Изобретение относится к органической химии, конкретно к способу получения 4,4-диметокси-2,3,5-трихлорциклопент-2-ен-1-она формулы (I),
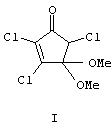
который может найти применение в качестве полупродукта в синтезе природных хлорсодержащих циклопентенонов, в частности высокоактивного антибиотика и фунгицида криптоспориопсина [1, 2], а также хлорвулонов и пунагландинов, эффективных простаноидов антивирусного и антиракового действия, выделенных из морских кораллов [3-5].
Известны способы получения соединения (I), исходящие из гексахлорциклопентадиена (ГХЦПД) [6-10].
Двухстадийный путь получения циклопентенона (I), предложенный авторами [6, 7], включает фотореакцию ГХЦПД в растворе этилацетата (EtOAc). При конверсии 70-80% ГХЦПД выход ацетоксипроизводного (II), выделенного хроматографическим путем из сложной смеси продуктов, составляет лишь 5-15% [6]. Вторая стадия этого процесса представляет перемешивание 0.001 М производного (II) с 5-кратным избытком гидроксида калия (КОН) в метаноле (МеОН) (3 ч, 20°С). Последующая обработка реакционной смеси (упаривание, экстракция диэтиловым эфиром, концентрирование раствора) и хроматографическая очистка на силикагеле (SiO2) приводят к целевому соединению (I) с выходом 92% [7] и общим выходом 4-13% в расчете на исходный ГХЦПД.
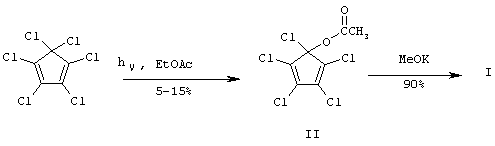
Основным недостатком этой схемы является крайне низкие селективность и выход на фотохимической стадии процесса. Кроме этого, к недостаткам следует отнести и необходимость использования специальной аппаратуры и усложняющее технологию процесса хроматографическое выделение как целевого (I), так промежуточного (II) соединения.
Второй известный путь - 3-стадийного получения (I) [8] основан на взаимодействии диметоксипроизводного (III) [9] с натриевым производным борнеола. Вначале, согласно методу [8] взаимодействием ГХЦПД с КОН (1:2.6) в МеОН при 30-40°С (12 ч) получают 5,5-диметокси-1,2,3,4-тетрахлорциклопентадиен (III) (ДМТХЦПД) с выходом 62% после вакуумной перегонки. Далее, согласно схеме синтеза [8] к алкоголяту натрия, полученному при действии на раствор изоборнеола гидрида натрия (NaH) в диметилсульфоксиде (ДМСО), прибавляют раствор ДМТХЦПД в ДМСО, перемешивают 5 ч, разлагают водой и экстрагируют этилацетатом. После промывки органического экстракта насыщенным раствором хлористого натрия (NaCl), осушки сульфатом магния (MgSO4), упаривания и хроматографической очистки на SiO2 с выходом 60% был получен виниловый эфир (IV). Последний медленно при комнатной температуре распадается (стадия фрагментации, 30 суток) с образованием целевого соединения (I) с выходом 80% и камфена, отделение которого от целевого (I) также проводят хроматографическим путем. Выход соединения (I) в расчете на диметоксипроизводное (III) составляет 48%, на ГХЦПД ˜30%.
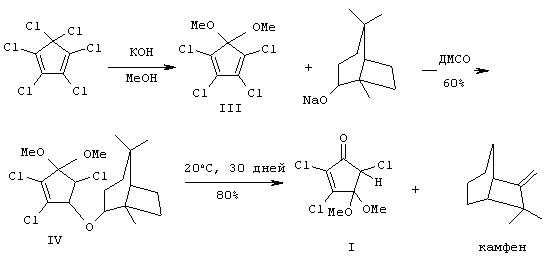
Недостатки данной трехстадийной схемы включают использование дорогостоящих ДМСО и NaH (и связанное с использованием этого металлорганического реагента применение инертных газов - азота или аргона), малодоступного борнеола, необходимость двойной хроматографической очистки на колонке с SiO2 (как промежуточного винилового эфира (IV), так и целевого соединения (I)). Кроме того, важным недостатком данного подхода является большая продолжительность во времени (˜30 суток) стадии фрагментации эфира (IV) и невысокий общий выход (˜30% из ГХЦПД).
