Область техники
Настоящее изобретение относится к новым замещенным производным кислоты, которые модулируют уровень глюкозы в крови, уровень триглицеридов, уровень инсулина и уровень неэтерифицированных жирных кислот (NEFA) и, следовательно, особенно полезны для лечения диабета и ожирения, а также к способу лечения диабета, особенно диабета типа 2, так же как и гипергликемии, гиперинсулинемии, гиперлипидемии, ожирения, атеросклероза и родственных болезней, при применении таких замещенных производных кислоты отдельно или в сочетании с другим антидиабетическим агентом и/или гиполипидемическим агентом.
Описание изобретения
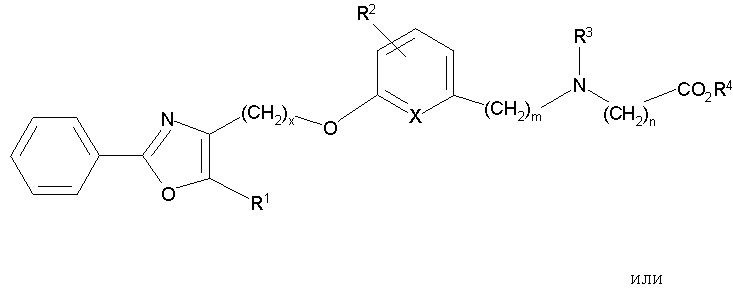
В соответствии с настоящим изобретением предложены замещенные производные кислоты формулы I
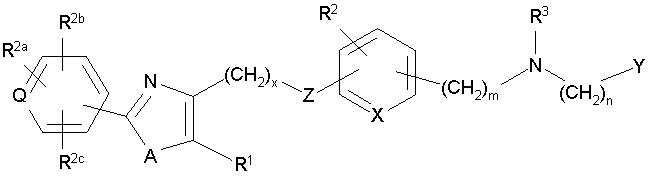
где x имеет значение 1, 2, 3 или 4; m имеет значение 1 или 2; n имеет значение 1 или 2;
Q представляет собой С или N;
А представляет собой О или S;
Z представляет собой О или связь;
R1 представляет собой низший алкил;
X представляет собой СН;
R2 представляет собой Н;
R2a, R2b и R2c могут быть одинаковыми или различными и их выбирают из Н, алкокси, галогена;
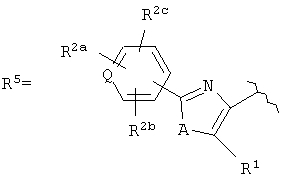
R3 представляет собой арилоксикарбонил, алкилоксикарбонил, алкил(галоген)арилоксикарбонил, алкилокси(галоген)арилоксикарбонил, циклоалкиларилоксикарбонил, циклоалкилоксиарилоксикарбонил, арилкарбониламино, алкилсульфонил, циклогетероалкилоксикарбонил, гетероарилалкенил, алкоксиарилоксикарбонил, арилалкилоксикарбонил, алкиларилоксикарбонил, галогеналкоксиарилоксикарбонил, алкоксикарбониларилоксикарбонил, арилалкенилоксикарбонил, арилоксиарилалкилоксикарбонил, арилалкенилсульфонил, гетероарилсульфонил, арилсульфонил, арилалкениларилалкил, арилалкоксикарбонилгетероарилалкил, гетероарилоксиарилалкил;
циклоалкил обозначает насыщенные или частично ненасыщенные (содержащие 1 или 2 двойные связи) циклические углеводородные группы, содержащие от 1 до 3 колец, включая моноциклоалкил, бициклоалкил и трициклоалкил, содержащие в общем от 3 до 20 атомов углерода, образующих кольца, предпочтительно от 3 до 10 атомов углерода, образующих кольца;
арил обозначает моноциклические или бициклические ароматические группы, содержащие от 6 до 10 атомов углерода в кольце, выбранном из фенила или нафтила, включая 1-нафтил и 2-нафтил и могут необязательно включать от одного до трех дополнительных колец, сконденсированных с карбоциклическим или гетероциклическим кольцом, выбранным из арильного, циклоалкильного, гетероарильного или циклогетероалкильного кольца;
гетероарил обозначает 5- или 6-членное ароматическое кольцо, которое содержит 1, 2, 3 или 4 гетероатома, выбранных из азота, кислорода или серы, и такие кольца сконденсированы с арильным кольцом;
циклогетероалкил обозначает циклогетероалкильную группу, 5- или 6-членное насыщенное или частично ненасыщенное гетероциклическое кольцо, которое содержит 1, 2 или 3 гетероатома, выбранных из N, О или S, связанное через С атом или гетероатом с (СН2)p цепью; р имеет значение 0 или 1;
гетероарилалкенил обозначает гетероарильную группу, связанную через С атом или гетероатом с алкиленом или алкениленом;
Y представляет собой CO2R4, где R4 представляет собой Н или алкил,
где во всех указанных группах алкил представляет собой С1-8алкил, алкенил представляет собой С2-8алкенил, алкилен представляет собой C1-8алкилен, алкенилен представляет собой С2-8алкенилен,
включая все их стереоизомеры и фармацевтически приемлемые соли, при условии, что если А представляет собой О, тогда R3 не является арилоксикарбонилом или алкоксиарилоксикарбонилом.
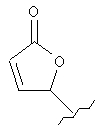
Соединения формулы I по изорбетению могут иметь структуру
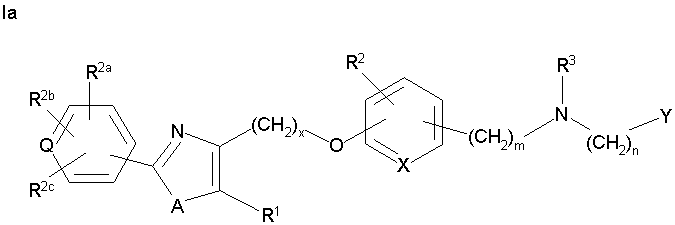
или
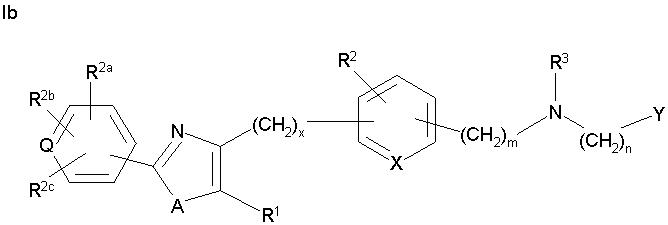
Предпочтительными являются соединения формулы I по изобретению, имеющие структуру
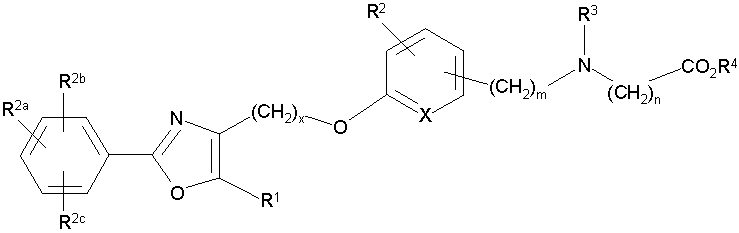
Более предпочтительными являются соединения формулы I по изобретению, имеющие структуру

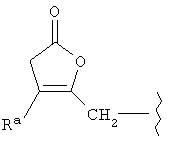
В указанных выше соединениях наиболее предпочтительно, когда R2а представляет собой алкокси, но более предпочтительно Н, Z представляет собой связь, но более предпочтительно О, (СН2)x представляет собой СН2, (СН2)2, (СН2)3 или 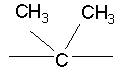 , (СН2)m представляет собой СН2, или
, (СН2)m представляет собой СН2, или 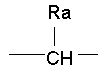 (где Ra представляет собой алкил, такой как метил, или алкенил, такой как
(где Ra представляет собой алкил, такой как метил, или алкенил, такой как 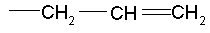 или
или  , (СН2)n представляет собой СН2, R1 представляет собой низший алкил, предпочтительно -СН3, R2 представляет собой Н, R2a представляет собой Н, R4 представляет собой Н, X представляет собой СН и R3 представляет собой арилалкилоксикарбонил, арилгетероарилалкил, арилоксиарилалкил, арилалкил, арилоксикарбонил, галогенарилоксикарбонил, алкоксиарилоксикарбонил, алкиларилоксикарбонил, арилоксиарилоксикарбонил, гетероарилоксиарилалкил, гетероарилоксикарбонил, арилоксиарилкарбонил, арилалкенилоксикарбонил, циклоалкиларилоксикарбонил, арилалкиларилкарбонил, гетероарил-гетероарилалкил, циклоалкилоксиарилоксикарбонил, гетероарил-гетероарилкарбонил, алкилоксиарилоксикарбонил, арилалкилсульфонил, арилалкенилсульфонил, алкоксиарилалкил, арилтиокарбонил, циклогетероалкилалкилоксикарбонил, циклогетероалкилоксикарбонил или полигалогеналкиларилоксикарбонил, где указанные выше предпочтительные группы могут быть необязательно замещены.
, (СН2)n представляет собой СН2, R1 представляет собой низший алкил, предпочтительно -СН3, R2 представляет собой Н, R2a представляет собой Н, R4 представляет собой Н, X представляет собой СН и R3 представляет собой арилалкилоксикарбонил, арилгетероарилалкил, арилоксиарилалкил, арилалкил, арилоксикарбонил, галогенарилоксикарбонил, алкоксиарилоксикарбонил, алкиларилоксикарбонил, арилоксиарилоксикарбонил, гетероарилоксиарилалкил, гетероарилоксикарбонил, арилоксиарилкарбонил, арилалкенилоксикарбонил, циклоалкиларилоксикарбонил, арилалкиларилкарбонил, гетероарил-гетероарилалкил, циклоалкилоксиарилоксикарбонил, гетероарил-гетероарилкарбонил, алкилоксиарилоксикарбонил, арилалкилсульфонил, арилалкенилсульфонил, алкоксиарилалкил, арилтиокарбонил, циклогетероалкилалкилоксикарбонил, циклогетероалкилоксикарбонил или полигалогеналкиларилоксикарбонил, где указанные выше предпочтительные группы могут быть необязательно замещены.
Предпочтительные соединения по изобретению включают следующие:

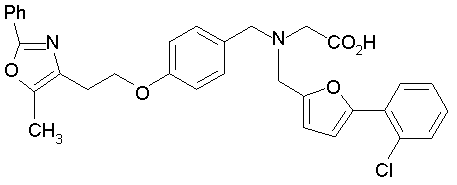
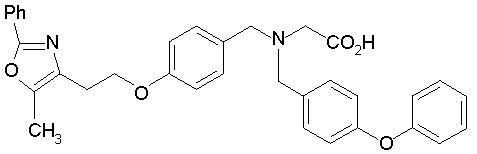

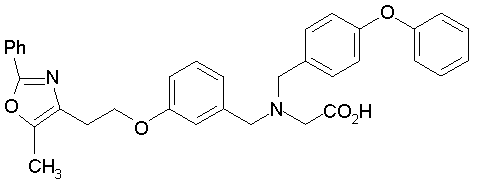
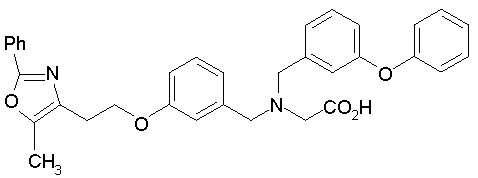
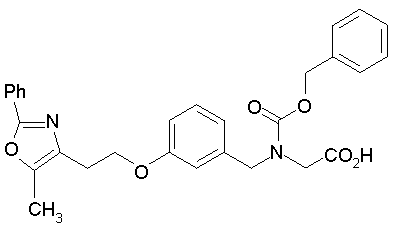
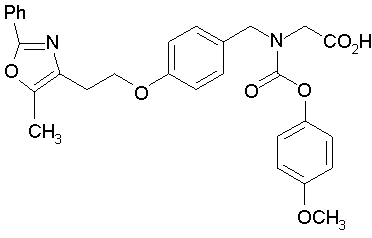
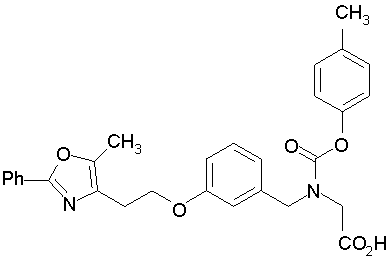
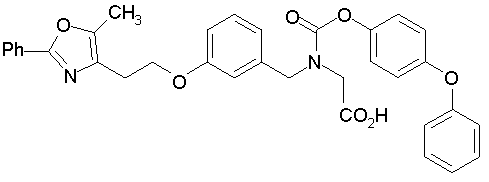
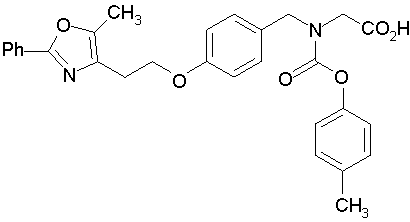

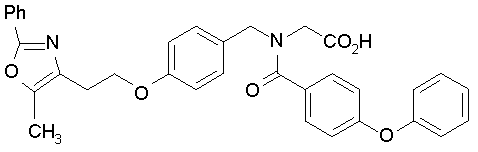
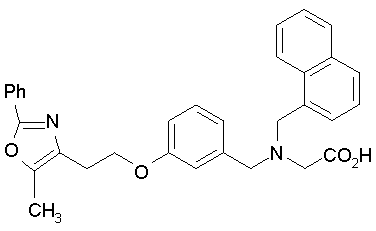
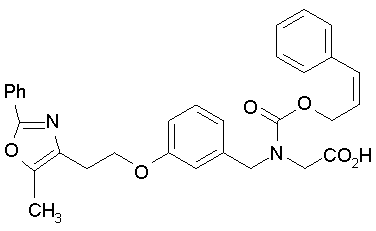
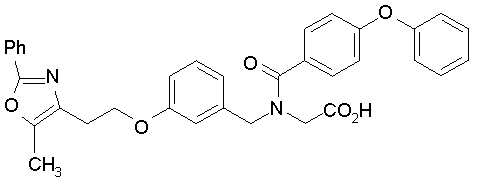
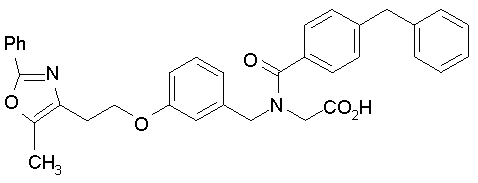
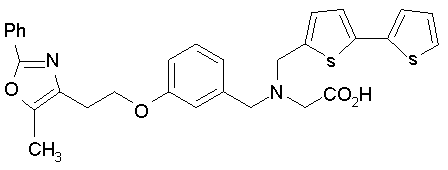
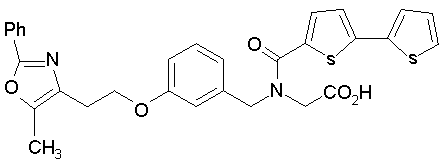
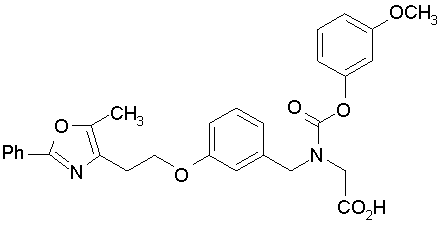
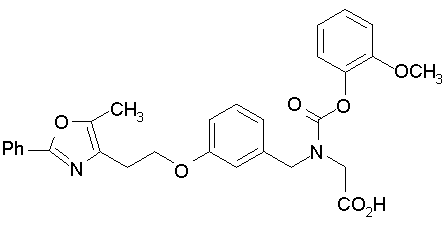
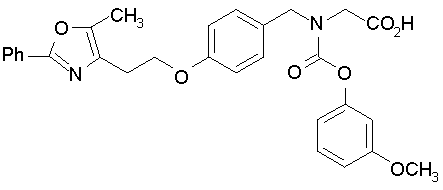
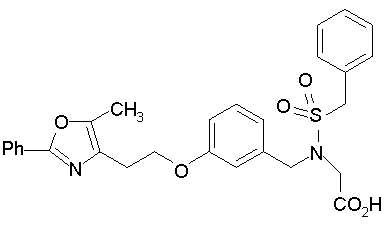
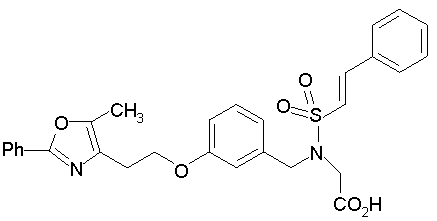

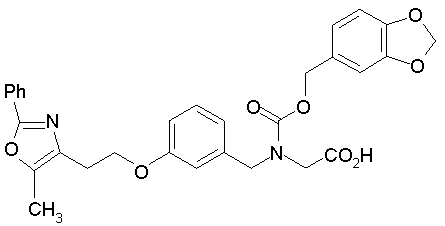

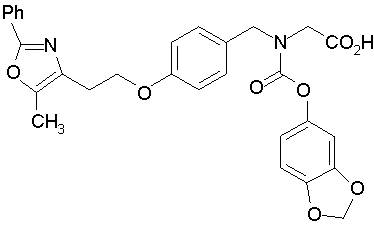
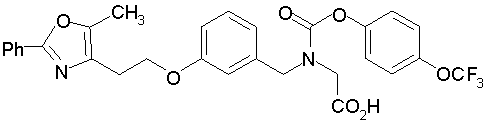
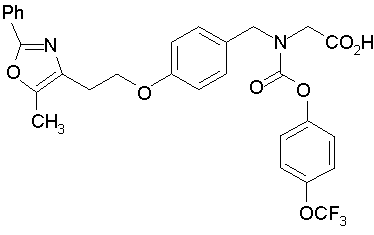
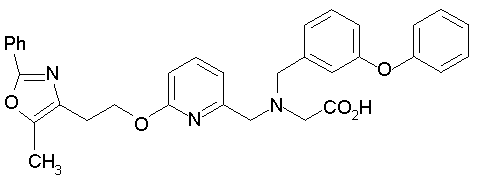
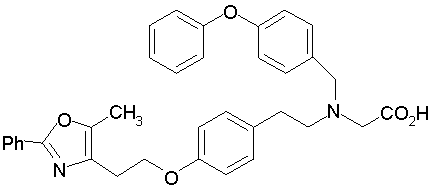
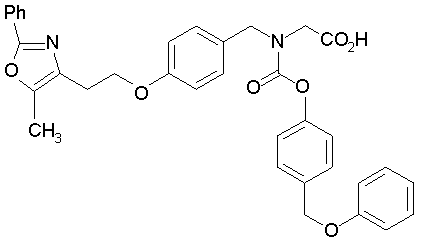
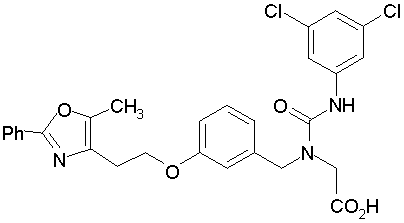
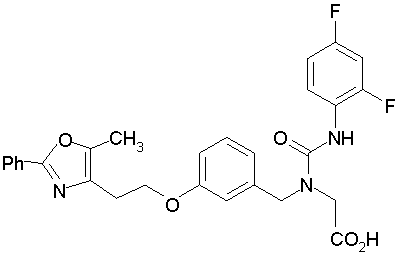
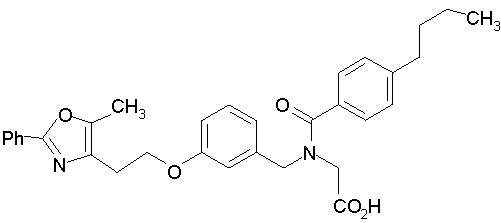
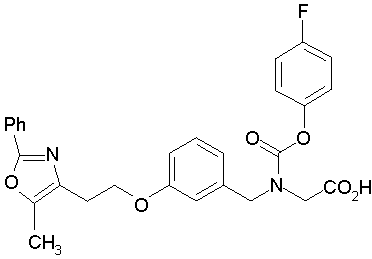
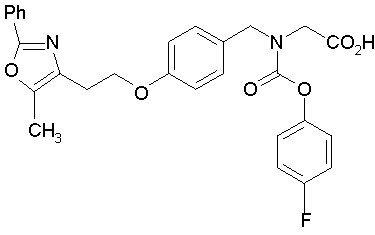
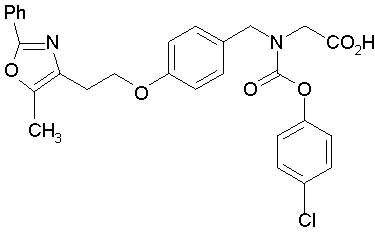
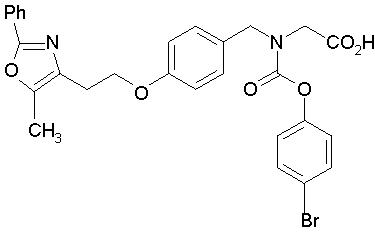
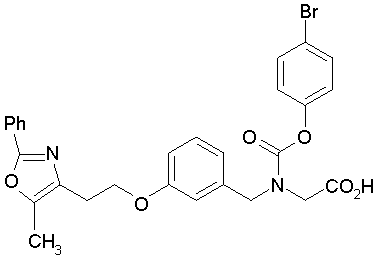

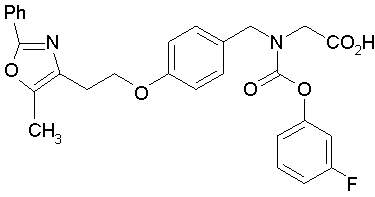
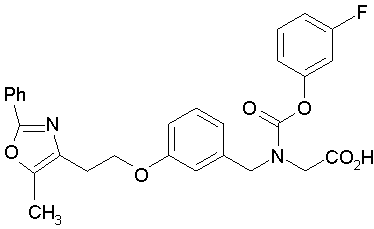
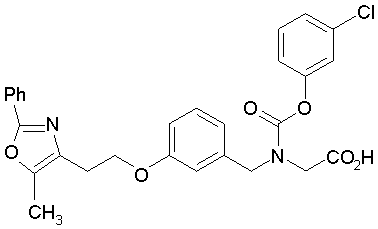
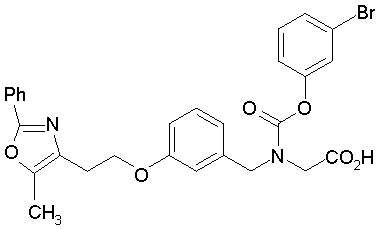
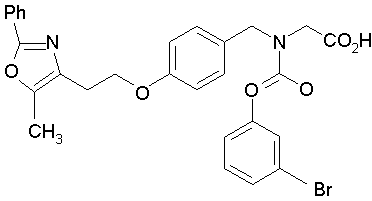
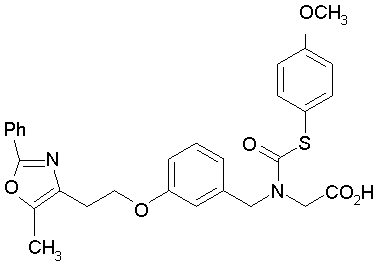
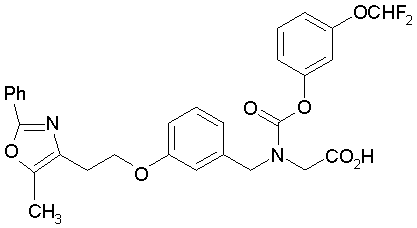
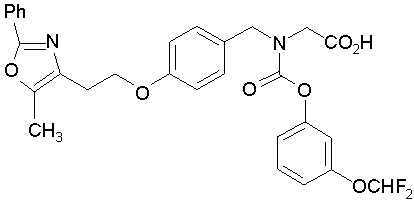
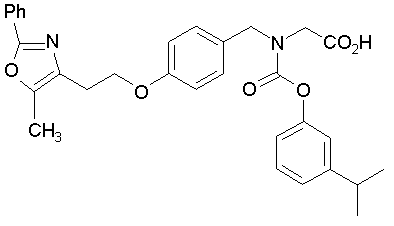
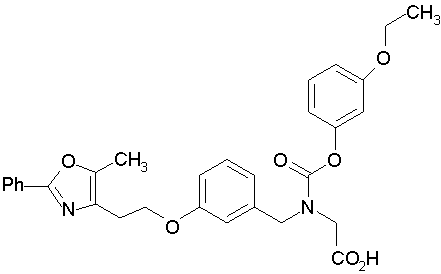
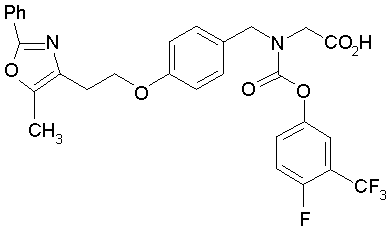
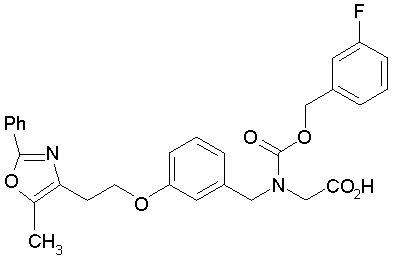
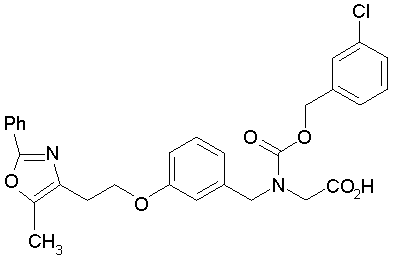


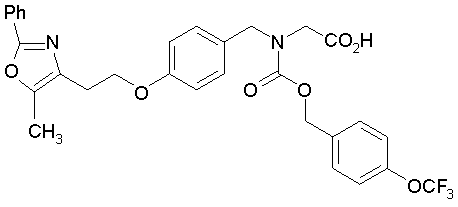
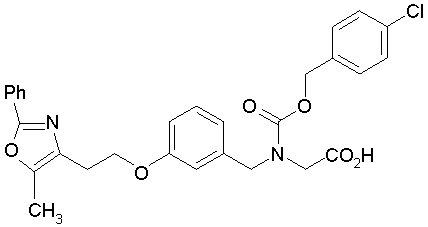
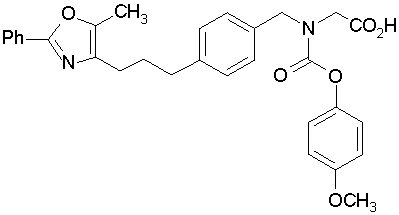

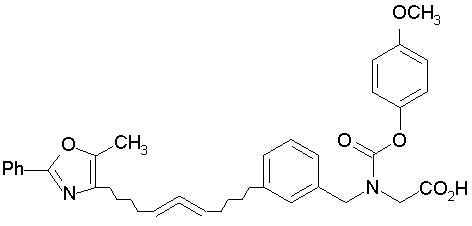

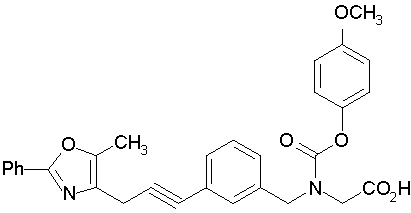
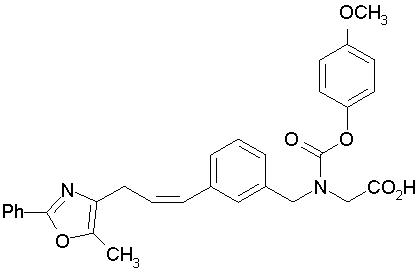
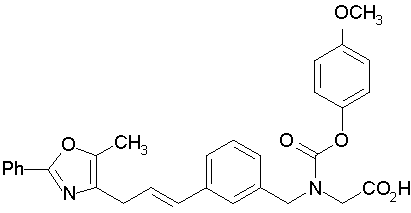
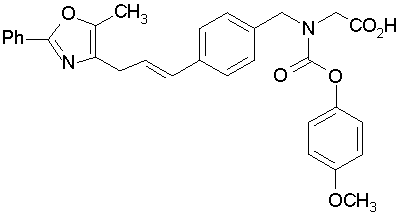
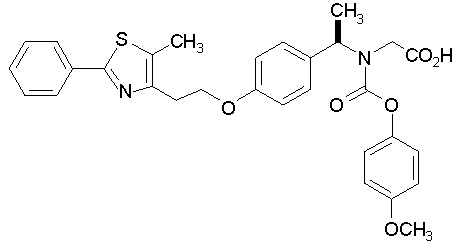



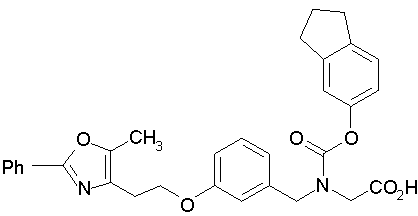

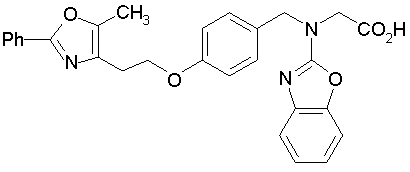
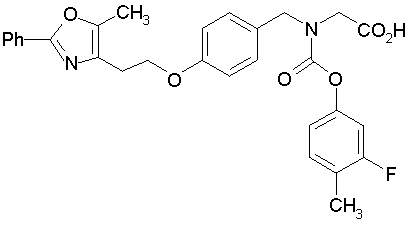
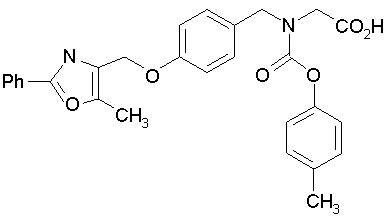
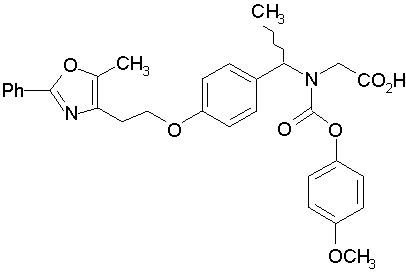
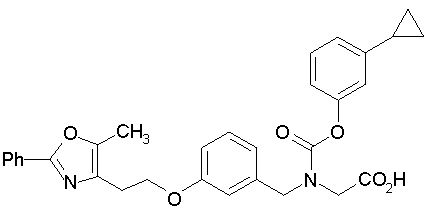
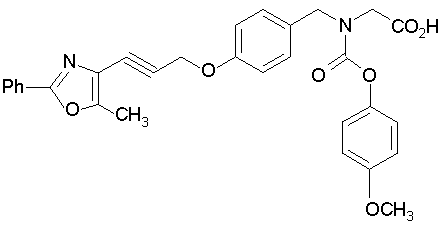
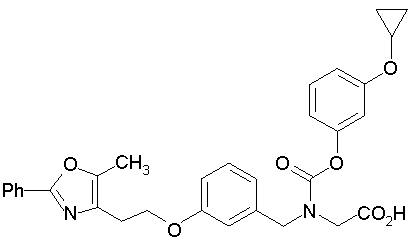
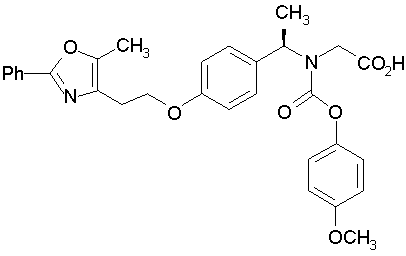

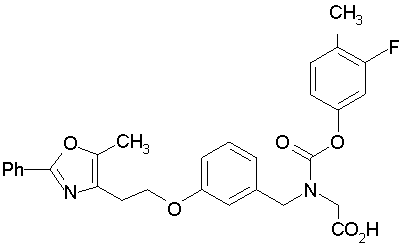
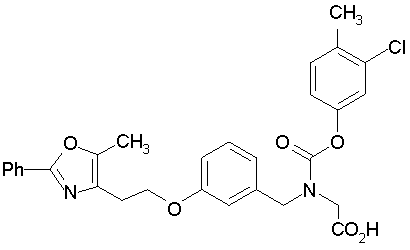
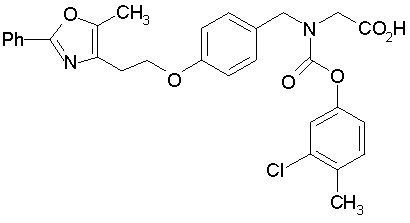
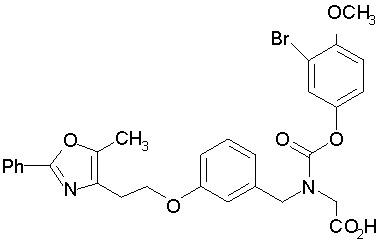

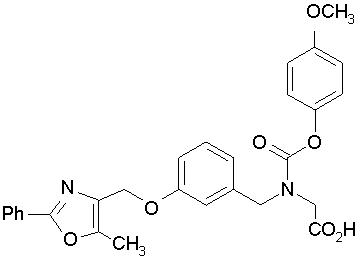
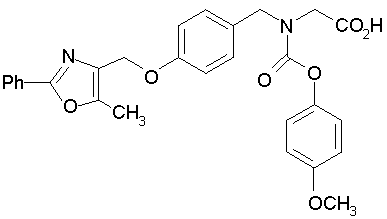
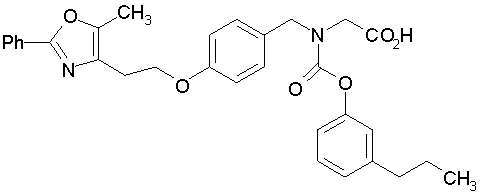

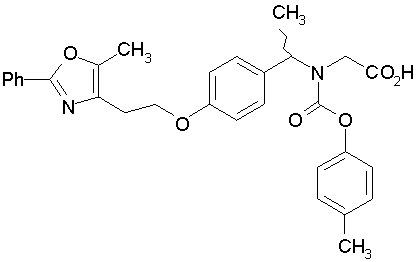
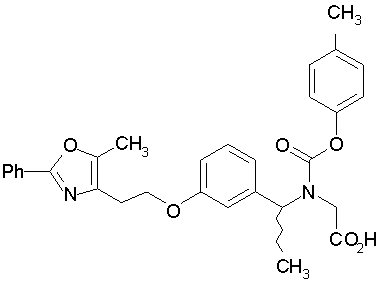

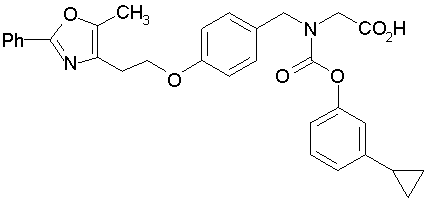
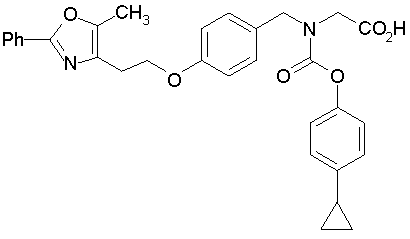
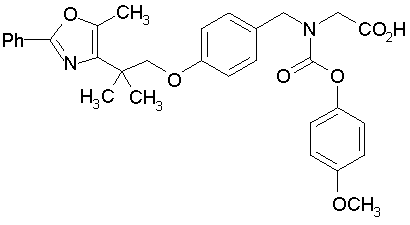
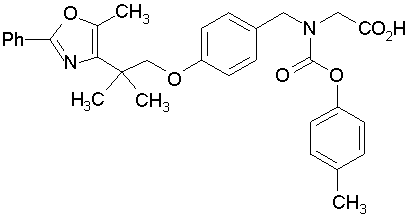
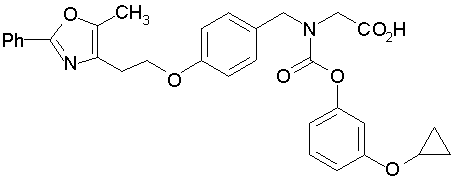
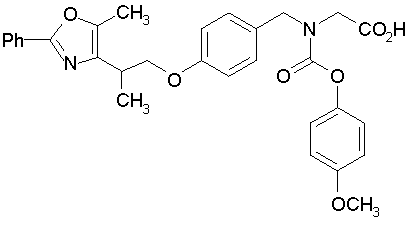
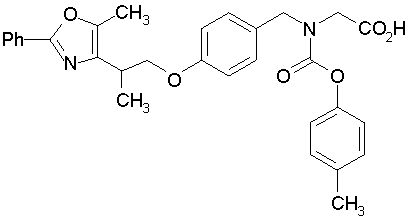
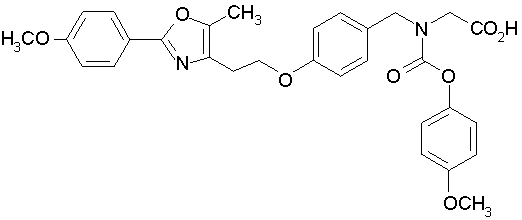
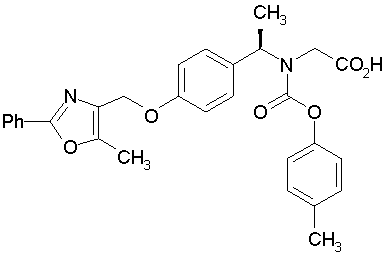
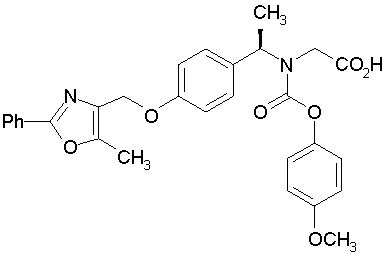
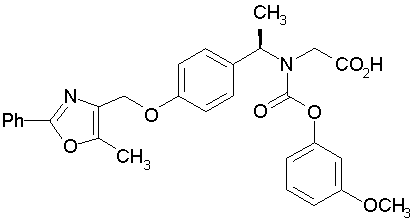
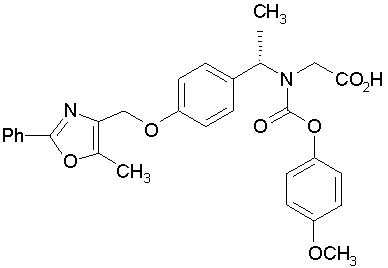
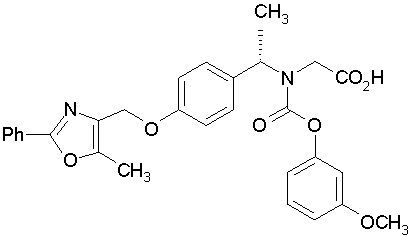

