Перекрестная ссылка на родственные заявки
Настоящая заявка имеет приоритет и испрашивает приоритет заявки на патент Японии № 2004-169188, поданной 7 июня 2004; и заявки на патент Японии № 2004-375769, поданной 27 декабря 2004; которые приведены здесь в качестве ссылки в полном объеме.
Предшествующий уровень техники
1. Область, к которой относится изобретение
Настоящее изобретение относится к новой альдолазе, продуцирующей 4-(индол-3-илметил)-4-гидрокси-2-оксоглутаровую кислоту (IHOG), которая является предшественником монатина, и к способам получения 4R-IHOG и 4R-монатина с использованием указанной кислоты.
2. Описание прототипов
4-(Индол-3-илметил)-4-гидрокси-2-глутаминовая кислота (3-(1-амино-1,3-дикарбокси-3-гидроксибутан-4-ил)индол) (называемая далее монатином), представленная нижеследующей структурной формулой, содержится в корнях растений, Schlerochiton ilicifolius, и представляет собой соединение, которое ранее использовали как низкокалорийный подсластитель, поскольку оно обладает высокой степенью сладости (см. JP-Р-64-25757-А).
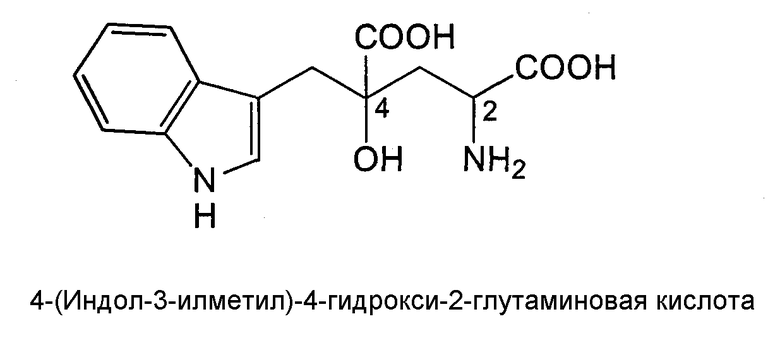
Монатин имеет два асимметрических атома углерода (в положениях 2 и 4), и сообщалось, что его стереоизомером является (2S,4S)-изомер. Были идентифицированы еще три стереоизомера, и было подтверждено, что каждый из них обладает высокой степенью сладости, которая в несколько десятков, а то и в несколько тысяч раз превышает сладость сахарозы (таблица 1).
Как показано в таблице 1, высокий показатель сладости имеет не только природный (2S,4S)-монатин, но также и все другие стереоизомеры. В частности, (2R,4R)-монатин имеет сильно выраженную сладость, которая в 2700 раз превышает сладость сахарозы, а поэтому его использование в качестве подслащивающего агента или ингредиента подслащивающего агента (подсластителя) является очень перспективным. Таким образом, было бы желательно разработать способ эффективного получения монатина с высоким содержанием (2R,4R)-монатина.
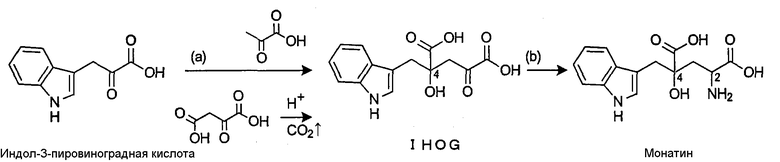
Авторами настоящего изобретения был разработан новый способ синтеза монатина, предусматривающий проведение нижеследующих реакций (а) и (b) с использованием коммерчески доступной индолпировиноградной кислоты и пировиноградной кислоты в качестве реагентов (патентный документ 1).
(а) Реакция синтеза предшественника кетокислоты (IHOG) путем альдольной конденсации индолпировиноградной кислоты и пировиноградной кислоты (или щавелево-уксусной кислоты).
(b) Реакция аминирования IHOG в положении 2.
В патентном документе 1 описаны альдолазы, полученные из Pseudomonas taetrolens и Pseudomonas coronafaciens, являющиеся ферментами, способными продуцировать предшественник кетокислоты (IHOG) из индолпировиноградной кислоты и пировиноградной кислоты (или щавелево-уксусной кислоты) в реакции альдольной конденсации (а) в вышеупомянутом пути синтеза монатина. Было обнаружено, что помимо реакции получения IHOG, эти альдолазы катализируют реакцию получения кетокислоты, такой как 4-фенилметил-4-гидрокси-2-оксоглутаровая кислота (PHOG).
IHOG имеет два изомера 4R-изомер и 4S-изомер. Для эффективного получения (2R,4R)-монатина, который представляет собой изомер, обладающий наиболее высокой степенью сладости, желательно, чтобы в реакции альдольной конденсации (а) в вышеупомянутом пути синтеза монатина предпочтительно продуцировался 4R-изомер IHOG (4R-IHOG) (далее 4S-изомер обозначается 4S-IHOG) и была получена обогащенная 4R-изомером IHOG. Хиральная молекула часто обнаруживает физиологическую активность, которая является разной для каждого изомера, а поэтому, очевидно, что каждый изомер IHOG обладают различными свойствами. Таким образом, отдельно полученные 4R- и 4S-изомеры могут быть использованы не только как предшественники монатина, но также и в других целях. Следовательно, разработка способа получения преимущественно одного изомера из изомеров 4R-IHOG и 4S-IHOG имеет особенно важное промышленное значение.
Патентный документ 1: проспект публикации Международной заявки 03/056026.
Патентный документ 1: проспект публикации Международной заявки 04/018672.
Описание сущности изобретения
Однако в ранее описанных химических системах синтеза, полученная IHOG представляет собой смесь 4R- и 4S-изомеров (рацематов). Авторами настоящего изобретения была получена альдолаза, из Pseudomonas taetrolens, которая является подходящей для синтеза IHOG, но при этом было продемонстрировано, что IHOG, получаемая указанной альдолазой, является слегка обогащенной 4S-изомером и не содержит большое количество 4R-изомера, в зависимости реакционных условий (патентные документы 1 и 2). До настоящего времени еще не было обнаружено альдолазы, которая продуцировала бы преимущественно 4R-IHOG. То есть в настоящее время пока еще не существует способа эффективного получения 4R-IHOG, а в частности, IHOG, обогащенной 4R-изомером.
На основании вышеуказанных исследований было разработано настоящее изобретение, целью которого является получение новой альдолазы, продуцирующей PHOG и IHOG, и в частности, 4R-IHOG, а также были разработаны способы получения IHOG и монатина с их использованием.
В результате интенсивных исследований, проводимых для достижения вышеуказанной цели, авторами настоящего изобретения было обнаружено, что альдолаза, которая может быть использована для синтеза нужной 4R-IHOG, присутствует в некоторых микроорганизмах, и такое использование этих микроорганизмов было положено в основу настоящего изобретения, которое относится к способам получения 4R-IHOG и 4R-монатина.
Таким образом, настоящее изобретение относится по крайней мере к:
[1] Способу получения (4R)-4-(индол-3-илметил)-4-гидрокси-2-оксоглутаровой кислоты (4R-IHOG) формулы (I) или ее соли:
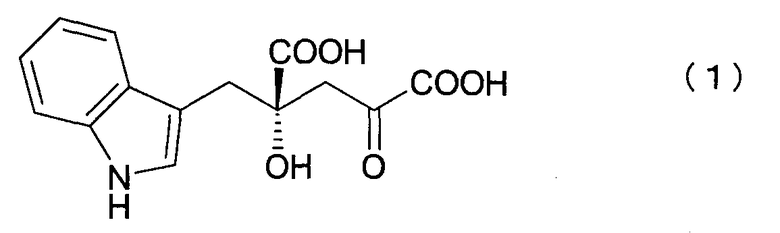
который заключается в проведении реакции взаимодействия индол-3-пировиноградной кислоты с пировиноградной кислотой или щавелево-уксусной кислотой в присутствии белка или микроорганизма, содержащего такой белок, с получением 4R-IHOG, имеющего оптическую чистоту 70% или выше,
где указанный белок выбран из группы, включающей (а) и (b):
(а) белок с аминокислотной последовательностью SEQ ID NO:2,
(b) белок, который на 70% или более гомологичен аминокислотной последовательности SEQ ID NO:2 и который обладает 4R-альдолазной активностью.
[2] Способу получения 4R-монатина или его соли, который включает:
первую стадию, в которой проводят реакцию взаимодействия индол-3-пировиноградной кислоты с пировиноградной кислотой или щавелево-уксусной кислотой в присутствии белка или микроорганизма, содержащего такой белок, с получением преимущественно 4R-IHOG,
где указанный белок выбран из группы, включающей:
(а) белок с аминокислотной последовательностью SEQ ID NO:2,
(b) белок который на 70% или более гомологичен аминокислотной последовательности SEQ ID NO:2 и который обладает 4R-альдолазной активностью, и
вторую стадию, в которой осуществляют превращение карбонильной группы 4R-IHOG или его соли, полученной в первой стадии, в аминогруппу, с образованием 4R-монатина формулы (2) или его соли, где указанный 4R-монатин или его соль имеют оптическую чистоту 90% или более,
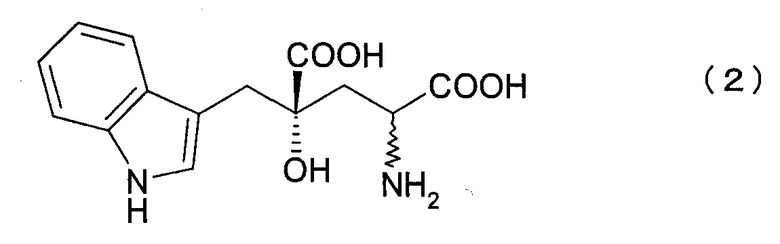
где связь, обозначенная волнистой линией, может иметь как R-, так и S-конфигурацию.
[3] Способу получения 4R-монатина или его соли по пункту [2],
где, во второй стадии, карбонильную группу преобразовывают в аминогруппу путем аминирования в присутствии фермента, воздействующего на 4R-IHOG.
[4] Способу получения 4R-монатина или его соли по пунктам [2] или [3],
где, во второй стадии, карбонильную группу преобразуют в аминогруппу способами, предусматривающими:
проведение реакции взаимодействия 4-(индол-3-илметил)-4-гидрокси-2-оксоглутаровой кислоты, содержащейся в реакционной смеси, с аминовым соединением нижеприведенной формулы (3) или с его солью:

где R представляет собой атом водорода, алкильную, арильную или аралкильную группу, в нейтральных или щелочных условиях с получением 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (IHOG-оксима) нижеследующей формулы (4) или его соли:
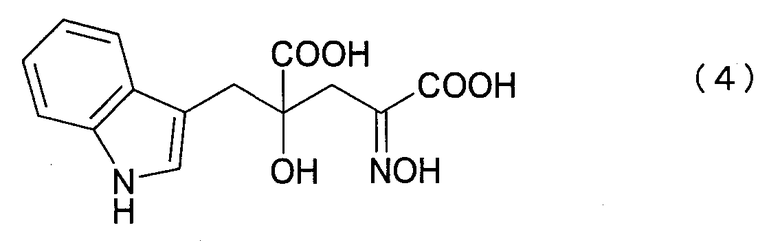
и кристаллизацию 4R-изомера полученного IHOG-оксима или его соли с последующим восстановлением и получением 4R-монатина или его соли, имеющих оптическую чистоту 90% или выше:
[5] Способу получения 4R-монатина по пункту [4],
где аминовое соединение формулы (3) представляет собой, по крайней мере, одно аминовое соединение, выбранное из группы, включающей гидроксиламин, метоксиамин и бензилоксиамин.
[6] Способу получения 4R-монатина по пунктам [4] или [5],
где 4R-изомер IHOG-оксима или его соль восстанавливают в присутствии водорода и гидрированного катализатора.
[7] Способу получения 4R-монатина или его соли в соответствии с любым из пунктов [4]-[6],
где (2R, 4R)-монатин выделяют путем кристаллизации во второй стадии.
[8] Способу получения 4R-монатина или его соли в соответствии с любым из пунктов [4] или [7],
где во второй стадии кристаллизацию осуществляют с использованием кристаллизующего растворителя, выбранного из группы, включающей воду, спиртовой растворитель и водный спиртовой растворитель.
[9] Способу по любому из пунктов [1]-[8],
где белок, используемый в этом способе, получают из микроорганизма, выбранного из бактерий, принадлежащих к роду Sphingomonas или Burkholderia.
[10] Способу по пункту [9],
где микроорганизм выбран из группы, включающей штамм Sphingomonas sp. AJ110329, штамм Sphingomonas sp. AJ110372 и штамм Burkholderia sp. AJ110371.
[11] Белку, выбранному из группы, включающей белок (а)-(с):
(а) белок с аминокислотной последовательностью SEQ ID NO:2;
(b) белок, который, по крайней мере, на 70% или более гомологичен аминокислотной последовательности SEQ ID NO:2 и который обладает 4R-альдолазной активностью, и
(с) белок с аминокислотной последовательностью, содержащей мутацию, выбранную из группы, включающей замену, делецию, инсерцию, добавление и инверсию одного или нескольких аминокислотных остатков в аминокислотной последовательности SEQ ID NO:2, и обладает альдолазной активностью.
[12] Белку по пункту [11],
где указанный белок, который по крайней мере на 70% или более гомологичен аминокислотной последовательности SEQ ID NO:2 и который обладает 4R-альдолазной активностью, представляет собой белок с аминокислотной последовательностью, представленной в SEQ ID NO:13 или 15.
[13] ДНК, кодирующей белок, описанный в пункте [11] или [12].
[14] ДНК, выбранной из (d) и (е):
(d) ДНК, с нуклеотидной последовательностью SEQ ID NO:1 или последовательности нуклеотидов 210-1004 в той же самой последовательности SEQ ID NO; и
(е) ДНК, которая в жестких условиях гибридизуется с ДНК, содержащей нуклеотидную последовательность, комплементарную нуклеотидной последовательности SEQ ID NO:1 или последовательности нуклеотидов №№ 210-1004 в той же самой последовательности SEQ ID NO, и которая кодирует белок, обладающей альдолазной активностью.
[15] ДНК по пункту [14],
где ДНК, которая в жестких условиях гибридизуется с ДНК, содержащей нуклеотидную последовательность, комплементарную нуклеотидной последовательности SEQ ID NO:1 или последовательности нуклеотидов 210-1004 в той же самой последовательности SEQ ID NO, которая кодирует белок, обладающей альдолазной активностью, и представляет собой любую из следующих ДНК: (f) ДНК с нуклеотидной последовательностью SEQ ID NO:12 или последовательностью нуклеотидов 399-1253 в той же самой последовательности SEQ ID NO, или (g) ДНК, с нуклеотидной последовательностью SEQ ID NO:14 или последовательностью нуклеотидов 531-1385 в той же самой последовательности SEQ ID NO.
[16] Рекомбинантной ДНК, полученной путем присоединения ДНК, выбранной из группы, состоящей из ДНК по пунктам [14] и [15], к векторной ДНК.
[17] Клетке, трансформированной рекомбинантной ДНК по пункту [16].
[18] Способу получения белка, обладающего альдолазной активностью, который заключается:
в культивировании клеток по пункту [17] в среде; и
в аккумуляции указанного белка, обладающего альдолазной активностью, в среде и/или в клетках.
С использованием альдолазы по настоящему изобретению 4R-IHOG может быть преимущественно получена из индолпировиноградной кислоты и пировиноградной кислоты (или щавелево-уксусной кислоты). Поскольку 4R-монатин может быть синтезирован путем аминирования полученной 4R-IHOG, то полученная альдолаза может быть использована преимущественно для получения монатина, обладающего высокой степенью сладости.
Обычно, если 4R-изомер выделяют из рацемической IHOG (4R, 4S-IHOG), то необходимо, чтобы эта рацемическая IHOG подвергалась реакции оксимирования и полученная 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровая кислота (IHOG-оксим) взаимодействовала с хиральными аминами для кристаллизации IHOG-оксима 4R-изомера (4R-IHOG-оксима). Напротив, в соответствии с настоящим изобретением после кристаллизации не требуется какого-либо оптического разделения с использованием хиральных аминов, поскольку обогащенный 4R-изомером IHOG может быть получен в стадии альдольной конденсации. После оксимирования 4R-IHOG-оксим может быть непосредственно кристаллизован. Таким образом, это дает возможность минимизировать процесс очистки 4R-IHOG.
Краткое описание чертежей
На фиг.1 проиллюстрировано сравнение, проводимое в целях выявления гомологии альдолаз настоящего изобретения;
на фиг.2 представлена блок-схема, иллюстрирующая стадии получения альдолазы по настоящему изобретению;
на фиг.3A представлен график, иллюстрирующий скорости реакции SpALD с использованием PHOG в качестве субстрата;
на фиг.3B представлен график, иллюстрирующий скорости реакции SpALD в зависимости от концентраций MgCl2;
на фиг.4 представлен график, иллюстрирующий результаты измерения рН, при котором SpALD обладает стабильностью;
на фиг.5 представлен график, иллюстрирующий результаты измерения термостабильности SpALD;
на фиг.6 представлен график, иллюстрирующий рН SpALD, при котором SpALD обладает оптимальной активностью в реакции альдольной деградации;
на фиг.7 представлен график, иллюстрирующий рН SpALD, при котором SpALD обладает оптимальной активностью в реакции альдольной конденсации;
на фиг.8A представлен график, иллюстрирующий скорости реакции BuALD с использованием PHOG в качестве субстрата;
на фиг.8B представлен график, иллюстрирующий скорости реакции BuALD в зависимости от концентраций MgCl2;
на фиг.9 представлен график, иллюстрирующий результаты измерения рН, при котором BuALD является стабильным;
на фиг.10 представлен график, иллюстрирующий результаты измерения термостабильности BuALD.
Подробное описание предпочтительных вариантов осуществления изобретения
Исследования, проведенные авторами настоящего изобретения, подтвердили, что существуют некоторые бактериальные штаммы, которые продуцируют альдолазу с активностью, направленной на преимущественный синтез 4R-IHOG, и на основании этих исследований были разработаны способы получения 4R-IHOG и 4R-монатина.
В нижеследующем описании настоящего изобретения будут подробно и по порядку описаны следующие способы:
[I] способ получения оптически активной IHOG и
[II] способ получения оптически активного монатина
со ссылками на прилагаемый графический материал.
[I] Способ получения оптически активной IHOG
(1) Взаимодействие
Ниже описан способ получения 4R-IHOG по настоящему изобретению. Способ получения 4R-IHOG по настоящему изобретению заключается в преимущественном получении 4R-IHOG, представленного нижеследующей формулой (1):
путем взаимодействия реакции индолпировиноградной кислоты, представленной нижеследующей формулой (5):
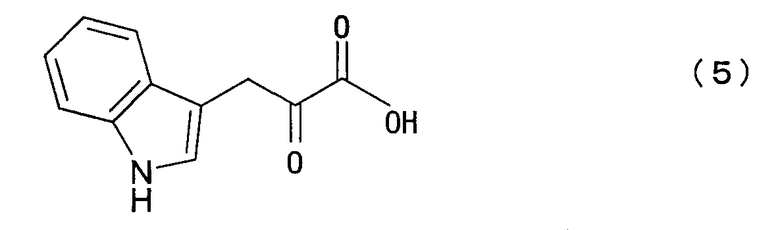
с пировиноградной кислотой или щавелево-уксусной кислотой, представленной нижеследующей формулой (6):
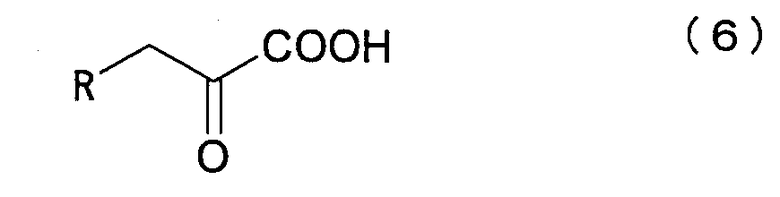
где R представляет собой атом водорода или карбоксильную группу и отличается тем, что указанную реакцию проводят в присутствии белка, который катализирует эту реакцию.
Вышеупомянутый "белок, катализирующий реакцию" предпочтительно представляет собой белок с 4R-альдолазной активностью и который может быть получен из микроорганизмов или химическим синтезом. Термин "4R-альдолазная активность" означает активность, способную катализировать реакцию, в которой 4R-IHOG, представленный формулой (1), преимущественно получают путем альдольной конденсации индолпировиноградной кислоты, представленной формулой (5), с пировиноградной кислотой или щавелево-уксусной кислотой, представленной формулой (6), и/или реакцию, в которой 4R-PHOG преимущественно получают из фенилпировиноградной кислоты и пировиноградной кислоты. Такой белок конкретно не ограничен и может быть использован в настоящем изобретении. Используемый здесь термин "преимущественное получение 4R-IHOG" обозначает, что оптическая чистота R-изомера превышает оптическую чистоту S-изомера в положении 4 и что может быть достигнута такая эффективность реакции, при которой оптическая чистота R-изомера будет предпочтительно составлять 70% или более, а особенно предпочтительно 90% или более. В зависимости от условий реакции величина оптической чистоты может изменяться, однако специалист в данной области может легко определить оптимальные условия такой реакции. Таким образом, настоящее изобретение включает любой способ, в котором достигается вышеупомянутая оптическая чистота в условиях, близких к оптимальным, даже в том случае, когда вышеуказанная оптическая чистота не достигается, если изменяются условия реакции. Взаимодействие, в котором используется белок, подходящий для осуществления настоящего изобретения, может проводиться для получения такой же или меньшей оптической чистоты путем регулирования реакционных условий для установки нужного соотношения смеси 4R- и 4S-изомеров. Такой вариант также входит в рамки способа по настоящему изобретению. Оптическая чистота 4R-IHOG может быть определена как энантиомерный избыток (% э.и.) по формуле: ([4R-IHOG]-[4S-IHOG])/([4R-IHOG]+[4S-IHOG])×100.
Альдолаза как белок, катализирующий указанную реакцию, описана ниже в пункте (2). При этом предпочтительным является белок, обладающий альдолазной активностью. В данной реакции может быть использована альдолаза, полученная путем культивирования микробных клеток, продуцирующих белок (альдолазу), который катализирует данную реакцию; или альдолаза, полученная путем создания трансформанта методами рекомбинантных ДНК, продуцирующего белок, катализирующий указанную реакцию, с последующим культивированием этого трансформанта.
Белок, катализирующий такую реакцию, может быть добавлен в реакционную систему в любой форме при условии, что этот белок может катализировать реакцию, в которой преимущественно синтезируется 4R-IHOG. То есть белок, катализирующий такое взаимодействие, может быть добавлен в реакционную систему отдельно, либо в эту систему может быть добавлена композиция, обладающая альдолазной активностью и включающая белок (альдолазу), катализирующий данную реакцию.