Ближайшим прототипом предлагаемого изобретения выбран метод получения целевого соединения (I) из (ДМТХЦПД) (III) действием Na-производных третичных спиртов [10]. Приготовленный отдельно растворением металлического натрия (Na) в соответствующем спирте (трет-бутиловом спирте, 2-метил-2-деканоле, 1-винилциклогексан-1-оле) Na-алкоголят смешивают с раствором диметоксипроизводного (III) в ДМСО (соотношение ДМТХЦПД:RONa=1:0.8), полученного взаимодействием ГХЦПД с КОН в МеОН (аналогично описанному выше, [8]), и массу перемешивают при комнатной температуре в течение 24 ч. После обработки реакционной массы, включающей нейтрализацию 10%-ной соляной кислотой и экстракцию хлороформом (CHCl3) (3×300 мл), целевое соединение (I) выделяют из смеси с соответствующим олефином вакуумной перегонкой (для третбутилата натрия) или хроматографированием на SiO2 (для высококипящих спиртов) с выходами 70-80%. Суммарный выход (I) из ГХЦПД составляет 40-50%.
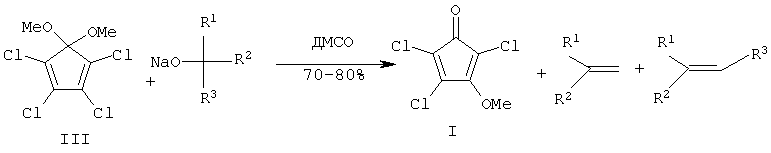
R1=R2=СН3, R2=C8H17 (а); R1=R2=R3=СН3 (б); R1=R2=циклогексил, R3=винил (в)
Недостатками известного двухстадийного способа-прототипа являются, во-первых, необходимость отдельного получения диметоксипроизводного (III) и натриевых производных третичных спиртов; во-вторых, использование дорогостоящих растворителя ДМСО, металлического Na и третичных спиртов и, в-третьих, получение целевого ссоединения (I) сопровождается образованием одновременно с целевым (I) соответствующих олефинов - продуктов дегидратации используемых спиртов, и выделение целевого соединения (I) требует проведения сложных технологических приемов, в частности, необходимость хроматографической очистки на финальной стадии (кроме варианта использования трет-бутанола).
Задача, на решение которой направлено заявляемое изобретение, заключается в упрощении процесса и повышении выхода целевого соединения (I).
По предлагаемому двухстадийному «одногоршковому» способу, смесь ГХЦПД и КОН в МеОН, взятых в молярном соотношении 1:(8-10), перемешивают при 20-22°С в течение 24 ч и промежуточное соединение (V) без выделения гидролизуют путем добавления 50%-ного водного раствора серной кислоты (Н2SO4) до рН 1 и перемешивания при 20-22°С в течение 10 ч. Затем МеОН упаривают на роторном испарителе, продукт экстрагируют EtOAc (3×100 мл). Объединенные органические экстракты промывают водой, насыщенным раствором NaCl, сушат MgSO4 и после перегонки в вакууме выделяют целевое соединение с выходами 75-80% в расчете на ГХЦПД и чистотой 96-98% (по данным газожидкостной хроматографии).
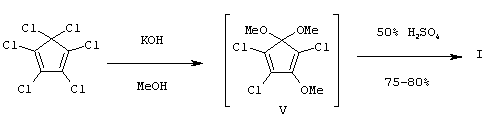
Преимущества предлагаемого метода, по сравнению с прототипом, состоят, во-первых, в исключении при осуществлении процесса малодоступных и дорогостоящих реагентов, а также металлорганических реагентов (Na, NaH), что позволяет исключить необходимость применения инертных газов, во-вторых, в возможности осуществления двух последовательных стадий в одном реакторе (колбе) («одногоршковость»), в-третьих, в технологической простоте очистки продукта (однократная вакуумная перегонка), в-четвертых, в более высоком выходе целевого продукта (75-80%).
