) Оценка ингибирования фермента в сгустках. После удаления физиологического раствора, сгусток плазмы инкубировали при 37°C с 400 мкл ингибитора (или буфером А в качестве отрицательного контроля) в течение 5 мин и с 100 мкл субстрата S1 или S3 в течение 30 мин, затем реакцию обрывали 100 мкл 50% уксусной кислоты. 150-мкл порции реакционных смесей помещали в лунки титрационного микропланшета и считывали при 405 нм (ELISA READER 800, Bio-Tek Instruments Inc. Winooski, VT, USA). Значения IC50 получали из данных экстинкции графически.
Результаты представлены в Колонке B Таблицы 1. Соединения 1, 2, 3 и 4 оказываются единственными аналогами, которые могут превзойти Эфегатран в ингибировании связанного со сгустком тромбина, однако, в ингибировании связанного со сгустком фактора Ха, каждый аналог оказывается лучше, чем Эфегатран. Таким образом, соединения 1, 2, 3 и 4 обладают наибольшим ингибирующим действием в отношении обоих связанных со сгустком ферментов.
Пример 7
Противофибринолитическая активность
Ингибирующие действия соединений изобретения на плазмин (PL) и генерацию плазмина активаторами плазминогена (Plogen), как например тканевый активатор плазминогена (tPA), и урокиназу (UK), исследовали посредством чашечного теста определения гемолиза. (Смотри, например, Bagdy, D., Barabas, E., Bajusz, S., and Szell, E., Thromb. Haemostas., 67: 325-330 (1992); Barabas, E. Szell, E. and Bajusz, S, Blood Coagulation and Fibrinolysis, 4: 243 (1993)). Результаты представлены в Таблице 1, Колонка C. За редким исключением, аналоги являются несколько более ингибирующими, чем Эфегатран по отношению к трем фибринолитическим ферментам. Исключения представляют: C1 в отношении PL, и C1, C2 в отношении UK, в то время как 3 обладает активностью, почти равной Эфегатрану, в отношении UK.
Пример 8
Модель кролика для диссеминированного внутрисосудистого свертывания
Соединения 1 и 3 изобретения и другие пептидиларгинали Таблицы 1, а также С3 исследовали на их ДВС-ингибирующую активность на кроликах, обработанных эндотоксином (липополисахариды, ЛПС, LPS), как описано [Scherer, M. U. et al: Lab. Anim Sci 45, 538 (1995)]. Процедура испытания продолжалась на протяжении 4 часов. Эндотоксин вводили при дозах 80 и 40 мкг/кг путем быстрой в.в. (i.v. bolus)) инъекции для получения немедленной ответной реакции (в дальнейшем, в.в. инъекция ударной дозы) на 0 и 120 мин, соответственно, в то время как пептидиларгинали 1, 3 и С-С3 (0,25 и/или 0,5 мг/кг/ч) вливали на протяжении всего эксперимента. Контрольную группу животных обрабатывали 0,9% солевым раствором (физиологическим раствором). Гемостатические параметры определяли на момент, равный 0, 120 и 240 мин.
Несмотря на осторожную обработку, летальность составляла 31%, наиболее вероятно из-за высокой чувствительности к эндоксину кроликов [Semerano, N. et al.: Int. J. Clin. Lab. Res. 21, 214 (1992)].
число умерших/обработанные животные и %
Как следует из Таблицы 2, соединения 1 и 3 существенно снижают летальность обработанных эндотоксином кроликов, в то время как другие антикоагулянты либо не оказывают влияния на летальность (С1), либо вызывают некоторое увеличение в летальности, наивысшее значение, в ˜1,8 раза, получено в случае использования С3. Стоит отметить, исходя из данных Таблицы 1, что снижающие летальность (соединения) 1 и 3 обладают и наиболее ингибирующим действием в отношении связанного со сгустком тромбина, а также фактора Ха, и, кроме этого, эффективно ингибируют как свертывание плазмы, так и фибринолитические ферменты, плазмин, и активаторы плазминогена.
Пример 9
Крысиная модель для диссеминированного внутрисосудистого свертывания
Среди наиболее серьезных последствий ДВС - отложение фибрина в различных органах, изменения клеток крови (гемоцитов), например, уменьшение числа тромбоцитов, и изменения в продуктах деградации фибрина. Влияние соединения 1 изобретения и двух контрольных антикоагулянтов, Эфегатрона (С) и гепарина (Н) на вышеупомянутые явления исследовали у обработанных эндотоксином крыс [Ford, A. J. and Longridge, D.J.: Br. J. Pharmacol. 110, Suppl. 131P (1993); Hasegawa, N.; et al: Am. J. Resp. Crit. Care Med. 153, 1831 (1996); Dichneite, G. et al: Thromb. Res. 77, 357 (1995)].
