Настоящее изобретение относится к способу получения производных тиено[3,2-с]пиридина и используемых при этом промежуточных соединений.
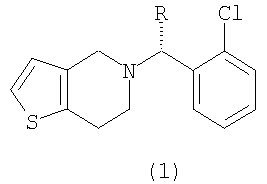
Известно, что производные тиено[3,2-с]пиридина (1) проявляют высокую ингибирующую активность в отношении агрегации тромбоцитов крови и противотромбозную активность и могут оказаться полезными при использовании в качестве лекарственного средства, способствующего улучшению кровообращения при лечении заболеваний периферических артерий, таких как апоплексия мозга, тромб и эмболия, или заболеваний коронарных артерий, таких как инфаркт миокарда и стенокардия
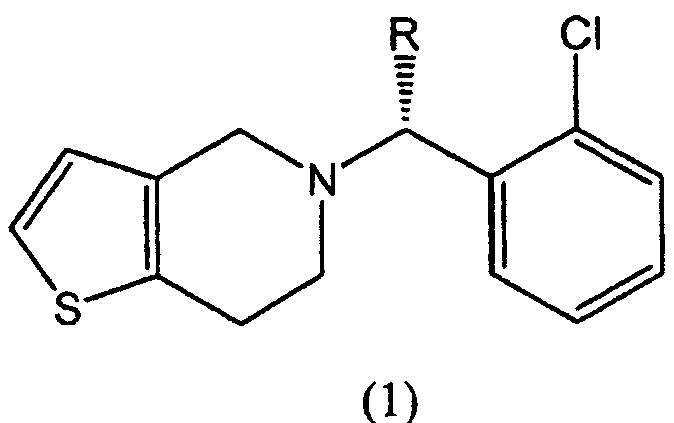
в которой R представляет собой водород или метоксикарбонил.
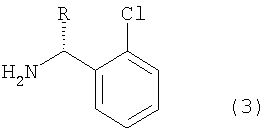
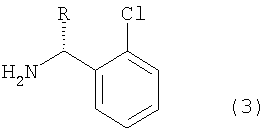
Соединение формулы (1), в котором R представляет собой водород, называют тиклопидин, а соединение формулы (1), в котором R представляет собой метоксикарбонил, называют клопидогрель (смотри патенты США №№ 4051141, 4529596 и 4847265).
Ранее тиклопидин и клопидогрель синтезировали способами, представленными совместно на схеме реакции 1 (смотри патенты США №№ 4127580, 4174448, 6043368, 4529596, 4847265 и 5204469, британский патент № 2166730, европейскую патентную публикацию № 0522956 А и международные публикации №№ WO 98/51689 и WO 02/59128).
Схема реакции 1
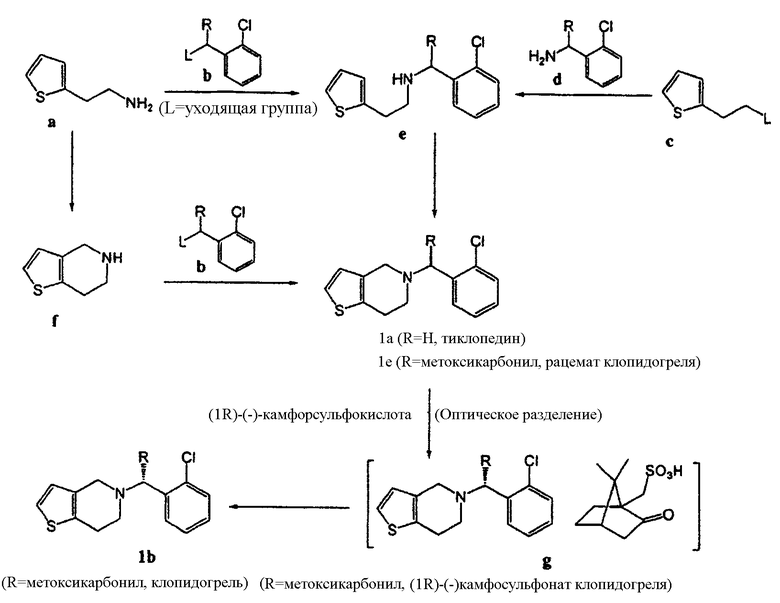
Как показано на схеме реакции 1, тиклопидин формулы (1а) можно получить путем взаимодействия 2-(2-аминоэтил)тиофена формулы (а) с соединением формулы (b) (в котором L представляет собой уходящую группу, такую как хлор), или путем взаимодействия соединения формулы (с) (в котором L представляет собой уходящую группу, такую как п-толуолсульфонил) с производным о-хлорбензиламина формулы (d), получая соединение формулы (е), а затем циклизуя соединение (е) с формилирующим агентом, таким как формальдегид, А-СН2-В (где А представляет собой галоген, алкокси, алкилтио или амино-группу, а В представляет собой алкокси, алкилтио, амино или алкоксикарбонилокси-группу), или гетероциклическим соединением формулы
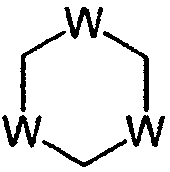
в которой W представляет собой O, NH или S.
Альтернативным образом, тиклопидин формулы (1а) можно также получить прямой циклизацией 2-(2-аминоэтил)тиофена формулы (а) с формилирующим агентом, получая 4,5,6,7-тетрагидротиено[3,2-c]пиридин формулы (f), а затем путем взаимодействия соединения (f) с соединением формулы (b).
Аналогичным образом, рацемат клопидогреля формулы (1с) можно получить описанным выше способом, но данный рацемат необходимо перевести в оптически чистый клопидогрель формулы (1b) при помощи сложного способа оптического разделения, который включает в себя введение во взаимодействие рацемата формулы (1с) с оптически активной кислотой, например (1R)-(-)камфорсульфокислотой, получая диастереомерную соль формулы (g), подвергая данную диастереомерную соль ряду процессов дробной кристаллизации для повышения оптической чистоты, а затем удаляя оставшуюся оптически активную кислоту из продукта. Таким образом, в результате данного способа соединение формулы (1b) образуется с низким выходом.
Соответственно, авторы настоящего изобретения провели исследования с целью разработки простого способа получения оптически чистого клопидогреля и обнаружили, что когда определенное производное тиофена вводят во взаимодействие с оптически чистым производным 2-хлорбензиламина, можно получить оптически чистый клопидогрель с высоким выходом простым способом наряду с тиклопидином.
Основной задачей настоящего изобретения является разработка простого нового способа получения оптически чистого клопидогреля.
Задачей настоящего изобретения является также создание новых промежуточных соединений, используемых в способе изобретения.
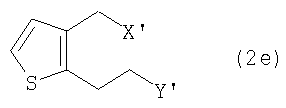
В соответствии с одним из аспектов настоящего изобретения предложен способ получения производного тиено[3,2-c]пиридина формулы (1), включающий взаимодействие соединения формулы (2е) с соединением формулы (3):
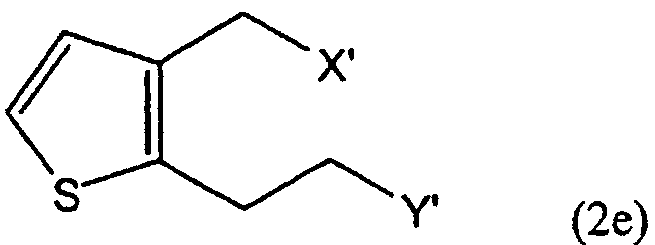
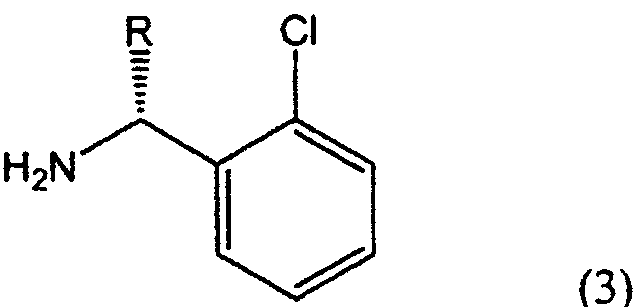
в которых R представляет собой водород или метоксикарбонил, а каждый из X' и Y' независимо представляет собой хлор, бром, метансульфонил или п-толуолсульфонил.