Подробное описание изобретения
Соединения формулы I настоящего изобретения могут быть получены в соответствии со следующими общими схемами синтеза, также как и с опубликованными в литературе релевантными способами, которые используют специалисты данного уровня техники. Примеры реагентов и методики этих реакций описаны здесь далее, а также в рабочих примерах. Защита и снятие защиты в схемах, приведенных ниже, может осуществляться способами, хорошо известными из уровня техники (см., например, Greene, Т. W. и Wuts, P. G. M., Protecting Groups in Organic Synthesis, 3rd Edition, 1999 [Wiley]).
Схема 1 описывает общий синтез аминокислот, описанных в настоящем изобретении. Спирт II (R5(CH2)xOH) (наиболее предпочтительным является 2-фенил-5-метилоксазол-4-этанол) конденсируют с гидроксиарил- или гетероарилальдегидом III (предпочтительно 3- или 4-гидроксибензальдегидом) в стандартных условиях реакции Митсунобу (Mitsunobu) (например, Mitsunobu, О., Synthesis, 1981, 1). Образующийся альдегид IV затем подвергают восстановительному аминированию, используя методики, известные из уровня техники (например, Abdel-Magid и др., J. Org. Chem. 1996, 61, 3849), с гидрохлоридом α-аминоэфира V. PG на схеме 1 обозначает предпочтительную защитную группу для карбоновой кислоты, такую, как метиловый или трет-бутиловый эфир. Образующийся вторичный аминоэфир VI затем подвергают повторному восстановительному аминированию, используя способы, известные из уровня техники (например, Abdel-Magid и др., J. Org. Chem. 1996, 61, 3849), с R3a альдегидом VII. Конечное снятие защитных групп с эфира карбоновой кислоты в стандартных условиях, известных из уровня техники (Greene), при применении основных условий (для метиловых эфиров) или кислых условий (для трет-бутиловых эфиров) затем приводит к получению желаемых продуктов, которыми являются аминокислоты ID.
Альтернативный путь к альдегиду IV показан на схеме 1А. Спирт II (R5(CH2)хОН) (из которых наиболее предпочтительньм является 2-[2-фенил-5-метилоксазол-4-ил]этанол) обрабатывают метансульфонилхлоридом с получением соответствующего мезилата VIII. Мезилат затем алкилируют в стандартных основных условиях с помощью гидроксиарил- или гидроксигетероарилальдегида III с получением альдегида IV.
Альтернативный путь к аминокислотам IF показан на схеме 2. Со вторичного аминоэфира VI снимают защитные группы в стандартных условиях (основные условия, если защитной группой (PG) является метил; кислые условия, если PG является трет-бутилом) с получением соответствующей аминокислоты IE. Восстановительное аминирование с R3a альдегидом в аналогичных условиях, как описано на схеме 1, приводит к получению желаемых третичных продуктов аминокислот IF в качестве продуктов.
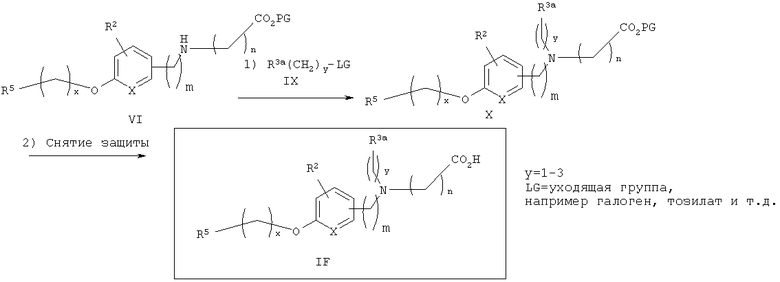
Альтернативно, как показано на схеме 3, третичные аминокислоты IF также могут быть получены алкилированием вторичного аминоэфира VI алкилирующим агентом IX (с подходящей уходящей группой (LG), такой, как галоген, мезилат или тозилат) в стандартных условиях, известных из уровня техники, с последующим стандартным снятием защитных групп с эфира карбоновой кислоты Х с получением аминокислот IF.
Как показано на схеме 4, третичная аминокислота IF может быть образована через восстановительное аминирование вначале R3a альдегида XI с подходящим гидрохлоридом аминоэфира V. Образующийся вторичный аминоэфир XII затем подвергают восстановительному аминированию с подходящим альдегидом алкила, арила или гетероарила IV (как на схеме 1) с последующим снятием защитных групп с эфира карбоновой кислоты с получением желаемых аналогов аминокислоты IF.
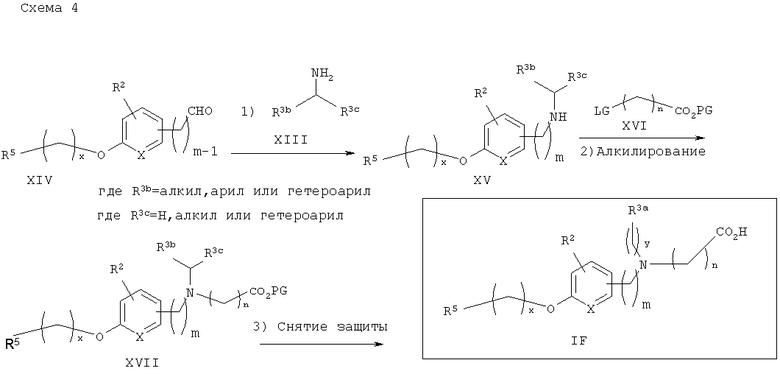
Дальнейшее замещение аминокислот показано на общей схеме синтеза 5. Восстановительное аминирование подходящего амина XIII арил- или гетероарилальдегидом XIV в стандартных условиях приводит к получению соответствующего вторичного амина XV, который затем подвергают реакции с галогенэфиром XVI (например, трет-бутилбромацетатом) с получением соответствующего α-аминоэфира XVII. У полученного аминоэфира XVII затем снимают защитные группы в стандартных условиях с получением желаемых аналогов аминокислоты IF.
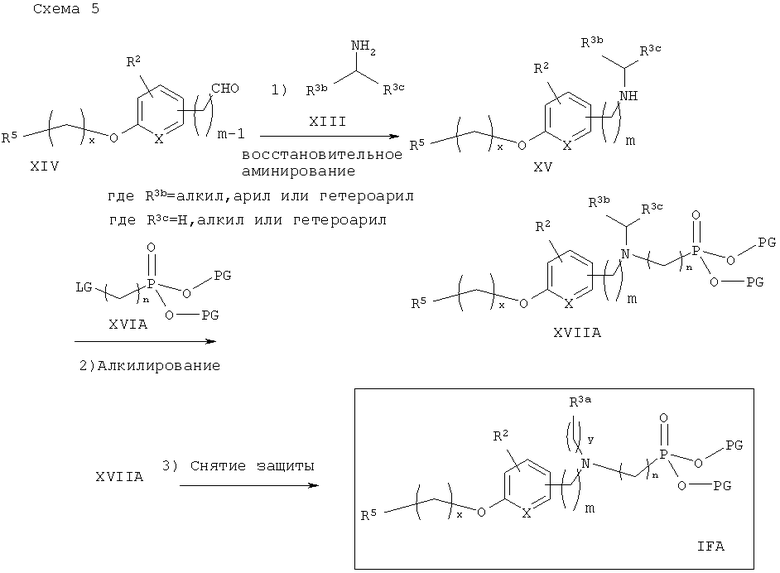
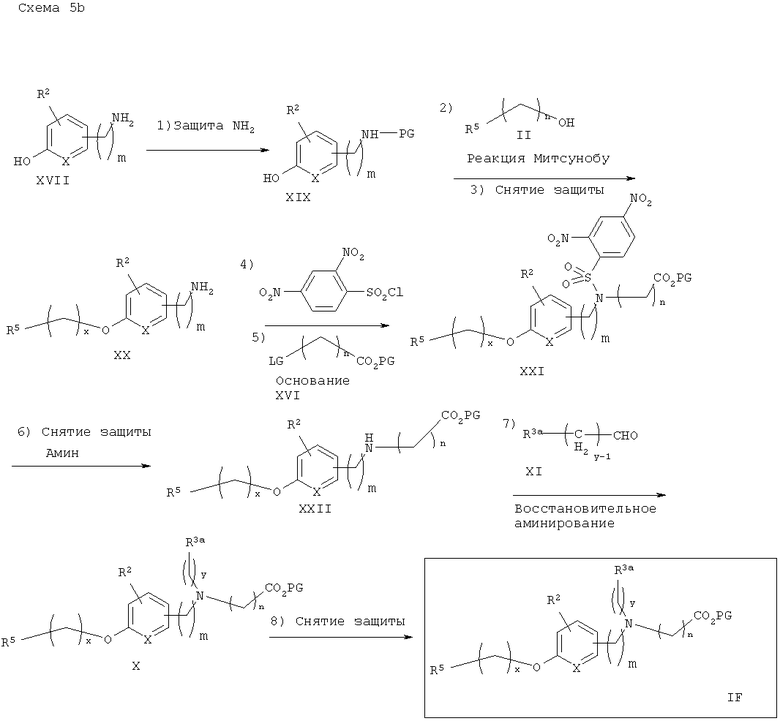
Способ синтеза на схеме 5 также показывает общую схему синтеза соответствующих аминофосфорных кислот IFA, как показано на схеме 5а. Вторичный амин XV подвергают реакции с подходящим защищенным галогенфосфатом XVIA с получением соответствующего аминофосфатного эфира XVIIA, с которого затем снимают защитные группы в стандартных условиях (Greene & Wuts) с получением аминофосфорной кислоты IFA. На схеме 5b показан синтез аминофосфиновых кислот IFB, который также включают реакцию подходяще защищенного галогенфосфинатного эфира XVIB со вторичным амином XV. Снятие защитных групп образующегося аминофосфинатного эфира затем приводит к получению фосфиновой кислоты IFB.
Альтернативный способ к последовательности на схеме 5 показан на схеме 6. Гидроксиарил- или гетероариламин XVIII селективно защищают по азоту с получением защищенного амина XIX. Предпочтительный R5(CH2)nOH (II) затем подвергают реакции с XIX в условиях реакции Митсунобу (Mitsunobu) с получением соответствующего простого эфира, с последующим снятием защитных групп амина, с получением свободного амина XX. Свободный амин XX затем активируют стандартной активирующей группой (2,4-динитробензолсульфонамид; Т.Fukuyama и др., Tetrahedron Lett. 1997, 38, 5831) и затем обрабатывают α-галогенэфиром XVI, как показано на схеме 5. С 2,4-динитробензолсульфонамида XXI снимают защитные группы в условиях, известных из уровня техники (Т. Fukuyama и др., Tetrahedron Lett., 1997, 38, 5831) с получением вторичного α-аминоэфира XXII, который затем подвергают восстановительному аминированию с R3a альдегидом XI с последующим снятием защитных групп с эфира Х с получением желаемых аналогов IF.
Схема 7 описывает альтернативный общий путь к аналогам аминокислоты IG. Гидроксиарил или гетероарилальдегид III подвергают обычным условиям восстановительного аминирования с подходящим гидрохлоридом аминоэфира V. Образующийся вторичный аминоэфир XXIII функционализируют в данном случае повторным восстановительным аминированием с R3a альдегидом VII с получением соответствующего третичного гидроксиаминоэфира XXTV. Его можно подвергнуть реакции Митсунобу с соответствующим спиртом II (R5(CH2)nOH) с последующим снятием защитных групп эфира XXV, что приводит к получению желаемых аналогов IG.
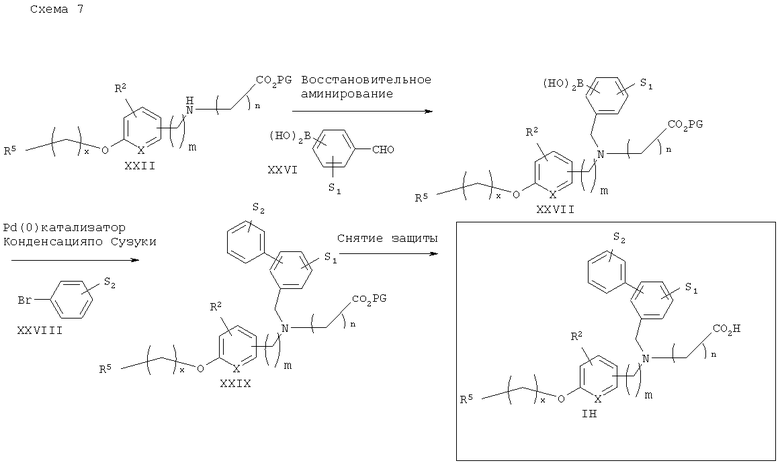
Схема 8 описывает общий синтез диарил- и арилгетероарилзамещенных аналогов аминокислоты IH. Вторичный аминоэфир XXII подвергают восстановительному аминированию с подходяще замещенной формилфенилбороновой кислотой XXVI в стандартных условиях с получением соответствующего третичного аминоэфира бороновой кислоты XXVII. Арилбороновая кислота XXVII может затем подвергаться конденсации по Сузуки (Suzuki) (например, условия описаны в Gibson, S.E., Transition Metals in Organic Synthesis, A Practical Approach, pp.47-50, 1997) с арил- или гетероарилгалогенидами XXVIII (особенно бромидами) с получением подходящих межконденсированных диарильных продуктов XXIX. Снятие защитных групп с аминоэфира XXIX приводит к получению желаемых аналогов аминокислоты IH.
Схема 9 описывает общий синтез диарил- и арилгетероарилэфирозамещенных аналогов аминокислоты IJ. Третичный аминоэфир бороновой кислоты XXVII, который описан на схеме 8, может быть подвергут конденсации с подходяще замещенными фенолами XXX в условиях, известных из уровня техники (D.A.Evans и др., Tetrahedron Lett., 1998, 39, 2937) с получением соответствующих диарил- или арилгетероарилэфиров XXXI, которые после снятия защитных групп приводят к получению аналогов аминокислот IJ.
Альтернативно, как показано на схеме 10, восстановительное аминирование вторичного аминоэфира XXII с подходяще замещенным гидроксиарил- или гидроксигетероарилальдегидом XXXII приводит к получению соответствующего третичного феноламиноэфира XXXIII. Фенол XXXIII может далее подвергаться связыванию с подходящими арильными или гетероарильными бороновными кислотами XXXIV в условиях, известных из уровня техники (D.A.Evans и др., Tetrahedron Lett., 1998, 39, 2937) с получением соответствующих диарил- или арилгетероарилэфироамино сложных эфиров XXXI. Желаемые аналоги IJ могут затем быть получены снятием защитных групп аминоэфира XXXI.
На схеме 11 показан синтез карбаматокислотных аналогов IK. Вторичный аминоэфир XXII может реагировать с подходящими хлорформиатами XXXV в условиях, известных из уровня техники (оптимально в CH2Cl2 или CHCI3 в присутствии основания, такого, как Et3N) с получением соответствующих карбаматоэфиров. Желаемые аналоги IK затем получают после снятия защитных групп с карбаматоэфира. Альтернативно, вторичный аминоэфир XXII может реагировать с фосгеном с получением соответствующего карбамоилхлорида XXXVI. Указанный промежуточный карбамоилхлорид XXXVI может реагировать с R3a-OH (XXXVII) (оптимально замещенными фенолами) с получением соответствующих карбамат-кислот IK после снятия защитных групп.
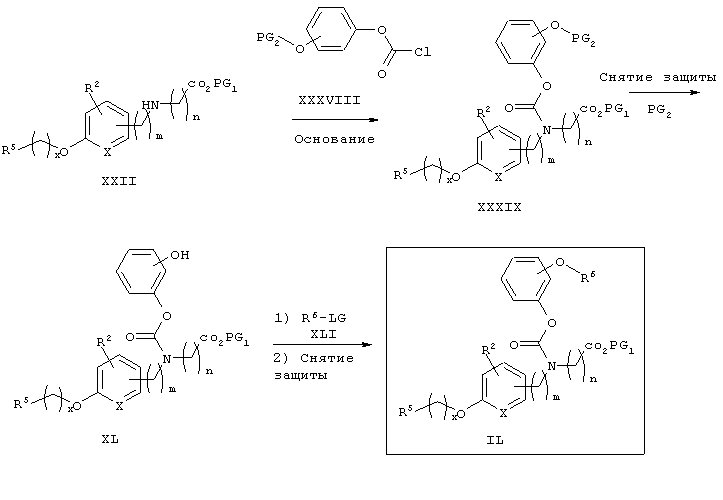
На схеме 12 показана дальнейшая функционализация арильных карбаматокислотных аналогов IK. Вторичный аминоэфир XXII подвергают реакции с арилхлорформиатом XXXVIII (содержащим защищенную гидроксигруппу) с получением XXXIX. С гидроксильной группы затем селективно снимают защиту в присутствии эфира с получением XL, затем алкилируют подходящим R6-LG (XLI) (где LG представляет собой галонид, мезилат или тозилат и R6 представляет собой наиболее предпочтительно CHF2- или СН3СН2-) в присутствии основания. Снятие защитных групп с эфира затем приводит к получению желаемых карбаматокислотных аналогов IL.
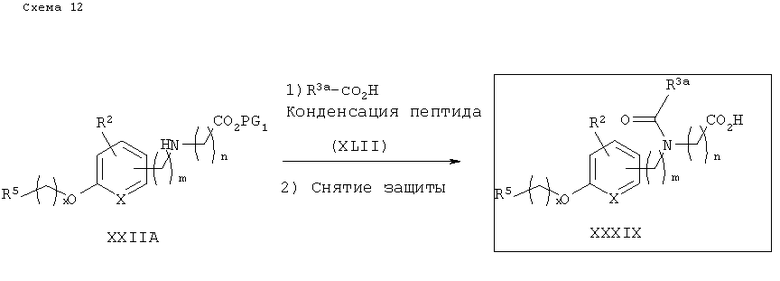
Вторичный аминоэфир XXIIA может быть функционирован замещенными арильными или алифатическими карбоновыми кислотами XLII, в стандартных условиях получения пептида, как показано на схеме 13. Реакции образования амидной связи проводят в соответствии со стандартными методиками получения пептида, известными из уровня техники. Оптимально реакцию осуществляют в растворителе, таком, как ДМФА, при температуре от 0°С до комнатной, используя 1-этил-3-(3-диметиламинопропил)карбодиимид (EDAC или EDCI или WSC), 1-гидроксибензотриазол (НОВТ) или 1-гидрокси-7-азабензотриазол (НОАТ) и основание, например основание Ханига (Hunig) (диизопропилэтиламин), N-метилморфолин или триэтиламин. Снятие защитных групп с амидоэфира затем приводит к получению желаемых амидокислотных аналогов IM.
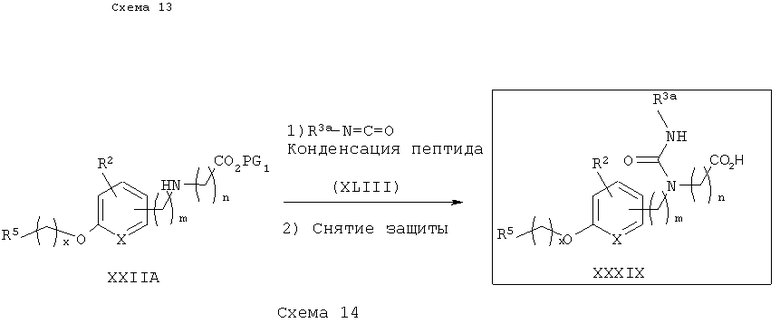
Вторичный аминоэфир XXIIA может также реагировать с алифатическими или арильными изоцианатами XLIII с получением соответствующих уреа-эфиров. Снятие защитных групп с уреа-эфиров приводит к получению желаемых уреа-кислотных аналогов IN, как показано на схеме 14. Альтернативно, как показано на схеме 15, промежуточный карбамоилхлорид XXXVI, описанный на схеме 11, может реагировать с подходящими алифатическими или арильньми аминами XLIV в присутствии третичного амина (например, Et3N) с получением три- или тетразамещенных мочевинокислотных аналогов IO или IP после снятия защитных групп с эфира.
Вторичный аминоэфир XXIIA может также реагировать с подходящими сульфонилхлоридами XL VI в стандартных условиях, известных из литературы (оптимально в присутствии основания, такого, как пиридин, чистого или в смеси с хлороформом в качестве сорастворителя), с последующим снятием защитных групп, с получением соответствующих сульфонамидных кислот IQ, как показано на схеме 16.
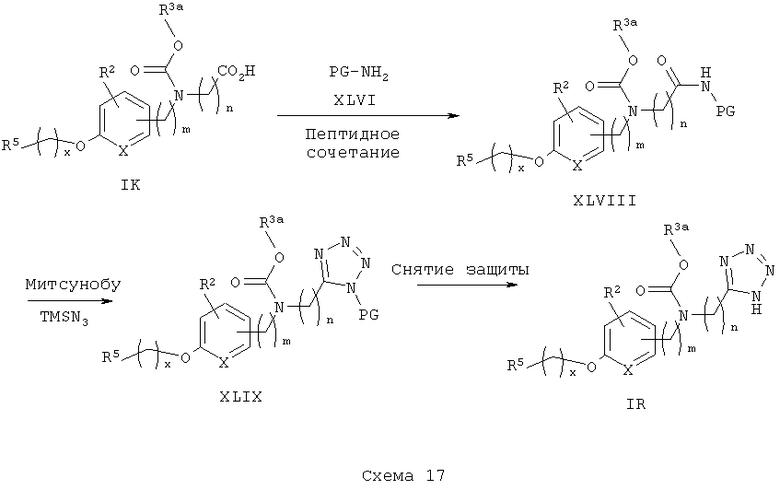
Замещение функциональной группы карбоновой кислоты в этих аналогах на тетразол может осуществляться, как показано на схеме 17. Кислотный аналог IK конденсируют с амином (содержащим подходящую тетразольную защитную группу) XLVII (предпочтительно 3-аминопропионитрил) в стандартных условиях получения пептида. Образующийся вторичный амид XLVIII затем подвергают реакции Митсунобу в стандартных условиях с триметилсилилазидом (TMSN3) с получением защищенного тетразола XLIX. Снятие защитных групп с цианоэтильной группы проводят предпочтительно в присутствии основания с получением желаемого свободного аналога тетразола IR.
Схема 18 описывает общий синтез гидразидокислотных аналогов IS. Замещенную арилкарбоновую кислоту 1 обрабатывают метансульфонилхлоридом в присутствии основания; промежуточное соединение затем подвергают реакции с защищенным гидразиноэфиром VA с получением соответствующего ацилированного гидразина 1а (см. Synthesis, 1989, 745-747).
Ацилгидразин 1а конденсируют с подходящим замещенным арилальдегидом IV в условиях восстановительного аминирования с получением соответствующего защищенного гидразида эфира 3 (см. Can. J. Chem., 1998, 76, 1180-1187).
Снятие защитных групп с эфира 3 затем приводит к получению гидразид-кислотных аналогов IS.
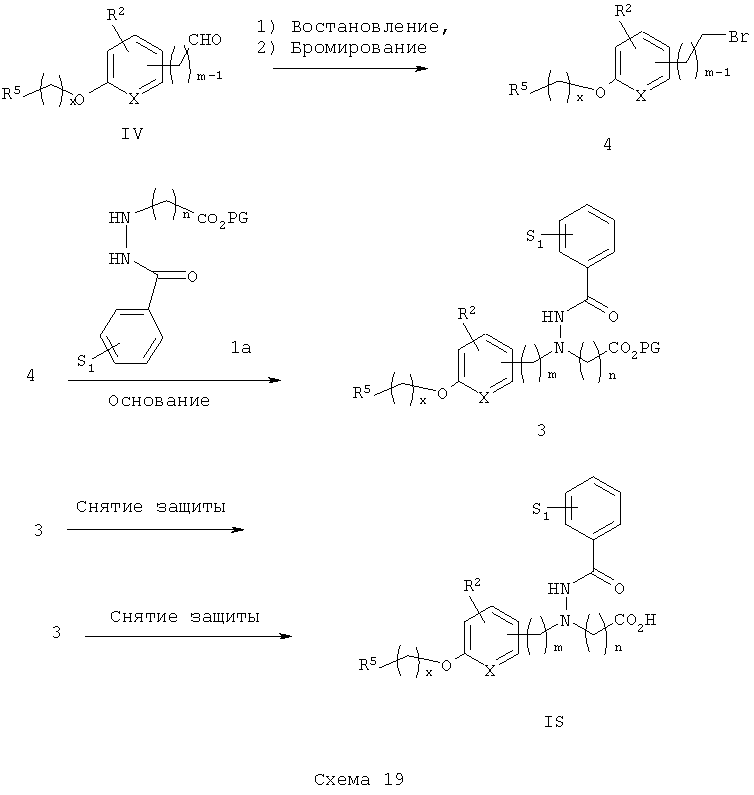
Альтернативный синтез, относящийся к гидразидокислотам IS, показан на схеме 19. Арилальдегид IV может быть восстановлен до соответствующего спирта в стандартных условиях (например, NaBH4), Указанный спирт затем превращают в соответствующий бромид 4, используя стандартные условия (например, Ph3Р/CBr4 или PBr3). Бромид 4 затем подвергают реакции с гидразинэфиром 1а (см. Tetrahedron Lett., 1993, 34, 207-210) с получением защищенного гидразидоэфира 3, с которого затем снимают защитные группы с получением гидразидокислотных аналогов IS.
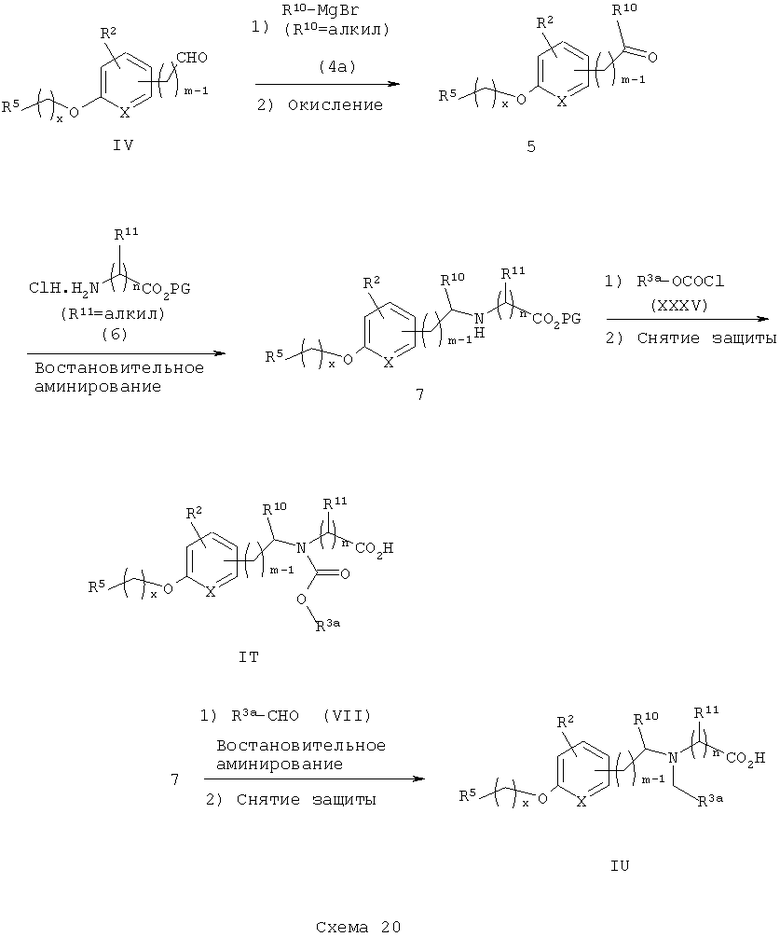
Различные подходы к получению аналогов α-алкилбензиламинокислоты и карбамат-кислоты IT и IU представлены на следующих схемах синтеза. На схеме 20 подходящий замещенный арилальдегид IV обрабатывают подходящим органометаллическим реагентом (например, реактивом Гриньяра R10MgBr) в стандартных условиях с получением соответствующего вторичного спирта, который затем окисляют в стандартных условиях (например, окисление по Swern с (COCl)2/DMSO/Et3N или используя хлорхромат пиридиния) с получением соответствующего кетона 5. Восстановительное аминирование кетона 5 с подходяще замещенным аминоэфиром 6 приводит к получению соответствующего α-алкилбензиламиноэфира 7. Ясно, что в аминоэфире 6 остаток  не обязательно представляет собой две повторяющееся единицы.
не обязательно представляет собой две повторяющееся единицы.
Ацилирование аминоэфира 7 с подходящим замещенным арил- или гетероарилхлорформиатом XXXV с последующим снятием защитных групп приводит к получению рацемических карбаматокислотных аналогов IT. Восстановительное аминирование алкилбензиламиноэфира 7 с арилальдегидом VII с последующим снятием защитных групп приводит к получению рацемических аминокислотных аналогов IU.
Альтернативно, как показано на схеме 21, асимметричное восстановление (например, с использованием способа оксазаборолидинового восстановления Corey; см. E.J. Corey & С. Helal, Angew. Chem. bit. Ed. Engl., 1998, 37, 1986-2012) арилкетона 5 приводит к получению любых желаемых энантиомерных спиртов 8 (хотя только один энантиомер показан на схеме). Обработка хирального спирта 8 азидом в реакции, подобной реакции Митсунобу (см. A.S. Thompson et. al, J. Org. Chem. 1993, 58, 5886-5888), приводит к получению соответствующего хирального азида (с обращением стереохимии в отношении исходного спирта). Азид затем превращают в амин 9 стандартными методами восстановления (например, гидрогенирование или Ph3Р/ТГФ/Н2О). Обработка хирального амина 9 эфиром XVIA (содержащим подходящую уходящую группу) приводит к получению вторичного аминоэфира 10. Ацилирование аминоэфира 10 с арил- или гетероарилхлорформиатом XXXV с последующим снятием защитных групп приводит к получению хиральных карбаматокислотных аналогов ITa (которые могут быть любыми энантиомерами в зависимости от стереохимии соединения 8). Восстановительное аминирование алкиламиноэфира 10 с арилальдегидами VII с последующим снятием защитных групп приводит к получению хиральных аминокислотных аналогов IIIa (которые могут быть любьми энантиомерами в зависимости от стереохимии соединения 8).
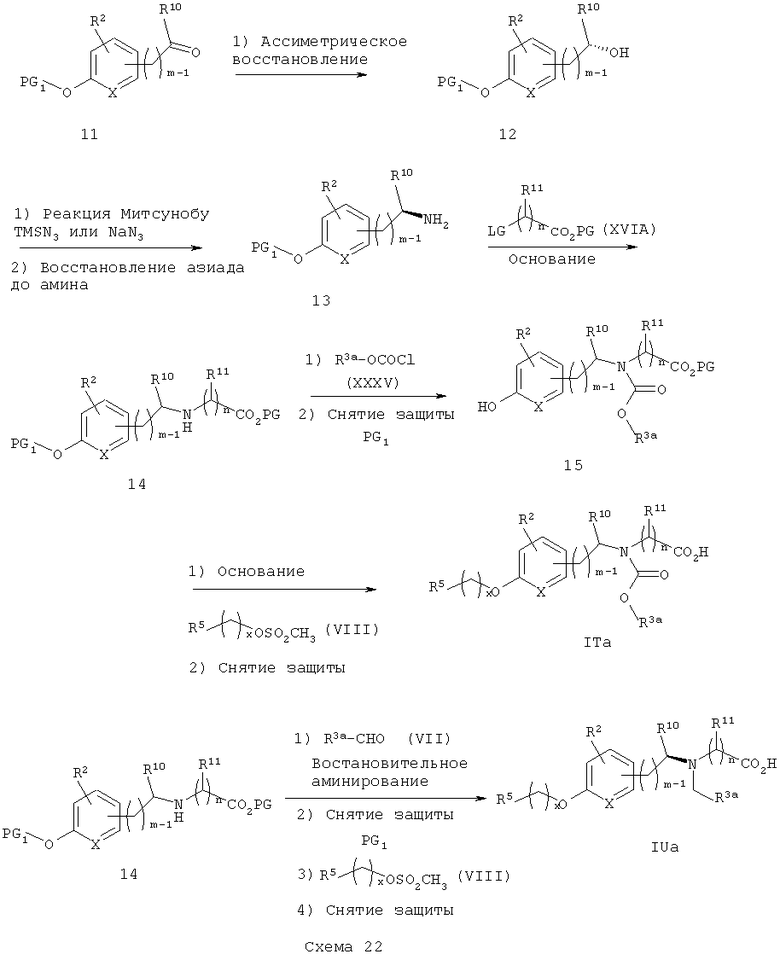
Альтернативный путь к схеме 21 показан на схеме 22. Подходяще защищенный оксиарилкетон 11 подвергают асимметричному восстановлению с получением хирального спирта 12. Его превращают в хиральный амин 13 в соответствии с методикой, аналогичной описанной на схеме 21 (через хиральный азид). Обработка хирального амина 13 эфиром XVIA (LG = галоген или мезилат) приводит к получению соответствующего вторичного аминоэфира 14. Ацилирование 14 арил- или гетероарилхлорформиатом XXXV приводит к получению соответствующего карбаматоэфира. Селективное снятие защитных групп приводит к получению свободного фенолкарбаматоэфира 15. Алкилирование фенола 15 галогенидом или мезилатом VIII с последующим снятием защитных групп приводит к получению карбаматокислотных аналогов ITa. Аналогичная последовательность (включающая восстановительное аминирование вторичного аминоэфира 14 с арил- или гетероарилальдегидом VII, затем селективное снятие защитных групп, алкилирование с VIII и конечное снятие защитных групп) приводит к получению аминокислотных аналогов IUa.
Очевидно, что (R)- или (S)-энантиомеры ITa или IIIa могут быть получены в соответствии со схемами 21 и 22 в зависимости от хиральности применяемого восстановительного агента.
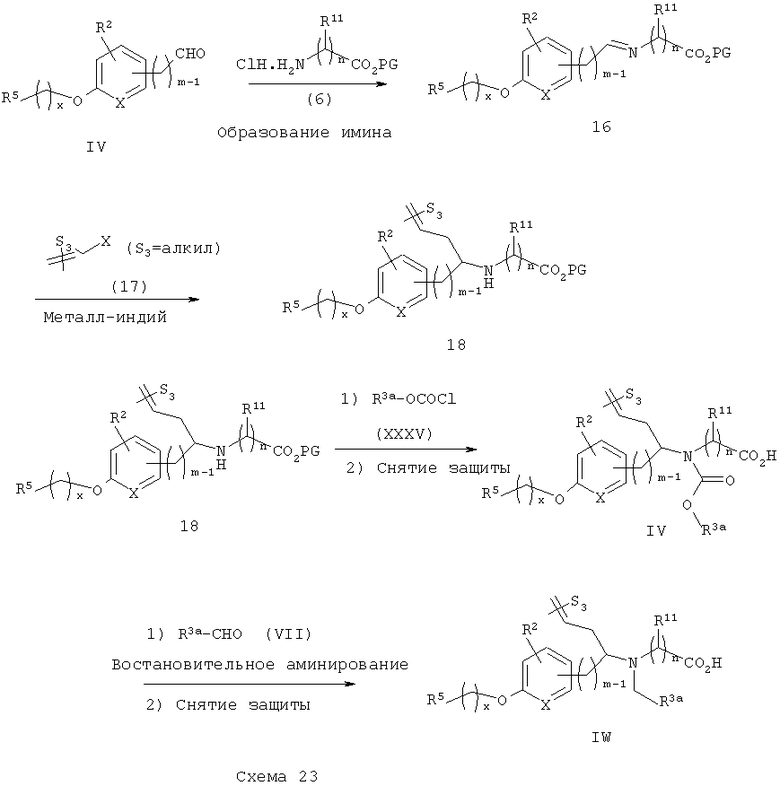
Четвертая последовательность синтеза показана на схеме 23. Замещенный альдегид IV конденсируют с гидрохлоридом аминоэфира 6 с получением соответствующего имина 16, который затем обрабатывают in situ подходящим замещенным аллилгалогенидом 17 в присутствии металлического индия (см. Loh, T.-P. и др., Tetrahedron Lett., 1997, 38, 865-868) с получением α-аллилбензиламиноэфира 18. Ацилирование амина 18 с арил- или гетероарилхлорформиатом XXXV, с последующим снятием защитных групп приводит к получению карбаматокислотных аналогов I. Восстановительное аминирование алкиламиноэфира 18 с арил- или гетероарилальдегидом VII с последующим снятием защитных групп приводит к получению аминокислотных аналогов IW.
На схеме 24 показано получение желаемого промежуточного 2-арил-5-метилоксазол-4-илметилхлорида 21 (следуя общей процедуре, описанной в Malamas, М.S. и др., J. Med. Chem., 1996, 39, 237-245). Замещенный арилальдегид 19 конденсируют с бутан-2,3-дионмонооксимом в кислых условиях с получением соответствующего оксазол-N-оксида 20. Деоксигенирование оксазол-N-оксида 20 с сопутствующим хлорированием приводит к получению желаемых хлорметиларилоксазолов 21. Гидролиз хлорметилоксазола 21 в основных условиях приводит к получению соответствующего оксазолметанола 22. Окисление спирта 22 до соответствующего альдегида проводят с последующим превращением в соответствующий дибромалкен 23 (например, Ph3Р/CBr4). Дибромид 23 превращают в соответствующий алкиниллитиевый остаток (используют органолитиевый реагент, такой, как n-BuLi), который может быть подвергнут реакции in situ с подходящим электрофилом, таким, как формальдегид, с получением соответствующего ацетиленового спирта (см. Corey, E.J. и др., Tetrahedron Lett. 1972, 3769, или Gangakhedkar, К.К., Synth. Commun. 1996, 26, 1887-1896). Указанный спирт затем может быть превращен в соответствующий мезилат 24 и алкилирован подходящим фенолом 25 с получением аналогов 1Х. Дальнейшее стереоселективное восстановление (например. Н2/катализатор Линдлара) приводит к получению Е- или Z-алкениланалогов IV.
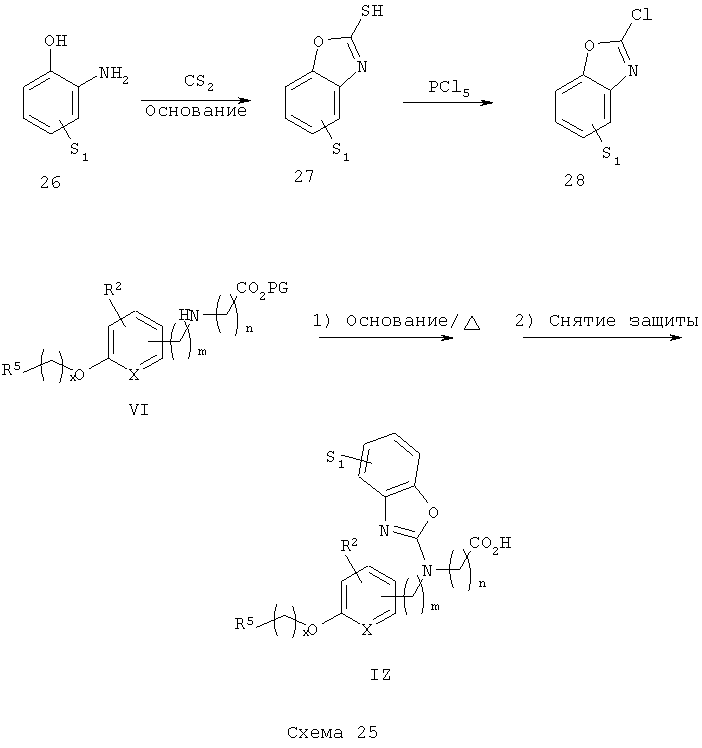
Схема 25 описывает общий синтез аминобензоксазольных аналогов IZ (общие ссылки: Sato, Y. и др., J. Med. Chem. 1998, 41, 3015-3021). Подходящий замещенный орто-аминофенол 26 обрабатывают с помощью CS2 в присутствии основания с получением соответствующего меркаптобензоксазола 27. Обработка полученного тиола 27 подходящим хлорирующим агентом (например, РСУ) приводит к получению ключевого промежуточного хлорбензоксазола 28, который подвергают реакции со вторичным аминоэфиром VI с получением, после снятия защитных групп, аминобензоксазолкислотных аналогов IZ.
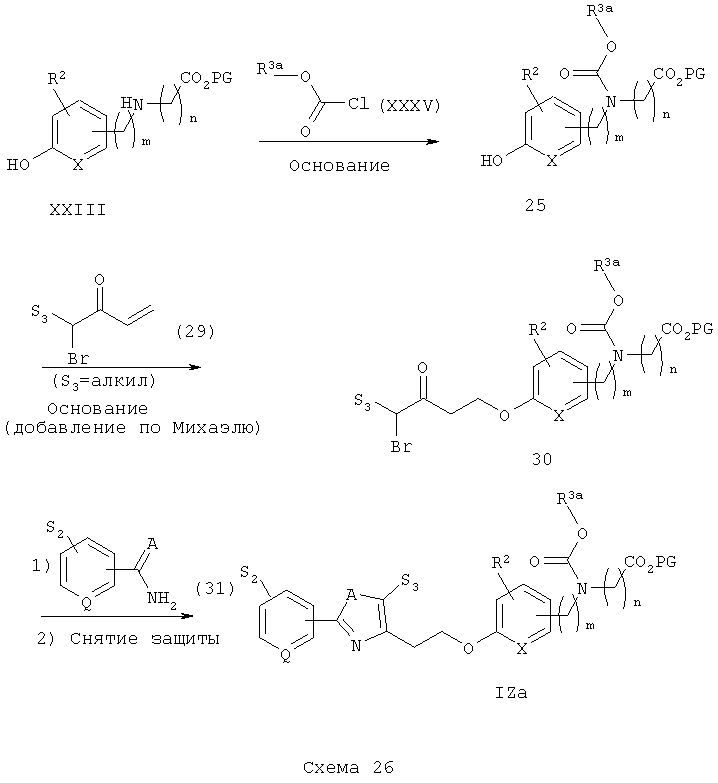
Тиазоланалоги IZa синтезируют в соответствии с общей схемой синтеза, приведенной на схеме 26 (см. Collins, J.L. и др., J. Med. Chem. 1998, 41, 5037). Вторичный аминоэфир XXIII подвергают реакции с арил- или гетероарилхлорформиатом XXXV в присутствии подходящего основания (например, пиридина или триэтиламина) с получением соответствующего гидроксиарилкарбаматоэфира 29. Гидроксиарилэфир 29 затем подвергают реакции с подходящим замещенным α-бромвинилкетоном 29а (для S3=СН3, например, Weyerstahl, Р. и др., Flavour Fragr. J., 1998, 23, 177, или Sokolov, N. А. и др., Zh. Org. Khim., 1980, 16, 281-283) в присутствии подходящего основания (например, К2СО3) с получением соответствующего аддукта реакции Михаэля, α-бромкетонкарбаматэфира 30. α-Бромкетон 30 затем подвергают реакции конденсации с подходящим замещенным ариламидом 31 (А=О) или арилтиоамидом 31 (А=S) с получением или соответствующего оксазола (из амида) или тиазола (из тиоамида) (см. Malamas, M. S. и др., J. Med. Chem., 1996, 39, 237-245). Наконец, снятие защитных групп с эфиров 31 затем приводит к получению замещенных оксазол- и тиазолкарбаматокислотных аналогов IZa.
Очевидно, что в следующих схемах, где получают карбаматокислотные аналоги, соответствующие аналоги аминокислот могут также быть получены замещением реакции с хлорформиатом на альдегид в восстановительном аминировании (как на схеме 20 с промежуточным амином 7).
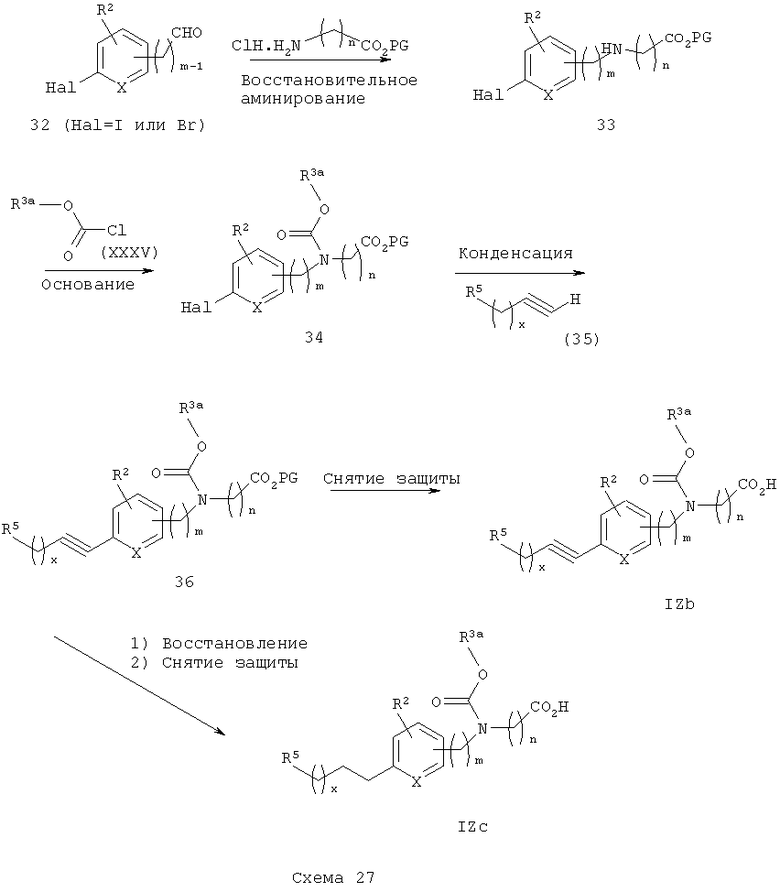
Схема 27 описывает общий синтез кислот IZb и IZc. Галогензамещенный арилальдегид 32 (предпочтительно йодид или бромид) подвергают восстановительному аминированию, используя методику, известную из уровня техники (например, Abdel-Magid и др., J. Org. Chem. 1996, 61, 3849) с гидрохлором α-аминокислотного эфира V. Образующийся вторичный аминоэфир 33 затем подвергают реакции с арил- или гетероарилхлорформиатом XXXV в присутствии подходящего основания (например, пиридина или триэтиламина) с получением соответствующего галогенарилкарбаматоэфира 34. Арилгалогенид 34 затем подвергают реакции с подходящим арил- или гетероарилзамещенным ацетиленом 35 (предпочтительным ацетиленом является 5-фенил-2-метилоксазол-4-илметилацетилен) в присутствии подходящего палладиевого катализатора (например, (Ph3Р)2PdCl2) и соли меди (I) (например, Cul) в реакции конденсации по Соногашира (Sonogashira) (см. Organocopper Reagents, a Practical Approach, R. J. К. Taylor, Ed., Chapter 10, pp 217-236, Campbell, I.B., Oxford University Press, 1994) с получением ключевого промежуточного арилацетиленкарбаматного эфира 36.
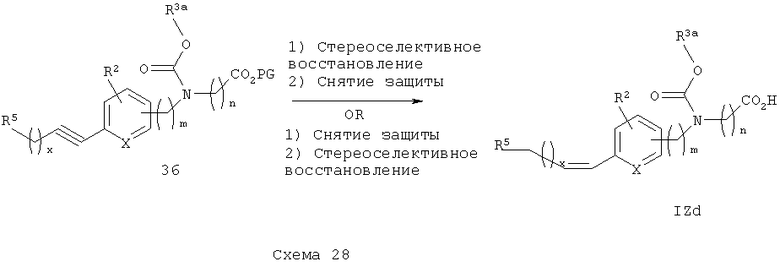
С арилацетиленового эфира 36 снимают защитные группы с получением соответствующих арилацетиленкислотных аналогов IZb. Ацетиленовая группа 36 может быть восстановлена стандартными методами (например, гидрогенированием, см. М. Hudlicky, Reductions in Organic Chemistry, 2nd Edition, ACS, 1996, Chapter 1) с получением соответствующего полностью насыщенного алкиларилкарбаматного эфира, с которого затем снимают защитные группы с получением алкиларилкарбаматокислотных аналогов IZc. Стереоселективное восстановление ацетиленового эфира 36 стандартными способами (например, катализатор Линдлара; см. Preparation of Alkenes, A Practical Approach, J.J. Williams, Ed., Chapter 6, pp 117-136, Oxford University Press, 1996) может быть проведено с получением соответствующего цис-алкениларилкарбаматоэфира, с которого затем снимают защитные группы с получением Z-алкениларилкарбаматкислотных аналогов IZd (схема 28). Альтернативно, эта последовательность может быть обращена, т.е. начальным шагом будет снятие защитных групп с ацетиленового эфира 36 с получением ацетиленовой кислоты с последующим стереоселективным восстановлением ацетиленовой группы с получением Z-алкенкислотных аналогов IZd.
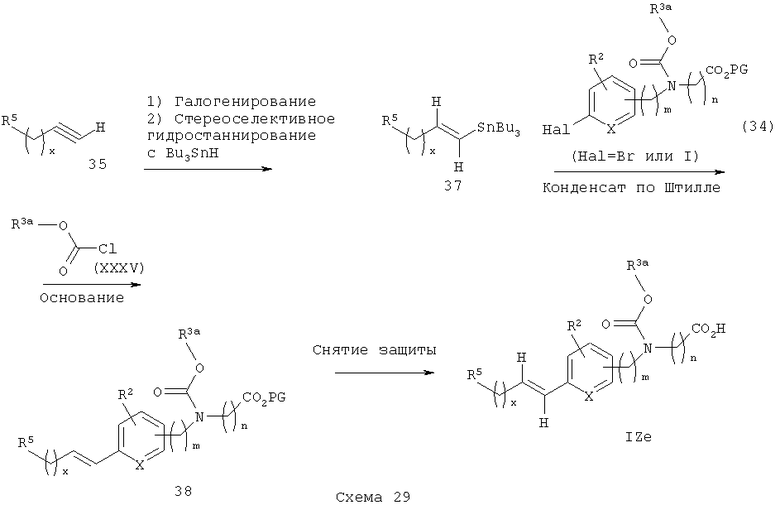
Соответствующие транс-алкениларилкарбаматные кислоты IZe могут быть получены в соответствии с общим способом по схеме 29. Арил- или гетероарилацетилен 35 (предпочтительной группой опять является 5-фенил-2-метилоксазол-4-илметилацетилен) галогенируют в стандартных условиях (см. Boden, С. D. J. и др., J. Chem. Soc. Perkin Trans. I, 1996, 2417, или Lu, W. et. al., Tetrahedron Lett. 1998, 39, 9521) с получением соответствующего галогенацетилена, который затем превращают в соответствующий транс-алкенилстаннан 37 (см. Boden, С. D.J., J. Chem. Soc., Perkin Trans. I, 1996, 2417). Полученный арил- или гетероарилзамещенный транс-алкенилстаннан 37 затем связывают с галогенарилкарбаматным эфиром 34 в стандартных условиях сочетания конденсации по Штилле (Stille) (см. Farina, V. Е. и др., "The Stille Reaction", Organic Reactions, 1997, 50, 1) с получением соответствующего транс-алкениларилкарбаматного эфира 38. С указанного карбаматоэфира затем снимают защитные группы в стандартных условиях с получением желаемых транс-алкениларилкарбаматокислотных аналогов IZe.
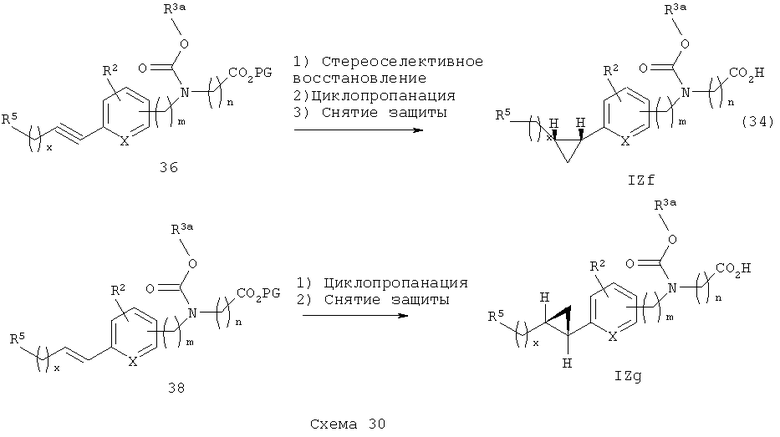
Соответствующие циклопропильные аналоги IZf и IZg получают в соответствии со Схемой 30. Для цис- или (Z-) циклопропильных аналогов используют стереоселективное восстановление (Н2/ катализатор Линдлара) алкинильной группы промежуточного алкинильного эфира 36 (как для аналогов IZd) с последующим циклопропанированием в стандартных условиях (Zhao, Y. и др., J. Org. Chem. 1995, 60, 5236-5242), и последующее снятие защитных групп приводит к получению цис-циклопропилкарбаматкислотных аналогов IZf. Для транс-циклопропильных аналогов IF применяют аналогичное циклопропанирование Е-алкеновой группы промежуточного соединения 38 с последующим снятием защитных групп, что приводит к получению транс-циклопропилкарбаматокислотных аналогов IZg.
 Схема 1
Схема 1
В этой и следующих реакционных схемах
Альтернативная схема 1а для получения альдегида IV
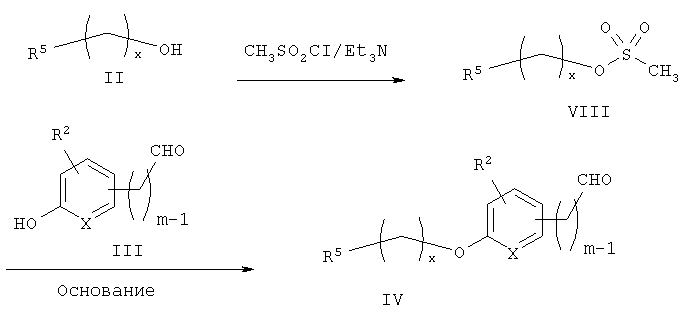
Схема 1а
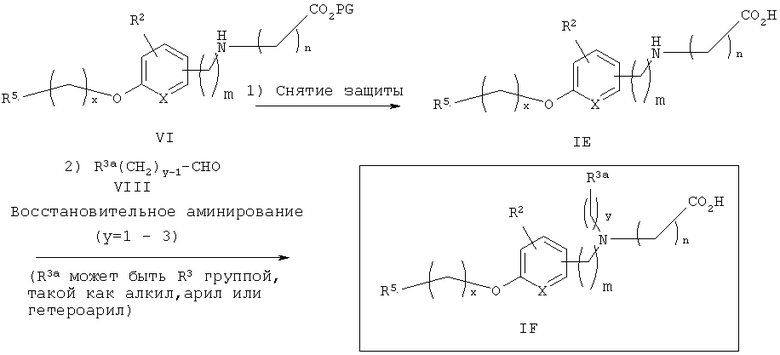 Схема 2
Схема 2