Используемый здесь термин "композиция, обладающая альдолазной активностью" может означать композицию, содержащую белок (альдолазу), катализирующий указанную реакцию, и включает, в частности, культуру, среду (культуру, из которой были удалены микробные клетки), микробные клетки (включая культивированные микробные клетки и отмытые микробные клетки), обработанный продукт микробных клеток, полученный путем разрушения или лизиса микробных клеток, и композицию (неочищенный раствор фермента, очищенный фермент), обладающую альдолазной активностью и полученную путем очистки среды и/или клеток. Так, например, если оптически активную IHOG получают с использованием продуцирующих альдолазу микроорганизмов или клеток, трансформированных рекомбинантной ДНК, то субстрат может быть добавлен в среду непосредственно при культивировании, и могут быть использованы микробные клетки, собранные из такой среды, или отмытые микробные клетки. Обработанные микробные клетки, полученные путем разрушения или лизиса микробных клеток, могут быть использованы непосредственно, либо альдолаза может быть собрана из обработанных продуктов микробных клеток и использована в качестве неочищенного раствора фермента, либо, помимо этого, может быть использован очищенный фермент. То есть в способе получения 4R-IHOG настоящего изобретения может быть использована фракция, обладающая альдолазной активностью, в любой форме.
Для проведения альдольной реакции с использованием альдолазы или композиции, обладающей альдолазной активностью, реакционная смесь, содержащая индолпировиноградную кислоту и пировиноградную кислоту или щавелево-уксусную кислоту, и белок, катализирующий такую реакцию, или содержащий альдолазу продукт, могут быть подвергнуты статическому инкубированию, встряхиванию или перемешиванию при соответствующей температуре 20-50°С в течение 30 минут - 5 дней при поддержании рН от 6 до 12.
В настоящем изобретении для более стереоселективного получения IHOG интересующий 4R-IHOG может быть получен с более высокой стереоселектиностью путем ингибирования спонтанной альдольной конденсации. В настоящем изобретении в качестве примера рассматривается случай, когда IHOG получают путем альдольной конденсации индол-3-пировиноградной кислоты и пировиноградной кислоты. В этой реакции, альдольная конденсация происходит спонтанно путем установки рН до щелочного значения, например, примерно до рН 9-12. IHOG, получаемая посредством такой спонтанной альдольной конденсации, представляет собой смесь 4R- и 4S-изомеров (рацемат), и в этом случае стереоселективность для положения 4 является низкой. Таким образом, в одном из аспектов настоящего изобретения, в реакции с использованием белка, катализирующего данную реакцию, рН устанавливают со значениями 9-7, а предпочтительно примерно со значениями 8,7-8, в целях предотвращения спонтанного получения IHOG, и, таким образом, под действием вышеупомянутого белка осуществляют реакцию альдольной конденсации, которая является селективной по отношению к 4R-IHOG. Следовательно, 4R-селективность полученной IHOG может быть увеличена. Каждый специалист в данной области может определить подходящие условия реакции путем простого предварительного экспериментирования.
Альдолаза, рассматриваемая в настоящем изобретении, предположительно принадлежит к так называемой альдолазе класса II, и ферментативная активность этого фермента увеличивается при добавлении двухвалентного катиона. Двухвалентный катион, добавленный в реакционную систему, иногда влияет на спонтанную альдольную конденсацию, а поэтому он иногда влияет на стереоселективность полученной IHOG в положении 4. Каждый специалист в данной области может определить подходящие реакционные условия путем простой предварительной оценки типа и концентрации двухвалентного катиона, добавляемого в реакционную систему.
Скорость реакции может быть также увеличена путем добавления в реакционную смесь двухвалентного катиона, такого как Mg2+, Mn2+, Ni2+ и Co2+. С экономической точки зрения иногда предпочтительно использовать Mg2+. При добавлении в реакционную смесь двухвалентного катиона могут быть использованы любые соли при условии, что такая соль не ингибирует реакцию, при этом предпочтительно используют MgCl2, MgSO4 и MnSO4. Каждый специалист в данной области может определить концентрацию добавляемого двухвалентного катиона путем простого предварительного экспериментирования. Так, например, при добавлении Mg2+ скорость спонтанной конденсации IHOG может быть уменьшена путем поддержания концентрации добавленного Mg2+ на уровне 1 мм или менее, предпочтительно 0,5 мМ или менее, а более предпочтительно, 0,1 мМ или менее. Следовательно, 4R-селективность IHOG, получаемой под действием альдолазы, может быть увеличена.
Один из примеров предпочтительных реакционных условий осуществления способа получения замещенного 4R-IHOG настоящего изобретения описан ниже. 4-(Индол-3-илметил)-4-гидрокси-2-оксоглутаровую кислоту (IHOG) получают путем добавления промытых экспрессирующих альдолазу микробных клеток E.coli в качестве источника фермента с концентрацией 10% (мас./об.) в реакционную смесь, состоящую из 100 мМ буфера, 300 мМ индол-3-пировиноградной кислоты, 600 мМ пировиноградной кислоты, 0,1 мМ MgCl2 и 1% (об/об) толуола с последующим проведением реакции со встряхиванием указанной при 37°С в течение 4 часов.
Полученная IHOG может быть выделена и очищена общеизвестными способами. Таким способом может быть, например, способ, в котором IHOG подвергают контакту с ионообменной смолой для абсорбции основных аминокислот с последующим элюированием и кристаллизацией, или способ, в котором элюент обесцвечивают путем фильтрации через активированный уголь с последующей кристаллизацией. Реакционная смесь, содержащая полученную IHOG, может быть использована непосредственно в следующей стадии.
Что касается соотношения 4S-IHOG и 4R-IHOG, то в одном из примеров получения IHOG способом по настоящему изобретению было установлено, что, в случае использования SpALD в присутствии 0,1 мМ MgCl2 при рН 8,7-8,0 с добавлением 300 мМ индол-3-пировиноградной кислоты и 600 мМ пировиноградной кислоты в качестве субстрата, 4S-IHOG и 4R-IHOG получают в отношении примерно 4:96 (см. пример 12). Кроме того, оптическая чистота, составляющая 90% или более в положении 4 4R-IHOG-оксима, может быть достигнута путем кристаллизации реакционной смеси, содержащей изомер 4R-IHOG, полученный указанным способом, после проведения реакции оксимирования, описанной ниже. Измерение оптической чистоты является эквивалентным при измерении в любой форме, поскольку реакция, в которой IHOG превращают в IHOG-оксим с использованием гидроксиламина, не является оптически селективной по положению 4.
4R-IHOG, полученный указанным способом, может быть с успехом использован в качестве промежуточного соединения для получения 4R-монатина.
(2) Белок (альдолаза) с альдолазной активностью
Этот белок (иногда называемый просто "4R-альдолазой"), обладающий 4R-альдолазной активностью и используемый в способе по настоящему изобретению, отличается тем, что катализирует вышеупомянутую реакцию. 4R-Альдолаза может быть также получена из микроорганизма, обладающего 4R-альдолазной активностью, и такой микроорганизм может быть выявлен методом скрининга, описанным ниже.
(i) Микроорганизм, обладающий 4R-альдолазной активностью
(а) Метод скрининга для выявления микроорганизма, обладающего 4R-альдолазной активностью
Микроорганизм, обладающий 4R-альдолазной активностью, может быть получен из окружающей среды, такой как почва и вода. То есть для этого в данную среду желательно добавить монатин, IHOG, IHOG-оксим, PHG, PHOG или PHOG-оксим, которые являются субстратом для рассматриваемой альдолазы, в качестве источника углерода или источника азота, а предпочтительно в качестве единственного источника углерода или единственного источника азота, и инокулировать образец в качестве источника микроорганизма с последующим его культивированием. В качестве добавки может быть использована рацемическая смесь, но предпочтительно использовать 4R-изомер, а более предпочтительно (2R- и 4R)-монатин. Органические микроэлементы, не являющиеся источником углерода, могут быть соответствующим образом выбраны из обычных ингредиентов среды. В качестве источника азота могут быть использованы аммониевая соль органической кислоты, нитратная соль, органические азотистые соединения, такие как пептон, дрожжевой экстракт и мясной экстракт или их смеси. Кроме того, обычно используемые микроэлементы, такие как неорганические соли, следовые количества металлов и витамины, могут быть соответствующим образом смешаны. Микроорганизмы, способные расти в такой обогащенной культуральной среде, в избытке содержатся в бактериях, обладающих альдолазной активностью.
Затем из обогащенных микроорганизмов в вышеупомянутой среде получают одиночную колонию, и эту колонию снова культивируют в культуральном планшете с использованием соответствующего субстрата в качестве единственного источника углерода, а затем оценивают на альдолазную активность. После скрининга в качестве условий для культивирования, кроме источника углерода, могут быть использованы стандартные условия культивирования. В качестве примеров могут служить условия, описанные в приведенном ниже разделе (с) "Метод культивирования микроорганизмов, обладающих альдолазной активностью".
При проведении оценки альдолазной активности, получаемой микроорганизмом, желательно выделить данный фермент из микробных клеток и провести оценку ферментной реакции с использованием очищенного фермента. Конкретными примерами такого метода являются: (i) метод детекции выделенной пировиноградной кислоты, служащей в качестве субстрата, из IHOG или PHOG (детекция активности деградации) и (ii) метод детекции IHOG или PHOG, получаемых в реакции альдольной конденсации, проводимой с использованием индолпировиноградной кислоты или фенилпировиноградной кислоты и пировиноградной кислоты (или щавелево-уксусной кислоты) в качестве субстратов, с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) (детекция эффективности синтеза). Кроме того, желательно провести оценку 4R-селективности путем подтверждения с помощью ВЭЖХ молекулярной асимметрии в положении 4 в IHOG или PHOG, получаемых посредством альдольной конденсации, как описано в ii).
В частности, альдолазная активность может быть оценена путем добавления альдолазы в реакционную смесь, состоящую из 100 мМ буфера, 300 мМ индол-3-пировиноградной кислоты, 600 мМ пировиноградной кислоты, 0,1 мМ MgCl2 и 1% (об./об.) толуола, с последующим проведением реакции со встряхиванием при 37°С в течение 4 часов и количественной оценки полученной IHOG с помощью ВЭЖХ.
Количество IHOG может быть оценено с использованием ВЭЖХ-системы, снабженной, например, колонкой "Inertsil ODS-2" (5 мкм, 4,6×250 мм), поставляемой GL Sciences Inc. Один из примеров условий анализа приводится ниже.
Подвижная фаза: 40% (об./об.) ацетонитрил/5 мМ раствор дигидрофосфата тетрабутиламмония.
Скорость потока: 1 мл/мин.
Температура колонки: 40°С.
Детекция: УФ 210 нм.
(b) Микроорганизмы, выявленные путем скрининга
В результате скрининга обогащенных микроорганизмов авторами настоящего изобретения были отобраны микроорганизмы, принадлежащие к роду Sphingomonas, и микроорганизмы, принадлежащие к роду Burkholderia, и было обнаружено, что альдолазу, используемую в настоящем изобретении, получают микроорганизмами, принадлежащими к обоим указанным видам микроорганизмов, и родственными микроорганизмами. Поэтому микроорганизмами, обладающими альдолазной активностью, используемой в настоящем изобретении, являются микроорганизмы, принадлежащие к роду Sphingomonas, роду Burkholderia или к родственным родам. Примерами родственных видов Sphingomonas могут служить микроорганизмы рода Rhizomonas, Blastomonas, Erythromicrobium, Porphyrobacter, Agrobacterium и Erythrobacter. Недавно была предложена переклассификация микроорганизмов рода Sphingomonas, вследствие чего такие микроорганизмы иногда также относят к микроорганизмам рода Sphingobium, Novosphingobium или Sphingopixis (International Journal of Systematic and Evolutionary Microbiology (2001), 51, 1405-1417), однако используемый здесь термин "Sphingomonas" охватывает микроорганизмы этих родов.
Примерами микроорганизмов, принадлежащих к роду Sphingomonas, могут служить микроорганизмы Sphingomonas sp., Sphingomonas trueperi, Sphingomonas parapaucimobilis, Sphingomonas sanguinis, Sphingomonas paucimobilis, Sphingomonas adhaesiva, Sphingomonas pruni, Sphingomonas mali, Sphingomonas asaccharolytica, Sphingomonas echinoids, Sphingomonas yanoikuyae, Sphingomonas herbicidovorans, Sphingomonas chlorophenolica, Sphingomonas agrestis, Sphingomonas rosa, Sphingomonas subarctica, Sphingomonas stygia, Sphingomonas subterranean, Sphingomonas aromaticivorans, Sphingomonas capsulate, Sphingomonas macrogoltabidus, Sphingomonas terrae, Rhizomonas suberifaciens, Blastomonas natatoria, Blastomonas ursincola, Agrobacterium sanguineum, Erythrobacter longus, Erythrobacter litoralis.
Примерами микроорганизмов, принадлежащих к роду Burkholderia, могут служить микроорганизмы Burkholderia sp., Burkholderia phenazinium, Burkholderia caribensis, Burkholderia graminis, Burkholderia kururiensis, Burkholderia brasilensis, Burkholderia caryophylli, Burkholderia glathei, Burkholderia plantarii, Burkholderia vandii, Burkholderia glumae, Burkholderia cocovenenans, Burkholderia gladioli, Burkholderia vietnamiensis, Burkholderia multivorans, Burkholderia cepacia, Burkholderia pyrrocinia, Burkholderia thailandensis, Burkholderia pseudomallei, Burkholderia mallei, Burkholderia andropogonis (Current Microbiology Vol. 42 (2001), pp. 269-275).
В частности, предпочтительно в настоящее изобретение включены нижеследующие микроорганизмы. Адреса депозитариев этих микроорганизмов приводятся ниже.
Штамм AJ110329 Sphingomonas sp. (штамм С77)
(i) Регистрационный номер: FERM BP-10027
(ii) Дата принятия первичной заявки на депонирование: 21 мая, 2004.
(iii) Адрес депозитария: International Patent Organism Depositary, National Institute of Advanced Industrial Science and Technology (Central № 6, 1-1-1 Higashi, Tsukuba-shi, Ibaraki Prefecture, Japan).
Штамм AJ110372 Sphingomonas sp. (штамм С43)
(i) Регистрационный номер: FERM BP-10156
(ii) Дата принятия первичной заявки на депонирование: 28 октября, 2004.
(iii) Адрес депозитария: International Patent Organism Depositary, National Institute of Advanced Industrial Science and Technology (Central № 6, 1-1-1 Higashi, Tsukuba-shi, Ibaraki Prefecture, Japan).
Штамм AJ110371 Burkholderia sp. (штамм С24)
(i) Регистрационный номер: FERM BP-10155
(ii) Дата принятия первичной заявки на депонирование: 28 октября, 2004.
(iii) Адрес депозитария: International Patent Organism Depositary, National Institute of Advanced Industrial Science and Technology (Central № 6, 1-1-1 Higashi, Tsukuba-shi, Ibaraki Prefecture, Japan).
Штамм AJ110329 (штамм С77, FERM BP-10027) был идентифицирован как вышеупомянутый Sphingomonas sp. в результате проведения нижеследующих экспериментов для классификации.
Область примерно в 500 п.н. у 5'-конца гена рибосомной РНК 16S (рДНК 16S) амплифицировали с помощью ПЦР из геномной ДНК штамма AJ110329 и секвенировали (SEQ ID NO:16). Гомологию полученной последовательности анализировали с использованием бактериальной библиотеки Microseq Bacterial 500 Library v.0023 (Apllied Biosystems, СА, USA) в качестве базы данных и с использованием системного программного обеспечения для идентификации микроорганизмов (MicroSeq Microbial Identification System Software V. 1.4.1). В результате этого анализа не было обнаружено какой-либо известной последовательности, которая соответствовала бы нуклеотидной последовательности рДНК 16S штамма AJ110329 и которая обнаруживала бы наиболее высокую гомологию, составляющую 96,6%, с последовательностью рДНК 16S Sphingomonas capsulata. В молекулярном филогенетическом древе рДНК 16S штамма AJ110329 была включена в кластер, образованный рДНК 16S Sphingomonas. Его бактериальные характеристики, представленные в таблице 2, также совпадали с результатом анализа нуклеотидной последовательности рДНК 16S. В соответствии с этим, штамм AJ110329 был определен как штамм Sphingomonas sp.
Штамм AJ110372 (штамм С43, FERM BP-10156) был идентифицирован как вышеупомянутый штамм Sphingomonas sp. в результате проведения нижеследующего эксперимента по классификации.
Область примерно в 500 п.н. у 5'-конца гена рибосомной РНК 16S (рДНК 16S) амплифицировали с помощью ПЦР из геномной ДНК штамма AJ110372 и секвенировали (SEQ ID NO:17). Гомологию полученной последовательности анализировали с использованием бактериальной библиотеки Microseq Bacterial 500 Library v.0023 (Apllied Biosystems, СА, USA) в качестве базы данных и с использованием системного программного обеспечения для идентификации микроорганизмов (MicroSeq Microbial Identification System Software V. 1.4.1). В результате этого анализа не было обнаружено какой-либо известной последовательности, которая соответствовала бы нуклеотидной последовательности рДНК 16S штамма AJ110372 и которая имела бы наиболее высокую гомологию, составляющую 98,94%, с последовательностью рДНК 16S Sphingomonas yanoikuyae. В молекулярном филогенетическом древе рДНК 16S штамма AJ110372 была включена в кластер, образованный рДНК 16S Sphingomonas. В соответствии с этим, штамм AJ110372 был определен как штамм Sphingomonas sp.
Штамм AJ110371 (штамм С24, FERM BP-10155) был идентифицирован как вышеупомянутый штамм Burkholderia sp. в результате проведения нижеследующих экспериментов по классификации.
Область примерно в 500 п.н. у 5'-конца гена рибосомной РНК 16S (рДНК 16S) амплифицировали с помощью ПЦР из геномной ДНК штамма AJ110371 и секвенировали (SEQ ID NO:18). Гомологию полученной последовательности анализировали с использованием бактериальной библиотеки Microseq Bacterial 500 Library v.0023 (Apllied Biosystems, СА, USA) в качестве базы данных и с использованием системного программного обеспечения для идентификации микроорганизмов (MicroSeq Microbial Identification System Software V. 1.4.1). В результате этого анализа не было обнаружено какой-либо известной последовательности, которая соответствовала бы нуклеотидной последовательности рДНК 16S штамма AJ110371 и которая имела бы наиболее высокую гомологию, составляющую 95,21%, с последовательностью рДНК 16S Burkholderia phenazinium. В молекулярном филогенетическом древе рДНК 16S штамма AJ110371 была включена в кластер, образованный рДНК 16S Burkholderia. Его бактериальные характеристики, представленные в таблице 3, также совпадали с результатом анализа нуклеотидной последовательности рДНК 16S. В соответствии с этим, штамм AJ110371 был определен как штамм Burkholderia sp.
Бактериальные характеристики штамма AJ110329 Sphingomonas (FERM ВР-10027) описаны в нижеследующей таблице 2-1
температура
Бактериальные характеристики штамма AJ110371 Burkholderia (FERM ВР-10155) описаны в нижеследующей таблицах 3-1 и 3-2.
Литература и используемый набор:
1) BARROW (G.I.) and FELTHAM (R.K.A): Cowan & Steel's Manual for the Identification of Medical Bacteria. 3rd edition. 1993, Cambridge University Press.
2) Toshikazu Sakazaki, Etsuro Yoshizaki and Kanji Miki: Shin Saikin Baichigaku Kouza II (2nd edition), 1988, Kindai Shuppan, Tokyo.
3) Набор для идентификации бактерий: API20, NE (bioMerieux, France: http://www.biomerieux.fr/home_en.htm).
c) Метод культивирования микроорганизма, обладающего альдолазной активностью
Культивирование микроорганизма, являющегося источником альдолазы, может быть осуществлено в жидкой культуре или на твердой культуре, но для промышленных целей, предпочтительным методом является культивирование взвешенной культуры с перемешиванием и аэрацией. Для культивирования микроорганизмов могут быть использованы обычно используемые для этих целей источники азота в питательной среде, источники углерода, источники азота, неорганические соли и микроэлементы. Все они могут быть использованы при условии, если они утилизуются данным штаммом.
В качестве условий аэрации используются аэробные условия. Температура культивирования может варьироваться в пределах, приемлемых для бактериального роста и получения альдолазы. Поэтому эти условия не являются жесткими, и температура обычно составляет от 10 до 50°С, а предпочтительно 30-40°С. Время культивирования варьируется в зависимости от других условий культивирования. Так, например, культивирование может осуществляться до тех пор, пока происходит наиболее интенсивное получение альдолазы, обычно в период времени от 5 часов до 7 дней, а предпочтительно примерно от 10 часов до 3 дней.
d) Метод выделения альдолазы из микроорганизма
После культивирования микроорганизма, обладающего альдолазной активностью, микробные клетки собирают путем центрифугирования (например, при 10000×g, 10 мин). Альдолазу солюбилизируют путем разрушения или лизиса микробных клеток, поскольку наибольшее количество альдолазы присутствует в микробных клетках. Микробные клетки разрушают путем ультразвукового разрушения, разрушения с использованием Френч-пресса или разрушения стеклянными шариками. Микробные клетки подвергают лизису путем обработки лизоцимом яичного белка или пептидазой или их комбинацией.
Очистку альдолазы, выделенной из продуцирующих альдолазу бактерий, осуществляют с использованием ферментного раствора в качестве исходного материала. Если присутствуют неразрушенный или нелизированный клеточный дебрис, то этот осажденный дебрис желательно удалить путем повторного центрифугирования данного раствора.
Альдолаза может быть очищена любыми стандартными методами, обычно применяемыми для очистки этого фермента, например высаливанием аммониевым сульфатом, гель-хроматографией, ионообменной хроматографией, гидрофобной хроматографией и хроматографией на гидроксиапатите. В результате такой очистки может быть получена фракция, содержащая альдолазу с более высокой удельной активностью.
Аминокислотная последовательность в аминоконцевом домене может быть определена путем помещения очищенного препарата альдолазы в секвенатор для определения последовательностей белков методом деградации по Эдману (Edman, P., Acta Chem. Acand. 4, 227 (1950)). Внутренняя аминокислотная последовательность может быть определена путем получения и очистки пептидного препарата с помощью обращенно-фазовой хроматографии после его обработки пептидазой и с последующим его помещением в секвенатор для определения последовательности белка методом деградации по Эдману.
На основании представленной аминокислотной последовательности может быть определена нуклеотидная последовательность кодирующей ДНК. Нуклеотидная ДНК-последовательность может быть выведена путем использования универсальных кодонов.
На основании определенной нуклеотидной последовательности может быть синтезирована молекула ДНК, содержащая примерно 30 пар оснований. Способ синтеза молекулы ДНК описан в Tetrahedron Letters, 22:1859 (1981). Молекула ДНК может быть также синтезирована с использованием синтезатора, поставляемого Apllied Biosystems. Эта молекула ДНК может быть использована в качестве зонда при выделении кодирующей альдолазу полноразмерной ДНК из хромосомной генной библиотеки бактерий, продуцирующих альдолазу. Альтернативно, молекула ДНК может быть использована в качестве праймера, если ДНК, кодирующая альдолазу настоящего изобретения, амплифицирована посредством ПЦР. Однако, если ДНК, амплифицированная с помощью ПЦР, не содержит полноразмерную ДНК, кодирующую альдолазу, то кодирующую альдолазу полноразмерную ДНК выделяют из хромосомной генной библиотеки бактерий, продуцирующих альдолазу, с использованием ПЦР-амплифицированной ДНК в качестве зонда.