В соответствии с другим аспектом настоящего изобретения предложено соединение формулы (2), используемое при получении соединения формулы (1) в качестве промежуточного соединения:
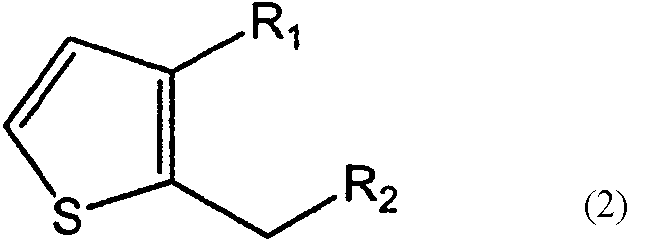
в которой
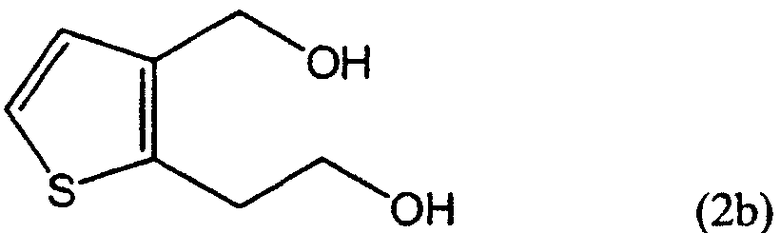
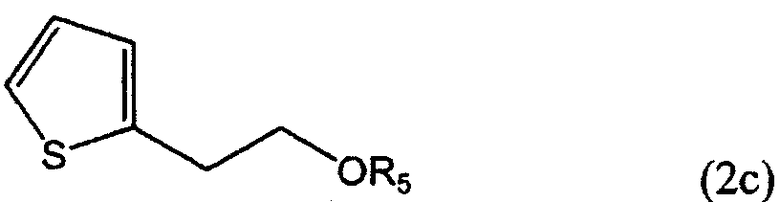
R1 представляет собой водород, CH2X или CO2R3, а R2 представляет собой CH2Y, CO2R4 или CO2R5, или R1 и R2 связаны друг с другом с образованием -СН2-О-СН2-, где каждый из Х и Y независимо представляет собой гидрокси, хлор, бром, метансульфонил или п-толуолсульфонил, каждый из R3 и R4 независимо представляет собой водород или линейный или разветвленный С1-6 алкил, R5 представляет собой С1-4 алкоксиметил, такой как метоксиметил, этоксиметил или 2-метоксиэтоксиметил, при условии, что когда R1 представляет собой водород, R2 не является гидроксиметилом.
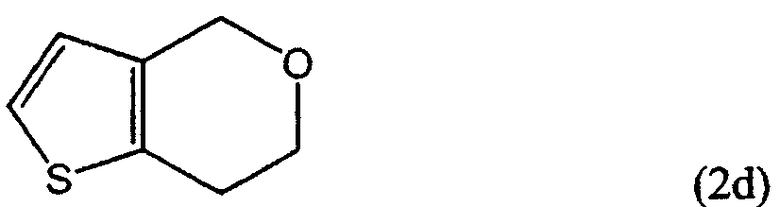
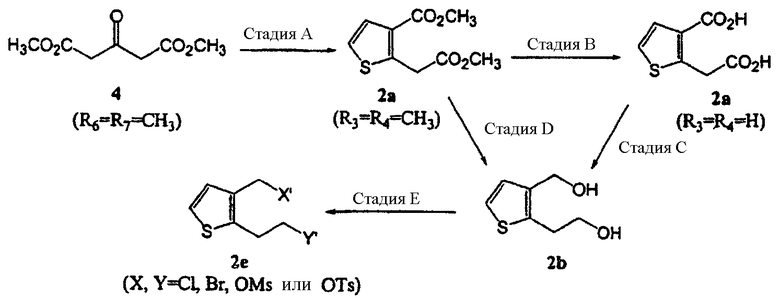
В настоящем изобретении соединение формулы (2е) можно получить (i) циклизацией соединения формулы (4), получая соединение формулы (2а), восстановлением соединения формулы (2а) восстанавливающим агентом, получая соединение формулы (2b), и введением во взаимодействие соединения формулы (2b) с галоидирующим или сульфонилирующим агентом, или (ii) прямой циклизацией 2-тиофенэтанола с формилирующим агентом, получая соединение формулы (2d), или введением во взаимодействие 2-тиофенэтанола с диалкоксиметаном, получая соединение формулы (2с), и циклизацией соединения формулы (2с), получая соединение формулы (2d), а затем введением во взаимодействие соединения формулы (2d) с галогенирующим агентом:
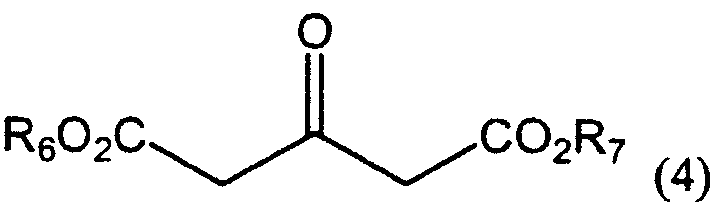
в которых
каждый из R3 и R4 независимо представляет собой водород или линейный или разветвленный С1-6 алкил,
R5 представляет собой С1-4 алкоксиметил, такой как метоксиметил, этоксиметил или 2-метоксиэтоксиметил, а
каждый из R6 и R7 независимо представляет собой линейный или разветвленный С1-6 алкил.
Подробное описание изобретения
В целом настоящее изобретение можно представить схемами реакций со 2 по 4. На схемах реакций 2 и 3 представлен вариант осуществления синтеза соединения формулы (2), используемого в качестве промежуточного соединения в способе изобретения, тогда как на схеме реакции 4 представлен метод синтеза соединения формулы (1).
Схема реакции 2
<Стадия А>
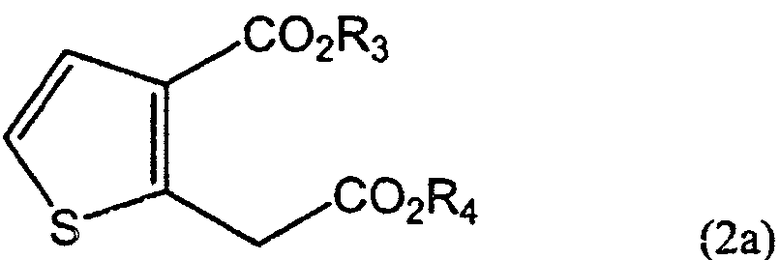
Сначала на стадии А дикарбоксилат ацетона формулы (4) вводят во взаимодействие с 2,5-дигидрокси-1,4-дитианом, который является димером меркаптоацетальдегида, в присутствии катализатора - кислоты Льюиса, и растворителя, получая соединение тиофен-диэфира формулы (2а-1), соединение формулы (2), в котором R1 и R2 представляют собой CO2R3 и CO2R4 соответственно, а R3 и R4 являются метилом.
Используемый на данной стадии растворитель может быть протонным или апротонным растворителем или их смесью, а конкретные их примеры включают воду, С1-4 низший спирт, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, ацетонитрил, пропионитрил, бутиронитрил, метилбутират, изопропилацетат, бутилацетат и толуол. Особенно предпочтительными являются диоксан, ацетонитрил и пропионитрил.
Представительные примеры катализатора - кислоты Льюиса, могут представлять собой хлорид индия(III), бромид индия(III), трифлат индия(III), бромид магния, бромид лития, хлорид лития и хлорид олова(IV), а особенно предпочтительными являются бромид лития и хлорид лития. Данный катализатор можно использовать в количестве от 0,01 до 1 молярного эквивалента, предпочтительно от 0,1 до 0,2 молярного эквивалента, из расчета на количество дикарбоксилата ацетона формулы (4).
2,5-дигидрокси-1,4-дитиан можно использовать в количестве от 0,5 до 1 молярного эквивалента из расчета на количество дикарбоксилата ацетона формулы (4).
Данную реакцию можно проводить при температуре в интервале от комнатной температуры до температуры кипения используемого растворителя, предпочтительно от 50°С до температуры кипения растворителя.
<Стадия В>
Затем на стадии В соединение тиофен-диэфира формулы (2а-1), полученное на стадии А, гидролизуют в присутствии основания, получая тиофен-дикарбоновую кислоту формулы (2а-2), соединение формулы (2), в котором R1 и R2 представляют собой CO2R3 и CO2R4 соответственно, а R3 и R4 являются водородом.
Используемый на данной стадии растворитель может представлять собой смесь воды и органического растворителя, такого как С1-4 спирт и ацетон.