Сущность изобретения подтверждается следующим примером.
Пример. Смесь 27.3 г (0.1 м) ГХЦПД, 56.0 г (1.0 м) КОН в 140 мл МеОН перемешивают при 20-22°С 24 ч. Затем реакционную массу охлаждают до 0°С и по каплям при перемешивании прибавляют 50%-ный водный раствор Н2SO4 до рН 1. Полученную массу перемешивают при 20-22°С в течение 10 ч, выделившуюся соль (KCl) отфильтровывают, МеОН упаривают на роторном испарителе и продукт экстрагируют EtOAc (3×100 мл). Объединенные органические экстракты промывают водой, насыщенным раствором NaCl. После осушки органического экстракта MgSO4 растворитель упаривают на роторном испарителе и остаток перегоняют в вакууме. Получают 18.4 г (75%) кристаллизующегося при стоянии маслообразного соединения (I), т.кип. 100-103°С при 0.2 мм рт.ст.
4,4-Диметокси-2,3,5 трихлорциклопент-2-ен-1-он (I). Бесцветные кристаллы с т.пл. 52-54°С (Rf=0.75, петролейный эфир - этилацетат, 1:1). ИК-спектр, ν, см-1: 1596, 1620, 1636, 1696, 1752. Спектр ЯМР 1Н, δ, м.д.: 3.30 с (3Н, ОСН3), 3.50 с (3Н, ОСН3), 4.64 с (1Н, С5H). Спектр ЯМР 13С, δ, м.д.: 52.03 (ОСН3), 52.07 (ОСН3), 62.19 (С5), 100.49 (С4), 133.77 (С2), 157.65 (С3), 186.17 (С1). Масс-спектр, m/z (Iотн, %): 249 (7.5), 247 (22.3), 245 (27.3) [М]+ 218 (31.4), 216 (93.6), 214 (100) [М-ОСН3]+, 212 (19.8), 210 (30.9) [М-Cl]+ 190 (4.9), 188 (14.0), 186 (15.5) [М-СН3О-СО]+ 136 (6.1), 134 (13.8), 112 (2.5), 110 (9.7), 108 (13.3), 97 (31.9), 89 (7.4), 87 (22.8), 69 (11.9), 59 (17.7) [ОСОСН3]+, 55 (11.1), 41 (8.0), 38 (5.5), 36 (14.8) [HCl]+ 28 (23.9). Найдено, %: С 34.10; Н, 2.82; Cl 43.40. С7Н7Cl3О3. Вычислено, %: С 34.25; Н 2.87; Cl 43.60.
Литература
1. W.J. MeGahren, J.H. van der Honde, L.A. Mitscher. J. Am. Chem. Soc. 1969, 91, 157.
2. S. Kabanyane, A. Decken, Chao-Mei Yu, J.M. Struns. Can. J. Chem. 2000, 78, 270.
3. K. Iguchi, S. Kaneta, K. Mori, Y, Yamada, A. Honde, Y. Mori. Tetrahedron Lett. 1985, 26, 5787.
4. M. Suzuki, Y. Morita, A. Yanagisawa, B. Bacer, P.J. Scheuer, R. Noyori. J. Org. Chem. 1988, 53, 286.
5. M. Fukushima. Anti-Cancer Drugs. 1994, 5, 131.
6. Н.С. Зефиров, М.А. Кирпиченок, Т.Г. Шестакова. Ж. Орг. химии. 1983, 19, 535.
7. Н.С. Зефиров, М.А. Кирпиченок, Т.Г. Шестакова. ДАН СССР. 1982, 262, 890.
8. О.М. Кузнецов, С.А. Торосян, Н.С. Востриков, М.С. Мифтахов. Изв. АН, Сер. Хим. 1997, 2799.