Процедура ПЦР описана White, T.J. et al., Trends Genet., 5:185 (1989). Метод получения хромосомной ДНК и метод выделения рассматриваемой молекулы ДНК из генной библиотеки с использованием молекулы ДНК в качестве зонда, описан в Molecular Cloning: 2nd edition, Cold Spring Harbor press (1989).
Способ определения нуклеотидной последовательности выделенной ДНК, кодирующей альдолазу, описан в A Practical Guide to Molecular Cloning, John Wiley & Sons, Inc. (1985). Альтернативно нуклеотидная последовательность может быть определена с использованием ДНК-севенатора, поставляемого Apllied Biosystems.
Метод получения кодирующей альдолазу ДНК из продуцирующих альдолазу бактерий также включает метод получения ДНК, которая гибридизуется с полноразмерной или неполной последовательностью ДНК, кодирующей альдолазу настоящего изобретения, в качестве зонда.
Для получения ДНК, кодирующей альдолазу, из продуцирующих альдолазу бактерий, ДНК-последовательность, выведенная из высококонсервативной области аминокислотной последовательности путем сопоставления аминокислотной последовательности настоящего изобретения методом выравнивания, может быть использована в качестве зонда или праймера в ПЦР. Если ДНК, амплифицированная с помощью ПЦР, не содержит полноразмерную ДНК, кодирующую альдолазу, то полноразмерную ДНК, кодирующую альдолазу, выделяют из хромосомной генной библиотеки бактерий, продуцирующих альдолазу, с использованием ПЦР-амплифицированной ДНК в качестве зонда.
Таким образом получают микроорганизм, обладающий альдолазной активностью, и из полученного микроорганизма могут быть выделены альдолаза и ДНК, кодирующая альдолазу.
(ii) Альдолаза
Авторами настоящего изобретения была получена альдолаза из штамма С77 (штамм AJ110329 Sphingomonas sp.), выделенного вышеупомянутым методом скрининга, и эта альдолаза была обозначена как SpALD. ДНК-последовательность, кодирующая SpALD по настоящему изобретению и определенная вышеупомянутыми методами, представлена в SEQ ID NO:1 (CDS: нуклеотиды 210-1004), и аминокислотная последовательность SpALD представлена в SEQ ID NO:2.
Кроме того, авторами настоящего изобретения был проведен скрининг микроорганизмов (подробно описанный в примерах), а также были получены новые гены альдолазы из штамма С43 (штамм AJ110372 Sphingomonas sp.) и штамма С24 (штамм AJ110371 Burkholderia sp.) (SEQ ID NO:12 и 14) с помощью Саузерн-анализа и путем гибридизации колоний с использованием неполных нуклеотидных последовательностей SEQ ID NO:1 в качестве зондов, и был получен белок (SpALD2), имеющий аминокислотную последовательность SEQ ID NO:13, и белок (BuALD), имеющий аминокислотную последовательность SEQ ID NO:15. Сравнение последовательностей SpALD, SpALD2 и BuALD проиллюстрировано на фиг.1. Коровая последовательность (SEQ ID NO:23) альдолазы, которая является общей для всех трех альдолаз, показана на верхней дорожке данной фигуры. Число аминокислотных остатков, общих для этих трех альдолаз, равно 200, что составляет 70,2% от всех остатков. Число аминокислотных остатков, общих для двух из трех указанных альдолаз, равно 76, что вместе с коровой последовательностью составляет 96,8% от всех остатков. Гомология между SpALD и SpALD2 составляет 88,4%, а гомология между SpALD и BuALD составляет 74,7%.
Кроме того, была проанализирована гомология с другими последовательностями, известными из публикаций. В результате было установлено, что аминокислотная последовательность SEQ ID NO:2 на 20% гомологична последовательности (2,4-дигидроксигепт-2-ен-1,7-дионовая кислота)-альдолазы (НрсН), происходящей от штамма С Escherichia coli (Stringfellow J.M. et al., Gene. 1995 166(1):73-6). Гомология с другими известными альдолазами, такими как IHOG-альдолаза (название гена: PtALD), происходящая от Pseudomonas taetrolens, IHOG-альдолаза (название гена: PcALD), происходящая от Pseudomonas coronafaciens (проспект публикации Международной заявки № 03/056026), 4-гидрокси-4-метил-2-оксоглутарат-альдолаза (название гена: proA), происходящая от Comamonas testeroni ATCC 49249 или 4-гидрокси-4-метил-2-оксоглутарат-альдолаза (название гена: proA), происходящая от Pseudomonas straminea, составляет, по меньшей мере, 10% или менее, то есть гомология почти отсутствует, что указывает на то, что альдолазы настоящего изобретения являются абсолютно новыми белками. Три альдолазы, полученные авторами настоящего изобретения, также составляют часть настоящего изобретения, и иногда будут обозначаться общим термином "альдолаза настоящего изобретения".
Гомология была проанализирована с использованием программного обеспечения для анализа генов "Genetyx ver.6" (Genetyx Corporation), а величины были вычислены с использованием параметров по умолчанию.
Любой из этих белков, имеющих гомологию 70% или более, предпочтительно 74% или более, более предпочтительно 80% или более, еще более предпочтительно 85% или более и особенно предпочтительно 95% или более по отношению к аминокислотной последовательности SEQ ID NO:2, обладает аналогичной ферментативной активностью и может быть использован в целях настоящего изобретения. Для этих целей особенно подходящим является белок, который на 70% или более гомологичен аминокислотной последовательности SEQ ID NO:2 и который содержит коровую последовательность, соответствующую SEQ ID NO:23.
Поэтому, белок, обладающий 4R-альдолазной активностью и используемый в способе настоящего изобретения, включает альдолазу настоящего изобретения, и таким белком являются белки (а) и (b), а именно:
(а) белок, имеющий аминокислотную последовательность SEQ ID NO:2; и
(b) белок, являющийся, по крайней мере, на 70% или более гомологичным аминокислотной последовательности SEQ ID NO:2 и обладающий 4R-альдолазной активностью.
Кроме того, белок (b) включает следующий белок (с):
(с) белок, имеющий любую аминокислотную последовательность из SEQ ID NO:13 или 15.
Альдолаза настоящего изобретения включает следующие белки:
d) белок, имеющий любую аминокислотную последовательность из SEQ ID NO:2, 13 или 15, и
(е) белок, имеющий аминокислотную последовательность, содержащую мутацию, выбранную из группы, состоящей из замены, делеции, инсерции, добавления и инверсии одного или нескольких аминокислотных остатков в любой из аминокислотных последовательностей SEQ ID NO:2, 13 или 15.
Используемый здесь термин "один или несколько" означает число аминокислотных остатков в белке, которое существенно не влияет на его трехмерную структуру и альдолазную активность, а в частности, такое число составляет от 1 до 90, предпочтительно 1-75, более предпочтительно 1-57, еще более предпочтительно 1-43, а особенно предпочтительно 1-15. При этом желательно, чтобы белок, имеющий аминокислотную последовательность, содержащую один или несколько замененных, делетированных, инсертированных, добавленных или инвертированных аминокислотных остатков в любой из аминокислотных последовательностей SEQ ID NO:2, 13 или 15, сохранял не менее чем 10%, предпочтительно не менее чем 30%, более предпочтительно не менее чем 50%, а еще более предпочтительно не менее чем 70% альдолазной активности, преимущественно 4R-альдолазной активности белка, имеющего любую из аминокислотных последовательностей SEQ ID NO:2, 13 или 15 при температуре 33°С и рН 9.
Вышеуказанными заменами, делециями, инсерциями или добавлениями являются консервативные мутации, при которых сохраняется альдолазная активность. Такой консервативной мутацией обычно является консервативная замена. Замена аминокислот, рассматриваемая как консервативная замена, включает замену Ala на Ser или Thr, замену Arg на Gln, His или Lys, замену Asn на Glu, Gln, Lys, His или Asp, замену Asp на Asn, Glu или Gln, замену Cys на Ser или Ala, замену Gln на Asn, Glu, Lys, His, Asp или Arg, замену Glu на Asn, Gln, Lys или Asp, замену Gly на Pro, замену His на Asn, Lys, Gln, Arg или Tyr, замену Ile на Leu, Met, Val или Phe, замену Leu на Ile, Met, Val или Phe, замену Lys на Asn, Glu, Gln, His или Arg, замену Met на Ile, Leu, Val или Phe, замену Phe на Trp, Tyr, Met, Ile или Leu, замену Ser на Thr или Ala, замену Thr на Ser или Ala, замену Trp на Phe или Tyr, замену Tyr на His, Phe или Trp и замену Val на Met, Ile или Leu.
Альдолазная активность, используемая в способе настоящего изобретения, означает 4R-альдолазную активность, причем к оптической селективности альдолазы (белка) настоящего изобретения не предъявляется строгих требований, однако более предпочтительной является 4R-альдолазная активность. Используемый в нижеследующем описании способа настоящего изобретения термин "альдолазная активность" означает 4R-альдолазную активность, которая может катализировать реакцию альдольной конденсации индолпировиноградной кислоты и пировиноградной кислоты или щавелево-уксусной кислоты, описанную в нижеследующем разделе "(1) Реакция", в которой преимущественно получают 4R-IHOG. Используемый здесь термин "преимущественное получение 4R-IHOG" означает, что оптическая чистота R-изомера превышает оптическую чистоту S-изомера в положении 4 получаемой IHOG, а предпочтительно означает, что в данной реакции эффективно получают R-изомер с оптической чистотой 70% или более, а особенно предпочтительно 90% или более.
При этом, если "альдолазная активность" означает активность белка настоящего изобретения, то такая активность означает альдолазную активность, которая не требует оптической селективности. Такой альдолазной активностью, не требующей оптической селективности, является активность, которая стимулирует синтез IHOG из индолпировиноградной кислоты и пировиноградной кислоты (или щавелево-уксусной кислоты) и для которой не требуется оптической селективности, поскольку альдолаза настоящего изобретения является эффективной независимо от уровня ее оптической селективности.
Химическая природа очищенных ферментов SpALD и BuALD описана ниже.
SpALD и BuALD катализируют реакцию, в которой при взаимодействии индолпировиноградной кислоты с пировиноградной кислотой или щавелево-уксусной кислотой преимущественно получают 4R-IHOG.
Оптимальный рН для SpALD составляет примерно 7,1-8,0 при 37°С. SpALD обладает стабильностью при рН 8 или ниже и при температуре 50°С или ниже, а при температуре 30°С или ниже, он обладает высокой стабильностью.
Молекулярная масса SpALD составляет примерно 155 кДа, как было определено с помощью гель-фильтрации, и примерно 30 кДа, как было определено с помощью электрофореза в ДСН-ПААГ. Таким образом, было установлено, что SpALD имеет гексамерную структуру с субъединицей, имеющей молекулярную массу примерно 30 кДа.
Поэтому, в другом аспекте изобретения, белок настоящего изобретения отличается тем, что
(А) обладает катализирующей реакцию активностью, в которой 4-(индол-3-илметил)-4-гидрокси-2-оксоглутаровую кислоту получают путем альдольной конденсации индол-3-пировиноградной кислоты и пировиноградной кислоты, и/или активностью, катализирующей реакцию, в которой 4-фенилметил-4-гидрокси-2-оксоглутаровую кислоту получают путем альдольной конденсации фенилпировиноградной кислоты и пировиноградной кислоты;
(В) обладает активностью, определенной в (А) при оптимальном рН примерно 7,5-8,0 и при 37°С;
(С) обладает стабильностью при рН 8 или ниже;
(D) обладает стабильностью при температуре 50°С или ниже; и
(Е) имеет молекулярную массу примерно 155 кДа, как было определено с помощью гель-фильтрации, и молекулярную массу примерно 30 кДа, как было определено с помощью электрофореза в ДСН-ПААГ.
BuALD имеет оптимальный рН примерно 6,5-7,5 при 37°С и обладает стабильностью при рН 7,5 или ниже и при температуре 37°С или ниже, а при температуре 30°С или ниже он обладает высокой стабильностью.
Молекулярная масса BuALD составляет примерно 160 кДа, как было определено с помощью гель-фильтрации, и примерно 30 кДа, как было определено с помощью электрофореза в ДСН-ПААГ. Таким образом, было установлено, что BuALD имеет гексамерную структуру с субъединицей, имеющей молекулярную массу примерно 30 кДа.
Поэтому в другом аспекте изобретения, белок настоящего изобретения отличается тем, что
(А) обладает катализирующей реакцию активностью, в которой 4-(индол-3-илметил)-4-гидрокси-2-оксоглутаровую кислоту получают путем альдольной конденсации индол-3-пировиноградной кислоты и пировиноградной кислоты, и/или активностью, катализирующей реакцию, в которой 4-фенилметил-4-гидрокси-2-оксоглутаровую кислоту получают путем альдольной конденсации фенилпировиноградной кислоты и пировиноградной кислоты;
(В) обладает активностью, определенной в (А) при оптимальном рН примерно 6,5-7,5 и при 37°С;
(С) обладает стабильностью при рН 8,5 или ниже;
(D) обладает стабильностью при температуре 37°С или ниже; и
(Е) имеет молекулярную массу примерно 160 кДа, как было определено с помощью гель-фильтрации, и молекулярную массу примерно 30 кДа, как было определено с помощью электрофореза в ДСН-ПААГ.
(iii) ДНК, кодирующая альдолазу
Авторами настоящего изобретения были получены ген альдолазы настоящего изобретения, имеющий нуклеотидную последовательность SEQ ID NO:1, ген альдолазы настоящего изобретения, имеющий нуклеотидную последовательность SEQ ID NO:12, и ген альдолазы настоящего изобретения, имеющий нуклеотидную последовательность SEQ ID NO:14, из штамма AJ110372 Sphingomonas sp. (штамма С77), штамма AJ110329 Sphingomonas sp. (штамма С43) и штамма AJ110371 Burkholderia sp. (штамма С24), соответственно, которые представляют собой продуцирующие альдолазу бактерии, выделенные авторами настоящего изобретения, как описано выше в (i) и (ii). Эти гены кодируют альдолазы настоящего изобретения, катализирующие реакцию, в которой IHOG синтезируется из индолпировиноградной кислоты и пировиноградной кислоты (или щавелево-уксусной кислоты), и эти гены входят в объем настоящего изобретения.
ДНК, кодирующая альдолазу, представляет собой не только ДНК, указанную в SEQ ID NO:1, 12 и 14. То есть это объясняется различиями в нуклеотидных последовательностях, наблюдаемыми для каждого вида и штамма бактерий, принадлежащих к роду Sphingomonas и Burkholderia, которые продуцируют альдолазу, катализирующую реакцию синтеза IHOG из индолпировиноградной кислоты и пировиноградной кислоты (или щавелево-уксусной кислоты).
Таким образом, настоящее изобретение относится к ДНК, кодирующей белок, который не менее чем на 70%, предпочтительно не менее чем на 74%, более предпочтительно не менее чем на 80%, еще более предпочтительно не менее чем на 85%, а особенно предпочтительно не менее чем на 95%, гомологичен аминокислотной последовательности SEQ ID NO:2 и который обладает альдолазной активностью, а предпочтительно 4R-альдолазной активностью, или белок, который не менее чем на 70% гомологичен аминокислотной последовательности SEQ ID NO:2 и который включает коровую последовательность альдолазы SEQ ID NO:23 и обладает альдолазной активностью, а предпочтительно 4R-альдолазной активностью.
ДНК настоящего изобретения представляет собой не только ДНК, кодирующую выделенную альдолазу, но очевидно также, что настоящее изобретение включает ДНК, кодирующую альдолазу и выделенную из хромосомной ДНК искусственно мутированных и продуцирующих альдолазу микроорганизмов при условии, что эта ДНК кодирует альдолазу. Наиболее часто используемым методом внесения искусственной мутации является метод сайт-направленного мутагенеза, описанный в "Method in Enzymol., 154 (1987)".
ДНК, которая гибридизуется с ДНК, содержащей нуклеотидную последовательность, комплементарную нуклеотидной последовательности SEQ ID NO:1, 12 или 14, в жестких условиях и которая кодирует белок, обладающей альдолазной активностью, а предпочтительно 4R-альдолазной активностью, также представляет собой ДНК настоящего изобретения. Используемый здесь термин "жесткие условия" означает условия, при которых образуется так называемый "специфический гибрид", а не "неспецифический гибрид". Хотя такие условия трудно поддаются количественному определению, однако их примерами могут служить условия, в которых гибридизуется пара ДНК-последовательностей с высокой степенью гомологии, например ДНК-последовательности, имеющие гомологию 50% или более, более предпочтительно 80% или более, еще более предпочтительно 90% или более, а особенно предпочтительно 95% или более, тогда как ДНК с более низкой степенью гомологии не гибридизуются; и условия промывки в стандартной Саузерн-гибридизации, то есть гибридизации при концентрациях соли, эквивалентных 0,1 × SSC и 0,1% ДСН при 37°С, предпочтительно 0,1 × SSC и 0,1% ДСН при 60°С, а более предпочтительно 0,1 × SSC и 0,1% ДСН при 65°С. Используемые здесь термины "альдолазная активность" или "4R-альдолазная активность" являются такими, как они были определены выше в разделе "(ii) Альдолаза". Однако в случае нуклеотидной последовательности, которая гибридизуется с нуклеотидной последовательностью, комплементарной нуклеотидной последовательности SEQ ID NO:1, 12 или 14, в жестких условиях, желательно, чтобы белок, происходящий от такой последовательности, примерно на 10% или более, предпочтительно на 30% или более, более предпочтительно на 50% или более, а еще более предпочтительно на 70% или более сохранял альдолазную активность, а предпочтительно 4R-альдолазную активность белка, имеющего аминокислотную последовательность SEQ ID NO:2, 13 или 15, при 33°С и при рН 9.
Кроме того, молекулой ДНК, кодирующей, в основном, такой же белок, как и альдолаза, кодируемая ДНК SEQ ID NO:1, 12 или 14, также является ДНК настоящего изобретения. То есть ДНК настоящего изобретения представляет собой:
(а) ДНК, включающую последовательность нуклеотидов №№ 210-1004 в нуклеотидной последовательности SEQ ID NO:1;
(b) ДНК, включающую последовательность нуклеотидов №№ 399-1253 в нуклеотидной последовательности SEQ ID NO:12;
(с) ДНК, включающая последовательность нуклеотидов №№ 531-1385 в нуклеотидной последовательности SEQ ID NO:14;
(d) ДНК, которая гибридизуется с ДНК, содержащей нуклеотидную последовательность, комплементарную нуклеотидной последовательности SEQ ID NO:1 или последовательности нуклеотидов №№ 210-1004 в той же самой последовательности; комплементарную нуклеотидной последовательности SEQ ID NO:12 или последовательности нуклеотидов №№ 399-1253 в той же самой последовательности, или комплементарную нуклеотидной последовательности SEQ ID NO:14 или последовательности нуклеотидов №№ 531-1385 в той же самой последовательности, и которая кодирует белок, обладающий альдолазной активностью;
(е) молекулу ДНК, кодирующую белок, имеющий аминокислотную последовательность SEQ ID NO:2, 13 или 15;
(f) молекулу ДНК, кодирующую белок, имеющий аминокислотную последовательность, содержащую мутацию, выбранную из группы, состоящей из замен, делеций, инсерций, добавлений или инверсий одного или нескольких аминокислотных остатков в аминокислотной последовательности SEQ ID NO:2, 13 или 15, и обладающий альдолазной активностью, и
(g) молекулу ДНК, кодирующую белок, который имеет аминокислотную последовательность, на 70% или более гомологичную аминокислотной последовательности SEQ ID NO:2, 13 или 15, и который обладает альдолазной активностью.
Используемый здесь термин "один или более" имеет значение, определенное в вышеописанном разделе "(ii) Альдолаза".
(3) Способ получения альдолазы
Способ получения альдолазы описан ниже. Альдолаза настоящего изобретения может быть получена двумя способами, а именно (i) способом, предусматривающим получение и аккумуляцию альдолазы путем культивирования продуцирующего альдолазу микроорганизма, и (ii) способом, предусматривающим получение продуцирующего альдолазу трансформанта методами рекомбинантных ДНК с последующим получением и аккумуляцией альдолазы путем культивирования такого трансформанта. Описанный способ (i) представляет собой способ получения продуцирующего альдолазу микроорганизма и способ культивирования такого продуцирующего альдолазу микроорганизма, описанные в вышеуказанном разделе (2) (i). Способ (ii) описан ниже.
(ii) Способ с применением техники рекомбинантных ДНК
В литературе описано множество примеров получения нужных белков, таких как ферменты и физиологически активные вещества, методами рекомбинантных ДНК. Применение методов рекомбинантных ДНК дает возможность осуществлять крупномасштабное получение нужного белка, который присутствует в природе в очень малых количествах.
На фиг.2 представлена блок-схема, иллюстрирующая стадии получения альдолазы настоящего изобретения.
Сначала получают молекулу ДНК, кодирующую альдолазу настоящего изобретения (стадия S1). Затем полученную молекулу ДНК лигируют с ДНК-вектором и получают рекомбинантную ДНК (стадия S2), после чего клетки трансформируют указанной рекомбинантной ДНК и получают трансформанты (стадия S3). После этого, при культивировании трансформантов в среде, альдолаза продуцируется и аккумулируется в этой среде и/или в клетках (стадия S4).
Затем очищенную альдолазу получают в стадии S5 путем выделения и очистки фермента.
Рассматриваемая оптически активная IHOG может быть получена в промышленном масштабе с использованием очищенной альдолазы, полученной в стадии S5, либо она может быть получена в среде и/или в клетках, в которых альдолаза аккумулируется (в стадии S4) для проведения альдольной реакции (в стадии S6).
Молекула ДНК, лигированная с ДНК-вектором, может экспрессировать альдолазу настоящего изобретения.
В качестве гена альдолазы, лигированного с ДНК-вектором, может быть использована любая из молекул ДНК, описанных в вышеуказанном разделе "(2) Альдолаза (iii) ДНК".
В случае крупномасштабного получения белка с применением техники рекомбинантных ДНК предпочтительный способ может включать получение телец включения указанного белка. К преимуществам такого способа экспрессии можно отнести защиту рассматриваемого белка от расщепления протеазами, присутствующими в микробных клетках, и простоту очистки рассматриваемого белка, которая может быть осуществлена путем разрушения микробных клеток и последующего центрифугирования.
Тельца включения белка, полученные этим способом, могут быть солюбилизированы с использованием агента, денатурирующего белок, после чего активность этих телец включения восстанавливают, главным образом, путем удаления денатурирующего агента, с получением белка, имеющего правильную повторную укладку и обладающего физиологической активностью. Имеется много примеров таких процедур, например регенерация активности человеческого интерлейкина 2 (JP 61-257931 A).