Основание может представлять собой гидроксид натрия и гидроксид калия и используется в количестве от 2 до 4 молярных эквивалентов из расчета на соединение тиофен-диэфира формулы (2а-1).
Данную реакцию можно проводить при температуре в интервале от 0°С до температуры кипения используемого растворителя, предпочтительно от комнатной температуры до температуры кипения растворителя.
<Стадия С>
На стадии С производное тиофен-дикарбоновой кислоты формулы (2а-2), полученное на стадии В, восстанавливают восстанавливающим агентом в растворителе, получая тиофен-диол формулы (2b), соединение формулы (2), в котором R1 и R2 представляют собой CH2X и CH2Y, соответственно а Х и Y являются гидроксилами.
Растворитель может предпочтительно представлять собой тетрагидрофуран, а предпочтительный пример восстанавливающего агента представляет собой боран в виде комплекса с диметилсульфидом или тетрагидрофураном, и он может быть использован в количестве от 1 до 10 молярных эквивалентов, предпочтительно от 4 до 8 молярных эквивалентов, из расчета на количество производного тиофен-дикарбоновой кислоты формулы (2а-2).
Данную реакцию можно проводить при температуре в интервале от -20°С до температуры кипения используемого растворителя, предпочтительно от комнатной температуры до температуры кипения растворителя.
<Стадия D>
В качестве альтернативы проведению стадий В и С производное тиофен-диэфира формулы (2а-1), полученное на стадии А, можно непосредственно восстановить действием восстанавливающего агента в растворителе, получая тиофен-диол формулы (2b), как показано на стадии D.
На данной стадии в качестве растворителя можно использовать диэтиловый эфир, тетрагидрофуран, диоксан, н-гексан, бензол и толуол, а особенно предпочтительными являются диэтиловый эфир и тетрагидрофуран.
В качестве восстанавливающего агента можно использовать боргидрид лития, смесь боргидрида натрия и хлорида лития или бромида лития, алюмогидрид лития, а когда используют боргидрид лития или боргидрид натрия, дополнительно можно использовать триметилборат. Алюмогидрид лития можно использовать в количестве от 0,5 до 2 молярных эквивалентов, а боргидрид лития или боргидрид натрия можно использовать в количестве от 1 до 5 молярных эквивалентов из расчета на количество производного тиофен-диэфира формулы (2а-1). Количество используемого хлорида лития или бромида лития может находиться в интервале от 1 до 2 молярных эквивалентов на моль используемого боргидрида натрия. Кроме того, триметилборат можно использовать в количестве от 0,05 до 0,2 молярных эквивалентов на моль используемого боргидрида натрия.
Реакцию можно проводить при температуре в интервале от комнатной температуры до температуры кипения растворителя.
<Стадия Е>
На стадии Е производное тиофен-диола формулы (2b) обрабатывают галогенирующим или сульфонилирующим агентом в растворителе, получая замещенное производное тиофена формулы (2е), соединение формулы (2), в котором R1 и R2 являются CH2X и CH2Y соответственно, а Х и Y представляют собой хлор, бром, метансульфонил или п-толуолсульфонил.
Растворитель, используемый при галогенировании, может представлять собой апротонный растворитель, такой как тетрагидрофуран, диоксан, 1,2-диметоксиэтан, хлористый метилен, хлороформ, 1,2-дихлорэтан, ацетонитрил, пропионитрил и бутиронитрил, из которых предпочтительными являются хлористый метилен, хлороформ и ацетонитрил. Представительные примеры галогенирующего агента включают в себя трифенилдибромфосфоран, трифенилдихлорфосфоран, хлористый тионил, хлористый сульфурил, хлорокись фосфора, трехбромистый фосфор, треххлористый фосфор, пентабромид фосфора и пентахлорид фосфора, из которых предпочтительными являются трифенилдибромфосфоран и трифенилдихлорфосфоран. Галогенирующий агент можно использовать в количестве от 2 до 3 молярных эквивалентов из расчета на производное тиофен-диола формулы (2b).
В реакцию галогенирования можно ввести основание, и его представительные примеры включают в себя пиридин, пиколин, триэтиламин, диизопропилэтиламин и трибутиламин. Основание можно использовать в количестве, достаточном для нейтрализации свободной хлористо-водородой или бромисто-водородной кислоты, образующейся в ходе реакции.
Реакцию галогенирования можно проводить при температуре в интервале от -40°С до температуры кипения используемого растворителя, предпочтительно от 0°С до комнатной температуры.
В случае проведения реакции сульфонилирования растворитель может представлять собой апротонный растворитель, выбранный из тетрагидрофурана, диоксана, 1,2-диметоксиэтана, хлористого метилена, хлороформа, 1,2-дихлорэтана и ацетонитрила или их смеси с водой. Предпочтительными растворителями являются хлористый метилен, хлороформ и 1,2-дихлорэтан. Представительные примеры сульфонилирующего агента могут включать в себя метансульфонилхлорид, бензолсульфонилхлорид, п-толуолсульфонилхлорид и другой замещенный бензолсульфонилхлорид. Сульфонилирующий агент можно использовать в количестве от 1 до 2 молярных эквивалентов из расчета на количество производного тиофен-диола формулы (2b).
В реакции сульфонилирования можно использовать основание, примеры которого включают в себя органическое основание, такое как триэтиламин, диизопропилэтиламин, трибутиламин, пиридин и пиколин, и неорганическое основание, такое как гидрид натрия, гидроксид натрия, гидрокарбонат натрия, карбонат натрия, гидрокарбонат калия, гидроксид калия, карбонат калия, гидрофосфат натрия и гидрофосфат калия. Основание предпочтительно используют в количестве от 1 до 2 молярных эквивалентов из расчета на количество используемого сульфонилирующего агента.
Реакцию сульфонилирования можно ускорить за счет присутствия каталитического количества четвертичной аммониевой соли, такой как хлорид трибутиламмония. Сульфонилирование можно проводить при температуре в интервале от 30°С до температуры кипения используемого растворителя, предпочтительно от -10°С до комнатной температуры.
Кроме того, соединение формулы (2), используемое в качестве промежуточного соединения в способе согласно изобретению, можно получить по схеме реакции 3:
Схема реакции 3
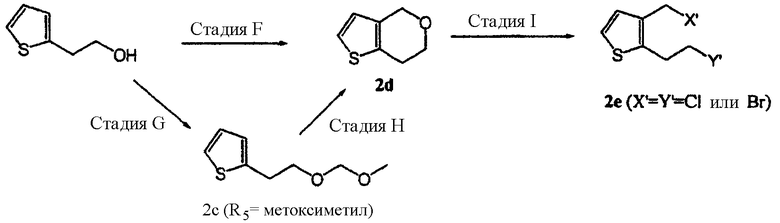
<Стадия F>
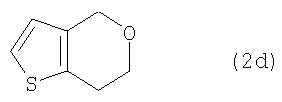
Сначала 2-тиофенэтанол вводят во взаимодействие с формилирующим агентом в растворителе в присутствии кислоты Льюиса, получая 6,7-дигидротиено[3,2-c]пиран формулы (2d), соединение формулы (2), в котором R1 и R2 связаны друг с другом, образуя -СН2-О-СН2-.
В данной реакции растворитель может представлять собой апротонный растворитель, такой как тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 1,2-дихлорэтан, ацетонитрил, пропионитрил и бутиронитрил, из которых предпочтительными являются ацетонитрил и пропионитрил. Представительные примеры кислоты Льюиса включают в себя хлорид индия(III), бромид индия(III), трифлат индия(III), бромид магния и хлорид олова(IV), а особенно предпочтительными являются хлорид индия(III) и бромид магния. Кислоту Льюиса можно использовать в количестве от 0,01 до 1 молярного эквивалента в случае применения соли индия и в количестве от 1 до 5 молярных эквивалентов в случае применения других солей из расчета на количество 2-тиофенэтанола. Кроме того, к реагентам в каталитическом количестве можно добавить п-толуолсульфокислоту.
Формилирующий агент может представлять собой раствор формальдегида, параформ, 1,3-диоксолан, 1,3,5-триоксан, диметоксиметан и диэтоксиметан, из которых предпочтительным является параформ. Его можно использовать в количестве от 1 до 3 молярных эквивалентов из расчета на количество 2-тиофенэтанола.
Реакцию можно проводить при температуре в интервале от комнатной температуры до температуры кипения используемого растворителя, предпочтительно от 50°С до температуры кипения растворителя.
