Заявка претендует на приоритет Патентной заявки США №10/007877, поданной 13 ноября 2001 года и преобразованной в предварительную заявку США №__, которая включена в настоящую заявку во всей ее полноте для всех целей путем отсылки.
Область техники, к которой относится изобретение
Настоящее изобретение относится к новым пероральным составам с замедленным высвобождением, предназначенным для доставки активного агента (например, лекарственного средства), в особенности хорошо растворимого в воде лекарственного средства. Более конкретно, настоящее изобретение относится к новым составам, включающим мицеллообразующее лекарственное средство, имеющее заряд, и по меньшей мере один полимер, имеющий противоположный заряд.
Уровень техники
Введение лекарств с помощью традиционных перорального и внутривенного способов резко ограничивает эффективность большинства лекарственных средств. Вместо того чтобы поддерживать уровни лекарств в пределах терапевтических диапазонов, эти способы приводят к быстрому начальному росту уровней концентрации в плазме с последующим быстрым падением ниже терапевтических уровней по мере того, как лекарственные средства метаболизируются организмом. По этой причине для поддержания на терапевтических уровней лекарств в течение периода времени, достаточного для достижения терапевтического эффекта, требуются повторные дозы. Для решения этой проблемы с целью устранения начального эффекта "взрыва" и обеспечения высвобождения лекарств на постоянных уровнях были разработаны многочисленные препараты длительного высвобождения.
Для достижения продолжительного высвобождения лекарственных средств обычно применяют полимерные составы (см. Langer et al., Nature 392:6679 supp.(1998)). Для высвобождения лекарств с различными физическими свойствами были разработаны разнообразные успешно действующие полимерные препараты длительного высвобождения. Такие препараты оказались чрезвычайно эффективными для увеличения времени высвобождения в тех случаях, когда используются относительно гидрофобные и нерастворимые в воде лекарственные средства.
Однако вследствие быстрой диффузии лекарств через полимерные матрицы оказалось затруднительным достичь продолжительного высвобождения для хорошо растворимых лекарств с использованием современных технологий замедленного высвобождения. Таким образом, существует потребность в новых составах и способах, которые могли бы понизить диффузию лекарства и устранить эффект "взрыва" для хорошо растворимых в воде лекарств. Настоящее изобретение решает эти и другие задачи.
Раскрытие изобретения
Настоящее изобретение обеспечивает, inter alia, пероральный состав с замедленным высвобождением, включающий мицеллообразующее лекарство и противоположно заряженный полимер. Хотя представление о мицеллах в области поверхностно-активных веществ или носителей лекарств хорошо известно, применение мицеллообразующего лекарства для состава замедленного высвобождения совершенно не известно. Более того, то, что такой состав является чрезвычайно эффективным в отношении продолжительного высвобождения активных агентов, в частности водорастворимых лекарственных средств, оказалось в действительности неожиданным. Другим преимуществом состава является его способность обеспечить медленное высвобождение даже тогда, когда состав содержит большие дозы лекарства.
В таком качестве настоящее изобретение обеспечивает пероральный фармацевтический состав с замедленным высвобождением, включающий мицеллообразующее лекарственное средство, имеющее заряд, и по меньшей мере один полимер, имеющий противоположный заряд, и, кроме того, при необходимости, включает образующее гидрогель полимерное вещество и гидрофильную основу. Мицеллообразующее лекарственное средство может иметь при физиологическом рН либо положительный, либо отрицательный заряд.
В другом воплощении настоящее изобретение обеспечивает способ модулирования профиля высвобождения мицеллообразующего лекарства, включающий варьирование молярного отношения, имеющего заряд мицеллообразующего лекарства и по меньшей мере одного полимера, имеющего противоположный заряд, варьирование дополнительного количества полимера, несущего противоположный заряд, модулируя тем самым профиль высвобождения мицеллообразующего лекарства. В число подходящих мицеллообразующих лекарств входят, например, антидепрессанты, блокирующие β-адренорецептор агенты, анестетики, антигистаминные агенты и т.п. Предпочтительно мицеллообразующее лекарство является водорастворимым лекарственным средством.
В еще одном воплощении настоящее изобретение обеспечивает способ продления высвобождения мицеллообразующего лекарства, включающий: пероральное введение фармацевтического состава, содержащего мицеллообразующее лекарство, имеющее заряд, и по меньшей мере один полимер, имеющий противоположный заряд, увеличивая тем самым продолжительность высвобождения мицеллообразующего лекарства.
В еще одном воплощении настоящее изобретение обеспечивает способ продления продолжительности высвобождения мицеллообразующего лекарства, включающий: пероральное введение фармацевтического состава, содержащего мицеллообразующее лекарство, имеющее заряд, и по меньшей мере один полимер, имеющий противоположный заряд, и, кроме того, при необходимости, образующее гидрогель полимерное вещество и гидрофильную основу, продлевая тем самым высвобождение мицеллообразующего лекарственного средства.
Другие цели и преимущества станут более очевидными при прочтении приведенного ниже детального описания с чертежами.
Краткое описание чертежей
Фиг.1 иллюстрирует высвобождение растворимого лекарства (10 мас.%) из 400 мг матрицы ПАК/ПЭО в искусственной кишечной среде (SIF).
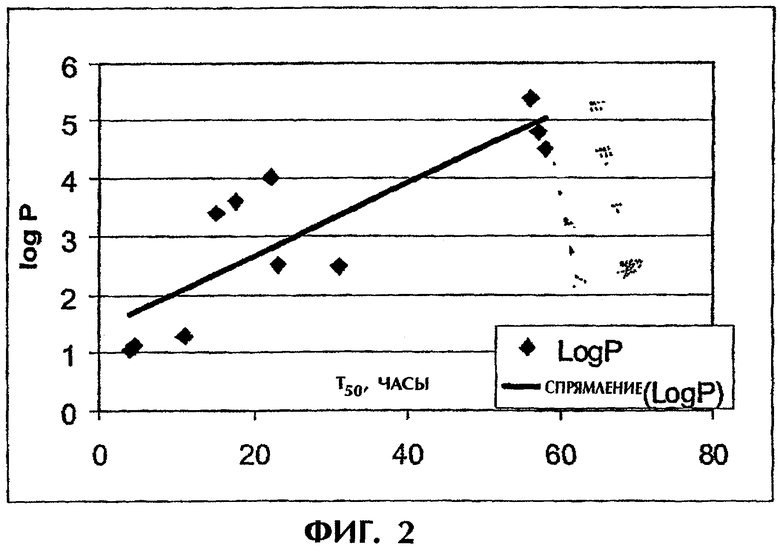
Фиг.2 иллюстрирует корреляцию между Т50 и logP для щелочных хорошо растворимых лекарств, высвобождаемых из таблетки ПАК/ПЭО (1:1,5) массой 400 мг.
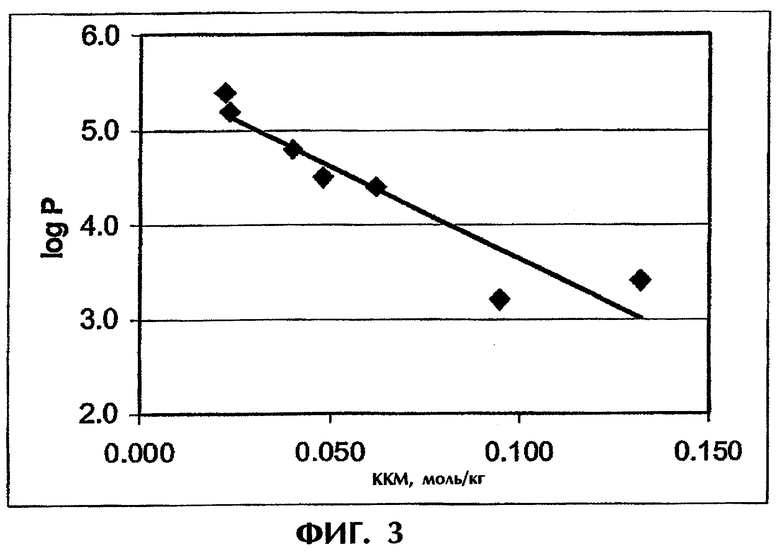
Фиг.3 иллюстрирует корреляцию между критической концентрацией мицеллообразования (ККМ) и logP.
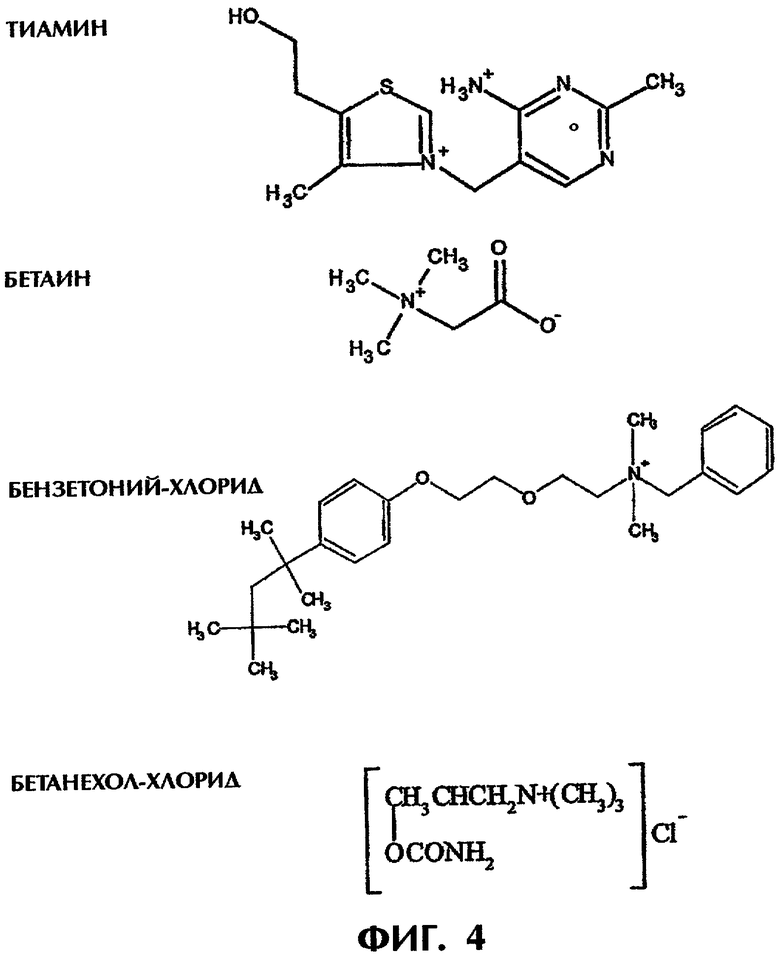
Фиг.4 иллюстрирует примеры заряженных (положительно или отрицательно) лекарств, пригодных для использования в экспериментах по высвобождению.
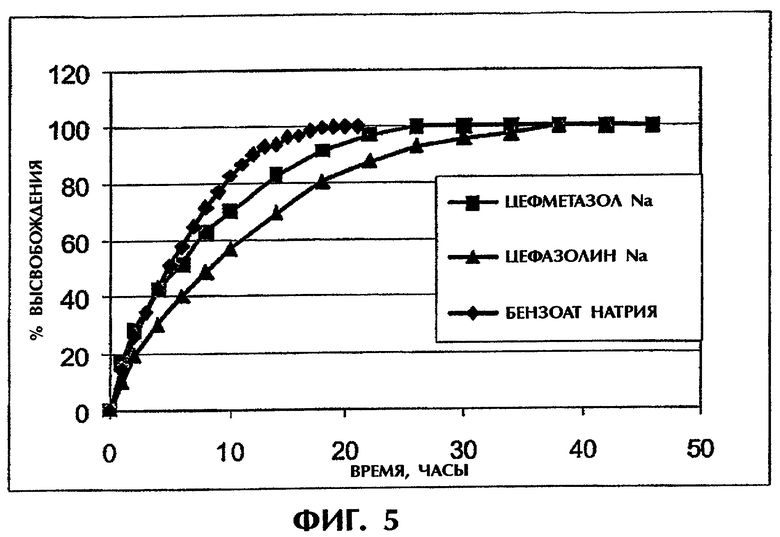
Фиг.5 иллюстрирует высвобождение отрицательно заряженных лекарств из матрицы ПАК/ПЭО.