Для получения активного белка из телец включения белка требуется проведение серии манипуляций, таких как солюбилизация и регенерация активности, и такие манипуляции являются более сложными, чем манипуляции, проводимые в случае прямого получения активного белка. Однако, если белок, который влияет на рост микробных клеток, продуцируется в микробных клетках в большом количестве, то такое влияние может быть ингибировано путем аккумуляции такого белка в виде неактивных телец включения в микробных клетках.
Способами крупномасштабного получения рассматриваемого белка в виде телец включения могут быть способы экспрессии отдельного белка под контролем сильного промотора, а также способы экспрессии рассматриваемого белка в виде гибрида с белком, о котором известно, что он экспрессируется в большом количестве.
Этот способ является эффективным для введения последовательности, распознаваемой рестриктирующей протеазой, в соответствующее положение, и для отщепления рассматриваемого белка после экспрессии гибридного белка.
В случае крупномасштабного получения белка методами рекомбинантных ДНК трансформируемыми клетками-хозяевами могут быть бактериальные клетки, клетки актиномицетов, дрожжевые клетки, клетки грибов, клетки растений и клетки животных. Микроорганизмом, используемым в системе хозяин-вектор, может быть микроорганизм рода Escherichia, рода Pseudomonas, рода Corynebacterium и рода Bacillus, а предпочтительно Escherichia coli, поскольку существует множество технологий крупномасштабного получения белка с использованием Escherichia coli. Способ получения альдолазы с использованием трансформированной Escherichia coli (E.coli) описан ниже.
В качестве промоторов экспрессии кодирующей ДНК могут быть использованы промоторы, обычно применяемые для получения чужеродных белков в E.coli, и примерами таких промоторов могут быть сильные промоторы, такие как промотор Т7, промотор trp, промотор lac, промотор tac и промотор PL.
Для получения альдолазы в виде телец включения гибридного белка ген, кодирующий другой белок, а предпочтительно гидрофильный пептид, лигируют с выше- или нижерасположенным геном альдолазы, в результате чего может быть получен гибридный белок. Таким геном, кодирующим другой белок, может быть ген, обеспечивающий увеличение количества аккумулируемого гибридного белка и повышение растворимости указанного гибридного белка после проведения стадий модификации и регенерации, и примерами таких генов-кандидатов являются ген 10 Т7, ген β-галактозидазы, ген дегидрофолат-редуктазы, ген интерферона-γ, ген интерлейкина-2, ген прохимозина и тому подобное.
Эти гены подвергают лигированию с геном, кодирующим альдолазу, так, чтобы сохранялись рамки считывания кодонов. Такое лигирование может быть осуществлено путем встраивания данного гена в соответствующем рестрикционном сайте, или с использованием синтетической ДНК, имеющей соответствующую последовательность.
Для увеличения количества получаемой альдолазы предпочтительно, чтобы терминатор, то есть последовательность терминации транскрипции, был лигирован ниже гена гибридного белка. Таким терминатором может быть терминатор Т7, терминатор фага fd, терминатор Т4, терминатор гена резистентности к тетрациклину и терминатор гена trpA Escherichia coli.
В качестве вектора для введения гена, кодирующего альдолазу или гибридный белок альдолазы с другими белками в E.coli, предпочтительно использовать векторы так называемого многокопийного типа. Предпочтительными плазмидами могут служить плазмиды, имеющие сайт инициации репликации, происходящий от Со1Е1, такие как плазмиды типа pUC, плазмиды типа pВR322 или их производные. Используемый здесь термин "производные" может включать производные плазмиды, в которые были внесены модификации путем замены, делеции, инсерции, добавления и/или инверсии нуклеотидов. Используемый здесь термин "модификация" может также включать модификацию, внесенную путем мутации мутагенами и УФ-облучением, или спонтанной мутации.
При этом предпочтительно, чтобы данный вектор имел маркер, такой как ген резистентности к ампициллину для отбора трансформанта. Такой плазмидой может быть экспрессирующий вектор, несущий сильный промотор и являющийся коммерчески доступным (типа pUC (поставляемый Takara Shuzo Co, Ltd.), типа pPROK (поставляемый Clontech), рКК233-2 (поставляемый Clontech), и т.п.).
Рекомбинантную ДНК получают путем лигирования ДНК-фрагмента, где промотор, ген, кодирующий альдолазу или гибридный белок альдолазы и другого белка, и терминатор последовательно присоединены друг к другу, с ДНК-вектором.
E.coli трансформируют с использованием рекомбинантной ДНК, эту трансформированную E.coli культивируют, в результате чего экспрессируется и получается альдолаза или гибридный белок альдолазы с другим белком. Для получения трансформированного хозяина может быть использован микроорганизм, обычно применяемый для экспрессии чужеродного гена, а в частности штамм JM109 E.coli (DЕ3), при этом штаммы JM109 являются предпочтительными. Методы трансформации и отбора трансформанта описаны в Molecular Cloning: 2nd edition, Cold Spring Harbor Press (1989).
При экспрессии альдолазы в виде гибридного белка эта альдолаза может быть гидролизована с использованием рестриктирующей протеазы, распознающей последовательность, которая обычно не присутствует в альдолазе. Рестриктирующими протеазами являются фактор свертывания крови Ха, калликреин или т.п.
Средой для получения может быть среда, обычно используемая для культивирования E.coli, например среда, содержащая М9-казаминокислоты и среда LB. Условия культивирования и условия стимуляции получения могут быть соответствующим образом выбраны в зависимости от типа маркера вектора, промотора вектора, микроорганизма-хозяина и т.п.
Альдолаза или гибридный белок альдолазы и другого белка могут быть собраны следующим способом, а именно: если альдолаза или ее гибридный белок солюбилизированы в микробных клетках, то микробные клетки могут быть собраны, а затем подвергнуты разрушения или лизису с получением неочищенного раствора фермента. Если это необходимо, то альдолаза или гибридный белок могут быть затем подвергнуты очистке стандартными методами, такими как преципитация, фильтрация и колоночная хроматография. В этом случае очистка может быть также осуществлена методами с использованием антитела против альдолазы или гибридного белка.
В том случае, если образуются тельца включения белка, то они могут быть солюбилизированы с использованием денатурирующего агента. Такие тельца включения могут быть солюбилизированы вместе с микробными клетками. Однако при проведении описанного ниже способа очистки предпочтительно осуществлять сбор телец включения до проведения солюбилизации. Сбор телец включения из микробных клеток может быть проведен стандартными и хорошо известными методами. Так, например, микробные клетки разрушают и тельца включения выделяют путем центрифугирования и т.п. Денатурирующим агентом, который солюбилизирует тельца включения белка, может быть смесь "гуанидин-соляная кислота" (например, 6 М, рН 5-8) и мочевина (например, 8 М).
В результате удаления денатурирующего агента путем диализа и т.п. белок может быть регенерирован с получением белка, обладающего активностью. Растворами для диализа могут быть Трис-HCl-буфер, фосфатный буфер и т.п. Их концентрация может составлять 20 мМ-0,5 М, а рН может составлять 5-8.
При этом предпочтительно, чтобы концентрация белка в стадии регенерации составляла примерно 500 мкг/мл или менее. Для ингибирования самосшивания регенерированной альдолазы предпочтительно, чтобы диализ проходил при температуре 5°С или ниже. Методами удаления денатурирующего агента, помимо метода диализа, являются метод разведения и метод ультрафильтрации. Регенерация активности может быть осуществлена с использованием любого из этих методов.
Если в качестве кодирующей альдолазу ДНК используют ДНК, представленную в SEQ ID NO:1, 12 или 14, то в этом случае получают альдолазу, имеющую аминокислотную последовательность SEQ ID NO:2, 13 или 15 соответственно.
[II] Способ получения оптически активного монатина
В способе получения оптически активного монатина настоящего изобретения оптически активную IHOG получают способом, описанным в разделе [I] "Способ получения оптически активной IHOG", а затем IHOG превращают в монатин. При получении IHOG способом настоящего изобретения преимущественно получают 4R-IHOG. Поэтому оптически активный 4R-монатин, то есть (2R,4R)-монатин и (2S,4R)-монатин преимущественно получаются из IHOG, полученной в соответствии с настоящим изобретением [(2R,4R)-монатин и (2S,4R)-монатин вместе называются 4R-монатином].
Для эффективного получения (2R,4R)-монатина, то есть изомера, который из 4 типов изомеров монатина обладает самой высокой степенью сладости, предпочтительно использовать IHOG, обогащенную 4R-изомером. В этом случае процент 4R-IHOG от всего количества IHOG составляет предпочтительно более чем 55%, более предпочтительно более чем 60%, еще более предпочтительно более чем 70%, а особенно предпочтительно более чем 80%.
Способ превращения IHOG в монатин не имеет конкретных ограничений, и для этой цели может быть использован общеизвестный метод, такой как метод химической реакции или ферментативный метод.
(1) Метод химической реакции
Методом получения оптически активного монатина из оптически активной IHOG с помощью химической реакции может быть метод, в котором оптически активную IHOG подвергают оксимированию, и соответствующий IHOG-оксим, представленный нижеследующей формулой (4), или его соль подвергают химическому восстановлению с получением оптически активного монатина.
Предпочтительно IHOG, обогащенную 4R-изомером, подвергают оксимированию, а затем 4R-IHOG-оксим или его соль выделяют путем кристаллизации раствора, содержащего эту обогащенную 4R-изомером IHOG, и выделенный 4R-IHOG-оксим или его соль подвергают химическому восстановлению с получением 4R-монатина.
IHOG подвергают оксимированию путем реакции взаимодействия IHOG в нейтральных или щелочных условиях с аминовым соединением, представленным нежеследующей формулой (3):
где R представляет собой атом водорода, алкильную группу, арильную группу или аралкильную группу, или с его солью. С точки зрения кристаллизации, если в используемом здесь аминовом соединении R представляет собой алкильную, арильную или аралкильную группу, то предпочтительно, чтобы R представлял собой алкильную группу, имеющую 1-3 атома углерода, арильную или аралкильную группу, которая может иметь заместитель на боковой цепи, а более предпочтительно, чтобы R был выбран из метильной, этильной или бензильной группы.
Реакция оксимирования может быть осуществлена непосредственно путем добавления вышеуказанного амина формулы (3) в альдолазную реакционную смесь, содержащую IHOG. 4R-изомер может быть выделен путем кристаллизации 4R-IHOG-оксима или его соли из раствора, содержащего IHOG-оксим, обогащенный 4R-изомером. Предпочтительным растворителем для кристаллизации может быть вода, спиртовой растворитель или водный спиртовой растворитель.
4R-монатин может быть получен путем восстановления 4R-IHOG-оксима или его соли, полученных посредством кристаллизации. 4R-IHOG-оксим или его соль восстанавливают в присутствии водорода и гидрированного катализатора. В качестве гидрированного катализатора предпочтительно использовать катализатор на основе металла, где этот катализатор, такой как платина, родий, палладий, никель и кобальт, наносят на носитель, такой как двуокись кремния, окись алюминия, окись титана, окись магния, окись циркония и активированный уголь.
Обычно эффективного получения оптически активной IHOG не происходит. Поэтому для выделения 4R-IHOG из рацемата IHOG (4R,4S-IHOG) необходимо провести реакцию оксимирования 4R,4S-IHOG, а затем реакцию взаимодействия с хиральным амином с последующей кристаллизацией 4R-IHOG-оксима. И наоборот, в соответствии с настоящим изобретением обогащенная 4R-изомером IHOG может быть получена в стадии альдольной конденсации. Поэтому перед кристаллизацией не требуется оптического разделения с использованием хирального амина, и после оксимирования обогащенной 4R-изомером IHOG, 4R-IHOG-оксим может быть сразу кристаллизован. Таким образом, это позволяет снизить материальные затраты на очистку 4R-IHOG.
4R-монатин, полученный путем химического восстановления, представляет собой рацемическую смесь (2R,4R)-монатина и (2S,4R)-монатина. Для выделения (2R,4R)-монатина, (2R,4R)-монатин может быть кристаллизован. В частности, может быть использован метод, описанный в проспекте публикации Международной заявки WO 03/059865.
(2) Ферментативный метод
Если 4R-монатин получают из 4R-IHOG ферментативным методом, то положение 2 4R-IHOG может быть аминировано в присутствии фермента, который аминирует положение 2 4R-IHOG. Примерами фермента, катализирующего такую реакцию, может служить аминотрансфераза, которая катализирует реакцию переноса аминогруппы в 4R-IHOG, или дегидрогеназа, которая катализирует реакцию восстановительного аминирования 4R-IHOG. Более предпочтительно использовать аминотрансферазу.
При использовании аминотрансферазы 4R-монатин может быть получен посредством реакции 4R-IHOG в присутствии аминотрансферазы и донора аминогруппы. В частности, может быть использован метод, описанный в проспекте публикации Международной заявки WO 03/056026.
В данном случае может быть использована либо L-аминотрансфераза, либо D-аминотрансфераза. При использовании L-аминотрансферазы, 2S-монатин может быть селективно получен путем переноса аминогруппы L-аминокислоты в положение 2 IHOG. При использовании D-аминотрансферазы 2R-монатин может быть селективно получен путем переноса аминогруппы D-аминокислоты в положение 2 IHOG. Поэтому для селективного получения (2R,4R)-монатина, обладающего высокой степенью сладости, предпочтительно проводить реакцию взаимодействия 4R-IHOG с D-аминотрансферазой.
Реакция получения монатина с использованием аминотрансферазы может быть осуществлена после альдольной конденсации с последующим выделением полученного 4R-IHOG, либо она может быть осуществлена в том же самом реакционном растворе в присутствии альдолазы и аминотрансферазы. При осуществлении реакций в том же самом реакционном растворе может быть использован микроорганизм, в котором совместно экспрессируется ДНК, кодирующая альдолазу, и ДНК, кодирующая аминотрансферазу, либо эти ферменты могут быть получены отдельно и добавлены в реакционный раствор. Микроорганизм (клетка-хозяин), в котором совместно экспрессируется ДНК, кодирующая альдолазу, и ДНК, кодирующая аминотрансферазу, может быть получен путем ко-трансфекции экспрессирующего вектора, содержащего функциональную ДНК, кодирующую альдолазу, и экспрессирующего вектора, содержащего функциональную ДНК, кодирующую аминотрансферазу, либо путем трансформации экспрессирующим вектором, содержащим ДНК, кодирующую альдолазу, и ДНК, кодирующую аминотрансферазу, в форме, способной эффективно экспрессироваться в клетке-хозяине.
ПРИМЕРЫ
Настоящее изобретение подробно описано в нижеследующих примерах, но не ограничивается этими примерами. IHOG, PHOG и (2R,4R)-монатин, используемые в примерах, были синтезированы способами, описанными в сравнительных примерах 1, 2 и 3.
В приведенных ниже примерах количество IHOG и PHOG оценивали с помощью ВЭЖХ-анализа с использованием колонки "Inertsil ODS-2" (5 мкм, 4,6×250 мм), поставляемой GL Sciences Inc. Условия анализа приводятся ниже.
Подвижная фаза: 40% (об./об.) ацетонитрил/5 мМ раствор дигидрофосфата тетрабутиламмония.
Скорость потока: 1 мл/мин.
Температура колонки: 40°С.
Детекция: УФ 210 нм.
Асимметрия полученных IHOG или PHOG была проанализировано с помощью ВЭЖХ-анализа, где колонка "Inertsil ODS-2" (5 мкм, 4,6×250 мм), поставляемая GL Sciences Inc., непосредственно соединена с колонкой "SUMICHIRAL OA-7100" (5 мкм, 4,6×250 мм), поставляемой Sumika Chemical Analysis Service Ltd., в указанном порядке. Условия анализа приводятся ниже.
Подвижная фаза А: 5% (об./об.) ацетонитрил, 20 мМ калийфосфатный буфер (рН 6,8).
Подвижная фаза В: 50% (об./об.) ацетонитрил, 20 мМ калийфосфатный буфер (рН 6,8).
Элюирование подвижной фазой А, 0-90 минут и 90-120 минут, и промывка подвижной фазой В.
Скорость потока: 0,4 мл/мин.
Температура колонки: 17°С.
Детекция: УФ 210 нм.
Пример 1. Скрининг бактерий, обладающих IHOG-альдолазной активностью, из окружающей среды
Бактериальный штамм, обладающий альдолазной активностью с оптической селективностью по отношению к 4R-IHOG, выделяли и получали из природной среды путем осуществления обогатительного культивирования с использованием среды, в которой (2R,4R)-монатин (RR-монатин) является единственным источником углерода.
Образец почвы инокулировали в тест-пробирку, содержащую 1 мл среды, с использованием RR-монатина в качестве единственного источника углерода (0,4 г/л монокалиевой соли RR-монатина, 0,67 г/л азотистого основания дрожжей без аминокислоты (поставляемого Difco)), и культивировали со встряхиванием при 37°С в течение 7 дней. И снова 0,1 мл полученного культурального бульона инокулировали в тест-пробирку, содержащую 1 мл среды, с использованием RR-монатина в качестве единственного источника углерода, и снова культивировали со встряхиванием при 37°С в течение 7 дней. Полученный культуральный бульон соответствующим образом разводили стерильным физиологическим раствором, а затем наносили на плотную среду СМ2G (5 г/л глюкозы, 10 г/л дрожжевого экстракта, 10 г/л пептона, 5 г/л NaCl, 20 г/л агарового порошка, рН 7,0) и культивировали при 30°С в течение 24 часов для выделения колонии.
Штамм, обладающий альдолазной активностью, отбирали из аккумулированных микробных штаммов. Полученные микроорганизмы инокулировали в чашки с агаровым бульоном (поставляемым Eiken Chemical Co., Ltd.) и культивировали при 30°С в течение 24 часов. Этот микроорганизм инокулировали в чашки, содержащие среду, продуцирующую фермент (5 г/л глицерина, 5 г/л фумаровой кислоты, 3 г/л дрожжевого экстракта, 2 г/л пептона, 3 г/л сульфата аммония, 3 г/л К2НРО4, 1 г/л КН2РО4, 0,5 г/л MaSO4 7H2O, 2,5 г/л фталата натрия, 20 г/л агарового порошка, рН 6,5) и культивировали при 30°С в течение 24 часов. Полученные микробные клетки инокулировали в реакционную смесь, состоящую из 100 мМ Трис-HCl (pH 8,0), 50 мМ PHOG, 1 мМ MgCl2, 5 мМ раствора фосфата калия (KPi) и 1% (об./об.) толуола. Концентрация инокулированных клеток составляла примерно 1% от сырой массы микробных клеток. Смесь подвергали реакции при 30°С в течение 24 часов. Концентрацию свободной пировиноградной кислоты в реакционной смеси количественно определяли ферментативным методом с использованием лактатдегидрогеназы (LDH). 10 мкл реакционной смеси добавляли к 200 мкл раствора, состоящего из 100 мМ Трис-HCl (рН 8,0), 1,5 мМ NADH, 5 мМ MgCl2 и 25 ед./мл LDH. Перемешанный раствор инкубировали при 30°С в течение 10 минут. После проведения реакции измеряли оптическую плотность при 340 нм, и количество пировиноградной кислоты в образце определяли с использованием уменьшенного количества NADH.
Количество полученной фенилпировиноградной кислоты определяли с помощью ВЭЖХ-анализа с использованием колонки "Inertsil ODS-2" (5 мкм, 4,6×250 мм), поставляемой GL Sciences Inc. Условия анализа приводятся ниже.
Подвижная фаза: 20% (об./об.) ацетонитрил/водный раствор 0,05% (об./об.) трифторуксусной кислоты.
Скорость потока: 1 мл/мин.
Температура колонки: 40°C.
Детекция: УФ 210 нм.
В указанных условиях PHOG и фенилпировиноградную кислоту элюировали при времени удерживания примерно 9,8 мин и примерно 12 мин соответственно, после чего их можно было идентифицировать и количественно оценивать.
Количество, получаемое альдолазой, вычисляли путем вычитания количества, полученного контролем без микробных клеток, из количества пировиноградной кислоты или фенилпировиноградной кислоты, получаемых из PHOG с микробными клетками. В результате было обнаружено, что если в качестве субстрата использовался PHOG, то бактериальные штаммы обладали альдолазной активностью.
Затем синтезировали IHOG с использованием собранных микробных клеток, обладающих альдолазной активностью. Эти микробные клетки инокулировали в тест-пробирку, содержащую 3 мл среды, продуцирующей фермент (5 г/л глицерина, 5 г/л фумаровой кислоты, 3 г/л дрожжевого экстракта, 2 г/л пептона, 3 г/л сульфата аммония, 3 г/л К2НРО4, 1 г/л КН2РО4, 0,5 г/л MaSO4·7H2O, 2,5 г/л фталата натрия, рН 6,5). Среду культивировали при 30°С в течение 16 часов. Полученные микробные клетки суспендировали при концентрации 1% (мас./об.) в расчете на сырую массу микробных клеток в указанной ниже реакционной смеси для синтеза IHOG.
Реакционная смесь для синтеза IHOG содержала: 100 мМ Hepes-КОН (pH 8,5), 300 мМ индолпировиноградной кислоты, 750 мМ натриевой соли пировиноградной кислоты, 1 мМ MgCl2 и 5 мМ калийфосфатного буфера (рН 8,5).
Реакционную смесь инкубировали при 37°С в течение 16 часов, а затем определяли количество получаемой IHOG и анализировали хиральность реакционного продукта в положении 4. В результате анализа отобранного бактериального штамма С77 было обнаружено, что при альдольной конденсации продуцируется преимущественно 4R-IHOG, как показано в таблице 4.
Реакция синтеза IHOG в микробных клетках штамма С77
Пример 2. Очистка IHOG-продуцирующей альдолазы (SpALD), происходящей от штамма С77 (штамма AJ110329 Sphingomonas sp.)
IHOG-альдолазу очищали из растворимой фракции штамма С77 (штамма AJ110329 Sphingomonas sp.) следующим образом. Альдолазную активность в реакции альдольной деградации детектировали с использованием PHOG в качестве субстрата в указанных ниже условиях.
Реакционные условия: 50 мМ Hepes-КОН (pH 8,5), 2 мМ PHOG, 0,25 мМ NADH, 0,2 мМ KPi, 1 мМ MgCl2, 16 ед./мл лактат-дегидрогеназы, 3 мкл фермента на 600 мкл реакционной смеси, 30°С, оптическую плотность измеряли при 340 нм.