В качестве альтернативы проведению стадии F 2-тиофенэтанол можно обработать производными алкоксиметана, а затем подвергнуть продукт циклизации, получая соединение формулы (2d).
<Стадия G>
На стадии G 2-тиофенэтанол вводят во взаимодействие (i) с низшим алкоксиметаном, таким как диметоксиметан, в присутствии п-толуолсульфокислоты, или (ii) с низшим алкоксиметилгалогенидом, таким как метоксиметилхлорид, этоксиметилхлорид и 2-метоксиэтоксиметилхлорид, в присутствии основания, получая соединение формулы (2с), соединение формулы (2), в котором R1 представляет собой водород, а R2 является CH2OR5, при этом R5 представляет собой метоксиметил.
Реакцию (i) можно проводить в органическом растворителе, таком как бензол, толуол, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, хлороформ, 1,2-дихлорэтан и ацетонитрил, предпочтительно с избытком низшего диалкоксиметана в отсутствие растворителя.
В реакции (i) в каталитическом количестве используют п-толуолсульфокислоту, а для ускорения реакции (i) к реакционной смеси можно прибавить каталитическое количество бромида лития или хлорида лития.
Реакцию можно проводить при температуре в интервале от комнатной температуры до температуры кипения используемого растворителя, предпочтительно от 50°С до температуры кипения растворителя.
Кроме того, реакцию (ii) можно проводить в растворителе, таком как тетрагидрофуран, диоксан, 1,2-диметоксиэтан, хлороформ, 1,2-дихлорэтан и ацетонитрил. Можно использовать алкоксиметилгалогенид в количестве от 1 до 2 молярных эквивалентов из расчета на количество 2-тиофенэтанола.
Основание, которое можно использовать в реакции (ii), может представлять собой органическое основание, такое как триэтиламин, диизопропилэтиламин, трибутиламин, пиридин и пиколин, или неорганическое основание, такое как гидрокарбонат натрия, карбонат натрия, гидрокарбонат калия и карбонат калия. Основание можно использовать в количестве от 1 до 3 молярных эквивалентов из расчета на количество 2-тиофенэтанола.
Реакцию (ii) можно проводить при температуре в интервале от 0°С до температуры кипения используемого растворителя, предпочтительно от комнатной температуры до 70°С.
<Стадия Н>
На стадии Н, на которой происходит циклизация соединения формулы (2с), соединение формулы (2с), полученное на стадии G, циклизуют в растворителе в присутствии кислоты Льюиса, получая дигидротиено[3,2-с]пиран формулы (2d).
Растворитель, используемый на данной стадии, может представлять собой апротонный растворитель, такой как тетрагидрофуран, диоксан, 1,2-диметоксиэтан, хлористый метилен, хлороформ, 1,2-дихлорэтан, ацетонитрил, пропионитрил и бутиронитрил, а предпочтительными растворителями являются хлористый метилен, хлороформ и 1,2-дихлорэтан.
Представительные примеры кислоты Льюиса включают в себя хлорид индия(III), бромид индия(III), трифлат индия(III), бромид магния, бромид лития, хлорид лития и хлорид олова(IV), а особенно предпочтительными являются хлорид индия(III) и бромид магния. Соль индия можно использовать в количестве от 0,01 до 1 молярного эквивалента, а прочие можно использовать в количестве от 1 до 5 молярных эквивалентов из расчета на количество соединения формулы (2с).
Реакцию можно проводить при температуре в интервале от комнатной температуры до температуры кипения используемого растворителя, предпочтительно от 40°С до температуры кипения растворителя.
<Стадия I>
На стадии I дигидротиено[3,2-c]пиран формулы (2d) вводят в реакцию с галогенирующим агентом в присутствии или в отсутствие кислоты Льюиса, получая замещенное производное тиофена формулы (2е), соединение формулы (2), в котором R1 и R2 являются CH2X' и CH2Y', а X' и Y' представляют собой хлор или бром.
Данную реакцию можно проводить в растворителе, который может представлять собой апротонный растворитель, такой как тетрагидрофуран, диоксан, 1,2-диметоксиэтан, хлористый метилен, хлороформ, 1,2-дихлорэтан, ацетонитрил, пропионитрил и бутиронитрил, при этом предпочтительными являются хлористый метилен, хлороформ и ацетонитрил.
Представительные примеры галогенирующего агента включают в себя трифенилдибромфосфоран, трифенилдихлорфосфоран, трехбромистый фосфор, треххлористый фосфор, пентабромид фосфора и пентахлорид фосфора, при этом предпочтительными являются трифенилдибромфосфоран и трифенилдихлорфосфоран. Галогенирующий агент можно использовать в количестве от 1 до 2 молярных эквивалентов из расчета на количество производного тиенопирана формулы (2d).
В случае использования кислоты Льюиса она может представлять собой хлорид цинка или бромид цинка.
Реакцию галогенирования можно проводить при температуре в интервале от -40°С до температуры кипения используемого растворителя, предпочтительно от 0°С до комнатной температуры.
В соответствии с настоящим изобретением соединение замещенного тиофена формулы (2е) вводят во взаимодействие с производным 2-хлорбензиламина формулы (3), или его солью, в растворителе в присутствии основания, получая производное тиено[3,2-с]пиридина формулы (1), показанное на схеме реакции 4:
Схема реакции 4
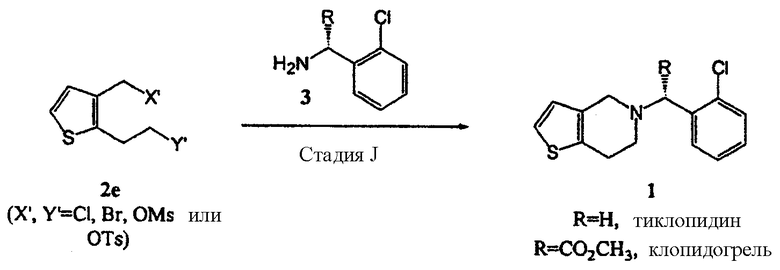
В данной реакции представительными примерами растворителя являются третичные спирты, такие как трет-бутанол и амиловый спирт, простые эфиры, такие как диизопропиловый эфир, тетрагидрофуран и диоксан, нитрилы, такие как ацетонитрил, пропионитрил и бутиронитрил, сложные эфиры, такие как метилацетат, этилацетат и изопропилацетат, углеводороды, N,N-диметилформамид и N,N-диметилацетамид, толуол и диметилсульфоксид, при этом предпочтительными являются ацетонитрил и пропионитрил.
Основание, которое можно использовать в указанной выше реакции, представляет собой органическое основание, такое как триэтиламин, диизопропилэтиламин, трибутиламин, пиридин и пиколин, или неорганическое основание, такое как гидрокарбонат натрия, карбонат натрия, гидрокарбонат калия, карбонат калия, гидрофосфат натрия и гидрофосфат калия. Основание можно предпочтительно использовать в количестве от 2 до 5 молярных эквивалентов из расчета на количество замещенного производного тиофена формулы (2е).
Производное 2-хлорбензиламина формулы (3) или его соль предпочтительно используют в количестве от 1 до 2 молярных эквивалентов из расчета на количество замещенного производного тиофена формулы (2е).
Указанную выше реакцию можно проводить при температуре в интервале от комнатной температуры до температуры кипения используемого растворителя, предпочтительно от 40°С до температуры кипения растворителя.
Производные тиено[3,2-c]пиридина по настоящему изобретению, то есть тиклопидин и клопидогрель, можно легко перевести в гидрохлорид тиклопидина и бисульфат клопидогреля соответственно, которые применимы в качестве активных ингредиентов в противотромбозных лекарственных препаратах.
Следующие далее получения и примеры приведены только в целях иллюстрации и не подразумевают ограничения объема изобретения.
Пример 1: Синтез метил 2-метоксикарбонилметилтиофен-3-карбоксилата (соединение формулы 2а)
100 г диметил 1,3-ацетондикарбоксилата растворяли в 2800 мл диоксана и добавляли 52,5 г 2,5-дигидрокси-1,4-дитиана и 5,0 г бромида лития. Полученную смесь кипятили 15 часов, охлаждали до комнатной температуры и концентрировали упариванием. К остатку добавляли 1000 мл н-гексана, перемешивали и отделяли гексановый слой. Данную процедуру экстракции повторяли дважды, используя порции по 500 мл н-гексана. Отделенные слои н-гексана объединяли и концентрировали при пониженном давлении, получая 60,9 г (выход 49%) указанного в заголовке соединения в виде масла, которое непосредственно использовали на следующей стадии без какой-либо очистки. Аналитический образец указанного в заголовке соединения можно было бы получить перегонкой в вакууме.