9. E.T. McBee, D.L. Crain, B.D. Crain, L.R. Belohlav, H.P. Braendlin. J.Am.Chem.Spc. 1962, 84, 3557.
10. Г.А. Толстиков, С.А. Исмаилов, Я.Л. Вельдер, М.С. Мифтахов. Ж. Орг. химии. 1990, 26, 672.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения 4,5,6,7,10,10-гексахлор-4,7-эндометилен-4,7,8,9-тетрагидрофталана | 1986 |
|
SU1482918A1 |
| ПРОТИВОВИРУСНОЕ СРЕДСТВО И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2015 |
|
RU2599792C1 |
| СПОСОБ ПОЛУЧЕНИЯ 7,7-ДИМЕТОКСИБИЦИКЛО[2.2.1]-ГЕПТАДИЕНА-2,5 | 2006 |
|
RU2331627C2 |
| СПОСОБ ПОЛУЧЕНИЯ 6-МЕТИЛЕНО-16α,17α-ЦИКЛОГЕКСАНОПРЕГН-4-ЕН-3,20-ДИОНА | 2014 |
|
RU2566368C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5,8-ДИГИДРОКСИ-2,6-7-ТРИМЕТОКСИ-3-ЭТИЛ-1,4-НАФТОХИНОНА | 2005 |
|
RU2277083C1 |
| Способ получения замещенных 1Н-имидазолов или их солей присоединения нетоксичных, фармацевтически приемлемых кислот | 1987 |
|
SU1662349A3 |
| СПОСОБ ПОЛУЧЕНИЯ 1,2,3,4,11,11-ГЕКСАХЛОР-7,8- | 1972 |
|
SU353937A1 |
| 3,6-ДИМЕТОКСИ-17-МЕТИЛ-7АЛЬФА-(ТРИФТОРАЦЕТИЛ)-4,5АЛЬФА-ЭПОКСИ-6АЛЬФА,14АЛЬФА-ЭТЕНОИЗОМОРФИНАН И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2012 |
|
RU2503677C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ ФОСФОРИЛЗАМЕЩЕННЫХ 1,4-ДИКАРБОНОВЫХ КИСЛОТ | 2008 |
|
RU2413733C2 |
| СПОСОБ ПОЛУЧЕНИЯ (4S,5S)-4,5-О-ИЗОПРОПИЛИДЕНЦИКЛОПЕНТ-2-ЕН-1-ОНА | 2009 |
|
RU2400478C1 |
Изобретение относится к способу получения 4,4-диметокси-2,3,5-трихлорциклопент-2-ен-1-она из гексахлорциклопентадиена. Способ заключается в проведении в одном реакторе двух последовательных стадий - взаимодействия гексахлорциклопентадиена с гидроокисью калия в метаноле при соотношении, равном 1:(8-10), при температуре 20-22°С в течение 24 ч и кислотного гидролиза 50%-ной водной серной кислотой образующегося промежуточного продукта без его выделения. Целевое соединение выделяют путем вакуумной перегонки с выходом 75-80% и чистотой 96-98%. Технический результат - исключение дорогостоящих и малодоступных реагентов, упрощение процесса и повышение выхода целевого соединения.
Способ получения 4,4-диметокси-2,3,5-трихлорциклопент-2-ен-1-она формулы I
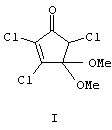
путем взаимодействия гексахлорциклопентадиена с гидроокисью калия в метаноле, отличающийся тем, что процесс проводят при мольном соотношении гексахлорциклопентадиен : гидроокись калия, равном 1:(8-10), при температуре 20-22°С в течение 24 ч с последующим кислотным гидролизом образующегося промежуточного продукта без его выделения и выделением целевого продукта вакуумной перегонкой.
| Толстиков Г.А | |||
| и др | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Кузнецов О.М | |||
| и др | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Зефиров Н.С | |||
| и др | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Двухкулачковый самоцентрирующий поворотный патрон | 1980 |
|
SU931316A1 |