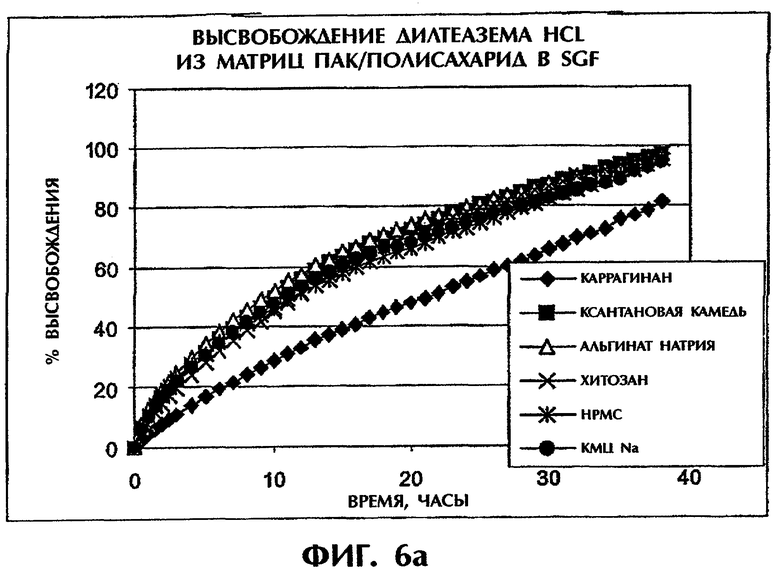
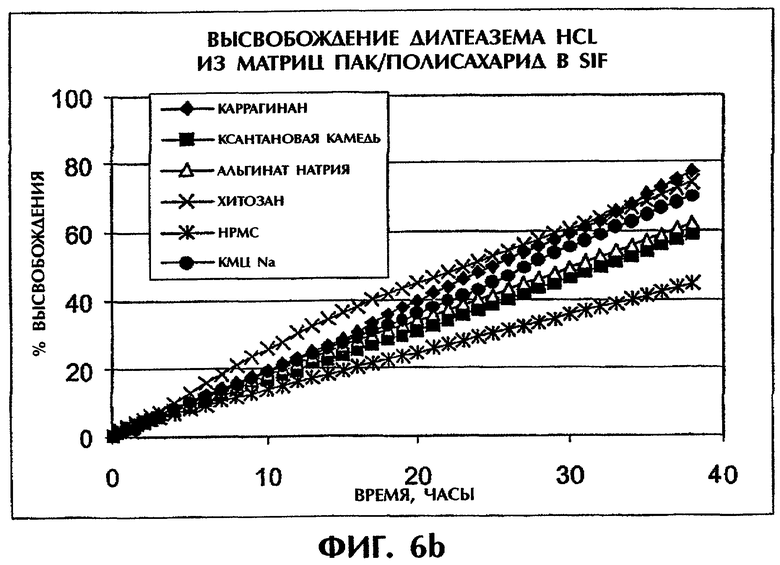
Фиг.6 иллюстрирует высвобождение Дилтиазема HCl из таблеток (400 мг) с матрицей ПАК/полисахарид в SGF (фиг.6а) и SIF (фиг.6b).
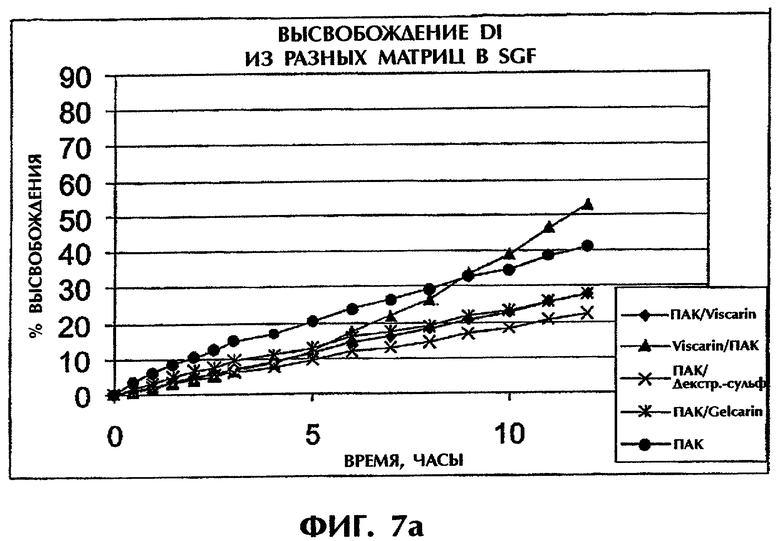
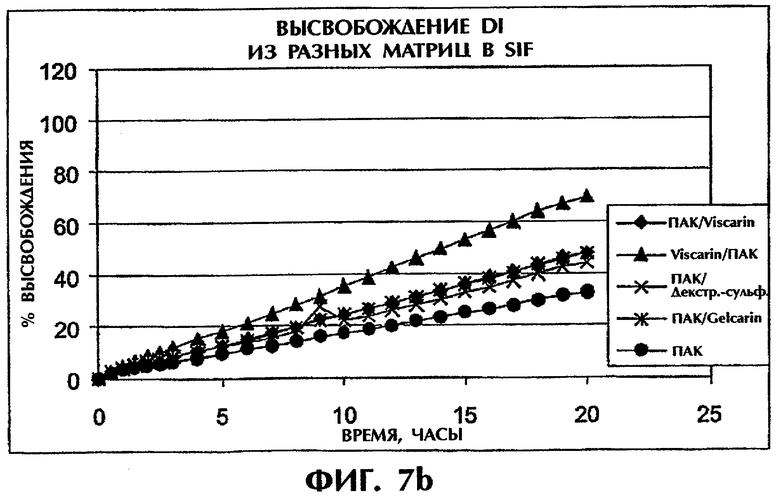
Фиг.7 иллюстрирует высвобождение Дилтиазема HCl из таблеток (400 мг) с матрицей ПАК/сульфатированный полимер в SGF (фиг.7а) и SIF (фиг.7b).
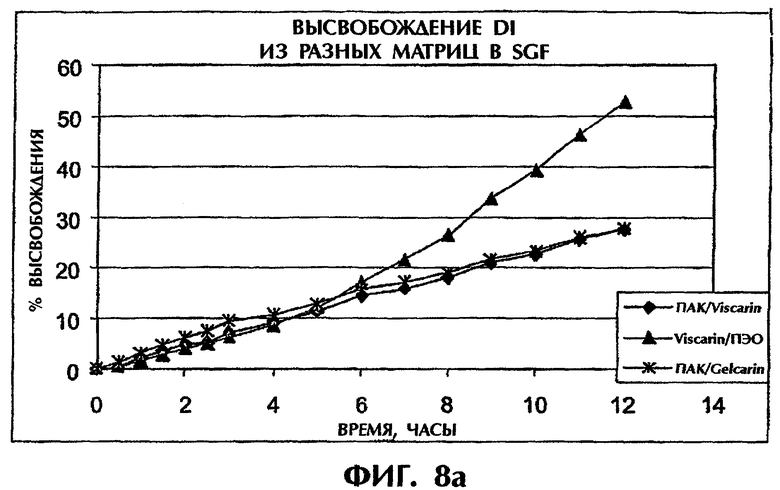
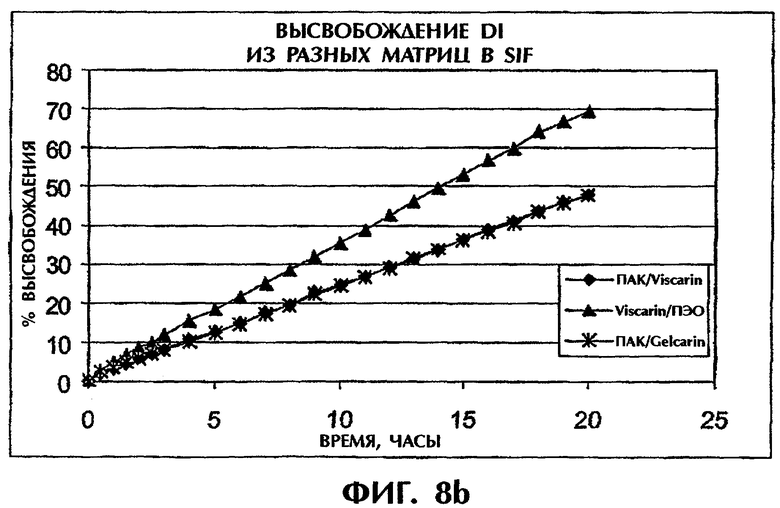
Фиг.8 иллюстрирует высвобождение Дилтиазема HCl из таблеток с различной матрицей в SGF (фиг.8а) и SIF (фиг.8b).
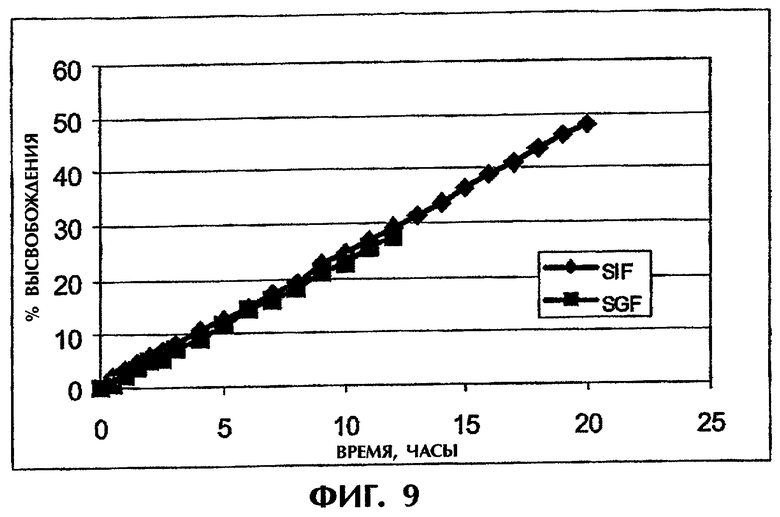
Фиг.9 иллюстрирует высвобождение Дилтиазема HCl (25 мас.%) из матрицы ПАК/каррагинан (1:1) в SGF и SIF.
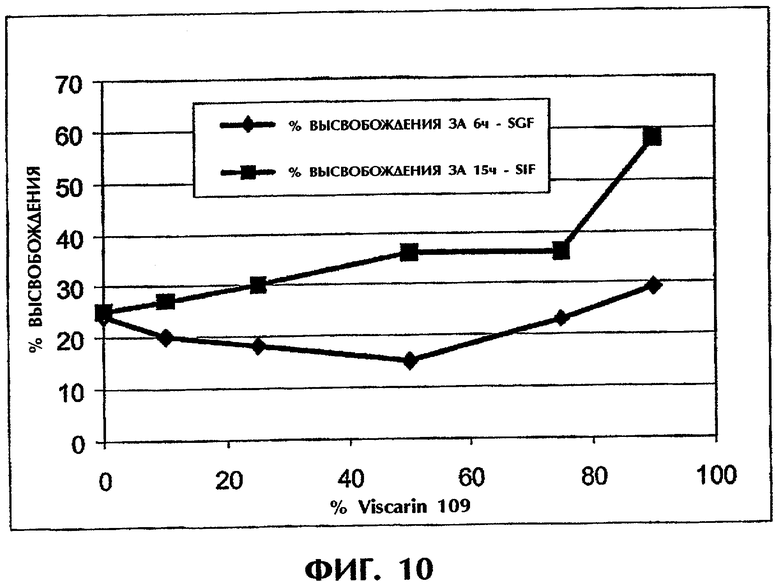
Фиг.10 иллюстрирует оптимизацию соотношения ПАК/каррагинан для состава с 25 мас.% Дилтиазема HCl.