Температура кипения: 120˜122°С (0,5 мм рт. ст.)
ЯМР 1H (CDCl3, м.д.): δ 3,73 (c, 3H), 3,83 (c, 3H), 4,22 (c, 2H), 7,15 (д, 1Н, J=5,6 Гц), 7,44 (д, 1Н, J=5,6 Гц)
ЯМР 13С (CDCl3, м.д.): δ 34,7; 52,0; 52,7; 123,7; 129,4; 129,9; 144,3; 164,0; 170,8.
Масс (электронный удар, m/z): 214(M+), 182.
Пример 2: Синтез 2-(3-гидроксиметилтиофен-2-ил)этанола (соединение формулы 2b)
В высушенный сосуд в атмосфере азота загружали 600 мг алюмогидрида лития, а затем добавляли 26 мл безводного тетрагидрофурана. К полученной суспензии примерно в течение 5 минут добавляли раствор 2,8 г соединения, полученного в примере 1, растворенного в 10 мл тетрагидрофурана. Полученную смесь перемешивали при комнатной температуре в течение 1 часа, а затем кипятили 1 час. Полученную реакционную смесь охлаждали до 0°С, а затем медленно по очереди прибавляли 0,5 мл воды, 0,5 мл 15%-ного водного раствора гидроксида натрия и 4 мл воды. Полученную смесь перемешивали примерно в течение 1 часа, а нерастворимые вещества отфильтровывали и промывали 30 мл тетрагидрофурана. Объединенный фильтрат концентрировали упариванием при пониженном давлении, получая 1,8 г (выход 87%) указанного в заголовке маслообразного соединения.
ЯМР 1H (CDCl3, м.д.): δ 2,98 (т, 2H, J=5,7 Гц), 3,69 (т, 2H, J=5,7 Гц), 3,93 (уш.с, 1H), 4,10 (уш.с, 1Н), 4,43 (с, 2Н,), 6,93 (д, 1Н, J=5,1 Гц) 7,08 (д, 1Н, J=5,1 Гц).
ЯМР 13С (CDCl3, м.д.): δ 31,2; 57,7; 63,3; 123,2; 129,4; 138,9; 139,1.
Масс (электронный удар, m/z): 158(M+), 110.
Пример 3: Синтез 2-(3-гидроксиметилтиофен-2-ил)этанола (соединение формулы 2b)
К 800 мл безводного тетрагидрофурана добавляли 31,0 г боргидрида натрия и 71,3 г бромида лития, а затем к нему по каплям прибавляли при 0°C 10,6 мл триметилбората. Полученную смесь перемешивали при комнатной температуре в течение 30 минут, а затем в течение 2 часов медленно прибавляли раствор, полученный растворением 70,4 г соединения, полученного в примере 1, в 200 мл тетрагидрофурана, при кипении образующейся смеси. Реакционную смесь кипятили еще 2 часа, затем охлаждали до комнатной температуры. К данному раствору медленно прибавляли 100 мл безводного метанола, а затем добавляли 700 мл этилового эфира и 700 мл воды. Органическую фазу отбирали, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая 47,1 г (выход 90%) указанного в заголовке соединения в виде масла.
Данные анализа полученного при этом соединения были аналогичны данным из примера 2.
Пример 4: Синтез 2-карбоксиметилтиофен-3-карбоновой кислоты (соединение формулы 2а)
13,0 г соединения, полученного в примере 1, растворяли в 260 мл метанола и прибавляли к нему раствор, полученный при растворении 4,84 г гидроксида натрия в 26 мл воды. Полученный раствор кипятили в течение 4 часов, охлаждали до комнатной температуры, концентрировали при пониженном давлении для удаления метанола. Полученный водный раствор промывали 10 мл диэтилового эфира и подкисляли до рН 2-3 концентрированной HCl. Выпавшие при этом осадки отфильтровывали, промывали небольшим количеством холодной воды и сушили при 40°С, получая 9,60 г (выход 85%) указанного в заголовке соединения в виде коричневого цвета твердого вещества.
Температура плавления: 212˜213°С
ЯМР 1H (CD3COCD3, м.д.): δ 4,26 (c, 2H), 7,39 (д, 1H, J=5,4 Гц), 7,42 (д, 1H, J=5,4 Гц).
Пример 5: Синтез 2-(3-гидроксиметилтиофен-3-ил)этанола (соединение формулы 2b)
0,45 г соединения, полученного в примере 4, растворяли в 10 мл безводного тетрагидрофурана и охлаждали полученный раствор до -20°С. К нему медленно прибавляли 3,7 мл 2 М раствора комплекса боран-диметилсульфид в тетрагидрофуране и давали полученной смеси прореагировать, по меньшей мере, в течение 1 часа при -10°С. Реакционную смесь нагревали до комнатной температуры, а затем добавляли к ней еще 3,7 мл 2 М раствора комплекса боран-диметилсульфид в тетрагидрофуране. Полученную смесь выдерживали при комнатной температуре в течение 2 часов и охлаждали до 0°С. После добавления к ней воды полученный раствор трижды экстрагировали этилацетатом порциями по 15 мл, а органические слои объединяли, промывали 20 мл воды, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая 0,22 г (выход 57%) указанного в заголовке соединения в виде масла.
Данные анализа полученного при этом соединения были аналогичны данным из примера 2.
Пример 6: Синтез 2-(3-гидроксиметилтиофен-2-ил)этанола (соединение формулы 2b)
0,50 г соединения, полученного в примере 4, растворяли в 10 мл безводного тетрагидрофурана и охлаждали полученный раствор до -20°С. К нему медленно прибавляли 4 мл 1,5 М раствора комплекса боран-диметилсульфид в тетрагидрофуране и давали полученной смеси прореагировать, по меньшей мере, в течение 1 часа при -10°С. Реакционную смесь нагревали до комнатной температуры, а затем добавляли к ней еще 4,5 мл 1,5 М раствора комплекса боран-диметилсульфид в тетрагидрофуране. Полученную смесь выдерживали при комнатной температуре в течение 1 часа. Потом повторяли методику примера 5, получая 0,37 г (выход 87%) указанного в заголовке соединения в виде масла.
Данные анализа полученного при этом соединения были аналогичны данным из примера 2.
Пример 7: Синтез 2-(2-бромэтил)-3-бромметилтиофена (соединение формулы 2е)
2,0 г соединения, полученного в примере 2, растворяли в 40 мл хлористого метилена и охлаждали полученный раствор до температуры ниже 5°С. К нему прибавляли 13,4 г трифенилдибромфосфорана и нагревали полученную смесь до комнатной температуры, перемешивали в течение 4 часов и концентрировали упариванием при пониженном давлении. К остатку добавляли 5 мл этилацетата, а затем 15 мл н-гексана. Полученную смесь перемешивали в течение 1 часа при комнатной температуре и в течение 2 часов при температуре ниже 5°С, фильтровали и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (элюент, н-гексан:этилацетат=10:1), получая 2,9 г (выход 80%) указанного в заголовке соединения в виде желтоватого масла.
ЯМР 1H (CDCl3, м.д.): δ 3,38 (т, 2H, J=7,2 Гц), 3,59 (т, 2H, J=7,2 Гц), 4,49 (c, 2H), 7,02 (д, 1Н, J=5,1 Гц), 7,18 (д, 1Н, J=5,1 Гц)
ЯМР 13С (CDCl3, м.д.): δ 25,8; 31,7; 32,0; 124,3; 129,5; 135,2; 139,7.
Масс (электронный удар, m/z): 286, 284, 282, 205, 203.
Пример 8: Синтез 2-(2-бромэтил)-3-бромметилтиофена (соединение формулы 2е)
17,8 г трифенилфосфина растворяли в 40 мл хлористого метилена и охлаждали полученный раствор до температуры ниже 5°С. К нему медленно, в течение 10 минут, прибавляли 10,6 г брома. Полученную смесь перемешивали в течение 30 минут при комнатной температуре и к ней медленно прибавляли раствор 5,0 г соединения, полученного в примере 2, в 20 мл хлористого метилена. Полученную смесь перемешивали при комнатной температуре в течение 4 часов и концентрировали упариванием при пониженном давлении. К остатку добавляли 40 мл этилацетата, а затем 120 мл н-гексана и перемешивали полученную смесь в течение 1 часа при комнатной температуре и в течение 2 часов при температуре ниже 5°С. Нерастворимые вещества отфильтровывали, а фильтрат концентрировали при пониженном давлении, получая 7,8 г (выход 87%) указанного в заголовке соединения в виде желтоватого масла.