(S1=H, алкил, галоген, алкокси, алкилтио, алкиламино, арилокси, арил, гетероарил, алкоксикарбонил, алкиламинокарбонил; S2=H, алкил, галоген, алкокси, алкилтио, алкиламино, арилокси, арил, гетероарил, алкоксикарбонил, алкиламинокарбонил)
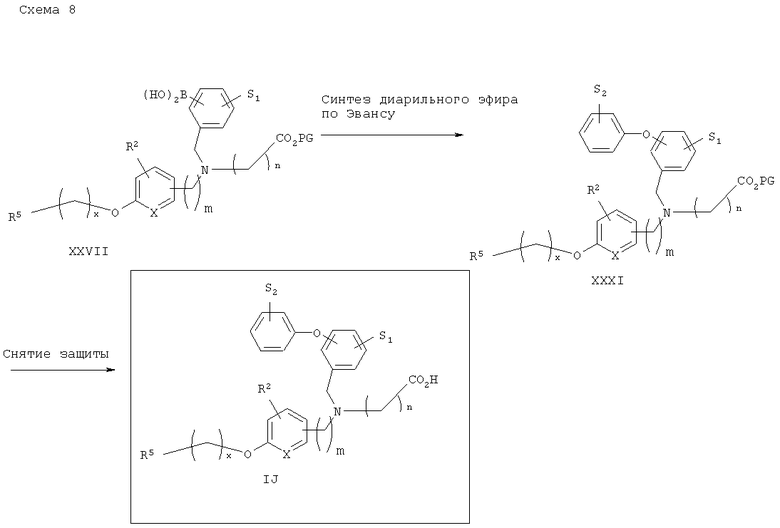
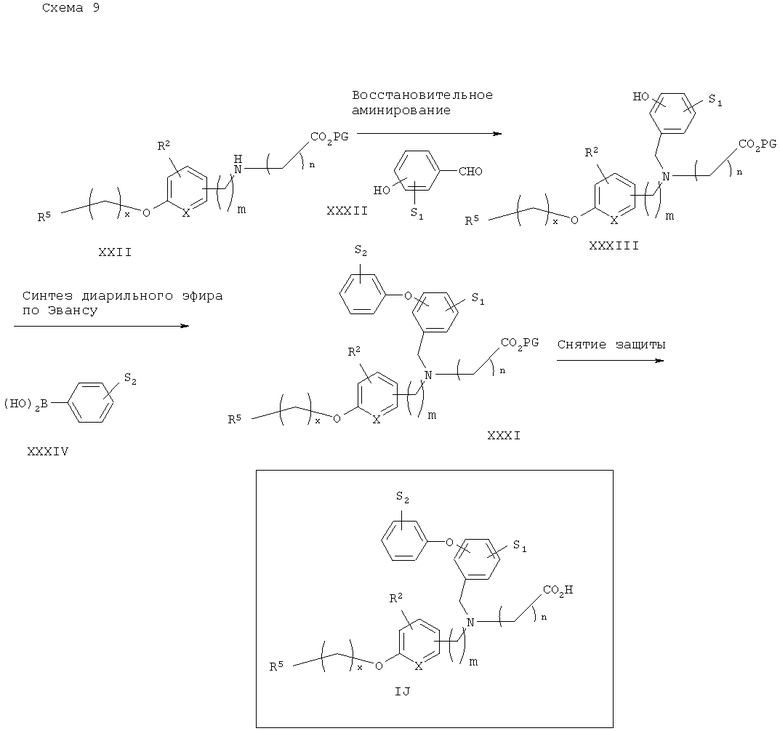
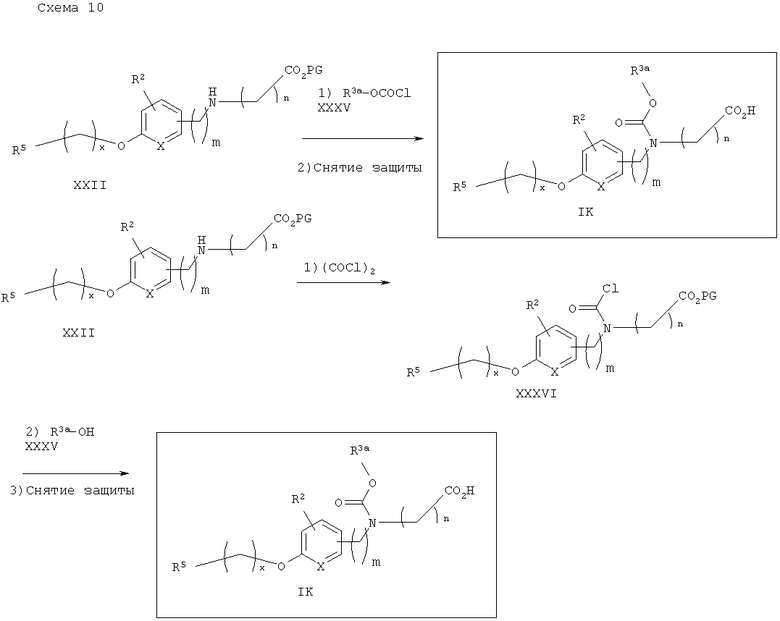
Схема 11