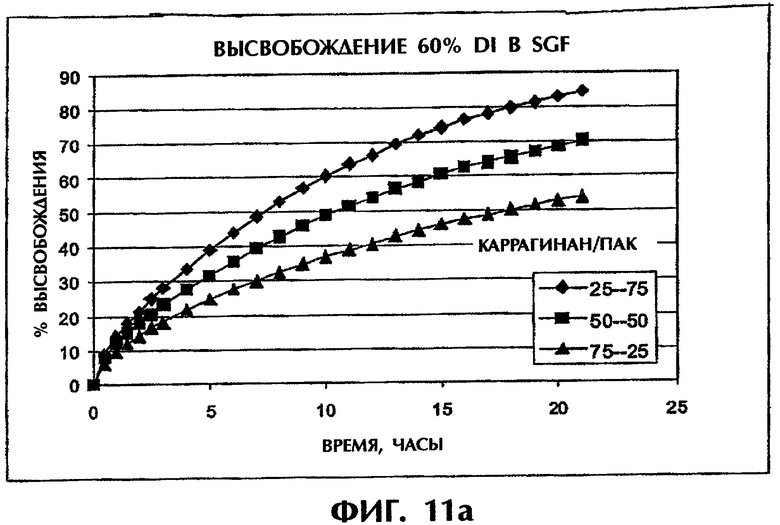
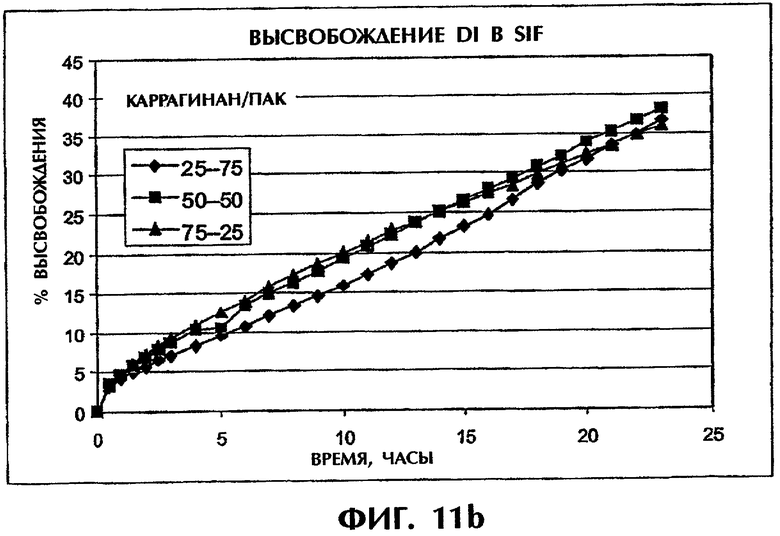
Фиг.11 иллюстрирует скорости высвобождения Дилтиазема HCl (60 мас.%) из таблеток с матрицами с различными соотношениями ПАК/каррагинан в SGF (фиг.11а) и SIF (фиг.11b).
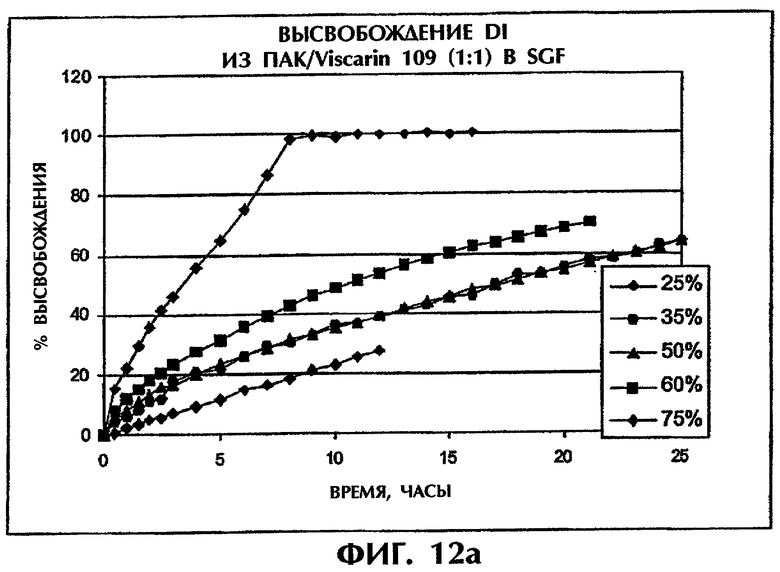
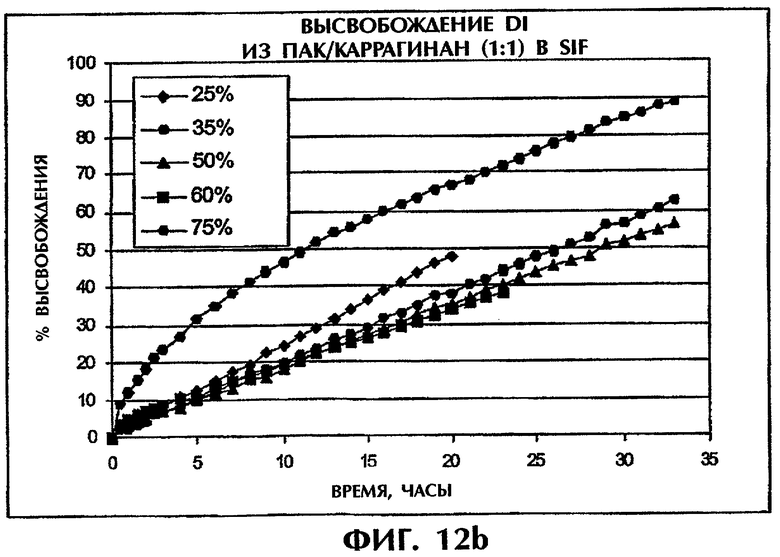
Фиг.12 иллюстрирует высвобождение Дилтиазема HCl из матрицы ПАК/Viscarin 109 при разном содержании лекарства в SGF (фиг.12а) и SIF (фиг.12b).
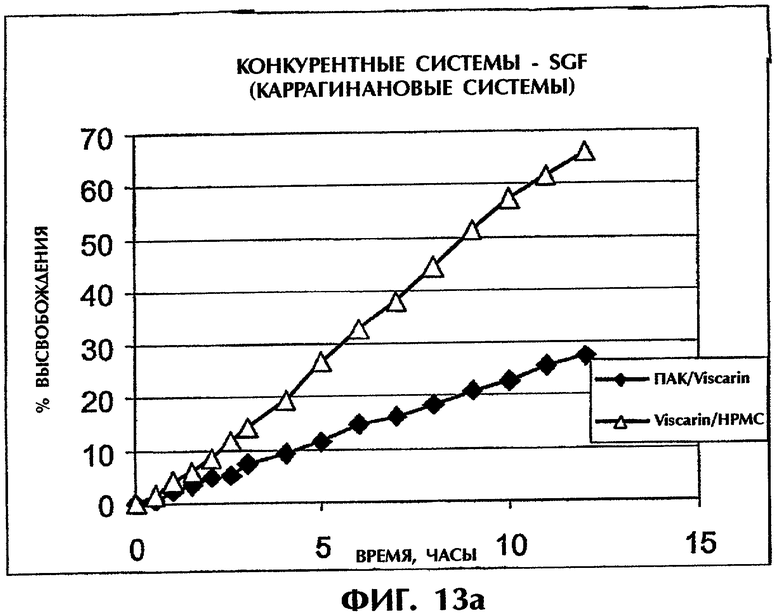
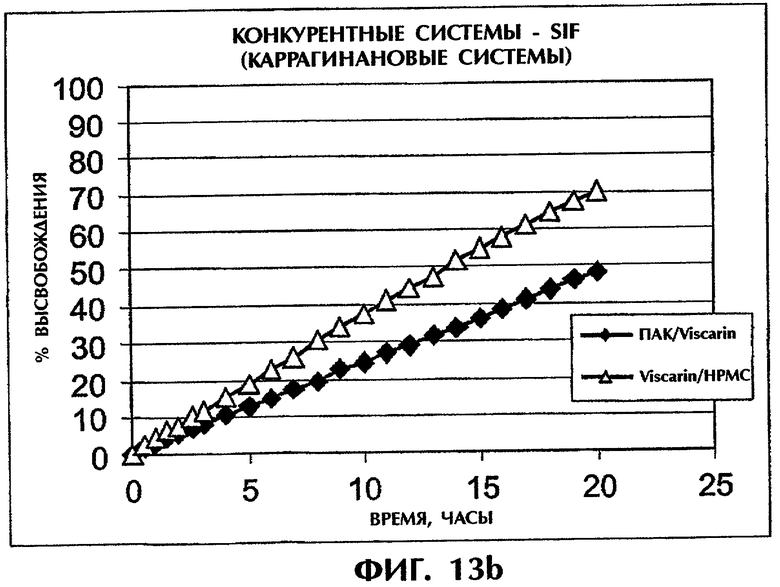
Фиг.13 иллюстрирует высвобождение Дилтиазема HCl (25 мас.%) из конкурирующих систем на основе каррагинана в SGF (фиг.13а) и SIF (фиг.13b).
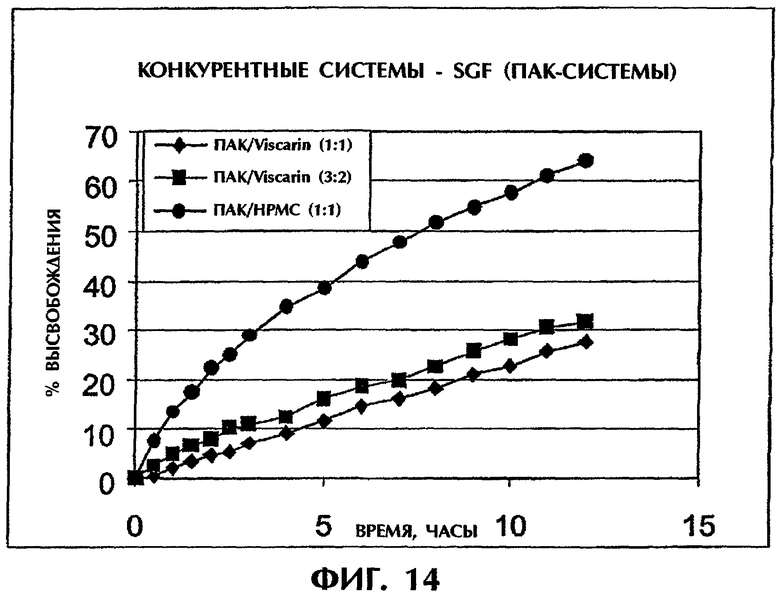
Фиг.14 иллюстрирует высвобождение Дилтиазема HCl (25 мас.%) из конкурирующих систем на основе ПАК в SGF.
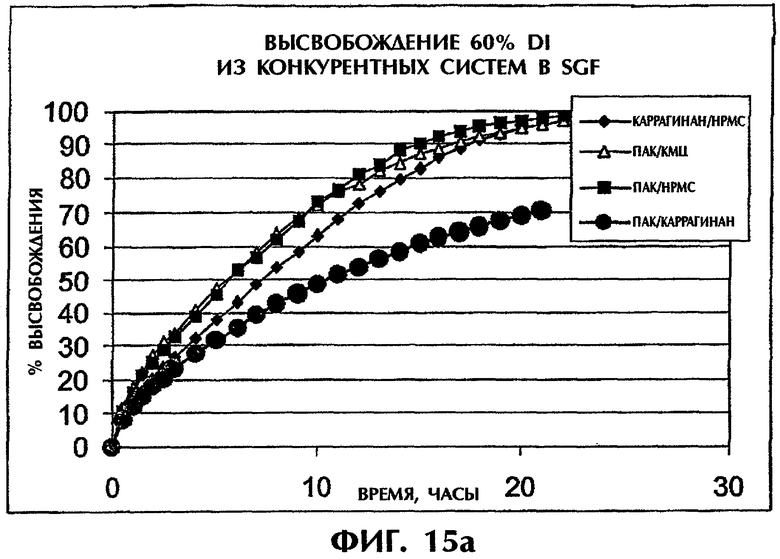
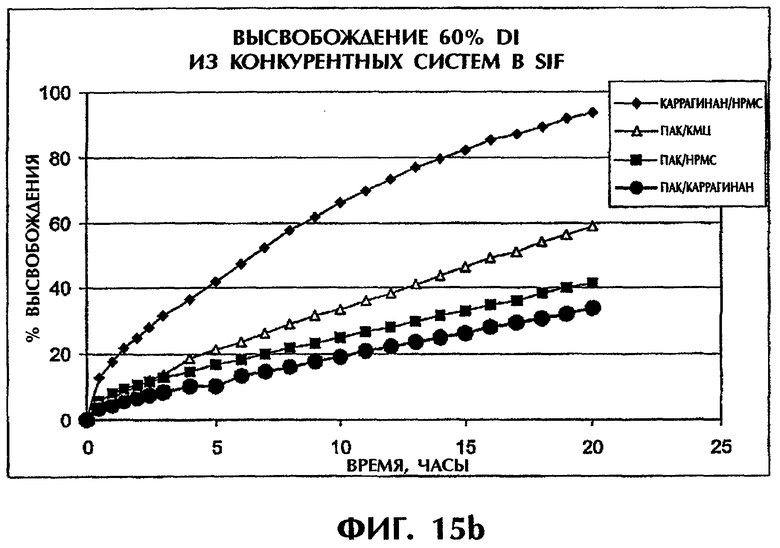
Фиг.15 иллюстрирует высвобождение Дилтиазема HCl (60 мас.%) из конкурирующих систем в SGF (фиг.15а) и SIF (фиг.15b).
