Область техники
Настоящее изобретение относится к новому бензамидному производному формулы 1, которое проиллюстрировано здесь и далее, или к его фармацевтически приемлемой соли, способу получения и к агонисту 5-HT4 рецептора, содержащему их в качестве активного ингредиента.
Предпосылки изобретения
Серотонин (5-HT) представляет собой нейромедиатор, широко распределенный по организму. В настоящее время известны семь подтипов серотонина. В частности, большое внимание было сосредоточено на исследовании 5-HT4 рецептора и подтверждении его фармацевтического действия.
Обычно, агонисты 5-HT4 рецептора оказываются терапевтически эффективными для лечения различных заболеваний, таких как гастроэзофагеальная рефлюксная болезнь, желудочно-кишечное заболевание, расстройство перистальтики желудка, неязвенная диспепсия, функциональная диспепсия, синдром раздраженной толстой кишки (IBS), констипация, диспепсия, эзофагит, желудочно-пищеводное заболевание, тошнота, заболевание центральной нервной системы, болезнь Альцгеймера, когнитивное нарушение, рвота, мигрень, неврологическое заболевание, боль, сердечно-сосудистые расстройства, сердечная недостаточность, сердечная аритмия, диабет и синдром апноэ (См., Tips, 13, 141; Ford A.P.D.W. et al., Med. Res. Rev., 1993, 13. 633; Gullikson G.W. et al., Drug Dev. Res., 1992, 26, 405; Richard M. Eglen et al., Tips, 1995, 16, 391; Bockaert J. et al., CNS Drugs, 1, 6; Romanelli M.N. et al., Arzheim Forsch./Drug Res., 43, 913; Kaumann A. et al., Naunyn-Schmiedeberg's. 1991, 344, 150; и Romanelli M.N. et al., Arzheim Forsch./Drug Res., 1993, 43, 913).
Несмотря на широкое использование агонистов 5-HT4 рецептора, существуют немного соединений агонистов 5-HT4 рецептора, которые в настоящее время используются клинически. В связи с этим существует необходимость в агонистах 5-HT4 рецептора, которые способны проявлять отличное лечебное действие, имея минимальные побочные эффекты.
Бензамидные производные обладают несколькими известными фармакологическими действиями. Эта превосходная фармакологическая активность бензамидных производных обусловлена их воздействием на нервную систему, которая контролируется серотонином, являющимся нейромедиатором. Роль серотонина, то есть фармакологическое действие бензамидных производных были широко вовлечены в различные заболевания и состояния на протяжении многих лет. Кроме того, большая часть изучений и исследований была сосредоточена на продукцию и участки хранения серотонина, а также участки нахождения рецепторов серотонина, чтобы определить связь между положением рецепторов серотонина и различными болезненными состояниями или состояниями у людей.
Цизаприд, который является типичным агонистом 5-HT4 рецептора, представляет собой одно из бензамидных производных. В патентах США №№ 4962115, 5057525 и 5137896 описаны N-(3-гидрокси-4-пиперидинил)бензамиды, включая цизаприд. Указанные соединения известны как стимулирующие желудочно-кишечную моторику. Кроме того, бензамидные производные описаны в патенте США № 5864039.
С этой целью авторам настоящего изобретения удалось синтезировать новые бензамидные производные, которые обладают агонистической активностью благодаря сильному связыванию с 5-HT4 рецептором и хорошему желудочно-кишечному всасыванию и которые способны свести к минимуму побочные эффекты. Настоящее изобретение было создано на основе этого открытия.
Раскрытие изобретения
Техническая задача
Настоящее изобретение касается нового бензамидного производного или его фармацевтически приемлемой соли и их способа получения.
Кроме того, настоящее изобретение касается агониста 5-HT4 рецептора, содержащего в качестве активного ингредиента новое бензамидное производное или его фармацевтически приемлемую соль или гидрат.
Решение задачи
Настоящее изобретение касается нового бензамидного производного, представленного формулой 1 (соединение, представленное формулой 1):
[Формула 1]

где m обозначает целое число от 1 до 10; и Q представляет собой гетероароматическое кольцо или фенил, где гетероароматическое кольцо или фенил независимо замещены 0, 1, 2 или 3 заместителями, выбранными из алкила, алкокси, гидрокси, циано, нитро и галогена; или его фармацевтически приемлемой соли.
Если не указано иного, в данном описании следующие термины имеют следующие значения.
Как здесь используется, термин "алкил" относится к линейному или разветвленному моновалентному насыщенному C1-C20 углеводородному радикалу, содержащему только атомы углерода и атомы водорода. Примеры алкильного радикала включают метил, этил, пропил, изопропил, 2,2-диметилпропил, бутил, изобутил, втор-бутил, трет-бутил, 3-метилбутил, пентил, 3-метилпентил, 4-метилпентил, н-гексил, 2-этилгексил, октил и додецил.
Как здесь используется, термин "алкокси" относится к радикалу -OR, где R представляет собой алкил, как описано выше. Примеры алкокси радикала включают метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, пентокси, 3-метилпентокси, 4-метилпентокси, н-гексокси и 2-этилгексокси.
Как здесь используется, термин "гетероароматическое кольцо" относится к ароматическому кольцу или бициклическому ароматическому кольцу, содержащему от 1 до 4 гетороатомов, выбранных из O, N или S. Примеры гетероароматического кольца включают пиррол, имидазол, триазол, тетразол, пиридин, пиримидин, оксазол, оксадиазол, изоксазол, индол, хинолин и бензофуран.
Далее, настоящее изобретение относится к новому бензамидному производному, представленному формулой 1, где m обозначает целое число от 1 до 5; и Q представляет собой гетероароматическое кольцо или фенил, где гетероароматическое кольцо или фенил независимо замещены 0, 1, 2 или 3 заместителями, выбранными из C1-C4 алкила, C1-C4 алкокси, гидрокси и галогена, где гетероароматическое кольцо представляет собой C1-C12 ароматическое кольцо или C1-C12 бициклическое ароматическое кольцо, независимо содержащее от 1 до 4 гетороатомов, выбранных их N, О или S; или его фармацевтически приемлемой соли.
В настоящем изобретении, фармацевтически приемлемая соль может представлять собой кислотно-аддитивную соль с приемлемой свободной кислотой. Свободная кислота может быть неорганической или органической кислотой. Примеры неорганической кислоты включают хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту и фосфорную кислоту. Примеры органической кислоты включают лимонную кислоту, уксусную кислоту, молочную кислоту, малеиновую кислоту, фумаровую кислоту, глюконовую кислоту, метансульфоновую кислоту, гликолевую кислоту, янтарную кислоту, 4-толуолсульфоновую кислоту, трифторуксусную кислоту, галактуроновую кислоту, эмбоновую кислоту, глютаминовую кислоту и аспарагиновую кислоту.
Кроме того, соединение формулы 1 или его фармацевтически приемлемая соль могут проявлять полиморфизм и могут быть также представлены в виде сольвата (например, гидрат и т.д.).
Далее, настоящее изобретение относится к новому бензамидному производному, выбранному из группы, состоящей из следующих соединений:
(1) N-((1-(3-(1,2,4-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(2) N-((1-(3-(тетразол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(3) N-((1-(3-(индол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(4) N-((1-(3-(2-метилимидазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(5) N-((1-(5-(индол-1-ил)пентил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(6) N-((1-(5-(1,2,3-триазол-1-ил)пентил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(7) N-((1-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(8) N-((1-(3-(1,2,3-триазол-2-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(9) N-((1-(пиридин-3-илметил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(10) N-((1-((1-метилиндол-3-ил)метил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(11) N-((1-(имидазол-2-илметил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(12) N-((1-((1-метилпиррол-2-ил)метил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(13) N-((1-(4-фторбензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(14) N-((1-(4-гидроксибензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(15) N-((1-(2-(индол-3-ил)этил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(16) гидрохлорид N-((1-(3-(тетразол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(17) гидрохлорид N-((1-(5-(1,2,3-триазол-1-ил)пентил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(18) гидрохлорид N-((1-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(19) гидрохлорид N-((1-((1-метилиндол-3-ил)метил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(20) гидрохлорид N-((1-(4-фторбензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(21) гидрохлорид N-((1-(4-гидроксибензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида, и его фармацевтически приемлемой соли.
Далее, настоящее изобретение относится к способу получения бензамидного производного формулы 1 или его фармацевтически приемлемой соли.
Настоящее изобретение относится к способу получения соединения формулы 1 или его фармацевтически приемлемой соли, который включает взаимодействие соединения формулы 2 или его фармацевтически приемлемой соли с соединением формулы 3 в присутствии основания для введения соединения формулы 3 на амин в 1-м положении пиперидинового кольца соединения формулы 2 или его фармацевтически приемлемой соли, получая таким образом соединение формулы 1 (здесь и далее, на способ ссылаются как на "способ получения 1").
[Формула 1]

[Формула 2]
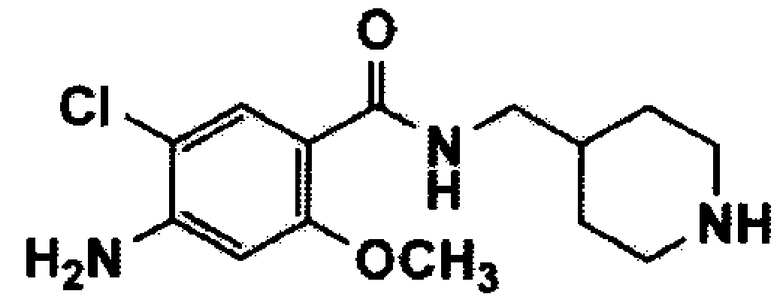
[Формула 3]
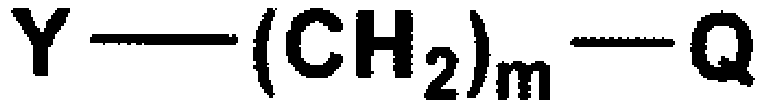
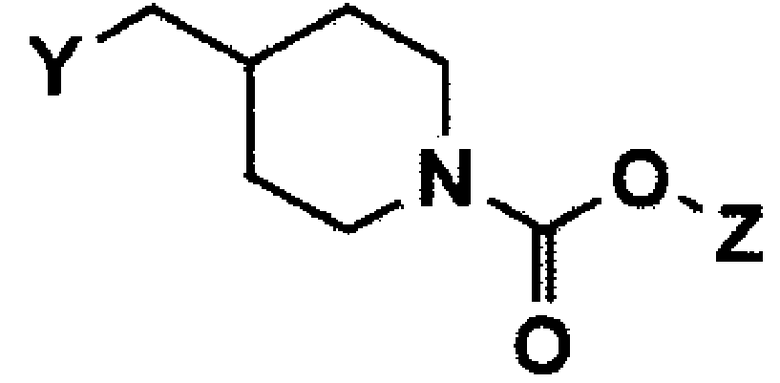
В вышеуказанных формулах m и Q имеют значения, указанные для формулы 1, и Y представляет собой атом галогена или C1-C4 алкилсульфонат.
В способе получения 1 по настоящему изобретению, основание, предпочтительно, выбрано из карбоната калия, йодида калия, триэтиламина, диизопропилэтиламина и их смеси, растворитель может представлять собой диметилформамид, диметилацетамид, ацетон, 1,4-диоксан или тому подобное, и реакция может быть осуществлена при температуре от 50°C до 140°C.
Настоящее изобретение относится к способу получения соединения формулы 1 или его фармацевтически приемлемой соли, который включает взаимодействие соединения формулы 2 или его фармацевтически приемлемой соли с соединением формулы 11 в присутствии восстанавливающего агента с получением соединения формулы 1 (здесь и далее, на способ ссылаются как на "способ получения 2").