Если не указано иначе, термин "низший алкил", "алкил" или "алк", который используется здесь сам по себе или как часть другой группы, включает линейные и разветвленные цепи углеводородов, содержащих от 1 до 20 атомов углерода, предпочтительно от 1 до 10 атомов углерода, более предпочтительно от 1 до 8 атомов углерода, в нормальной цепи и могут необязательно включать кислород или азот в нормальной цепи, такие, как метил, этил, пропил, изопропил, бутил, трет-бутил, изобутил, пентил, гексил, изогексил, гептил, 4,4-диметилпентил, октил, 2,2,4-триметилпентил, нонил, децил, ундецил, додецил, их различные разветвленные изомеры, и им подобные, также как и такие группы, которые включают от 1 до 4 заместителей, таких, как галоген, например F, Br, Cl или I или CF3, алкокси, арил, арилокси, арил(арил) или диарил, арилалкил, арилалкилокси, алкенил, циклоалкил, циклоалкилалкил, циклоалкилалкилокси, амино, гидрокси, гидроксиалкил, ацил, гетероарил, гетероарилокси, циклогетероалкил, арилгетероарил, арилалкоксикарбонил, гетероарилалкил, гетероарилалкокси, арилоксиалкил, арилоксиарил, алкиламидо, алканоиламино, арилкарбониламино, нитро, циано, тиол, галогеналкил, тригалогеналкил и/или алкилтио и/или любая из R3 групп.
Если не указано иначе, термин "циклоалкил", который используется здесь сам по себе или как часть другой группы, включает насыщенные или частично ненасыщенные (содержащие 1 или 2 двойные связи) циклические углеводородные группы, содержащие от 1 до 3 колец, включая моноциклоалкил, бициклоалкил и трициклоалкил, содержащие в общем от 3 до 20 атомов углерода, образующих кольца, предпочтительно от 3 до 10 атомов углерода, образующих кольца, и которые могут быть конденсированы с 1 или 2 ароматическими кольцами, как описано для арила, которые включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклодецил и циклододецил, циклогексенил,
 ,
,  ,
,  ,
,  ,
,  ,
,
любые из указанных групп могут быть необязательно замещены 1-4 заместителями, такими, как галоген, алкил, алкокси, гидрокси, арил, арилокси, арилалкил, циклоалкил, алкиламидо, алканоиламино, оксо, ацил, арилкарбониламино, амино, нитро, циано, тиол и/или алкилтио и/или любыми заместителями для алкила.
Термин "циклоалкенил", который используется здесь сам по себе или как часть другой группы, относится к циклическим углеводородам, содержащим от 3 до 12 атомов углерода, предпочтительно от 5 до 10 атомов углерода, и 1 или 2 двойные связи. Примеры циклоалкенильных групп включают циклопентенил, циклогексенил, циклогептенил, циклооктенил, циклогексадиенил и циклогептадиенил, которые могут быть необязательно замещены, как указано для циклоалкила.
Термин "циклоалкилен", который здесь используется, обозначает циклоалкильную группу, которая включает свободные связи и, следовательно, является линкерной группой, такой, как  ,
,  и им подобные, и может необязательно быть замещена, как указано выше для "циклоалкила".
и им подобные, и может необязательно быть замещена, как указано выше для "циклоалкила".
Термин "алканоил", который используется здесь сам по себе или как часть другой группы, относится к алкилу, связанному с карбонильной группой.
Если не указано иначе, термин "низший алкенил" или "алкенил", который используется здесь сам по себе или как часть другой группы, относится к линейным или разветвленным радикалам, содержащим от 2 до 20 атомов углерода, предпочтительно от 2 до 12 атомов углерода и более предпочтительно от 2 до 8 атомов углерода в нормальной цепи, которые включают от одной до шести двойных связей в нормальной цепи, и могут необязательно включать кислород или азот в нормальной цепи, такие, как винил, 2-пропенил, 3-бутенил, 2-бутенил, 4-пентенил, 3-пентенил, 2-гексенил, 3-гексенил, 2-гептенил, 3-гептенил, 4-гептенил, 3-октенил, 3-ноненил, 4-деценил, 3-ундеценил, 4-додеценил, 4,8,12-тетрадекатриенил и им подобные, и которые могут быть необязательно замещены от 1 до 4 заместителями, а именно галогеном, галогеналкилом, алкилом, алкокси, алкенилом, алкинилом, арилом, арилалкилом, циклоалкилом, амино, гидрокси, гетероарилом, циклогетероалкилом, алканоиламино, алкиламидо, арилкарбониламино, нитро, циано, тиолом, алкилтио и/или любым заместителем для алкила, указанным выше.
Если не указано иначе, термин "низший алкинил" или "алкинил", который используется здесь сам по себе или как часть другой группы, относится к линейным или разветвленным радикалам, содержащим от 2 до 20 атомов углерода, предпочтительно от 2 до 12 атомов углерода и более предпочтительно от 2 до 8 атомов углерода, в нормальной цепи, которые включают одну тройную связь в нормальной цепи и могут необязательно включать кислород или азот в нормальной цепи, такие, как 2-пропинил, 3-бутинил, 2-бутинил, 4-пентинил, 3-пентинил, 2-гексинил, 3-гексинил, 2-гептинил, 3-гептинил, 4- гептинил, 3-октинил, 3-нонинил, 4-децинил, 3-ундецинил, 4-додецинил и им подобные, и которые могут быть необязательно замещены 1-4 заместителями, а именно галогеном, галогеналкилом, алкилом, алкокси, алкенилом, алкинилом, арилом, арилалкилом, циклоалкилом, амино, гетероарилом, циклогетероалкилом, гидрокси, алканоиламино, алкиламидо, арилкарбониламино, нитро, циано, тиолом и/или алкилтио, и/или любым из заместителей для алкила, указанных выше.
Термины "арилалкенил" и "арилалкинил", которые используются сами по себе или как часть другой группы, относятся к алкенильным и алкинильным группам, как описано выше, имеющим арильный заместитель.
Там, где алкильные группы, которые определены выше, имеют простые связи для присоединения к другим группам и двух различных атомов углерода, они называются "алкиленовые" группы и могут необязательно быть замещены, как указано выше для "алкила".
Где алкенильные группы, как определено выше, и алкинильные группы, как определено выше, соответственно, имеют простые связи для присоединения к двум различным атомам углерода, они называются "алкениленовые группы" и "алкиниленовые группы", соответственно, и могут необязательно быть замещены, как указано выше для "алкенила" и "алкинила".
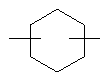
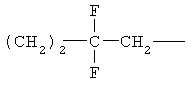
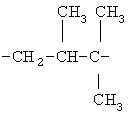
(СН2)х, (СН2)m, (СН2)n или (СН2)y включает алкилен, алленил, алкенилен или алкиниленовые группы, как здесь определено, каждая из которых может необязательно включать кислород или азот в нормальной цепи, которые могут необязательно включать 1, 2, или 3 заместителя, которые включают алкил, алкенил, галоген, циано, гидрокси, алкокси, амино, тиоалкил, кето, С3-С6циклоалкил, алкилкарбониламино или алкилкарбонилокси; алкильным заместителем может быть алкиленовая группа с 1-4 атомами углерода, которая может присоединяться к одному или двум атомам углерода в (СН2)х или (СН2)m или (СН2)n группе с образованием циклоалкильной группы.
Примеры (СН2)х, (СН2)m, (СН2)n, (СН2)y, алкилена, алкенилена и алкинилена включают
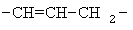 ,
,  ,
, 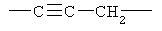 ,
,  ,
,
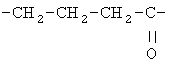 ,
,  ,
, 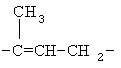 ,
,
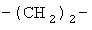 ,
, 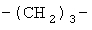 ,
, 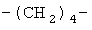 ,
, 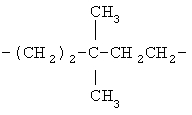 ,
, 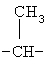 ,
,
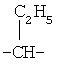 ,
, 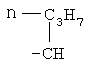 ,
, 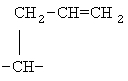 ,
, 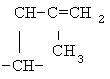 ,
,  ,
,
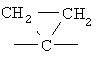 ,
, 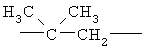 ,
, 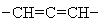 ,
, 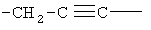 ,
, 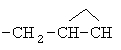 ,
,
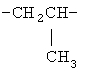 ,
, 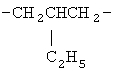 ,
,  ,
,  ,
,
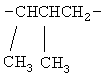 ,
, 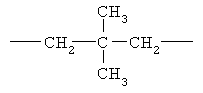 ,
, 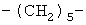 ,
,  ,
,
 ,
,  ,
,  ,
,
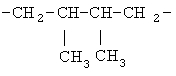 ,
, 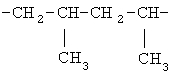 ,
, 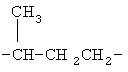 ,
,
 ,
, 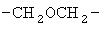 ,
, 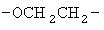 ,
,  ,
,
 ,
,  ,
, 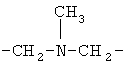 или
или 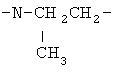 .
.
Термин «галоген» или «гало», который используется здесь сам по себе или как часть другой группы, обозначает хлор, бром, фтор и йод, также как и CF3, предпочтительно хлор или фтор.
Термин «ион металла» обозначает ионы щелочных металлов, таких, как натрий, калий или литий, и щелочно-земельных металлов, таких, как магний или кальций, также цинк и алюминий.
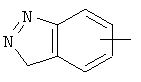
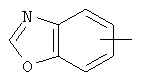
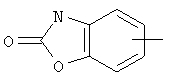
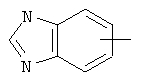
Если не указано иначе, термин «арил», который используется здесь сам по себе или как часть другой группы, обозначает моноциклические или бициклические ароматические группы, содержащие от 6 до 10 атомов углерода в кольце (такие, как фенил или нафтил, включая 1-нафтил и 2-нафтил) и они могут необязательно включать от одного до трех дополнительных колец, сконденсированных с карбоциклическим или гетероциклическим кольцом (такие, как арильные, циклоалкильные, гетероарильные или циклогетероарильные кольца, например
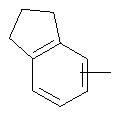 ,
, 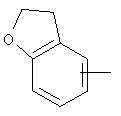 ,
, 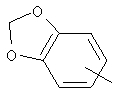 ,
,  ,
,  ,
,
 ,
,  ,
,  ,
,  ,
,
 ,
, 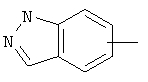 ,
, 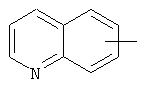 ,
,  ,
,
и они могут быть необязательно замещены через приемлемые атомы углерода 1, 2, или 3 группами, выбранными из водорода, галогена, галогеналкила, алкила, галогеналкила, алкокси, галогеналкокси, алкенила, трифторметила, трифторметокси, алкинила, циклоалкил-алкила, циклогетероалкила, циклогетероалкилалкила, арила, гетероарила, арилалкила, арилокси, арилоксиалкила, арилалкокси, алкоксикарбонила, арилкарбонила, арилалкенила, аминокарбониларила, арилтио, арилсульфинила, арилазо, гетероарилалкила, гетероарилалкенила, гетероарилгетероарила, гетероарилокси, гидрокси, нитро, циано, амино, замещенного амина, где амин включает 1 или 2 заместителя (которыми являются алкил, арил или любые другие арильные соединения, указанные в описании в разделе «определения»), тиола, алкилтио, арилтио, гетероарилтио, арилтиоалкила, алкоксиарилтио, алкилкарбонила, арилкарбонила, алкиламинокарбонила, ариламинокарбонила, алкоксикарбонила, аминокарбонила, алкилкарбонилокси, арилкарбонилокси, алкилкарбониламино, арилкарбониламино, арилсульфинила, арилсульфинилалкила, арилсульфониламино или арилсульфаминокарбонила и/или любого заместителя для алкила, указанные здесь.
Если не указано иначе, термин "низший алкокси", "алкокси", "арилокси" или "аралкокси", который используется здесь сам по себе или как часть другой группы, включает любую из указанных выше алкильную, аралкильную или арильную группы, связанные с атомом кислорода.
Если не указано иначе, термин "замещенный амино", который используется здесь сам по себе или как часть другой группы, относится к амину, замещенному одним или двумя заместителями, которые могут быть одинаковыми или различными, такими, как алкил, арил, арилалкил, гетероарил, гетероарилалкил, циклогетероалкил, циклогетероалкилалкил, циклоалкил, циклоалкилалкил, галогеналкил, гидроксиалкил, алкоксиалкил или тиоалкил. Эти заместители могут быть далее замещены карбоновой кислотой и/или любым заместителем для алкила, приведенным выше. Кроме того, аминозаместители могут быть взяты вместе с атомом азота, к которому они присоединены, с образованием 1-пирролидинила, 1-пиперидинила, 1-азепинила, 4-морфолинила, 4-тиоморфолинила, 1-пиперазинила, 4-алкил-1-пиперазинила, 4-арилалкил-1-пиперазинила, 4-диарилалкил-1-пиперазинила, 1-пирролидинила, 1-пиперидинила или 1-азепинила, необязательно замещеными алкилом, алкокси, алкилтио, галогеном, трифторметилом или гидрокси.
Если не указано иначе, термин "низший алкилтио", алкилтио", "арилтио" или "аралкилтио", который используется здесь сам по себе или как часть другой группы, включает любую указанную выше алкильную, аралкильную или арильную группы, связанные с атомом серы.
Если не указано иначе, термин "низший алкиламино", "алкиламино", "ариламино" или "арилалкиламино", который используется здесь сам по себе или как часть другой группы, включает любую указанную выше алкильную, арильную или арилалкильную группы, связанные с атомом азота.
Если не указано иначе, термин "ацил", который здесь используется сам по себе или как часть другой группы, как определено здесь, обозначает органический радикал, связанный с карбонильной ( ) группой; примеры ацильных групп включают любые R3 группы, присоединенные к карбонилу, такие, как алканоил, алкеноил, ароил, аралканоил, гетероароил, циклоалканоил, циклогетероалканоил и им подобные.
) группой; примеры ацильных групп включают любые R3 группы, присоединенные к карбонилу, такие, как алканоил, алкеноил, ароил, аралканоил, гетероароил, циклоалканоил, циклогетероалканоил и им подобные.
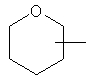
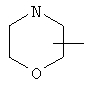
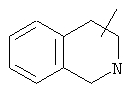
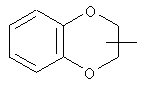
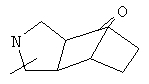
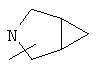
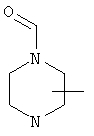
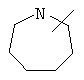
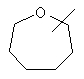
Если не указано иначе, термин «циклогетероалкил», который используется здесь сам по себе или как часть другой группы, обозначает 5-, 6- или 7-членное насыщенное или частично ненасыщенное кольцо, которое включает от 1 до 2 гетероатомов, таких, как азот, кислород и/или сера, связанное через атом углерода или гетероатом, где возможно, необязательно через линкер (СН2)p (где р обозначает 1, 2 или 3), такое, как
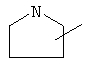 ,
, 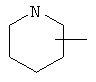 ,
,  ,
,  ,
,
 ,
,  ,
,  ,
,  ,
,
 ,
,  ,
,  ,
,
 ,
,  ,
,  ,
, 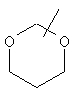
и им подобные. Указанные выше группы могут включать от 1 до 4 заместителей, таких, как алкил, галоген, оксо и/или любой из заместителей для алкила или арила, указанные здесь. Кроме того, любое из циклогетероалкильных колец может быть сконденсировано с циклоалкильным, арильным, гетероарильным или циклогетероалкильным кольцом.
Если не указано иначе, термин "гетероарил", который используется здесь сам по себе или как часть другой группы, обозначает 5- или 6-членное ароматическое кольцо, которое содержит 1, 2, 3 или 4 гетероатома, таких, как азот, кислород или сера, и такие кольца сконденсированы с арильным, циклоалкильным, гетероарильным или циклогетероалкильным кольцом (например, бензотиофенил, индолил), и включает возможно N-оксиды. Гетероарильная группа может необязательно включать от 1 до 4 заместителей, таких, как любой из заместителей для алкила или арила, указанных выше.
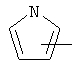
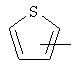
Примеры гетероарильных групп включают следующие:
 ,
,  ,
,  ,
, 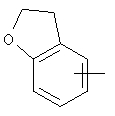 ,
,
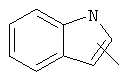 ,
, 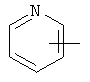 ,
, 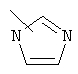 ,
, 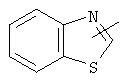 ,
,  ,
,  ,
,
 ,
, 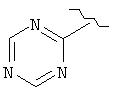 ,
, 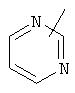 ,
,  ,
, 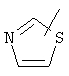 ,
,  ,
,
 ,
,  ,
,  ,
,  ,
, 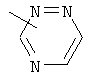 ,
,
и им подобные.
Термин "циклогетероалкилалкил", который используется здесь сам по себе или как часть другой группы, относится к циклогетероалкильным группам, как определено выше, связанным через С атом или гетероатом с (СН2)p цепью.
Термин "гетероарилалкил" или "гетероарилалкенил", который используется здесь сам по себе или как часть другой группы, обозначает гетероарильную группу, как определено выше, связанную через С атом или гетероатом с -(СН2)p- цепью, алкиленом или алкениленом, как определено выше.
Термин "полигалогеналкил", который здесь используется, обозначает "алкильную" группу, как определено выше, которая включает от 2 до 9, предпочтительно от 2 до 5, галогеновых заместителей, таких, как F или Cl, предпочтительно F, таких, как CF3СН2, CF3 или CF3CF2СН2.
Термин "полигалогеналкилокси", который здесь используется, обозначает группу "алкокси" или "алкилокси", как определено выше, которая включает от 2 до 9, предпочтительно от 2 до 5, галогеновых заместителей, таких, как F или Cl, предпочтительно F, таких, как CF3CH2O, CF3О или CF3CF2СН2О.
Термин "пролекарственные эфиры", который здесь используется, включает пролекарственные эфирные формы, которые известны из уровня техники для эфиров карбоновой и фосфорной кислот, такие, как метиловые, этиловые, бензиловые и им подобные. Другие примеры пролекарственного эфира R4 включают следующие группы:
(1-алканоилокси)алкил, такой, как
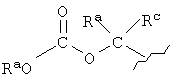 или
или 
где Ra, Rb и Rc представляют собой Н, алкил, арил или арилалкил;
однако RaO не может быть НО.
Примеры таких пролекарственных эфиров R4 включают
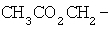 ,
, 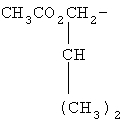 ,
, 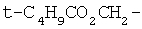 или
или
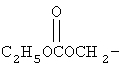 .
.
Другие примеры подходящих пролекарственных эфиров R4 включают
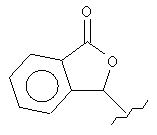 ,
, 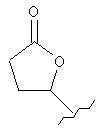 ,
,  ,
,  ,
,
 ,
, 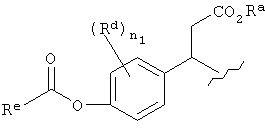
где Rа может быть Н, алкилом (таким, как метил или трет-бутил), арилалкилом (таким, как бензил) или арилом (таким, как фенил); Rd представляет собой Н, алкил, галоген или алкокси, Re представляет собой алкил, арил, арилалкил или алкоксил и n1 имеет значение 0, 1 или 2.
Там, где соединения структуры I находятся в кислотной форме, они могут образовывать фармацевтически приемлемую соль, такую, как соли щелочных металлов, таких, как литий, натрий или калий, соли щелочно-земельных металлов, таких, как кальций или магний, также соли цинка или алюминия и других катионов, таких, как аммоний, холин, диэтаноламин, лизин (D или L), этилендиамин, трет-бутиламин, трет-октиламин, трис-(гидроксиметил)аминометан (TRIS), N-метилглюкозамин (NMG), триэтаноламин и дегидроабиетиламин.
Все стереоизомеры соединений настоящего изобретения подразумеваются либо в смеси, либо в чистой или почти чистой форме. Соединения настоящего изобретения могут иметь асимметрические центры при любых атомах углерода, включая любой один или R заместителей. Следовательно, соединения формулы I могут существовать в виде энантиомерных или диастереомерных форм или в их смесях. Способы получения могут использовать рацематы, энантиомеры или диастереомеры в качестве исходных материалов. При получении диастереомерных или энантиомерных продуктов они могут быть разделены обычными методами, например хроматографически или фракционной кристаллизацией.
При необходимости соединения структуры I могут использоваться в комбинации с одним или более гиполипидемических агентов или липид-понижающих агентов и/или одним или более других типов терапевтических агентов, включая антидиабетические агенты, агенты против ожирения, антигипертензивные агенты, ингибиторы агрегации тромбоцитов и/или агенты против остеопороза, которые могут вводиться орально в той же дозовой форме, в отдельной оральной дозовой форме или путем инъекции.
Гиполипидемический агент или липид-понижающий агент, который может необязательно применяться в комбинации с соединениями формулы I по изобретению, может включать 1, 2, 3 или более МТР ингибитора, ингибитора HMG СоА-редуктазы, ингибитора скваленовых синтетаз, ингибитора производных фибриновых кислот, АСАТ ингибитора, ингибитора липоксигеназы, ингибитора абсорбции холестерина, ингибитора подвздошных сотранспортеров Na+/желчной кислоты, регулятора активности рецептора LDL, вещества, усиливающие экскрецию желчной кислоты и/или никотиновой кислоты и их производные.
МТР ингибиторы, применимые здесь, включают МТР ингибиторы, описанные в US 5595872, US 5739135, US 5712279, US 5760246, US 5827875, US 5885983 и заявке US 09/175180, поданной 20.10.1998, в настоящий момент US 5962440. Предпочтительными являются любой из предпочтительных МТР ингибиторов, описанных в каждом из указанных выше патентах и заявках.
Все указанные выше US патенты и заявки приведены здесь в качестве ссылок.
Наиболее предпочтительные МТР ингибиторы, применимые в соответствии с настоящим изобретением, включают предпочтительные МТР ингибиторы, которые описаны в US 5739135, 5712279 и US 5760246.
Наиболее предпочтительным МТР ингибитором является 9-[4-[4-[[2-(2,2,2-трифторэтокси)бензоил]амино]-1-пиперидинил]бутил]-N-(2,2,2-трифторэтил)-9Н-флуорен-9-карбоксамид
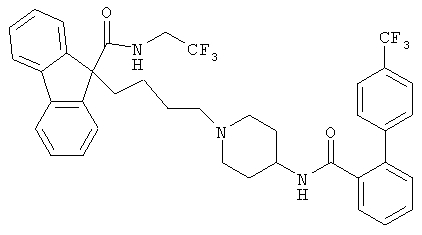
Гиполипидемическим агентом может быть HMG ингибитор СоА редуктазы, который включает, но не ограничивается такими соединениями, как мевастатин и родственные соединения, которые описаны в US 3983140, ловастатин (мевинолин) и родственные соединения, которые описаны в US 4231938, правастатин и родственные соединения, которые описаны в US 4346227, симвастатин и родственные соединения, которые описаны в US 4448784 и 4450171. Другие HMG ингибиторы СоА редуктазы, которые могут здесь использоваться, включают, но не ограничиваются, флувастатин, описанный в US 5354772, церивастатин, описанный в US 5006530 и 5177080, аторвастатин, описанный в US 4681893, 5273995, 5385929 и 5686104, итавастатин (Nissan/Sankyo нисвастатин (NK-104)), описанный в US 5011930, Shionogi-Astra/Zeneca визастатин (ZD-4522), описанный в US 5260440, и родственные соединения статина, описанные в US 5753675, пиразольные аналоги мевалонолактоновых производных, которые описаны в US 4613610, инденовые аналоги мевалонолактоновых производных, которые описаны в заявке РСТ WO 86/03488, 6-[2-(замещенный-пиррол-1-ил)алкил)пиран-2-оны и их производные, которые описаны в US 4647576, Searle SC-45355 (3-замещенное производное пентандионовой кислоты), дихлорапетатные, имидазольные аналоги мевалонолактона, которые описаны в заявке РСТ WO 86/07054, производные 3-карбокси-2-гидроксипропанфосфорной кислоты, которые описаны в FR 2596393, 2,3-дизамещенные пиррольные, фурановые и тиофеновые производные, которые описаны в заявке ЕР 0221025, нафтильные аналоги мевалонолактона, которые описаны в US 4686237, октагидронафталены, такие, как описаны в US 4499289, кетоаналоги мевинолина (ловастатина), которые описаны в заявке ЕР 0142146 А2, и хинолиновые и пиридиновые производные, описанные в US 5506219 и 5691322.
Кроме того, соединения фосфиновой кислоты, полезные для ингибирования HMG СоА редуктазы, пригодные для использования здесь, описаны в GB 2205837.
Ингибиторы скваленовых синтетаз, пригодные для использования здесь, включают, не ограничиваясь, α-фосфоносульфонаты, описанные в US 5712396, описанные Biller и др., J. Med. Chem., 1988, Vol.31, No.10, pp 1869-1871, включая изопреноид (фосфинилметил)фосфонаты, также как другие известные ингибиторы скваленовых синтетаз, например, которые описаны в US 4871721 и 4924024 и в Biller, S.A., Neuenschwander, К., Ponpipom, M.M. и Poulter, C.D., Current Pharmaceutical Design, 2, 1-40 (1996).
Кроме того, другие ингибиторы синтетазы, пригодные для использования в соответствии с изобретением, включают терпеноидные пирофосфаты, описанные Р. Ortiz de Montellang и др., J. Med. Chem., 1977, 20, 243-249, дифосфатный аналог фарнезила А и прескваленовые пирофосфатные (PSQ-PP) аналоги, которые описаны Согеу и Volante, J. Am. Chem. Soc., 1976, 98, 1291-1293, фосфинилфосфонаты, описанные McClard, R.W. и др., J.A.C.S., 1987, 109, 5544, и циклопропаны, описанные Capson, T.L., PhD dissertation, June. 1987, Dept. Med. Chem. U of Utah, Abstract, Table of Contents, pp 16, 17, 40-43, 48-51, Summary.
Другие липолипидемические агенты, пригодные для использования здесь, включают, но не ограничиваясь, производные фибриновой кислоты, такие, как фенофибрат, гемфиброзил, клофибрат, безафибрат, ципрофибрат, клинофибрат и им подобные, пробукол и родственные соединения, которые описаны в US 3674836, пробукол и гемфиброзил, являются предпочтительными веществами, усиливающими экскрецию желчной кислоты, такими, как холестирамин, колестипол и DEAE-Sephadex (Secholex®, Policexide®) и холестагель (Sankyo/Geltex), также как липостабил (Rhone-Poulenc), Eisai E-5050 (N-замещенное этаноламинное производное), иманиксил (НОЕ-402), тетрагидролипстатин (THL), истигмастанилфосфорилхолин (SPC, Roche), аминоциклодекстрин (Tanabe Seiyoku), Ajinomoto AJ-814 (азуленовое производное), мелинамид (Sumitomo), Sandoz 58-035, American Cyanamid CL-277082 и CL-283546 (дизамещенные производные мочевины), никотиновая кислота (ниацин), аципимокс, ацифран, неомицин, n-аминосалициловая кислота, аспирин, производные поли(диаллилметиламина), такие, которые описаны в US 4759923, четвертичный амин поли(диаллилдиметиламмонийхлорид) и ионены, такие, как описано в US 4027009, и другие известные агенты, снижающие уровень холестерина плазмы.
Гиполипидемический агент может быть ингибитором АСАТ, таким, как описано в Drugs of the Future 24, 9-15 (1999), (Avasimibe); "The АСАТ inhibitor, Cl-1011 is effective in the prevention и regression of aortic fatty streak area in hamsters", Nicolosi и др., Atherosclerosis (Shannon, Irel) (1998), 137 (1), 77-85;
"The pharmacological profile of FCE 27677: a novel АСАТ inhibitor with potent hypolipidemic activity mediated by selective suppression of the hepatic secretion of ApoB 100-containing lipoprotein", Ghiselli, Giancarlo, Cardiovasc. Drug Rev. (1998), 16 (1), 16-30; "RP 73163: a bioavailable alkylsulfinil-diphenylimidazole АСАТ inhibitor". Smith, C. и др., Bioorg. Med. Chem. Lett. (1996), 6 (1), 47-50; "АСАТ inhibitors: physiologic mechanisms for hypolipidemic и anti-atherosclerotic activities in experimental animals", Krause и др., Editor(s): Ruffolo, Robert R., Jr.; Hollinger, Mannfred A., Inflammation: Mediators Pathways (1995), 173-98, Publisher: CRC, Boca Raton, Fla.; "АСАТ inhibitors: potential anti-atherosclerotic agents", Sliskovic и др., Curr. Med. Chem. (1994), 1 (3), 204-25; "Inhibitors of acyl-CoA:cholesterol O-acyl transferase (АСАТ) as hypocholesterolemic agents. The first water-soluble АСАТ inhibitor with lipid-regulating activity. Inhibitors of acyl-CoA:cholesterol acyltransferase (АСАТ). Development of a series of substituted N-phenyl-N'-[(1-phenylcyclopentyl)methyl]ureas with enhanced hypocholesterolemic activity". Stout и др., Chemtracts: Org. Chem. (1995), 8 (6), 359-62, или TS-962 (Taisho Pharmaceutical Co. Ltd).
Гиполипидемический агент может быть регулятором активности рецептора LD2 таким, как MD-700 (Taisho Pharmaceutical Co. Ltd) и LY295427 (Eh Lilly).
Гиполипидемический агент может быть ингибитором абсорбции холестерина, предпочтительно Schering-Plough's SCH48461, также как описанный в Atherosclerosis 115, 45-63 (1995) и J. Med. Chem. 41, 973 (1998).
Гиполипидемический агент может быть ингибитором подвздошного сотранспортера Na+/желчной кислоты, такой, как описано в Drugs of the Future, 24, 425-430 (1999).
Предпочтительными гиполипидемическими агентами являются правастатин, ловастатин, симвастатин, аторвастатин, флувастатин, церивастатин, итавастатин и визастатин.
Указанные выше US патенты указаны здесь в качестве ссылок. Применяемые количества и дозы будут такими, как указано в Physician's Desk Reference и/или в патентах, указанных выше.
Соединения формулы I по изобретению применяют в массовом соотношении с гиполипидемическим агентом (присутствующим), которое лежит в области от около 500:1 до около 1:500, предпочтительно от около 100:1 до около 1:100.
Вводимая доза должна быть тщательно подобрана в соответствии с возрастом, весом и состоянием пациента, также как и способ введения, дозовая форма и режим, а также желаемый результат.
Дозы и составы для гиполипидемического агента являются такими, как описано в различных патентах и заявках, описанных выше.
Дозы и составы для других применяемых гиполипидемических агентов, где возможно, являются такими, как описано в последней редакции Physicians' Desk Reference.
В случае орального введения удовлетворительный результат может быть получен при использовании МТР ингибитора, взятого в количестве в пределах от около 0.01 мг до около 500 мг и предпочтительно от около 0.1 мг до около 100 мг, от одного до четырех раз ежедневно.
Предпочтительная оральная дозовая форма, такая, как таблетки или капсулы, может содержать МТР ингибитор, взятый в количестве от около 1 до около 500 мг, предпочтительно от около 2 до около 400 мг и более предпочтительно от около 5 до около 250 мг, от одного до четырех раз ежедневно.
Для орального введения может быть получен удовлетворительный результат, используя ингибитор HMG СоА редуктазы, например правастатин, ловастатин, симвастатин, аторвастатин, флувастатин или церивастатин, в применяемых дозах, которые указаны в Physician's Desk Reference, в количестве в пределах от около 1 до 2000 мг и предпочтительно от около 4 до около 200 мг.
Ингибитор скваленовых синтетаз может применяться в дозах в количестве в пределах от около 10 мг до около 2000 мг и предпочтительно от около 25 мг до около 200 мг.
Предпочтительная оральная дозовая форма, такая, как таблетки или капсулы, может содержать ингибитор HMG СоА редуктазы в количестве от около 0,1 до около 100 мг, предпочтительно от около 0,5 до около 80 мг и более предпочтительно от около 1 до около 40 мг.
Предпочтительная оральная дозовая форма, такая, как таблетки или капсулы, может содержать ингибитор скваленовых синтетаз в количестве от около 10 до около 500 мг, предпочтительно от около 25 до около 200 мг.
Гиполипидемическим агентом может также быть ингибитор липоксигеназы, включая ингибитор 15-липоксигеназы (15-LO), такой, как производные бензимидазола, которые описаны в WO 97/12615, 15-LO ингибиторы, которые описаны в WO 97/12613, изотиазолоны, которые описаны в WO 96/38144, и 15-LO ингибиторы, которые описаны Sendobry и др. "Attenuation of dietret-induced atherosclerosis in rabbits with a highly selective 15-lipoxygenase ingibitor lacking significant antioxidant properties", Brit. J. Pharmacology (1997) 120, 1199-1206, и Comicelli и др., «15-Lipoxygenase and its Inhibition: A Novel Therapeutic Target for Vascular Disease», Current Pharmaceutical Design, 1999,5,11-20.
Соединения формулы I и гиполипидемический агент могут применяться вместе в одной оральной дозовой форме или в отдельных оральных дозовых формах, принимаемых одновременно.
Композиции, описанные выше, могут вводиться в дозированных формах, как описано выше в разовой или раздельных дозах от одного до четырех раз ежедневно. Желательно начать лечение пациента с применения низкой дозы комбинации и постепенно достигать высокой дозы комбинации.
Предпочтительным гиполипидемическим агентом является правастатин, симвастатин, ловастатин, аторвастатин, флувастатин или церивастатин, также как ниацин и/или холестагель.
Антидиабетический агент, который может необязательно применяться в сочетании с соединением формулы I, может представлять собой 1, 2, 3 или более антидиабетических агентов или антигипергликемических агентов, включая вещества, усиливающие секрецию инсулина или инсульновые сенсибилизирующие вещества, которые могут включать бигуанидины, сульфонилмочевины, ингибиторы глюкозидазы, PPAR γ агонисты, такие, как тиазолидиндионы, аР2 ингибиторы, PPAR α/γ дуальные агонисты, дипептидил пептидазы IV (DP4) ингибиторы, SGLT2 ингибиторы и/или меглитиниды, также как и инсулин, и/или глюкагоноподобный пептид-1 (GLP-1).
Антидиабетический агент может орально вводиться с антигипергликемическим агентом, предпочтительно бигуанидином, таким, как метформин или фенформин или их соли, предпочтительно метформин HCl.
Там, где антидиабетическим агентом является бигуанидин, соединения структуры I могут применяться в массовом соотношении с бигуанидином, которое лежит в пределах от около 0.001:1 до около 10:1, предпочтительно от около 0.01:1 до около 5:1.
Антидиабетическим агентом может также предпочтительно быть сульфонилмочевина, такая, как глибурид (также известная как глибенкламид), глимепирид (описанный в US 4379785), глипизид, гликлазид или хлорпропамид, другие известные сульфонилмочевины или другие антигипергликемические агенты, который действуют на АТР-зависимый канал Р-клеток, предпочтительными являются глибурид и глипизид, которые могут вводиться в одной или в различных оральных дозовых формах.
Соединения структуры I могут применяться в массовом соотношении с сульфонилмочевиной, которое лежит в области от около 0.01:1 до около 100:1, предпочтительно от около 0.02:1 до около 5:1.
Орально вводимым антидиабетическим агентом может быть также ингибитор глюкозидазы, такой, как акарбоза (описанный в US 4904769) или миглитол (описанный в US 4639436), которые могут вводиться в одной или в отдельных оральных дозовых формах.
Соединения структуры I могут применяться в массовом соотношении с ингибитором глюкозидазы, которое лежит в пределах от около 0.01:1 до около 100:1, предпочтительно от около 0.05:1 до около 10:1.
Соединения структуры I могут применяться в сочетании с PPAR γ агонистом, таким, как тиазолидиндионовый оральный антидиабетический агент или другие инсулиновые сенсибилизирующие вещества (которые имеют инсулин-чувствительное действие у NIDDM пациентов), такие, как троглитазон (Warner-Lambert's Rezulin®, описанный в US 4572912), розиглитазон (SKB), пиоглитазон (Takeda), Mitsubishi's МСС-555 (описанный в US 5594016), Glaxo-Welcome's GL-262570, энглитазон (СР-68722, Pfizer) или дарглитазон (СР-86325, Pfizer, изаглитазон (MIT/J&J), JTTPET-501 (JPNT/P&U), L-895645 (Merck), R-119702 (Sankyo/WL), NN-2344 (Dr. Reddy/NN) или YM-440 (Yamanouchi), предпочтительно розиглитазон и пиоглитазон.
Соединения структуры I могут применяться в массовом соотношении с тиазолидиндионом в количестве, которое лежит в пределах от около 0.01:1 до около 100:1, предпочтительно от около 0.05 до около 10:1.
Сульфонилмочевина и тиазолидиндион в количестве менее чем около 150 мг орального антидиабетического агента могут вводиться в одной таблетке с соединениями структуры I.
Соединения структуры I могут также применяться в сочетании с антигипергликемическим агентом, таким, как инсулин или с глюкагоноподобным пептидом-1 (GLP-1) таким, как GLP-1 (1-36) амид, GLP-1 (7-36) амид, GLP-1 (7-37) (которые описаны в US 5614492 Habener, описание которого приведено здесь в качестве ссылки), также как АС2993 (амилин) и LY-315902 (Lilly), которые могут вводиться инъекцией, интраназально, ингаляционно или с помощью трансдермальных или буккальных устройств.
Там, где присутствуют метформин, сульфонилмочевины, такие, как глибурид, глимепирид, глипирид, глипизид, хлорпропамид и гликлазид, и ингибиторы глюкозидазы, акарбозы или миглитол или инсулин (инъекционно, внутрилегочно, буккально или орально) могут применяться в составах, которые определены выше, и в количестве и дозе, как указано в Physician's Desk Reference (PDR).
Там, где присутствуют метформин или его соль, его можно применять в количествах, лежащих в пределах от около 500 до около 2000 мг в день, которые могут вводиться в одной или раздельных дозах от одного до четырех раз ежедневно.
Там, где присутствует тиазолидиндионовый антидиабетический агент, его можно применять в количествах, лежащих в пределах от около 0.01 до около 2000 мг/день, который может вводиться в одной или раздельных дозах от одного до четырех раз ежедневно.
Там, где присутствует инсулин, его можно применять в составах, количестве и дозах, как указано в Physician's Desk Reference.
Там, где присутствуют GLP-1 пептиды, их можно вводить орально в буккальных составах, назальным введением или парентерально, как описано в US 5346701 (TheraTech), 5614492 и 5631224, которые приведены здесь в качестве ссылок.
Антидиабетическим агентом может также быть PPAR α/γ дуальный агонист, такой, как AR-H039242 (Astra/Zeneca), GW-409544 (Glaxo-Wellcome), KRP297 (Kyorin Merck), также как те, которые описаны Murakami и др., "A Novel Insulin Sensitizer Acts As a Coligand for Peroxusome Proliferation -Activated Receptor Alpha (PPAR alpha) and PPAR gamma. Effect on PPAR alpha Activation on Abnormal Lipid Metabolism in Liver of Zucker Fatty Rats", Diabetes 47, 1841-1847 (1998).
Антидиабетическим агентом может быть SGLT2 ингибитор, такой, как описано в US заявке 60/158773, опубликованной 12.10.1999 (attorney file LA49), применяя дозы, которые там указаны. Предпочтительными являются соединения, указанные в качестве, предпочтительных в приведенной заявке.
Антидиабетическим агентом может быть аР2 ингибитор, такой, как описано в заявке US 09/391053, опубликованной 7.09.1999, и в заявке US 60/127,745, опубликованной 05.04.1999 (attorney file LA27*), применяя дозы, которые там указаны. Предпочтительными являются соединения, обозначенные как предпочтительные в указанной заявке.
Антидиабетическим агентом может быть DP4 ингибитор, такой, как описано в заявке 60/188,555, опубликованной 10.03.2000 (attorney file LA50), WO 99/38501, WO 99/46272, WO 99/67279 (PROBIODRUG), WO 99/67278 (PROBIODRUG), WO 99/61431 (PROBIODRUG), NVP-DPP728A (1-[[[2-[(5-цианопиридин-2-ил)амино]этил]амино]ацетил]-2-циано-(S)-пирролидин) (Novartis) (предпочтительно), как описано Hughes и др., Biochemistry, 38 (36), 11597-11603, 1999, TSL-225 (триптофил-1,2,3,4-тетрагидроизохинолин-3-карбоновая кислота (описанная Yamada и др., Bioorg. & Med. Chem. Lett. 8 (1998) 1537-1540, 2-цианопирролидины и 4-цианопирролидиды, которые описаны Ashworth и др., Bioorg. & Med. Chem. Lett., Vol.6, No.22, pp. 1163-1166 и 2745-2748 (1996), применяя дозы, которые указаны в ссылках.
Меглитинидом, который может необязательно применяться в сочетании с соединением формулы I по изобретению, может быть репаглинид, натеглинид (Novartis) или KADI 229 (PF/Kissei), предпочтительно репаглинид.
Соединение формулы I может применяться в массовом соотношении с меглитинидом, PPAR γ агонистом, PPAR α/γ дуальным агонистом, аР2 ингибитором, DP4 ингибитором или SGLT2 ингибитором, которое лежит в пределах от около 0.01:1 до около 100:1, предпочтительно от около 0.05 до около 10:1.
Другим типом терапевтического агента, который может необязательно применяться с соединением формулы I, может быть 1, 2, 3 или более агентов против ожирения, включая бета-3 адренергический агонист, ингибитор липазы, ингибитор серотонина (и допамина), аР2 ингибитор, агонист тироидного рецептора и/или аноректический агент.
Бета-3 адренергическим агонистом, который может необязательно применяться в сочетании с соединением формулы I, может быть AJ9677 (Takeda/Dainippon), L750355 (Merck) или СР331648 (Pfizer) или другие известные бета-3 агонисты, которые описаны в US 5541204, 5770615, 5491134, 5776983 и 5488064, предпочтительными являются AJ9677, L750355 и СР331648.
Ингибитором липазы, который может необязательно применяться в сочетании с соединением формулы I, может быть орлистат или ATL-962 (Alizyme), предпочтительным является орлистат.
Ингибитором серотонина (и допамина), который может необязательно применяться в сочетании с соединением формулы I, может быть сибутрамин, топирамат (Johnson & Johnson) или аксокин (Regeneron), предпочтительными являются сибутрамин и топирамат.
Агонистом тироидного рецептора, который может необязательно применяться в сочетании с соединением формулы I, может быть лиганд тироидного рецептора, который описан в WO 97/21993 (U. Cal SF), WO 99/00353 (KaroBio), GB 98/284425 (KaroBio) и заявке US 60/183223, опубликованной 17.02.2000, предпочтительными являются соединения KaroBio заявок и указанной US заявки.
Аноректическим агентом, который может необязательно применяться в сочетании с соединением формулы I, может быть дексамфетамин, фентермин, фенилпропаноламин или мазиндол, предпочтительным является дексамфетамин.
Различные агенты против ожирения, описанные выше, могут применяться в одной дозированной форме с соединением формулы I или в различных дозированных формах, в дозах и режимах, которые хорошо известны из уровня техники или из PDR.
Антигипертензивные агенты, которые могут применяться в сочетании с соединением формулы I по изобретению, включают АСЕ ингибиторы, антагонисты рецептора ангиотензина II, NEP/ACE ингибиторы, также как блокаторы кальциевых каналов, β-адренергические блокаторы и другие виды антигипертензивных агентов, включая диуретики.
Ингибитор ангиотензинтрансформирующего фермента, который может применяться здесь, включает содержащий меркаптогруппу (-S-), такой, как замещенные пролиновые производные, такие, как любой из описанных в US 4046889 Ondetti и др., указанном выше, предпочтительным является каптоприл, которым является 1-[(2S)-3-меркапто-2-метилпропионил]-L-пролин и меркаптоацильные производные замещенных пролинов, такие, как любой из описанных в US 4316906, предпочтительным является зофеноприл.
Другие примеры меркаптосодержащих АСЕ ингибиторов, которые могут здесь использоваться, включают рентиаприл (фентиаприл, Santen), описанный в Clin. Exp. Pharmacol. Physiol. 10: 131 (1983); также как пивоприл и YS980.
Другие примеры ингибиторов ангиотензин-трансформирующего фермента, которые могут здесь использоваться, включают любой из описанных в US 4374829, упомянутом выше, предпочтительным является N-(1-этоксикарбонил-3-фенилпропил)-L-аланил-L-пролин, или эналаприл, любой из фосфонатов замещенных амино- или иминокислот или соли, описанные в US 4452790, предпочтительным является (S)-1-[6-амино-2-[[гидрокси-(4-фенилбутил)фосфинил]окси]-1-оксогексил]-L-пролин или (церонаприл), фосфинилалканоилпролины, описанные в US 4168267, указанный выше, предпочтительным является фозиноприл, любой из фосфинилалканоилзамещенных пролинов, описанных в US 4337201, и фосфонамидаты, описанные в US 4432971, указанные выше.
Другие примеры АСЕ ингибиторов, которые могут здесь использоваться, включают Beecham's BRL 36378, которые описаны в ЕР 80822 и 60668; Chugai's MC-838, описанный в СА 102:72588V и Jap. J. Pharmacol. 40:373 (1986); Ciba-Geigy's CGS 14824 (3-([1- этоксикарбонил-3-фенил-(1S)-пропил]амино)-2,3,4,5-тетрагидро-2-оксо-1-(3S)-бензазепин-1 уксусная кислота HCl), описанная в UK 2103614 и CGS 16617, (3(S)-[[(1S)-5-амино-1-карбоксипентил]амино]-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-этановая кислота), описанная в US 4473575; цетаприл (алацеприл, Dainippon), описанный в Eur. Therap. Res. 39:671 (1986); 40:543 (1986); рамиприл (Hoechst), описанный в ЕР 79-022 и Curr. Ther. Res. 40:74 (1986); Ru 44570 (Hoechst), описанный в Arzneimittelforschung 34: 1254 (1985), цилазаприл (Hoffman-LaRoche), описанный в J. Cardiovasc. Pharmacol. 9:39 (1987); R 31-2201 (Hoffman-LaRoche), описанный в FEBS Lett. 165: 201 (1984); лизиноприл (Merck), индадаприл (делаприл), описанный в US 4385051; индолаприл (Schering), описанный в J. Cardiovasc. Pharmacol. 5:643655 (1983), спираприл (Schering), описанный в Acta. Pharmacol. Toxicol. 59 (Supp.5):173 (1986); периндоприл (Servier), описанный в Eur. J. Clin. Pharmacol. 31:519 (1987); куинаприл (Warner-Lambert), описанный в US 4344949 и СI925 (Warner-Lambert) ([3S-[2[R(*)R(*)]]3R(*)]-2-[2-[[1-(этоксиоксикарбонил)-3-фенилпропил]амино]-1-оксопропил]-1,2,3,4-тетрагидро-6,7-диметокси-3-изохинолинкарбоновая кислота HCl), описанная в Pharmacologist 26:243, 266 (1984), WY-44221 (Wyeth), описанный в J. Med. Chem. 26:394 (1983).
Предпочтительными АСЕ ингибиторами являются каптоприл, фозиноприл, эналаприл, лизиноприл, куинаприл, беназеприл, фентиаприл, рамиприл и моэксиприл.
NEP/ACE ингибиторы, которые могут также здесь использоваться, включают те, которые обладают ингибиторной активностью нейтральной эндопептидазы (NEP) и ингибиторной активностью ангиотензинтрансформирующего фермента (АСЕ). Примеры NEP/ACE ингибиторов, пригодные для использования здесь, включают описанные в US 5362727, 5366973, 5225401, 4722810, 5223516, 4749688, US 5552397, US 5504080, US 5612359, US 5525723, EP 0599444, 0481522, 0599444, 0595610, EP 0534363 A2, 534396 и 534492, и Е 0629627 А2.
Предпочтительными являются те NEP/ACE ингибиторы и их дозы, которые указаны как предпочтительные в указанных патентах/заявках US, приведенные здесь в качестве ссылок; наиболее предпочтительным является омапатрилат, BMS 189921 ([S-(R*,R*)]-гексагидро-6-[(2-меркапто-1-оксо-3-фенилпропил)амино]-2,2-диметил-7-охо-1Н-азепин-1-уксусная кислота (гемопатрилат)) и CGS 30440.
Антагонист рецептора ангиотензина II (также называется здесь как антагонист ангиотензина II или AII антагонист), пригодный для использования здесь, включает, но не ограничивается, ирбесартаном, лосартаном, валсартаном, кандесартаном, телмисартаном, тазосартаном или эпросартаном, предпочтительными являются ирбесартан, лосартан или валсартан.
Предпочтительная оральная дозированная форма, такая, как таблетки или капсулы, может содержать АСЕ ингибитор или AII антагонист в количестве, которое находится в пределах от около 0.1 до около 500 мг, предпочтительно от около 5 до около 200 мг и более предпочтительно от около 10 до около 150 мг.
Для парентерального введения ингибитор АСЕ, антагонист ангиотензина II или NEP/ACE ингибитор могут применяться в количестве, которое находится в пределах от около 0.005 мг/кг до около 10 мг/кг и предпочтительно от около 0.01 мг/кг до около 1 мг/кг.
При введении лекарственного внутривенно средства оно может находиться в обычном растворителе, таком, как дистиллированная вода, рассол, соляной раствор, раствор Рингера или другие обычные носители.
Учитывалось, что предпочтительные дозы АСЕ ингибитора и AII антагониста, также как других антигипертензивных средств, описываемых здесь, могут быть такими, как указано в последней редакции Physician's Desk Reference (PDR).
Другие примеры предпочтительных антигипертензивных агентов, пригодных для использования здесь, включают омапатрилат (Vanlev®), амлодипин безилат (Norvasc®), празосин HCl (Minipress®), верапамил, нифедипин, надолол, дилтиозем, фелодипин, низолдипин, израдипин, никардипин, атенолол, карведиол, соталол, теразозин, доксазосин, пропранолол и клонидин HCl (Catapres®).
Диуретики, которые могут применяться в сочетании с соединениями формулы I, включают гидрохлортиазид, торасемид, фуросемид, спиронолактоно и индапамид.
Антитромботические агенты, которые могут применяться в сочетании с соединениями формулы I по изобретению, включают аспирин, клопидогрель, тиклопидин, дипиридамол, абсиксимаб, тирофибан, эптифибатид, анагрелид и ифетробан, предпочтительными являются клопидогрел и аспирин.
Антитромботические лекарства могут применяться в количествах, которые указаны в PDR. Ифетробан может применяться в количествах, как указано в US 5100889.
Антиостеопорозные агенты, пригодные для использования здесь в сочетании соединениями формулы I по изобретению, включают паратиероидный гормон или бисфосфонаты, такие, как МК-217 (алендронат) (Fosamax®). Применяемые дозы могут быть такими, как указано в PDR.
При осуществлении способа по изобретению применяют фармацевтическую композицию, которая содержит соединения структуры I, с или без другого терапевтического агента, в сочетании с фармацевтическим носителем или растворителем. Фармацевтическая композиция может быть приготовлена с использованием обычных твердых или жидких наполнителей или растворителей и фармацевтических добавок подходящего типа для способа желаемого введения. Соединения могут вводиться млекопитающим, включая людей, обезьян, собак и т.д., оральным путем, например, в виде таблеток, капсул, гранул или порошков или их могут вводить парентеральным путем в виде инъекционных препаратов. Доза для введения находится предпочтительно между 50 и 2000 мг в день и ее можно вводить в одной дозе или в виде индивидуальных доз от 1-4 раз в день.
Типичные капсулы для орального введения включают соединения структуры I (250 мг), лактозу (75 мг) и стеарат магния (15 мг). Смесь пропускают через 60-ячеечное сито и помещают в желатиновую капсулу No. 1.
Типичный инъекционный препарат получают асептическим наполнением 250 мг соединений структуры I в ампулу, асептическим замораживанием-высушиванием и запаиванием. Для использования содержимое ампул смешивают с 2 мл физиологического рассола с получением инъекционного препарата.
Следующие примеры представляют предпочтительные воплощения изобретения.
В примерах используются следующие сокращения:
Ph=фенил
Bn=бензил
трет-Bu=трет-бутил
Me=метил
Et=этил
TMS=триметилсилил
TMSN3=триметилсилилазид
TBS=трет-бутилдиметилсилил
FMOC=флуоренилметоксикарбонил
Boc=трет-бутоксикарбонил
Cbz=карбобензилокси или карбобензокси или бензилоксикарбонил
THF=тетрагидрофуран (ТГФ)
Et2O=диэтиловый эфир
hex=гексаны
EtOAc=этилацетат
DMF=диметилформамид
МеОН=метанол
EtOH=этанол
i-PrOH=изопропанол
DMSO=диметилсульфоксид (ДМСО)
DME=1,2-диметоксиэтан
DCE=1,2-дихлорэтан
НМРА=гексаметилтриамид фосфорной кислоты
НОАс или АсОН=уксусная кислота
TFA=трифторуксусная кислота
i-Pr2NEt=диизопропилэтиламин
Et3N=триэтиламин
NMM=N-метилморфолин
DMAP=4-диметиламинопиридин
NaBH4=борогидрид натрия
NaBH(ОАс)3=триацетоксиборогидрид натрия
DIBALH=диизобутилалюмогидрид
LiAlH4=литийалюмогидрид
n-BuLi=н-бутиллитий
Pd/C=палладий на угле
PtO2=оксид платины
КОН=гидроксид калия
NaOH=гидроксид натрия
LiOH=гидроксид лития
К2СО3=карбонат калия
NaHCO3=бикарбонат натрия
DBU=1,8-диазабицикло[5.4.0]ундек-7-ен
EDC (или EDC·HCl) или EDCI (или EDCI·HCl) или EDAC=3-этил-3'-(диметиламино)пропилкарбодиимида гидрохлорид (или 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид)
НОВТ или HOBT·H2O=1-гидроксибензотриазола гидрат
BOAT=1-гидрокси-7-азабензотриазол
ВОР реагент=бензотриазол-1-илокси-трис(диметиламино)фосфоний гексафторфосфат
NaN(TMS)2=гексаметилдисилазид натрия или бис(триметилсилил)амид натрия
Ph3Р=трифенилфосфин
Pd(OAc)2=ацетат палладия
(Ph3P)4Pd=тетракистрифенилфосфинпалладий
DEAD=диэтилазодикарбоксилат
DIAD=диизопропилазодикарбоксилат
Cbz-Cl=бензилхлорформиат
CAN=цериевый нитрат аммония
SAX=сильный анионообменник
SCX=сильный катионообменник
Ar=аргон
N2=азот
min=минута(ы)
h или hr=час(ы)
L=литр
mL=миллилитр
μL=микролитр (мкл)
g=грамм(ы)
мг=миллиграмм(ы)
mol=моли
mmol=миллимоль(и) (ммоль)
meq=миллиэквивалент
RT=комнатная температура
sat или sat'd=насыщенный
aq.=водный
TLC=тонкослойная хроматография
HPLC=высокоэффективная жидкостная хроматография
LC/MS=высокоэффективная жидкостная хроматография/масс-спектрометрия
MS или Mass Spec=масс-спектрометрия
NMR=ядерно-магнитный резонанс
NMR спектральные данные: s(с)=синглет; d(д)=дуплет; m(м)=мультиплет; br=размытый; t(т)=триплет
mp=точка плавления (т.пл.)
Пример 1
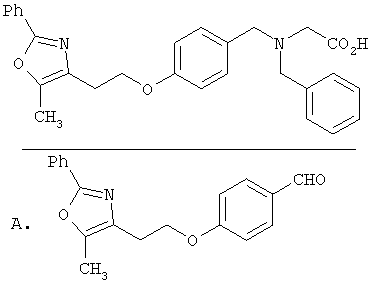
К охлажденному до 0°С раствору 4-гидроксибензальдегида (1.70 г, 12.3 ммоль), 5-фенил-2-метилоксазол-4-этанола (Maybridge; 2.50 г, 14.0 ммоль) и Ph3Р (4.20 г, 16.0 ммоль) в сухом ТГФ (30 мл) добавляют по каплям DEAD (3.20 г, 15.0 ммоль). Раствор перемешивают при 0°С в течение 0.5 ч, затем оставляют нагреваться до комнатной температуры и перемешивают в течение ночи. Оранжево-красный раствор упаривают в вакууме и остаток хроматографируют (постепенный градиент от 5:1 до 5:2, гексан:EtOAc) с получением соединения, часть А (2.47 г, 65%) в виде прозрачного светло-желтого вязкого масла.
A1. Альтернативный способ получения альдегида, часть А.
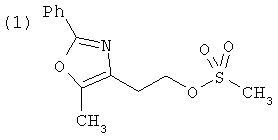
К охлажденному до -5°С раствору 5-фенил-2-метилоксазол-4-этанола (20.00 г, 0.098 моль) в СН2Cl2 (100 мл) одной порцией добавляют метансульфонилхлорид (12.40 г, 0.108 моль) (экзотермическая реакция). После повторного охлаждения до -5°С медленно добавляют Et3N (11.1 г, 0.110 моль) в течение более 30 минут (температура внутри <3°С). Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение 1 часа (реакцию отслеживают с помощью аналитической ВЭЖХ), после чего исходный продукт исчезает. Реакционную смесь промывают водным раствором HCl (2×50 мл 3 н. раствора). Объединенные водные слои экстрагируют с помощью СН2Cl2 (50 мл). Полученные объединенные органические экстракты последовательно промывают насыщенным водным раствором NaHCO3 и рассолом (50 мл каждый), сушат (Na2SO4) и концентрируют до объема ˜30 мл. Добавляют метил-трет-бутиловый эфир (120 мл) и смесь перемешивают; образуется белое твердое вещество. Смесь охлаждают до -20°С для завершения кристаллизации. Продукт фильтруют и сушат под вакуумом, что дает мезилат (23.3 г, 85%) в виде белого твердого вещества. Маточную жидкость концентрируют под вакуумом и перекристаллизовывают из метил-трет-бутилового эфира(МТВЕ)/гептана, что дает вторую порцию продукта, такого как мезилат (3.3 г, 12%; общий выход 97%).