Крыс-самцов обрабатывали в.в. (i.v.) инъекцией ударной дозы 10 мг/кг эндотоксина. Вслед за этим следовало в.в. (i.v.) вливание физиологического раствора или испытываемых соединений в течение четырех часов. В случае соединений 1 и 3, 0,25 мг/кг давали в виде начальной инъекции ударной дозы с последующим в.в. (i.v.) вливанием 0,25 мг/кг/ч в течение четырех часов. Аналогично применяли гепарин (Н), 50 МЕ/кг в виде начальной инъекции ударной дозы с последующим в.в. (i.v.) вливанием 50 МЕ/кг на протяжении четырех часов. Контрольную группу животных обрабатывали 0,9% солевым раствором (физиологическим раствором).
Отложение 125I-фибрина исследовали в выбранных органах (печень и почки). 125I-фибриноген инъецировали за 30 мин до инъекции эндотоксина. Радиоактивности в образцах тканей измеряли гамма-счетчиком (Wallac Wizard 1470). Образование микротромбов в органах оценивали, используя отношение активности 125I органа к общей инъецированной активности 125I, определяемое как индекс (образования) микротромбов. Изменения в этом параметре выражают в процентах по сравнению с группой, получавшей физиологический раствор.
Число счета тромбоцитов определяли в автоматическом приборе (Sysmex F-800) и соотносили с контрольными значениями.
Определение ПДФ (FDP) (продукты деградации фибрина) осуществляли согласно Aggristin (Ristocetin) преципитационному анализу. Животных убивали через четыре часа после введения эндотоксина.
Полученные данные суммированы в Таблице 3.
+ 1, 0,25 мг/кг/чb
13%
26%
48%
1,66
+ H, 50 NIH Е/кг/чb
22%
28%
43%
2,37
bВ.В. инъекция ударной дозы + В.В. вливание.
Данные Таблицы 3 указывают на то, что ДВС-ингибирующий потенциал соединения 1 оказался более резко выраженным, чем ДВС-ингибирующий потенциал как гепарина, так и Эфегатрана.
Пример 10
Исследование выживаемости на крысиной модели для диссеминированного внутрисосудистого свертывания
Крыс-самцов обрабатывали в.в. (i.v.) инъекцией ударной дозы 30 мг/кг ЛПС (LPS) (эндотоксин). Пептиды при дозах 0,5 и/или 0,75, 1,0 и 1,5 мг/кг вводили животным в виде первичной инъекции ударной дозы с последующим вливанием в течение восьми часов сразу же после введения ЛПС (LPS). Летальность (смертность) регистрировали через 4, 5, 6, 7, 8 часов после введения ЛПС.
Данные Таблицы 4 указывают на то, что обработка ЛПС снижала коэффициент выживаемости крыс до 20% к концу эксперимента (8 часов). Все исследованные пептидиларгинали пролонгировали время выживания и снижали смертность по сравнению с ЛПС-группой. Соединения 1, 2, 3 и 4 оказались более эффективными, чем эталонное соединение С.
ТАБЛИЦА 4
Влияние соединений 1, 2, 3 и 4 изобретения и родственного соединения С на летальность ЛПС-обработанных крыс
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения D-фенилаланил-L-пролил-L-аргининальдегид сульфата | 1982 |
|
SU1442078A3 |
| Способ получения сульфатов пептидил-аргининальдегидов | 1985 |
|
SU1384203A3 |
| ПРОИЗВОДНЫЕ БЕНЗАЗЕПИНА ИЛИ БЕНЗОТИАЗЕПИНА | 1989 |
|
RU2090562C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2026296C1 |
| Способ получения ариламидов аминокислот или пептидов или их солей | 1986 |
|
SU1512484A3 |
| ПРОИЗВОДНЫЕ ТРИПЕПТИДОВ В ВИДЕ R- ИЛИ RS-ФОРМЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ НЕТОКСИЧНЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2077538C1 |
| Способ получения полипептидов или их солей | 1977 |
|
SU910116A3 |
| 6-(ТОЗИЛГЛИЦИЛ-L-ПРОЛИЛ-L-АРГИНИЛ)-АМИНОНАФТАЛИН-1-ИЗОБУТИЛ-СУЛЬФАМИД В КАЧЕСТВЕ СУБСТРАТА ДЛЯ ФЛЮОРЕСЦЕНТНОГО ОПРЕДЕЛЕНИЯ ТРОМБИНА | 1991 |
|
RU2087481C1 |
| Способ получения октапептида | 1983 |
|
SU1147010A1 |
| Способ получения трипептидамидов | 1980 |
|
SU963463A3 |
Изобретение относится к новым пептидиларгиналям формулы (I)
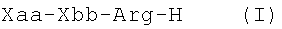
где Хаа означает остаток альфа-замещенной угольной кислоты формулы(II)
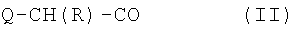
где Q означает C1-3алкилоксикарбониламино группу, метиламино группу или гидроксильную группу, и R означает C7-9циклоалкилметильную группу или С5-7циклоалкильную группу, Xbb представляет остаток L-пролина или L-азетидин-2-карбоновой кислоты и его аддитивные соли кислоты, образованные с органической или неорганической кислотой. Описаны промежуточные соединения. Соединения формулы I обладают ингибирующим действием на свободный тромбин, связанный со сгустком тромбин, фактор Ха, плазмин и активаторы плазминогена, что позволяет использовать их в фармацевтической композиции для лечения пациента, страдающего диссеминированным внутрисосудистым свертыванием. 6 н. и 11 з.п. ф-лы, 4 табл.