[Формула 1]
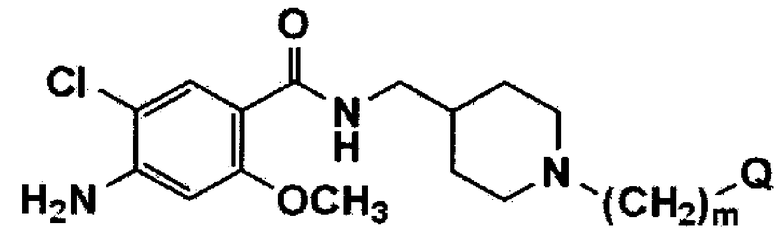
[Формула 2]
[Формула 11]
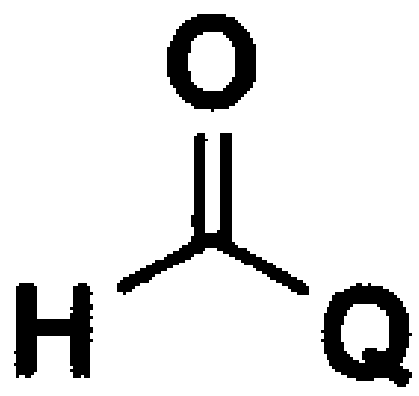
В вышеуказанных формулах Q имеет значения, указанные для формулы 1, и m обозначает целое число 1.
В способе получения 2 по настоящему изобретению восстанавливающим агентом, предпочтительно, является цианборгидрид натрия и уксусная кислота, или боргидрид натрия, растворителем может быть C1-C6 низший спирт, предпочтительно, этанол или метанол, и реакция может быть осуществлена при температуре от 50°C до 100°C.
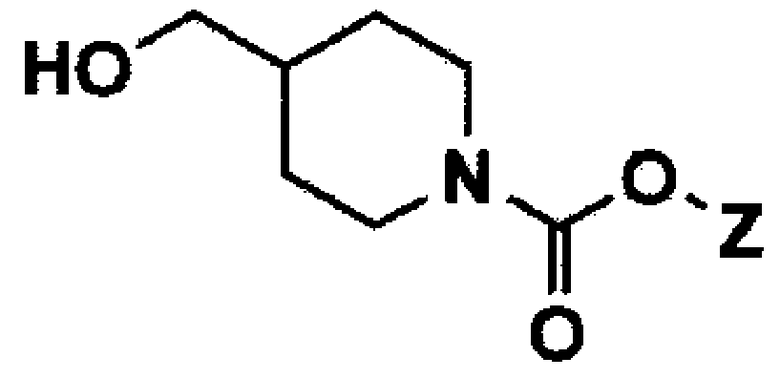
Соединение формулы 2 или его фармацевтически приемлемая соль в способе получения 1 или способе получения 2 по настоящему изобретению может быть получено с помощью следующих стадий:
(1) взаимодействие соединения формулы 4 с реагентом для введения аминозащитной группы по амину в 1-м положении пиперидинового кольца соединения формулы 4, с получением таким образом соединения формулы 5;
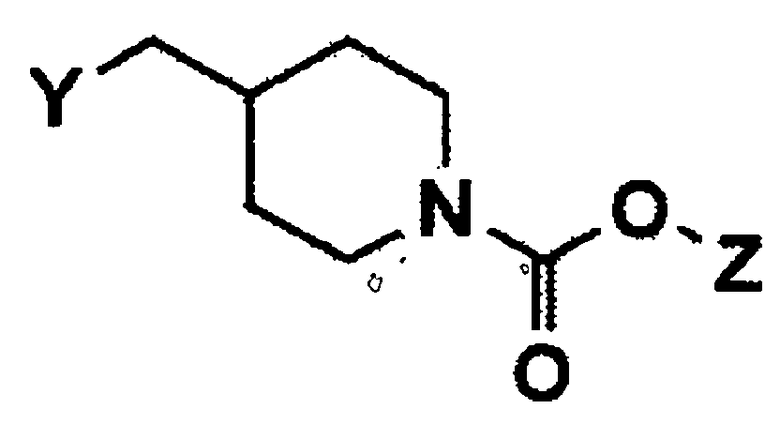
(2) взаимодействие гидрокси соединения формулы 5 с N-бромсукцинимидом и тетрабромидом углерода, или с C1-C4 алкилсульфонилгалогенидом в присутствии основания с получением соединения формулы 6;
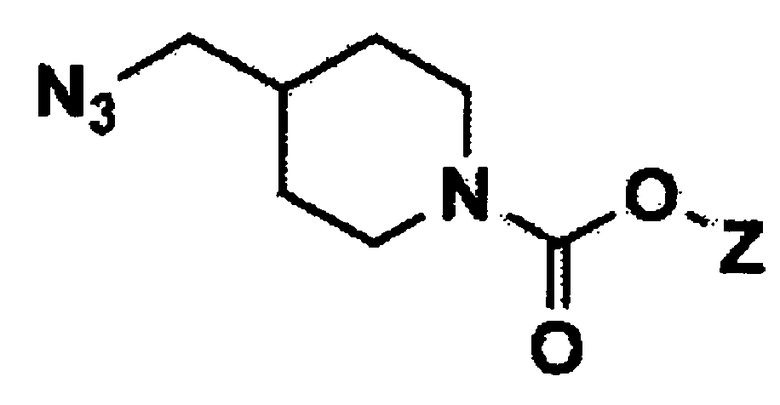
(3) взаимодействие заместителя Y соединения формулы 6 с азидом натрия с получением соединения формулы 7;
(4) восстановление азидо заместителя соединения формулы 7 до амина в присутствии восстановителя с получением соединения формулы 8;
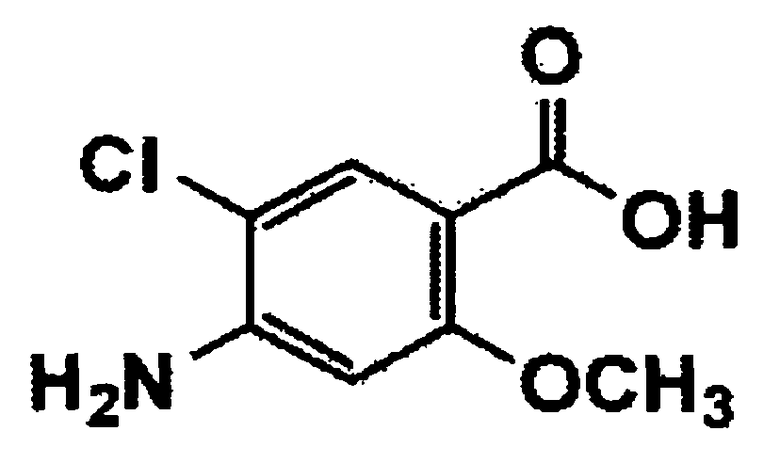
(5) взаимодействие соединения формулы 8 с соединением формулы 9 в присутствии реагента, индуцирующего образование амидной связи, с получением соединения формулы 10; и
(6) удаление аминозащитной группы пиперидинового кольца соединения формулы 10 в присутствии основания или кислоты.
[Формула 2]
[Формула 4]

[Формула 5]
[Формула 6]
[Формула 7]
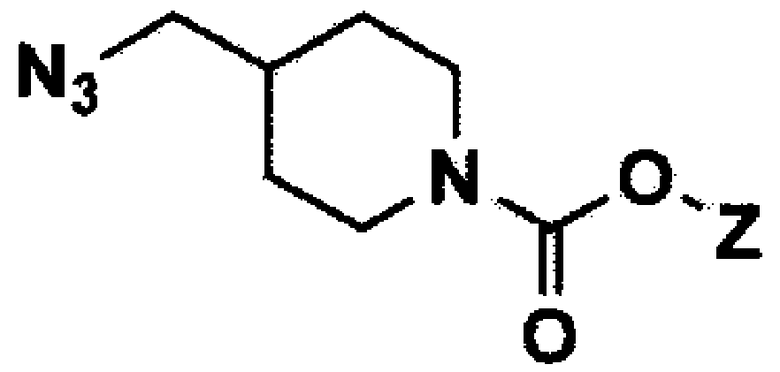
[Формула 8]
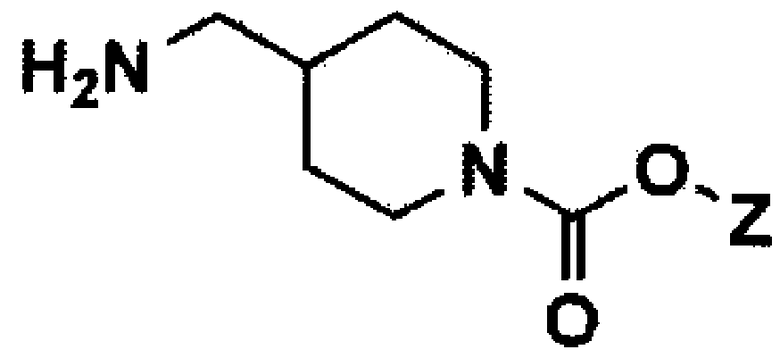
[Формула 9]
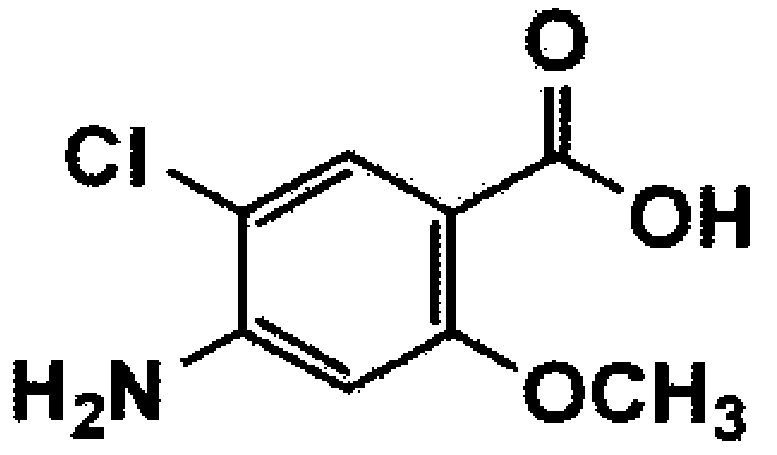
[Формула 10]

В вышеуказанных формулах Y представляет собой атом галогена или C1-C4 алкилсульфонат, и Z представляет собой C1-C4 алкил.
В способе получения соединения формулы 2 в соответствии с настоящим изобретением, реагент, для введения аминозащитной группы на стадии (1), относится к реагенту, обычно используемому для защиты амина с целью предохранения аминогруппы от участия в реакции. Например, такой реагент, предпочтительно, выбирают из ди-трет-бутил дикарбоната или этил хлорформиата в присутствии третичного амина, такого как триэтиламин. Растворителем может быть C1-C6 низший спирт. Реакция может быть осуществлена при ступенчатом повышении температуры от 0°C до комнатной температуры.
В способе получения соединения формулы 2 в соответствии с настоящим изобретением, C1-C4 алкилсульфонилгалогенид со стадии (2), предпочтительно, представляет собой метансульфонилхлорид, метансульфонилбромид или метансульфонилфторид, третичным амином может быть триэтиламин, диизопропилэтиламин или тому подобное, и растворителем может быть дихлорметан, хлороформ или тому подобное. Реакция может быть осуществлена при ступенчатом повышении температуры от 0°C до комнатной температуры.
В способе получения соединения формулы 2 в соответствии с настоящим изобретением, растворителем, используемым на стадии (3), может быть диметилформамид, диметилацетамид или тому подобное, и температура реакции может быть в интервале от 80 до 140°C.