Смесь полученного выше мезилата (13.6 г, 0.048 моль), 4-гидроксибензальдегида (7.09 г, 0.058 моль) и К2СО3 (9.95 г, 0.072 моль) в ДМФА (110 мл) нагревают до 100°С в течение 2 часов (реакция завершена, как показывает аналитическая ВЭЖХ). Смеси дают охладиться до комнатной температуры, затем выливают в ледяную воду (400 мл) и перемешивают в течение 30 минут. Полученное твердое вещество фильтруют, промывают ледяной водой (3×25 мл) и сушат под вакуумом при 50-60°С в течение ночи. Сырой продукт кристаллизуют из МТВЕ-гексана, что дает (12.2 г, 82%; 2 порции) альдегида (соединение, часть А1 ) в виде белого твердого вещества.
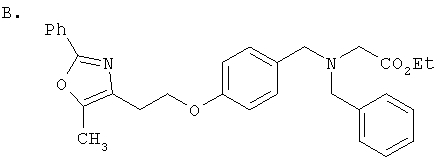
К раствору N-бензилглицинэтилового эфира (43 мг; 0.22 ммоль) и соединения из части А1 (52 мг; 0.17 ммоль) в DCE (10 мл) добавляют NaBH(OAc)3 (56 мг; 0.26 ммоль). Реакционную смесь энергично перемешивают в течение ночи (12 часов). Добавляют насыщенный водный раствор NaHCO3 (10 мл) и смесь экстрагируют с помощью EtOAc (3×10 мл). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4), концентрируют под вакуумом и хроматографируют (гексан:EtOAc, 4:1), что дает соединение из части В (45 мг; 55%) в виде бледно-желтого масла, кроме того исходный продукт (14 мг; 27%).
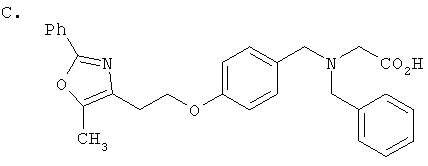
К раствору соединения из части В (45 мг) в СН3ОН (2 мл) добавляют водный раствор NaOH (3 мл 1 M раствора). Раствор перемешивают в течение ночи 14 часов и затем подкисляют до рН 5 избытком водного раствора HCl (1 M раствор). Смесь экстрагируют с помощью EtOAc (2×10 мл); объединенные органические экстракты промывают рассолом, сушат (Na2SO4), концентрируют под вакуумом, что дает желаемую кислоту, которая еще загрязнена исходным материалом. Указанную смесь растворяют в СН3ОН (2 мл) и водном растворе NaOH (3.0 мл 1 M раствора), полученный раствор нагревают с обратным холодильником в течение 1.5 часов. Экстрактивная обработка кислотой, как описано выше, дает желаемое названное соединение в виде бесцветного твердого вещества (28 мг; 71%). [М+Н]+=457.2
Пример 2
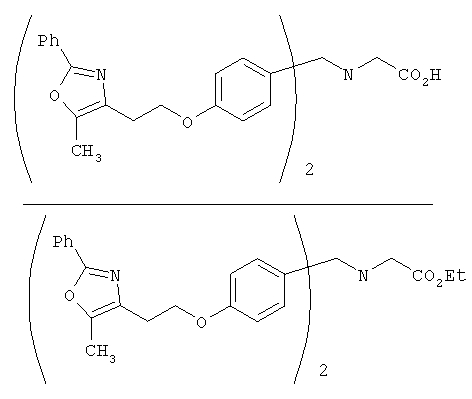
К раствору соединения из примера 1, часть А (147 мг; 0.479 ммоль) и гидрохлорида глицинэтилового эфира (73 мг; 0.52 ммоль) в DCE (2 мл) добавляют Et3N и NaBH(OAc)3 (156 мг; 0.74 ммоль), реакционную массу перемешивают в течение ночи при комнатной температуре. Флэш-хроматография (постепенный градиент от 7:3 до 2:3 гексан:EtOAc) дает 35 мг (21%) дибензилглицинового эфира (соединение по примеру 2, часть А). Кроме того, получают 127 мг (67%) монобензилглицинового эфира (соединение по примеру 3, часть А).
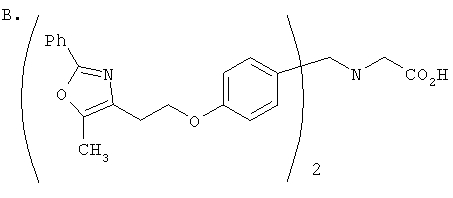
Раствор соединения по примеру 1, часть А (35 мг; 0.051 ммоль) в СН3ОН (2 мл) и водном растворе NaOH (3 мл 1 М раствора) нагревают при кипении с обратным холодильником в течение 12 часов. Раствор доводят до рН 5 водным раствором 1 М HCl и водным раствором 1 М NaOH, затем экстрагируют с помощью EtOAc (3х). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом, что дает названное соединение (13 мг) в виде бесцветного твердого вещества. [М+Н]+=658.2
Пример 3
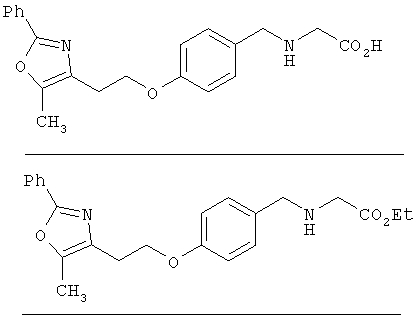
К раствору соединения по примеру 1, часть А (147 мг; 0.479 ммоль) и гидрохлорида глицинэтилового эфира (73 мг; ммоль) в DCE добавляют Et3Н и NaBH(ОАс)3 (156 мг; 0.74 ммоль). Флэш-хроматография (постепенный градиент от 7:3 до 2:3, гексан:EtOAc) дает 127 мг (67%) названного соединения. Кроме того, в качестве побочного продукта получают 35 мг (21%) бисбензилглицинового эфира (пример 2,часть А).
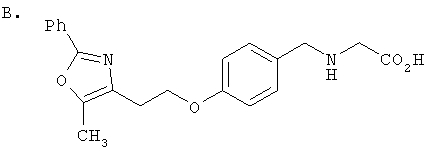
Раствор соединения, часть А (72 мг; 0.18 ммоль) в водном растворе NaOH (2 мл 1 М раствора) и метаноле (2 мл) нагревают с обратньм холодильником в течение 3 часов. Реакционную массу доводят до рН 5 водным раствором 1 М HCl и твердое вещество отфильтровывают. Фильтрат экстрагируют с помощью EtOAc (3х). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом, что дает бесцветное твердое вещество, которое очищают препаративной ВЭЖХ (используя YMC S5 ODS 20 мм × 100 мм колонку с непрерывным градиентом от 70% А: 30% В до 100% В в течение 10 минут при скорости потока 20 мл/мин, где А=90:10:0.1 H2O:метанол:ТФК и где В=90:10:0.1 метанол:Н2O:ТФК), что дает названное соединение(10 мг; 15%) в виде бесцветного твердого вещества. [М+Н]+=367.2
Пример 4

Раствор амино-трет-бутилового эфира (0.040 г, 0.095 ммоль; получают, как раскрыто в примере 7, часть С, за исключением того, что альдегид, используемый в восстановительном аминировании, берут из примера 1, часть А, а не из примера 7, часть А)
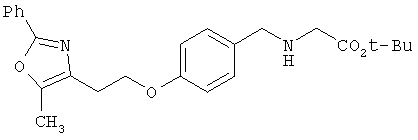
и пропаргилбромид (0.014 г, 0.120 ммоль), а также DBU (0.5 мл; 2.96 ммоль) в DCE (1 мл) перемешивают при 0°С в течение 5 часов. ТЖХ (тонкослойная жидкостная хроматография) показывает, что реакция в этот момент завершена. Добавляют EtOAc (10 мл), органическую фазу промывают Н2O и концентрируют в вакууме. Оставшееся масло растворяют в СН2Cl2/ТФК (1:1,1 мл) и перемешивают при комнатной температуре в течение 5 часов, затем концентрируют под вакуумом. Остаток очищают препаративной ВЭЖХ (YMC S5 ODS 30 мм × 250 мм колонка с обращенной фазой; скорость потока 25 мл/мин; 30 мин, непрерывный градиент от 70:30 А:В до 100% В; где А=90:10:0.1 Н2O:метанол:ТФК (трифторуксусная кислота) и где В=90:10:0.1 метанол:Н2O:ТФК), что дает названное соединение (34 мг, 92%) в виде масла. ЖХ/МС (электронапыление) дает точное значение [М+H]+=405.2 для названного соединения.
Пример 5
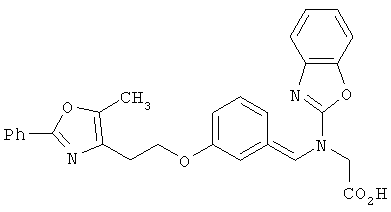
Раствор 2-хлорбензоксазола (20 мг; 0.131 ммоль), вторичного аминометилового эфира (52 мг; 0.146 ммоль)
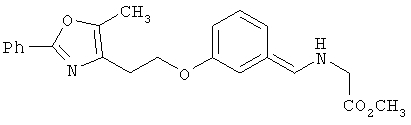
(получают, как раскрыто в примере 3, часть А, за исключением того, что глицинэтиловый эфир HCl замещают на глицинметиловый эфир·HCl и применяют альдегид по примеру 7, часть А) и избыток Et3N (0.5 мл) в ТГФ (2.0 мл) нагревают до 100°С в герметично закрываемой трубке и реакцию отслеживают с помощью ЖХ/МС. Через 4 дня исходный амин полностью вступает в реакцию. Реакционную смесь охлаждают до комнатной температуры и к раствору добавляют водный раствор LiOH (0.50 мл 1 М раствора). Полученный раствор перемешивают при комнатной температуре в течение 5 часов, после чего гидролиз оказывается завершенным. Смесь концентрируют под вакуумом, что дает сырую кислоту в виде масла, которую очищают препаративной ВЭЖХ (30 минут, непрерывный градиент от 70:30 А:В до 100% В, где А=90:10:0.1 H2O:метанол:ТФК и В=90:10:0.1 метанол:H2O:ТФК; скорость потока=25 мл/мин; YMC S5 ODS 30×250 мм колонка с обращенной фазой ), что дает названное соединение (52 мг; 82%) в виде твердого вещества после лиофилизации из (метанола/Н2О). [М+Н]+=484.2.
Пример 6
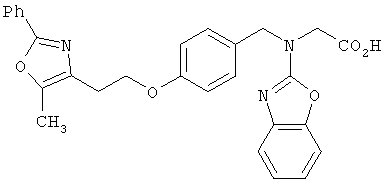
Названное соединение (13 мг; 21%) получают по аналогии с методикой примера 5, используя соответствующий вторичный аминометиловый эфир.

(указанное соединение получают, как раскрыто в примере 3, часть А, за исключением того, что глицинэтиловый эфир HCl заменяют на глицинметиловый эфир·HCl). [М+Н]+=484.2.
Пример 7
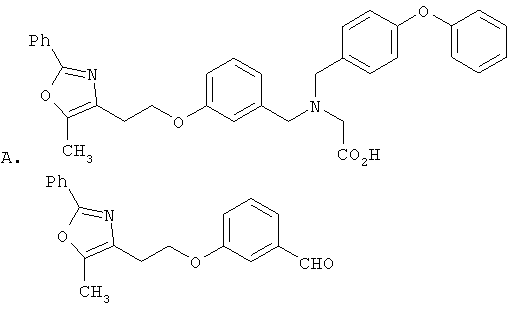
К охлажденному до 0°С раствору 3-гидроксибензальдегида (3.00 г; 24.6 ммоль), 2-фенил-5-метилоксазол-4-этанола (5.00 г; 24.6 ммоль) и Ph3Р (7.10 г; 27.1 ммоль) в сухом ТГФ (75 мл) добавляют по каплям DEAD (4.27 мл; 27.1 ммоль) в течение более 10 минут. Коричнево-оранжевому раствору дают нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 24 часов. Раствор концентрируют под вакуумом и хроматографируют (SiO2; постепенный градиент: 100% гексан до гексана:EtOAc, 3:1), что дает соединение, часть А, в виде бледно-желтого вязкого масла (4.01 г; 53%).
А.1. Альтернативный процесс получения альдегида, часть А.
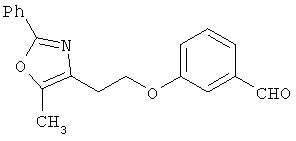
К раствору 3-гидроксибензальдегида (9.1 г; 0.074 ммоль) в CH3CN (206 мл) добавляют К2СО3 (10.3 г). Смесь нагревают до 90°С на масляной бане и перемешивают в течение 18 часов при 90°С (в этот момент реакция завершена, как показала аналитическая ВЭЖХ). Реакционную массу охлаждают до комнатной температуры, затем разбавляют EtOAc (500 мл), промывают Н2О, водным раствором NaOH (2×100 мл 1 М раствора) и рассолом. Органическую фазу сушат (MgSO4) и концентрируют под вакуумом. Оставшееся масло хроматографируют (SiO2; гексан:EtOAc, от 9:1 до 4:1), что дает альдегид, часть А (12.7 г; 67%) в виде вязкого прозрачного бледно-желтого масла.
В.
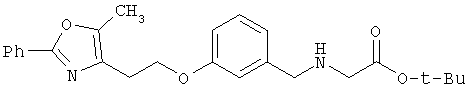
Раствор соединения, часть А1 (4.00 г; 13.0 ммоль), гидрохлорида глицин -трет-бутилового эфира (2.40 г; 14.3 ммоль) и Et3N (2.18 мл; 15.7 ммоль) в метаноле (30 мл) перемешивают при комнатной температуре в течение 6 часов и затем охлаждают до 0°С. К раствору NaBH4 (594 мг; 15.7 ммоль) в метаноле (10 мл) добавляют порциями при 0°С раствор сырого имина в течение более 15 минут. Раствор перемешивают при 0°С в течение 3 часов, затем при комнатной температуре 3 часа, затем концентрируют под вакуумом без нагревания, чтобы удалить метанол. Остаток разделяют между насыщенным водным раствором NaCl и EtOAc (50 мл каждый). Водный слой экстрагируют EtOAc (2×50 мл). Объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом, что дает желтое масло, которое хроматографируют на SiO2 (постепенный градиент; гексан:EtOAc, от 4:1 до 2:3) и получают соединение, часть В, в виде вязкого бледно- желтого масла (4.82 г; 88%).
С.
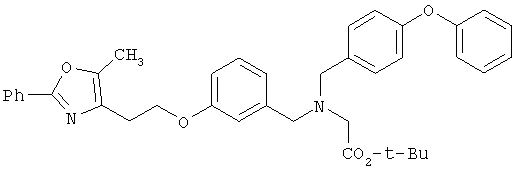
К раствору соединения, часть В (0.400 г; 0.95 ммоль) и 4-феноксибензальдегида (0.216 г; 1.09 ммоль) в DCE (5 мл) добавляют NaBH(OAc)3 (0.300 г; 1.42 ммоль), а затем НОАс (25 мкл). Реакционную массу перемешивают при комнатной температуре в течение 24 часов. Как показала аналитическая ВЭЖХ 10% непрореагировавший исходный амин все еще присутствует. Вводят дополнительное количество альдегида (30 мг) и NaBH(ОАс)3 (60 мг) и реакцию перемешивают при комнатной температуре в течение последующих 18 часов, после чего она завершается. Раствор разделяют между водным раствором NaHCO3 (50 мл 10% раствора) и EtOAc (50 мл). Водный слой экстрагируют EtOAc (2×25 мл). Объединенные органические экстракты промывают водным раствором NaHCO3 (2×15 мл 10% раствора), сушат (Na2SO4) и концентрируют под вакуумом, что дает соединение, часть С (521 мг сырой продукт) в виде прозрачного бесцветного масла.
D.
Соединение, часть С, растворяют в CHCl3 (2 мл) и ТФК (1.5 мл) и раствор перемешивают при комнатной температуре в течение 24 часов. Раствор концентрируют под вакуумом и остаток очищают препаративной ВЭЖХ (YMC S5 ODS 20×250 мм колонка; непрерывный градиент от 40:60 растворителя А:В до 100% растворителя В, где растворитель А=90:10:0.1 H2O:метанол:ТФК; растворитель В=90:10:0.1 метанол:H2O:ТФК). Очищенный продукт лиофилизуют из метанола/Н2О, что дает названную аминокислоту (312 мг; 48% в 2 стадии) в виде ее соли с ТФК (грязно-белый лиофилат). [М+Н]+(электронапыление)=549.3.
Пример 8
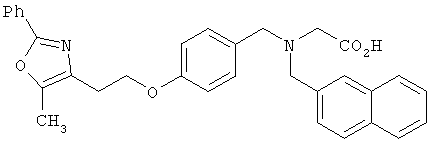
Смесь аминоэфира (39 мг; 0.092 ммоль)
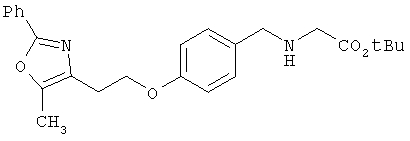
(получают, как раскрыто в примере 4), 2-нафтальдегида (29 мг; 0.185 ммоль) и NaBH(OAc)3 (100 мг; 0.472 ммоль) в DCE (1.5 мл) перемешивают при комнатной температуре в течение 16 часов. Затем к смеси добавляют ТФК (1.0 мл) и перемешивают при комнатной температуре в течение последующих 12 часов. Летучие продукты удаляют под вакуумом. Полученный остаток разбавляют метанолом (1,5 мл), фильтруют и очищают препаративной ВЭЖХ (YMC S5 ODS 30 мм × 250 мм колонка; непрерывный 30 мин градиент @ 25 мл/мин, от 100% А до 100% В; растворитель А=90:10:0.1 H2O:метанол:ТФК; В=90:10:0.1 метанол:H2O:ТФК), что дает желаемый названный продукт (39 мг; 68%) в виде прозрачного вязкого масла. [М+Н]+=507.3.
Пример 9
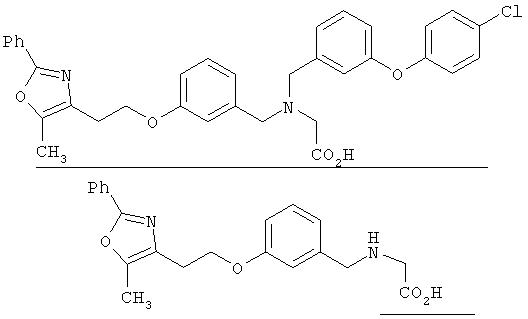
Раствор трет-бутилового эфира аминокислоты (1.8 г, 4.27 ммоль)
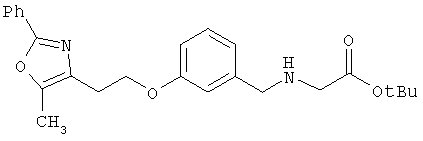
(получают, как раскрыто в примере 7, часть В) и ТФК (20 мл) в СН2Cl2 (40 мл) перемешивают при комнатной температуре в течение ночи. Раствор концентрируют под вакуумом и остаток растворяют в CH2Cl2, а затем элюируют через твердый NaHCO3 (чтобы удалить избыток ТФК) избытком СН2Cl2. Объединенные фильтраты концентрируют под вакуумом, что обеспечивает желаемую аминокислоту, часть А (1.48 г; 95%). [M+H]+=457.2.
В.

Названное соединение получают как часть библиотеки в потоке растворимой фазы, используя следующий примерный процесс.
К раствору аминокислоты, часть А (27 мг, 0.074 ммоль; в 2 мл СН2Cl2)
добавляют (4-хлорфенокси)-3-бензальдегид (86 мг; 0.37 ммоль), NaBH(OAc)3 (79 мг, 0.37 ммоль) и НОАс (0.1 мл). Реакционную массу перемешивают при комнатной температуре в течение 15 часов.
Продукт очищают с помощью твердофазной экстракции, используя Varian SAX картридж (3 г сорбента в 6 мл колонке, 0.3 meg/г) с помощью процесса, описанного ниже:
1) колонку заполняют метанолом (10 мл) и CH2Cl2 (20 мл);
2) реакционную смесь загружают в SAX колонку;
3) колонку промывают СН2Cl2 (10 мл);
4) колонку промывают 1% ТФК в метаноле (3 мл);
5) продукт элюируют с помощью 1% ТФК в метаноле (20 мл).
Раствор продуктов реакции (объединяют фракции после 5 стадий) концентрируют, используя Speed Vac (быстрый вакуум) в течение 16 часов, что приводит к сырому продукту (25 мг; 49%) в виде твердого вещества. Анализ ВЭЖХ с обращенной фазой (YMC S5 ODS 4.6×33 мм колонка, непрерывный градиент от 100% А до 100% В в течение 2 мин при скорости потока 5 мл/мин [растворитель А=10% метанола/90% H2O/0.2% Н3PO4; растворитель В=90% метанола/10% Н2O/0.2% Н3PO4]) показывает, что чистота продукта составляет 92%. Кроме того, ЖХ/МС (электронапыление) определяет точный молекулярный ион [(М+Н)+=583] для названного соединения.
Пример 10 (процесс, использующий гетероциклический альдегид)
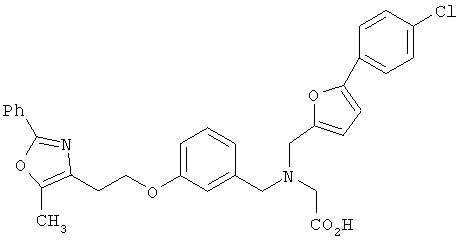
Названное соединение получают как часть библиотеки в потоке растворимой фазы, используя следующий примерный процесс. Смесь аминокислоты (14 мг; 0.038 ммоль)
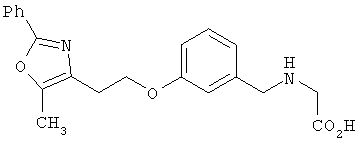
(получают, как раскрыто в примере 9, часть А), 5-(4-хлорфенил)-2-фурфураля (16 мг; 0.076 ммоль) и NaBH(OAc)3 (72 мг; 0.34 ммоль) в DCE (1.5 мл) перемешивают при комнатной температуре в течение 16 часов. Затем к смеси добавляют ТФК (1.0 мл), которую перемешивают при комнатной температуре в течение последующих 12 часов. Летучие продукты удаляют под вакуумом. Оставшийся остаток разбавляют метанолом (1.5 мл), фильтруют и очищают препаративной ВЭЖХ (YMC S5 ODS 30 мм × 250 мм колонка; непрерывный 30-минутный градиент @ 25 мл/мин, от 100% А до 100% В; растворитель А=90:10:0.1 H2O:метанол:ТФК; В=90:10:0.1 метанол:H2O:ТФК), что дает желаемый названный продукт (39 мг; 68%) в виде прозрачного вязкого масла.
Пример 10А
Альтернативный процесс очистки препаративной ВЭЖХ используют следующим образом.
Сырой продукт восстановительного аминирования очищают с помощью твердофазной экстракции, используя SAX картридж (United Chemicals; 3 г сорбента в 6 мл колонке, 0.3 meg/г) с помощью процесса, раскрытого ниже:
1) колонку заполняют метанолом (5 мл) и CH2Cl2 (5 мл);
2)реакционную смесь, разбавленную 2 мл СН2Cl2, загружают в SAX колонку;
3) колонку промывают СН2Cl2 (8 мл);
4) продукт элюируют с помощью 1% ТФК в метаноле (20 мл).
Содержащие продукт фракции концентрируют под вакуумом, используя Speed Vac в течение 16 часов, что дает сырой продукт. Его растворяют в СН2Cl2:метаноле (95:5) и загружают на силикагельный картридж (1.5 г SiO2), после чего продукт элюируют с помощью СН2Cl2:метанола (95:5; 8 мл). Продукт, содержащий фракции, концентрируют под вакуумом, используя Speed Vac, что дает желаемый названный продукт.
Анализ ВЭЖХ с обращенной фазой (YMC S5 ODS 4.6×33 мм колонка, непрерывный градиент от 100% А до 100% В в течение 2 минут при скорости потока 5 мл/мин, [растворитель А=10% метанола/90% Н2O/0.2% Н3PO4; растворитель В=90% метанола/10% H2O/0.2% Н3PO4]) показывает, что чистота продукта составляет 92%. Кроме того, ЖХ/МС (электронапыление) определяет точный молекулярный ион [(М+Н)+=583] для названного соединения.
Пример 11

К смеси амино-трет-бутилового эфира (0.339 г, 0.80 ммоль)
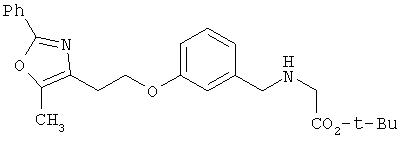
(получают, как раскрыто в примере 7, часть В), 4-гидроксибензальдегида (0.127 г, 1.03 ммоль) и NaBH(ОАс)3 (0.510 г, 2.4 ммоль) добавляют 7 капель НОАс. Реакционную массу перемешивают при комнатной температуре в течение 16 часов. Смесь разбавляют EtOAc, затем промывают водным раствором NaHCO3. Органическую фазу сушат (MgSO4) и концентрируют под вакуумом. Сырой продукт хроматографируют (SiO2; гексан/EtOAc, 3:1 до 1:4), что обеспечивает 4-гидроксибензиламиноэфир, названное соединение (0.381 г, 90%).
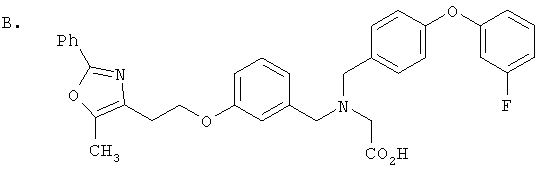
Названное соединение получают как часть библиотеки в потоке растворимой фазы, используя следующий примерный процесс.
К раствору фенольного соединения, часть А (30 мг, 0.057 ммоль) в CH2Cl2 (1 мл) добавляют 3-фторфенилбороновой кислоты (12 мг; 0.086 ммоль) и 4Å молекулярные сита (их предварительно сушат при 400°С в течение ночи) при комнатной температуре.
После перемешивания в течение 5 минут к смеси добавляют Cu(ОАс)2 (1 экв), Et3N (5 экв) и пиридин (5 экв). Сосуд вскрывают и дают воздуху пройти через реакционную массу. Затем ее перемешивают при комнатной температуре в течение 60 часов, после чего реакция оказывается завершенной, как показывает аналитическая ВЭЖХ и ЖХ/МС. (Для других реакций, которые не завершаются за это время, вводят дополнительную бороновую кислоту (1.5 экв), чтобы образовать дополнительный желаемый продукт). Реакционную смесь фильтруют и концентрируют под вакуумом. Продукт очищают с помощью твердофазной экстракции, используя United Technology SCX колонку (2 г сорбента в 6 мл колонке) с помощью процесса, раскрытого ниже:
1) колонку заполняют метанолом (10 мл) и СН2Cl2 (10 мл);
2) остаток растворяют в минимальном количестве CH2Cl2 и загружают в SCX колонку;
3) картридж последовательно промывают СН2Cl2 (20 мл); СН2Cl2/метанолом (20% метанол, 20 мл) и метанолом (20 мл);
4) продукт элюируют раствором 0.5 н. NH3 в метаноле.
Содержащие продукт фракции концентрируют под вакуумом, что дает желаемый трет-бутиловый эфир. (Некоторые незавершенные реакции требуют проведения хроматографии (на SiO2) сырого продукта, что дает эфиры требуемой чистоты). трет-Бутиловый эфир обрабатывают раствором 30% ТФК в СН2Cl2 в течение ночи. Летучие продукты удаляют и остаток вновь растворяют в СН2Cl2 (1 мл) и концентрируют под вакуумом на Speed Vac (быстром вакууме), что позволяет получить желаемый названный продукт (30 мг; 77%). Анализ ВЭЖХ с обращенной фазой показывает, что чистота продукта составляет 90%. Кроме того, ЖХ/МС определяет точный молекулярный ион [(М+Н)+=567] для желаемого названного соединения.
Пример 12
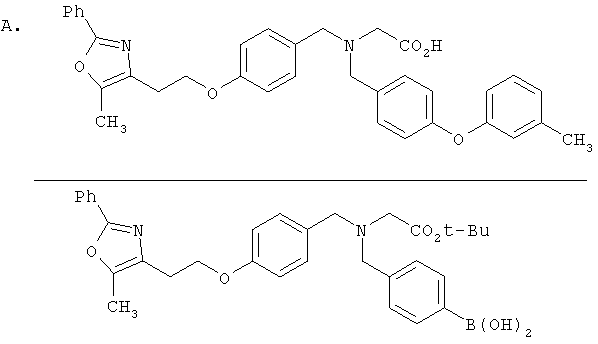
К раствору вторичного амино-трет-бутилового эфира (110 мг; 0.26 ммоль)
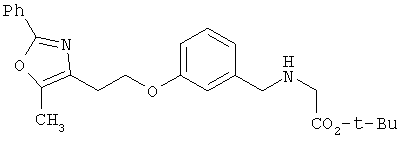
(получают, как раскрыто в примере 7, часть В) в 1,2-дихлорэтане (4 мл) последовательно добавляют 4-формилфенилбороновую кислоту (47 мг; 0.31 ммоль) и NaBH(ОАс)3 (165 мг; 0.78 ммоль). Смесь перемешивают при комнатной температуре в течение 3 часов. Анализ ВЭЖХ и ЖХ/МС показывает что реакция завершена именно в этот момент. Летучие продукты удаляют под вакуумом и остаток хроматографируют (SiO2; постепенный градиент от 3:1 до 1:1, гексан:EtOAc), что обеспечивает названное соединение (133 мг; 91%) в виде белой пены.
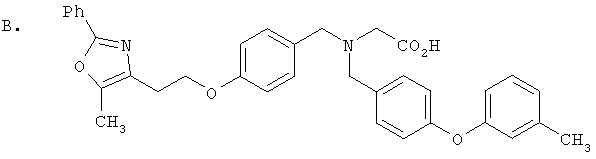
Названное соединение получают как часть библиотеки в потоке растворимой фазы, используя следующий процесс. К раствору соединения бороновой кислоты, часть А (40 мг, 0.072 ммоль) в CH2Cl2 (1 мл) добавляют мета-крезол (23 мг; 0.22 ммоль) и 4Å молекулярные сита (150 мг; предварительно сушат при 400°С в течение ночи). После перемешивания в течение 5 минут к смеси добавляют Cu(ОАс)3 (1 экв), Et3N (5 экв) и пиридин (5 экв). Сосуд открывают и воздуху дают войти в реакционную массу, после чего ее перемешивают при комнатной температуре в течение 24 часов. Реакционную смесь фильтруют через подушку из Целита и концентрируют под вакуумом. Продукт очищают с помощью твердофазной экстракции, используя United Technology SCX колонку (2 г сорбента в 6 мл колонке) с помощью процесса, описанного ниже:
1) колонку заполняют метанолом (10 мл) и CH2Cl2 (10 мл);
2) остаток растворяют в минимальном объеме CH2Cl2 и загружают в SCX колонку;
3) картридж последовательно промывают СН2Cl2 (20 мл) и метанолом (20 мл);
4) продукт элюируют раствором 0.5 н. NH3 в метаноле;
5) содержащие продукт фракции концентрируют под вакуумом;
6) остаток растворяют в минимальном количестве СН2Cl2 и загружают на силикагельный картридж (2 мл);
7) картридж элюируют гексаном:EtOAc (3:1; 20 мл);
8) содержащие продукт фракции собирают и концентрируют под вакуумом, что дает очищенный трет-бутиловый эфир.
трет-Бутиловый эфир обрабатывают раствором (1:1) ТФК в СН2Cl2 в течение ночи. Летучие продукты удаляют и остаток вновь растворяют в CH2Cl2 (1 мл) и концентрируют под вакуумом на Speed Vac, что дает желаемый названный продукт (25 мг; 48%) в виде слегка желтоватого масла. Анализ ВЭЖХ с обращенной фазой показывает, что чистота продукта составляет 91%. Кроме того, ЖХ/МС определяет точный молекулярный ион [(М+H)+=563.2] для желаемого соединения.
Пример 13
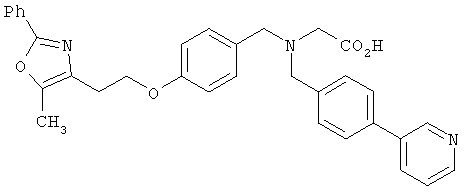
Названное соединение получают как часть библиотеки в потоке растворимой фазы, используя следующий примерный процесс.
К раствору 3-бромпиридина (32 мг; 0.2 ммоль) в DME (1 мл) последовательно добавляют (Ph3Р)4Pd (5 мг; 0.05 моль экв) и из примера 12, часть А, бороновую кислоту (50 мг; 0.09 ммоль)
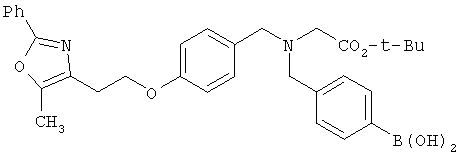
Под конец добавляют водный раствор Na2CO3 (19 мг в 0.3 мл Н2О) и смесь нагревают на масляной бане при 85°С в течение 5 часов; ЖХ/МС показывает, что на этот момент реакция завершена.
Реакционную смесь фильтруют и фильтрат хроматографируют на силикагельном картридже (2 мл; EtOAc). Содержащие продукт фракции концентрируют под вакуумом и остаток хроматографируют на другом силикагельном картридже (2 мл; постепенный градиент гексана, гексан:EtOAc 3:1 и EtOAc). Содержащие продукт фракции концентрируют под вакуумом и остаток элюируют через SCX (2 г) картридж (20 мл каждого СН2Cl2 и метанола; затем продукт элюируют с 2 М аммиака в метаноле). Содержащие продукт фракции концентрируют под вакуумом, что дает желаемый биарил амино- трет-бутилового эфира как продукта реакции. Его обрабатывают раствором СН2Cl2/ТФК (7:3; 1 мл) в течение ночи 14 часов. Летучие продукты удаляют, что дает названное соединение (39 мг; 67%) в виде масла. [М+Н]+=534.3.
Примеры с 14 по 124
Следуя одному из перечисленных выше процессов, получают следующие соединения по изобретению:
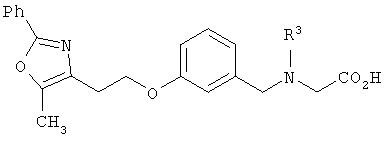





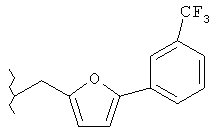
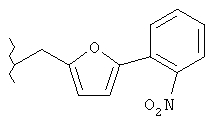
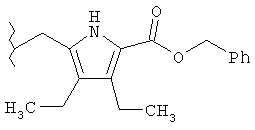
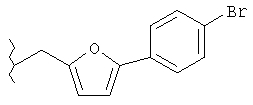
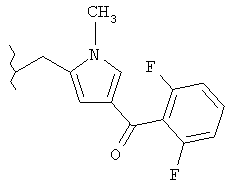
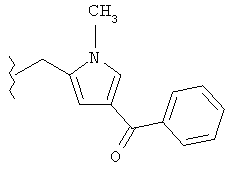
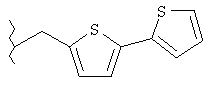
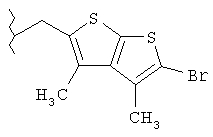

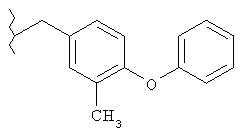
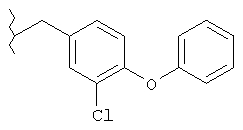

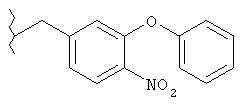
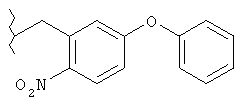

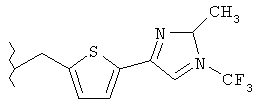
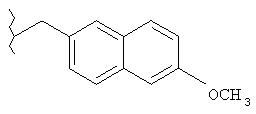
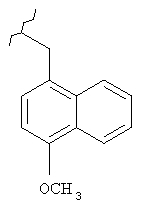
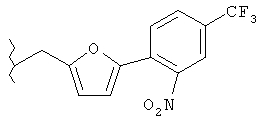
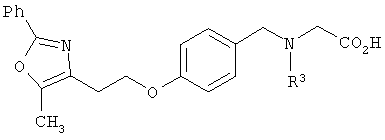













Пример 125
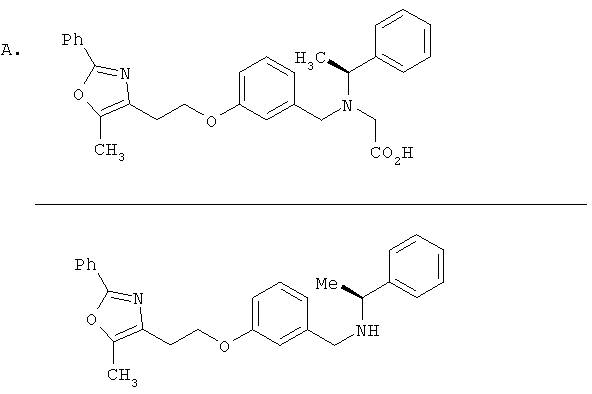
Раствор альдегида по примеру 7, часть А (60 мг; 0.20 ммоль) и (S)-α-метилбензиламина (30 мг; 0.24 ммоль) в метаноле (1 мл) перемешивают при комнатной температуре в течение 6 часов. Раствор охлаждают до 0°С и добавляют порциями предварительно полученный раствор NaBH4 (9 мг; 0.24 ммоль) в метаноле (0.5 мл). Реакционную массу перемешивают при комнатной температуре в течение ночи, затем концентрируют под вакуумом без нагревания. Остаток разделяют между водным раствором NaHCO3 и EtOAc (5 мл каждый). Водный раствор (слой) экстрагируют EtOAc (2×5 мл). Объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом, что дает названное соединение в виде оранжево-желтого масла (81 мг сырой).
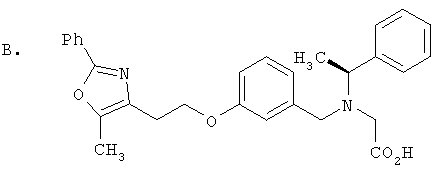
Раствор соединения из части А (70 мг; 0.17 ммоль), трет-бутилбромацетата (66 мг; 0.34 ммоль) и изо-Pr2NEt в ДМФА (0.5 мл) перемешивают при комнатной температуре в течение 2 дней. ЖХ/МС показывает что реакция завершена и прошла нацело.
Сырую реакционную смесь разделяют между H2O (30 мл) и EtOAc (20 мл). Водный слой экстрагируют с помощью Et2O (2×10 мл); объединенные органические экстракты сушат (MgSO4) и концентрируют под вакуумом, что дает сырой амино-трет-бутиловый эфир.
Полученный сырой продукт перемешивают в растворе (1:1) CHCl3 и ТФК (2 мл) в течение 18 часов при комнатной температуре. Затем раствор концентрируют под вакуумом и очищают препаративной ВЭЖХ с обращенной фазой (как в примере 10). Очищенный материал лиофилизуют из метанола-H2O, что дает названное соединение (71 мг; 71%) в виде белого лиофилата. [М+Н]+=471.2.
Пример 126
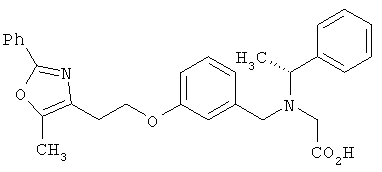
Названное соединение синтезируют, следуя тому же процессу, что раскрыт выше в примере 125 за исключением того, что (S)-α-метилбензиламин заменяют на (R)-α-метилбензиламин в синтезе соединения, часть А. Названное соединение получают с 67% общим выходом (66 мг). [М+Н]+=471.2.
Пример 127
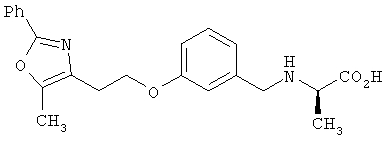
Смесь соединения по примеру 7, часть А (30 мг, 0.098 ммоль), гидрохлорида трет-бутилового эфира D-аланина (23 мг; 0.127 ммоль), Et3N (5 капель) и 4Å молекулярных сит в метаноле (2 мл) перемешивают при комнатной температуре в течение 4 часов. Вводят NaBH4 (12 мг, 0.0294 ммоль) и вновь перемешивают при комнатной температуре в течение 30 минут. Затем реакционную смесь концентрируют под вакуумом, разбавляют СН2Cl2 (2 мл) и фильтруют через ткань. К фильтрату вновь добавляют ТФК (1 мл) и реакционную массу перемешивают при комнатной температуре в течение ночи. Затем ее концентрируют под вакуумом, разбавляют EtOAc, промывают несколько раз указанным водным раствором NaHCO3, затем рассолом. Органическую фазу сушат (MgSO4) и концентрируют под вакуумом. Остаток очищают препаративной ВЭЖХ (YMC ODS 30 мм × 250 мм колонка с обращенной фазой; скорость потока 25 мл/мин; 30 мин непрерывный градиент от 50:50 А:В до 100% В, где А=90:10:0.1 H2O:метанол:ТФК и В=90:10:0.1 метанол:H2O:ТФК), что обеспечивает названное соединение (7.8 мг, 21%) в виде белого лиофилата. [М+Н]+=381.1.
Пример 128