(1) Получение растворимой фракции
Одну платиновую петлю микробных клеток штамма С77 (штамма AJ110329 Sphingomonas sp.), полученного путем культивирования в чашках с агаровым бульоном при 30°С в течение 24 часов, инокулировали в 500 мл колбу, содержащую 50 мл среды, продуцирующей фермент (5 г/л глицерина, 5 г/л фумаровой кислоты, 5 г/л сульфата аммония, 3 г/л К2НРО4, 1 г/л КН2РО4, 0,5 г/л MaSO4·7H2O, 3 г/л дрожжевого экстракта, 2 г/л пептона, 2,5 г/л фталата натрия (поставляемого Sigma), рН доводили до 6,5 с использованием КОН), и культивировали со встряхиванием при 30°С в течение 24 часов. Затем 0,5 мл культурального бульона инокулировали в 20 колб емкостью 500 мл, содержащих 50 мл среды, продуцирующей фермент, и культивировали со встряхиванием при 30°C в течение 24 часов. Микробные клетки собирали из полученного культурального бульона путем центрифугирования. Собранные микробные клетки суспендировали в буфере А (20 мМ Hepes-КОН, рН 7,6) и промывали этим буфером. Затем микробные клетки снова собирали путем центрифугирования. Полученные промытые микробные клетки суспендировали в 80 мл буфера А, и подвергали разрушению путем обработки ультразвуком при 4°С в течение 30 минут. Дебрис микробных клеток удаляли путем центрифугирования для разрушения суспензии (×8000 об/мин, 10 мин, два раза) и получали супернатант в виде растворимой фракции.
(2) Анионообменная хроматография: Q-сефароза FF
Растворимую фракцию (40 мл) наносили на анионообменную колонку с Q-сефарозой FF 26/10 (поставляемой Pharmacia, CV=20 мл), уравновешенную буфером А для абсорбции на носителе. Белки, которые не абсорбировались на носителе (неабсорбированные белки), отмывали с использованием буфера А. Затем абсорбированный белок элюировали линейным градиентом концентраций KCl от 0 М до 0,7 М (всего 140 мл). PHOG-альдолазную активность измеряли для каждой элюированной фракции, а затем пик PHOG-альдолазной активности детектировали во фракции, соответствующей примерно 0,4 М. Эту хроматографическую процедуру повторяли два раза.
(3) Гидрофобная хроматография: фенилсефароза НР HR 16/10
Раствор, в котором была детектирована альдолазная активность, диализовали против буфера В (20 мМ Hepes-КОН, 1 М сульфата аммония, рН 7,6) при 4°С в течение ночи и фильтровали через фильтр с размером пор 0,45 мкм. Полученный фильтрат наносили на гидрофобную хроматографическую колонку с фенилсефарозой НР HR 16/10 (поставляемой Pharmacia), предварительно уравновешенную буфером В. В результате этой процедуры альдолаза абсорбировалась на носителе.
Белки, не абсорбированные на носителе, отмывали с использованием буфера В, а затем альдолазу элюировали линейным градиентом концентраций сульфата аммония от 1 М до 0 М. Альдолазную активность измеряли для каждой элюированной фракции, и эта альдолазная активность детектировалась в тех положениях элюирования, где концентрация сульфата аммония составляла примерно 0,4-0,5 М.
(4) Гель-хроматография: сефадекс 200 НР 16/60
Фракции, содержащие альдолазу, собирали, диализовали против буфера А и фильтровали через фильтр с размером пор 45 мкм. Полученный фильтрат концентрировали с использованием ультрафильтрационной мембраны, Centriprep 10. Полученный концентрированный раствор наносили на гель-фильтрационную колонку с сефадексом 200 НР 16/60 (поставляемым Pharmacia), предварительно уравновешенную буфером С (20 мМ Hepes-КОН, 0,1 М KCl, рН 7,6), и элюировали при скорости потока 1 мл/мин. В этой процедуре альдолаза элюировалась во фракциях примерно 66 мл. По положению пика активности при элюировании было установлено, что молекулярная масса альдолазы составляет примерно 155 кДа.
(5) Анионообменная хроматография: Mono Q HR 5/5
Полученную фракцию фильтровали через фильтр с размером пор 0,45 мкм. Полученный фильтрат наносили на анионообменную хроматографическую колонку с Mono Q HR 5/5 (поставляемой Pharmacia), предварительно уравновешенную буфером А. В результате этой процедуры, альдолаза абсорбировалась на носителе. Неабсорбированные белки отмывали буфером А, а затем белок элюировали линейным градиентом концентраций KCl от 0 мМ до 700 мМ (всего 24 мл). Альдолазную активность измеряли для каждой фракции, и альдолазная активность детектировалась в тех положениях элюирования, где концентрация KCl составляла примерно 0,4 М.
Полученные фракции подвергали электрофорезу в ДСН-ПААГ, в результате чего в активной фракции наблюдалось примерно 10 полос. Из них полосу примерно 30 кДа, где профиль активности соответствовал профилю интенсивности ДСН-ПААГ-полосы, вырезали из ДСН-ПААГ как кандидат на альдолазу, и секвенировали для определения аминокислотной последовательности.
Неполная очистка IHOG-альдолазы из штамма С77 (штамма AJ110329 Sphingomonas sp.)
Пример 3. Определение внутренней аминокислотной последовательности IHOG-альдолазы
Очищенную фракцию, содержащую альдолазу, подвергали электрофорезу в ДСН-ПААГ, а затем полосу, соответствующую 30 кДа, вырезали, и образец в ДСН-ПААГ-геле обрабатывали трипсином (рН 8,5, 35°С, 20 часов), а затем подвергали обращенно-фазовой ВЭЖХ фрагментированных пептидов. Из этих фракций анализировали две фракции, и были определены 17 и 12 остатков аминокислотных последовательностей (SEQ ID NO:3 и 4), показанные в нижеследующей таблице 6.
Таблица 6
Определенные внутренние аминокислотные последовательности
Пример 4. Клонирование гена IHOG-альдолазы, происходящего от штамма С77 (штамма AJ110329 Sphingomonas sp.)
(1) Получение хромосомной ДНК
Штамм С77 (штамм AJ110329 Sphingomonas sp.) культивировали с использованием 50 мл среды CM2G при 30°С в течение ночи (предварительное культивирование). Основное культивирование проводили с использованием 5 мл этой культуры в качестве инокулята и с использованием 50 мл среды. После культивирования до достижения поздней логарифмической фазы роста 50 мл культуры подвергали центрифугированию (12000×g, 4°С, 15 минут), и клетки собирали. С использованием этих микробных клеток получали хромосомную ДНК стандартными методами.
(2) Определение внутренней последовательности с помощью ПЦР
На основе определенных внутренних аминокислотных последовательностей IHOG-альдолазы были синтезированы смешанные праймеры (SEQ ID NO:5 и 6), описанные в таблице 7.
Таблица 7
Синтезированные смешанные праймеры, сконструированные из внутренних аминокислотных последовательностей
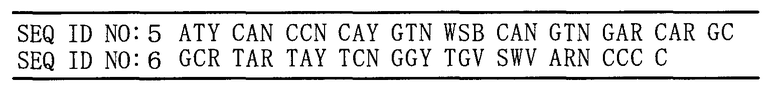
С использованием смешанных праймеров, полученных как описано выше, и с использованием хромосомной ДНК штамма С77 (штамма AJ110329 Sphingomonas sp.) в качестве матрицы проводили ПЦР-амплификацию. ПЦР-реакцию осуществляли в 30 циклов с использованием термоячейки для проведения ПЦР PCR Thermal PERSONEL (поставляемой TaKaRa Holdings Inc.) при следующих условиях.
94°С - 30 секунд
55°С - 30 секунд
72°С - 1 минута
ПЦР-продукт подвергали электрофорезу в агарозном геле, после чего наблюдалась амплификация фрагмента размером примерно 200 п.н. ДНК-фрагмент клонировали в вектор рТ7Blue (поставляемый от Novagen) и определяли нуклеотидную последовательность. В результате было обнаружено, что аминокислотная последовательность, происходящая от полученного ДНК-фрагмента, соответствовала внутренней аминокислотной последовательности IHOG-альдолазы. Таким образом было подтверждено получение нужного гена альдолазы.
(3) Определение полноразмерного гена путем гибридизации колоний
С использованием ДНК-фрагмента, амплифицированного с помощью ПЦР, был получен полноразмерный ген путем проведения Саузерн-анализа и гибридизации колоний. ДНК-зонд получали с использованием праймера DIGHigh (поставляемого Roche Diagnostics) и метили при инкубировании в течение ночи при 37°С в соответствии с инструкциями. Саузерн-анализ проводили путем полного гидролиза 1 мкг хромосомной ДНК различными рестриктирующими ферментами, электрофореза на 0,8% агарозном геле и блоттинга на найлоновой мембране с проведением последующих стадий в соответствии с инструкциями. Гибридизацию осуществляли с использованием набора DIG Easy Hyb (поставляемого Roche Diagnostics), при этом предварительную гибридизацию осуществляли при 50°С в течение одного часа, а затем добавляли зонд и осуществляли гибридизацию в течение ночи. Полосы детектировали с использованием набора для детекции нуклеотидов (DIG Nucleotide Detection Kit). В результате был детектирован PstI/BamHI-фрагмент размером примерно 3,2 т.п.н., который интенсивно гибридизовался с указанным зондом для ПЦР-фрагмента. Затем получали PstI/BamHI-фрагмент путем гибридизации колоний. Хромосомную ДНК (20 мкг) обрабатывали PstI/BamHI и подвергали электрофорезу на агарозном геле с получением фрагмента размером примерно 3,2 т.п.н. Этот фрагмент лигировали в pUC19 и создавали библиотеку в JM109 E.coli. Колонии переносили на найлоновый мембранный фильтр (Hybond-N, поставляемый Amersham), а затем подвергали щелочной модификации, нейтрализации и фиксации. Гибридизацию осуществляли с использованием набора DIG Easy Hyb. Фильтр погружали в буфер, и предварительно гибридизовали при 42°С в течение одного часа. Затем добавляли меченый зонд и проводили гибридизацию при 42°С в течение 16 часов. После промывки SSC, колонию, которая гибридизовалась с зондом, детектировали с использованием набора для детекции нуклеотидов (DIG Nucleotide Detection Kit) (поставляемого Roche Diagnostics). В результате был получен клон, который интенсивно гибридизовался с зондом.
Затем определяли нуклеотидную последовательность плазмидной ДНК полученного клона, в результате чего было продемонстрировано, что такая плазмидная ДНК имеет нуклеотидную последовательность SEQ ID NO:1. Была также обнаружена ОРС размером 855 п.н., включая нуклеотидные последовательности, соответствующие определенным внутренним аминокислотным последовательностям (№№ 519-569 и 666-701 в SEQ ID NO:1), и таким образом, была получена нужная полноразмерная альдолаза.
Пример 5. Крупномасштабная экспрессия IHOG-альдолазы (SpALD) в E.coli
(1) Конструирование плазмиды рTrp4, содержащей промотор trp и терминатор rrnB
Промоторную область оперона trp на хромосомной ДНК W3110 E.coli подвергали ПЦР-амплификации с использованием олигонуклеотидов, представленных в таблице 8 (комбинация SEQ ID NO:7 и 8) в качестве праймеров, и полученный ДНК-фрагмент лигировали с вектором pGEM-Teasy (поставляемым Promega). JM109 E.coli трансформировали раствором, содержащим этот продукт лигирования, и штаммы, содержащие рассматриваемую плазмиду, в которой промотор trp был встроен в направлении, противоположном направлению промотора lac, отбирали из штаммов, резистентных к ампициллину. Затем ДНК-фрагмент, содержащий промотор trp, полученный путем обработки этой плазмиды ферментами EcoO109i/EcoRI, лигировали в плазмиду pUC19 (поставляемую Takara), обработанную EcoO109i/EcoRI. JM109 E.coli трансформировали раствором, содержащим этот продукт лигирования, и штамм, содержащий рассматриваемую плазмиду, отбирали из штаммов, резистентных к ампициллину. Эту плазмиду обозначали рTrp1. Затем плазмиду рКК223-3 (поставляемую Amersham Pharmacia) обрабатывали HindIII/HincII, и полученный ДНК-фрагмент, содержащий терминатор rrnB, лигировали в плазмиду рTrp1, обработанную HindIII/HincII. JM109 E.coli трансформировали раствором, содержащим этот продукт лигирования, и штамм, содержащий рассматриваемую плазмиду, отбирали из штаммов, резистентных к ампициллину. Эту плазмиду обозначали рTrp2. Затем промоторную область trp подвергали ПЦР-амплификации с использованием рTrp2 в качестве матрицы и олигонуклеотидов, представленных в таблице 8 (комбинация SEQ ID NO:7 и 9) в качестве праймеров. Этот ДНК-фрагмент обрабатывали EcoO109i/NdeI, и лигировали в плазмиду рTrp2, обработанную EcoO109i/NdeI. JM109 E.coli трансформировали раствором, содержащим этот продукт лигирования, и штамм, содержащий рассматриваемую плазмиду, отбирали из штаммов, резистентных к ампициллину. Эту плазмиду обозначали рTrp4.
Таблица 8
Последовательности праймеров

(2) Конструирование плазмиды ptrpSpALD, экспрессирующей ген альдолазы и его экспрессия в E.coli
Фрагмент, полученный путем амплификации из хромосомной ДНК штамма С77 (штамма AJ110329 Sphingomonas sp.) с использованием праймеров (SEQ ID NO:10 и 11), указанных в таблице 9, гидролизовали ферментами NdeI/PstI, и встраивали в NdeI/PstI-сайт плазмиды рTrp4 с получением плазмиды ptrpSpALD. Эта плазмида экспрессировала ген альдолазы, кодирующий аминокислотную последовательность SEQ ID NO:2, полученную путем трансляции области, простирающейся от ATG в положении нуклеотида 210, который представляет собой кодон инициации трансляции, до нуклеотида 1004 в нуклеотидной последовательности SEQ ID NO:1.
Таблица 9
Последовательность праймеров для конструирования ptrpSpALD

JM109 E.coli трансформировали сконструированной экспрессионной плазмидой, и одну платиновую петлю полученного трансформанта инокулировали в 50 мл среды LB, содержащей 100 мкг/мл ампициллина и проводили реакцию со встряхиванием при 37°С в течение 16 часов. После культивирования микробные клетки собирали из 1 мл полученной культуры, промывали и суспендировали в 1 мл 20 мМ Hepes-КОН (Рн 7,6), а затем микробные клетки разрушали ультразвуком. Супернатант получали путем центрифугирования при 150000 об/мин в течение 10 минут для разрушения суспензии и использовали в качестве неочищенного ферментного раствора.
Альдолазную активность измеряли с использованием неочищенного ферментного раствора. Эту альдолазную активность измеряли как активность альдольного расщепления, осуществляемого с использованием PHOG в качестве субстрата в нижеследующих условиях.
Реакционные условия: 50 мМ Hepes-КОН (pH 8,5), 2 мМ PHOG, 0,25 мМ NADH, 0,2 мМ KPi, 1 мМ MgCl2, 16 ед./мл лактат-дегидрогеназы, 3 мкл фермента на 600 мкл реакционной смеси, 30°С, оптическую плотность измеряли при 340 нм.
В результате измерения какой-либо альдолазной активности в E.coli (контроль), трансформированной плазмидой рTrp4, не наблюдалось, тогда как в штамме, трансформированном плазмидой ptrpSpALD, было детектировано 33,6 ед./мг белка, обладающего альдолазной активностью. Это свидетельствовало о том, что был действительно клонирован нужный ген альдолазы, и что была сконструирована плазмида, содержащая в высокой степени экспрессирующуюся SpALD.
Пример 6. Очистка рекомбинантного фермента альдолазы, происходящего от штамма С77 (штамма AJ110329 Sphingomonas sp.)
Рекомбинантную SpALD выделяли из растворимой фракции E.coli, обнаруживающей высокую степень экспрессии альдолазы (SpALD), происходящей от штамма С77 (штамма AJ110329 Sphingomonas sp.), следующим образом. Альдолазную активность измеряли как активность в альдольном расщеплении, осуществляемом с использованием PHOG в качестве субстрата в следующих условиях.
Реакционные условия: 50 мМ Hepes-КОН (pH 8,5), 2 мМ PHOG, 0,25 мМ NAD, 1 мМ MgCl2, 16 ед./мл лактат-дегидрогеназы, 3 мкл фермента на 600 мкл реакционной смеси, 30°С, оптическую плотность измеряли при 340 нм.
(1) Получение растворимой фракции
Одну платиновую петлю микробных клеток E.coli/ptrpSpALD, культивированных в чашках с агаровой средой LB-amp при 37°С в течение 16 часов, инокулировали в 10 колб емкостью 500 мл, содержащих 50 мл среды LB-amp. Затем осуществляли культивирование со встряхиванием при 37°С в течение 16 часов. Микробные клетки собирали из полученной культуры путем центрифугирования, суспендировали в буфере А (20 мМ Hepes-КОН, рН 7,6) и промывали этим буфером, а затем снова центрифугировали и бактериальные клетки собирали. Полученные промытые микробные клетки суспендировали в 20 мл буфера А, и подвергали разрушению путем обработки ультразвуком при 4°С в течение 30 минут. Дебрис микробных клеток удаляли путем центрифугирования для разрушения суспензии (×8000 об/мин, 10 мин, два раза), и полученный супернатант использовали в качестве неочищенной экстрагированной фракции.
(2) Анионообменная хроматография: Q-сефароза FF
Неочищенную экстрагированную фракцию (20 мл), полученную как описано выше, наносили на анионообменную колонку с Q-сефарозой FF 26/10 (поставляемой Pharmacia CV=20 мл), предварительно уравновешенную буфером А для абсорбции белков на носителе. Белки, которые не абсорбировались на носителе (неабсорбированные белки), отмывали с использованием буфера А. Затем абсорбированный белок элюировали линейным градиентом концентраций KCl от 0 М до 0,7 М (всего 140 мл). PHOG-альдолазную активность измеряли для каждой элюированной фракции, а затем пик PHOG-альдолазной активности детектировали во фракции, соответствующей примерно 0,4 М.
(3) Гидрофобная хроматография: фенилсефароза НР HR 16/10
Раствор, в котором была детектирована альдолазная активность, диализовали против буфера В (20 мМ Hepes-КОН, 1 М сульфата аммония, рН 7,6) при 4°С в течение ночи, и фильтровали через фильтр с размером пор 0,45 мкм. Полученный фильтрат наносили на гидрофобную хроматографическую колонку с фенилсефарозой НР HR 16/10 (поставляемой Pharmacia), предварительно уравновешенную буфером В. В результате этой процедуры альдолаза абсорбировалась на носителе.
Белки, не абсорбированные на носителе, отмывали с использованием буфера В, а затем альдолазу элюировали линейным градиентом концентраций сульфата аммония 1 М → 0 М. Альдолазную активность измеряли для каждой элюированной фракции, и такая альдолазная активность детектировалась в тех положениях элюирования, где концентрация сульфата аммония составляла примерно 0,4-0,5 М.
(4) Гель-хроматография: сефадекс 200 НР 16/60
Фракции, содержащие альдолазу, собирали, диализовали против буфера А и фильтровали через фильтр с размером пор 45 мкм. Полученный фильтрат концентрировали с использованием ультрафильтрационной мембраны, Centriprep 10. Полученный концентрированный раствор наносили на гель-фильтрационную колонку с сефадексом 200 НР 16/60 (поставляемым Pharmacia), предварительно уравновешенную буфером С (20 мМ Hepes-КОН, 0,1 М KCl, рН 7,6), и элюировали при скорости потока 1 мл/мин. В этой процедуре, альдолаза элюировалась во фракциях примерно 66 мл.
(5) Анионообменная хроматография: Mono-Q HR 5/5
Полученную фракцию фильтровали через фильтр с размером пор 0,45 мкм. Полученный фильтрат наносили на анионообменную хроматографическую колонку с Mono Q HR 5/5 (поставляемой Pharmacia), предварительно уравновешенную буфером А. В результате этой процедуры альдолаза абсорбировалась на носителе. Неабсорбированные белки отмывали буфером А, а затем белок элюировали линейным градиентом концентраций KCl от 0 мМ до 700 мМ (всего 24 мл). Альдолазную активность измеряли для каждой фракции, и эта альдолазная активность детектировалась в тех положениях элюирования, где концентрация KCl составляла примерно 0,4 М.
Фракции, очищенные с помощью описанной выше колоночной хроматографии, подвергали электрофорезу в ДСН-ПААГ, а затем одну полосу в положении, соответствующем примерно 30 кДа, детектировали путем окрашивания в СВВ. Полученный раствор рекомбинантной SpALD диализовали против буфера А при 4°С в течение ночи. В результате этой процедуры получали 1,5 мл раствора, содержащего 452 ед./мл SpALD.
Пример 7. Синтез 4-(индол-3-илметил)-4-гидрокси-2-оксоглутаровой кислоты (IHOG) из индол-3-пировиноградной кислоты и пировиноградной кислоты с использованием SpALD
4-(Индол-3-илметил)-4-гидрокси-2-оксоглутаровую кислоту (IHOG) синтезировали из индол-3-пировиноградной кислоты и пировиноградной кислоты с использованием SpALD, полученной как описано в примере 6, в качестве источника фермента. Реакционную смесь, состоящую из 100 мМ Hepes-КОН (рН 8,5), 300 мМ индол-3-пировиноградной кислоты, 750 мМ пировиноградной кислоты и 1 мМ MgCl2, подвергали реакции при 37°С в течение 3 минут путем добавления 7,9 мг/мл SpALD. Ферментную реакционную смесь соответствующим образом разбавляли и анализировали с помощью ВЭЖХ для определения количества получаемой IHOG. В результате было подтверждено получение IHOG с помощью альдолазы. Было определено, что начальная активность синтеза IHOG в данных условиях составляла 451 ед./мг.
Пример 8. Ферментативные свойства SpALD
С использованием SpALD, полученной как описано в примере 6, были исследованы нижеследующие параметры.
Условия предварительных измерений: 50 мМ Hepes-КОН (pH 8,5), 2 мМ PHOG, 5 мМ MgCl2, 16 ед./мл лактат-дегидрогеназы, при этом наблюдалась пониженная оптическая плотность, измеренная при 340 нм и при 30°С.
(1) Кинетическая константа, определяемая с использованием PHOG в качестве субстрата
Исходя из результатов, представленных на фиг.3, были определены следующие параметры: Vmax (для PHOG)=979 мкмоль/мин/мг, Km (для PHOG)=0,06 мМ, Km (для MgCl2)=0,034 мМ. Какого-либо увеличения альдолазной активности при добавлении раствора КРВ не наблюдалось.
(2) рН-стабильность
Было определено, что рН-стабильность составляет в пределах от 3 до 10. Для измерения были использованы следующие буферы: натрийцитратный буфер (рН 3; 4; 4,5), натрийацетатный буфер (рН 4; 4,5; 5; 5,5; 6; 6,5), калийфосфатный буфер (рН 6; 6,5; 7; 7,5; 8; 8,5), буфер Hepes-KOH (рН 7,5; 8; 8,5; 9; 9,5), буфер CAPS-NaOH (рН 9; 9,5; 10). После инкубирования SpALD в 100 мМ каждого из этих буферов при 37°С в течение 30 минут, остаточную активность измеряли в условиях предварительных измерений. Результаты представлены на фиг.4.
(3) Термостабильность
После инкубирования SpALD в 100 мМ калийфосфатного буфера (рН 7,0) и 100 мМ буфера Hepes-KOH (рН 8,5) при 25-70°С в течение 30 минут, остаточную активность измеряли в условиях для предварительных измерений. Результаты представлены на фиг.5.