В способе получения соединения формулы 2 в соответствии с настоящим изобретением, восстановитель, используемый на стадии (4), предпочтительно, представляет собой трифенилфосфин или литийалюминийгидрид, и используемым растворителем может быть тетрагидрофуран. Реакция может быть осуществлена при ступенчатом повышении температуры от 0°C до комнатной температуры или может быть осуществлена в интервале от 60 до 80°C.
В способе получения соединения формулы 2 в соответствии с настоящим изобретением, реагент, индуцирующий образование амидной связи, используемый на стадии (5), относится к известному реагенту, который используется обычными специалистами и который применяется для удаления воды, образуемой в результате реакции, с целью облегчения образования амидной связи между карбоновой кислотой и амином, или используется для активации амина или карбоновой кислоты. Примеры реагента, индуцирующего образование амидной связи, включают гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида и 1-гидроксибензотриазол в присутствии основания, этил хлорформиата в присутствии основания и карбодиимидазола в отсутствие основания. В данном описании, примеры основания, используемого с гидрохлоридом N-(3-диметиламинопропил)-N'-этилкарбодиимида и 1-гидроксибензотриазолом, или этилхлорформиатом, включают триэтиламин и диизопропилэтиламин. В данном описании, используемым реакционным растворителем может быть диметилформамид, диметилацетамид, дихлорметан или тому подобное. Реакция может быть осуществлена при ступенчатом повышении температуры от 0°C до комнатной температуры.
В способе получения соединения формулы 2 в соответствии с настоящим изобретением, основание или кислота на стадии (6) относится к основанию или кислоте, которые обычно используются для удаления карбаматной группы у амина, и их примеры включают хлористоводородную кислоту, трифторуксусную кислоту и гидроксид калия. Используемым реакционным растворителем может быть 1,4-диоксан, дихлорметан, C1-C6 низший спирт или тому подобное. Реакция может быть осуществлена при ступенчатом повышении температуры от 0°C до комнатной температуры.
Соединение формулы 3 по настоящему изобретению может быть получено в соответствии со способом, представленным на реакционной схеме 1 или на реакционной схеме 2, ниже.
[Реакционная схема 1]
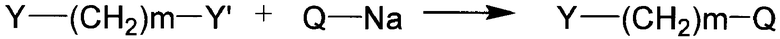
На реакционной схеме 1, заместители Q и m имеют значения, указанные для формулы 1, и оба Y и Y' представляют собой атомы галогена, которые, предпочтительно, отличаются друг от друга, например, один из Y и Y' представляет собой хлор (Cl), а другой из Y и Y' представляет собой бром (Br).
Взаимодействие в соответствии с реакционной схемой 1 может быть осуществлено в присутствии сильного основания, такого как гидрид лития, гидрид натрия или гидрид калия, в органическом растворителе, таком как диметилформамид, диметилацетамид или тетрагидрофуран, при температуре реакции от 0 до 40°C в течение от 1 до 24 часов.
[Реакционная схема 2]

На реакционной схеме 2, заместители Q и m имеют значения, указанные для формулы 1, Y представляет собой C1-C4 алкилсульфонилгалогенид или атом галогена, Y" представляет собой гидроксильную группу, и Y”' представляет собой атом галогена, выбранный из Cl, Br или I.
Первая стадия реакции по реакционной схеме 2 может быть осуществлена в присутствии основания, такого как карбонат калия и йодид калия, в растворителе, таком как 1,4-диоксан или ацетон, при температуре от 60 до 120°C в течение от 1 до 12 часов.
Когда Y в формуле 3 представляет собой C1-C4 алкилсульфонилгалогенид, вторая стадия реакции по реакционной схеме 2 может быть осуществлена путем взаимодействия  с C1-C4 алкилсульфонилгалогенидом (например, метансульфонилхлоридом, метансульфонилбромидом или метансульфонилфторидом) в присутствии основания, такого как триэтиламин или диизопропилэтиламин, в органическом растворителе, таком как дихлорметан или хлороформ, при температуре реакции от 0 до 40°C в течение от 1 до 24 часов.
с C1-C4 алкилсульфонилгалогенидом (например, метансульфонилхлоридом, метансульфонилбромидом или метансульфонилфторидом) в присутствии основания, такого как триэтиламин или диизопропилэтиламин, в органическом растворителе, таком как дихлорметан или хлороформ, при температуре реакции от 0 до 40°C в течение от 1 до 24 часов.
Когда заместитель Y в формуле 3 представляет собой атом галогена, вторая стадия реакции по реакционной схеме 2 может быть осуществлена в общеизвестных условиях реакции замещения гидроксильной группы галогеном. Например, когда заместитель Y в
в формуле 3 представляет собой бром (Br), реакция может быть осуществлена в присутствии одного из агентов, выбранных из N-бромсукцинимида или тетрабромида углерода и трифенилфосфина, в органическом растворителе, таком как дихлорметан, при температуре реакции от 0 до 40°C в течение от 1 до 24 часов.
Кислотно-аддитивная соль соединения, представленного формулой 1, в форме свободного основания может быть получена с использованием традиционного способа, известного в данной области, например, путем смешивания свободного основания соединения формулы 1 с соответствующей кислотой в подходящем растворителе, который затем упаривают с получением соли, или к которому добавляют анти-растворитель для осаждения соли. Например, можно упомянуть способ, который включает обработку раствора или суспензии свободного основания желаемой кислотой в инертном по отношению к реакции растворителе, с последующим концентрированием при пониженном давлении, или кристаллизацией, или проведением любых стандартных методов для получения желаемой соли. В одном варианте осуществления изобретения, гидрохлорид соединения формулы 1 может быть получен путем растворения свободного основания соединения формулы 1 в C1-C4 спиртовом растворителе, таком как этанол или метанол, с добавлением хлористоводородной кислоты, и последующего перемешивания смеси при комнатной температуре.
Далее, настоящее изобретение касается агониста 5-HT4 рецептора, содержащего соединение формулы 1 или его фармацевтически приемлемую соль или гидрат в качестве активного ингредиента.
В настоящем изобретении, агонист 5-HT4 рецептора может представлять собой композицию для предупреждения или лечения заболевания, выбранного из гастроэзофагеальной рефлюксной болезни, желудочно-кишечного заболевания, расстройства перистальтики желудка, неязвенной диспепсии, функционального диспепсита, синдрома раздраженной толстой кишки (IBS), констипации, диспепсии, эзофагита, желудочно-пищеводного заболевания, тошноты, заболевания центральной нервной системы, болезни Альцгеймера, когнитивного нарушения, рвоты, мигрени, неврологического заболевания, боли, сердечно-сосудистого расстройства, сердечной недостаточности, сердечной аритмии, диабета или синдрома апноэ.
В настоящем изобретении, агонист 5-HT4 рецептора по настоящему изобретению может, помимо соединения формулы 1 в соответствии с настоящим изобретением или его фармацевтически приемлемой соли, кроме того, содержать один или несколько активных ингредиентов, проявляющих идентичное или подобное действие.
Для целей желаемого пути введения, агонист или композиция по настоящему изобретению могут быть приготовлены в виде различных лекарственных форм путем включения одного или нескольких фармацевтически приемлемых носителей в сочетании с вышеуказанным активным ингредиентом. Примеры фармацевтически приемлемого носителя включают физиологический раствор, стерильную воду, раствор Рингера, буферный раствор, раствор декстрозы, раствор мальтодекстрина, глицерин и этанол. Указанные вещества могут быть использованы самостоятельно или в любом сочетании. Если необходимо, могут быть добавлены другие обычные добавки, такие как антиоксидант, буфер и бактериостатическое средство. Кроме того, могут быть дополнительно добавлены разбавитель, диспергатор, поверхностно-активное вещество, связующее и смазывающее средство для получения инъекционного препарата, такого как водный раствор, суспензия или эмульсия, или для получения перорального препарата, такого как пилюля, капсула, гранула или таблетка. Кроме того, желаемая лекарственная форма, предпочтительно, может быть приготовлена в зависимости от заболеваний, подвергаемых лечению, и ингредиентов, используя любой подходящий способ, известный в данной области, как описано в обзоре Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, PA.
Далее, когда агонист 5-HT4 рецептора по настоящему изобретению предназначен для перорального введения, содержание соединения формулы 1 в соответствии с настоящим изобретением или его фармацевтически приемлемой соли в препарате может быть в интервале от 1 до 95% по массе и, предпочтительно, от 1 до 70% по массе.
Агонист или композиция по настоящему изобретению могут быть введены парентерально (например, внутривенно, подкожно, интраперитонеально или местно) или перорально, в зависимости от желаемого применения. Доза активного ингредиента может изменяться в зависимости от различных факторов, таких как масса, возраст, пол, состояние здоровья и пищевые привычки пациентов, время введения и пути введения, скорость экскреции и серьезность заболеваний. Бензамидное производное по настоящему изобретению может быть введено в дозе от 1 до 1000 мкг/кг, предпочтительно, от около 10 до 500 мкг/кг и, более предпочтительно, от около 83 до 167 мкг/кг, один или несколько раз в сутки.
Далее, настоящее изобретение относится к способу предупреждения, лечения или облегчения заболевания, обусловленного ослабленной эффективностью 5-HT4 рецептора, включающему введение агониста 5-HT4 рецептора, содержащего в качестве активного ингредиента соединение формулы 1 в соответствии с настоящим изобретением или его фармацевтически приемлемую соль, млекопитающему, включая человека, нуждающегося в агонистическом действии на 5-HT4 рецептор. В данном документе, агонист 5-HT4 рецептора может представлять собой композицию для предупреждения или лечения заболевания, выбранного из гастроэзофагеальной рефлюксной болезни, желудочно-кишечного заболевания, расстройства перистальтики желудка, неязвенной диспепсии, функциональной диспепсии, синдрома раздраженной толстой кишки (IBS), констипации, диспепсии, эзофагита, желудочно-пищеводного заболевания, тошноты, заболевания центральной нервной системы, болезни Альцгеймера, когнитивного нарушения, рвоты, мигрени, неврологического заболевания, боли, сердечно-сосудистого расстройства, сердечной недостаточности, сердечной аритмии, диабета или синдрома апноэ.
Для предупреждения или лечения заболевания, обусловленного ослабленной эффективностью 5-HT4 рецептора, агонист 5-HT4 рецептора по настоящему изобретению может быть использован самостоятельно или в сочетании со способами с использованием хирургических операций, гормональной терапии, медикаментозной терапии и модификаторов биологического ответа.
Положительные эффекты изобретения
Бензамидные производные по настоящему изобретению обладают превосходным сродством к 5-HT4 рецепторам, способностью сокращать время опорожнения желудка и низкой токсичностью, и, следовательно, являются терапевтически эффективными для лечения различных заболеваний, связанных с 5-HT4 рецепторами.
Способ осуществления изобретения
Здесь и далее, настоящее изобретение описано более подробно с отсылками к следующим примерам. Однако следующие примеры представлены только для иллюстрации настоящего изобретения и не должны рассматриваться как ограничивающие объем и сущность настоящего изобретения.
Если не указано иного, реагенты, используемые здесь и далее, были закуплены у фирм Aldrich Korea, Acros, Lancaster, TCI, и т.д., 1H ЯМР анализ осуществляли, используя спектрометр Varian 400 МГц.