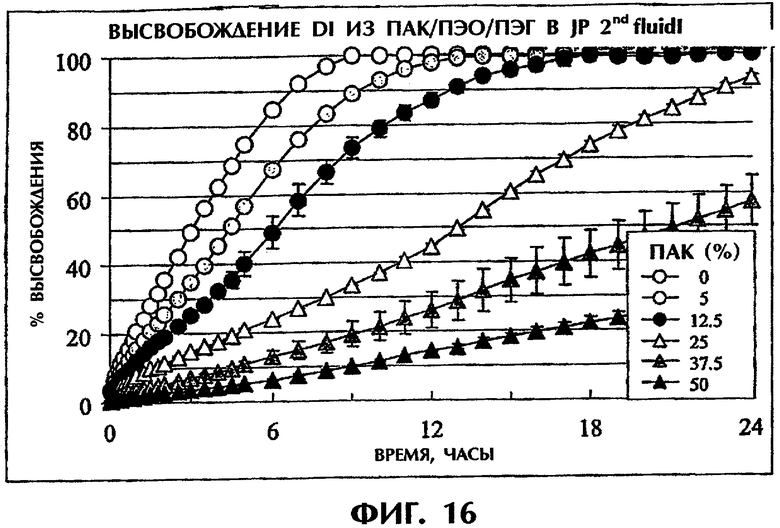
Фиг.16 иллюстрирует влияние дополнительного количества ПАК на высвобождение Дилтиазема HCl (50 мас.%) в среде JP 2nd fluid.
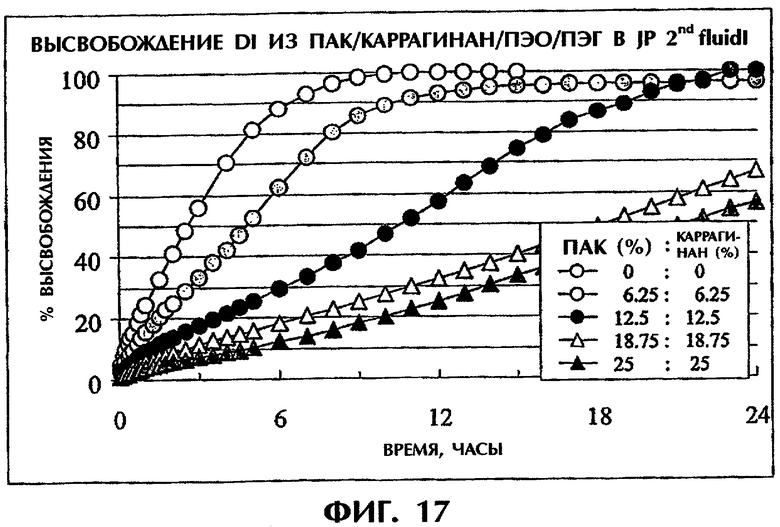
Фиг.17 иллюстрирует влияние дополнительного количества ПАК/каррагинан на высвобождение Дилтиазема HCl (50 мас.%) в среде JP 2nd fluid.
Осуществление изобретения
Определения
Термин "активный агент" означает любое лекарственное средство, которое может находиться в физиологически приемлемой таблетке для перорального применения. В число предпочтительных активных агентов входят мицеллообразующие активные агенты, способные образовывать электрически заряженные коллоидные частицы.
Термин "сПз", или "сантипуаз", является единицей вязкости, равной мПа·с. Вязкость измеряют на вискозиметрах Брукфильда или других вискозиметрах. См., например, Wang et al., Clin. Hemorheol. Microcirc. 19:25-31 (1988); Wang et al., J. Biochem. Biophys. Methods 28:251-61 (1994); Cooke et al., J. Clin. Pathol. 41:1213-1216 (1998).
Упоминаемый здесь термин "каррагинан" относится ко всем формам водорастворимого экстракта каррагинана, ирландского мха, морской водоросли с атлантического побережья Европы и Северной Америки. Источниками являются, например, Viscarin®109 и Gelcarin® такой как GP-911, GP-812, GP-379, GP-109, GP-209, коммерчески доступные от фирмы FMC. Каррагинаны являются высокомолекулярными, высокосульфатированными линейными молекулами с галактозным скелетом. Они состоят из сульфатированных и несульфатированных повторяющихся звеньев галактозы и 3,6-ангидрогалактозы, соединенных между собой чередующимися α-(1-3) и β-(1-4)-гликозидными связями. Другим коммерческим источником каррагинанов является фирма Sigma and Hercules Inc.
Используемый здесь термин "полиакриловая кислота", или "ПАК", включает все формы и молекулярные массы полимеров ПАК. В число источников входит, например, Carbopol 971 фирмы B.F.Goodrich.
Используемый здесь термин "полиэтиленоксидный полимер", или "ПЭО", включает все формы и молекулярные массы полимеров ПЭО. В число источников полимеров ПЭО входят, например, Polyox WSR-303™ (средняя MM: 7·106, вязкость 7500-10000 сПз, 1% в Н2O, 25°С); Polyox WSR Coagulant™ (средняя MM: 5·106, вязкость 5500-7500 сПз, в тех же условиях, что и выше); Polyox WSR-301™ (средняя MM: 4·106, вязкость 1650-5500 сПз, в тех же условиях, что и выше); Polyox WSR-N-60K™ (средняя MM: 2·106, вязкость 2000-4000 сПз, 2% в Н2О, 25°С), все из которых являются коммерческими названиями фирмы Union Carbide Со. См. также WO 94/06414, который включен в настоящую заявку путем отсылки.
Используемый здесь термин "полиэтиленгликоль", или "ПЭГ", включает все формы и молекулярные массы полимеров ПЭГ. В число источников ПЭГ входят Macrogol 400, Macrogol 1500, Macrogol 4000, Macrogol 6000, Macrogol 20000, все из которых являются коммерческими названиями фирмы Nippon Oil and Fats Co.
Термины "гидроксипропилметилцеллюлоза", "карбоксиметилцеллюлоза натрия", "гидроксиэтилцеллюлоза" и "карбоксивиниловый полимер" включают их обычные употребления. В число источников входят: для гидроксипропилметилцеллюлозы (НРМС), например Metolose 90SH100000™ (вязкость 2900-3900 сПз, в тех же условиях, как выше); Metolose 90SH30000™ (вязкость 25000-35000 сПз, 2% в Н2O, 20°С), все из которых являются торговыми названиями фирмы Shin-Etsu Chemicals Co. Для карбоксиметилцеллюлозы натрия (CMC-Na, или КМЦ-Na), например Sanlose F-150MC™ (средняя ММ 2×105; вязкость 1200-1800 сПз, 1% в Н2О, 25°С), Sanlose F-1000MC™ (средняя ММ 4.2×104; вязкость 8000-12000 сПз, в тех же условиях, как выше); Sanlose F-300МС™ (средняя ММ 3×105; вязкость 2500-3000 сПз, в тех же условиях, как выше), все из которых являются торговыми названиями фирмы Nippon Seishi Co., Ltd. Для гидроксиэтилцеллюлозы (НЕС), например НЕС Daicel SE850™ (средняя ММ 1,48×106; вязкость 2400-3000 сПз, 1% в Н2O, 25°С); НЕС Daicel SE900™ (средняя ММ 1,56×106, вязкость 4000-5000 сПз, в тех же условиях, как выше), все из которых являются торговыми названиями фирмы Daicel Chemical Industries. Для карбоксивинильных полимеров, например Carbopol 940™, средняя ММ равна приблизительно 25×105, B.F. Goodrich Chemical Co.
Используемый здесь термин "терапевтическое лекарственное средство" означает любое лекарственное средство, которое может доставляться в виде применяемой перорально физиологически приемлемой таблетки.
Термин "мицеллообразование" относится к любому соединению, способному образовывать электрически заряженные коллоидные частицы, ионы, состоящие из ориентированных молекул или агрегатов некоторого числа соединений (молекул), непрочно удерживаемых вместе с помощью вторичных связей.
Настоящее изобретение обеспечивает, inter alia, пероральный препарат с замедленным высвобождением, включающий мицеллообразующий активный агент (т.е. лекарство) и противоположно заряженный полимер, образующий гидрогелевую матрицу. Состав обычно изготовляют путем непосредственного прессования лекарства с полимерным наполнителем.
Состав преимущественно обеспечивает чрезвычайно низкую скорость высвобождения активного агента. В предпочтительном аспекте связанные водородными связями комплексы между противоположно заряженными полимерами и мицеллами лекарства препятствуют быстрой диффузии лекарства. Не будучи связанными какой-либо специальной теорией, предполагается, что высвобождение лекарственного средства происходит тогда, когда заряд полимера нейтрализуется ионами ОН в зоне матрица/граница растворения и названные выше связи разрываются.
В одном из воплощений количество введений состава может быть уменьшено, улучшая тем самым соблюдение больным предписанного режима. Далее побочные эффекты лекарства могут быть уменьшены путем подавления быстрых возрастаний концентрации лекарства в крови (что наблюдается в случае обычных составов). Еще одним преимуществом состава является то, что содержание в нем больших количеств лекарства не влияет существенным образом на скорости высвобождения.
В настоящей заявке обсуждаются факторы и события, которые создают теоретическую основу для осуществления изобретения. Однако приведенные обсуждения ни в коем случае не следует рассматривать как обязывающие или ограничивающие настоящее изобретение. Специалистам должно быть понятно, что различные воплощения изобретения могут быть реализованы вне зависимости от модели, используемой для описания теоретических основ изобретения.
I. Активные агенты изобретения
Активными агентами изобретения могут быть любые лекарства, которые образуют мицеллы. Мицеллообразование наблюдалось ранее для антидепрессантов, агентов, блокирующих β-адренорецепторы, анестетиков, антигистаминных препаратов, фенотиазинов, антиацетилхолинов, транквилизаторов, антибактериальных средств и антибиотиков (см. Attwood et al., J. Pharm. Pharmac., 30, 176-180 (1978); Attwood et al., J. Pharm. Pharmac., 31, 392-395 (1979); Attwood et al., J. Pharm. Pharmac., 38, 494-498 (1986); Attwood J. Pharm. Pharmac., 24, 751-752 (1972); Attwood et al. J. Pharm Sci. v.63, по. 6, 988993 (1974); Attwood, J. Pharm. Pharmacol., 28, 407-409 (1976)). В число репрезентативных мицеллообразующих лекарственных средств - антидепрессантов - входят имипрамин HCl, омипрамол HCl и амитриптилин HCl. В число репрезентативных мицеллообразующих блокаторов β-адренорецепторов входят окспренолол HCl, ацебутолол HCl и солатол HCl. В число репрезентативных мицеллообразующих анестетиков входят прокаин HCl, лидокаин HCl и аметокаин HCl. В число репрезентативных мицеллообразующих антигистаминов входят дифенгидрамин HCl, хлорциклизин HCl, дифенилпиралин HCl, прометазин HCl, бромдифенгидрамин HCl, трипеленамин HCl и мепирамин малеат. В число репрезентативных мицеллообразующих фенотиазинов входят хлорпромазин HCl и прометазин HCl. В число других мицеллообразующих лекарств входят транквилизаторы, антибактериальные препараты и антибиотики.