Данные анализа полученного при этом соединения были аналогичны данным из примера 7.
Пример 9: Синтез 2-(2-бромэтил)-3-бромметилтиофена (соединение формулы 2е)
10,7 г трифенилфосфина растворяли в 40 мл ацетонитрила и охлаждали полученный раствор до температуры ниже 5°С и медленно, в течение 10 минут, прибавляли к нему 6,4 г брома. Полученную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа, а затем добавляли раствор 3,0 г соединения, полученного в примере 2, в 15 мл ацетонитрила. Полученную смесь перемешивали при комнатной температуре в течение 4 часов и концентрировали упариванием при пониженном давлении. К остатку добавляли 20 мл этилацетата, а затем 40 мл диизопропилового эфира и перемешивали полученную смесь в течение 1 часа при комнатной температуре и в течение 2 часов при температуре ниже 5°С. Нерастворимые вещества отфильтровывали, а фильтрат концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (элюент, н-гексан:этилацетат=10:1), получая 3,5 г (выход 65%) указанного в заголовке соединения в виде желтоватого масла.
Данные анализа полученного при этом соединения были аналогичны данным из примера 7.
Пример 10: Синтез 2-(2-бромэтил)-3-бромметилтиофена (соединение формулы 2е)
5,0 г соединения, полученного в примере 2, растворяли в 95 мл хлороформа и прибавляли к нему 17,1 г трехбромистого фосфора при температуре ниже 5°С. Полученную смесь нагревали до комнатной температуры, перемешивали в течение 15 часов и дважды промывали водой порциями по 100 мл. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (элюент, н-гексан:этилацетат=10:1), получая 4,3 г (выход 48%) указанного в заголовке соединения в виде желтоватого масла.
Данные анализа полученного при этом соединения были аналогичны данным из примера 7.
Пример 11: Синтез 2-(2-хлорэтил)-3-хлорметилтиофена (соединение формулы 2е)
3,0 г соединения, полученного в примере 2, растворяли в 57 мл хлористого метилена и охлаждали полученный раствор до -30°С. К данному раствору прибавляли 6,1 г диизопропилэтиламина и 3,1 г хлористого сульфурила и перемешивали полученную смесь в течение 30 мин при той же температуре и в течение часа при комнатной температуре. К реакционной смеси добавляли 20 мл хлористого метилена и дважды промывали полученную смесь водой порциями по 40 мл. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (элюент, н-гексан:этилацетат=10:1), получая 1,0 г (выход 27%) указанного в заголовке соединения в виде бесцветного масла.
ЯМР 1H (CDCl3, м.д.): δ 3,30 (т, 2H, J=7,3 Гц), 7,27 (т, 2H, J=7,3 Гц), 4,59 (c, 2H), 7,02 (д, 1Н, J=5,2 Гц), 7,18 (д, 1Н, J=5,2 Гц).
ЯМР 13С (CDCl3, м.д.): δ 32,0; 39,4; 45,3; 124,7; 135,7; 139,2.
Масс (электронный удар, m/z): 194(M+), 159, 145.
Пример 12: Синтез 3-хлорметил-2-(2-метансульфонилоксиэтил)тиофена (соединение формулы 2е)
2,0 г соединения, полученного в примере 2, растворяли в 35 мл хлористого метилена и охлаждали полученный раствор до температуры ниже 5°С. К данному раствору прибавляли 3,0 г метансульфонилхлорида и 4,1 г диизопропилэтиламина и перемешивали полученную смесь в течение 30 минут. К реакционной смеси добавляли 30 мл хлористого метилена и дважды промывали полученную смесь водой порциями по 30 мл. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая 2,9 г (выход 91%) указанного в заголовке соединения в виде масла.
ЯМР 1H (CDCl3, м.д.): δ 3,03 (с, 3Н), 3,38 (т, 2H, J=6,6 Гц), 4,05 (т, 2H, J=6,6 Гц), 4,66 (c, 2H), 7,11 (д, 1Н, J=5,2 Гц), 7,27 (д, 1Н, J=5,2 Гц).
ЯМР 13С (CDCl3, м.д.): δ 28,1; 37,7; 38,9; 69,8; 124,6; 129,4; 135,6; 136,8.
Пример 13: Синтез 3-хлорметил-2-(2-п-толуолсульфонилоксиэтил)тиофена (соединение формулы 2е)
1,5 г соединения, полученного в примере 2, растворяли в 40 мл хлористого метилена и охлаждали полученный раствор до температуры ниже 5°С. К данному раствору прибавляли по каплям 3,6 г п-толуолсульфонилхлорида и 1,9 г триэтиламина и нагревали полученную смесь до комнатной температуры, перемешивали в течение 6 часов и дважды промывали водой порциями по 20 мл. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (элюент, н-гексан:этилацетат=10:1), получая 1,5 г (выход 48%) указанного в заголовке соединения в виде масла.
ЯМР 1H (CDCl3, м.д.): δ 2,41 (с, 3Н), 3,16 (т, 2H, J=6,8 Гц), 4,18 (т, 2H, J=6,8 Гц), 4,46 (c, 2H), 6,94 (д, 1Н, J=5,2 Гц), 7,10 (д, 1Н, J=5,2 Гц), 7,29 (д, 2Н, J=8,2 Гц), 7,71 (д, 2Н, J=8,2 Гц).
ЯМР 13С (CDCl3, м.д.): δ 22,0; 27,9; 38,8; 70,1; 124,4; 128,2; 129,2; 130,3; 133,0; 135,5; 136,7; 145,9.
Пример 14: Синтез 6,7-дигидро-4Н-тиено[3,2-c]пирана (соединение формулы 2d)
К 1500 мл ацетонитрила прибавляли 20,0 г 2-тиофенэтанола, 6,1 г параформа и 1,7 г хлорида индия(III) и кипятили полученную смесь в течение 2 часов. Полученный раствор охлаждали до комнатной температуры и концентрировали упариванием при пониженном давлении. Полученный при этом остаток перегоняли при пониженном давлении, получая 14,4 г (выход 66%) указанного в заголовке соединения в виде масла.
Температура кипения: 88˜90°С (8 мм рт. ст.)
ЯМР 1H (CDCl3, м.д.): δ 2,88 (т, 2Н, J=5,5 Гц), 3,97 (т, 2H, J=5,5 Гц), 4,73 (с, 2H), 6,73 (д, 1H, J=5,1 Гц), 7,11 (д, 1Н, J=5,1 Гц).
ЯМР 13С (CDCl3, м.д.): δ 26,3; 65,7; 67,3; 123,5; 124,4; 133,0; 134,5.
Масс (электронный удар, m/z) 140(M+), 110.
Пример 15: Синтез 6,7-дигидро-4Н-тиено[3,2-c]пирана (соединение формулы 2d)
К 780 мл ацетонитрила прибавляли 10,0 г 2-тиофенэтанола, 2,8 г параформа и 2,2 г хлорида индия(III) и кипятили полученную смесь в течение 12 часов. Раствор продукта реакции охлаждали до комнатной температуры и концентрировали упариванием при пониженном давлении. Остаток перегоняли при пониженном давлении, получая 4,7 г (выход 43%) указанного в заголовке соединения, данные анализа которого были аналогичны данным, полученным в примере 14.
Пример 16: Синтез 6,7-дигидро-4Н-тиено[3,2-c]пирана (соединение формулы 2d)
К 780 мл ацетонитрила прибавляли 10,0 г 2-тиофенэтанола, 5,9 г параформа и 28,7 г бромида магния и кипятили полученную смесь в течение 24 часов. Раствор продукта реакции охлаждали до комнатной температуры и концентрировали упариванием при пониженном давлении. К остатку добавляли 100 мл волы и 150 мл н-гексана и перемешивали полученную смесь в течение 5 минут. Органический слой отделяли и сушили над безводным сульфатом магния, а затем концентрировали при пониженном давлении. Остаток перегоняли при пониженном давлении, получая 4,7 г (выход 43%) указанного в заголовке соединения, данные анализа которого были аналогичны данным, полученным в примере 14.