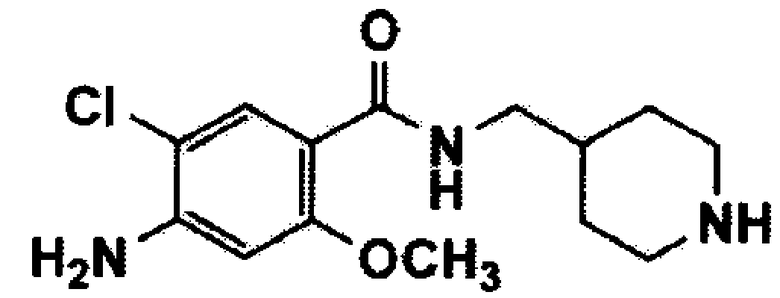
Пример 1: Получение N-((1-(3-(1,2,4-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
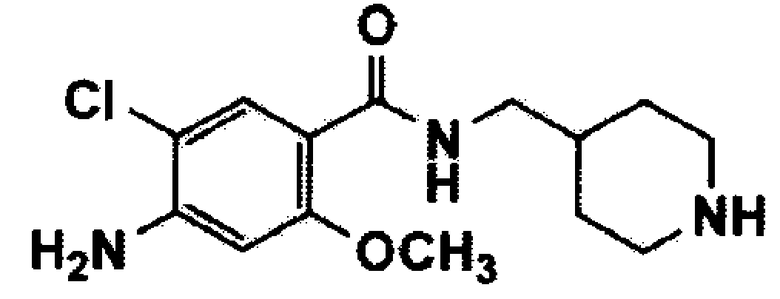
1-1. Получение гидрохлорида 4-амино-5-хлор-2-метокси-(пиперидин-4-илметил)бензамида (соединение формулы 2)
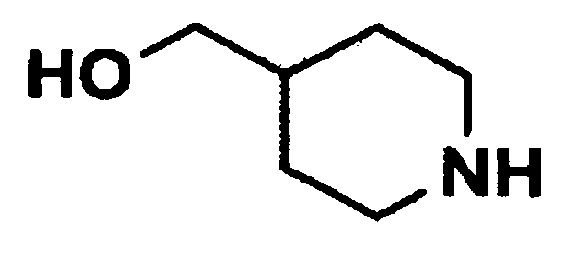
Стадия 1: Получение третичного бутила 4-(гидроксиметил)пиперидин-1-карбоксилата (соединение формулы 5)
4-Пиперидинметанол (соединение формулы 4) (20 г, 174 ммоль) растворяли в метаноле (30 мл), и раствор охлаждали до температуры 0°C. Затем добавляли триэтиламин (48,8 мл, 347 ммоль) и ди-третичный-бутил дикарбонат (56,8 г, 260 ммоль). Реакционную смесь нагревали до комнатной температуры, перемешивали в течение 2 часов и концентрировали при пониженном давлении для удаления растворителя. После экстракции с помощью дихлорметана и воды органический слой промывали раствором лимонной кислоты, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением указанного в заголовке соединения (35,25 г, 94%).
1H ЯМР (CDCl3, 400 МГц): δ 4,18-4,04 (м, 2H), 3,47 (т, J=5,6 Гц, 2H), 2,70-2,64 (м, 2H), 1,70-1,56 (м, 3H), 1,42 (с, 9H), 1,16-1,09 (м, 2H).
Стадия 2: Получение третичного бутила 4-((метилсульфонилокси)метил)пиперидин-1-карбоксилата (соединение формулы 6)
Третичный бутил 4-(гидроксиметил)пиперидин-1-карбоксилат (35,25 г, 164 ммоль) растворяли в дихлорметане (300 мл), и раствор охлаждали до температуры 0°C. Затем добавляли триэтиламин (46,01 мл, 327 ммоль) и метансульфонилхлорид (19,14 мл, 246 ммоль). Реакционную смесь нагревали до комнатной температуры, перемешивали в течение 2 часов и экстрагировали дихлорметаном и водой. Органический слой промывали раствором лимонной кислоты, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением указанного в заголовке соединения (48 г, 100%).
1H ЯМР (CDCl3, 400 МГц): δ 4,18-4,06 (м, 2H), 4,04 (д, J=6,4 Гц, 2H), 2,99 (с, 3H), 2,78-2,62 (м, 2H), 1,92-1,85 (м, 1H), 1,73-1,70 (м, 2H), 1,43 (с, 9H), 1,24-1,13 (м, 2H).
Стадия 3: Получение третичного бутила 4-(азидометил)пиперидин-1-карбоксилата (соединение формулы 7)
Третичный бутил 4-((метилсульфонилокси)метил)пиперидин-1-карбоксилата (48 г, 163,6 ммоль) растворяли в диметилформамиде (500 мл) и добавляли азид натрия (19,15 г, 295 ммоль), затем перемешивали при температуре 120°C в течение 6 часов. После завершения реакции реагенты охлаждали до комнатной температуры, экстрагировали этилацетатом и водой, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением указанного в заголовке соединения (35 г, 89%).
1H ЯМР (CDCl3, 400 МГц): δ 4,18-4,00 (м, 2H), 3,16 (д, J=6,0 Гц, 2H), 2,72-2,58 (м, 2H), 1,70-1,64 (м, 3H), 1,43 (с, 9H), 1,20-1,08 (м, 2H).
Стадия 4: Получение третичного бутила 4-(аминометил)пиперидин-1-карбоксилата (соединение формулы 8)
Третичный бутил 4-(азидометил)пиперидин-1-карбоксилат (30 г, 124,8 ммоль) растворяли в тетрагидрофуране (300 мл) и добавляли трифенилфосфин (39,3 г, 149,8 ммоль), затем перемешивали при кипячении с обратным холодильником в течение 2 часов. Добавляли воду (120 мл), затем перемешивали при кипячении с обратным холодильником в течение еще 3 часов и концентрировали при пониженном давлении. Полученный остаток экстрагировали этилацетатом и 1н. раствором соляной кислоты, и водный слой нейтрализовали с помощью 2н. раствора гидроксида натрия, затем повторно экстрагировали дихлорметаном. Полученный органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением указанного в заголовке соединения (21,16 г, 79%).
1H ЯМР (CDCl3, 400 МГц): δ 4,16-3,98 (м, 2H), 2,69-2,63 (м, 2H), 2,56 (д, J=6,8 Гц, 2H), 1,69-1,66 (м, 2H), 1,46-1,38 (м, 10H), 1,10-1,04 (м, 2H).
Стадия 5: Получение третичного бутила 4-((4-амино-5-хлор-2-метоксибензамидо)метил)пиперидин-1-карбоксилата (соединение формулы 10)
4-Амино-5-хлор-2-метоксибензойную кислоту (соединение формулы 9) (16,6 г, 82,28 ммоль) растворяли в диметилформамиде (166 мл), и раствор охлаждали до температуры 0°C. Затем добавляли третичный бутил 4-(аминометил)пиперидин-1-карбоксилат (21,16 г, 98,74 ммоль), триэтиламин (11,56 мл, 246,84 ммоль), гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (EDC, 20,51 г, 106,96 ммоль) и 1-гидроксибензотриазол (HOBT, 16,68 г, 123,42 ммоль). Реакционную смесь нагревали до комнатной температуры, перемешивали в течение 4 часов и экстрагировали этилацетатом и водой. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением указанного в заголовке соединения (31,4 г, 96%).
1H ЯМР (CDCl3, 400 МГц): δ 8,08 (с, 1H), 7,76-7,70 (м, 1H), 6,27 (с, 1H), 4,37 (с, 2H), 4,16-4,04 (м, 2H), 3,88 (с, 3H), 3,34-3,26 (м, 2H), 2,74-2,60 (м, 2H), 1,80-1,63 (м, 3H), 1,42 (с, 9H), 1,20-1,10 (м, 2H).
Стадия 6: Получение гидрохлорида 4-амино-5-хлор-2-метокси-(пиперидин-4-илметил)бензамида (соединение формулы 2)
Третичный бутил 4-((4-амино-5-хлор-2-метоксибензамидо)метил)пиперидин-1-карбоксилат (30,7 г, 77,16 ммоль) растворяли в 1,4-диоксане (300 мл) и добавляли 4M раствор хлористого водорода (220 мл) в 1,4-диоксане. Смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали при пониженном давлении с получением указанного в заголовке соединения (24 г, 93%).
1H ЯМР (CD3OD, 400 МГц): δ 7,84 (с, 1H), 6,84 (с, 1H), 3,95 (с, 3H), 3,42-3,29 (м, 6H), 3,01-2,95 (м, 2H), 1,98-1,93 (м, 3H), 1,54-1,42 (м, 2H).
1-2. Получение 1-(3-хлорпропил)-1,2,4-триазола (соединение формулы 3)
Натриевую соль 1,2,4-триазола (5 г, 54,91 ммоль) растворяли в диметилформамиде (50 мл), и раствор охлаждали до температуры 0°C. Затем добавляли гидрид натрия (60%, 2,86 г, 71,38 ммоль), далее перемешивали в течение 30 минут. Добавляли 1-бром-3-хлорпропан (6,5 мл, 65,89 ммоль), затем перемешивали при комнатной температуре в течение 12 часов, и реакцию останавливали путем добавления насыщенного раствора хлорида аммония. После экстракции с помощью этилацетата и воды органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии с получением указанного в заголовке соединения (2,77 г, 35%).
1H ЯМР (CDCl3, 400 МГц): δ 8,08 (с, 1H), 7,94 (с, 1H), 4,36 (т, J=6,4 Гц, 2H), 3,45 (т, J=6 Гц, 2H), 2,35-2,30 (м, 2H).
1-3: Получение N-((1-(3-(1,2,4-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида (соединение формулы 1)
Гидрохлорид 4-амино-5-хлор-2-метокси(пиперидин-4-илметил)бензамида (соединение формулы 2) (500 мг, 1,5 ммоль) растворяли в диметилформамиде (10 мл) и добавляли 1-(3-хлорпропил)-1,2,4-триазол (соединение формулы 3) (306 мг, 2,1 ммоль), карбонат калия (497 мг, 3,6 ммоль) и йодид калия (50 мг, 0,3 ммоль). Реагенты перемешивали при температуре 100°C в течение 8 часов, и реакцию останавливали путем добавления воды. После экстракции с помощью этилацетата и воды органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии с получением указанного в заголовке соединения (55 мг, 9%).
1H ЯМР (CDCl3, 400 МГц): δ 8,09 (с, 1H), 8,04 (с, 1H), 7,91 (с, 1H), 7,78-7,70 (м, 1H), 6,27 (с, 1H), 4,35 (с, 2H), 4,22 (т, J=6,4 Гц, 2H), 3,88 (с, 3H), 3,30 (т, J=6,4 Гц, 2H), 2,85-2,81 (м, 2H), 2,24 (т, J=6,4 Гц, 2H), 2,06-2,00 (м, 2H), 1,97-1,90 (м, 2H), 1,50 (м, 3H), 1,39-1,28 (м, 2H).
Пример 2: Получение N-((1-(3-(тетразол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
1-(3-Хлорпропил)тетразол получали с использованием 1H-тетразола и 1-бром-3-хлорпропана в качестве исходных продуктов для <примеров 1-2>, и далее указанное в заголовке соединение (186 мг) получали, используя 1-(3-хлорпропил)тетразол и гидрохлорид 4-амино-5-хлор-2-метокси(пиперидин-4-илметил)бензамида в качестве исходных продуктов для <примеров 1-3>.
1H ЯМР (CDCl3, 400 МГц): δ 8,60 (с, 1H), 8,07 (с, 1H), 7,75-7,68 (м, 1H), 6,28 (с, 1H), 4,49 (т, J=6,8 Гц, 2H), 4,39 (с, 2H), 3,88 (с, 3H), 3,30 (т, J=6,4 Гц, 2H), 2,79-2,76 (м, 2H), 2,23 (т, J=6,8 Гц, 2H), 2,10-2,05 (м, 2H), 1,92-1,86 (м, 2H), 1,75-1,69 (м, 2H), 1,62-1,50 (м, 1H), 1,35-1,23 (м, 2H).