Названное соединение (20% общий выход) синтезируют, следуя тому же процессу, что описан в примере 125, используя гидрохлорид трет-бутилового эфира D-фенилаланина вместо гидрохлорида трет-бутилового эфира D-аланина. [М+Н]+=457.2.
Пример 129
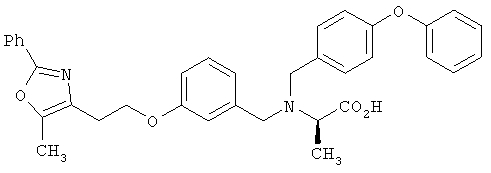
Смесь соединения по примеру 7, часть А (40 мг, 0.13 ммоль), гидрохлорида трет-бутилового эфира D-аланина (31 мг, 0.17 ммоль), Et3N (6 капель) и 4Å молекулярных сит в метаноле (2 мл) перемешивают при комнатной температуре в течение 4 часов. Вводят NaBH4 (15 мг, 3 экв) и смесь перемешивают при комнатной температуре в течение 30 мин, затем концентрируют под вакуумом. Остаток растворяют в СН2Cl2 (2 мл) и фильтруют. В пробирку с фильтратом добавляют 4-феноксибензальдегид (77 мг, 0.39 ммоль) и NaBH(ОАс)3 (138 мг, 0.65 ммоль). Реакционную массу перемешивают при комнатной температуре в течение 18 часов. После этого полученную реакционную смесь хроматографируют на SiO2, используя гексан/EtOAc (9:1 - 4:1), чтобы получить чистый трет-бутиловый эфир. Указанный продукт растворяют в CH2Cl2 (2 мл) и медленно добавляют ТФК (1 мл). Раствор перемешивают при комнатной температуре в течение ночи, затем концентрируют под вакуумом. Остаток вновь растворяют в СН2Cl2 и фильтруют через твердый NaHCO3, чтобы удалить остаточную ТФК. Этот раствор в дальнейшем разбавляют CH2Cl2, промывают 1 М водным раствором NaHSO4 и рассолом, сушат (MgSO4), фильтруют и концентрируют под вакуумом, чтобы получить названное соединение (9.1 мг, 12%). [М+Н]+=563.2.
Пример 130

Названное соединение (13% общий выход) синтезируют, следуя тому же процессу, который раскрыт в примере 127, используя гидрохлорид трет-бутилового эфира D-фенилаланина вместо гидрохлорида трет-бутилового эфира D-аланина. [М+Н]+=639.2.
Примеры с 131 по 135
Другие аналоги в этих сериях, полученные аналогичными процессами, приведены в следующей таблице.
Таблица 3
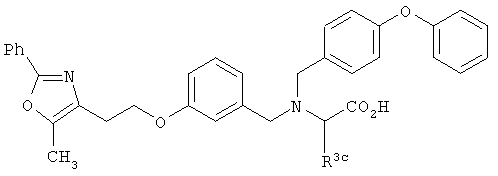

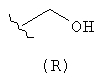
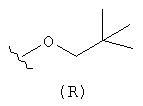
Пример 136
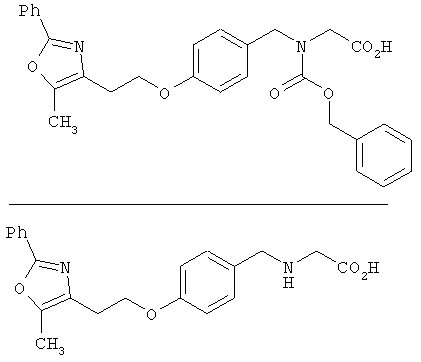
Раствор вторичного аминоэтилового эфира (72 мг; 0.183 ммоль)
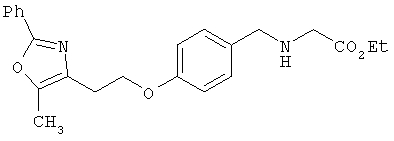
(получают, как раскрыто в примере 3, часть А) в метаноле (2 мл) и водном растворе NaOH (2 мл 1 М раствор) нагревают при кипении с обратным холодильником в течение 12 часов. Значение рН раствора доводят до 5 ( водный раствор 1 М NaOH и 1 М HCl), после чего бесцветное твердое вещество выпадает в осадок. Его отфильтровывают и фильтрат экстрагируют EtOAc (3х); объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом, что дает названную сырую аминокислоту в виде бесцветного твердого вещества (97 мг).
B.
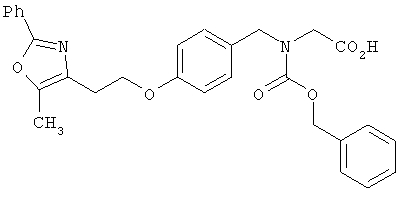
К раствору аминокислоты, часть А (15 мг; 0.04 ммоль) в диоксане:H2O (1:1, 8 мл) добавляют К2СО3 (22 мг; 0.16 ммоль), а затем бензилхлорформиат (15 мг; 0.09 ммоль).Реакционную массу перемешивают в течение ночи, затем концентрируют под вакуумом и подкисляют избытком водного раствора 1 М HCl. Проводят экстракцию EtOAc (3x); объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом, что дает названное соединение (13 мг; 63%) в виде бесцветного твердого вещества. [М+Н]+=501.3.
Пример 137

К охлажденному до 0°С раствору амино-трет-бутилового эфира (75 мг; 0.18 ммоль)
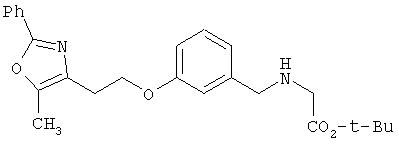
(получают, как раскрыто в примере 7, часть В) в СН2Cl2 (1 мл) добавляют CbzCl (28 мкл; 0.20 ммоль), затем Et3N (54 мкл; 0.39 ммоль). Реакционной смеси дают нагреться до комнатной температуры и затем перемешивают при этой температуре в течение ночи (18 часов). Добавляют водный раствор NaHCO3 (2 мл 10% раствора) и водный слой экстрагируют EtOAc (2×2 мл). Объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом. Сырой карбаматоэфир растворяют в CHCl3 (3 мл) и ТФК (1 мл); раствор перемешивают при комнатной температуре в течение 24 часов, затем концентрируют под вакуумом. Сырую карбаматокислоту очищают препаративной ВЭЖХ с обращенной фазой на С-18 колонке (непрерывный градиент более чем 14 мин; 4 мин время выдержки; скорость потока=20 мл/мин, от 1:1 А:В до 100% В; растворитель А=90:10:0.1 H2O:метанол:ТФК; растворитель В=90:10:0.1 метанол:H2O: ТФК). Продукт лиофилизуют из MeOH/H2O, что дает названное соединение в виде белого лиофилата. [М+H]+=501.3.
Пример 138
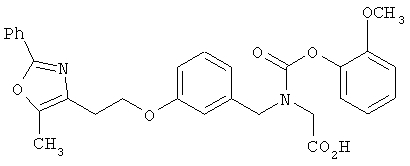
А. Требуемые арилформиаты (если они не являются коммерчески доступными) получают согласно следующему общему процессу, который раскрыт на примере синтеза 2-метоксифенилхорформиата.
Раствор 2-метоксифенола (2 г, 16.1 ммоль), N,N-диметиламина (1.95 г, 16.1 ммоль), фосгена (8.34 мл 1.93 М раствор в толуоле, 16.1 ммоль) и каталитического количества ДМФА в хлорбензоле (5 мл) перемешивают в трубке под давлением течение 2 часов при температуре 80°С. Органический слой отделяют и концентрируют в вакууме. Остаток перегоняют (Buchi Kugelrohr; т.кип.=115°С @ 10 мм рт.ст.), чтобы получить 2-метоксифенилхлорформиат (1.5 г; 50%) в виде прозрачного масла.
B.

Раствор амино-трет-бутилового эфира (20 мг, 0.05 ммоль)
(приготовленного, как описано в примере 7, часть В), 2-метоксифенилхлорформиата (8 мг, 0.05 ммоль) полученного выше, и поливинилпиридина (Aldrich; 16 мг, 0.3 ммоль) в СН2Cl2 (1 мл) перемешивают в течение 30 мин при комнатной температере. Добавляют аминосмолу WA21J (Supeico; 200 мг) и смесь вновь перемешивают при комнатной температуре в течение 30 мин для того, чтобы удалить непрореагировавший хлорформиат. Реакционную массу фильтруют и концентрируют в вакууме, что дает желаемый 2-метоксифенилкарбамат-эфир.
Эфир обрабатывают раствором 30% ТФК в CH2Cl2 (5 мл) в течение ночи. Летучие продукты удаляют в вакууме, что дает сырую кислоту. Этот продукт очищают с помощью твердофазной экстракции, используя колонку с анионообменником (CHQAX13M6 колонка; United Technologies; 3 г сорбента в 6 мл колонке) по процессу, описанному ниже:
1) колонку заполняют метанолом (10 мл) и СН2Cl2 (10 мл);
2) сырую кислоту растворяют в минимальном объеме CH2Cl2 и загружают в SAX колонку;
3) картридж промывают CH2Cl2 (10 мл), СН2Cl2/метанолом (10 мл 4:1 CH2Cl2:СН3ОН раствор);
4) продукт элюируют с помощью СН2Cl2/CH3ОН (10 мл раствора 4:1 CH2Cl2:СН3ОН). Содержащие продукт фракции концентрируют в вакууме на Speed Vac, что позволяет получить названное соединение в виде масла. Аналитическая HPLC с обращенной фазой (стандартные условия) определяет чистоту продукта как 90%. Кроме того, ЖХ/МС определяет точный молекулярный ион [(М+Н)+=517.3] для желаемого названного соединения.
Пример 139
A.
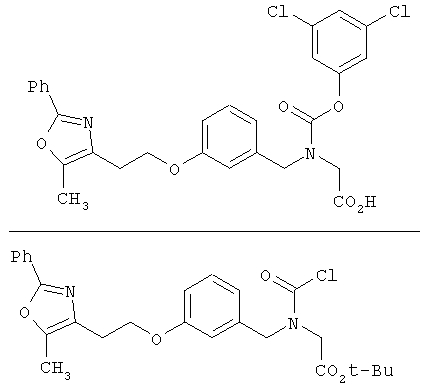
Фосген (0.21 мл 1.93 М раствора в толуоле; 0.40 ммоль) прибавляют по каплям к раствору амино-трет-бутилового эфира (100 мг, 0.24 ммоль)
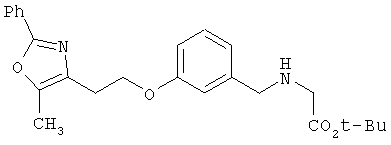
(получают, как раскрыто в примере 7, часть В) и Et3N (30.3 мг; 0.30 ммоль) в 3 мл СН2Cl2 при -5°С. Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Смесь концентрируют под вакуумом, что дает сырой продукт, который хроматографируют (SiO2; гексан/EtOAc, 1:5), что обеспечивает названное соединение (0.105 г, 91%).
B.
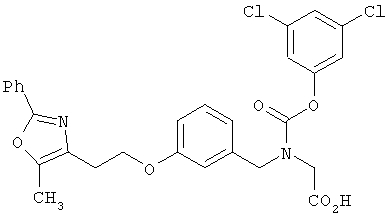
Названное соединение получают как часть библиотеки в потоке растворимой фазы, используя следующий примерный процесс.
Смесь карбамоилхлорида, часть А (20 мг; 0.045 ммоль), 3,5-дихлорфенола (16 мг; 0.07 ммоль) и пиридина (0.5 мл) перемешивают при 80°С в течение 16 часов. Пиридин удаляют под вакуумом и остаток очищают с помощью твердофазной экстракции, используя CHQAX1 картридж (2 г сорбента в 6 мл колонке, 0.3 мг/г), с помощью процесса, описанного ниже:
1) колонку заполняют метанолом (10 мл) и CH2Cl2(20 мл);
2) реакционную смесь в CH2Cl2 загружают в SAX колонку;
3) продукт элюируют с помощью СН2Cl2 (10 мл).
Содержащие продукт фракции концентрируют под вакуумом, используя Speed Vac в течение более 16 часов, что дает чистый арилкарбамат-трет-бутиловый эфир, который обрабатывают раствором 30% ТФК в СН2Cl2 в течение ночи. Летучие продукты удаляют, используя Speed Vac в течение 16 часов, что дает сырую кислоту как конечный продукт. Продукт вначале очищают с помощью твердофазной экстракции, используя Varian SAX картридж (2 г сорбента в 6 мл колонке, 0.3 meg/г), с помощью процесса, описанного ниже:
1) колонку заполняют метанолом (10 мл) и CH2Cl2 (20 мл);
2) реакционную смесь в CH2Cl2 загружают в SAX колонку;
3) колонку промывают CH2Cl2 (10 мл);
4) колонку промывают 10% метанолом в CH2Cl2 (10 мл);
5) продукт элюируют с помощью 2% ТФК в CH2Cl2 (10 мл).
Содержащие продукт фракции концентрируют под вакуумом, используя Speed Vac, в течение 16 часов, что дает очищенный продукт (20 мг, 80%) в виде твердого вещества. Анализ ВЭЖХ с обращенной фазой (YMC S5 ODS 4.6×33 мм колонка, непрерывный градиент от 50% А до 100% В в течение 2 мин при скорости потока 5 мл/мин. [растворитель А=10% метанола/90%H2O/0.2% Н3PO4; растворитель В=90% метанола/10% H2O/0.2% Н3PO4]) показывает, что чистота продукта составляет 96%.
Кроме того, ЖХ/МС определяет точный молекулярный ион [(М+Н)+=555.2] (электронапыление) для названного соединения.
Пример 140
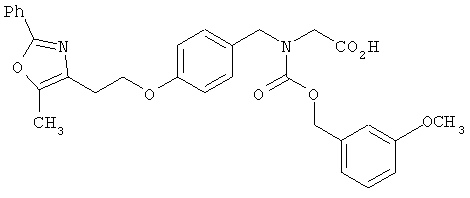
Бензилхлорформиаты синтезируют в соответствии со следующим общим процессом, как показано на примере метоксибензилхлорформиата.
A.
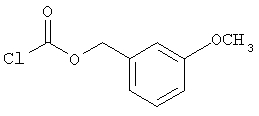
К раствору 3-метоксибензилового спирта (2.0 г; 7.24 ммоль), N,N-диметиланилина (0.877 г; 7.24 ммоль) в безводном эфире (5 мл) добавляют по каплям фосген (3.8 мл 1.93 М раствор в толуоле; 7.3 ммоль) при 0°С. Реакционную смесь перемешивают при 0°С в течение 2 часов, после чего твердое вещество отфильтровывают. Фильтрат концентрируют под вакуумом при комнатной температуре. Сырой хлорформиат отгоняют от безводного Et2O (2×2 мл) и используют без дальнейшей очистки в следующей реакции. Последовательно получают также другие хлорформиаты, используя указанный стандартный процесс.
B.

Названное соединение получают как часть библиотеки растворимой фазы, которую перегоняют, используя следующий стандартный процесс.
К суспензии аминокислоты (сольТФК) по примеру 3

(25 мг, 0.05 ммоль) в CH2Cl2 (1 мл) добавляют соединение, часть А (10 мг; 0.05 ммоль) и изо-Pr2NEt (19.4 мг; 0.15 ммоль). После перемешивания в течение 30 мин при комнатной температуре реакционную смесь концентрируют под вакуумом.
Продукт очищают с помощью твердофазной экстракции, используя Varian CHQAX13M6 (анионный обмен) колонку (3 г сорбента в 6 мл колонке) с помощью процесса, раскрытого ниже:
1) колонку заполняют метанолом (10 мл) и CH2Cl2 (10 мл);
2) остаток растворяют в минимальном объеме CH2Cl2 и загружают в SAX колонку;
3) картридж промывают последовательно CH2Cl2 (10 мл), 20% метанолом/СН2Cl2 (10 мл);
4) продукт элюируют раствором 20% метанолом/СН2Cl2 (10 мл).
Содержащие продукт фракции концентрируют под вакуумом, используя Speed Vac, что позволяет получить названное соединение. Анализ ВЭЖХ с обращенной фазой с использованием стандартного состояния показывает, что чистота продукта составляет 90%. Кроме того, ЖХ/МС (электронапыление) определяет точный молекулярный ион [(М+Н)+=531.3] для желаемого названного соединения.
Пример 141
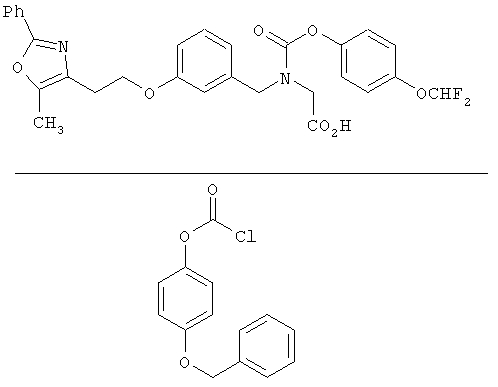
Раствор 4-(бензилокси)фенола (2.0 г; 9.99 ммоль), N,N-диметиланилина (1.21 г; 9.99 ммоль), фосгена (5.2 мл 1.95 М раствора в толуоле; 10 ммоль) и каталитическое количество ДМФА в хлорбензоле (5 мл) нагревают до 80°С в трубке под давлением в течение 2.5 часов. Смеси дают охладиться до комнатной температуры. Верхний прозрачный раствор отделяют и концентрируют под вакуумом, что дает сырой названный арилхлорформиат в виде кристаллов (2 г сырого продукта).
B.
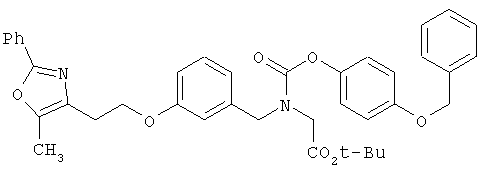
К смеси хлорформиата, часть А (184 мг, 0.70 ммоль) в СН2Cl2 (5 мл) и поливинилпиридина (Aldrich; 315 мг, 1 ммоль) добавляют раствор амино-трет-бутилового эфира (280 мг, 0.66 ммоль)

(получают, как раскрыто в примере 7, часть В) в CH2Cl2 (5 мл). Реакционную массу перемешивают при комнатной температуре в течение 15 мин. К смеси добавляют связанный со смолой амин (WA21J, Supeico; 150 мг). Реакционную смесь перемешивают в течение еще 15 минут. Связанный со смолой амин и поливинилпиридин отфильтровывают и фильтрат концентрируют под вакуумом, что дает сырой продукт. Его хроматографируют (SiO2; гексан/EtOAc, 1:4), что обеспечивает названное соединение (0.30 г, 70%).
С.
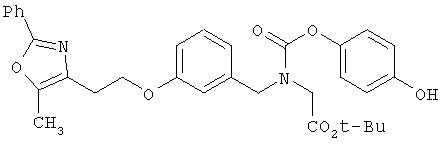
Раствор соединения, часть В (75 мг; 0.42 ммоль) в 20 мл метанола гидрируют в присутствии 20 мг 10% Pd/C в атмосфере Н2 (баллон) в течение 24 часов.
Палладиевый катализатор удаляют фильтрованием и фильтрат концентрируют под вакуумом, что дает сырой названный трет-бутиловый эфир (240 мг, 90%), который используют без дальнейшей очистки на следующей стадии.
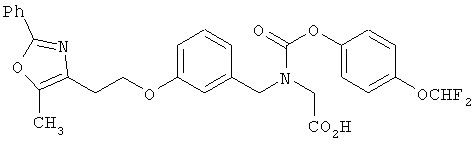
Раствор фенол-трет-бутилового эфира, часть С (50 мг; 0.089 ммоль), каталитического количества Bu4NBr (1.5 мг, 0.0047 ммоль), водного раствора NaOH (0.7 мл 1 М раствор) и изопропанола (2 мл) в трубке под давлением охлаждают до -50°С. В раствор в течение 1 мин пробулькивают фреон. Трубку герметизируют и нагревают до 80°С в течение 12 часов. Смесь экстрагируют EtOAc (3×10 мл). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом, что дает масло, которое затем обрабатывают раствором 30% ТФК в СН2Cl2 в течение ночи. Летучие продукты удаляют под вакуумом и остаток очищают, используя препаративную ВЭЖХ (YMC S5 ODS 30×250 мм колонка с обращенной фазой; 30 мин непрерывный градиент от 70:30 А:В до 100% В, где А=90:10:0.1 H2O:метанол:ТФК и В=90:10:0.1 метанол:H2O:ТФК), что приводит к желаемому названному продукту (14 мг; 28%). Анализ ВЭЖХ с обращенной фазой показывает, что чистота продукта составляет 97%. Кроме того, ЖХ/МС (электронапыление) определяет точный молекулярный ион [(М+Н)+=553.1] для желаемого соединения.
Пример 142
Следующий пример 142 раскрывает процесс получения аналогичных соединений [(M+H)+=553.2].
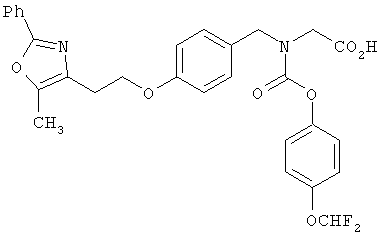
С промежуточных соединений, соответствующих по примеру 141 частям В и С, снимают защиту, используя тот же самый ТФК/CHCl3 процесс, как описано выше, и очищают, как обычно, что дает следующие аналоги:
Пример 143: [M+H]+=593.4

Пример 144: [М+Н]+=503.1
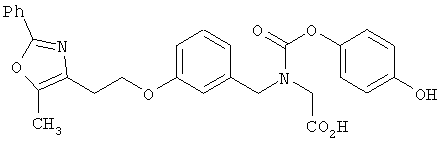
Примеры с 145 по 305.
Следующие аналоги карбаматокислоты, сведенные в таблицы 4 и 5, синтезируют согласно одной из приведенных выше методик.
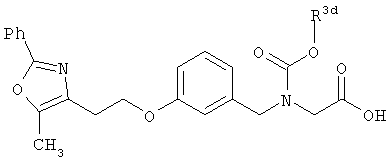












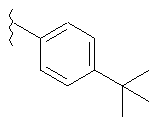
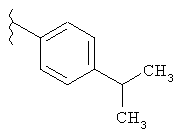
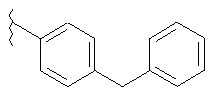
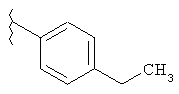


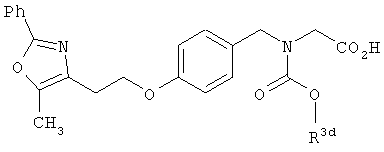
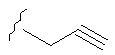
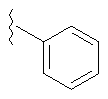
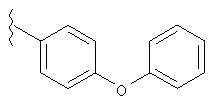
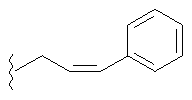

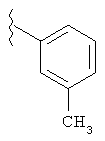

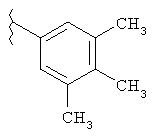

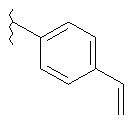
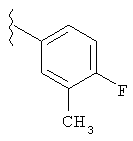

Пример 149
К раствору вторичного аминоэфира (2.1 г; 5.52 ммоль)
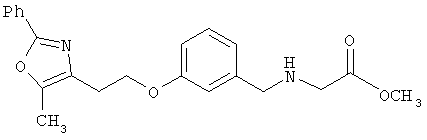
в СН2Cl2 (10 мл) добавляют 4-метилфенилхлорформиат (0.79 мл; 5.52 ммоль) и поливинилпиридин (Aldrich; 1.74 г; 16.5 ммоль). Смесь перемешивают при комнатной температуре в течение 15 минут; в этот момент ТСХ показывает, что исходный продукт вступил в реакцию. Раствор фильтруют, концентрируют под вакуумом и остаток хроматографируют (SiO2; гексан:EtOAc, 4:1), что обеспечивает чистый карбаматоэфир (2 г). Его растворяют в смеси ТГФ (10 мл), метанола (1 мл) и водного раствора LiOH (8 мл 1 М раствор). Раствор перемешивают при комнатной температуре в течение ночи, затем подкисляют до рН 3 избытком водного раствора 1 М HCl. Раствор экстрагируют EtOAc (2×50 мл). Объединенные органические экстракты промывают H2O (2×50 мл) и рассолом (50 мл), сушат (Na2SO4) и концентрируют под вакуумом, что обеспечивает названное соединение в виде белого твердого вещества (1.75 г; 63%). [М+Н]+=501.2.
[М+Н]+=501.2.
1H ЯМР (400 МГц; CDCl3): δ 7.93-7.99 (м, 2Н), 7.38-7.43 (м, 3H), 7.23 (кв, 1Н, J=8 Гц), 7.02-7.12 (м, 3Н), 6.98-7.02 (м, 2Н), 6.82-6.89 (м, 2Н), 4.71 (с, 1Н), 4.64 (с, 1Н), 4.25 (т, 2Н, J=7 Гц), 4.07 (с, 2Н), 2.90-2.98 (м, 2Н), 2.37 (с, 3H), 2.29 (с. 3Н).
Пример 230
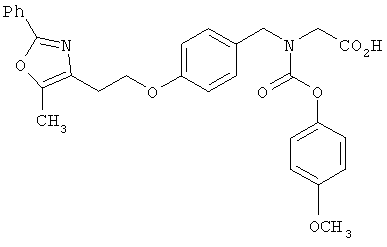
К охлажденному до 0°С раствору вторичного амина (3.0 г; 7.9 ммоль)
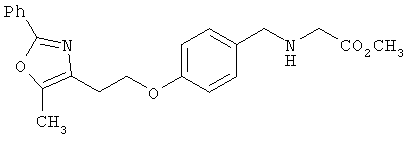
в СН2Cl2 (45 мл) последовательно добавляют пиридин (0.8 мл; 9.9 ммоль) и 4-метоксифенилхлорформиат (1.3 мл; 8.7 ммоль). Реакцию перемешивают при 0°С в течение 3 часов, к этотму моменту исходный продукт вступил в реакцию (аналитическая ВЭЖХ). Реакционный раствор промывают водным раствором HCl (2×25 мл 1 М раствора), рассолом (2х), сушат (Na2SO4) и концентрируют под вакуумом. Сырой продукт хроматографируют (SiO2; постепенный градиент от 4:1 до 3:7 гексан:EtOAc), что обеспечивает желаемый карбаматоэфир (4.2 г; 100%). Эфир растворяют в смеси ТГФ:метанол:H2O (50 мл, 3:1:1 раствора) и добавляют LiOH·H2O (0.5 г; 11.9 ммоль). Раствор перемешивают в течение ночи при комнатной температуре. Исходный продукт еще присутствует, как показывает ВЭЖХ. Вводят дополнительное количество LiOH.H2O (0.2 г; 4.8 ммоль) и смесь осторожно нагревают до солюбилизации LiOH, затем перемешивают при комнатной температуре в течение 4 часов. Реакция завершается именно в этот момент и смесь подкисляют до рН 3 избытком водного раствора 1 М HCl, затем органический растворитель удаляют под вакуумом. Оставшуюся водную фазу экстрагируют EtOAc (3×50 мл). Объединенные органические экстракты последовательно промывают H2O и рассолом (50 мл каждый), сушат (Na2SO4), фильтруют и концентрируют под вакуумом, что дает названное соединение в виде бесцветного твердого вещества (3.07 г; 75%). [М+Н]+=517.2.
1H ЯМР (400 МГц; CDCl3): δ 7.96-7.98 (м, 2Н), 7.4-7.45 (м, 3H), 7.2-7.3 (м, 2Н), 7.0-7.1 (м, 2Н), 6.8-7.0 (м, 4 Н), 4.65 (с, 1Н), 4.55 (с, 1Н), 4.20-4.24 (м, 2Н), 4.02 (с, 2Н), 3.77 (с, 3H), 3.00 (с, 2Н), 2.38 (с, 3Н).
Соединения в следующих примерах (167, 187, 216, 229, 247 и 263) синтезируют согласно методикам, раскрытым в примерах 149 и 230.
Пример 167
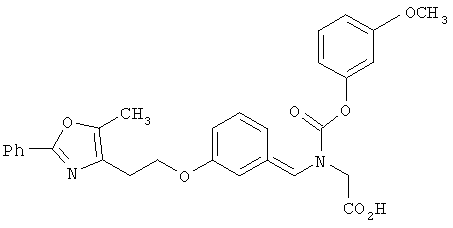
1H ЯМР (ДМСО-d6; 500 МГц): δ 2.37 (с, 3H), 2.94 (м, 2Н), 3.73 (2с, 3H), 4.06 (д, J=4.8 Гц, 2Н), 4.25 (т, J=7.2 Гц, 2Н), 4.66 (2с, 2Н), 6.71 (м, 3H), 6.85 (м, 2Н), 7.06 (д, J=16 Гц, 1Н), 7.22 (м, 2Н), 7.39 (м, 3H), 7.96 (м, 2Н).
Пример 187
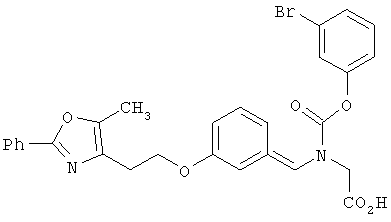
1H ЯМР (ДМСО-d6; 500 МГц): δ 2.36 (с, 3H), 2.93 (т, J=6.6 Гц, 2Н), 4.02 (2с, 2Н), 4.21 (т, J=6.6 Гц, 2Н), 4.55 (2с, 2Н), 6.94 (м, 3H), 7.48 (м, 8Н), 7.90 (м, 2Н).
Пример 216
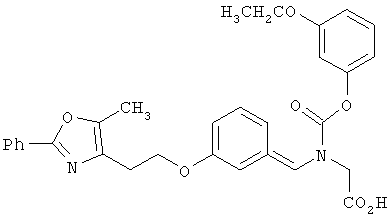
1H ЯМР (CDCl3; 400 МГц): δ 1.3-1.4 (м, 3H), 2.39 (с, 3H), 2.9-3.05 (м, 2Н), 3.9-4.05 (м, 2Н), 4.06 (расширенный с, 2Н), 4.25 (т, J=7.0 Гц, 2Н), 6.85 (дд, J=11.4, 8.8 Гц, 2Н), 6.99 (дд, J=15.8, 8.8 Гц, 2Н), 7.18 (дд, J=8.4, 2.6 Гц, 2Н), 7.38-7.50 (м, 5Н), 7.99 (расширенный д, J=7.9 Гц, 2Н).
Пример 229
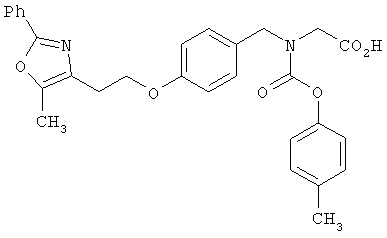
1H ЯМР (CDCl3; 400 мГц): δ 2.30 (2 пика, 3H), 2.38 (2 пика, 3H), 3.03 (дд, J=5.7, 5.7 Гц; 2Н), 3.99 (с, 2Н), 4.21 (дд, J=6.1, 6.1 Гц; 2Н), 4.63 (2 пика, 2Н), 6.82-6.87 (м, 2Н), 6.96-7.01 (м, 2Н), 7.09-7.14 (м, 2Н), 7.18-7.20 (м, 2Н), 7.43-7.45 (м, 3H), 7.96-7.98 (м, 2Н).
Пример 247
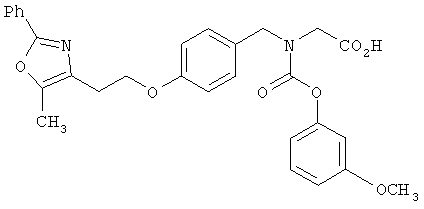
1H ЯМР (ДМСО-d6; 500 МГц): δ 2.36 (с, 3H), 2.93 (т, J=6.6 Гц, 2Н), 3.74 (с, 3H), 3.96 (2с, 2Н), 4.20 (т, J=6.6 Гц, 2Н), 4.55 (2с, 2Н), 6.65 (м, 2Н), 6.94 (м, 3H), 7.27 (м, 3H), 7.48 (м, 3H), 7.91 (д, J=6.1 Гц, 2Н).
Пример 263
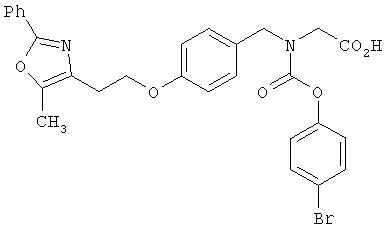
1H ЯМР (CDCl3; 400 МГц): δ 2.42 (2с, 3Н; ротамеры); 3.0-3.5 (м, 2Н), 3.99 (расширенный с, 2Н), 4.15-4.25 (м, 2Н), 4.57 (АВ дуплет, J=38.2 Гц, 2Н), 6.85 (дд, J=11.4, 8.8 Гц, 2Н), 6.99 (дд, J=15.8, 8.8 Гц, 2Н), 7.18 (дд, J=8.4, 2.6 Гц, 2Н), 7.38-7.50 (м, 5Н), 7.99 (расширенный д, J=7.9 Гц, 2Н).
Пример 306
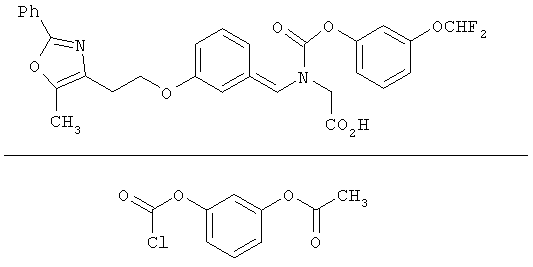
Раствор моноацетата резорцина (2 г; 13.14 ммоль), N,N-диметиланилина (1.6 г; 13.14 ммоль), фосгена (6.8 мл 1.95 М раствор в толуоле; 13.1 ммоль) и каталитическое количество ДМФА в хлорбензоле (5 мл) нагревают при 80°С в трубке под давлением в течение 2.5 часов и затем дают охладиться до комнатной температуры. Прозрачный супернатантный раствор отделяют и концентрируют под вакуумом. Остаток очищают отгонкой под вакуумом (140-150°С @ 1.0 мм рт.ст.), что дает названное соединение в форме прозрачного масла (2 г; 71%).
B.
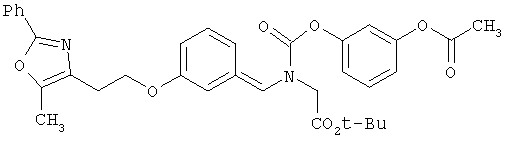
К смеси хлорформиата, часть А (50 мг, 0.237 ммоль)и поливинилпиридина (PVP) (75 мг, 0.70 ммоль) добавляют CH2Cl2 раствор (2 мл) амино-трет-бутилового эфира (100 мг, 0.237 ммоль),
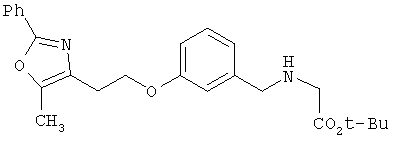
(получают, как раскрыто в примере 7, часть В).
Реакционную массу перемешивают при комнатной температуре в течение 15 мин. Связанный со смолой амин (WA21J, Supeico; 150 мг) добавляют к смеси. Реакционную смесь перемешивают в течение дополнительных 15 минут. Связанный со смолой амин и PVP удаляют фильтрованием и фильтрат концентрируют под вакуумом, что дает сырой продукт. Сырой продукт хроматографируют (SiO2; гексан/EtOAc, 1:4), что обеспечивает названное соединение (0.1 г, 70%).
С.
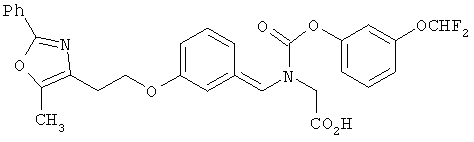
Раствор фенол-трет- бутилового эфира, часть В (60 мг; 0.10 ммоль), Bu4NBr (0.32 мг, 0.001 ммоль), водного раствора NaOH (0.5 мл 1 М раствора; 0.5 ммоль) и изопропанола (1 мл) в трубке под давлением охлаждают до -50°С. Через раствор в течение 1 минуты пробулькивают газообразный фреон. Трубку герметизируют и нагревают до 80°С в течение 12 часов. Смесь экстрагируют EtOAc (3×10 мл). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом, что дает сложный сырой дифторметокси-трет-бутиловый простой эфир в виде масла. Сырой эфир затем обрабатывают раствором 30% ТФК в СН2Cl2 в течение ночи. Летучие продукты удаляют под вакуумом и остаток очищают, используя препаративную ВЭЖХ с обращенной фазой (как в примере 127, за исключением того, что используют непрерывный градиент от А:В 70:30 до 100% В), чтобы получить два продукта, желаемую названную дифторметоксиэфир-кислоту (13 мг; 23%) и фенол-кислоту, определенную ниже (32 мг; 63%). Анализ ВЭЖХ с обращенной фазой, используя стандартные условия показывает, что чистота продукта составляет >92%. Кроме того, ЖХ/МС (электронапыление) определяет точный молекулярный ион [(М+Н)+=553.2 и 503.2 соответственно] для двух соединений.
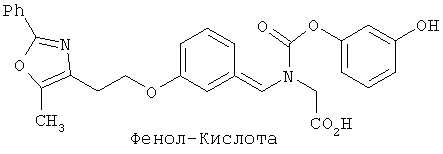
Примеры 307 и 308
Следуя общему процессу, приведенному выше по примеру 306, получают следующие соединения:
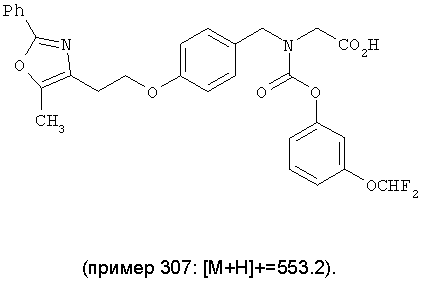
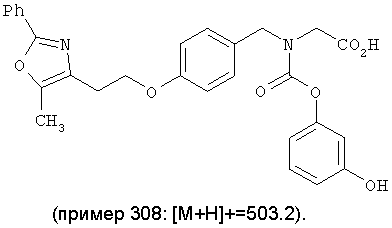
Пример 309

К смеси фенилхлортионоформиата (11 мг, 0.063 ммоль) и триэтиламина (6.5 мг, 0.063 ммоль) добавляют раствор амино-трет-бутилового эфира (20 мг, 0.053 ммоль),
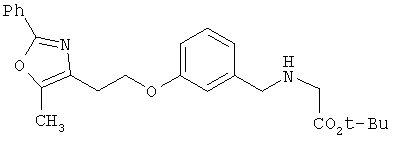
(получают, как раскрыто в примере 7, часть В) в СН2Cl2 (1 мл). Реакционную массу перемешивают при комнатной температуре в течение 15 минут и смесь концентрируют под вакуумом, что дает сырой тионокарбаматовый трет-бутиловый эфир. Указанный продукт растворяют в водном растворе LiOH (0.50 мл 1.0 М раствора) и ТГФ (2 мл) и перемешивают при комнатной температуре в течение 5 часов. Раствор концентрируют под вакуумом, что дает сырую кислоту в виде масла. Сырой продукт очищают, используя препаративную ВЭЖХ, чтобы получить желаемый названный продукт (10 мг; 38%). [М+Н]+=503.2.
Пример 310
Соответствующий тиокарбамат в 1,4 сериях получают по методике, которая раскрыта в примере 309.
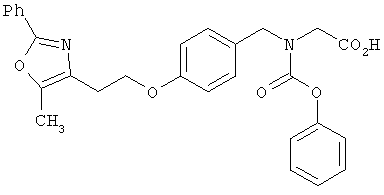
Пример 311