В некоторых аспектах в число активных агентов входят, но не ограничивают их, бетакаин гемисульфат, цинхокаин гидрохлорид ВР и лигнокаин гидрохлорид (Sigma); прилокаин гидрохлорид ВР, бупивакаин гидрохлорид (Astra Pharmaceuticals), мепивакаин гидрохлорид (Leo), пропакаин гидрохлорид (Squibb) и аметокаин гидрохлорид ВР (Smith and Nephew Pharmaceuticals). В ряде аспектов для настоящего изобретения оказываются полезными следующие активные ингредиенты. В их число входят, но не ограничивают их тем самым, (4'-(1-гидрокси-2-изопропил-аминоэтил)метансульфонанилид) (Duncan, Flockhart); лабетолол [5-(1-гидрокси-2-(1-метил-3-фенил-пропиламино)этил)салициламид] (Allen and Hanburys); ацебуртолол ((±)-3'-ацетил-4'-(2-гидрокси-3-изопропиламинопропокси)бутиранилид) (May and Baker); пропранолол {(±)-1-изопропиламино-3-нафт-1'-илоксипропан-2-ол} (ICI) и окспренолол {(±)-1-(о-аллилоксифенокси)-3-изопропиламинопропан-2-ол)} (Ciba); тимолол-малеат {малеат (-)-1-бутиламино-3-(4-морфолино-1,2,5-тиедиазол-3-ил-окси)пропан-2-ола} (Merck, Sharp and Dohme); метропророл-тартрат [тартрат (±)-1-изопропиламино-3-р-(2-метоксиэтил)феноксипропан-2-ола] (Geigy Pharmaceuticals). В другом воплощении в число активных компонентов входят, но не ограничивают их тем самым, адифенин гидрохлорид (Ciba); полдин метилсульфат В.Р. (Beecham Research); лахесино хлорид В.Р.С. (Vestric); хлорфеноксамин гидрохлорид (Evans Medical); пипериодолат гидрохлорид и пипензолат бромид (М.С.Р. Pharmaceutical); орфенадрин гидрохлорид В.Р. (Brocades, Gt Britain); бензтропин мезилат В.Р. (Merck Sharp and Dohme); клидиний бромид (Roche); амбутоний бромид (Wyeth) и бензилонитум бромид (Parke Davis). Дифенилгидрамин гидрохлорид В.Р. (2-дифенилметокси-NN-диметилэтиламин гидрохлорид) и хлорциклизин гидрохлорид В.Р. [1-(р-хлордифенилметил)-4-метилпиперазин гидрохлорид], полученные от Parke-Davis and Company и Burroughs Welcome and Company, соответственно. Бромдифенгидрамин гидрохлорид [2-(α-p-бромфенил-α-фенилметокси)-NN-диметиламин гидрохлорид] и дифенилпиралин гидрохлорид (4-дифенилметокси-1-метилпиперидин гидрохлорид), соответственно. Специалистам известны другие активные ингредиенты, пригодные для использования в настоящем изобретении.
В предпочтительных воплощениях активными агентами настоящего изобретения являются хорошо растворимые в воде лекарственные средства. В еще более предпочтительных воплощениях активными агентами настоящего изобретения являются основные лекарственные средства. Изобретение особенно хорошо подходит для таких лекарств, которые проявляют сильный эффект "взрыва", обусловленный быстрой диффузией сквозь полимерные матрицы. В число хорошо растворимых в воде лекарств входят соли, образуемые с неорганическими и органическими кислотами (положительно заряженные за счет нековалентно связанных протонов), постоянно положительно (или отрицательно) заряженные молекулы и отрицательно заряженные молекулы (соли слабых и сильных кислот). Хорошо растворимое в воде лекарственное средство означает, например, что его растворимость превышает 10 мг/мл, предпочтительно выше 100 мг/мл.
Конкретные активные агенты, пригодные для использования в составах настоящего изобретения, заряженные мицеллообразующие лекарственные средства, отбирают на основании критической концентрации мицеллообразования (ККМ) и/или logP, которые тесно между собой связаны (см. пример 3). logP (коэффициент распределения лекарства между октанолом и водой) характеризует гидрофобные свойства незаряженной формы лекарственного средства. ККМ, представляющая собой меру концентрации, при которой конкретное соединение образует мицеллу, является функцией гидрофобности, а также стереохимии молекулы, способности вращения групп и противоионов. Присутствие мицеллоподобных заряженных агрегатов лекарства внутри гидрогелевой матрицы, содержащей противоположно заряженные полимеры, приводит к кооперативному взаимодействию. Именно это кооперативное взаимодействие определяет скорость высвобождения из полимерной матрицы. Следовательно, ККМ и logP могут быть использованы для предсказания скорости высвобождения лекарственного средства и, таким образом, идентифицировать те лекарства, которые будут иметь продолжительное высвобождение в составах настоящего изобретения. Лекарства с низкой ККМ и/или высоким logP в составах настоящего изобретения будут высвобождаться медленно, в то время как лекарства с меньшей склонностью к образованию мицелл будут высвобождаться в соответствии с профилями, аналогичными профилям для стандартных пероральных составов.
Соответственным образом, профиль высвобождения лекарства можно модулировать, используя стандартные способы, известные специалистам для модулирования критической концентрации мицеллообразования и/или степени кооперативного взаимодействия между мицеллообразующим лекарством и противоположно заряженными полимерами. Способы модулирования ККМ и/или степени кооперативного взаимодействия включают изменение гидрофобности лекарственного средства путем введения функциональных групп и любые другие способы изменения электростатического взаимодействия между лекарством и полимерным наполнителем. В некоторых аспектах настоящее изобретение обеспечивает способ уширения профиля высвобождения мицеллообразующего лекарства, который включает понижение критической концентрации мицеллообразования мицеллообразующего лекарства, в результате чего уширяется профиль высвобождения мицеллообразующего лекарства.
В некоторых других аспектах настоящее изобретение предоставляет дополнительные способы уширения профиля высвобождения мицеллообразующего лекарства. Эти способы включают, например, варьирование полимерных композиций, изменение отношения полимер/лекарство, варьирование дополнительного количества имеющего противоположный заряд полимера, а также варьирование размеров и формы таблетки.
Один из способов установления существования мицелл состоит в измерении изменения светорассеяния под углом 90°, т.е. S90, как функции концентрации в подходящем растворе. Полученные таким образом графики рассеяния могут быть затем проанализированы. Если рассеяние монотонно возрастает с возрастанием концентрации, никакого образования мицелл не происходит. Если графики обнаруживают четко выраженный излом зависимости S90 от концентрации, его приписывают образованию мицелл. Критическую концентрацию мицеллообразования определяют по точке излома на графиках рассеяния под углом 90° к падающему лучу, S90, как функции молярной концентрации. Специалистам известны и другие способы установления мицеллообразования.
Преимуществом является то, что содержание лекарства в составах настоящего изобретения может быть экстремально высоким. При этом скорость высвобождения не повышается в значительной мере с ростом содержания лекарства (например, до 60 мас.%) в SGF и реально снижается с ростом содержания лекарства в SIF (см. пример 8).
В некоторых предпочтительных аспектах мицеллообразующее лекарство имеет положительный заряд или отрицательный заряд при физиологическом рН. Как используется в настоящей заявке, физиологический рН составляет от примерно 0,5 до примерно 8, предпочтительнее от примерно 0,5 до примерно 5,5. Положительный заряд или отрицательный заряд при физиологическом рН подразумевает полный заряд на молекуле. Иными словами, если полный заряд является положительным или отрицательным, возможно наличие более одной функциональной группы, вносящей свой вклад в заряд.
Один из способов анализа для определения того, имеет ли мицеллообразующее лекарство или полимер положительный или отрицательный заряд при физиологическом рН, состоит в эмпирическом определении заряда на молекуле. Например, приготовляют подходящий буферный раствор или гель, имеющий определенный рН. В забуференный раствор помещают катод и анод или, в альтернативном варианте, используют гель-электрофорез. В том случае, когда мицеллообразующее лекарство заряжено положительно, оно мигрирует к катоду. Если же мицеллообразующее лекарство заряжено отрицательно, оно мигрирует к аноду. Полимер, обладающий в фармацевтическом составе противоположным зарядом, будет мигрировать к противоположному электроду. Например, если мицеллообразующее лекарство заряжено положительно, оно будет мигрировать к катоду. Имеющий противоположный заряд полимер будет при этом мигрировать к аноду.
В другом оценочном способе заряд на мицеллообразующем лекарстве и/или полимере устанавливают с использованием уравнения Гендерсона-Хассельбаха. Уравнение Гендерсона-Хассельбаха является математической формулировкой, которая определяет рН раствора сопряженной пары кислота/основание на основе константы диссоциации слабой кислоты и равновесных концентраций кислоты и сопряженного основания. Когда рК=рН, [На] равна [А]. Значения рК дают количественную информацию относительно силы кислоты, причем очень сильные кислоты характеризуются неопределенными значениями рК (рК=-log0, пример HCl), кислоты средней силы характеризуются малыми значениями рК и слабые кислоты характеризуются высокими значениями рК. При использовании уравнения Гендерсона-Хассельбаха заряд мицеллообразующего лекарства и/или полимера устанавливают с целью определения находящегося на них заряда.
II. Заряженные полимерные наполнители изобретения
Состав настоящего изобретения включает также по меньшей мере один полимерный наполнитель или полимер с зарядом, противоположным заряду мицеллообразующего лекарственного средства изобретения. В одном из предпочтительных аспектов кооперативное взаимодействие заряженного носителя с мицеллообразующим лекарством является основой для свойств продолжительного высвобождения настоящего изобретения.
Состав может включать отрицательно заряженные полимеры, такие как полимеры с карбоксильной группой или сульфатной группой. В число этих полимеров входят, но не ограничивают их тем самым, сульфатированные полимеры, полиакриловая кислота, полиметакриловая кислота, сополимер метилметакрилата с метакриловой кислотой, альгинаты, ксантановая камедь, геллановая камедь, гуаровая камедь, карбоксиметилцеллюлоза, смола рожкового дерева и гиалуроновая кислота.
К числу особо предпочтительных полимеров с отрицательным зарядом относятся полиакриловая кислота и сульфатированные полимеры. Сульфатированные полимеры включают в себя каррагинан (например, Viscarin® и/или Gelcarin®) и декстран-сульфат. Если в качестве одного полимера выбрана полиакриловая кислота, предпочтительно, чтобы другими полимерами были сульфатированные полимеры.
Предпочтительно, состав может также включать образующий гидрогель полимер с такими физическими характеристиками, как высокая вязкость при гелеобразовании, которая позволяет препарату настоящего изобретения быть устойчивым к сократительным силам пищеварительного тракта, возникающим при переваривании пищи, и в той или иной степени сохранять свою форму в процессе его продвижения в направлении нижнего отдела пищеварительного тракта, а именно толстой кишки. Например, особенно предпочтителен полимер, обладающий вязкостью не менее 1000 сПз в 1%-ном водном растворе (25°С).
Свойства полимера зависят от его молекулярной массы. Образующим гидрогель полимером, который может быть использован в настоящем изобретении, преимущественно является вещество со сравнительно высокой молекулярной массой, а именно полимер, имеющий среднюю молекулярную массу по меньшей мере 2·106 и более предпочтительно не менее 4-10. При этом полимеры могут быть с разветвленной цепью, прямой цепью, быть поперечно сшитыми или сочетать в себе названные особенности.
Примерами названного полимерного вещества являются полиэтиленоксид, такой как POLYOX® WSR 303 (средневязкостная молекулярная масса 7000000, вязкость от 7500 до 10000 сПз (1%-ный водный раствор при 25°С)), POLYOX® WSR Coagulant (средневязкостная молекулярная масса 5000000, вязкость от 5500 до 7500 сПз (1%-ный водный раствор при 25°С)), POLYOX® WSR 301 (средневязкостная молекулярная масса 4000000, вязкость 1650-5500 сПз (1%-ный водный раствор при 25°С)), POLYOX® WSR N-60К (средневязкостная молекулярная масса 2000000, вязкость от 2000 до 4000 сПз (2%-ный водный раствор при 25°С)) (все произведены фирмой Union Carbide); ALKOX® E-75 (средневязкостная молекулярная масса от 2000000 до 2500000, вязкость от 40 до 70 сПз (0,5%-ный водный раствор при 25°С)), ALKOX® Е-100 (средневязкостная молекулярная масса от 2500000 до 3000000, вязкость от 90 до 110 сПз (0,5%-ный водный раствор при 25°С)), ALKOX® E-130 (средневязкостная молекулярная масса от 3000000 до 3500000, вязкость от 130 до 140 сПз (0,5%-ный водный раствор при 25°С)), ALKOX® Е-160 (средневязкостная молекулярная масса от 3600000 до 4000000, вязкость от 150 до 160 сПз (0,5%-ный водный раствор при 25°С)), ALKOX® Е-240 (средневязкостная молекулярная масса от 4000000 до 5000000, вязкость от 200 до 240 сПз (0,5%-ный водный раствор при 25°С)) (все произведены фирмой Meisei Cagaku Co., Ltd.); PEO-8 (средневязкостная молекулярная масса от 1700000 до 2200000, вязкость от 20 до 70 сПз (0,5%-ный водный раствор при 25°С)), РЕО-15 (средневязкостная молекулярная масса от 3300000 до 3800000, вязкость от 130 до 250 сПз (0,5%-ный водный раствор при 25°С)), РЕО-18 (средневязкостная молекулярная масса от 4300000 до 4800000, вязкость от 250 до 480 сПз (0,5%-ный водный раствор при 25°С)) (все произведены фирмой Seitetsu Cagaku Kogyo., Ltd.) и т.д.