Пример 3: Получение N-((1-(3-(индол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
1-(3-Хлорпропил)индол получали с использованием индола и 1-бром-3-хлорпропана в качестве исходных продуктов для <примеров 1-2>, и затем указанное в заголовке соединение (26 мг) получали, используя 1-(3-хлорпропил)индол и гидрохлорид 4-амино-5-хлор-2-метокси-(пиперидин-4-илметил)бензамида в качестве исходных продуктов для <примера 1-3>.
1H ЯМР (CDCl3, 400 МГц): δ 8,09 (с, 1H), 7,85-7,76 (м, 1H), 7,60 (д, J=7,6 Гц, 1H), 7,34 (д, J=8,0 Гц, 1H), 7,15 (т, J=7,2 Гц, 1H), 7,10-7,07 (м, 2H), 6,45 (д, J=3,2 Гц, 1H), 6,27 (с, 1H), 4,34 (с, 2H), 4,18 (т, J=7,2 Гц, 2H), 3,88 (с, 3H), 3,32 (т, J=6,4 Гц, 2H), 2,88-2,85 (м, 2H), 2,26 (т, J=7,2 Гц, 2H), 2,03-1,97 (м, 2H), 1,90-1,80 (м, 2H), 1,73-1,60 (м, 3H), 1,40-1,32 (м, 2H).
Пример 4: Получение N-((1-(3-(2-метилимидазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
1-(3-Хлорпропил)-2-метилимидазол получали с использованием 2-метилимидазола и 1-бром-3-хлорпропана в качестве исходных продуктов для <примеров 1-2>, и затем указанное в заголовке соединение (226 мг) получали, используя 1-(3-хлорпропил)-2-метилимидазол и гидрохлорид 4-амино-5-хлор-2-метокси-(пиперидин-4-илметил)бензамида в качестве исходных продуктов для <примеров 1-3>.
1H ЯМР (CDCl3, 400 МГц): δ 8,06 (с, 1H), 7,78-7,69 (м, 1H), 6,85 (с, 1H), 6,78 (с, 1H), 6,27 (с, 1H), 4,46 (с, 2H), 3,87-3,83 (м, 5H), 3,29 (т, J=6,0 Гц, 2H), 2,85-2,79 (м, 2H), 2,34 (с, 3H), 2,22 (т, J=6,8 Гц, 2H), 1,90-1,83 (м, 4H), 1,72-1,67 (м, 2H), 1,63-1,50 (м, 1H), 1,32-1,26 (м, 2H).
Пример 5: Получение N-((1-(5-(индол-1-ил)пентил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
1-(5-Хлорпентил)индол получали с использованием индола и 1-бром-5-хлорпентана в качестве исходных продуктов для <примеров 1-2>, и затем указанное в заголовке соединение (250 мг) получали, используя 1-(5-хлорпентил)индол и гидрохлорид 4-амино-5-хлор-2-метокси(пиперидин-4-илметил)бензамида в качестве исходных продуктов для <примеров 1-3>.
1H ЯМР (CDCl3, 400 МГц): δ 8,09 (с, 1H), 7,75-7,69 (м, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,31 (д, J=7,6 Гц, 1H), 7,19-7,16 (м, 1H), 7,09-7,05 (м, 2H), 6,47-6,44 (м, 1H), 6,26 (с, 1H), 4,34 (с, 2H), 4,11-4,08 (м, 2H), 3,87 (с, 3H), 3,30 (т, J=6,0 Гц, 2H), 2,92-2,86 (м, 2H), 2,28-2,20 (м, 2H), 1,91-1,80 (м, 4H), 1,72-1,48 (м, 5H), 1,39-1,26 (м, 4H).
Пример 6: Получение N-((1-(5-1,2,3-триазол-1-ил)пентил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
1-(5-Хлорпентил)-1,2,3-триазол получали с использованием 1,2,3-триазола и 1-бром-5-хлорпентана в качестве исходных продуктов для <примеров 1-2>, и затем указанное в заголовке соединение (225 мг) получали, используя 1-(5-хлорпентил)-1,2,3-триазол и гидрохлорид 4-амино-5-хлор-2-метокси(пиперидин-4-илметил)бензамида в качестве исходных продуктов для <примера 1-3>.
1H ЯМР (CDCl3, 400 МГц): δ 8,06 (с, 1H), 7,79-7,70 (м, 1H), 7,67 (с, 1H), 7,52 (с, 1H), 6,27 (с, 1H), 4,41 (с, 2H), 4,35 (т, J=6,8 Гц, 2H), 3,86 (с, 3H), 3,28 (т, J=6,0 Гц, 2H), 2,92-2,88 (м, 2H), 2,28 (т, J=7,6 Гц, 2H), 1,94-1,84 (м, 4H), 1,72-1,68 (м, 2H), 1,66-1,46 (м, 3H), 1,34-1,21 (м, 4H).
Пример 7: Получение N-((1-(3-(1,2,3-триазол-1-ил)пропил)иперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Стадия 1: Получение 3-(1,2,3-триазол-1-ил)пропанола
1,2,3-Триазол (2 г, 28,96 ммоль) растворяли в 1,4-диоксане (40 мл) и добавляли карбонат калия (8 г, 57,92 ммоль), йодид калия (962 мг, 5,79 ммоль) и 3-бромпропанол (3,3 мл, 43,43 ммоль), затем перемешивали при температуре 100°C в течение 3 часов. После завершения реакции реагенты охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии с получением указанного в заголовке соединения (2,5 г, 68%).
1H ЯМР (CDCl3, 400 МГц): δ 7,68 (с, 1H), 7,59 (с, 1H), 4,54 (т, J=6,8 Гц, 2H), 3,62 (кв, J=5,6 Гц, 2H), 2,44 (т, J=5,2 Гц, 1H), 2,15-2,10 (м, 2H).
Стадия 2: Получение 3-(1,2,3-триазол-1-ил)пропилметансульфоната
3-(1,2,3-Триазол-1-ил)пропанол (2,5 г, 19,66 ммоль) растворяли в дихлорметане (50 мл), и раствор охлаждали до температуры 0°C. Затем добавляли триэтиламин (5,53 мл, 39,32 ммоль) и метансульфонилхлорид (2,3 мл, 29,5 ммоль). Реакционную смесь нагревали до комнатной температуры, перемешивали в течение 2 часов, экстрагировали дихлорметаном и водой, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии с получением указанного в заголовке соединения (3,01 г, 75%).
1H ЯМР (CDCl3, 400 МГц): δ 7,72 (с, 1H), 7,63 (с, 1H), 4,55 (т, J=6,8 Гц, 2H), 4,22 (т, J=6 Гц, 2H), 3,03 (с, 3H), 2,40-2,36 (м, 2H).
Стадия 3: Получение N-((1-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Гидрохлорид 4-амино-5-хлор-2-метокси(пиперидин-4-илметил)бензамида (соединение формулы 2) (400 мг, 1,2 ммоль) растворяли в диметилформамиде (10 мл) и добавляли 3-(1,2,3-триазол-1-ил)пропил метансульфонат (соединение формулы 3) (344 мг, 1,68 ммоль), триэтиламин (0,5 мл, 3,591 ммоль), карбонат калия (232 мг, 1,68 ммоль) и йодид калия (40 мг, 0,24 ммоль), затем перемешивали при температуре 120°C в течение 4 часов. После завершения реакции реагенты охлаждали до комнатной температуры, экстрагировали этилацетатом и водой, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии с получением указанного в заголовке соединения (185 мг, 38%).
1H ЯМР (CDCl3, 400 МГц): δ 8,09 (с, 1H), 7,74-7,70 (м, 1H), 7,67 (с, 1H), 7,54 (с, 1H), 6,27 (с, 1H), 4,43 (т, J=6,8 Гц, 2H), 4,35 (с, 2H), 3,88 (с, 3H), 3,31 (т, J=6,4 Гц, 2H), 2,85-2,82 (м, 2H), 2,27 (т, J=6,8 Гц, 2H), 2,07-2,04 (м, 2H), 1,92-1,86 (м, 2H), 1,73-1,69 (м, 2H), 1,62-1,50 (м, 1H), 1,34-1,22 (м, 2H).
Пример 8: Получение N-((1-(3-(1,2,3-триазол-2-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Используя в качестве исходных продуктов 3-(1,2,3-триазол-2-ил)пропанол, полученный в качестве побочного продукта на стадии 1 примера 7, и гидрохлорид 4-амино-5-хлор-2-метокси(пиперидин-4-илметил)бензамида, указанное в заголовке соединение (250 мг) получали тем же способом, что и в <примере 7>.
1H ЯМР (CDCl3, 400 МГц): δ 8,03 (с, 1H), 7,83-7,75 (м, 1H), 7,53 (с, 2H), 6,25 (с, 1H), 4,50-4,42 (м, 4H), 3,82 (с, 3H), 3,26 (т, J=6,0 Гц, 2H), 2,86-2,83 (м, 2H), 2,31-2,27 (м, 2H), 2,12-2,05 (м, 2H), 1,89-1,83 (м, 2H), 1,67-1,64 (м, 2H), 1,60-1,50 (м, 1H), 1,31-1,20 (м, 2H).
Пример 9: Получение N-((1-(пиридин-3-илметил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Гидрохлорид 4-амино-5-хлор-2-метокси(пиперидин-4-илметил)бензамида (1 г, 2,99 ммоль) растворяли в метаноле (40 мл) и добавляли 3-пиридинкарбоксальдегид (0,42 мл, 4,49 ммоль), уксусную кислоту (1 мл) и цианборгидрид натрия (470 мг, 7,48 ммоль), затем перемешивали при температуре 60°C в течение 5 часов. После завершения реакции реакционную смесь концентрировали при пониженном давлении для удаления растворителя, экстрагировали дихлорметаном и водой и далее промывали раствором бикарбоната натрия. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии с получением указанного в заголовке соединения (355 мг, 30%).
1H ЯМР (CDCl3, 400 МГц): δ 8,07 (с, 1H), 7,74-7,69 (м, 1H), 7,65 (д, J=7,6 Гц, 1H), 7,37 (с, 1H), 7,30 (д, J=8,4 Гц, 1H), 7,25-7,22 (м, 1H), 7,19-7,15 (м, 1H), 6,26 (с, 1H), 4,42 (с, 2H), 3,83 (с, 3H), 3,77 (с, 3H), 3,28 (т, J=5,6 Гц, 2H), 2,95-2,82 (м, 4H), 2,19-2,14 (м, 4H), 1,91-1,85 (м, 2H), 1,80-1,72 (м, 2H), 1,69-1,63 (м, 2H), 1,61-1,53 (м, 1H), 1,32-1,15 (м, 5H).
Пример 10: Получение N-((1-((1-метилиндол-3-ил)метил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Используя в качестве исходных продуктов 1-метилиндол-3-карбоксальдегид и гидрохлорид 4-амино-5-хлор-2-метокси(пиперидин-4-илметил)бензамида, указанное в заголовке соединение (60 мг) получали тем же способом, что и в <примере 9>.
1H ЯМР (CDCl3, 400 МГц): δ 8,02 (с, 1H), 7,82-7,75 (м, 1H), 7,56 (д, J=8,4 Гц, 1H), 7,33-7,22 (м, 3H), 7,12 (т, J=7,2 Гц, 1H), 6,27 (с, 1H), 4,38 (с, 2H), 4,01 (с, 2H), 3,87 (с, 3H), 3,79 (с, 3H), 3,30-3,23 (м, 2H), 2,46-2,37 (м, 2H), 1,88-1,75 (м, 3H), 1,59-1,50 (м, 2H).