К смеси амино-трет-бутилового эфира (306 мг, 0.73 ммоль)
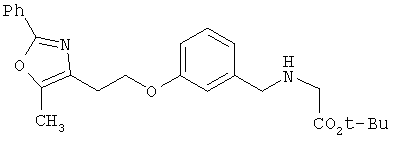
(получают, как раскрыто в примере 7, часть В) и п-феноксибензойной кислоты (220 мг; 1.02 ммоль; 1.4 экв) в CH3CN (20 мл) добавляют ВОР реагент (372 мг, 0.84 ммоль, 1.15 экв) одной порцией, а затем изо-Pr2NEt (0.5 мл; 2.9 ммоль; 3.9 экв) по каплям. Реакционную массу перемешивают в течение ночи при комнатной температуре, после чего летучие продукты удаляют под вакуумом. Остаток растворяют в EtOAc и промывают водным раствором 1 н. HCl. Водный раствор (слой) экстрагируют EtOAc (2х) и объединенные органические экстракты промывают H2O, насыщенным водным раствором NaHCO3 и рассолом, сушат (Na2SO4) и концентрируют под вакуумом, что дает желаемый продукт. Полученный сырой амидоэфир используют на следующей стадии без дальнейшей очистки.
Раствор сырого амидоэфира в 40% ТФК- CH2Cl2 (25 мл) перемешивают в течение 5 часов при комнатной температуре. Летучие продукты удаляют под вакуумом и сырую кислоту очищают препаративной ВЭЖХ (YMC S5 ODS 30 мм × 250 мм колонка с обращенной фазой; скорость потока=25 мл/мин; 30 мин непрерывный градиент от 70:30 А:В до 100% В; растворитель А=90:10:0.1 H2O:метанол:ТФК; растворитель В=90:10:0.1 метанол:H2O:ТФК), получая названное соединение (238 мг; 58% выход после 2 стадий) в виде белого твердого вещества. Анализ ВЭЖХ с обращенной фазой: время задержки=7.53 мин (непрерывный градиент растворитель система: от 50% А:50% В до 0% А:100% В (А=90% H2O/10% метанол/0.2% Н3PO4; В=90% метанол/10% H2O/0.2% Н3PO4) в течение 8 мин; определение при 220 нм; YMC S3 ODS 4.6×50 мм колонка). [М+Н]+=563.3.
Пример 311 А
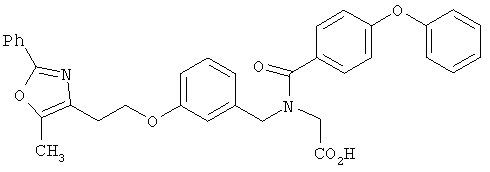
(альтернативный процесс синтеза)
К раствору вторичного амино-трет-бутилового эфира (35 мг, 0.083 ммоль), (получают как раскрыто в примере 7, часть В).
4-феноксибензойной кислоты (30 мг, 0.14 ммоль) и НОАТ (30 мг, 0.22 ммоль) в ТГФ/ДМФА (1 мл/0.05 мл) добавляют EDCI (20 мг, 0.10 ммоль) и смесь перемешивают при комнатной температуре в течение ночи. Реакционную массу разбавляют EtOAc, промывают водным раствором 1 н. HCl, H2O, насыщенным водным раствором NaHCO3 и рассолом, сушат (Na2SO4) и концентрируют под вакуумом. Сырой амид-трет-бутиловый эфир растворяют в ТФК/СН2Cl2 (5 мл, 1:1 раствор). Полученный розовый раствор перемешивают в течение ночи и концентрируют под вакуумом, что обеспечивает сырой амид кислоты в виде темно-коричневого масла. Сырой продукт очищают препаративной ВЭЖХ (YMC S5 ODS 20×100 мм колонка, 10 мин непрерывный градиент от 60:40 А:В до 100% В; растворитель А=90:10:0.1 H2O:метанол:ТФК; растворитель В=90:10:0.1 метанол:H2O:ТФК; скорость потока 20 мл/мин), что обеспечивает названное соединение (32 мг, 69%). [M+H]+=563.3.
Пример 312
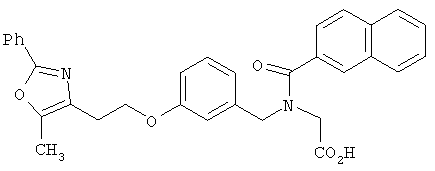
1) К раствору вторичного амино-трет-бутилового эфира (25 мг; 006 ммоль)
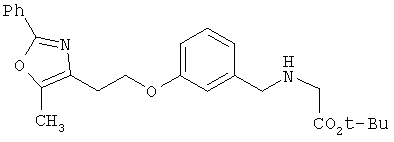
(получают, как раскрыто в примере 7, часть В) в ТГФ (0.5 мл) добавляют 2-нафталинкарбоновую кислоту (25 мг; 0.15 ммоль; 2.5 экв);
2) добавляют НОАТ (48 мг; 0.35 ммоль; 5.8 экв);
3) добавляют ДМФА (50 мкл);
4) добавляют EDCI (20 мг, 0.1 ммоль, 1.8 мэкв);
5) реакционный сосуд в течение 24 часов при комнатной температуре;
6) реакции разбавляют метанолом (2 мл) и фильтруют;
7) амид-трет-бутиловый эфир очищают препаративной ВЭЖХ (YMC S5 ODS 20×100 мм колонка; скорость потока 25 мл/мин; 10 мин непрерывный градиент от 70:30 А:В до 100% В; растворитель А=90:10:0.1 H2O:метанол:ТФК; растворитель В=90:10:0.1 метанол:H2O:ТФК);
8) фракции, содержащие очищенный амидоэфир, обрабатывают раствором ТФК в CH2Cl2 (0.5 мл, 1:1 раствор) в течение ночи. Реакционную массу концентрируют под вакуумом (Speed Vac), что дает названное соединение (8 мг; 25%). Анализ ВЭЖХ с обращенной фазой показывает, что чистота составляет >88%; ЖХ/МС (электронапыление) определяет точный ион [М+Н]+=521.2 для названного соединения.
Пример 313

Смесь аминоэфира (20 мг; 0.0474 ммоль)

(получают, как раскрыто в примере 7, часть В), тиофен-2-карбоновой кислоты (9.1 мг, 0.71 ммоль), EDCI (26 мг, 1.4 ммоль) и DMAP (каталитическое количество) растворяют в CH2Cl2 (2 мл) и перемешивают при комнатной температуре в течение ночи. Реакционный раствор последовательно промывают водным раствором 1 н. HCl (2 мл) и насыщенным водным раствором NaHCO3 (2 мл). К органической фазе затем добавляют 0.5 г безводного Na2SO4 и 0.2 г WA21J аминосвязанной смолы (Supeico). Смесь встряхивают в течение 0.5 часов и твердое вещество отфильтровывают. ТФК (2.0 мл) добавляют к фильтрату и раствор встряхивают при комнатной температуре в течение ночи. Реакционный раствор концентрируют под вакуумом, используя Speed Vac в течение 16 часов, что позволяет получить названное соединение в виде желтого масла. Анализ ВЭЖХ с обращенной фазой (YMC S5 ODS 4.6×33 мм колонка, непрерывный градиент от 100% A до 100% B в течение 2 мин при скорости потока 5 мл/мин [растворитель А=10% метанол/90% H2O/0.2% Н3PO4; растворитель В = 90% метанол/10% H2O/0.2% Н3PO4]) показывает, что чистота продукта составляет 92.7%. Кроме того, ЖХ/МС (электронапыление) дает точный молекулярный ион [(М+Н)+=477.2] для желаемого названного соединения.
Пример 314
Другую методику очистки, используя аминосвязанную смолу для амидокислоты, иллюстрирует следующий синтез.
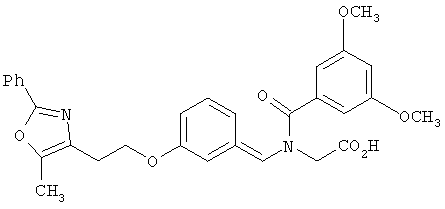
К смеси аминоэфира (20 мг; 0.0474 ммоль)
(получают, как раскрыто в примере 7, часть В) и 3,5-диметоксибензойной кислоты (13 мг, 0.071 ммоль) в безводном СН3CN (0.5 мл) добавляют раствор ВОР реагента (31 мг, 0.071 ммоль) в СН3CN (0.5 мл), а затем DIEA (41 мкл, 0.23 ммоль) в СН3CN (0.5 мл). Реакционную массу встряхивают при комнатной температуре в течение ночи. Летучие продукты удаляют под вакуумом, используя Speed Vac, и добавляют СН2Cl2 (2 мл). Раствор промывают последовательно водным раствором 1 н. HCl (2 мл) и насыщенным водным раствором NaHCO3 (2 мл). В органическую фазу добавляют 0.5 г безводного Na2SO4 и 0.2 г WA21J связанного со смолой амина (Supeico). Смесь встряхивают в течение 0.5 часов и твердое вещество отфильтровывают. Затем к фильтрату добавляют ТФК (2 мл) и раствор встряхивают при комнатной температуре в течение ночи. Реакционный раствор концентрируют под вакуумом, используя Speed Vac, в течение 16 часов, что позволяет получить конечный продукт в виде желтой смолы. Анализ ВЭЖХ с обращенной фазой (YMC S5 ODS 4.6×33 мм колонка, непрерывный градиент от 100% A до 100% В в течение 2 мин при скорости потока 5 мл/мин [растворитель А=10% метанола/90% Н2O/0.2% Н3PO4; растворитель В=90% метанола/10% H2O/0.2% Н3PO4]) показывает, что чистота продукта составляет 90%. Кроме того, ЖХ/МС (электронапыление) определяет точный молекулярный ион [(М+Н)+=531.3] для названного соединения.
Примеры с 315 по 391
Следуя описанным выше процессам, получают следующие соединения, сведенные в таблицы 6 и 7 по изобретению.










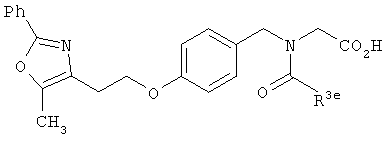







Пример 392
A.
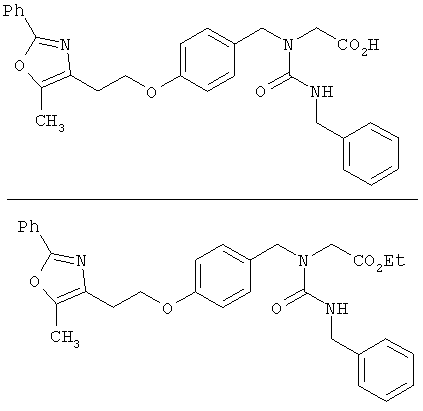
К раствору амина (47 мг; 0.12 ммоль)
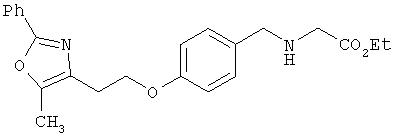
(получают, как раскрыто в примере 3, часть А) в СН3Cl2 (5 мл) добавляют изо-Pr2NEt (0.1 мл; 0.57 ммоль) и DMAP (14 мг; 0.12 ммоль), а затем бензилизоцианат (24 мг; 0.18 ммоль). Реакционную массу перемешивают в течение 14 часов, затем пропускают через SCX картридж [3 г; SCX картридж предварительно промывают последовательно метанолом (10 мл) и СН2Cl2 (5 мл)], элюируя СН2Cl2 (15 мл). Фильтрат концентрируют под вакуумом, что дает сырую мочевину, соединение, часть А (53 мг; 84%), которое является по существу чистым и может быть использовано на следующей стадии без дальнейшей очистки.
B.
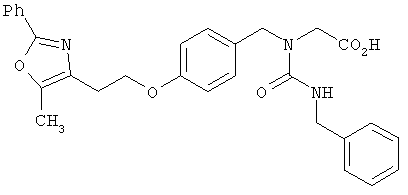
Раствор сырого уреа-этилового эфира (53 мг) и LiOH.H2O (12 мг) в ТГФ:метаноле:H2О (3:1:1; 5 мл) перемешивают при комнатной температуре в течение 2 дней. Раствор подкисляют до рН 3 водным раствором 1 М HCl, концентрируют под вакуумом и очищают препаративной ВЭЖХ (используя YMC S5 ODS 20 мм × 100 мм колонку; непрерывный градиент от 70% А:30% В до 100% В в течение 10 минут скорость потока 20 мл/мин, где А 90:10:0.1 H2O:метанол:ТФК и где В=90:10:0.1 метанол:H2O:ТФК), что дает названное соединение (12 мг; 24%) в виде грязно-белого твердого вещества. [М+Н]+=500.2.
Пример 393

A.
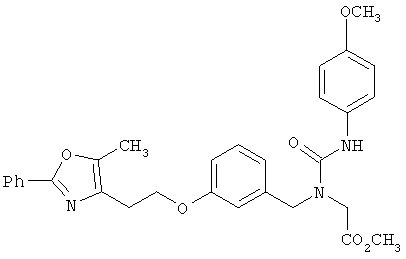
К раствору амина (0.25 г, 0.66 ммоль)

(получают, как раскрыто в Примере 6) в CH2Cl2 (5 мл) одной порцией добавляют 4-метоксифенилизоцианат (0.20 г, 1.32 ммоль) и полученный раствор перемешивают в течение 1 часа при комнатной температуре. Реакционную смесь затем концентрируют под вакуумом, что дает масло, которое хроматографируют (SiO2; 1.5% метанол/CH2Cl2), что обеспечивает названное соединение (0.34 г; 97%) в виде бесцветного масла.
B.
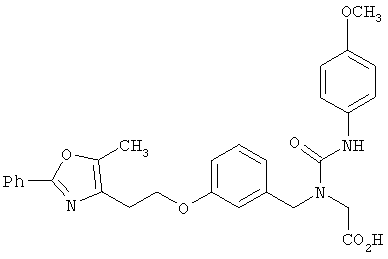
Раствор соединения, часть А (0.14 г, 0.26 ммоль) и LiOH (0.1 г, 4.3 ммоль) в H2O/ТГФ (5 мл, 40:60 раствор) перемешивают в течение 12 часов при 25 °С. Реакционную смесь подкисляют НОАс и экстрагируют EtOAc (2x). Объединенные органические экстракты сушат (MgSO4) и концентрируют под вакуумом, что обеспечивает названное соединение (0.12 г; 90%) в виде бесцветного масла. [М+Н]+=516.
1H ЯМР (CD3OD): δ 7.94 (м, 2Н), 7.45 (м, 3H), 7.23 (м, 3H), 6.80 (м, 2Н), 6.80 (м, 3H), 4.58 (с, 2Н), 4.23 (т, J=7.9 Гц, 2Н), 3.81 (с, 2Н), 3.73 (с, 3H), 2.98 (т, J=7.9 Гц, 2Н), 2.36 (с, 3Н).
Пример 394
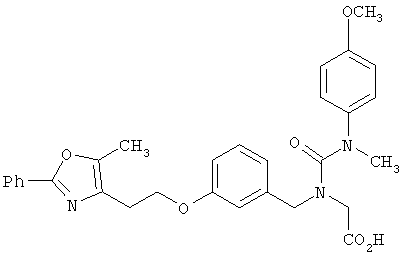
A.
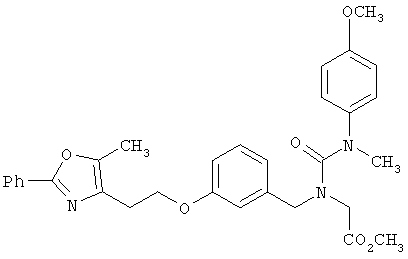
Раствор ранее описанного карбамоилхлорида (соединение по примеру 139, часть А; 0.15 г; 0.34 ммоль)

N-метил-п-анизидина (0.14 г, 1.0 ммоль) и К2СО3 (0.15 г, 1.1 ммоль) в 5 мл ацетона перемешивают при 25°С в течение 12 часов. Реакционную смесь концентрируют под вакуумом, что приводит к маслянистому остатку, который хроматографируют (SiO2; 1.5% метанол/СН2Cl2), что, в свою очередь, обеспечивает названное соединение (0.12 г; 65%) в виде бесцветного масла.
В.
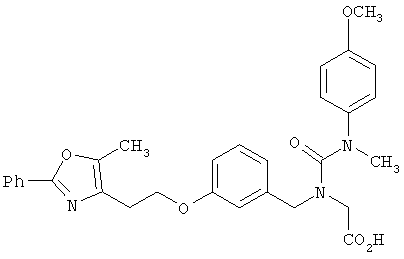
Раствор соединения, часть А (0.12 г, 0.22 ммоль) и LiOH (0.050 г, 2.1 ммоль) в H2O/ТГФ (5 мл 40:60 раствора) перемешивают при комнатной температуре в течение 12 часов. Реакционную смесь концентрируют под вакуумом, что приводит к маслянистому остатку, который очищают препаративной ВЭЖХ (YMC S5 ODS 30×250 мм колонка; скорость потока 25 мл/мин. 30 мин непрерывный градиент от А:В=50:50 до 100% В; растворитель А=90:10:0.1 H2O:метанол:ТФК; растворитель В=90:10:0.1 метанол:H2O:ТФК), что обеспечивает названное соединение (59 мг, 50%) в виде бесцветного масла. [М+Н]+=530.3.
1H ЯМР (CDCl3): 7.99 (д, 6.2 Гц, 2Н), 7.45 (м, 3H), 7.24 (м, 3H), 6.82 (д, 6.2 Гц, 2Н), 6.79 (м, 1Н), 6.63 (м, 1Н), 6.55 (с, 1Н), 4.24 (с, 2Н), 4.16 (т, 7.8 Гц, 2Н), 3.72 (с, 3H), 3.59 (с, 2Н), 3.16 (с, 2Н), 3.02 (т, 7.8 Гц, 2Н), 2.40 (с, 3Н).
Примеры с 395 по 409
Используя приведенные выше процессы, синтезируют аналоги, сведенные в таблицы 8 и 9.


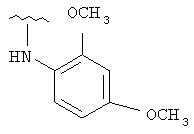
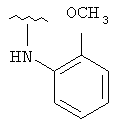
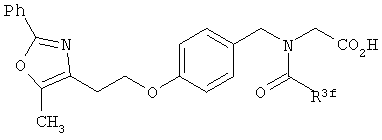

Пример 410
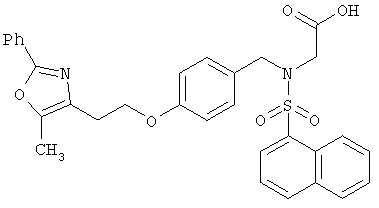
Названное соединение получают как часть библиотеки в потоке растворимой фазы, используя следующий процесс.
A.
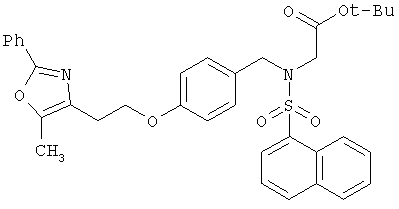
К смеси 1-нафтилсульфонилхлорида (26.8 мг, 0.12 ммоль) и DMAP (2 мг, 0.016 ммоль) в пиридине (2 мл) добавляют раствор амино-трет-бутилового эфира
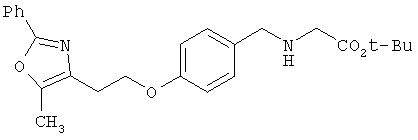
(получают, как раскрыто в примере 8) (20 мг, 0.05 ммоль) в пиридине (0.6 мл). Реакционную массу перемешивают при комнатной температуре в течение 20 часов. Связанный со смолой амин (WA21J, Supeico; 5.8 ммоль/г загрузка; 150 мг) добавляют к смеси. Реакционную массу перемешивают в течение последующих 4 часов. Смолу отфильтровывают и фильтрат концентрируют под вакуумом, что дает сырой продукт, который хроматографируют (CUSIL12M6 колонка; United technology; 2 г сорбента в 6 мл колонке) с помощью приведенного ниже процесса:
1) колонку заполняют гексаном (20 мл);
2) остаток растворяют в минимальном объеме EtOAc и загружают в колонку с силикагелем;
3) картридж элюируют гексаном/EtOAc (3:1), гексаном/EtOAc (1:1). Желаемую фракцию (идентифицируют по ТСХ) собирают и концентрируют, что дает названное соединение в виде вязкого масла, которое используют на следующей стадии без дополнительной очистки.
B.
Et3N (0.3 мл 1 М раствора в CH2Cl2) и TMSI (0.3 мл 1 М раствор в CH2Cl2) последовательно добавляют к раствору соединения из части А в СН2Cl2. Реакционную смесь перемешивают при комнатной температуре в течение 12 часов и затем концентрируют под вакуумом, что дает сырой продукт. Продукт очищают твердофазной экстракцией, используя CHQAX12M6 колонку (United technology; 2 г сорбента в 6 мл колонке) с помощью приведенного ниже процесса:
1) колонку заполняют CH2Cl2 (25 мл);
2) остаток растворяют в минимальном объеме CH2Cl2 и загружают в SAX колонку;
3) картридж промывают последовательно CH2Cl2 (25 мл), СН2Cl2/метанолом (5% метанол, 15 мл), СН2Cl2/метанолом (50% метанол, 15 мл), метанол (20 мл);
4) продукт элюируют раствором 1% ТФК в метаноле (20 мл).
Конечный продукт, содержащий фракции, собирают и концентрируют под вакуумом, используя Speed Vac, что позволяет получить BMS-329075 (16 мг; 62%). Анализ ВЭЖХ с обращенной фазой показывает, что чистота продукта составляет 90%. Кроме того, ЖХ/МС (электронапыление) определяет точный молекулярный ион [(М+Н)+=557.1] для желаемого соединения.
Пример 411
А.
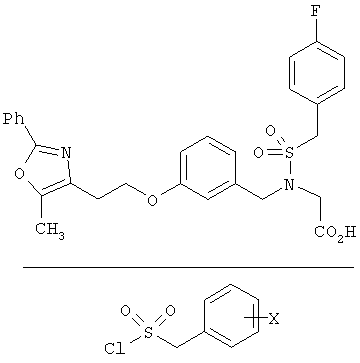
X = галоген, алкил, CF3, CF3О и т.п.
Следующий общий процесс используют для приготовления требуемых замещенных бензилсульфонилхлоридов.
Газообразный Cl2 пропускают в охлажденный до 0°С раствор 4-фторбензилмеркаптана (1.0 г, Lancaster) в ледяной уксусной кислоте (100 мл) и H2O (5.0 мл) в течение 1 часа. Реакционную смесь затем выливают в смесь льда-Н2О и немедленно экстрагируют CH2Cl2 (200 мл); органическую фазу тщательно промывают последовательно H2O (200 мл), водным раствором насыщенного NaHCO3 (2×100 мл) и наконец рассолом (200 мл). Органическую фазу сушат (MgSO4) и концентрируют под вакуумом, что приводит к 4-фторбензилсульфонилхлориду в виде бесцветного твердого вещества (1.3 г; 89%).
B.
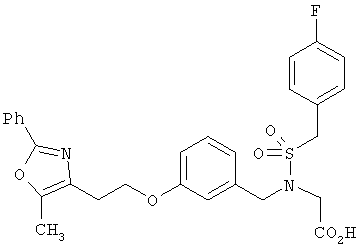
К раствору вторичного аминометилового эфира (25 мг; 0.066 ммоль)
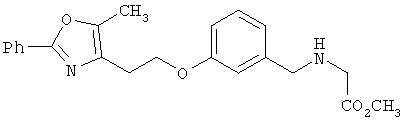
(получают, как раскрыто в примере 6) в пиридине (0.8 мл) добавляют 4-фторбензилсульфонилхлорид (68 мг; 0.33 ммоль; 5 экв).
Смесь нагревают до 75°С, перемешивают в течение ночи при 75°С и затем концентрируют под вакуумом. Черный остаток обрабатывают водным раствором LiOH (1.0 мл 0.3 М раствора) в H2O/метаноле/ТГФ в течение 18 часов, затем концентрируют под вакуумом. Остаток подкисляют 1.0 М водным раствором HCl до рН=1-2 и экстрагируют EtOAc (2x), сушат (Na2SO4) и концентрируют под вакуумом, что дает сырой продукт. Очистка препаративной ВЭЖХ (YMC S5 CDS 20 мм × 250 мм колонка с обращенной фазой; 15-минутный непрерывный градиент от 60:40 А:В до 100% В с 10-минутным временем выдержки, где А=90:10:0.1 H2O:метанол:ТФК и В=90:10:0.1 метанол:H2O:ТФК; скорость потока 25 мл/мин) дает названное соединение (12 мг; 34%) в виде белого твердого вещества. [М+Н]+(ЖХ/МС)=539.1.
Примеры с 412 по 456
Используя приведенные выше процессы, синтезируют аналоги, сведенные в таблицы 10 и 11.
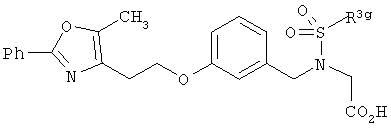


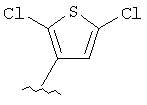

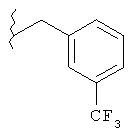
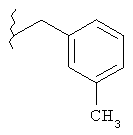
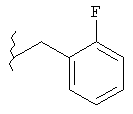




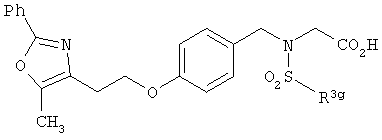







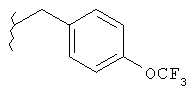
Пример 457
A.
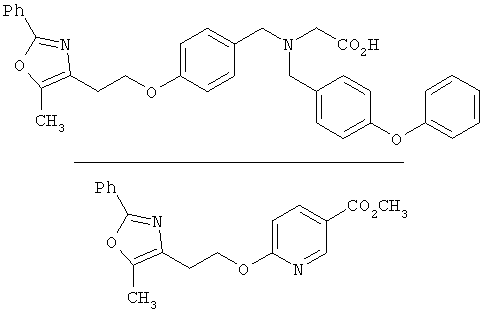
К охлажденному до 0°С раствору метил-2-гидроксипиридин-5-карбоксилату (0.2 г, 1.3 ммоль), 2- (5-метил-2-фенилоксазол-4-ил)этанолу (0.32 г, 1.56 ммоль) и Ph3Р (0.38 г, 1.56 ммоль) в CH2Cl2 (10 мл) добавляют DEAD (0.2 мл, 1.95 ммоль) по каплям и реакционную массу перемешивают при 25°С в течение 12 часов. Раствор концентрируют под вакуумом и хроматографируют на SiO2 (4:1, гексан:EtOAc), что обеспечивает названное соединение (0.28 г, 63%) в виде масла.
B.
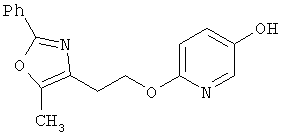
К охлажденному до -78°С раствору соединения из части А (0.28 г., 0.82 ммоль) в ТГФ (10 мл) добавляют DIBALH (2.0 мл 1 М раствора в CH2Cl2; 1.95 ммоль) и реакцию перемешивают при -78°С в течение 4 часов. ТСХ аликвоты реакционной массы показывает наличие соответствующих альдегида и спирта. Реакционную массу нагревают до 25°С и перемешивают при комнатной температуре в течение 1 часа, после которого по ТСХ наблюдают только спирт. Реакцию захолаживают водой и разбавляют EtOAc. Органический слой промывают рассолом, сушат (MgSO4) и концентрируют в вакууме до образования названного соединения в виде масла. Полученный сырой продукт используют в следующей реакции без дальнейшей очистки.
C.

К охлажденному до -78°С раствору оксалилхлорида (0.22 мл, 2.6 ммоль) и ДМСО (0.37 мл, 5.2 ммоль) в СН2Cl2 (15 мл) по каплям добавляют раствор соединения из части В (0.42 г сырой продукт в 5 мл CH2Cl2). Реакционную смесь перемешивают в течение 2 часов при -78°С и затем (1 мл) добавляют по каплям Et3N. Реакционную смесь перемешивают в течение дополнительных 0.5 часов при -78°С и затем медленно нагревают до 25°С. Затем реакционную смесь разбавляют EtOAc (200 мл) и промывают последовательно водным раствором NaHCO3 и рассолом. Органический слой сушат (MgSO4), затем концентрируют под вакуумом, что обеспечивает названное соединение (0.40 г; 95%) в виде масла, которое используют на следующей стадии без дальнейшей очистки.
D.
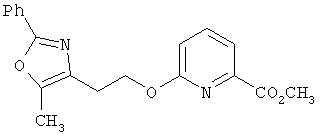
Смесь соединения, часть С (<0.82 ммоль), гидрохлорида глицинметилового эфира (0.5 г, 4.0 ммоль), NaBH(OAc)3 (0,85 г, 4.0 ммоль) и DCE (10 мл) перемешивают при 25°С в течение 12 часов. Реакционную смесь затем разбавляют EtOAc (50 мл) и промывают последовательно водным раствором NaHCO3 и рассолом. Органический слой сушат (MgSO4), затем концентрируют под вакуумом, что дает названное соединение (0.31 г; 82%) в виде масла (>95% чистота, как показала аналитическая ВЭЖХ с обращенной фазой), который используют на следующей стадии без дальнейшей очистки.
E.
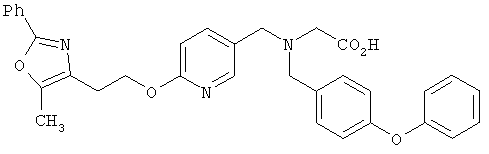
Смесь соединения, часть D (0.050 г; 0.13 ммоль), 4-феноксибензальдегида (0.048 г; 0.26 ммоль), NaBH(ОАс)3 (0.082 г; 0.39 ммоль) в DCE (10 мл) перемешивают при 25°С в течение 12 часов. Реакционную смесь разбавляют EtOAc (50 мл) и промывают последовательно водным раствором NaHCO3 и рассолом. Органический слой сушат (MgSO4), затем концентрируют под вакуумом, что дает третичный аминометиловый эфир в виде маслянистого остатка. К указанному остатку добавляют LiOH (0.050 г) и Н2О/ТГФ (2 мл 60:40 раствора) и реакционную массу перемешивают при комнатной температуре в течение 12 часов. Препаративной ВЭЖХ (YMC S5 ODS 30×250 мм колонка, непрерывный градиент более 30 мин; скорость потока 25 мл/мин, от 30:70 А:В до 100% В; А=90:10:0.1 H2O:метанол:CF3CO2Н; В=90:10:0.1 метанол:H2O:CF3CO2Н) обеспечивается получение названного соединения (0.021 г; 30%) в виде соли с ТФК.
1H ЯМР (CDCl3): δ 8.18 (с, 1Н), 7.94 (д, 6.6 Гц, 2Н), 7.86 (д. 8.8 Гц, 1Н), 7.45 (м, 3H), 7.34 (м, 3H), 7.14 (т, 7.4 Гц, 1Н), 7.02-6.92 (м, 5Н), 6.81 (т, 8.8 Гц, 1Н), 4.51 (м, 6Н), 3.59 (с,2Н), 3.06 (т, 6.2 Гц, 2Н).
Пример 458
А.
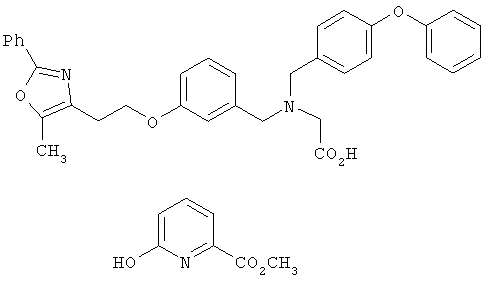
Смесь 2-гидроксипиридин-6-карбоновой кислоты (1.30 г, 9.4 ммоль), концентрированной H2SO4 (0.5 мл) и метанола (20 мл) нагревают при кипении с обратньм холодильником в течение 12 часов. Реакция завершается именно в этот момент, как показывает аналитическая ВЭЖХ. Реакционную смесь концентрируют под вакуумом, что дает легкое желтое масло, которое разбавляют EtOAc и промывают водным раствором NaHCO3. Органическую фазу сушат (MgSO4) и концентрируют под вакуумом, чтобы получить названное соединение в виде твердого вещества (0.43 г, 30%).
B.
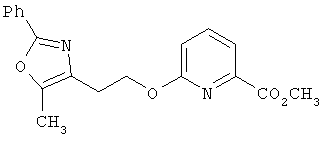
К раствору соединения, часть А (0.43 г, 2.8 ммоль), 2-(5-метил-2-фенилоксазол-4-ил)этанола (0.68 г, 3.3 ммоль) и Ph3Р (1.0 г, 4.07 ммоль) в ТГФ (10 мл) добавляют DEAD (0.66 мл, 4.2 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 12 часов. Раствор концентрируют под вакуумом и остаток хроматографируют (SiO2; 20% ацетона в гексане), что обеспечивает названное соединение в виде масла (0.92 г; 97%).
C. 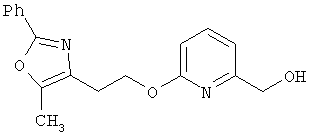
К раствору соединения, часть В (0.92 г, 2.7 ммоль) в ТГФ (50 мл) добавляют LiAlH4 (5 мл 1.0 М раствора в ТГФ, 5 ммоль) по каплям при -78°С и конечную реакционную массу нагревают до 0°С в течение 2 часов. Затем реакцию захолаживают, добавляя в смесь несколько кусочков льда. После этого реакционную смесь разделяют между EtOAc (200 мл) и рассолом (50 мл). Органическую фазу сушат (MgSO4) и концентрируют под вакуумом, что дает масло (0.92 г; 95%) которое используют в следующей реакции без дальнейшей очистки.
D. 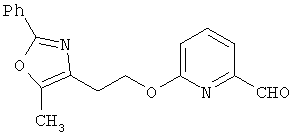
К раствору оксалилхлорида (0.47 мл, 5.4 ммоль) и ДМСО (0.36 мл, 10.8 ммоль) в СН2Cl2 (15 мл) добавляют по каплям раствор соединения, часть С (0.92 г; >2.7 ммоль) в СН2Cl2 (10 мл) при -78°С. Реакционную смесь перемешивают в течение 2 часов и затем по каплям добавляют Et3N (1 мл). Реакционной смеси дают перемешиваться в течение дополнительных 0.5 часов при -78°С и затем медленно нагревают до 25°С. Реакционную смесь затем разбавляют EtOAc (200 мл) и промывают последовательно водным раствором NaHCO3 и рассолом. Органический слой сушат (MgSO4) и затем концентрируют под вакуумом, чтобы получить названное соединение (0.90 г; чистота >90%, 1H ЯМР анализ) в виде масла. Полученный продукт затем используют без последующей очистки.
E. 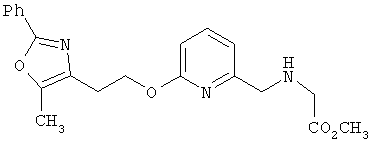
К раствору соединения, часть D (0.90 г; 2.7 ммоль), гидрохлорида глицинметилового эфира (1.7 г, 13.5 ммоль) в 1,2 дихлорэтане (10 мл) добавляют NaBH(ОАс)3 (1.7 г, 8.1 ммоль) одной порцией. Полученный раствор перемешивают при 25°С в течение 12 часов. Реакционную смесь концентрируют под вакуумом, что дает масло, которое хроматографируют (SiO2; 30% ацетона в гексане), что обеспечивает названное соединение (0.86 г; 83%) в виде бесцветного масла.
F.
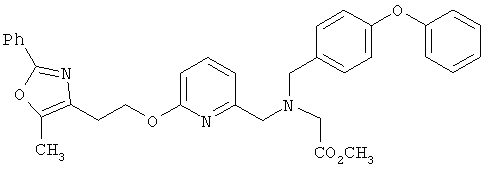
Раствор соединения, часть Е (0.040 г, 0.1 ммоль), 4-феноксибензальдегида (0.030 г, 0.15 ммоль) и NaBH(OAc)3 (0.060 г, 0.3 ммоль) в DCE (10 мл) перемешивают при комнатной температуре в течение 12 часов. Реакционную смесь концентрируют под вакуумом и маслянистый остаток хроматографируют (SiO2; 30% ацетона в гексане), что обеспечивает аминоэфирное названное соединение (56 мг; >95%) в виде бесцветного масла.
G. 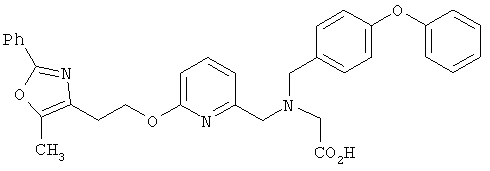
Раствор соединения, часть F (56 мг; 0.1 ммоль) и LiOH (0.050 г; 0.21 ммоль) в H2O/ТГФ (2 мл 6:4 раствор) перемешивают при комнатной температуре в течение 12 часов. Реакционную смесь концентрируют под вакуумом, что дает белое твердое вещество, которое растворяют в метаноле и очищают препаративной ВЭЖХ (YMC S5 ODS 30×250 мм колонка; непрерывный градиент более 30 мин; скорость потока 25 мл/мин, от 30:70 А:В до 100% В; А = 90:10:0.1 H2O:метанол:ТФК; В=90:10:0.1 метанол:H2O:ТФК). Названное соединение (41 мг; 72%) получают в виде соли с ТФК.
1Н ЯРМ (метанол-D4): 7.90 (м, 2Н), 7.71 (т, 8.4 Гц, 1Н), 7.51 (д, 8.7 Гц, 2Н), 7.44 (м, 3H), 7.36 (т, 8.7 Гц, 2Н). 7.17 (т, 8.4 Гц, 1Н), 6.96 (м, 5Н), 6.82 (д, 8.4 Гц, 1Н), 4.62 (т, 6.2 Гц, 2Н), 4.56 (с, 2Н), 4.50 (с, 2Н), 4.17 (с, 2Н), 3.00 (т, 6.2 Гц, 2Н), 2.36 (с, 3Н). С34Н31N3O5. 550.23 (М+H+) по ЖХ/МС (электронапыление).
Пример 459
A.
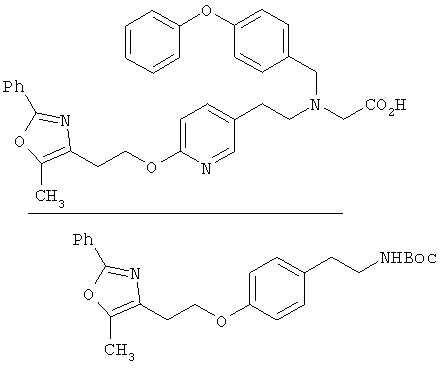
К охлажденному до 0°С раствору 2-(5-метил-2-фенилоксазол-4-ил)этанола (1.07 г, 5.25 ммоль), Ph3Р (1.38 г, 5.25 ммоль) и N-Boc-4-гидроксифенилэтиламина (1.24 г, 5.25 ммоль) в ТГФ (36 мл) добавляют DEAD (0.83 мл, 5.25 ммоль), дают нагреться до комнатной температуры и перемешивают в течение 15 часов. Реакционную смесь концентрируют под вакуумом и остаток хроматографируют (SiO2; постепенный градиент от 95:5 до 4:1, гексан:EtOAc) для получения названного соединения (1.43 г, 65%).
В.
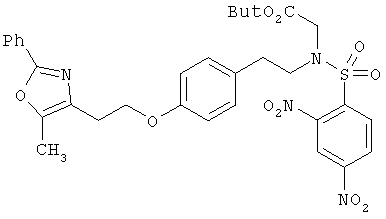
Раствор соединения, часть А (1.01 г, 2.37 ммоль) и ТФК (8 мл) в CH2Cl2 (30 мл) перемешивают при комнатной температуре в течение 4.5 часов. Раствор концентрируют под вакуумом и остаток растворяют в СН2Cl2, а затем фильтруют через подушку твердого К2СО3. Фильтрат концентрируют под вакуумом, что дает соответствующий сырой амин. К раствору сырого амина в ТГФ (11.9 мл) добавляют пиридин (0.383 мл, 4.74 ммоль) и 2,4-динитробензолсульфонилхлорид (0.85 г, 3.19 ммоль) и раствор перемешивают при комнатной температуре в течение 15 часов. Поскольку в этот момент еще остается некоторое количество исходного продукта, вводят дополнительное количество сульфонилхлорида (0.32 г, 1.2 ммоль). После последующих 4 часов анализ ВЭЖХ показывает, что весь исходный продукт вступил в реакцию. Реакционную смесь разбавляют Et2O, промывают 1 н. водной HCl, насыщенным водным раствором NaHCO3 и рассолом, сушат (MgSO4), фильтруют и концентрируют под вакуумом, что обеспечивает сырой 2,4-динитробензолсульфонамид.
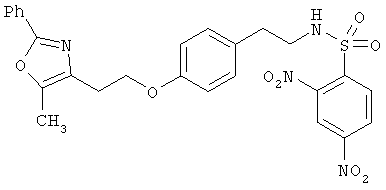
К раствору сырого 2,4-динитробензолсульфонамида в СН3CN (3 мл) добавляют К2СО3 (избыток) и трет-бутиловый бромацетат (7.11 ммоль). Реакционную массу перемешивают при комнатной температуре в течение ночи. ВЭЖХ анализ показывает, что соотношение продукта и исходного материала составляет 2:1. К реакционной смеси добавляют дополнительное количество ДМФА (3 мл), К2СО3 и трет-бутилбромацетат. Реакцию завершают за 2 часа. Реакционную смесь разбавляют Et2O, промывают 1 н. водным раствором HCl, насыщенным раствором NaHCO3 и рассолом, сушат (MgSO4) и концентрируют под вакуумом, что обеспечивает сырой трет-бутиловый эфир. Указанный сырой продукт хроматографируют (SiO2; гексан/EtOAc; постепенный градиент от 9:1 до 2:1), что дает названное соединение (0.663 г, 42% общий).
C. 
К раствору соединения, часть В (0.663 г, 0.995 ммоль) в ТГФ (2.5 мл) добавляют Et3N (0.208 мл, 1.49 ммоль) и меркаптоуксусную кислоту (0.090 мл, 1.29 ммоль). Реакционную массу перемешивают при комнатной температуре в течение ночи. Реакционную смесь затем разбавляют Et2O, промывают 1 н. водным раствором HCl, насыщенным раствором NaHCO3 и рассолом, сушат (MgSO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; гексан/EtOAc; постепенный градиент от 9:1 до 2:1), что дает названное соединение (0.265 г, 61%).
D. 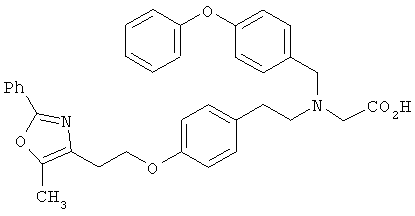
К раствору соединения, часть С (0.015 г, 0.0344 ммоль) в DCE (1 мл) добавляют 4-феноксибензальдегид (0.103 ммоль) и NaBH(ОАс)3 (0.0365 г, 0.172 ммоль). Реакционную массу перемешивают при комнатной температуре в течение 15 часов. Затем ее фильтруют через тканевую подушку, что обеспечивает прозрачный раствор, который разбавляют CH3Cl2, промывают насыщенный водным раствором NaHCO3 и рассолом, сушат (MgSO4) и концентрируют под вакуумом. Сырой продукт очищают препаративной ВЭЖХ (YMC S5 ODS 30×250 мм колонка; скорость потока 25 мл/мин, градиент от 20% В до 100% В более 25 мин, 100% В, выдержка в течение 15 мин время задержки=29.1 мин), чтобы получить трет-бутиловый эфир. Полученный эфир растворяют в CH2Cl2 (1.3 мл) и медленно добавляют ТФК (0.5 мл). Реакционную массу перемешивают при комнатной температуре в течение ночи и затем концентрируют под вакуумом. Остаток затем растворяют в СН2Cl2, промывают H2O, насыщенным водным раствором NaHCO3 и рассолом, сушат (MgSO4) и концентрируют под вакуумом, что дает названное соединение (0.012 г, 61%). ЖХ/МС дает правильный [М+Н]+=563.3.
Другие аналоги (как показано в таблице) синтезируют тем же процессом восстановительного аминирования, как раскрыто в примере 459, часть D, используя соединение по примеру 459, часть С, и различные ароматические альдегиды Кроме того, карбаматокислоты, такие как описаны в примере 461, также синтезируют, используя общий процесс, раскрытый ранее для синтеза соединения по примеру 136.