Для получения пригодного для замедленного высвобождения препарата гидрогелевого типа обычно предпочтительно, чтобы препарат содержал от примерно 10 до примерно 95 мас.% и более предпочтительно от примерно 15 до примерно 90 мас.% образующего гидрогель полимера в препарате массой менее 600 мг. Предпочтительно, чтобы препарат включал в себя не менее 70 и предпочтительно не менее 100 мг образующего гидрогель полимера на препарат. Названные уровни будут обеспечивать то, чтобы состав выдерживал эрозию в пищеварительном тракте в течение достаточно длительного времени, чтобы достичь достаточного замедленного высвобождения.
Названный выше образующий гидрогель полимер может быть использован один, либо же может быть использована смесь двух или более типов названных выше образующих гидрогель полимеров.
Конкретное сочетание и относительные пропорции полимерных наполнителей предпочтительно таковы, что они обеспечивают как можно более низкую скорость высвобождения в условиях как желудка, так и кишечника, независимо от рН. Оптимальные сочетание и пропорция могут варьироваться в зависимости от конкретного активного агента и процентного содержания активного агента.
Предпочтительные комбинации наполнителей включают ПАК/ПЭО, ПАК/каррагинан и ПАК/декстран-сульфат. Предпочтительно, чтобы соотношение полимеров составляло 1:0,5, 1:1 или 1:5 и наиболее предпочтительно соотношение полимеров составляет 1:1,5.
В число предпочтительных комбинаций наполнителей входит также ПАК/каррагинан/ПЭО. Предпочтительно, чтобы отношение ПАК к каррагинану составляло 1:0,5, 1:1 или 1:5 и наиболее предпочтительно соотношение полимеров 1:1,5. Предпочтительно, чтобы отношение суммы ПАК и каррагинана к ПЭО составляло 1:0,5, 1:1 или 1:2 и наиболее предпочтительно соотношение полимеров 1:1,5.
Для обеспечения замедленного высвобождения лекарства как в нижнем отделе пищеварительного тракта, так и в верхнем отделе пищеварительного тракта человека препарат должен претерпевать гелеобразование в течение не менее чем 2 ч после введения, после чего таблетка должна эродировать при ее продвижении через нижний отдел пищеварительного тракта, в результате чего будет осуществляться высвобождение.
Используемый в настоящем изобретении термин "степень гелеобразования состава" подразумевает выраженную в процентах долю таблетки, которая претерпела гелеобразование, после того как спрессованная таблетка была подвергнута увлажнению в течение предусмотренного времени, и определяется способом определения степени гелеобразования, который описан ниже (см. Метод тестирования 2). Поскольку при нахождении в верхнем отделе пищеварительного тракта препарат поглощает воду, тем самым почти полностью превращаясь в гель (т.е. степень гелеобразования по меньшей мере 70%, предпочтительно не ниже 75% и наиболее предпочтительно не ниже 80%), и перемещается к нижнему отделу пищеварительного тракта, в процессе чего поверхность препарата подвергается эрозии и по мере продолжающегося разрушения происходит высвобождение лекарства, лекарство непрерывно и полностью высвобождается и всасывается. В результате этого осуществляется замедленное высвобождение, даже в нижнем отделе пищеварительного тракта, где содержится малое количество воды. В конкретном случае, если степень гелеобразования ниже приблизительно 70%, достаточное высвобождение лекарства достигнуто не будет и при этом появляется вероятность уменьшения биодоступности лекарственного препарата (ЕР 1205190 А1).
Используемый в настоящем изобретении термин "верхний отдел пищеварительного тракта" подразумевает часть от желудка до двенадцатиперстной кишки и тощей кишки, а "нижний отдел пищеварительного тракта" подразумевает часть от подвздошной кишки до толстой кишки.
С целью достижения более высокой степени гелеобразования состав может также включать гидрофильную основу. Особых ограничений в отношении этой гидрофильной основы не существует до тех пор, пока она может раствориться, прежде чем названное выше образующее гидрогель полимерное вещество образует гель. В частности, количество воды, необходимое для растворения 1 г этой гидрофильной основы, составляет предпочтительно 5 мл или меньше (при 20±5°С), предпочтительнее 4 мл или меньше (при той же температуре).
Примерами такой гидрофильной основы являются водорастворимые полимеры, такие как полиэтиленгликоль (например Macrogol 4000, Macrogol 6000 и Macrogol 20000, все из которых являются торговыми названиями фирмы Nippon Oil and Fats Co.), поливинилпирролидон (например PVP® К30, что является торговым названием фирмы BASF), сахара-спирты, такие как D-сорбит, ксилит и т.п., сахариды такие как сахароза, мальтоза, лактулоза, D-фруктоза, декстран (например Dextran 40), глюкоза и т.п., ПАВ такие как полиоксиэтиленированное гидрогенизованное касторовое масло (например, Cremophor® RH40 (производства фирмы BASF), НСО-40, НСО-60 (производства Nikko Chemicals), полиоксиэтилен-полиоксипропиленгликоль (например, Pluronic® F68, что является торговым названием фирмы Asahi Denka) и т.д. Предпочтительными из них являются полиэтиленгликоль, сахароза и лактулоза, из которых наиболее предпочтителен полиэтиленгликоль (в частности, Macrogol 6000). Такого рода гидрофильная основа может быть использована одна, или же может быть использована смесь двух или более типов названной гидрофильной основы.
Когда в настоящем изобретении добавляют гидрофильную основу, доля в расчете на весь препарат составляет предпочтительно от примерно 5 до примерно 80 мас.%, предпочтительнее от 5 до 60 мас.% в расчете на весь препарат.
Предпочтительные комбинации наполнителей включают ПАК/ПЭО/ПЭГ. Отношение ПАК к ПЭО предпочтительно составляет 1:0,5, 1:1 или 1:5. Более предпочтительное количество ПЭГ составляет от 5 до 60 мас.% в расчете на весь препарат.
Предпочтительные комбинации наполнителей включают также ПАК/каррагинан/ПЭО/ПЭГ. Отношение ПАК к каррагинану предпочтительно составляет 1:0,5, 1:1 или 1:5. Отношение суммы ПАК и каррагинана к ПЭО составляет предпочтительно 1:0,5, 1:1 или 1:2. Более предпочтительное количество ПЭГ составляет от 5 до 60 мас.% в расчете на весь препарат.
Состав может также включать один положительно заряженный полимер или комбинации таких полимеров, в число которых входят, но не ограничивают их тем самым, полиэтиленимин, хитозан, поливинилпиридиний-бромид и полидиметиламиноэтилметакрилат.
В зависимости от вязкости полимера(ов) полимерный материал может образовывать матрицу, включающую активный ингредиент. Например, полимер, обладающий вязкостью не менее 1000 сПз в 1%-ном водном растворе, является особенно предпочтительным благодаря его способности образовывать матрицу.
При применении способа перорального введения состава настоящего изобретения может быть достигнуто продолжительное высвобождение мицеллообразующего лекарственного средства.
III. Другие модификации таблеток
Модификация высвобождения лекарственного средства через матрицу таблетки настоящего изобретения может быть осуществлена любым известным способом, таким, например, как нанесение различных покрытий, например ионообменных комплексов, например, Amberlite IRP-69. Таблетки изобретения могут также включать или вводиться вместе с уменьшающими подвижность желудочно-кишечного тракта (GI) средствами. Активный агент может быть также модифицирован с получением пролекарства в результате химической модификации биологически активного соединения, которое будет высвобождать активное соединение in vivo путем ферментативного или гидролитического расщепления и т.д. Дополнительные слои или покрытие могут действовать как диффузионные барьеры, обеспечивая дополнительные средства для регулирования скорости и координации во времени высвобождения лекарства.
IV. Добавки к составу
При желании препарат настоящего изобретения может включать подходящие количества других фармацевтически приемлемых добавок, таких как индифферентные наполнители (например, лактоза, маннит, картофельный крахмал, рисовый крахмал, кукурузный крахмал и кристаллическая целлюлоза), связующие агенты (например, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, метилцеллюлоза и гуммиарабик), агенты набухания (например, карбоксиметилцеллюлоза и карбоксиметилцеллюлоза кальция), смазочные агенты (например, стеариновая кислота, стеарат кальция, стеарат магния, тальк, метасиликат-алюминат магния, гидрофосфат кальция и безводный гидрофосфат кальция), пластификаторы (например, водный кремнезем, светлая безводная кремневая кислота и высушенный гель гидроксида алюминия), красители (например, желтый полуторный оксид железа и полуторный оксид железа), ПАВ (например, лаурилсульфат натрия и эфир жирной кислоты с сахарозой), покровные агенты (например, зеин, гидроксипропилметилцеллюлоза и гидроксипропилцеллюлоза), буферные агенты (например, хлорид натрия, хлорид магния, лимонная кислота, винная кислота, двухосновный фосфат натрия, одноосновный фосфат натрия, гидрофосфат кальция, аскорбиновая кислота), отдушки (например, 1-ментол, масло перечной мяты и фенхелевое масло) и консерванты (например, сорбат натрия, сорбат калия, метиловый эфир п-бензойной кислоты и этилбензоат).
V. Изготовление
Препарат настоящего изобретения представляет собой твердый препарат определенной формы, который может быть изготовлен с использованием любых традиционных способов. К числу типичных способов относятся, например, способы изготовления с применением таблетирования прессованием. Эти способы включают смешивание и, при необходимости, гранулирование активного агента, заряженных полимеров и, при необходимости, дополнительных добавок и прессование-формование образовавшейся композиции (состава). В число альтернативных способов входят, например, способ капсульного компрессионного наполнения, экструзионный способ формования, включающий расплавление смеси и схватывание расплавленной смеси, способ литьевого формования и т.п. Может также проводиться любая обработка с целью создания покрытия, например покрытия сахаром.
Приведенные ниже примеры предназначены проиллюстрировать, но не ограничить настоящее изобретение.
ПРИМЕРЫ
Метод тестирования 1
Данный метод тестирования иллюстрирует основную методику изготовления составов настоящего изобретения, а также измерение высвобождения лекарства.
Были изготовлены несколько разных составов с различными лекарственными средствами. Лекарства вручную смешивают с наполнителями в ступке и прессуют в 400-мг таблетки, используя для этого пресс Carver или пресс Oil с прилагаемым давлением 70 кг/см2. Использовали округлую инструментальную оснастку с плоской поверхностью диаметром 11 мм.
Материалы
Carbopol 971 (BF Goodrich); Polyox 303 (Union Carbide); два типа каррагинана, Viscarin P 109 и Gelcarin (FMC); Xantural™ 180 (Monsanto Pharmaceutical Ingredients), ксантановая камедь Keltone LVCR (Monsanto Pharmaceutical Ingredients); альгинат натрия Chitosan (M.W. International, Inc.); Macrogol 6000 (Nippon Oil and Fats Co.); Methocel K100M (The Dow Chemical Company); гидроксипропилметилцеллюлоза (НРМС); Cellulose Gum 12M31P TP (Hercules); карбоксиметилцеллюлоза натрия (CMC); и декстран сульфат (Sigma).
Методы
Высвобождение лекарства in vitro измеряют в экспериментах с растворением in vitro. Эти исследования проводят с использованием устройства USP (United State Pharmacopoeia) II при скорости лопастей 100 об/мин в 1000 мл растворяющей среды для примеров 1-10. Высвобождение лекарства измеряют с использованием или искусственной желудочной среды (SGF), рН 1,2, либо искусственной кишечной среды (SIF), рН 7,5, которые готовят в соответствии с USP без добавления ферментов. Во всех экспериментах использовали грузила для таблеток. Через определенные промежутки времени образец отбирают из емкости и анализируют на УФ-видимом спектрофотометре при длине волны 240 нм.
Пример 1
Этот пример иллюстрирует то, что скорость высвобождения лекарства не коррелируется с растворимостью лекарства, указывая на то, что на скорость высвобождения влияет специфическое взаимодействие.
Поведение высвобождения большой группы основных хорошо растворимых лекарственных средств (10 мас.% лекарства) из полученных прямым прессованием матричных таблеток с использованием в качестве наполнителя смеси полиакриловая кислота/полиэтиленоксид (ПАК/ПЭО в соотношении 1:1,5) изучают в условиях модифицированной искусственной кишечной среды (SIF). Скорость высвобождения характеризуют с помощью т50 (время высвобождения 50% лекарства из матрицы в раствор) (фиг.1). Результаты исследований представлены в табл.1, в которой суммированы свойства лекарства и скорости высвобождения.
Одинаково заряженные лекарственные средства обладают значительно различающимися профилями высвобождения в модифицированной SIF, которые не коррелируются с растворимостью лекарства (фиг.1, табл.1). Следовательно, можно заключить, что одно только электростатическое взаимодействие не приводит само по себе к продолжительному высвобождению растворимых лекарственных средств.