Пример 11: Получение N-((1-(имидазол-2-илметил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Используя в качестве исходных продуктов 2-имидазол карбоксальдегид и гидрохлорид 4-амино-5-хлор-2-метокси(пиперидин-4-илметил)бензамида, указанное в заголовке соединение (123 мг) получали тем же способом, что и в <примере 9>.
1H ЯМР (CDCl3, 400 МГц): δ 8,07 (с, 1H), 7,75-7,66 (м, 1H), 6,96 (с, 2H), 6,27 (с, 1H), 4,38 (с, 2H), 3,87 (с, 3H), 3,61 (с, 2H), 3,31 (т, J=6,8 Гц, 2H), 2,85-2,80 (м, 2H), 2,11-2,04 (м, 2H), 1,74-1,69 (м, 2H), 1,63-1,53 (м, 1H), 1,34-1,23 (м, 2H).
Пример 12: Получение N-((1-((1-метилпиррол-2-ил)метил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Используя в качестве исходных продуктов 1-метил-2-пиррол карбоксальдегид и гидрохлорид 4-амино-5-хлор-2-метокси(пиперидин-4-илметил)бензамида, указанное в заголовке соединение (50 мг) получали тем же способом, что и в <примере 9>.
1H ЯМР (CDCl3, 400 МГц): δ 8,08 (с, 1H), 7,75-7,64 (м, 1H), 6,57-6,53 (м, 1H), 6,26 (с, 1H), 6,00-5,96 (м, 1H), 5,95-5,93 (м, 1H), 4,38 (с, 2H), 3,85 (с, 3H), 3,60 (с, 2H), 3,28 (т, J=6,8 Гц, 2H), 2,86-2,82 (м, 2H), 1,92-1,84 (м, 2H), 1,68-1,63 (м, 2H), 1,61-1,53 (м, 1H), 1,30-1,19 (м, 2H).
Пример 13: Получение N-((1-(4-фторбензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Используя в качестве исходных продуктов 4-фторбензальдегид и гидрохлорид 4-амино-5-хлор-2-метокси(пиперидин-4-илметил)бензамида, указанное в заголовке соединение (215 мг) получали тем же способом, что и в <примере 9>.
1H ЯМР (CDCl3, 400 МГц): δ 8,07 (с, 1H), 7,75-7,68 (м, 1H), 7,27-7,21 (м, 2H), 7,00-6,94 (м, 2H), 6,26 (с, 1H), 4,36 (с, 2H), 3,86 (с, 3H), 3,46-3,43 (м, 2H), 3,28 (т, J=6,4 Гц, 2H), 2,87-2,84 (м, 2H), 2,01-1,91 (м, 2H), 1,70-1,56 (м, 3H), 1,36-1,20 (м, 2H).
Пример 14: Получение N-((1-(4-гидроксибензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Используя в качестве исходных продуктов 4-гидроксибензальдегид и гидрохлорид 4-амино-5-хлор-2-метокси(пиперидин-4-илметил)бензамида, указанное в заголовке соединение (75 мг) получали тем же способом, что и в <примере 9>.
1H ЯМР (CDCl3, 400 МГц): δ 7,81 (с, 1H), 7,19 (д, J=8,8 Гц, 2H), 6,75 (с, J=8,8 Гц, 2H), 6,20 (с, 1H), 4,40 (с, 2H), 3,76 (с, 2H), 3,32-3,24 (м, 5H), 3,12-3,05 (м, 2H), 2,68-2,59 (м, 2H), 1,80-1,72 (м, 3H), 1,27-1,12 (м, 2H).
Пример 15: Получение N-((1-(2-(индол-3-ил)этил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Стадия 1: Получение 2-(индол-3-ил)этанола
Индол-3-уксусную кислоту (5 г, 28,54 ммоль) растворяли в диэтиловом эфире (100 мл), и раствор охлаждали до температуры 0°C. Добавляли литийалюминийгидрид (1,19 г, 31,39 ммоль), затем перемешивали в течение 4 часов. Реакцию останавливали путем добавления воды и 10%-ного раствора гидроксида натрия. Реакционную смесь фильтровали через целит и концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии с получением указанного в заголовке соединения (1,24 г, 27%).
1H ЯМР (CDCl3, 400 МГц): δ 8,10-7,96 (ушир. с, 1H), 7,61 (д, J=7,6 Гц, 1H), 7,36 (дд, J=8 Гц, 0,8 Гц, 1H), 7,24-7,17 (м, 1H), 7,15-7,07 (м, 2H), 3,92-3,87 (м, 2H), 3,05-3,00 (м, 2H).
Стадия 2: Получение 3-(2-бромэтил)индола (соединение формулы 3)
2-(Индол-3-ил)этанол (623 мг, 3,86 ммоль) растворяли в дихлорметане (20 мл), и раствор охлаждали до температуры 0°C. Затем добавляли трифенилфосфин (1,12 г, 4,25 ммоль) и тетрабромметан (1,41 г, 4,25 ммоль), далее перемешивали в течение 1 часа. После завершения реакции реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Полученный остаток очищали путем колоночной хроматографии с получением указанного в заголовке соединения (765 мг, 88%).
1H ЯМР (CDCl3, 400 МГц): δ 8,10-7,88 (ушир. с, 1H), 7,58 (д, J=8 Гц, 1H), 7,38-7,34 (м, 1H), 7,24-7,20 (м, 1H), 7,19-7,08 (м, 2H), 3,65-3,60 (м, 2H), 3,35-3,30 (м, 2H).
Стадия 3: Получение N-((1-(2-(индол-3-ил)этил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Используя в качестве исходных продуктов 3-(2-бромэтил)индол и гидрохлорид 4-амино-5-хлор-2-метокси(пиперидин-4-илметил)бензамида, указанное в заголовке соединение (190 мг) получали тем же способом, что и в <примерах 1-3>.
1H ЯМР (CDCl3, 400 МГц): δ 8,09 (с, 1H), 8,05 (с, 1H), 7,81-7,75 (м, 1H), 7,59 (д, J=7,6 Гц, 1H), 7,33 (д, J=8,4 Гц, 1H), 7,16 (т, J=7,2 Гц, 1H), 7,09 (т, J=7,6 Гц, 1H), 7,00 (д, J=1,6 Гц, 1H), 6,27 (с, 1H), 4,36 (с, 2H), 3,88 (с, 3H), 3,32 (т, J=6,4 Гц, 2H), 3,12-3,07 (м, 2H), 3,01-2,96 (м, 2H), 2,73-2,69 (м, 2H), 2,11-2,04 (м, 2H), 1,79-1,75 (м, 2H), 1,74-1,63 (м, 1H), 1,49-1,38 (м, 2H).
Пример 16: Получение гидрохлорида N-((1-(3-(тетразол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
N-((1-(3-(Тетразол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид (пример 2) (517 мг, 1,27 ммоль) растворяли в этаноле (10 мл) и добавляли 12н. раствор хлористого водорода (0,16 мл, 1,90 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и затем фильтровали с получением указанного в заголовке соединения (417 мг, 74%).
1H ЯМР (ДМСО-d 6 , 400 МГц): δ 10,52 (с, 1H), 9,47 (с, 1H), 8,02-7,99 (м, 1H), 7,64 (с, 1H), 6,47 (с, 1H), 5,95 (с, 2H), 4,59-4,56 (м, 2H), 3,81 (с, 3H), 3,45-3,42 (м, 2H), 3,18-3,08 (м, 2H), 3,04-2,98 (м, 2H), 2,87-2,79 (м, 2H), 2,35-2,31 (м, 2H), 1,90-1,89 (м, 1H), 1,78-1,75 (м, 2H), 1,58-1,52 (м, 2H).
Пример 17: Получение гидрохлорида N-((1-(5-1,2,3-триазол-1-ил)пентил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Используя в качестве исходного продукта N-((1-(5-(1,2,3-триазол-1-ил)пентил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид (пример 6), указанное в заголовке соединение (112 мг, 35%) получали тем же способом, что и в <примере 16>.
1H ЯМР (ДМСО-d 6 , 400 МГц): δ 10,06 (с, 1H), 8,14 (с, 1H), 8,02-7,99 (м, 1H), 7,71 (с, 1H), 7,64 (с, 1H), 6,47 (с, 1H), 5,95 (с, 2H), 4,40-4,37 (м, 2H), 3,81 (с, 3H), 3,40-3,32 (м, 2H), 3,18-3,12 (м, 2H), 2,96-2,90 (м, 2H), 2,84-2,75 (м, 2H), 1,87-1,70 (м, 7H), 1,59-1,48 (м, 2H), 1,25-1,17 (м, 2H).
Пример 18: Получение гидрохлорида N-((1-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Используя в качестве исходного продукта N-((1-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид (пример 7), указанное в заголовке соединение (291 г, 93%) получали тем же способом, что и в <примере 16>.
1H ЯМР (ДМСО-d 6 , 400 Гц): δ 10,67 (с, 1H), 8,19 (с, 1H), 8,02-7,99 (м, 1H), 7,74 (с, 1H), 7,64 (с, 1H), 6,48 (с, 1H), 5,93 (ушир.с, 2H), 4,51-4,47 (м, 2H), 3,81 (с, 3H), 3,44-3,41 (м, 2H), 3,34-3,31 (м, 2H), 2,98-2,96 (м, 2H), 2,87-2,79 (м, 2H), 2,34-2,29 (м, 2H), 1,96-1,86 (м, 1H), 1,77-1,74 (м, 2H), 1,60-1,54 (м, 2H).
Пример 19: Получение гидрохлорида N-((1-((1-метилиндол-3-ил)метил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Используя в качестве исходного продукта N-((1-((1-метилиндол-3-ил)метил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид (пример 10), указанное в заголовке соединение (237 мг, 44%) получали тем же способом, что и в <примере 16>.
1H ЯМР (ДМСО-d 6 , 400 МГц): δ 10,34 (с, 1H), 7,96-7,93 (м, 1H), 7,79 (д, J=8 Гц, 1H), 7,64-7,59 (м, 2H), 7,47 (д, J=8 Гц, 1H), 7,22-7,19 (м, 1H), 7,14-7,10 (м, 1H), 6,47 (с, 1H), 4,36 (д, J=4,4 Гц, 2H), 3,81 (с, 3H), 3,79 (с, 3H), 3,40-3,37 (м, 2H), 3,14-3,11 (м, 2H), 2,90-2,82 (м, 2H), 1,77-1,71 (м, 3H), 1,53-1,48 (м, 2H).
Пример 20: Получение гидрохлорида N-((1-(4-фторбензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Используя в качестве исходного продукта N-((1-(4-фторбензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид (пример 13), указанное в заголовке соединение (569 г, 87%) получали тем же способом, что и в <примере 16>.
1H ЯМР (ДМСО-d 6 , 400 Гц): δ 10,78 (с, 1H), 7,98-7,96 (м, 1H), 7,73-7,63 (м, 3H), 7,30-7,25 (м, 2H), 6,47 (с, 1H), 5,94 (с, 2H), 4,22 (д, J=4,8 Гц, 2H), 3,80 (с, 3H), 3,29-3,26 (м, 2H), 3,14-3,07 (м, 2H), 2,86-2,78 (м, 2H), 1,89-1,85 (м, 1H), 1,77-1,74 (м, 2H), 1,59-1,53 (м, 2H).
Пример 21: Получение гидрохлорида N-((1-(4-гидроксибензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида
Используя в качестве исходного продукта N-((1-(4-гидроксибензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид (пример 14), указанное в заголовке соединение (158 г, 97%) получали тем же способом, что и в <примере 16>.