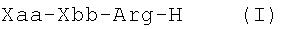
где Хаа представляет остаток альфа-замещенной угольной кислоты формулы (II)
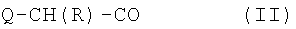
где Q представляет С1-3алкилоксикарбониламино группу, метиламино группу или гидроксильную группу, и R представляет С7-9циклоалкилметильную группу или С5-7циклоалкильную группу, Xbb представляет остаток L-пролина или L-азетидин-2-карбоновой кислоты, и его аддитивные соли кислоты, образованные с органической или неорганической кислотой.
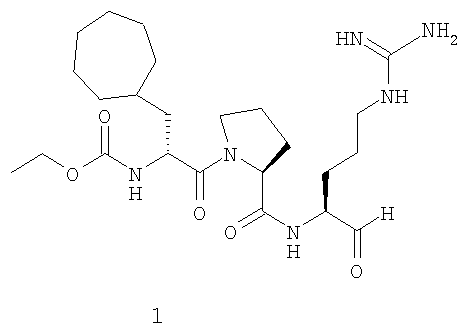
и его аддитивные соли кислоты.
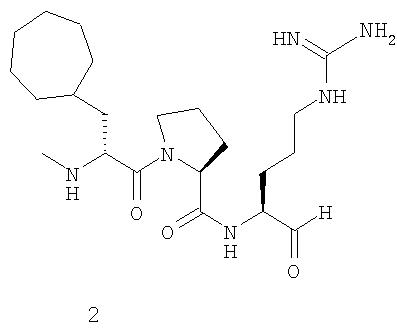
и его аддитивные соли кислоты.
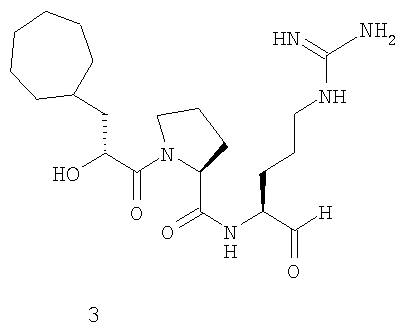
и его аддитивные соли кислоты.
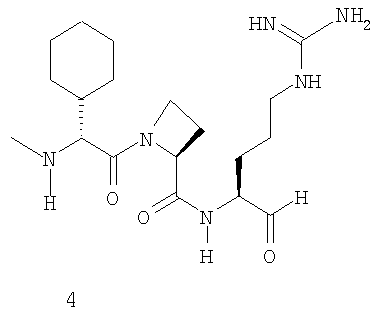
и его аддитивные соли кислоты.
где Хаа представляет остаток альфа-замещенной угольной кислоты формулы (II)
где Q представляет C1-3алкилоксикарбониламино группу, метиламино группу или гидроксильную группу, и R представляет C7-9циклоалкилметильную группу или C5-7циклоалкильную группу, и Xbb представляет остаток L-пролина или L-азетидин-2-карбоновой кислоты, или его фармацевтически приемлемых аддитивных солей кислоты.
или его фармацевтически приемлемых аддитивных солей кислоты.
или его фармацевтически приемлемых аддитивных солей кислоты.
или его фармацевтически приемлемых аддитивных солей кислоты.
или его фармацевтически приемлемых аддитивных солей кислоты.
| Установка для газожидкостной экстракции | 1977 |
|
SU670310A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| RU 2055078 C1, 27.02.1996. | |||