(4) Оптимальный рН
PHOG-разлагающую активность при 30°С измеряли колориметрическим методом (фиг.6). В результате было показано, что оптимальный рН в ретро-альдоной реакции расщепления PHOG составляет примерно рН 7,5.
Активность синтеза IHOG измеряли с использованием 300 мМ IPA и 750 мМ пировиноградной кислоты в качестве субстратов при каждом рН (реакционные условия: 50 мМ Hepes-КОН (pH 8,5), 300 мМ IPA, 750 мМ РА, 1 мМ MgCl2, 37оС, 16 ч). Величины, полученные путем вычитания количества полученной IHOG в отсутствие фермента из количества полученной IHOG в присутствии фермента, приводятся на фиг.7. В результате было показано, что оптимальный рН реакции альдольной конденсации IHOG, проводимой с использованием SpALD, составляет примерно 8,0.
Пример 9. Синтез IHOG с использованием SpALD
Одну платиновую петлю микробных клеток E.coli/ptrpSpALD, культивированных в чашках с агаровой средой LB-amp при 37°С в течение 16 часов, инокулировали в 12 колб емкостью 500 мл, содержащих 50 мл среды LB-amp. Затем проводили культивирование со встряхиванием при 37°С в течение 16 часов. Микробные клетки собирали из полученной культуры путем центрифугирования, суспендировали в буфере А (20 мМ Hepes-КОН, рН 7,6) и промывали этим буфером, а затем снова центрифугировали и микробные клетки собирали. Микробные клетки (сырая масса: примерно 3 г), собранные путем центрифугирования, суспендировали в 300 мл реакционной смеси, имеющей нижеследующий состав.
Реакционная смесь для синтеза IHOG содержала: 50 мМ Hepes-КОН (pH 8,5), 300 мМ индолпировиноградной кислоты, 750 мМ натриевой соли пировиноградной кислоты, 1 мМ MgCl2, 5 мМ калийфосфатного буфера (рН 8,5) (рН доводили до 8,5 добавлением 6 н. КОН).
Реакционную смесь, в которой были суспендированы микробные клетки, барботировали газом аргоном, а затем реакцию проводили в атмосфере аргона. Реакцию осуществляли с перемешиванием при 37°С в течение 20 часов. После завершения реакции и после центрифугирования для удаления микробных клеток получали 300 мл альдольной реакционной смеси.
Пример 10. Оксимирование альдольной реакционной смеси и выделение 4R-IHOG-оксима
При поддержании величины рН 9 путем добавления водного 8 н. раствора гидроксида натрия, к альдольной реакционной смеси, полученной как описано в примере 9, добавляли 18,8 г (270 ммоль) гидрохлорида гидроксиламина, а затем смесь перемешивали при 25°С в течение 3 часов и при 10°С в течение ночи. Концентрацию IHOG-оксима в полученной реакционной смеси количественно определяли с помощью ВЭЖХ-анализа. В результате, в этой реакционной смеси получалось 25 ммоль 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (IHOG-оксима). Затем проводили анализ на асимметрию в положении 4, и этот анализ подтвердил получение 21,4 ммоль 4R-IHOG-оксима и 3,6 ммоль 4S-IHOG-оксима. Таким образом, в этой реакции преимущественно получался 4R-изомер с оптической чистотой 71,3% э.и.
рН полученной реакционной смеси доводили до значения 2 с использованием концентрированной соляной кислоты, и органические вещества экстрагировали этилацетатом. Органический слой концентрировали и получали остаток. К этому остатку добавляли 12 мл 28% водного раствора аммиака и 25 мл воды, и эту смесь кристаллизовали путем добавления 138 мл 2-пропанола, в результате чего получали 9,76 г (сырой массы) диаммониевой соли 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты в виде кристаллов. Полученные кристаллы растворяли в 60 мл воды, а затем добавляли 50 мл 2-пропанола при 50°С, после чего в течение 3 часов по каплям добавляли еще 150 мл 2-пропанола при 50°С. Затем путем фильтрации полученных кристаллов и их сушки при пониженном давлении получали 5,38 г (15,8 ммоль) диаммониевой соли IHOG-оксима. После этого проводили анализ асимметрии полученных кристаллов в положении 4, и оптическая чистота 4R-изомера составляла 99,0% э.и. Таким образом, аммониевая соль 4R-IHOG-оксима с высокой степенью чистоты может быть выделена и получена путем кристаллизации из 2-пропанола.
Пример 11. Получение 4R-монатина путем химического восстановления 4R-IHOG-оксима
Аммониевую соль (4R)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (5,38 г, 15,8 ммоль), полученную как описано в примере 10, растворяли в 60 мл 28% водного раствора аммиака, а затем добавляли 2,84 г 5%-ного родия на угле (50% продукт в виде гидрата) и реакцию осуществляли при комнатной температуре под давлением водорода 1 МПа. Через 17 часов, катализатор отфильтровывали (фильтр 0,2 мкм), и в полученном фильтрате растворяли 1,04 г (7,5 ммоль) карбоната калия. Растворенный раствор концентрировали, затем к 15,4 г полученного концентрата добавляли 20 мл этанола и смесь перемешивали при 25°С. После этого в течение 3 часов по каплям добавляли еще 30 мл этанола, и смесь перемешивали при 25°С в течение 17 часов.
Полученные сырые кристаллы (2,93 г) растворяли в 4 мл воды, добавляли 8 мл этанола при 35°С, а затем в течение 3 часов по каплям добавляли 17 мл этанола при 35°С. Полученный этаноловый раствор охлаждали до 10°С в течение 4 часов и перемешивали при 10°С в течение одного часа. Полученные сырые кристаллы (2,30 г) сушили при пониженном давлении с получением нужного вещества, а именно 1,99 г калиевой соли (2R,4R)-монатина. Оптическая чистота полученного монатина составляла 99,6% д.и.
1ЯМР (400 МГц, D2O) δ: 2,06 (дд, J=11,8, 15,3 Гц, 1H), 2,67 (дд, J=2,0, 15,2 Гц, 1H), 3,08 (д, J=14,4 Гц, 1H), 3,28 (д, J=14,4 Гц, 1H), 3,63 (дд, J=2,2, 12,2 Гц, 1H), 7,12-7,16 (м, 1H), 7,20-7,24 (м, 2H), 7,48-7,49 (м, 1H), 7,71-7,73 (м, 1H).
ESI-МС вычисленное значение: C14H16N2O5=292,29, аналитическое значение: 291,28 [M-H]-
Пример 12. Усовершенствованный синтез IHOG с использованием SpALD
Одну платиновую петлю микробных клеток E.coli/ptrpSpALD, культивированных в чашках с агаровой средой LB-amp при 37°С в течение 16 часов, инокулировали в 12 колб емкостью 500 мл, содержащих 50 мл среды LB-amp. Затем проводили культивирование со встряхиванием при 37°С в течение 16 часов. Микробные клетки собирали из полученной культуры путем центрифугирования, суспендировали в буфере А (20 мМ Hepes-КОН, рН 7,6) и промывали этим буфером, а затем снова центрифугировали и микробные клетки собирали. Микробные клетки (сырая масса: примерно 3 г), собранные путем центрифугирования, суспендировали в 300 мл реакционной смеси, имеющей нижеследующий состав.
Реакционная смесь для синтеза IHOG содержала: 50 мМ фосфатного буфера (pH 8,7), 300 мМ индолпировиноградной кислоты, 600 мМ натриевой соли пировиноградной кислоты, 1 мМ MgCl2 (рН доводили до 8,5 добавлением 6 н. КОН).
Суспензию, в которой были суспендированы микробные клетки, барботировали газом аргоном, и последующую реакцию проводили в атмосфере аргона. Реакцию осуществляли с перемешиванием при 37°С в течение 20 часов. После завершения реакции и после центрифугировали для удаления микробных клеток получали 300 мл альдольной реакционной смеси. Полученную реакционную смесь подвергали оксимированию в соответствии с процедурой, описанной в примере 10, а затем количество IHOG оценивали с помощью ВЭЖХ. В результате получали 63,6 мМ 4R-IHOG с оптической чистотой 92,2% э.и., что свидетельствовало о том, что использование SpALD приводит к повышению 4R-селективности.
Пример 13. Клонирование гена альдолазы (buald), происходящего от штамма С24 (штамм AJ110371 Burkholderia sp.), и его крупномасштабная экспрессия в E.coli
Ген, кодирующий 4R-IHOG-альдолазу (BuALD), был получен из штамма С24 собранных обогащенных микробных клеток, и было обнаружено, что он обладает 4R-IHOG-альдолазной активностью, как описано в примере 1. Ген buald лигировали в рtrp4 для крупномасштабной экспрессии в JM109 E.coli.
(1) Получение хромосомной ДНК
Штамм С24 (штамма AJ110371 Burkholderia sp.) культивировали с использованием 50 мл среды CM2G при 30°С в течение ночи (предварительное культивирование). Массовое культивирование проводили с использованием 5 мл этой культуры в качестве инокулята и с использованием 50 мл бульонной среды. После культивирования до достижения поздней логарифмической фазы роста, 50 мл культуры подвергали центрифугированию (12000×g, 4°С, 15 минут), и микробные клетки собирали. С использованием этих микробных клеток получали хромосомную ДНК стандартными методами.
(2) Определение последовательности гена buald с помощью Саузерн-анализа и гибридизации колоний
Ген buald получали с использованием ДНК-фрагмента, кодирующего полноразмерный ген SpALD в качестве зонда, путем проведения Саузерн-анализа и гибридизации колоний. С использованием праймеров (SEQ ID NO:10 и 11), представленных в таблице 9, полноразмерный ген spald подвергали ПЦР-амплификации из хромосомной ДНК штамма С77 (штамма AJ110329 Sphingomonas sp.). ДНК-зонд получали с использованием амплифицированного фрагмента, метили с использованием праймера DIG High (поставляемого Roche Diagnostics) и инкубировали в течение ночи при 37°С в соответствии с инструкциями. Саузерн-анализ проводили путем полного гидролиза 1 мкг хромосомной ДНК, полученной из штамма С24, различными рестриктирующими ферментами, электрофореза на 0,8% агарозном геле, и блоттинга на найлоновой мембране с проведением последующих стадий в соответствии с инструкциями. Гибридизацию осуществляли с использованием набора DIG Easy Hyb (поставляемого Roche Diagnostics), при этом предварительную гибридизацию осуществляли при 50°С в течение одного часа, а затем добавляли зонд для проведения гибридизации в течение ночи. Полосы детектировали с использованием набора для детекции нуклеотидов (DIG Nucleotide Detection Kit). В результате был детектирован PstI/SmaI-фрагмент размером примерно 2,3 т.п.н., который интенсивно гибридизовался с зондом для ПЦР-фрагмента. Затем получали PstI/BamHI-фрагмент путем гибридизации колоний. Хромосомную ДНК (20 мкг) обрабатывали PstI/SmaI и подвергали электрофорезу на агарозном геле с получением фрагмента размером примерно 2,3 т.п.н. Этот фрагмент лигировали в pUC18, и создавали библиотеку в JM109 E.coli. Колонии переносили на найлоновый мембранный фильтр (Hybond-N, поставляемый Amersham), затем подвергали щелочной модификации, нейтрализации и фиксации. Гибридизацию осуществляли с использованием набора DIG Easy Hyb. Фильтр погружали в буфер и предварительно гибридизовали при 42°С в течение одного часа. Затем добавляли меченый зонд и проводили гибридизацию при 42°С в течение 16 часов. После промывки SSC колонию, которая гибридизовалась с зондом, детектировали с использованием набора для детекции нуклеотидов (DIG Nucleotide Detection Kit) (поставляемого Roche Diagnostics). В результате был получен клон, который интенсивно гибридизовался с зондом.
Затем определяли нуклеотидную последовательность плазмидной ДНК, выделенной из полученного клона, в результате чего было продемонстрировано, что эта плазмидная ДНК имеет нуклеотидную последовательность SEQ ID NO:11. Таким образом, был получен нужный полноразмерный ген buald.
(3) Конструирование плазмиды ptrpBuALD, экспрессирующей ген альдолазы и ее экспрессия в E.coli
Фрагмент, амплифицированный из хромосомной ДНК штамма С24 (штамма AJ110371 Burkholderia sp.) с использованием праймеров (SEQ ID NO:19 и 20), указанных в таблице 10, гидролизовали ферментами NdeI/PstI, и встраивали в NdeI/PstI-сайт плазмиды рTrp4, в результате чего получали плазмиду ptrpBuALD. Эта плазмида экспрессировала ген альдолазы, кодирующий аминокислотную последовательность SEQ ID NO:15, полученную путем трансляции области, простирающейся от ATG в положении нуклеотида № 531, который представляет собой кодон инициации трансляции, до нуклеотида № 1385 в нуклеотидной последовательности SEQ ID NO:14.
Таблица 10
Последовательность праймеров для конструирования ptrpBuALD
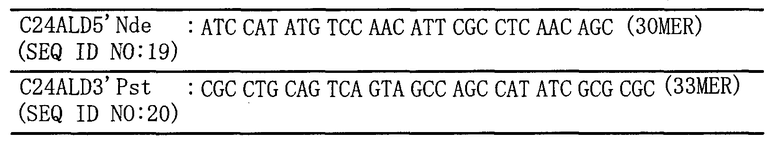
JM109 E.coli трансформировали сконструированной экспрессионной плазмидой, и одну платиновую петлю полученного трансформанта инокулировали в 50 мл среды LB, содержащей 100 мкг/мл ампициллина, проводили реакцию со встряхиванием при 37°С в течение 16 часов. После культивирования, микробные клетки собирали из 1 мл полученной культуры, промывали и суспендировали в 1 мл 20 мМ Hepes-КОН (рН 7,6), а затем микробные клетки подвергали разрушению ультразвуком. Супернатант получали путем центрифугирования при 15000 об/мин в течение 10 минут для разрушения суспензии и использовали в качестве неочищенного ферментного раствора.
Альдолазную активность измеряли с использованием неочищенного раствора фермента. Эту альдолазную активность измеряли как активность альдольного расщепления, осуществляемого с использованием PHOG в качестве субстрата в нижеследующих условиях.
Реакционные условия: 50 мМ Hepes-КОН (pH 8,5), 2 мМ PHOG, 0,25 мМ NADH, 0,2 мМ KPi, 1 мМ MgCl2, 16 ед./мл лактат-дегидрогеназы, 3 мкл фермента на 600 мкл реакционной смеси, 30°С, оптическую плотность измеряли при 340 нм.
В результате измерения какой-либо альдолазной активности в E.coli, трансформированной плазмидой рTrp4 (контроль), не наблюдалось, тогда как в штамме, трансформированном плазмидой ptrpBuALD, было детектировано 231 ед./мг белка, обладающего альдолазной активностью. Это свидетельствовало о том, что был действительно клонирован нужный ген альдолазы и что была сконструирована плазмида, содержащая в высокой степени экспрессирующуюся BuALD.
Пример 14. Очистка рекомбинантного фермента альдолазы, происходящего от штамма С24 (штамма AJ110371 Burkholderia sp.)
Рекомбинантную BuALD выделяли из растворимой фракции E.coli, обнаруживающей высокую степень экспрессии альдолазы (BuALD), происходящей от штамма С24 (штамма AJ110371 Burkholderia sp.), следующим образом. Альдолазную активность измеряли как активность в альдольном расщеплении, осуществляемом с использованием PHOG в качестве субстрата в следующих условиях.
Реакционные условия: 50 мМ Hepes-КОН (pH 8,5), 2 мМ PHOG, 0,25 мМ NAD, 1 мМ MgCl2, 16 ед./мл лактат-дегидрогеназы, 3 мкл фермента на 600 мкл реакционной смеси, 30°С, оптическую плотность измеряли при 340 нм.
(1) Получение растворимой фракции
Одну платиновую петлю микробных клеток E.coli/ptrpBuALD, культивированных в чашках с агаровой средой LB-amp при 37°С в течение 16 часов, инокулировали в 10 колб емкостью 500 мл, содержащих 50 мл среды LB-amp. Затем осуществляли культивирование со встряхиванием при 37°С в течение 16 часов. Микробные клетки собирали из полученной культуры путем центрифугирования, суспендировали в буфере А (20 мМ Hepes-КОН, рН 7,6) и промывали этим буфером, а затем снова центрифугировали и микробные клетки собирали. Полученные промытые микробные клетки суспендировали в 20 мл буфера А и подвергали разрушению путем обработки ультразвуком при 4°С в течение 30 минут. Дебрис микробных клеток удаляли путем центрифугирования (×8000 об/мин, 10 мин, два раза), для разрушения суспензии и полученный супернатант использовали в качестве неочищенной экстрагированной фракции.
(2) Анионообменная хроматография: Q-сефароза FF
Растворимую фракцию (20 мл), полученную как описано выше, наносили на анионообменную колонку с Q-сефарозой FF 26/10 (поставляемой Pharmacia CV=20 мл), предварительно уравновешенную буфером А для абсорбции белков на носителе. Белки, которые не абсорбировались на носителе (неабсорбированные белки), отмывали с использованием буфера А. Затем абсорбированные белки элюировали линейным градиентом концентраций KCl от 0 М до 0,7 М (всего 140 мл). PHOG-альдолазную активность измеряли для каждой элюированной фракции, а затем пик PHOG-альдолазной активности детектировали во фракции, соответствующей примерно 0,35 М.
(3) Гидрофобная хроматография: фенилсефароза НР HR 16/10
Раствор, в котором была детектирована альдолазная активность, диализовали против буфера В (20 мМ Hepes-КОН, 1 М сульфата аммония, рН 7,6) при 4°С в течение ночи, и фильтровали через фильтр с размером пор 0,45 мкм. Полученный фильтрат наносили на гидрофобную хроматографическую колонку с фенилсефарозой НР HR 16/10 (поставляемой Pharmacia), предварительно уравновешенную буфером В. В результате этой процедуры, альдолаза абсорбировалась на носителе.
Белки, не абсорбированные на носителе, отмывали с использованием буфера В, а затем альдолазу элюировали линейным градиентом концентраций сульфата аммония 1 М-0 М. Альдолазную активность измеряли для каждой элюированной фракции, и такая альдолазная активность детектировалась в тех положениях элюирования, где концентрация сульфата аммония составляла примерно 0,4-0,5 М.
(4) Гель-хроматография: сефадекс 200 НР 16/60
Фракции, содержащие альдолазу, собирали, диализовали против буфера А и фильтровали через фильтр с размером пор 45 мкм. Полученный фильтрат концентрировали с использованием ультрафильтрационной мембраны, Centriprep 10. Полученный концентрированный раствор наносили на гель-фильтрационную колонку с сефадексом 200 НР 16/60 (поставляемым Pharmacia), предварительно уравновешенную буфером С (20 мМ Hepes-КОН, 0,1 М KCl, рН 7,6), и элюировали при скорости потока 1 мл/мин. В этой процедуре, альдолаза элюировалась во фракциях примерно 66 мл.
(5) Анионообменная хроматография: Mono-Q HR 5/5
Полученную фракцию фильтровали через фильтр с размером пор 0,45 мкм. Полученный фильтрат наносили на анионообменную хроматографическую колонку с Mono-Q HR 5/5 (поставляемой Pharmacia), предварительно уравновешенную буфером А. В результате этой процедуры, альдолаза абсорбировалась на носителе. Неабсорбированные белки отмывали буфером А, а затем белок элюировали линейным градиентом концентраций KCl от 0 мМ до 700 мМ (всего 24 мл). Альдолазную активность измеряли для каждой фракции, и эта альдолазная активность детектировалась в элюированных положениях, где концентрация KCl составляла примерно 0,4 М.
Фракции, очищенные с помощью описанной выше колоночной хроматографии, подвергали электрофорезу в ДСН-ПААГ, а затем одну полосу в положении, соответствующем примерно 30 кДа, детектировали путем окрашивания СВВ. Полученный раствор рекомбинантной BuALD диализовали против буфера А при 4°С в течение ночи. В результате этой процедуры получали 1,5 мл раствора, содержащего 1241 ед./мл BuALD.
Пример 15. Ферментативные свойства BuALD
С использованием BuALD, полученной как описано в примере 14, были исследованы нижеследующие параметры:
Условия предварительных измерений: 50 мМ Hepes-КОН (pH 8,5), 2 мМ PHOG, 5 мМ MgCl2, 16 ед./мл лактат-дегидрогеназы, при этом наблюдалась пониженная оптическая плотность, измеренная при 340 нм и при 30°С.
(1) Кинетическая константа, определяемая с использованием PHOG в качестве субстрата
Исходя из результатов, представленных на фиг. 8A и 8B, были определены следующие параметры: Vmax (для PHOG)=483 мкмоль/мин/мг, Km (для PHOG)=0,66 мМ, Km (для MgCl2)=0,021 мМ. Какого-либо увеличения альдолазной активности при добавлении раствора КРВ не наблюдалось.
(2) рН-стабильность
Было определено, что рН-стабильность составляет в пределах от 3 до 10. Для измерения были использованы следующие буферы: натрийцитратный буфер (рН 3; 4; 4,5), натрийацетатный буфер (рН 4; 4,5; 5; 5,5; 6; 6,5), калийфосфатный буфер (рН 6; 6,5; 7; 7,5; 8; 8,5), буфер Hepes-KOH (рН 7,5; 8; 8,5; 9; 9,5), буфер CAPS-NaOH (рН 9; 9,5; 10). После инкубирования BuALD в 100 мМ каждого из этих буферов при 37°С в течение 30 минут, остаточную активность измеряли в условиях для предварительного измерения. Результаты представлены на фиг.9.
(3) Термостабильность
После инкубирования BuALD в 100 мМ калийфосфатного буфера (рН 7,0) и 100 мМ буфера Hepes-KOH (рН 8,5) при 25-70°С в течение 30 минут, остаточную активность измеряли в условиях для предварительных измерений. Результаты представлены на фиг.10.
Пример 16. Синтез и оксимирование IHOG с использованием BuALD и SpALD
Одну платиновую петлю микробных клеток JM109 E.coli/ptrpBuALD или JM109 E.coli/ptrpSpALD, культивированных в чашках с агаровой средой LB-amp при 37°С в течение 16 часов, инокулировали в 4 колбы емкостью 500 мл, содержащих 50 мл среды LB-amp. Затем осуществляли культивирование со встряхиванием при 37°С в течение 16 часов. Микробные клетки собирали из полученной культуры путем центрифугирования, суспендировали в буфере А (20 мМ Hepes-КОН, рН 7,6) и промывали этим буфером, а затем снова центрифугировали и собирали микробные клетки. Микробные клетки (сырая масса микробных клеток: примерно 1 г), собранные путем центрифугирования, суспендировали в 100 мл реакционной смеси, имеющей нижеследующий состав.
Реакционная смесь для синтеза IHOG содержала: 50 мМ КРВ (pH 8,0), 300 мМ индолпировиноградной кислоты, 600 мМ натриевой соли пировиноградной кислоты, 0,1 мМ MgCl2 (рН доводили до 8,0 добавлением 6 н. КОН).