1H ЯМР (ДМСО-d 6 , 400 Гц): δ 10,25 (с, 1H), 7,98-7,96 (м, 1H), 7,63 (с, 1H), 7,34 (д, J=6,4 Гц, 2H), 6,80 (д, J=6,4 Гц, 2H), 6,47 (с, 1H), 4,08 (д, J=3,6 Гц, 2H), 3,80 (с, 3H), 3,28-3,25 (м, 2H), 3,15-3,12 (м, 2H), 2,82-2,74 (м, 2H), 1,77-1,62 (м, 3H), 1,54-1,48 (м, 2H).
Экспериментальный пример 1: Сродство связывания соединений с 5-HT 4 рецептором
Сродство связывания соединений с человеческим 5-HT4 рецептором оценивали в соответствии со способом, описанным в литературе [Wyngaert et al., Journal of Neurochemistry, (1997) 69, 1810-1819]. С этой целью были получены и гомогенизированы клетки COS-7, экспрессирующие человеческий 5-HT4 рецептор, с получением мембранных гомогенатов, которые затем были использованы в экспериментах по анализу связывания. Для анализа связывания, мембранные гомогенаты были соответственно смешаны и инкубированы с различными концентрациями исследуемых веществ и [H3]-GR113808 (Amersham Biosciences). Концентрации отдельных исследуемых веществ составляли 4 мкМ, 1 мкМ, 0,25 мкМ и 0,0625 мкМ, соответственно, и концентрация [H3]-GR113808 была 0,595 нМ. После завершения инкубирования, продукты реакции собирали на фильтрах из стекловолокна GF/B, используя харвестер клеток Packard, и связанную радиоактивность определяли, используя жидкий сцинтилляционный счетчик клеток (Packard TopCount NXT™, Perkin Elmer). Специфическое связывание радиолиганда с 5-HT4 рецептором определяли путем вычитания неспецифического связывания радиолиганда от общего связывания радиолиганда. IC50 рассчитывали по % ингибирования специфического связывания радиолиганда с 5-HT4 рецептором относительно изменяющихся концентраций исследуемых веществ. Результаты представлены в таблице 1.
Как можно видеть из таблицы 1, соединения по настоящему изобретению ингибируют специфическое связывание радиолиганда с 5-HT4 рецептором при концентрации подобной или меньше чем концентрация цисаприда в качестве контроля, таким образом демонстрируя то, что заявленные соединения обладают сильным сродством связывания с 5-HT4 рецептором.
Экспериментальный пример 2: Оценка скорости опорожнения желудка
Оценка скорости опорожнения желудка была проведена на основе метода, описанного Iwanaga Y, et al., Jpn J Pharmacol. 1991, 56(3), 261-269. В качестве экспериментальных животных использовали крыс SD (от 240 до 250 г), и их разделяли на группы (n = от 5 до 6). За день до эксперимента животных не кормили в течение 18 часов. Тестируемые вещества суспендировали в 0,5% (масс/об.) метилцеллюлозе и вводили животным перорально в дозе 5 мг/кг. Через 1 час после перорального введения исследуемых веществ, перорально вводили животным по 2 мл полутвердого корма. Полутвердым кормом была овсяная каша, полученная измельчением стандартного твердого корма вместе с дистиллированной водой в миксере. Спустя 50 минут после введения полутвердого корма, животных умерщвляли путем смещения шейных позвонков. Затем разрезали брюшную полость и у животных отделяли желудок. Измеряли массу корма, остающегося в желудке, определяя таким образом скорость опорожнения желудка (%). Скорость опорожнения желудка рассчитывали в соответствии со следующим уравнением. Результаты представлены в таблице 2.
Скорость опорожнения желудка (%) = (1-X/Y)*100
X: Масса корма, остающегося в желудке, отделенного через 50 минут после введения еды
Y: Масса корма, остающегося в желудке, отделенного сразу после введения еды
Как видно из таблицы 2, введение соединений по настоящему изобретению приводит к заметному улучшению опорожнения желудка, по сравнению с обработанной метилцеллюлозой группой в качестве контроля, демонстрируя, таким образом, то, что заявленные соединения облегчают моторику желудочно-кишечного тракта.
Экспериментальный пример 3: Определение острой пероральной токсичности соединений на мышах
С целью определения острой токсичности соединений в соответствии с настоящим изобретением был проведен следующий эксперимент.
200 мг каждого из соединений по примерам растирали с 1% гидроксипропилметилцеллюлозой, и полученный порошок вводили перорально самцам мышей ICR 5-недельного возраста (20 г ± 2 г, n = 5) в дозе 1 г/10 мл/кг. Минимальную летальную дозу (МЛД, мг/кг) отдельных соединений исследовали путем изучения смертности, массы тела, клинических симптомов и тому подобного у животных в течение всего экспериментального периода в течение 2 недель. Результаты представлены в таблице 3.
Как видно из результатов испытаний по определению острой токсичности, представленных в таблице 3, все соединения, используемые в тесте, проявляют МЛД более 1000 мг/кг, таким образом, демонстрируя, что заявленные соединения являются безопасными при применении.
Экспериментальный пример 4: Сродство связывания лекарственных соединений с hERG рецептором
Сродство связывания соединений c калиевым (K+) каналом гена human ether-a-go-go-related gene (hERG), который связан с удлинением интервала кардиального QT, анализировали в MDS Pharma Service (номер по каталогу 265900). Мембранные гомогенаты были получены из HEK-293 клеток млекопитающих, экспрессирующих в калиевый канал hERG, и затем использовали в эксперименте по анализу связывания. Для эксперимента анализа связывания мембранные гомогенаты соответственно смешивали и инкубировали с 10 мкМ исследуемых веществ и 1,5 нМ [H3]-Astemizole (Perkin Elmer). После завершения инкубирования подсчитывали радиоактивное связывание с K+ каналом hERG. Сродство каждого испытуемого вещества к K+ каналу hERG рассчитывали по % ингибирования специфического связывания радиолиганда к K+ каналу hERG, получаемого при действии испытуемого вещества. Результаты приведены в таблице 4.
Частота возникновения заболеваний сердечной аритмией, которая дает неизбежный негативный эффект, связана с удлинением интервала кардиального QT, что является результатом чрезмерно высокого сродства препарата к hERG рецептору. Соединения по настоящему изобретению проявляют скорость ингибирования менее чем 50% даже в дозе 10 мкМ и, следовательно, имеют низкое сродство связывания hERG рецептора, таким образом, предполагают, что соединения по изобретению проявляют значительно пониженный риск вызова аритмии.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИПЕРИДИНОВЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ПРИМЕНЕНИЕ | 2009 |
|
RU2514827C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 1,3-ДИГИДРО-5-ИЗОБЕНЗОФУРАНКАРБОНИТРИЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ ДЛЯ ЛЕЧЕНИЯ ПРЕЖДЕВРЕМЕННОЙ ЭЯКУЛЯЦИИ | 2008 |
|
RU2448963C2 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2833354C1 |
| НОВОЕ ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2009 |
|
RU2470934C1 |
| НОВОЕ ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ, ОБЛАДАЮЩЕЕ ЭФФЕКТОМ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2744168C1 |
| 1,3,4,-ОКСАДИЗОЛАМИДНОЕ ПРОИЗВОДНОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2700696C2 |
| НОВОЕ ЗАМЕЩЕННОЕ ДЕЙТЕРИЕМ ПРОИЗВОДНОЕ ПИРИМИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2020 |
|
RU2811770C1 |
| ПРОИЗВОДНЫЕ ИНДОЛА И ИНДАЗОЛА, ОБЛАДАЮЩИЕ КОНСЕРВИРУЮЩИМ ДЕЙСТВИЕМ ПО ОТНОШЕНИЮ К КЛЕТКАМ, ТКАНЯМ И ОРГАНАМ | 2009 |
|
RU2460525C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2017 |
|
RU2714206C1 |
| СОЕДИНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТА АДЕНОЗИНОВОГО РЕЦЕПТОРА A2a И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2022 |
|
RU2840068C2 |
Изобретение относится к новым соединениям, представленным формулой 1:
[Формула 1]

где m обозначает целое число от 1 до 5; и Q представляет собой гетероароматическое кольцо или фенил, где гетероароматическое кольцо выбрано из группы, состоящей из триазола, тетразола, индола, имидазола, пиридина и пиррола, и независимо замещено 0, 1, 2 или 3 заместителями, выбранными из С1-С4алкила, С1-С4алкокси, гидрокси и галогена, и где фенил независимо замещен 1, 2 или 3 заместителями, выбранными из гидрокси и фтора; способу их получения и агониста 5-HT4 рецептора, содержащего их в качестве активного ингредиента. 5 н. и 12 з.п. ф-лы., 4 табл., 24 пр.
1. Соединение, представленное формулой 1:
[Формула 1]
где m обозначает целое число от 1 до 5; и Q представляет собой гетероароматическое кольцо или фенил, где гетероароматическое кольцо выбрано из группы, состоящей из триазола, тетразола, индола, имидазола, пиридина и пиррола, и независимо замещено 0, 1, 2 или 3 заместителями, выбранными из С1-С4алкила, С1-С4алкокси, гидрокси и галогена, и где фенил независимо замещен 1, 2 или 3 заместителями, выбранными из гидрокси и фтора; или его фармацевтически приемлемая соль.
2. Соединение формулы 1 или его фармацевтически приемлемая соль по п.1, где соединение формулы 1 или его фармацевтически приемлемая соль выбраны из группы, состоящей из следующих соединений:
(1) N-((1-(3-(1,2,4-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(2) N-((1-(3-(тетразол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(3) N-((1-(3-(индол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(4) N-((1-(3-(2-метилимидазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(5) N-((1-(5-(индол-1-ил)пентил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(6) N-((1-(5-(1,2,3-триазол-1-ил)пентил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(7) N-((1-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(8) N-((1-(3-(1,2,3-триазол-2-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(9) N-((1-(пиридин-3-илметил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(10) N-((1-((1-метилиндол-3-ил)метил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(11) N-((1-(имидазол-2-илметил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(12) N-((1-((1-метилпиррол-2-ил)метил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(13) N-((1-(4-фторбензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(14) N-((1-(4-гидроксибензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(15) N-((1-(2-(индол-3-ил)этил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(16) гидрохлорид N-((1-(3-(тетразол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(17) гидрохлорид N-((1-(5-(1,2,3-триазол-1-ил)пентил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(18) гидрохлорид N-((1-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(19) гидрохлорид N-((1-((1-метилиндол-3-ил)метил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(20) гидрохлорид N-((1-(4-фторбензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(21) гидрохлорид N-((1-(4-гидроксибензил)пиперидин-4-ил) метил)-4-амино-5-хлор-2-метоксибензамида, и их фармацевтически приемлемые соли.
3. Способ получения соединения формулы 1 или его фармацевтически приемлемой соли, включающий взаимодействие соединения формулы 2 или его фармацевтически приемлемой соли с соединением формулы 3 в присутствии основания для введения соединения формулы 3 по амину в 1-м положении пиперидинового кольца соединения формулы 2 или его фармацевтически приемлемой соли, получая таким образом соединение формулы 1.