Пример 2
Этот пример иллюстрирует то, что logP лекарственного средства может быть использован для предсказания того, будет ли достигнуто продолжительное высвобождение при использовании состава настоящего изобретения. Возможность предсказания поведения высвобождения лекарства на основании его характеристики logP является одним из ключевых преимуществ настоящего изобретения.
Способность лекарства связываться с каким-либо полиэлектролитом зависит от его критической концентрации мицеллообразования (ККМ). Однако, поскольку для лекарств значение ККМ редко бывает известным, была сделана попытка связать скорость высвобождения со свойствами лекарства, которые обычно используют для его характеризации. Для лекарственных средств, которые были использованы в описанных выше экспериментах по высвобождению (табл.1), анализируют различные параметры, в частности молекулярная масса, растворимость, рКа, logP, logD и поверхностное натяжение, с точки зрения их корреляции с временем высвобождения. Оказалось, что logP (коэффициент распределения незаряженной формы лекарства между октанолом и водой) демонстрирует близкую к линейной зависимости с Т50 (фиг.2). logP тесно связан с ККМ. Действительно, между logP и ККМ была выявлена практически линейная зависимость (фиг.3). Значения logP и ККМ для разных лекарственных средств были взяты из публикаций Attwood.
Пример 3
Этот пример иллюстрирует то, что продолжительного высвобождения для постоянно положительно заряженных молекул можно достичь, используя смесь наполнителей ПАК/ПЭО (1:1,5).
Испытывают следующие положительно заряженные молекулы: бензетоний-хлорид и бетанехол-хлорид, имеющие по одному положительному заряду; тиамин мононитрат и тиамин гидрохлорид, имеющие по два положительных заряда; и бетаин, представляющий собой диполь (фиг.4). Хотя тиамин HCl показал относительно быстрое высвобождение, все другие лекарственные средства продемонстрировали продолжительное высвобождение с разными скоростями (табл.2).
Эти результаты показывают, что, даже если лекарство не имеет сильных гидрофобных групп, все еще остается возможным специфическое взаимодействие с заряженными полимерными наполнителями (см. в качестве примера бетанехол-хлорид), если только лекарство несет постоянный положительный заряд. С другой стороны, в способности взаимодействовать с полимерными носителями могут играть важную роль структура лекарства и положение заряда (см. тиамин HCl). Расположение в тиамине зарядов в центре молекулы (фиг.4) может вызывать мицеллообразование.
Пример 4
Этот пример иллюстрирует то, что противоположно заряженные лекарства и полимерные наполнители являются критическим фактором для продления высвобождения лекарства. Как показано на фиг.5, хорошо растворимые отрицательно заряженные лекарственные средства, цефазолин натрия и цефматазол натрия, диффундируют из отрицательно заряженной матрицы ПАК/ПЭО с Т50, равным приблизительно 5 ч, без достижения продолжительного высвобождения.
Пример 5
Этот пример демонстрирует влияние жидкого окружения на профили высвобождения лекарственных средств для смесей ПАК/ПЭО (1:1,5).
Предыдущие эксперименты, описанные в примерах 1-4, проводят в условиях искусственной кишечной среды (SIF), в которой ПАК ионизована. Для оценки кинетики высвобождения в желудочных условиях растворение различных типов растворимых лекарств проводят в модифицированной искусственной желудочной среде (SGF). Таблица 3 сравнивает значения Т50 в SGF и SIF для разных лекарственных средств.
Как показано в табл.3, время высвобождения в SIF значительно больше, чем в SGF. Является очевидным, что в средах с низким рН ионизация ПАК в значительной степени подавляется. Это может предотвращать образование кооперативных связей между ПАК/ПЭО и лекарством. Другой возможной причиной малых времен высвобождения в SGF является то, что образование связанного водородными связями полимерного комплекса между электроотрицательным атомом кислорода ПЭО и карбоксильной группой ПАК в условиях низкого рН блокирует взаимодействие карбоксильных групп с лекарственным средством.
Пример 6
Этот пример иллюстрирует комбинации полимерных наполнителей, которые обеспечивают замедленное высвобождение в условиях как SGF, так и SIF.
Оценка множества полисахаридов
Скорости высвобождения лекарственных средств испытывают для семи разных полисахаридов (каррагинана, ксантановой камеди, альгината натрия, хитозана, НРМС (гидроксипропилметилцеллюлозы), КМЦ-Na) в сочетании с ПАК, при содержании 25 мас.% Дилтиазема HCl (DI) (фиг.6). Результаты показывают, что комбинация ПАК с каррагинаном может обеспечить наиболее медленное высвобождение лекарства в SGF. Этот эффект вероятно обусловлен сильнокислотной природой функциональных групп каррагинана (SO4 -), который остается отрицательно заряженным даже в условиях низких рН и делает возможным взаимодействие между каррагинаном и лекарственным средством.
Оценка других сульфатированных полимеров
Используют разные типы каррагинана, так же как и декстран-сульфата, в сочетании с ПАК или ПЭО при отношении 1:1 и Дилтиаземом HCl (25 мас.%) и измеряют скорость высвобождения в SGF и SIF. Продолжительное высвобождение наблюдалось для всех комбинаций, содержащих сульфатированные полимеры (фиг.7).
Дополнительный анализ влияния замещения ПАК на ПЭО
Когда ПАК в составе ПАК/каррагинан (1:1) замещают на высокомолекулярный ПЭО, профили высвобождения для составов ПАК/каррагинан (1:1) и ПЭО/каррагинан (1:1) в SGF перекрываются в течение приблизительно 6 ч (фиг.8а). После этого отрезка времени эрозия матрицы вызывает более быстрое высвобождение из матрицы ПЭО/каррагинан. Напротив, в SIF состав ПАК/каррагинан демонстрирует более медленное высвобождение лекарственного средства по сравнению с составом ПЭО/каррагинан в течение всего периода времени (фиг.8b). Таким образом, сочетание ПАК и каррагинана может обеспечить наилучшие характеристики высвобождения лекарства in vitro как в SGF, так и в SIF.
Сравнение скоростей высвобождения для ПАК/каррагинан в SGF и SIF.
Фиг.9 показывает, что высвобождение DI (25 мас.%) из матрицы ПАК/каррагинан (1:1) является линейным как в SGF, так и в SIF и что скорости высвобождения в обеих средах одинаковы. Тест на растворимость для образцов со сменой сред через 2 часа обнаружил линейный профиль высвобождения, очень близкий к профилям на фиг.9.
Пример 7
Этот пример иллюстрирует то, что оптимальный состав полимерного наполнителя зависит от среды (фиг.10).
Для состава, содержащего 25 мас.% DI (дилтиазем HCl), наиболее низкая скорость высвобождения в SGF достигалась для состава ПАК/каррагинан (1:1). В SIF скорость высвобождения понижалась при возрастании количества ПАК в составе.
Интересно, что другие оптимальные составы наблюдались для состава с высоким содержанием DI 60 мас.%. В SGF скорость высвобождения лекарства понижалась с ростом содержания каррагинана, а в SIF скорость высвобождения лекарства практически не зависела от соотношения компонентов наполнителя (фиг.11).
На основании этих наблюдений мы полагаем, что характер высвобождения по всей видимости определяется стехиометрией комплекса лекарство-наполнитель в различных средах.
Пример 8
Этот пример иллюстрирует то, что увеличение содержания лекарственного средства оказывает несущественное влияние на скорость высвобождения в SIF для содержания лекарства вплоть до 60 мас.%. В SGF повышение скорости высвобождения относительно невелико при содержании лекарственного средства до 50 мас.% (фиг.12).
Пример 9
Этот пример иллюстрирует превосходную способность составов настоящего изобретения к продлению высвобождения лекарственного средства.
Высвобождение DI (25%) из матрицы ПАК/каррагинан (1:1) сравнивают с описанными ранее составами, содержащими ПАК и каррагинан (Bonferoni et al., AAPS Pharm. Sci. Tech, 1(2) статья 15 (2000); Bubnis et al., Proceed. 1nt'l. Symp. Control. Rel. Bioact. Mater., 25, p. 820 (1998); Devi et al., Pharm. Res., v.6. No 4, 313-317 (1989); Randa Rao et al., J. Contr. Rel., 12, 133-141 (1990); Baveja et al., Int. J. Pharm., 39, 39-45 (1987); Stockwel et al., J. Contr. Rel. 3, 167-175 (1986); Perez Marcos et al., J. Pharm. Sci., v.85, No.3 (1996); Perez-Marcos et al., Int. J. Pharm. 111, 251-259 (1994); Dabbagh et al., Pharm. Dev. Tech., 4(3), 313-324 (1999); Bonferoni et al., J. Contr. Rel. 25, 119-127 (1993); Bonferoni et al., J. Contr. Rel. 30, 175-182 (1994); Bonferoni et al., J. Contr. Rel. 51, 231-239 (1998); US Patent 4777033; EU Patent 0205336 B1).
В число описанных в литературе содержащих каррагинан систем входят каррагинан/гидроксипропилметилцеллюлоза (НРМС) и каррагинан/КМЦ. Все матрицы приготовляли так же, как смесь Viscarin 109/второй полимер (1:1). Составы с матрицей ПАК/каррагинан (1:1) демонстрировали значительно более медленное высвобождение DI как в SGF, так и в SIF (фиг.13).
Описана система замедленного высвобождения с ПАК/НРМС (гидроксипропилметилцеллюлоза) (патент США 4777033, патент EU 0205336 B1).
Составы с матрицей ПАК/каррагинан (1:1 и 3:2) демонстрировали значительно более медленное высвобождение DI по сравнению с ПАК/НРМС (1:1), служащим в качестве контроля, в SGF (фиг.14), хотя в SIG все препараты проявляли продолжительное высвобождение лекарственного средства с Т50 выше 20 ч.
Когда количество лекарства в системе повышают до 60 мас.%, скорость высвобождения из системы ПАК/каррагинан сохраняется наиболее низкой по сравнению со всеми другими конкурирующими системами (фиг.15).
Пример 10
В этом примере сравниваются скорости высвобождения различных лекарственных средств для исходного состава (ПАК/ПЭО) и новых составов ПАК/каррагинан.
Скорости высвобождения различных лекарств, которые ранее проявляли взаимодействие с матрицей ПАК/ПЭО, сравнивают со скоростями высвобождения из матрицы ПАК/каррагинан (1:1). Оказалось, что большинство лекарственных средств проявляет продолжительное, близкое к кинетике нулевого порядка высвобождение из матрицы ПАК/каррагинан. Высвобождение лекарств из матрицы ПАК/каррагинан, как правило, было более медленным как в SGF, так и в SIF в сравнении с высвобождением из матрицы ПАК/ПЭО, хотя это наблюдалось не для всех лекарств.
В качестве иллюстрации приведенная ниже табл.4 представляет значения Т50 (время высвобождения) в SIF. В этом исследовании состав ПАК/ПЭО (1:1.5) содержал 10% активного вещества, а состав ПАК/каррагинан (1:1) содержал 25% активного вещества.
Метод тестирования 2
Тест на растворение
Высвобождение лекарства in vitro измеряют в экспериментах с растворением in vitro. Эти исследования проводят с использованием метода тестирования на растворение II (лопастной метод) в соответствии с Фармакопеей Японии XIV (далее упоминается как "JP") при скорости лопастей 200 об/мин в 900 мл растворяющей среды. Высвобождение лекарственного средства оценивают с помощью либо жидкости для анализа дезинтеграции 1 по JP (далее "JP 1st fluid"), pH 1,2, либо жидкости для анализа дезинтеграции 2 по JP (далее "JP 2nd fluid"), pH 6,8. В экспериментах не используют грузила для таблеток. Через определенные промежутки времени образец отбирают из емкости и анализируют на УФ-видимом спектрофотометре при длине волны 250 нм.
Тест на гелеобразование
Тест на гелеобразование проводят с использованием JP 1st fluid и JP 2nd fluid следующим образом. Испытуемую таблетку увлажняют в течение 2 ч в среде для анализа при 37°С, удаляют гелевый слой и отбирают сердцевинную, не образовавшую гель часть, которую высушивают в течение 5 суток при 40°С в сушилке и высушенную сердцевину взвешивают (Wobs). Степень гелеобразования составов рассчитывают с помощью уравнения 1. Значение, получаемое путем вычитания веса сердцевины из начального веса таблетки (Winitial) и деления на начальный вес таблетки, умножают на 100 для расчета степени гелеобразования в % (G).