Реакционную смесь, в которой были суспендированы микробные клетки, барботировали газом аргоном, а затем проводили реакцию с перемешиванием при 37°С в течение 18 часов. После завершения реакции и после центрифугирования для удаления микробных клеток получали примерно 100 мл альдольной реакционной смеси.
При поддерживании величины рН 9 путем добавления водного 6 н. раствора гидроксида натрия, примерно к 96 мл полученной альдольной реакционной смеси добавляли 6,25 г (90 ммоль) гидрохлорида гидроксиламина, а затем смесь перемешивали при 25°С в течение 6 часов и при 10°С в течение ночи. Количество IHOG-оксима в полученной реакционной смеси определяли с помощью ВЭЖХ-анализа. В результате (таблица 11), в этой реакционной смеси группы BuALD и группы SpALD преимущественно продуцировался 4R-IHOG-оксим.
Пример 17. Клонирование гена альдолазы (SpALD2) из штамма С43 (штамма AJ110372 Sphingomonas sp.) и его крупномасштабная экспрессия в E.coli
Ген, кодирующий 4R-IHOG-альдолазу (SpALD), получали из штамма С43, который был отобран из бактерий в обогащенной культуре, и было обнаружено, что он обладает 4R-IHOG-альдолазной активностью, как описано в примере 1. Ген spald2 лигировали в рtrp4 для крупномасштабной экспрессии в JM109 E.coli.
(1) Получение хромосомной ДНК
Штамм С43 (штамм AJ110329 Sphingomonas sp.) культивировали с использованием 50 мл среды CM2G при 30°С в течение ночи (предварительное культивирование). Основное культивирование проводили с использованием 5 мл этой предварительной культуры в качестве инокулята и с использованием 50 мл бульонной среды. После культивирования до достижения поздней логарифмической фазы роста 50 мл культуры подвергали центрифугированию (12000×g, 4°С, 15 минут), и микробные клетки собирали. С использованием этих микробных клеток получали хромосомную ДНК стандартными методами.
(2) Определение последовательности гена spald2 с помощью Саузерн-анализа и гибридизации колоний
Ген spald2 получали с использованием ДНК-фрагмента, кодирующего полноразмерный ген SpALD в качестве зонда, путем проведения Саузерн-анализа и гибридизации колоний. С использованием праймеров (SEQ ID NO:10 и 11), представленных в таблице 9, полноразмерный ген spald подвергали ПЦР-амплификации из хромосомной ДНК штамма С77 (штамма AJ110329 Sphingomonas sp.). ДНК-зонд получали с использованием амплифицированного фрагмента, метили с использованием праймера DIGHigh (поставляемого Roche Diagnostics) и инкубировали в течение ночи при 37°С в соответствии с инструкциями. Саузерн-анализ проводили путем полного гидролиза 1 мкг хромосомной ДНК, полученной из штамма С43, различными рестриктирующими ферментами, электрофореза на 0,8% агарозном геле, и блоттинга на найлоновой мембране с проведением последующих стадий в соответствии с инструкциями. Гибридизацию осуществляли с использованием набора DIG Easy Hyb (поставляемого Roche Diagnostics), при этом, предварительную гибридизацию осуществляли при 50°С в течение одного часа, а затем добавляли зонд для проведения гибридизации в течение ночи. Полосы детектировали с использованием набора для детекции нуклеотидов (DIG Nucleotide Detection Kit). В результате был детектирован PstI-фрагмент размером примерно 3 т.п.н., который интенсивно гибридизовался с зондом для ПЦР-фрагмента. Затем этот PstI-фрагмент получали путем гибридизации колоний. Хромосомную ДНК (20 мкг) обрабатывали PstI и подвергали электрофорезу на агарозном геле с получением фрагмента размером примерно 3 т.п.н. Этот фрагмент лигировали в pUC18 и создавали библиотеку в JM109 E.coli. Колонии переносили на найлоновый мембранный фильтр (Hybond-N, поставляемый Amersham), а затем подвергали щелочной модификации, нейтрализации и фиксации. Гибридизацию осуществляли с использованием набора DIG Easy Hyb. Фильтр погружали в буфер и предварительно гибридизовали при 42°С в течение одного часа. Затем добавляли меченный зонд и проводили гибридизацию при 42°С в течение 16 часов. После промывки SSC колонию, которая гибридизовалась с зондом, детектировали с использованием набора для детекции нуклеотидов (DIG Nucleotide Detection Kit) (поставляемого Roche Diagnostics). В результате был получен клон, который интенсивно гибридизовался с зондом.
Затем определяли нуклеотидную последовательность плазмидной ДНК, выделенной из полученного клона, в результате чего было продемонстрировано, что эта плазмидная ДНК имеет нуклеотидную последовательность SEQ ID NO:12. Таким образом, был получен нужный полноразмерный ген spald2.
(3) Конструирование плазмиды pUCSрАLD2, экспрессирующей ген альдолазы, и ее экспрессия в E.coli
Фрагмент, амплифицированный из хромосомной ДНК штамма С43 Sphingomonas sp. с использованием праймеров (SEQ ID NO:21 и 22), указанных в таблице 12, гидролизовали ферментами EcoRI/PstI, и встраивали в EcoRI/PstI-сайт плазмиды pUC18, в результате чего получали плазмиду pUCSрАLD2. Эта плазмида экспрессировала ген альдолазы, кодирующий аминокислотную последовательность SEQ ID NO:13, полученную путем трансляции области, простирающейся от ATG в положении нуклеотида № 399, который представляет собой кодон инициации трансляции, до нуклеотида № 1253 в нуклеотидной последовательности SEQ ID NO:12.
Таблица 12
Последовательность праймеров для конструирования pUCSрАLD2
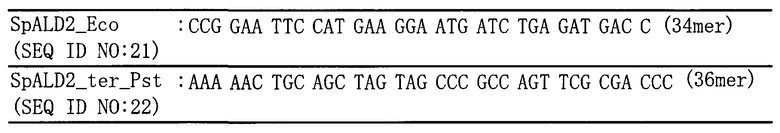
JM109 E.coli трансформировали сконструированной экспрессионной плазмидой и одну платиновую петлю полученного трансформанта инокулировали в 50 мл среды LB, содержащей 100 мкг/мл ампициллина и 0,1 мМ изопропил-b-D-тиогалактопиранозида (IPTG), и проводили реакцию со встряхиванием при 37°С в течение 16 часов. После завершения культивирования микробные клетки собирали из 1 мл полученной культуры, промывали и суспендировали в 1 мл 20 мМ Hepes-КОН (рН 7,6), а затем микробные клетки подвергали разрушения ультразвуком. Супернатант получали путем центрифугирования при 15000 об/мин в течение 10 минут для разрушения суспензии и использовали в качестве неочищенного ферментного раствора.
Альдолазную активность измеряли с использованием неочищенного раствора фермента. Эту альдолазную активность измеряли как активность в реакции альдольного расщепления, осуществляемой с использованием PHOG в качестве субстрата, в нижеследующих условиях.
Реакционные условия: Hepes-КОН (pH 8,5), 2 мМ PHOG, 0,25 мМ NADH, 1 мМ MgCl2, 16 ед./мл лактат-дегидрогеназы, 3 мкл фермента на 600 мкл реакционной смеси, 30°С, оптическую плотность измеряли при 340 нм.
В результате измерения какой-либо альдолазной активности в E.coli, трансформированной плазмидой pUC18 (контроль), не наблюдалось, тогда как в штамме, трансформированном плазмидой pUCSрАLD2, было детектировано 33,6 ед./мг белка, обладающего альдолазной активностью. Это свидетельствовало о том, что был действительно клонирован нужный ген альдолазы, и что была сконструирована плазмида, содержащая в высокой степени экспрессирующуюся SpALD2.
Пример 18. Синтез IHOG и IHOG-оксима с использованием SpALD2
Одну платиновую петлю микробных клеток JM109 E.coli/рCUSSрАLD2, культивированных в чашках с агаровой средой LB-amp при 37°С в течение 16 часов, инокулировали в 4 колбы емкостью 500 мл, содержащих 50 мл среды LB, включающей 100 мкг/мл ампициллина и 0,1 мМ IPTG. Затем осуществляли культивирование со встряхиванием при 34°С в течение 16 часов. Микробные клетки собирали из полученной культуры путем центрифугирования, суспендировали в буфере А (20 мМ Hepes-КОН, рН 7,6) и промывали этим буфером, а затем снова центрифугировали и микробные клетки собирали. Микробные клетки (сырая масса микробных клеток: примерно 1 г), полученные путем центрифугирования, суспендировали в 100 мл реакционной смеси, имеющей нижеследующий состав.
Реакционная смесь для синтеза IHOG содержала: 50 мМ КРВ (pH 8,0), 300 мМ индолпировиноградной кислоты, 600 мМ натриевой соли пировиноградной кислоты, 0,1 мМ MgCl2 (рН доводили до 8,0 добавлением 6 н. КОН).
Реакционную смесь, в которой были суспендированы микробные клетки, барботировали газом аргоном, а затем осуществляли реакцию с перемешиванием при 37°С в течение 18 часов. После завершения реакции и после центрифугирования для удаления микробных клеток получали примерно 100 мл альдольной реакционной смеси. Примерно к 96 мл полученной альдольной реакционной смеси добавляли водный раствор (5,95 мл, 90 ммоль) 50% гидроксиламина, а затем смесь перемешивали при 25°С в течение 6 часов и при 10°С в течение ночи. Количество IHOG-оксима в полученной реакционной смеси определяли с помощью ВЭЖХ-анализа. В результате было получено 3,84 ммоль 4R-IHOG-оксима и 0,15 ммоль 4S-IHOG-оксима, что указывало на то, что в этой реакции преимущественно продуцировался 4R-изомер с оптической чистотой 92,4% э.и.
Сравнительный пример 1. Синтез 4-гидрокси-4-(3-индолилметил)-2-кетоглутаровой кислоты (IHOG)
Индол-3-пировиноградную кислоту (7,50 г, 35,8 ммоль, содержание 97 мас.%) и 14,18 г (107,4 ммоль) щавелево-уксусной кислоты добавляли и растворяли в 64,45 мл воды, в которой было растворено 18,91 г гидроксида калия (286,5 ммоль, содержание 85 мас.%). Полученный смешанный раствор перемешивали при 35°С в течение 24 часов.
Затем смесь нейтрализовали 40,0 мл 3 н. соляной кислоты (рН 7,0) и получали 153,5 г нейтрализованной реакционной смеси. В этой нейтрализованной смеси содержалось 5,55 г 4-гидрокси-4-(3-индолил)метил-2-кетоглутаровой кислоты, а выход (по отношению к индолпировиноградной кислоте) составлял 53,3%.
К полученной нейтрализованной смеси добавляли воду до объема 168 мл, а затем эту смесь пропускали через колонку со смолой, заполненную 840 мл синтетического абсорбента (DIAION-SP207, поставляемого Mitsubishi Chemical Corporation). Водный раствор, содержащий 3,04 г 4-гидрокси-4-(3-индолилметил)-2-кетоглутаровой кислоты с высокой степенью чистоты, получали путем пропускания очищенной воды при скорости потока 23,5 мл/мин, и последующим сбором продукта 1,73/2,25 (L/L-R), выход которого (по отношению к количеству, нанесенному на смолу) составил 54,7%.
(ЯМР-измерения)
1H-ЯМР (400 МГц, D2O): 3,03 (д, 1H, J=14,6 Гц), 3,11 (д, 1H, J=14,6 Гц), 3,21 (д, 1H, J=18,1 Гц), 3,40 (д, 1H, J=18,1 Гц), 7,06-7,15 (м, 3H), 7,39 (д, 1H, J=7,8 Гц), 7,66 (д, 1H, J=7,8 Гц).
13C-ЯМР (100 МГц, D2O): 35,43, 47,91, 77,28, 109,49, 112,05, 119,44, 119,67, 121,91, 125,42, 128,41, 136,21, 169,78, 181,43, 203,58.
Сравнительный пример 2. Синтез 4-фенилметил-4-гидрокси-2-кетоглутаровой кислоты (PHOG)
Фенилпировиноградную кислоту (5,0 г, 30,5 ммоль) и 12,1 г (91,4 ммоль) щавелево-уксусной кислоты добавляли к 25 мл водного раствора, в котором растворяли 13,8 г гидроксида калия (чистота 85%), и проводили реакцию при комнатной температуре в течение 72 часов. рН реакционной смеси доводили до значения 2,2 с использованием концентрированной соляной кислоты, и реакционную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором NaCl, сушили над безводным сульфатом магния и концентрировали с получением остатка. Остаток перекристаллизовывали из этилацетата и толуола, в результате чего получали 2,8 г (11,3 ммоль) 4-фенилметил-4-гидрокси-2-кетоглутаровой кислоты в виде кристаллов.
(ЯМР-измерения)
1H ЯМР (D2O) δ: 2,48 (д, J=14,4 Гц, 0,18H), 2,60 (д, J=14,4 Гц, 0,18H), 2,85-3,30 (м, 3,64H), 7,17-7,36 (м, 5H).
(Измерения молекулярной массы)
ESI-MC: для С12Н12O6: вычисленное значение=252,23; аналитическое значение=251,22 (МН-).
Сравнительный пример 3. Получение 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты
Индол-3-пировиноградную кислоту (73,8 г, 352 ммоль) добавляли и растворяли в 917 г водного раствора 1,6 мас.% гидроксида натрия. Температуру реакционной смеси поддерживали при 35°С. При поддержании рН при 11,1 с использованием водного 30% раствора гидроксида натрия в течение 2 часов по каплям добавляли 310,2 г (1761 ммоль) водного 50%-ного раствора пировиноградной кислоты. Реакцию проводили в течение еще 4,5 часов и получали реакционную смесь, содержащую 4-гидрокси-4-(3-индолилметил)-2-кетоглутаровую кислоту. При поддержании рН при 7 путем добавления водного 30%-ного раствора гидроксида натрия, к смеси добавляли 367,2 г (2114 ммоль) водного 40% раствора гидрохлорида гидроксиламина, и смесь перемешивали при 5°С в течение 17,5 часов. рН реакционной смеси доводили до значения 2 с использованием концентрированной соляной кислоты, и органические вещества экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором NaCl и концентрировали с получением остатка. Остаток перекристаллизовывали из 60 мл 28% водного раствора аммиака и 350 мл 2-пропанола, в результате чего получали 43,4 г диаммониевой соли 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (142 ммоль, выход 40% по отношению к индол-3-пировиноградной кислоте) в виде кристаллов.
Сравнительный пример 4. Получение (R)-(+)-1-фенилэтиламиновой соли (4S)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты
Аммониевую соль (44,7 г, 0,131 моль) 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты растворяли в 500 мл воды при 25°С, а затем рН водного раствора доводили до значения 2 путем добавления 25,5 г 36% соляной кислоты. Кислотный раствор экстрагировали 1300 мл этилацетата, и этилацетатный раствор промывали 200 мл насыщенного водного раствора NaCl. К полученному этилацетатному раствору добавляли водный раствор (500 мл) карбоната натрия (13,9 г, 0,131 моль), и смесь перемешивали для отделения щелочного водного раствора от этилацетата. рН полученного щелочного водного раствора доводили до значения 2 путем добавления 23,1 г 36% соляной кислоты. В полученный кислотный водный раствор по каплям добавляли (R)-(+)-1-фенилэтиламин (6,99 г, 57,6 ммоль) и смесь перемешивали при 25°С в течение одного часа. Полученные кристаллы фильтровали и сушили при пониженном давлении, в результате чего получали 21,8 г (47,8 ммоль) (R)-(+)-1-фенилэтиламиновой соли (4S)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (выход: 72,7%, оптическая чистота: 87,4%).
(ЯМР-измерения)
1H-ЯМР (400 МГц, ДМСО-d6)δ: 1,48 (д, 3H, J=6,8 Гц), 2,63 (д, 1H, J=14,0 Гц), 2,70 (d, 1H, J=14,0 Гц), 2,90 (д, 1H, J=14,1 Гц), 3,06 (д, 1H, J=14,1 Гц), 4,40 (кв, 1H, J=6,8 Гц), 6,91-7,54 (м, 10H).
Сравнительный пример 5. Получение (S)-(-)-1-фенилэтиламиновой соли (4R)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты
(S)-(-)-1-Фенилэтиламин (7,12 г, 58,7 ммоль) по каплям добавляли в кристаллический фильтрат, полученный как описано в сравнительном примере 4, и перемешивали при 25°С в течение одного часа. Полученные кристаллы фильтровали и сушили при пониженном давлении с получением 23,8 г (53,3 моль) (S)-(-)-1-фенилэтиламиновой соли (4R)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (выход: 81,1%, оптическая чистота: 92,1%).
Сравнительный пример 6. (1) Получение аммониевой соли (4S)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты
При 25°С, 21,8 г (51,0 ммоль) (R)-(+)-1-фенилэтиламиновой соли (4S)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты добавляли к 200 мл воды и 18,5 г 28% водного раствора аммиака и растворяли, а затем к раствору добавляли 200 мл толуола и перемешивали. Водный слой, полученный после разделения слоев, нагревали до 60°С и в течение 2 часов по каплям добавляли 900 мл 2-пропанола. Этот 2-пропаноловый водный раствор охлаждали до 10°С в течение 5 часов, а затем перемешивали при 10°С в течение 10 часов. Полученные кристаллы фильтровали и сушили при пониженном давлении, в результате чего получали 14,75 г аммониевой соли (4S)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (выход: 85,1%, оптическая чистота: 99,0%).
Температура плавления: 205°С (разложение).
Удельное оптическое вращение: [α]D 20 + 13,4 (с=1,00, H2O).
(2) Получение аммониевой соли (4R)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты
Способом, описанным в приведенном выше сравнительном примере, получали 16,2 г аммониевой соли (4R)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (выход: 89,3%, оптическая чистота: 99,9%) из 23,8 г (55,3 ммоль) (R)-(+)-1-фенилэтиламиновой соли (4R)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты.
Удельное оптическое вращение: [α]D 20 - 13,6 (с=1,00, H2O).
Сравнительный пример 7. Получение (2R,4R)-монатина
Аммониевую соль (4R)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (13,2 г, 38,7 ммоль), полученную как описано в сравнительном примере 6, растворяли в 135 мл 28% водного аммиака, добавляли 6,93 г 5% родия на угле (готовый 50% водный раствор) и осуществляли реакцию при 25°С под давлением водорода 1 МПа. Через 24 часа катализатор отфильтровывали (фильтр 0,2 мкм) и в фильтрате растворяли 2,54 г (18,4 ммоль) карбоната калия. Растворенный раствор концентрировали, а затем к 32,7 г концентрата добавляли 20 мл воды и 45 мл этанола и смесь перемешивали при 25°С. После этого в течение 3 часов по каплям добавляли еще 60 мл этанола, и осуществляли кристаллизацию путем перемешивания при 25°С в течение 20 часов. Полученные сырые кристаллы (9,78 г) растворяли в 12 мл воды, добавляли 24 мл этанола, а затем в течение 3 часов по каплям добавляли еще 51 мл этанола. Полученный этаноловый раствор охлаждали до 15°С в течение 4 часов, а затем перемешивали при 15°С в течение 10 часов. Полученные сырые кристаллы (7,08 г) сушили при пониженном давлении и получали 5,7 г целевой калиевой соли (2R,4R)-монатина.
Как описано выше, благодаря использованию альдолазы настоящего изобретения стало возможным получать IHOG и PHOG с оптической селективностью. Альдолаза настоящего изобретения дает возможность эффективно вносить асимметрию в нужное положение в стадии проведения реакции альдольной конденсации синтеза монатина, и эта альдолаза может эффективно использована для получения оптически активных IHOG и монатина.
Другие преимущества и модификации будут очевидны для каждого специалиста. Поэтому настоящее изобретение в его более широких аспектах не ограничивается конкретными элементами и репрезентативными вариантами, представленными в настоящем описании. В соответствии с этим, в настоящее изобретение могут быть внесены различные модификации, не выходящие за рамки существа и объема общей идеи изобретения, определенной в прилагаемой ниже формуле изобретения и ее эквивалентах.