Пример 17: Синтез 6,7-дигидро-4Н-тиено[3,2-c]пирана (соединение формулы 2d)
К 1500 мл бутиронитрила прибавляли 10,0 г 2-тиофенэтанола, 3,5 г 1,3,5-триоксана и 862 мг хлорида индия(III) и кипятили полученную смесь в течение 6 часов. Раствор продукта реакции охлаждали до комнатной температуры и концентрировали упариванием при пониженном давлении. Остаток перегоняли при пониженном давлении, получая 6,6 г (выход 60%) указанного в заголовке соединения, данные анализа которого были аналогичны данным, полученным в примере 14.
Пример 18: Синтез 6,7-дигидро-4Н-тиено[3,2-c]пирана (соединение формулы 2d)
К 780 мл ацетонитрила прибавляли 10,0 г 2-тиофенэтанола, 11,5 г 1,3-диоксолана и 862 мг хлорида индия(III) и кипятили полученную смесь в течение 10 часов. Раствор продукта реакции охлаждали до комнатной температуры и концентрировали упариванием при пониженном давлении. К остатку добавляли 100 мл воды и 150 мл н-гексана и перемешивали полученную смесь в течение 5 минут. Органический слой отделяли и сушили над безводным сульфатом магния, а затем концентрировали при пониженном давлении. Остаток перегоняли при пониженном давлении, получая 6,6 г (выход 60%) указанного в заголовке соединения, данные анализа которого были аналогичны данным, полученным в примере 14.
Пример 19: Синтез 2-(2-метоксиметоксиэтил)тиофена (соединение формулы 2с)
В 2250 мл диметоксиметана растворяли 150 г 2-тиофенэтанола и добавляли к нему 22,3 г п-толуолсульфокислоты и 20,3 г хлорида лития. Полученную смесь кипятили в течение 5 часов и концентрировали упариванием при пониженном давлении. К остатку добавляли 1000 мл воды и 1500 мл н-гексана и перемешивали полученную смесь в течение 5 минут. Водный слой отбрасывали, а органический слой дважды промывали водой порциями по 700 мл, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая 193 г (выход 96%) указанного в заголовке соединения в виде бесцветного масла.
ЯМР 1H (CDCl3, м.д.): δ 3,15 (т, 2Н, J=6,6 Гц), 3,37 (c, 3H), 3,80 (т, 2H, J=6,6 Гц), 4,67 (с, 2H), 6,87-6,93 (м, 1Н), 6,96 (дд, 1Н, J=5,1; 3,4 Гц), 7,17 (дд, 1Н, J=5,1; 1,0 Гц).
Пример 20: Синтез 2-[2-(2-метоксиэтоксиметокси)этил]тиофена (соединение формулы 2с)
15,0 г 2-тиофенэтанола растворяли в 350 мл хлористого метилена и прибавляли к нему 30,2 г диизопропилэтиламина. Полученную смесь охлаждали до 0°С и добавляли к ней 17,5 г 2-метоксиэтоксиметилхлорида. Полученную смесь перемешивали в течение 2 часов и кипятили в течение 2 часов. Раствор продукта реакции охлаждали и концентрировали упариванием при пониженном давлении. К остатку добавляли 200 мл воды и 250 мл н-гексана и перемешивали полученную смесь в течение 5 минут. Водный слой отбрасывали, а органический слой дважды промывали водой порциями по 700 мл, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая 21,5 г (выход 85%) указанного в заголовке соединения в виде бесцветного масла.
ЯМР 1H (CDCl3, м.д.): δ 3,07 (т, 2Н, J=6,6 Гц), 3,34 (с, 3H), 3,44-3,52 (м, 2H), 3,58-3,64 (м, 2H), 3,76 (т, 2Н, J=6,6 Гц) 4,70 (с, 2Н), 6,80-6,83 (м, 1Н), 6,88 (дд, 1Н, J=5,1; 3,4 Гц), 7,09 (дд, 1Н, J=5,1; 1,1 Гц).
Пример 21: Синтез 6,7-дигидро-4Н-тиено[3,2-c]пирана (соединение формулы 2d)
К 850 мл хлористого метилена прибавляли 61,2 г 2-(2-метоксиметоксиэтил)тиофена, полученного в примере 19, и 130,8 г бромида магния и кипятили полученную смесь в течение 2 часов. Раствор продукта реакции охлаждали до комнатной температуры и концентрировали упариванием при пониженном давлении. К остатку добавляли 500 мл воды и 800 мл н-гексана и перемешивали полученную смесь в течение 5 минут. Водный слой отбрасывали, а органический слой дважды промывали водой порциями по 300 мл, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток перегоняли при пониженном давлении, получая 40,7 г (выход 82%) указанного в заголовке соединения, данные анализа которого были аналогичны данным, полученным в примере 14.
Пример 22: Синтез 6,7-дигидро-4Н-тиено[3,2-c]пирана (соединение формулы 2d)
К 750 мл ацетонитрила прибавляли 11,0 г 2-[2-(2-метоксиэтоксиметокси)этил]тиофена, полученного в примере 20, и 1,1 г хлорида индия(III) и кипятили полученную смесь в течение 5 часов. Раствор продукта реакции охлаждали до комнатной температуры и концентрировали упариванием при пониженном давлении. К остатку добавляли 100 мл воды и 150 мл н-гексана и перемешивали полученную смесь в течение 5 минут. Водный слой отбрасывали, а органический слой дважды промывали водой порциями по 70 мл, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток перегоняли при пониженном давлении, получая 2,9 г (выход 41%) указанного в заголовке соединения, данные анализа которого были аналогичны данным, полученным в примере 14.
Пример 23: Синтез 2-(2-бромэтил)-3-бромметилтиофена (соединение формулы 2е)
К раствору 9,5 г трифенилдибромфосфорана в 50 мл ацетонитрила прибавляли 2 г 6,7-дигидро-4Н-тиено[3,2-с]пирана, полученного в примере 14, и кипятили полученную смесь в течение 8 часов. Раствор продукта реакции концентрировали упариванием при пониженном давлении. К остатку добавляли 5 мл этилацетата, а затем 15 мл н-гексана. Полученную смесь перемешивали в течение 1 часа при комнатной температуре и в течение 2 часов при температуре ниже 5°С, фильтровали и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (элюент, н-гексан:этилацетат=10:1), получая 3,3 г (выход 80%) указанного в заголовке соединения в виде желтоватого масла, данные анализа которого были аналогичны данным, полученным в примере 7.
Пример 24: Синтез 5-[(2-хлорфенил)метил]-4,5,6,7-тетрагидротиено[3,2-c]пиридина (соединение формулы 1а; тиклопидин)
При температуре ниже 5°С 5,0 г 2-(2-бромэтил)-3-бромметилтиофена, полученного в примере 7, растворяли в 50 мл ацетонитрила и добавляли к нему раствор, полученный при растворении 2,7 г 2-хлорбензиламина и 6,8 г диизопропилэтиламина в 25 мл ацетонитрила. Полученную смесь кипятили в течение 5 часов и концентрировали упариванием при пониженном давлении. Остаток растворяли в 100 мл этилацетата и дважды промывали водой порциями по 70 мл. Органический слой промывали 50 мл насыщенного водного раствора хлорида натрия и концентрировали при пониженном давлении. Полученный при этом маслообразный остаток темно-желтого цвета подвергали колоночной хроматографии на силикагеле (элюент, н-гексан:этилацетат=5:1), получая 3,6 г (выход 78%) указанного в заголовке соединения в виде желтоватого масла.
ЯМР 1H (CDCl3, м.д.): δ 2,87-2,91 (м, 4Н), 3,66 (с, 2Н), 3,85 (с, 2Н), 6,73 (д, 1Н, J=5,0 Гц), 7,09 (д, 1H, J=0,5 Гц), 7,19-7,29 (м, 2H), 7,35-7,42 (м, 1H), 7,52-7,61 (м, 1Н).
Пример 25: Синтез метил (S)-(+)-α-(о-хлорфенил)-6,7-дигидротиено[3,2-c]пиридин-5(4Н)ацетата (соединение формулы 1b, клопидогрель)
В 50 мл ацетонитрила растворяли 5,0 г 2-(2-бромэтил)-3-бромметилтиофена, полученного в примере 7, и прибавляли к нему раствор, полученный при растворении 4,6 г гидрохлорида метилового эфира (S)-(+)-2-хлорфенилглицина и 6,8 г диизопропилэтиламина в 20 мл ацетонитрила в течение 30 минут. Полученную смесь кипятили в течение 8 часов и концентрировали упариванием при пониженном давлении. Остаток растворяли в 100 мл этилацетата и дважды промывали водой порциями по 70 мл. Органический слой промывали 50 мл насыщенного водного раствора хлорида натрия и концентрировали при пониженном давлении. Остаток растворяли в 100 мл этилацетата и дважды промывали водой порциями по 70 мл. Полученный при этом маслообразный остаток темно-желтого цвета подвергали колоночной хроматографии на силикагеле (элюент, н-гексан:этилацетат=5:1), получая 5,0 г (выход 88%) указанного в заголовке соединения в виде желтоватого масла.