[Формула 1]
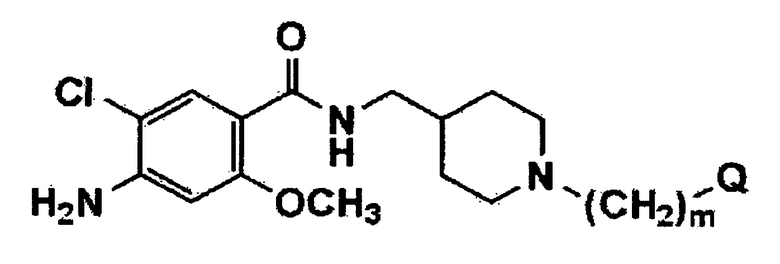
[Формула 2]
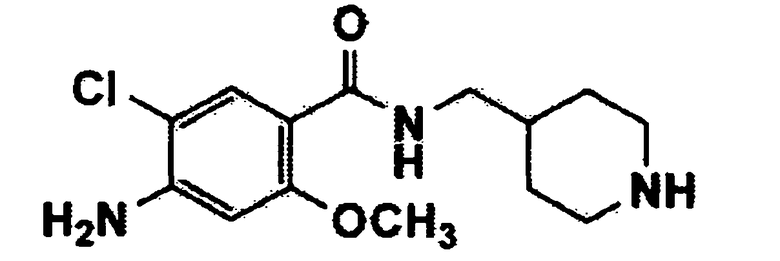
[Формула 3]

где m и Q имеют значения, указанные в п.1, и Y представляет собой атом галогена или C1-C4 алкилсульфонат.
4. Способ по п.3, где основание выбрано из карбоната калия, йодида калия, триэтиламина, диизопропилэтиламина и их смеси.
5. Способ получения соединения формулы 1 или его фармацевтически приемлемой соли, включающий взаимодействие соединения формулы 2 или его фармацевтически приемлемой соли с соединением формулы 11 в присутствии восстанавливающего агента с получением соединения формулы 1.
[Формула 1]
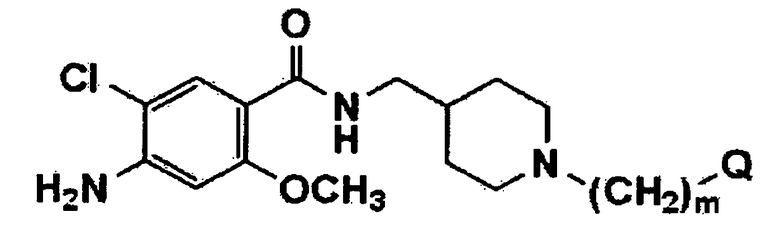
[Формула 2]
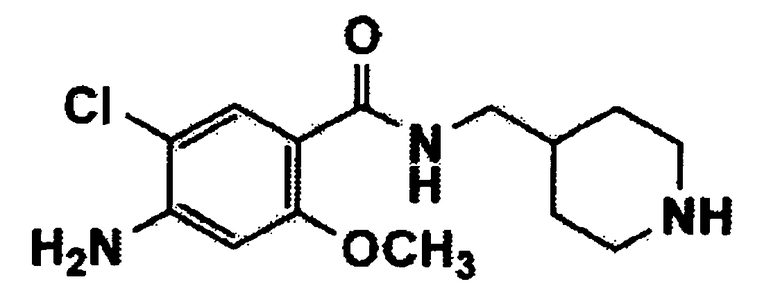
[Формула 11]

где Q имеет значения, указанные в п.1, и m обозначает целое число 1.
6. Способ по п.5 где восстанавливающий агент представляет собой цианборгидрид натрия и уксусную кислоту или боргидрид натрия.
7. Способ по п. 3 или 5, где соединение формулы 2 или его фармацевтически приемлемую соль получают путем:
(1) взаимодействия соединения формулы 4 с реагентом для введения аминозащитной группы по амину в 1-м положении пиперидинового кольца соединения формулы 4, с получением таким образом соединения формулы 5;
(2) взаимодействия гидрокси соединения формулы 5 с N-бромсукцинимидом и тетрабромидом углерода или с C1-C4 алкилсульфонилгалогенидом в присутствии основания с получением соединения формулы 6;
(3) взаимодействия заместителя Y соединения формулы 6 с азидом натрия с получением соединения формулы 7;
(4) восстановления азидо заместителя соединения формулы 7 до амина в присутствии восстановителя с получением соединения формулы 8;
(5) взаимодействия соединения формулы 8 с соединением формулы 9 в присутствии реагента, индуцирующего образование амидной связи, с получением соединения формулы 10; и
(6) удаления аминозащитной группы пиперидинового кольца соединения формулы 10 в присутствии основания или кислоты.
[Формула 2]

[Формула 4]
[Формула 5]

[Формула 6]
[Формула 7]
[Формула 8]

[Формула 9]
[Формула 10]
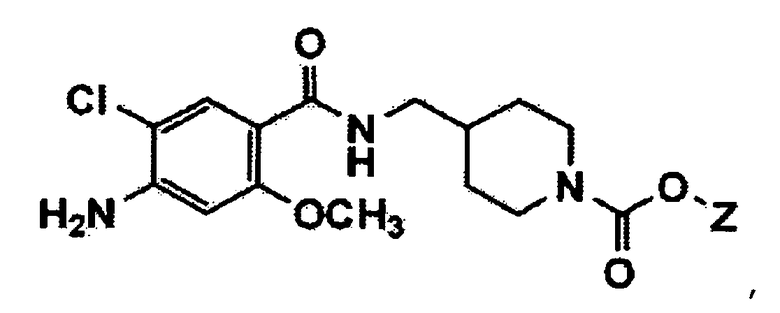
где Y представляет собой атом галогена или C1-C4 алкилсульфонат, и Z представляет собой C1-C4 алкил.
8. Способ по п.7, где реагент для введения аминозащитной группы на стадии (1) представляет собой ди-трет-бутил дикарбонат или этил хлорформиат в присутствии основания третичного амина.
9. Способ по п.7, где C1-C4 алкилсульфонилгалогенид на стадии (2) представляет собой метансульфонилхлорид, метансульфонилбромид или метансульфонилфторид.
10. Способ по п.7, где восстанавливающий агент на стадии (4) представляет собой трифенилфосфин или литийалюминийгидрид.
11. Способ по п.7, где реагент, индуцирующий образование амидной связи, используемый на стадии (5), выбран из гидрохлорида N-(3-диметиламинопропил)-N′-этилкарбодиимида и 1-гидроксибензотриазола в присутствии основания, этилхлорформиата в присутствии основания, или карбодиимидазола в отсутствие основания.
12. Способ по п.7, где основание или кислота на стадии (6) выбраны из хлористоводородной кислоты, трифторуксусной кислоты или гидроксида калия.
13. Агонист 5-НТ4 рецептора, включающий соединение формулы 1:
[Формула 1]
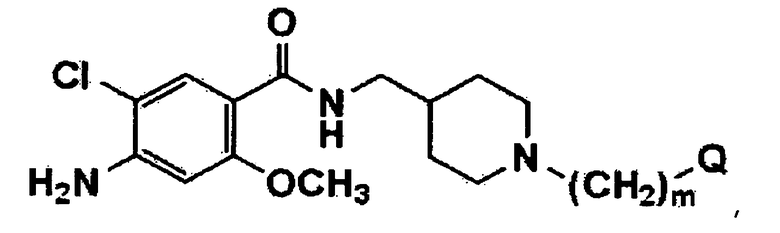
где m обозначает целое число от 1 до 5; и Q представляет собой гетероароматическое кольцо или фенил, где гетероароматическое кольцо выбрано из группы, состоящей из триазола, тетразола, индола, имидазола, пиридина и пиррола, и независимо замещено 0, 1, 2 или 3 заместителями, выбранными из С1-С4алкила, С1-С4алкокси, гидрокси и галогена, и
где фенил независимо замещен 1, 2 или 3 заместителями, выбранными из гидрокси и фтора; или его фармацевтически приемлемая соль, в качестве активного ингредиента.
14. Агонист по п.13, где соединение формулы 1 или его фармацевтически приемлемая соль представляет собой соединение, выбранное из группы, состоящей из следующих соединений:
(1) N-((1-(3-(1,2,4-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(2) N-((1-(3-(тетразол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(3) N-((1-(3-(индол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(4) N-((1-(3-(2-метилимидазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(5) N-((1-(5-(индол-1-ил)пентил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(6) N-((1-(5-(1,2,3-триазол-1-ил)пентил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(7) N-((1-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(8) N-((1-(3-(1,2,3-триазол-2-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(9) N-((1-(пиридин-3-илметил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(10) N-((1-((1-метилиндол-3-ил)метил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(11) N-((1-(имидазол-2-илметил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(12) N-((1-((1-метилпиррол-2-ил)метил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(13) N-((1-(4-фторбензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(14) N-((1-(4-гидроксибензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(15) N-((1-(2-(индол-3-ил)этил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамид,
(16) гидрохлорид N-((1-(3-(тетразол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(17) гидрохлорид N-((1-(5-(1,2,3-триазол-1-ил)пентил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(18) гидрохлорид N-((1-(3-(1,2,3-триазол-1-ил)пропил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(19) гидрохлорид N-((1-((1-метилиндол-3-ил)метил)пиперидин-4-ил) метил)-4-амино-5-хлор-2-метоксибензамида,
(20) гидрохлорид N-((1-(4-фторбензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида,
(21) гидрохлорид N-((1-(4-гидроксибензил)пиперидин-4-ил)метил)-4-амино-5-хлор-2-метоксибензамида, и их фармацевтически приемлемые соли.
15. Агонист по любому из пп. 13 или 14, где агонист 5-НТ4 рецептора представляет собой композицию, содержащую соединение формулы I по п.1 или 2 в качестве активного ингредиента и фармацевтически приемлемый носитель, и где указанная композиция предназначена для предупреждения или лечения заболевания, выбранного из гастроэзофагеальной рефлюксной болезни, желудочно-кишечного заболевания, расстройства перистальтики желудка, неязвенной диспепсии, функциональной диспепсии, синдрома раздраженной толстой кишки (IBS), констипации, диспепсии, эзофагита, желудочно-пищеводного заболевания, тошноты, заболевания центральной нервной системы, болезни Альцгеймера, когнитивного нарушения, рвоты, мигрени, неврологического заболевания, боли, сердечно-сосудистого расстройства, сердечной недостаточности, сердечной аритмии, диабета или синдрома апноэ.
16. Способ предупреждения, лечения или облегчения заболевания, вызванного ослабленной эффективностью 5-НТ4 рецептора, включающий введение агониста 5-НТ4 рецептора, содержащего в качестве активного ингредиента соединение формулы 1 по п.1 или 2 или его фармацевтически приемлемую соль, млекопитающему, включая человека, нуждающегося в агонистическом действии на 5-НТ4 рецептор.
17. Способ по п.16, где агонист 5-НТ4 рецептора представляет собой композицию, содержащую соединение формулы I по п.1 или 2 в качестве активного ингредиента и фармацевтически приемлемый носитель, и где указанная композиция предназначена для предупреждения или лечения заболевания, выбранного из гастроэзофагеальной рефлюксной болезни, желудочно-кишечного заболевания, расстройства перистальтики желудка, неязвенной диспепсии, функциональной диспепсии, синдрома раздраженной толстой кишки (IBS), констипации, диспепсии, эзофагита, желудочно-пищеводного заболевания, тошноты, заболевания центральной нервной системы, болезни Альцгеймера, когнитивного нарушения, рвоты, мигрени, неврологического заболевания, боли, сердечно-сосудистого расстройства, сердечной недостаточности, сердечной аритмии, диабета или синдрома апноэ.
| KATSUHIKO ITON et al., “Synthesis and pharmacological evaluation of carboxamide derivatives as selective serotoninergic 5-HT4 receptor agonists”, European Journal of Medicinal Chemistry, vol.34, no.4, 1999, pp.329-341 | |||
| SHUJI SONDA et al., “Design and synthesis of orally active benzamide derivatives as potent serotonin 4 receptor |