Используемый в настоящей заявке термин "степень гелеобразования" подразумевает выраженную в процентах долю таблетки, которая претерпела гелеобразование. Выбор способа расчета степени гелеобразования не является особенно существенным, и способ, который приводится ниже, можно рассматривать как пример.
Таким образом, таблетку увлажняют в течение предварительно заданного времени, после чего измеряют объем (или массу) не претерпевшей гелеобразования части и полученное значение вычитают из объема (или массы) таблетки перед началом теста.
Степень гелеобразования (G, %)=(1-(Wobs/Winitiai))×100 (уравнение 1)
Wobs - масса части, не претерпевшей гелеобразования после начала теста,
Winitial - масса препарата перед началом теста.
Пример 11
Этот пример иллюстрирует влияние дополнительного количества полимеров, имеющих заряд, противоположный заряду мицеллообразующего лекарственного средства, на профили высвобождения лекарств.
Используют разные количества ПАК в сочетании со смесью ПЭО/ПЭГ (1:1) при отношении 1:0 (мас.% ПАК к общему количеству равен 50), отношении 1:1 (мас.% ПАК к общему количеству равен 25), отношении 3:1 (мас.% ПАК к общему количеству равен 37,5), отношении 1:3 (мас.% ПАК к общему количеству равен 12,5) или отношении 1:9 (мас.% ПАК к общему количеству равен 5), при содержаний 50 мас.% Дилтиазема HCl. Состав, включающий ПЭО/ПЭГ при отношении 1:1 без ПАК, содержащий 50мас.% Дилтиазема HCl, был приготовлен в качестве контроля. Скорость высвобождения лекарства оценивали в JP 2nd fluid в соответствии с методом, описанным в Методе тестирования 2 (фиг.16). Продолжительное высвобождение лекарства достигалось для всех препаратов, содержащих ПАК, даже в случае содержания такого малого количества ПАК, как 5 мас.% от всего препарата. Результаты также показали, что скорость высвобождения лекарства снижается с увеличением количества ПАК за счет смеси ПЭО/ПЭГ (1:1).
Также изучают влияние дополнительного количества смеси ПАК с каррагинаном на профили высвобождения лекарств. Соотношение ПАК и каррагинана и отношение ПЭО/ПЭГ фиксируют равными 1:1. Разные количества ПАК/каррагинан (1:1) используют в комбинации с ПЭО/ПЭГ (1:1) при отношении 1:0 (мас.% ПАК и каррагинана к общему количеству составляет 25 и 25 соответственно), при отношении 3:1 (мас.% ПАК и каррагинана к общему количеству составляет 18,75 и 18,75 соответственно), 1:1 (мас.% ПАК и каррагинана к общему количеству составляет 12,5 и 12,5 соответственно), при отношении 1:3 (мас.% ПАК и каррагинана к общему количеству составляет 6,25 и 6,25 соответственно) при содержании 50 мас.% Дилтиазема HCl (фиг.7). В качестве контроля готовят состав, содержащий ПЭО/ПЭГ при отношении 1:1 без ПАК/каррагинана с содержанием 50 мас.% Дилтиазема HCl. Результаты показали также, что скорость высвобождения лекарства снижалась с увеличением количества смеси ПАК/каррагинан. Таким образом, скорость высвобождения лекарства можно регулировать путем варьирования дополнительного количества полимера(ов), имеющего(их) заряд, противоположный заряду мицеллообразующего лекарственного средства.
Пример 12
Этот пример иллюстрирует превосходную способность к гелеобразованию составов настоящего изобретения.
При проведении теста на гелеобразование для препаратов, содержащих ПАК/ каррагинан/ПЭО/ПЭГ при отношении 1:1:0:0, 1:1:1:1 или 1:1:3:3, и содержащих 50 мас.% Дилтиазема HCl, в соответствии со способом, описанным в Методе тестирования 2, степень гелеобразования названных составов в JP 1st fluid составила соответственно 75,0, 80,8 и 80,7%.
В случае препарата, содержащего ПАК/ПЭО/ПЭГ при отношении 1:9:9 степень гелеобразования в JP 1st fluid и в JP 2nd fluid составила соответственно 78,0 и 76,9%.
Пример 13
Фармакокинетические исследования на собаках-биглях
Девять самцов собак-биглей весом от 9,3 до 13,4 кг лишают пищи за 18 ч до введения. После перорального введения испытуемой таблетки, содержащей 200 мг Дилтиазема HCl, с 30 мл воды собакам позволяют свободный доступ к воде, но в пище отказывают до тех пор, пока не был произведен последний забор образца крови. Образцы крови забирают через 0,5, 1, 2, 3, 4, 6, 8, 10, 12 и 24 часа после введения таблетки. После этого отделяют на центрифуге плазму и подвергают ее количественному анализу в системе ВЭЖХ с УФ детектированием.
Пример 14
Этот пример иллюстрирует влияние степени гелеобразования препаратов на замедленное высвобождение лекарства in vivo.
Два препарата (Степень гелеобразования в JP 1st fluid: Препарат А: 63,4% и Препарат В: 77,6%), содержащие различные количества ПАК/ПЭО/ПЭГ, а также по 200 мг Дилтиазема HCl, использовали для фармакокинетического исследования на собаках-биглях. Результаты показали, что Препарат В давал замедленное высвобождение лекарства как в нижнем отделе пищеварительного тракта, так и в верхнем отделе пищеварительного тракта, в то время как Препарат А высвобождал мало лекарства в нижнем отделе пищеварительного тракта.
Для более детального сравнения высвобождения лекарства in vivo между двумя препаратами рассчитывают площадь под кривой зависимости концентрации лекарства в плазме от времени в пределах от 0 до 24 ч как функцию количества абсорбированного лекарства in vivo. Результаты показали, что эта площадь для Препарата В (7541,2±2153,7 нг·ч/мл) была значительно больше площади для Препарата А (4346,1±1811,6 нг·ч/мл), что подтвердило недостаточное высвобождение лекарства in vivo для препарата с более низкой степенью гелеобразования.
Все публикации, патенты и патентные заявки, упоминаемые в настоящей заявке, включены в описание путем отсылки во всем их объеме и для всех целей. Хотя изобретение описано с упоминанием предпочтительных воплощений и относящихся к ним примеров, объем настоящего изобретения не ограничивается только этими описанными воплощениями. Для специалистов должно быть очевидно, что модификации и адаптации описанного выше изобретения могут быть произведены без отступления от сути и объема изобретения, которое определено и описано в приложенной формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2323006C2 |
| ТРЕХБЛОЧНЫЕ ПОЛИМЕРЫ ВАВ, ОБЛАДАЮЩИЕ УЛУЧШЕННЫМИ ХАРАКТЕРИСТИКАМИ ВЫСВОБОЖДЕНИЯ | 2010 |
|
RU2575844C2 |
| Пероральная система доставки вещества белковой природы (варианты), защитная оболочка системы доставки (варианты) | 2014 |
|
RU2665367C2 |
| ТАБЛЕТКИ ТАМЗУЛОСИНА С МОДИФИЦИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ | 2002 |
|
RU2335280C2 |
| МАТРИКСНАЯ ТАБЛЕТКА С РЕГУЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ ТРИМЕТАЗИДИНА | 2006 |
|
RU2367438C2 |
| Новая лекарственная форма эхинохрома А, способ ее получения и применение | 2022 |
|
RU2800382C1 |
| ГИДРОГЕЛЕВЫЙ ПРЕПАРАТ С ДЛИТЕЛЬНЫМ ВЫСВОБОЖДЕНИЕМ ЛЕКАРСТВА | 1993 |
|
RU2121830C1 |
| ЛИПИДНО-ПОЛИМЕРНАЯ СИСТЕМА ОДНОВРЕМЕННОЙ ДОСТАВКИ ДВУХ АНТИБАКТЕРИАЛЬНЫХ СОЕДИНЕНИЙ | 2023 |
|
RU2838145C1 |
| СТАБИЛЬНАЯ ПРИ ОКИСЛЕНИИ, ПРОЧНАЯ НА ИЗЛОМ ЛЕКАРСТВЕННАЯ ФОРМА | 2010 |
|
RU2567723C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИОАДГЕЗИВНЫХ КОМПАКТИРОВАННЫХ МАТРИЦ, КОТОРЫЕ МОГУТ БЫТЬ ИСПОЛЬЗОВАНЫ КАК ТАКОВЫЕ ИЛИ ДЛЯ ЗАМЕДЛЕННОГО ВЫСВОБОЖДЕНИЯ АКТИВНЫХ ВЕЩЕСТВ, И КОМПАКТИРОВАННЫЕ МАТРИЦЫ, ПОЛУЧЕННЫЕ ТАКИМ СПОСОБОМ | 2009 |
|
RU2519224C2 |
Изобретение относится к лекарственным средствам и касается пероральных составов с замедленным высвобождением, включающих мицеллообразующее водорастворимое основное лекарственное средство, имеющее положительный заряд при физиологическом рН; полимер, имеющий противоположный заряд, выбранный из группы, состоящей из полиакриловой кислоты, карбоксиметилцеллюлозы, ксантановой камеди, геллановой камеди, гуаровой камеди, декстран-сульфата и каррагинана; полиэтиленоксид; и при необходимости включающих гидрофильную основу. Также предложен способ продления высвобождения мицеллообразующего лекарственного средства. 4 н. и 16 з.п. ф-лы, 24 ил., 4 табл.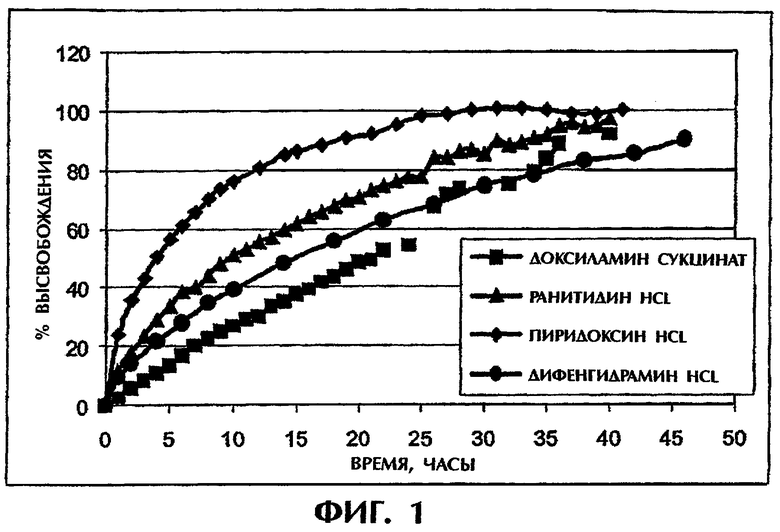
(I) мицеллообразующее водорастворимое лекарственное средство, причем указанное лекарственное средство является основным и имеет положительный заряд при физиологическом рН, и
(II) по меньшей мере один полимер, имеющий противоположный заряд, выбранный из группы, состоящей из полиакриловой кислоты, карбоксиметилцеллюлозы, ксантановой камеди, геллановой камеди, гуаровой камеди, декстран-сульфата и каррагинана;
(III) полиэтиленоксид; и при необходимости включающий
(IV) гидрофильную основу.
(I) мицеллообразующее лекарственное средство, являющееся водорастворимым лекарственным средством, имеющим положительный заряд при физиологическом рН,
(II) полиакриловую кислоту;
(III) полиэтиленоксид; и при необходимости включающий
(IV) гидрофильную основу.
(V) по меньшей мере один полимер, содержащий сульфатную группу.
(I) мицеллообразующее лекарственное средство, являющееся водорастворимым основным лекарственным средством, имеющим положительный заряд при физиологическом рН,
(II) полиакриловую кислоту;
(III) полиэтиленоксид, и дополнительно содержащего
(IV) гидрофильную основу,
продлевая таким образом высвобождение указанного мицеллообразующего лекарства.
(V) по меньшей мере один полимер, содержащий сульфатную группу;
продлевая таким образом высвобождение указанного мицеллообразующего лекарственного средства.
(I) мицеллообразующее водорастворимое лекарственное средство, которое является основным лекарственным средством и имеет положительный заряд при физиологическом рН,
(II) комбинацию двух полимеров, имеющих противоположный заряд, выбранных из группы, состоящей из полиакриловой кислоты, карбоксиметилцеллюлозы, ксантановой камеди, геллановой камеди, гуаровой камеди, декстран-сульфата и каррагинана, причем одним из полимеров является декстран-сульфат или каррагинан;
(III) полиэтиленоксид; и при необходимости включающий
(IV) гидрофильную основу.
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| US 5788959 A, 04.08.1998 | |||
| US 5219563 A, 15.06.1993. | |||