ЯМР 1H (CDCl3, м.д.): δ 2,89 (с, 4Н), 3,60-3,78 (м, 2Н), 3,73 (с, 3Н), 4,93 (с, 1Н), 6,67 (д, 1H, J=5,1 Гц), 7,06 (д, 1Н, J=5,1 Гц), 7,26-7,30 (м, 2H), 7,37-7,45 (м, 1H), 7,68-7,77 (м, 1Н).
Пример 26: Синтез метил (S)-(+)-α-(о-хлорфенил)-6,7-дигидротиено[3,2-c]пиридин-5(4Н)ацетата (соединение формулы 1b, клопидогрель)
Повторяли методику примера 25, за исключением того, что вместо ацетонитрила в качестве растворителя использовали трет-бутанол, получая указанное в заголовке соединение с выходом 85%.
Примеры 27 и 28: Синтез метил (S)-(+)-α-(о-хлорфенил)-6,7-дигидротиено[3,2-c]пиридин-5(4Н)ацетата (соединение формулы 1b, клопидогрель)
Повторяли методику примера 25, за исключением того, что вместо диизопропилэтиламина в качестве основания использовали триэтиламин и карбонат калия, соответственно, получая указанное в заголовке соединение с выходом 41% и 78% соответственно.
Примеры с 29 по 31: Синтез метил (S)-(+)-α-(о-хлорфенил)-6,7-дигидротиено[3,2-c]пиридин-5(4Н)ацетата (соединение формулы 1b, клопидогрель)
Повторяли методику примера 25, за исключением того, что вместо 2-(2-бромэтил)-3-бромметилтиофена, полученного в примере 7, в качестве исходного вещества использовали 2-(2-хлорэтил)-3-хлорметилтиофен, полученный в примере 12, 2-(2-метансульфонилоксиэтил)тиофен, полученный в примере 13, и 2-(2-п-толуолсульфонилоксиэтил)тиофен, полученный в примере 14, соответственно, получая указанное в заголовке соединение с выходом 85%, 79% и 58% соответственно.
Пример 32: Синтез метил (S)-(+)-α-(о-хлорфенил)-6,7-дигидротиено[3,2-c]пиридин-5(4Н)ацетата (соединение формулы 1b, клопидогрель)
К раствору 47,5 г трифенилдибромфосфорана в 250 мл ацетонитрила прибавляли 10 г 6,7-дигидро-4Н-тиено[3,2-с]пирана. Полученную смесь кипятили 24 часа, а затем прибавляли к ней по каплям раствор, полученный при растворении 14 г гидрохлорида метилового эфира (S)-(+)-2-хлорфенилглицина и 36 мл диизопропилэтиламина в 100 мл ацетонитрила в течение 30 минут, кипятя в то же время полученную смесь. Спустя 8 часов раствор продукта реакции концентрировали упариванием при пониженном давлении. К полученному при этом остатку добавляли 50 мл этилацетата и 150 мл н-гексана и отфильтровывали выпавшие при этом твердые вещества. Фильтрат дважды промывали водой порциями по 150 мл, а затем 50 мл насыщенного раствора хлорида натрия. Органическую фазу пропускали через слой активированного угля и концентрировали при пониженном давлении, получая 18,6 г (выход 81%) указанного в заголовке соединения в виде желтого масла.
Несмотря на то что варианты осуществления связанного изобретения были описаны и проиллюстрированы, очевидно, что в нем можно производить различные изменения, не выходя за рамки настоящего изобретения, которое может быть ограничено только рамками прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КЛОПИДОГРЕЛА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В СПОСОБЕ | 2005 |
|
RU2357970C1 |
| АКТИВАТОРЫ ГЛЮКОКИНАЗЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2008 |
|
RU2450001C2 |
| СОЕДИНЕНИЯ С ГИДРОКСИКАРБОНИЛЬНЫМИ-ГАЛОГЕНАЛКИЛЬНЫМИ БОКОВЫМИ ЦЕПЯМИ | 2000 |
|
RU2247106C2 |
| ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2326864C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭЗОМЕПРАЗОЛА И ЕГО СОЛЕЙ | 2006 |
|
RU2382777C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2711502C2 |
| НОВОЕ ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2009 |
|
RU2470934C1 |
| СПОСОБ ПОЛУЧЕНИЯ КЛОПИДОГРЕЛЯ И ЕГО ПРОИЗВОДНЫХ | 2009 |
|
RU2469039C2 |
| НОВЫЕ БЕНЗАМИДНЫЕ ПРОИЗВОДНЫЕ | 2011 |
|
RU2536688C2 |
Изобретение относится к новому способу получения производного тиено[3,2-с]пиридина формулы (1) (тиклопидина и клопидогреля), включающему взаимодействие соединения формулы (2е) с соединением формулы (3), или его солью:
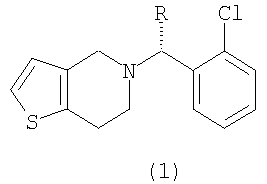
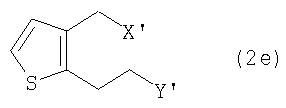
в которых R представляет собой водород или метоксикарбонил, а каждый из X' и Y' независимо представляет собой хлор, бром, метансульфонил или п-толуолсульфонил, а также к новым промежуточным соединениям и способам их получения. Тиклопидин и клопидогрель обладают высокой ингибирующей активностью в отношении агрегации тромбоцитов крови и противотромбической активностью. Технический результат - упрощение процесса. 6 н. и 9 з.п. ф-лы.
в которых R представляет собой водород или метоксикарбонил, а
каждый из X' и Y' независимо представляет собой хлор, бром, метансульфонил, или п-толуолсульфонил.
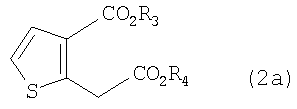
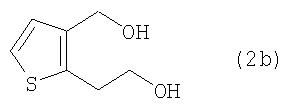
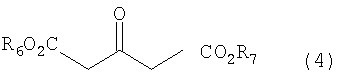
в которых каждый из R3 и R4 независимо представляет собой водород, или линейный или разветвленный С1-6алкил, а
каждый из R6 и R7 независимо представляет собой линейный или разветвленный С1-6алкил.
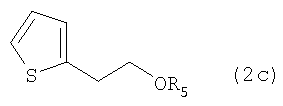
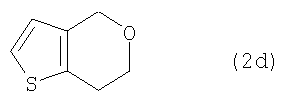
в которых R5 представляет собой С1-4алкоксиметил.
в которой каждый из R3 и R4 независимо представляет собой водород, или линейный или разветвленный С1-6алкил.
в которой R5 представляет собой С1-4алкоксиметил.
в которой каждый из X' и Y' независимо представляет собой хлор, бром, метансульфонил или п-толуолсульфонил.
| WO 02059128 А2, 01.08.2002 | |||
| WO 9851689 A1, 19.11.1998 | |||
| US 5204469 А, 20.04.1993 | |||
| СПОСОБ ПОЛУЧЕНИЯ РАЦЕМИЧЕСКИХ ИЛИ ОПТИЧЕСКИ АКТИВНЫХ МЕТИЛ(2-ГАЛОГЕНОФЕНИЛ)(6,7-ДИГИДРО-4Н-ТИЕНО[3,2-С]ПИРИДИН-5-ИЛ)АЦЕТАТОВ ИЛИ ИХ СОЛЕЙ | 1998 |
|
RU2172315C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ИНДИВИДУАЛЬНОЙ ХИМОЧУВСТВИТЕЛЬНОСТИ ОПУХОЛИ У БОЛЬНЫХ IN VITRO | 1994 |
|
RU2094802C1 |
| Приспособление для непрерывной очистки кардного покрова барабана чесальной машины | 1933 |
|
SU43397A1 |
| Устройство для непрерывного автоматического измерения среднего интервала времени между моментами выдачи изделий из проходной печи | 1972 |
|
SU555153A1 |