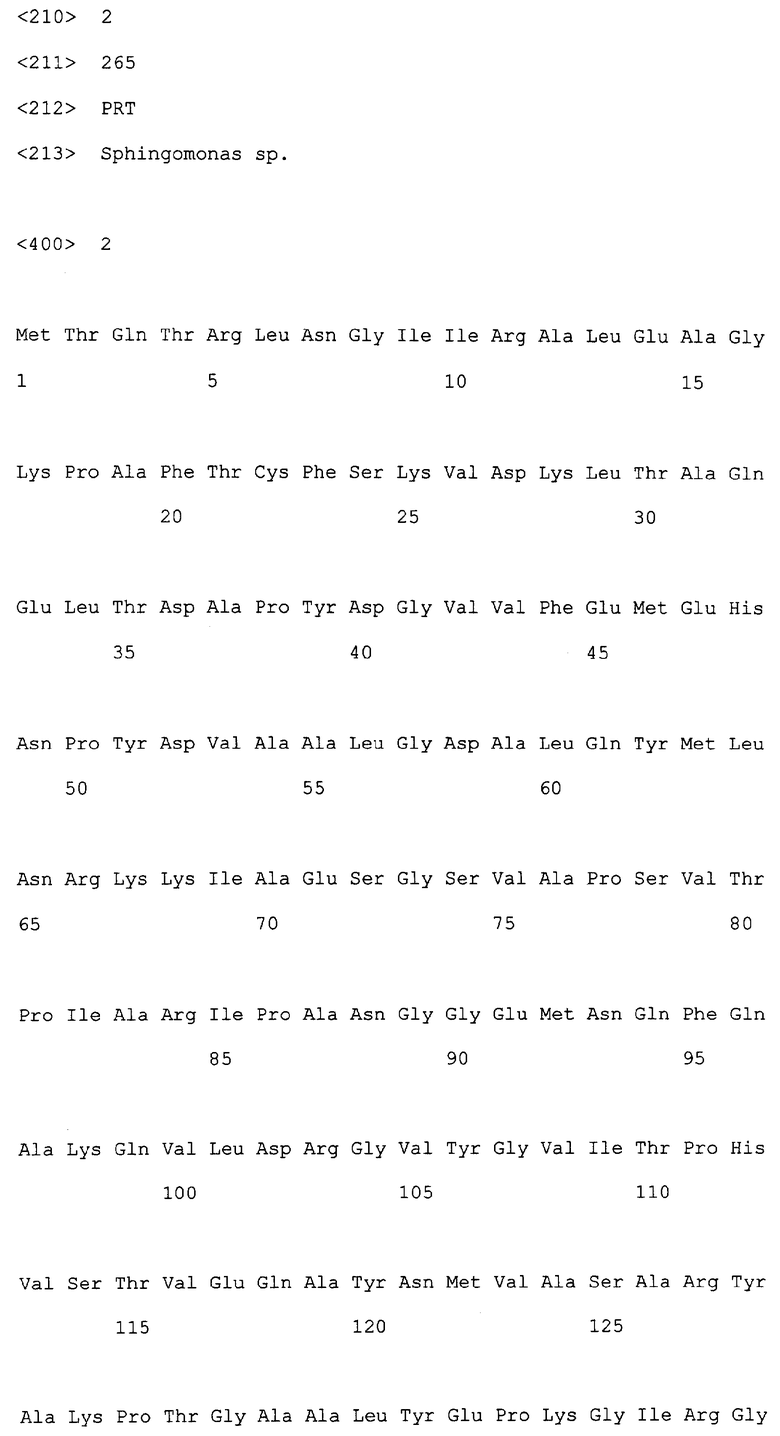



















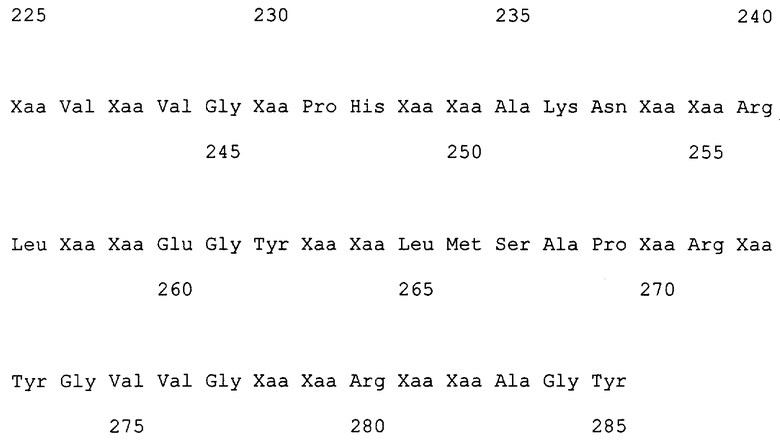
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВАЯ АЛЬДОЛАЗА И СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ α-КЕТОКИСЛОТ | 2003 |
|
RU2307871C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ГЛЮТАМАТА (ВАРИАНТЫ) И СПОСОБ ПОЛУЧЕНИЯ МОНАТИНА | 2002 |
|
RU2280078C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНДОЛ-3-ПИРОВИНОГРАДНОЙ КИСЛОТЫ И ЕЕ ПРОИЗВОДНЫХ | 2006 |
|
RU2325442C2 |
| ПИТЬЕВАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ МОНАТИН, И СПОСОБЫ ЕЕ ПОЛУЧЕНИЯ | 2004 |
|
RU2380989C2 |
| СПОСОБ ПОЛУЧЕНИЯ МОНАТИНА | 2011 |
|
RU2505606C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛИРОВАННОГО L-ЛЕЙЦИНА И БАКТЕРИЯ, ТРАНСФОРМИРОВАННАЯ ДНК, КОДИРУЮЩЕЙ ДИОКСИГЕНАЗУ | 2011 |
|
RU2468085C1 |
| НОВЫЙ ГЕН ПЕПТИДОБРАЗУЮЩЕГО ФЕРМЕНТА | 2003 |
|
RU2296798C2 |
| СТАБИЛЬНЫЙ ВЕКТОР КОНСТИТУТИВНО ВЫСОКОЙ ЭКСПРЕССИИ ДЛЯ ПОЛУЧЕНИЯ ВАКЦИНЫ ПРОТИВ ВПЧ И ТРАНСФОРМИРОВАННЫЕ ЭТИМ ВЕКТОРОМ РЕКОМБИНАНТНЫЕ МОЛОЧНОКИСЛЫЕ БАКТЕРИИ | 2010 |
|
RU2492240C2 |
| СПОСОБЫ И МАТЕРИАЛЫ ДЛЯ СОЗДАНИЯ И ПРИМЕНЕНИЯ ТРАНСГЕННЫХ ОРГАНИЗМОВ, ДИКАМБА-РАЗРУШАЮЩИХ | 2002 |
|
RU2292348C2 |
| ГЕН ПЕПТИДООБРАЗУЮЩЕГО ФЕРМЕНТА, ПЕПТИДООБРАЗУЮЩИЙ ФЕРМЕНТ И СПОСОБ ПОЛУЧЕНИЯ ДИПЕПТИДА | 2002 |
|
RU2280077C2 |
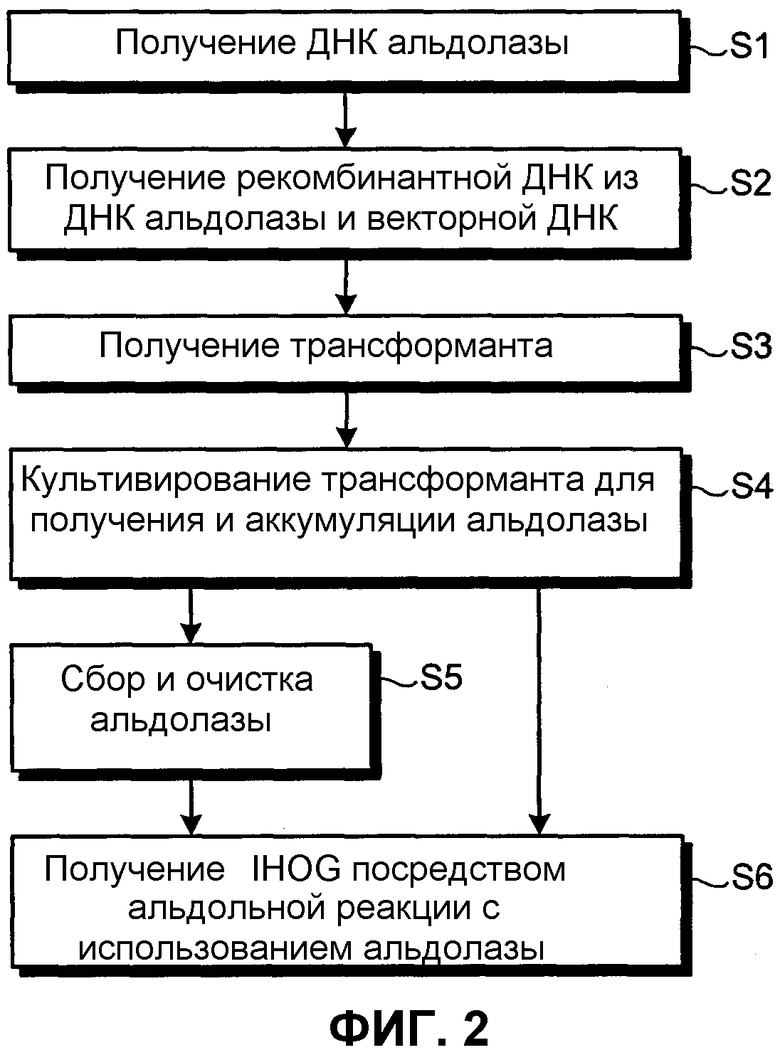
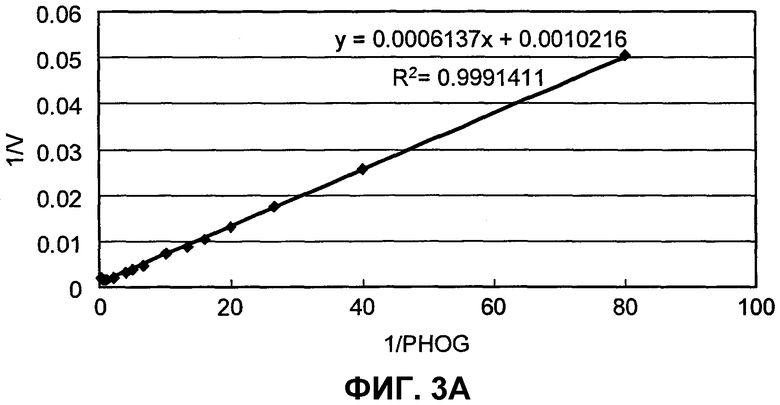
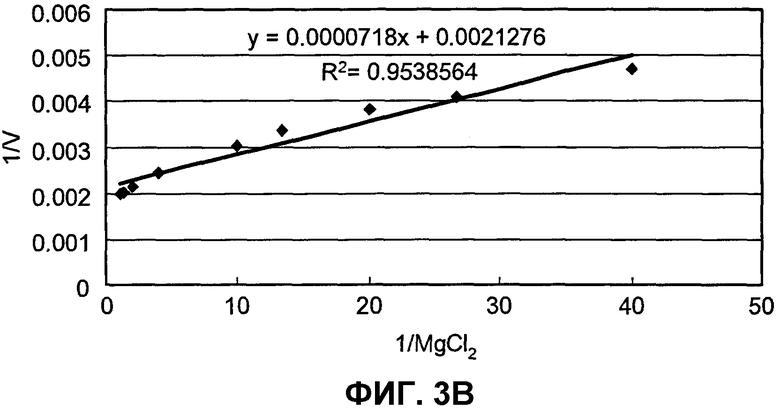
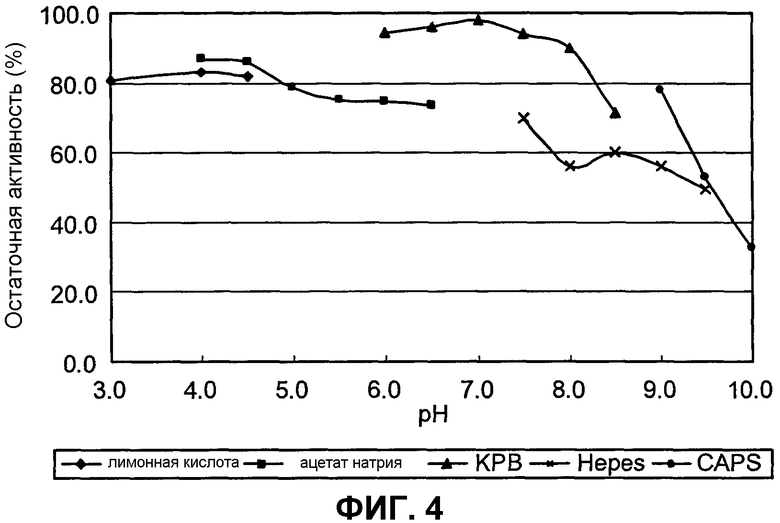
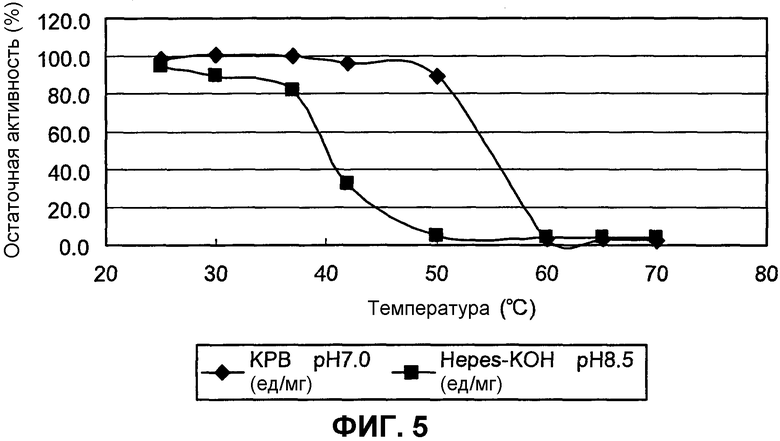
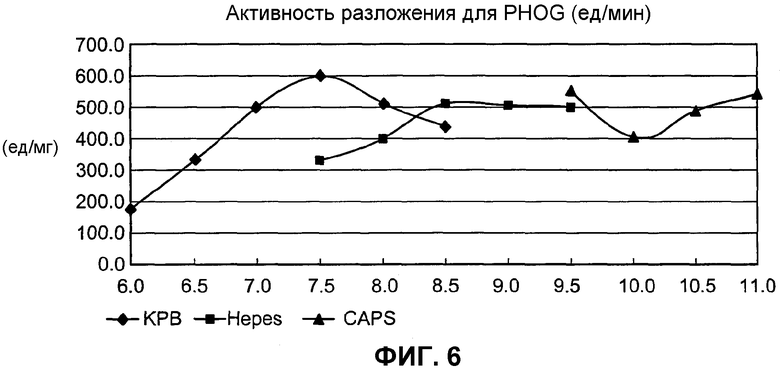
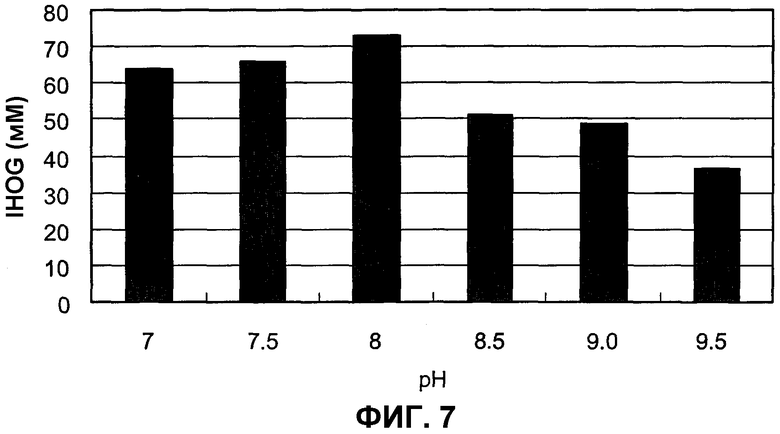
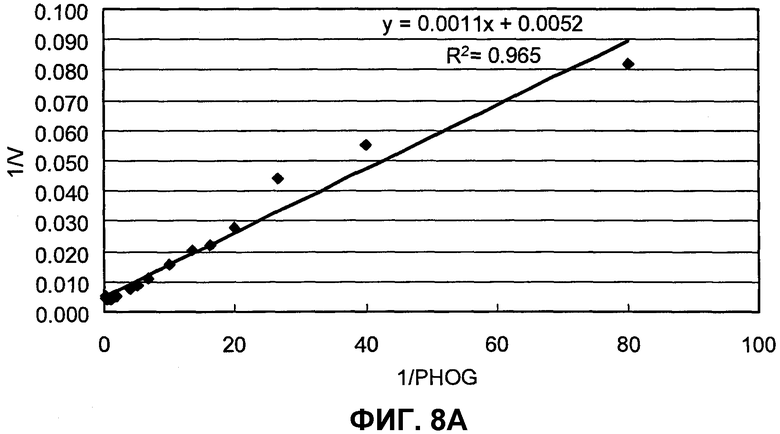
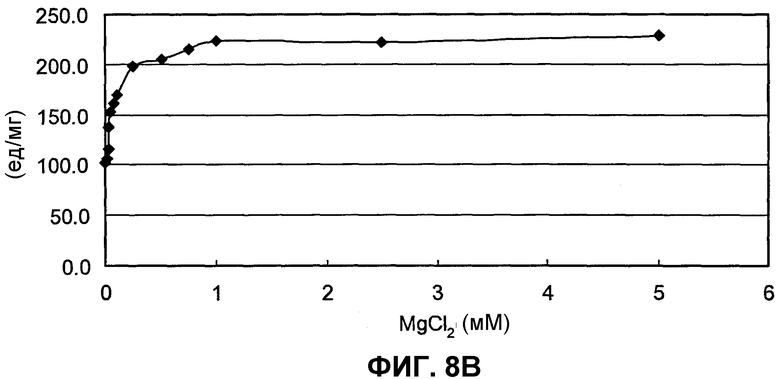
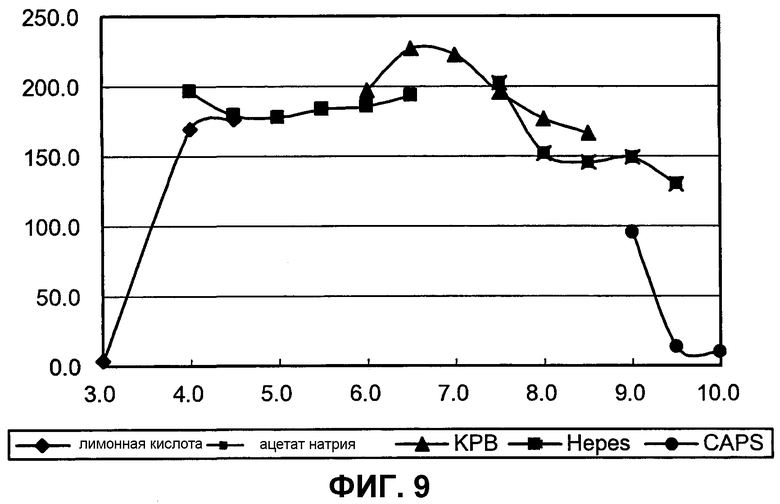
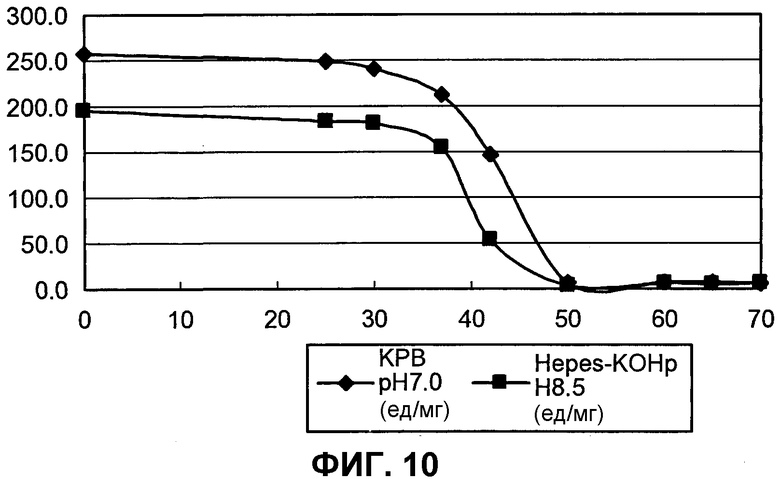
Изобретение относится к биотехнологии и представляет собой способ получения оптически активной 4-(индол-3-илметил)-4-гидрокси-2-оксоглутаровой кислоты, используемой для получения монатина, а также способ получения оптически активного монатина. Изобретение касается также и новой альдолазы, используемой в этих способах. 4-(Индол-3-илметил)-4-гидрокси-2-оксоглутаровая кислота с высокой оптической чистотой, являющаяся эффективной в качестве промежуточного соединения для синтеза оптически активного монатина, может быть синтезирована из индолпировиноградной кислоты и пировиноградной кислоты (или щавелево-уксусной кислоты). Изобретение позволяет получать 4-(индол-3-илметил)-4-гидрокси-2-оксоглутаровую кислоту и монатин с высокой степенью эффективности. 8 н. и 10 з.п. ф-лы, 12 ил., 12 табл.
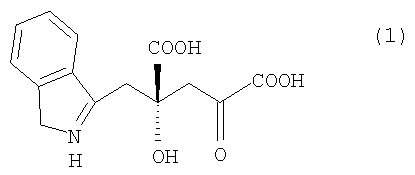
который предусматривает взаимодействие индол-3-пировиноградной кислоты с пировиноградной кислотой или щавелевоуксусной кислотой в присутствии белка или микроорганизма, содержащего такой белок, с получением 4R-IHOG, оптическая чистота которого достигает 70% или больше,
где указанный белок выбран из группы, состоящей из (а) и (b), а именно
(a) белок, содержащий аминокислотную последовательность SEQ ID NO:2, который обладает 4R-альдолазной активностью;
(b) белок, который на 70% или более гомологичен аминокислотной последовательности SEQ ID NO:2 и обладает 4R-альдолазной активностью.
первую стадию, в которой происходит взаимодействие индол-3-пировиноградной кислоты с пировиноградной кислотой или щавелевоуксусной кислотой в присутствии белка или микроорганизма, содержащего такой белок, с получением преимущественно 4R-IHOG,
где указанный белок выбран из группы, состоящий из (а) и (b), а именно
(a) белок, содержащий аминокислотную последовательность SEQ ID NO:2, который обладает 4R-альдолазной активностью;
(b) белок, который на 70% или более гомологичен аминокислотной последовательности SEQ ID NO:2, и который обладает 4R-альдолазной активностью, и
вторую стадию, в которой происходит преобразование карбонильной группы 4R-IHOG или его соли, полученной в первой стадии, в аминогруппу, с получением 4К-монатина формулы (2) или его соли, где оптическая чистота указанного 4К-монатина или его соли достигает 90% или более,
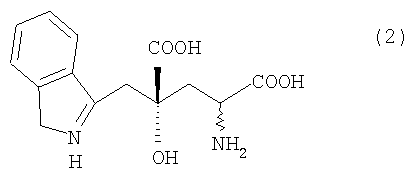
где связь, обозначенная волнистой линией, может иметь как R-, так и S-конфигурацию.
взаимодействие 4-(индол-3-илметил)-4-гидрокси-2-оксоглутаровой кислоты, содержащейся в реакционной смеси, с аминовым соединением формулы (3) или с его солью
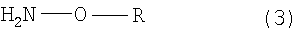
где R представляет собой атом водорода, алкильную, арильную или аралкильную группу, в нейтральных или щелочных условиях, с получением 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (IHOG-оксима) нижеследующей формулы (4) или ее соли
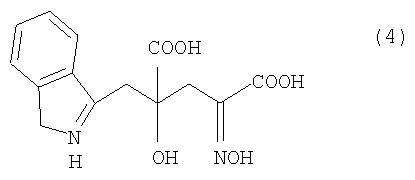
и кристаллизацию 4R-изомера полученного IHOG-оксима или его соли с последующим восстановлением и получением 4R-монатина или его соли, оптическая плотность которого составляет 90% или больше.
(a) белок, содержащий аминокислотную последовательность SEQ ID NO:2, который обладает 4R-альдолазной активностью;
(b) белок, который по крайней мере на 70% или более гомологичен аминокислотной последовательности SEQ ID NO:2 и обладает 4R-альдолазной активностью, и
(c) белок, который содержит аминокислотную последовательность, содержащую замену, делецию, инсерцию, добавление и инверсию одного или нескольких аминокислотных остатков в аминокислотной последовательности SEQ ID NO:2 и обладает альдолазной активностью.
(d) ДНК, состоящей из нуклеотидной последовательности SEQ ID NO:1 или из последовательности нуклеотидов 210-1004 той же самой последовательности SEQ ID NO; и которая кодирует белок, обладающий 4R-альдолазной активностью; и
(e) ДНК, которая в жестких условиях гибридизуется с ДНК, содержащей нуклеотидную последовательность, комплементарную нуклеотидной последовательности SEQ ID NO:1 или последовательности нуклеотидов 210-1004 той же самой последовательности SEQ ID NO, и которая кодирует белок, обладающий 4R-альдолазной активностью.
культивирование клеток по п.17 в среде; и
накопление белка, обладающего альдолазной активностью, в среде и/или в клетках.
Приоритет по пунктам:
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| WO 2004018672, 04.03.2004 | |||
| МАЙСТЕР А | |||
| Биохимия аминокислот | |||
| - М.: Издательство иностранной литературы, 1961, с.228-239. | |||