Пример 463
A.

К раствору кислоты по примеру 230 (240 мг, 0.47 ммоль) в ДМФА (2.0 мл) добавляют НОАТ (68 мг, 0.49 ммоль), EDAC (94 мг, 0.49 ммоль) и 2-цианоэтиламин (34 мг, 0.49 ммоль). Раствор перемешивают при комнатной температуре в течение 18 часов; анализ реакции по ЖХ/МС показывает, что исходный продукт еще присутствует. В реакционную массу вводят дополнительный 2-цианоэтиламин (34 мг, 0.49 ммоль) и ее перемешивают при комнатной температуре в течение 48 часов. Летучие продукты удаляют под вакуумом и остаток растворяют в CH2Cl2 (40 мл), а затем промывают последовательно водой (2×30 мл) и рассолом (30 мл). Органическую фазу сушат (MgSO4) и концентрируют под вакуумом. Полученный белый остаток растворяют в минимальном количестве СН2Cl2 (3 мл) и высаживают путем осторожного прибавления EtOAc, что приводит к амиду, который является названным соединением (184 мг; 70%) в виде белого твердого вещества.
B. 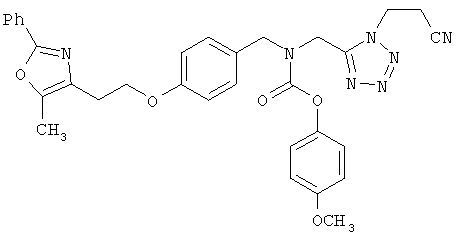
К охлажденному до 0°С раствору соединения, часть А (180 мг; 0.32 ммоль) в CH2Cl2 (1.5 мл) последовательно добавляют Ph3Р (83 мг; 0.32 ммоль), DEAD (100 мкл, 0.64 ммоль) и TMSN3 (85 мкл, 0.64 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 24 часов. ЖХ/МС анализ показывает, что все еще остается достаточное количество исходного продукта. Реакционную смесь затем концентрируют под вакуумом до 2/3 первоначального объема и вводят дополнительное количество Ph3Р, DEAD и TMSN3 (1 эквивалент каждого реагента). Реакционную смесь перемешивают при комнатной температуре в течение еще 24 часов и затем разбавляют с помощью EtOAc (40 мл). Раствор обрабатывают 5% водным раствором CAN (10 мл) и перемешивают в течение 15 минут. Реакционный раствор промывают водой (30 мл) и рассолом (30 мл), сушат (MgSO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; простой эфир: CH2Cl2, 3:7), что дает названное соединение (100 мг; 53%) в виде белого твердого вещества.
C. 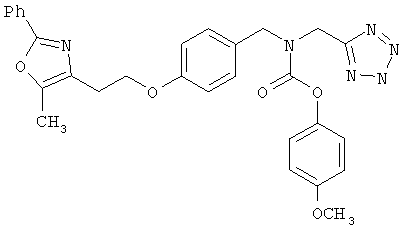
К раствору соединения, часть В (100 мг, 0.17 ммоль) в ТГФ/1,4-диоксане (6:1, 1.4 мл) добавляют водный раствор NaOH (0.6 мл 1.0 М раствора, 3.5 экв). Смесь перемешивают при комнатной температуре в течение 14 часов и затем подкисляют до рН ˜2 с помощью 1.0 М водного раствора Н3PO4. EtOAc (30 мл) добавляют и органическую фазу промывают водой (15 мл) и рассолом (15 мл), сушат (MgSO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; 4% метанол/CH2Cl2), что дает названный тетразол (35 мг; 38%) в виде белой пены. ЖХ/МС (электронапыление) дает правильный молекулярный ион: [М+Н]+=541.3.
Пример 464
A.

Смесь 2-гидроксибензальдегида (500 мг, 4.09 ммоль), гидрохлорида глицинметилового эфира (544 мг, 4.09 ммоль) и Et2N (495 мг, 4.9 ммоль) в сухом метаноле (5 мл) перемешивают при комнатной температуре в течение 3 часов. Затем тремя порциями добавляют NaBH4 (155 мг, 4.09 ммоль). Реакционную массу перемешивают при комнатной температуре в течение еще 30 минут. Насыщенный водный раствор Na2CO3 (1 мл) добавляют для того, чтобы разрушить оставшийся NaBH4, и затем добавляют водный раствор HCl (10 мл 1 н. раствора). Водную фазу промывают EtOAc (3×20 мл), затем осторожно подщелачивают 1 н. водным раствором NaOH до рН 7-8. Водную фазу затем экстрагируют с помощью EtOAc (3×20 мл). Красно-оранжевый раствор концентрируют под вакуумом, что дает названное соединение в виде желтого вязкого масла.
B.
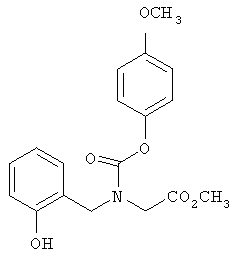
Соединение, часть А (38 мг, 0.195 ммоль), 4-метоксифенилхлорформиат и пиридин (39 мг, 5 ммоль) растворяют в 0.1 мл CH2Cl2 в течение 5 минут. Реакционную смесь затем промывают водным раствором HCl (2×2 мл 1 н. раствора). Органическую фазу промывают рассолом, сушат (Na2SO4), концентрируют под вакуумом и хроматографируют (SiO2; гексан:EtOAc, 7:3), что дает названное соединение (40 мг; 59%) в виде бледно-желтого масла.
C. 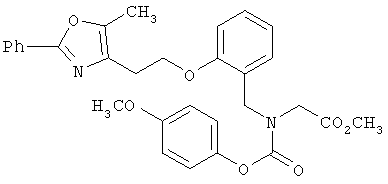
К раствору соединения, часть В (40 мг, 0.116 ммоль), 2-[2-фенил-5-метилоксазол-4-ил]этанола (Maybridge; 24 мг, 0.116 ммоль) и Ph3Р (40 мг, 0.151 ммоль) в сухом ТГФ (3 мл) добавляют по каплям DEAD (26 мг, 0.151 ммоль). Раствор перемешивают при комнатной температуре в течение ночи. Красно-оранжевый раствор концентрируют под вакуумом и остаток очищают препаративной ВЭЖХ (непрерывный градиент от 50% А : 50% В до 100%В; А = 90% H2O:10% метанола+0.1% ТФК); В=90% метанола/10% Н2O+0.1% ТФК) в течение 10 мин; YMC SH-343-5 ODS 20×100 мм (5 мкм) колонка), что обеспечивает названное соединение (30 мг, 47%) в виде желтого вязкого масла.
D. 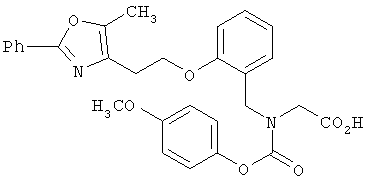
Соединение из части С растворяют в метаноле (3 мл) и H2O (0.3 мл). К полученному раствору добавляют LiOH (3 мг) и реакцию перемешивают при комнатной температуре в течение 3 часов. Летучие продукты удаляют под вакуумом и раствор подкисляют 1 н. водным раствором HCl до рН 3-4. Водную фазу экстрагируют EtOAc (3×10 мл). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом, что дает названное соединение в виде белого твердого вещества (18 мг; 64%). ЖХ/МС (электронапыление) определеяет точный молекулярный ион [(М+Н)+=516].
1H ЯМР: δ 2.27-2.32 (м, 3H), 2.96-2.98 (м, 2Н), 3.65-3.69 (д, 3H), 4.06-4.20 (м, 4Н), 4.55-4.63 (д, 2Н), 6.74-6.93 (м, 4Н), 7.19-7.35(м, 2h), 7.88-7.90 (м, 2Н).
Пример 465

A.
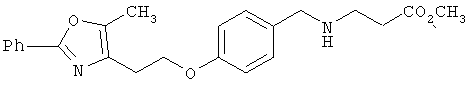
Смесь гидрохлорида п-аланинметилового эфира (51 мг; 0.363 ммоль), Et3N (50 мкл; 0.363 ммоль) и альдегида (100 мг; 0.33 ммоль)

в метаноле (1 мл) перемешивают при комнатной температуре в течение 3 часов. Добавляют NaBH4 (14 мг; 0.363 ммоль) и реакцию перемешивают при комнатной температуре в течение еще 1 часа. Летучие продукты удаляют под вакуумом и остаток разделяют между насыщенным водным раствором Na2СО3 и EtOAc (20 мл каждого). Органическую фазу концентрируют под вакуумом, что дает соединение, часть А, в виде желтого масла, которое используют на следующей стадии без дальнейшей очистки.
B.
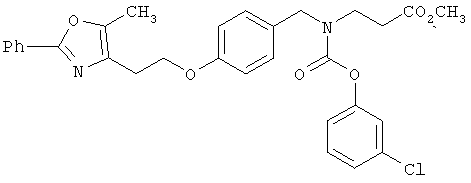
К раствору соединения из части А (20 мг; 0.050 ммоль) и пиридина (0.50 мл) в СН2Cl2 (2 мл) добавляют 3-хлорфенилхлорформиат (14 мг; 0.070 ммоль). Реакционную массу перемешивают при комнатной температуре в течение 2 часов, затем летучие продукты удаляют под вакуумом. Остаток очищают препаративной ВЭЖХ (YMC S5 ODS 20×75 мм колонка с обращенной фазой; непрерывный градиент от 50:50 А:В до 100% В, где А=90:10:0.1 H2O:метанол:ТФК и В=90:10:0.1 метанол:H2O:ТФК), что дает соединение, часть В.
C.
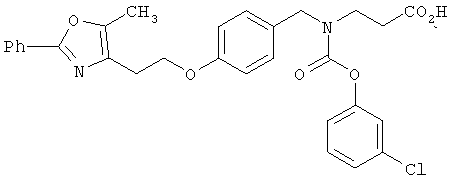
Раствор соединения из части В и LiOH·Н2О (5 мг) в ТГФ:H2O (4:1) перемешивают при комнатной температуре в течение 1 часа. Реакционный раствор подкисляют до рН 3 водным раствором HCl, затем экстрагируют EtOAc. Объединенные органические экстракты концентрируют под вакуумом, что дает названное соединение (5 мг; 18%) в виде белого твердого вещества. [М+Н]+=535.2; 537.2.
Пример 466
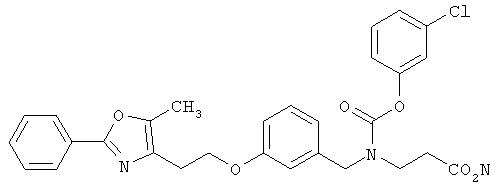
Названное соединение синтезируют, используя ту же последовательность, что в примере 465 за исключением того, что используют альдегид
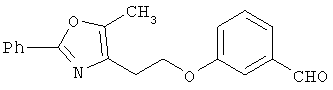
[M+H]+ = 535.2; 537.2.
Следуя процессам, раскрытым выше, получают соединения по примерам с 467 по 472.
Примеры с 467 по 469
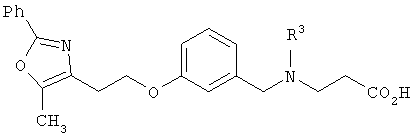
Примеры с 470 по 472
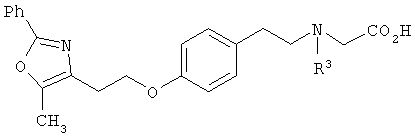
Пример 473

A.
Смесь 3-йодфенола (2.0 г; 9.1 ммоль), уксусного ангидрида (4.6 г; 45.5 ммоль) и пиридина (3.6 г; 45.5 ммоль) перемешивают в CH2Cl2 (20 мл) в течение 3 часов. Полученную смесь промывают насыщенным водным раствором NH4Cl (3×100 мл), сушат (MgSO4) и концентрируют под вакуумом, что дает соединение из части А (2.30 г; 97%) в виде желтого масла.
B.
Смесь соединения из части А (1.00 г; 4.0 ммоль), триметилсилилацетилена (780 мг; 8 ммоль), CuI (15 мг; 0.08 ммоль) и (Ph3Р)2Pd2Cl2 (28 мг; 0.04 ммоль) в диэтиламине (10 мл) перемешивают при комнатной температуре в течение 3 часов. Летучие продукты удаляют под вакуумом и остаток хроматографируют (SiO2; гексан:EtOAc, 4:1), что дает сырое соединение из части В, которое используют на следующей стадии без дальнейшей очистки.
C.

К раствору сырого соединения из части В в СН2Cl2 (2 мл) добавляют пиридин (3 мл; 37 ммоль) и уксусный ангидрид (4 мл; 42 ммоль). Реакционную массу перемешивают при комнатной температуре в течение 2 часов, затем разделяют между насыщенным водным раствором NH4Cl (30 мл) и СН2Cl2. Органическую фазу промывают дополнительньм количеством насыщенного водного раствора NH4Cl (30 мл) и H2O (100 мл), сушат (Na2SO4) и концентрируют под вакуумом, что дает соединение, часть С, которое используют на следующей стадии без дальнейшей очистки.
D. 
Раствор сырого соединения, часть С, и Bu4NF (1.1 г; 12 ммоль) в ТГФ (10 мл) перемешивают при комнатной температуре в течение 1.7 часов, после чего весь исходный продукт вступает в реакцию. Реакционный раствор промывают H2O, добавляют Celite® (Целит) и летучие продукты удаляют под вакуумом. Твердое вещество хроматографируют (SiO2; гексан:EtOAc, 9:1), что дает соединение, часть D (400 мг; 63% на 3 стадиях).
E. 
Смесь соединения из части D (400 мг; 2.5 ммоль) и катализатор Pd/СаСО3/Pb (40 мг; Aldrich) в метаноле (20 мл) перемешивают в атмосфере Н2 в течение 30 минут. Смесь фильтруют через Celite® и фильтрат концентрируют под вакуумом. Остаток хроматографируют (SiO2; гексан:EtOAc, 95:5), что дает соединение, часть Е (310 мг; 77%) в виде бесцветного масла.
F.
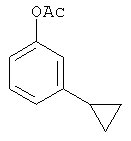
К охлажденному до 0°С раствору соединения из части Е (310 мг; 1.9 ммоль) в DCE (10 мл) последовательно добавляют по каплям чистый диэтилцинк (491 мкл; 4.8 ммоль; Aldrich) и ICH2Cl (700 мкл; 9.6 ммоль). Реакционной смеси дают нагреться до комнатной температуры и затем перемешивают при комнатной температуре в течение 3 часов, после чего ее разделяют между насыщенным водным раствором NH4Cl и EtOAc (50 мл каждый). Органическую фазу промывают насыщенным водным раствором NH4Cl и H2O (50 мл каждый) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; гексан:EtOAc, 9:1), что приводит к соединению, часть F (230 мг; 69%) в виде бесцветного масла.
G.

Смесь соединения из части F (100 мг; 0.57 ммоль) и К2СО3 (157 мг; 1.1 ммоль) в метаноле (5 мл) перемешивают при комнатной температуре в течение ночи (нет реакции). Добавляют водный раствор Li ОН (1.1 мл 1 М раствора; 1.1 ммоль) и раствор перемешивают при комнатной температуре в течение ночи. Летучие продукты удаляют под вакуумом и остаток распределяют между водным раствором 1 М HCl и EtOAc. Органическую фазу концентрируют под вакуумом и остаток хроматографируют (SiO2; гексан:EtOAc, 4:1), что приводит к соединению, часть G (70 мг; 92%) в виде желтого масла.
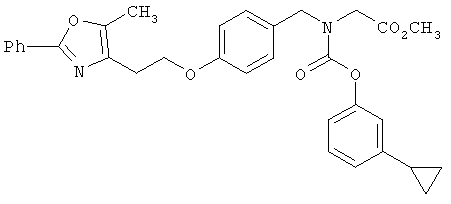
H.
К раствору соединения из части G (6 мг; 0.045 ммоль) в ДМФА (0.2 мл) добавляют трет-бутилат калия (5 мг; 0.05 ммоль). Реакцию перемешивают в течение 2 мин при комнатной температуре, после чего добавляют карбамоилхлорид (20 мг; 0.045 ммоль) 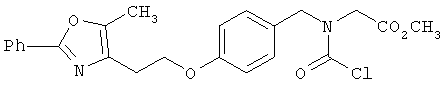
и реакционную массу перемешивают при комнатной температуре в течение последующих 15 минут. Летучие продукты затем удаляют под вакуумом и остаток хроматографируют (SiO2; гексан:EtOAc, 7:3), что приводит к соединению, часть Н (11 мг; 45%) в виде желтого масла.
I.
К раствору соединения из части Н и LiOH·H2O в метаноле/H2O (10 мл, 9:1 смеси) перемешивают при комнатной температуре в течение ночи. Затем раствор подкисляют до рН ˜3 водным раствором HCl и экстрагируют EtOAc. Объединенные органические экстракты концентрируют в вакууме и очищают препаративной ВЭЖХ, что дает названное соединение (10.1 мг; 95%) в виде грязно-белого лиофилата. [М+Н]+=527.3.
Пример 474
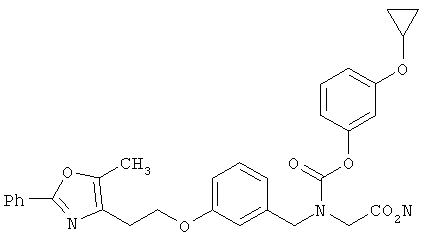
A.
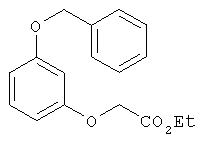
Смесь 3-бензилоксибензальдегида (2.00 г; 1.0 ммоль), этилбромацетата (1.67 г; 1.0 ммоль) и Cs2СО3 (3.25 г; 1.0 ммоль) в ДМФА (20 мл) перемешивают при комнатной температуре в течение 8 часов. Реакционную смесь разделяют между H2O (300 мл) и EtOAc (100 мл). Водную фазу экстрагируют EtOAc (2×100 мл). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; 85:15, гексан:EtOAc), чтобы получить соединение из части А (3.48 г; >100%) в виде бесцветного масла.
B.
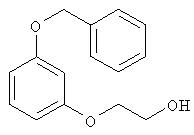
К раствору соединения из части А (3.4 г; 11.9 ммоль) в сухом ТГФ (50 мл) в атмосфере аргона добавляют LiAlH4 (36 мл 0.5 М раствора в ТГФ; 17.8 ммоль) по каплям. Реакционную массу перемешивают при комнатной температуре в течение 1 часа. Реакцию захолаживают, медленно добавляя насыщенный водный раствор NH4Cl (1 мл). Летучие продукты удаляют под вакуумом и остаток разделяют между EtOAc (100 мл) и 1 М водным раствором HCl. Органическую фазу сушат (Na2SO4) и концентрируют под вакуумом, что дает соединение, часть В (2.4 г; 98%) в виде белого твердого вещества.
C.
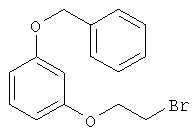
К раствору соединения из части В (2.4 г; 9.8 ммоль) и Ph3Р (3.1 г; 14.7 ммоль) в СН2Cl2 добавляют CBr4 (4.80 г; 14.7 ммоль). Реакционную массу перемешивают при комнатной температуре в течение ночи, затем концентрируют под вакуумом. Остаток хроматографируют (SiO2; 95:5, гексан:EtOAc), что дает соединение, часть С (2.8 г; 93%) в виде белого твердого вещества.
D.
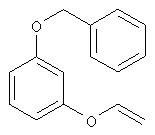
Смесь соединения из части С (310 мг; 1.0 ммоль) и трет-бутилата калия (113 мг; 2.0 ммоль) в толуоле (20 мл) нагревают до 105°С в течение 20 минут. Вводят дополнительное количество KO-t-Bu (56 мг; 1.0 ммоль) и реакцию нагревают при 105°С в течение еще 10 минут. Смесь охлаждают до комнатной температуры и разделяют между Н2О (100 мл) и EtOAc (100 мл). Органическую фазу промывают H2O (2×100 мл), сушат (Na2SO4) и концентрируют под вакуумом. Реакцию повторяют с дополнительным количеством соединения, часть С (500 мг; 1.63 ммоль) и KO-t-Bu (182 мг; 16 ммоль). Объединенные сырые продукты реакции хроматографируют (SiO2; гексан), что дает соединение, часть D (590 мг; 89%) в виде бесцветного масла.
E.
К охлажденному до 0°С раствору соединения из части D (1.4 г; 62 ммоль) в DCE (100 мл) добавляют по каплям чистый диэтиловый цинк (1.6 мл; 16 ммоль), а затем ICH2Cl (5.46 г; 31 ммоль).Реакционной смеси дают нагреться до комнатной температуры и перемешивают при комнатной температуре в течение ночи, затем промывают 1 М водным раствором HCl. Органическую фазу сушат (Na2SO4) и концентрируют под вакуумом. Сырой остаток хроматографируют дважды (SiO2; гексан), что дает соединение, часть Е (510 мг; 30%), кроме того, выделяют исходный продукт - соединение, часть D (250 мг; 18%).
F.

К охлажденному до -78°С раствору соединения из части Е (510 мг; 2.2 ммоль) в жидком NH3 (30 мл) добавляют Na (500 мг; 22 ммоль). Темно-голубой раствор перемешивают при -78°С в течение 4 часов, затем дают нагреться до комнатной температуры в течение ночи. Образовавшийся твердый остаток разделяют между 1 М водным раствором HCl и EtOAc (50 мл каждый). Органическую фазу сушат (Na2SO4) и концентрируют под вакуумом. Сырой продукт хроматографируют (SiO2; 9:1, гексан:EtOAc), что дает соединение, часть F (240 мг; 75%) в виде желтого масла.
G.
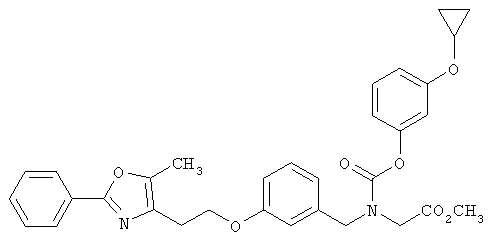
К раствору соединения из части F (150 мг; 1.0 ммоль) в ДМФА (10 мл) последовательно добавляют KO-t-Bu (112 мг; 1.0 ммоль) и раствор карбамоилхлорида (44 мг; 1.0 ммоль) в ДМФА (0.5 мл).
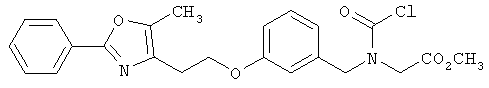
(получают, как раскрыто в примерах 5 и 139).
Реакционную массу перемешивают при комнатной температуре в течение 15 мин, после чего анализ ВЭЖХ показывает, что весь исходный продукт вступил в реакцию. Смесь разделяют между H2O и EtOAc (100 мл каждый). Органическую фазу промывают H2O (2×100 мл), сушат (Na2SO4) и концентрируют под вакуумом. Сырой продукт хроматографируют (SiO2; 9:1, гексан:EtOAc), что дает соединение, часть G, с примесями в виде желтого масла.
Н.
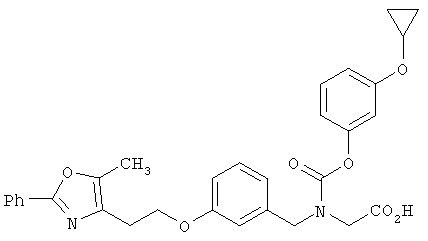
Раствор соединения из части G (556 мг; 1.0 ммоль) и LiOH·H2O (116 мг; 2.8 ммоль) в (10:1) метаноле: H2О (10 мл) перемешивают при комнатной температуре в течение 2 часов. Летучие продукты удаляют под вакуумом и остаток подкисляют до рН 2 водным раствором 1 М HCl, затем экстрагируют EtOAc (3×40 мл). Объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом. Сырой продукт очищают препаративной ВЭЖХ (YMC S5 ODS 50×250 мм колонка; скорость потока 25 мл/мин; непрерывный 20 мин градиент от 70:30 В:А до 100% В, где А=90:10:0.1 H2O:метанол:ТФК и В=90:10:0.1 метанол:H2O:ТФК), что дает продукт (120 мг; 30% на 2 стадиях) в виде бесцветного масла. [М+Н]+=543.2.
Пример 475
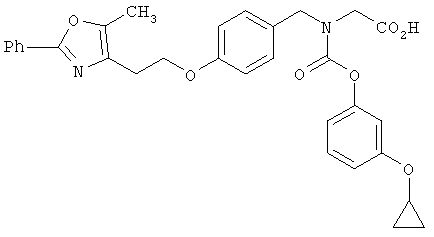
Названное соединение синтезируют из соединения примера 474, часть F (150 мг; 1.0 ммоль) и карбамоилхлорида (440 мг; 1.0 ммоль)
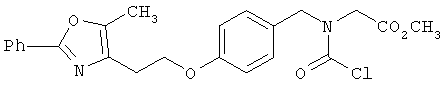
(получают, как раскрыто в примерах 6 и 139), с последующим гидролизом с помощью LiOH·H2O аналогично примеру 474. Названное соединение выделяют, очищают и получают в виде бесцветного масла (340 мг; 92% на 2 стадиях). [М+Н]+=543.3.
Следуя процессам, раскрытым выше, получают соединения по примерам с 476 по 494.
Примеры с 476 по 484
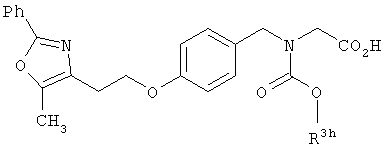


Примеры с 485 по 494
Пример 492
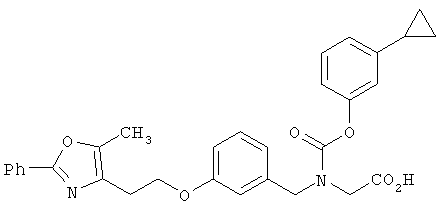
Соединение по примеру 492 синтезируют согласно процессам, раскрытым выше.
1H ЯМР (CDCl3; 400 МГц): δ 0.68 (т, J=4.4 Гц; 2Н), 0.94 (т, J=4.4 Гц; 2Н), 1.87 (м, 1Н), 2.42 (с, 3H), 3.06 (с, 2Н), 4.02 (т, J=5.2 Гц, 2Н), 4.22 (т, J=5.2 Гц, 2Н), 4.60 (2 пика, 2Н), 6.84-6.89 (м, 4Н), 7.15-7.26 (м, 4Н), 7.40-7.47 (м, 3H), 7.98-8.00 (м, 2Н).
Требуемые (коммерчески недоступные) фенолы и хлорформиаты для синтеза аналогов карбаматокислоты получают следующим образом.
3-Фтор-4-метилфенилхлорформиат
A.
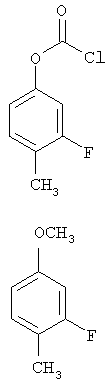
Смесь 5-метокси-2-метиланилина (5.0 г; 36 ммоль), HCl (7.6 мл 12 М раствора; 91 ммоль) и H2O (11 мл) нагревают до 60°С в течение 15 мин, пока завершается полное растворение. Реакцию охлаждают до 0°С и добавляют по каплям водный раствор NaNO2 (2.5 г; 36 ммоль) (внутренняя температура <7°С). Перемешивают при 0°С в течение 30 мин и осторожно добавляют охлажденный до 0°С раствор HBF4 (5.3 мл 48% раствора; 40 ммоль). Реакцию перемешивают при 0°С в течение 20 мин и полученное коричневое твердое вещество фильтруют, промывают льдом с водой (3×10 мл) и H2O (2×10 мл). Твердое вещество сушат под высоким вакуумом в течение 20 часов, затем нагревают (нагревательный пистолет), пока выделение BF3 (белый дым) не прекращается. Полученное коричневое масло разделяют между EtOAc и H2O. Органическую фазу сушат (Na2SO4), концентрируют под вакуумом и перегоняют (по Kugelrohr), что дает 3-фтор-4-метиланизол (1.6 г; 31%) в виде бесцветного масла.
B.
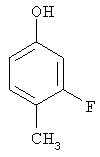
К охлажденному до -70°С раствору 3-фтор-4-метиланизола (1.62 г; 11.6 ммоль) в СН2Cl2 (10 мл) добавляют по каплям BBr3 (10 мл; 12 ммоль). Реакционную смесь перемешивают при -70°С в течение 10 мин, затем дают нагреться до 0°С и перемешивают при 0°С в течение 2 часов. Реакции дают нагреться до комнатной температуры и концентрируют под вакуумом, а остаток разделяют между H2O и EtOAc. Органическую фазу промывают H2O, сушат (Na2SO4) и концентрируют под вакуумом, что дает 3-фтор-4-метилфенол (1.1 г; 75%) в виде масла.
C.
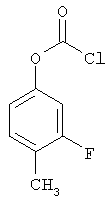
Раствор 3-фтор-4-метилфенола (1.1 г; 8.7 ммоль), фосгена (5.9 мл 1.93 М раствора в толуоле; 8.7 ммоль), ДМФА (10 мкл) и N,N-диметиланилина (1.27 г; 8.7 ммоль) в хлорбензоле (10 мл) в герметично закрываемой трубке перемешивают при комнатной температуре в течение 2 часов, затем при 80°С в течение 2 часов, охлаждают до комнатной температуры, перемешивают при комнатной температуре в течение 1 часа, затем концентрируют под вакуумом. Остаток перегоняют по Kugelrohr, чтобы получить 3-фтор-4-метилфенилхлорформиат (800 мг; 49%) в виде прозрачного масла.
3-Хлор-4-метилфенилхлорформиат
3-Хлор-4-метилфенилхлорформиат (600 мг; 45% общий для 2 стадий) синтезируют из 3-хлор-4-метиланизола (1.0 г), используя то же путь (BBr3-инициируемое расщепление метилового эфира, следующее за обработкой фосгеном), как описано выше.
3-Бром-4-метилфенилхлорформиат
К охлажденной до 0°С смеси 3-бром-4-метиланилина (5 г; 27 ммоль) и Н2SO4 (5.5 мл 16 М раствора) в H2O (7.5 мл) добавляют по каплям водный раствор NaNO2 (1.93 г; 28 ммоль в 7.5 мл H2O). Реакционную массу перемешивают при 0°С в течение 30 минут, затем нагревают до 50°С в течение 2 часов, затем охлаждают до комнатной температуры и экстрагируют EtOAc (2x). Объединенные органические экстракты промывают H2O, сушат (Na2SO4) и концентрируют под вакуумом, что дает 3-бром-4-метилфенол (1.72 г; 34%) в виде масла. Полученный фенол превращают в 3-бром-4-метилфенилхлорформиат (1.9 г; 82%), следуя тому же процессу (фосген/диметиланилин/нагревание), как и для 3-фтор-4-метилфенилхлорформиата, приведенного выше.
2-Метоксифенилхлорформиат (1.5 г) и 3-метоксифенилхлорформиат (1.5 г) оба синтезируют тем же путем, что и 3-фтор-4-метилфенилхлорформиат (фосген/диметиланилин/нагревание) из 2-метоксифенола (2 г) и 3-метоксифенола (2 г) соответственно.
3-Хлор-4-метоксифенол
К охлажденному до 0°С раствору 3-хлор-4-метоксианилина (1.0 г; 6.4 ммоль) в (1:1) H2O: конц. H2SO4 (100 мл) добавляют очень медленно раствор NaNO2 (0.5 г; 7.6 ммоль) в H2O (10 мл). Появляется густой желтый дым и черный раствор затем нагревают до кипения с обратньм холодильником в течение 30 мин. Смесь экстрагируют EtOAc (4×50 мл) и объединенные экстракты концентрируют под вакуумом. Остаток хроматографируют (SiO2; 4:1, гексан:EtOAc), чтобы получить 3-хлор-4-метоксифенол (300 мг; 30%) в виде желтого масла.
3-Фтор-4-метоксифенол
Раствор 3'-фтор-4'-метоксиацетофенона (10 г; 59 ммоль) и мета-хлорпербензойной кислоты (50% чистота; 30 г; 89 ммоль) в CH2Cl2 (300 мл) перемешивают при комнатной температуре в течение ночи. Раствор промывают насыщенным водным раствором Na2CO3, затем фильтруют через подушку из SiO2 (CH2Cl2 как элюент) и наконец хроматографируют (SiO2; гексан:EtOAc, 4:1), что дает сырой продукт (3'-фтор-4'-метоксифенилацетат; 63 г). Раствор указанного сырого продукта и LiOH.H2O (5 г; 120 ммоль) в метаноле:H2O (100 мл, смесь 9:1) перемешивают при комнатной температуре в течение ночи. Летучие продукты удаляют под вакуумом и остаток разделяют между избытком водного раствора 1 М HCl и EtOAc (водный раствор (слой) рН˜3). Водную фазу экстрагируют EtOAc (2х). Объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом, что дает 3-фтор-4-метоксифенол (6.1 г; 72%) в виде масла.
3-Бром-4-метоксифенол (4.39 г; 47% для 2 стадий) синтезируют, используя по сути аналогичную последовательность, исходя из 3-бром-4-метоксибензальдегида.
3-Пропилфенол
A.
Смесь 3-йоданизола (2 г; 8.5 ммоль), триметилсилилацетилена (1.67 г; 17 ммоль), Cul (32 мг; 0.17 ммоль) и (Ph3Р)2PdCl2 (59 мг; 0.085 ммоль) в диэтиламине (10 мл) перемешивают при комнатной температуре в течение 1 часа. Летучие продукты удаляют под вакуумом и остаток разделяют между EtOAc и рассолом. Органическую фазу промывают рассолом (2×10 мл) и затем фильтруют через подушку из SiO2. Летучие продукты удаляют под вакуумом, что дает сырой продукт (3-триметилсилилэтиниланизол) в виде светло- желтого масла. Раствор указанного сырого продукта и тетрабутиламмонийфторид (6.6 г; 26 ммоль) в ТГФ (10 мл) перемешивают при комнатной температуре в течение 15 минут. Летучие продукты удаляют под вакуумом и остаток хроматографируют (SiO2; 9:1, гексан:EtOAc), что приводит к соединению из части А (1.0 г; 89%) в виде желтого масла.
B.

К охлажденному до 0°С раствору соединения из части А (1.0 г; 7.6 ммоль) в безводном ТГФ (5 мл) добавляют по каплям n-BuLi (4.5 мл 2.0 М раствора в гексане; 9.1 ммоль). Полученный желтый раствор перемешивают при 0°С в течение 30 минут. Затем добавляют метилиодид (1.6 г; 11.4 ммоль) и дают нагреться до комнатной температуры, а затем перемешивают при комнатной температуре в течение 30 мин. Летучие продукты удаляют под вакуумом и остаток разделяют между водным раствором 1 н. HCl и EtOAc. Водную фазу экстрагируют EtOAc (3×20 мл), объединенные органические экстракты сушат (MgSO4) и концентрируют под вакуумом, что дает соединение, часть В (1,0 г; 92%) в виде желтого масла.
C. 
Раствор соединения из части В (1.0 г) в метаноле (5 мл) перемешивают над 10% Pd/C (10 мг) в атмосфере Н2 в течение ночи. Катализатор удаляют фильтрованием через подушку Celite® и фильтрат концентрируют под вакуумом, что дает соединение, часть С (1.0 г; 100%) в виде желтого масла.
D. 
К охлажденному до -78°С раствору соединения из части С (1.0 г; 6.6 ммоль) в СН2Cl2 (10 мл) добавляют BBr3 (4.8 мл 1 М раствора в СН2Cl2), дают нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 3 часов, после чего осторожно разделяют между водным раствором 1 М HCl и СН2Cl2. Органическую фазу промывают водным раствором NH4Cl, сушат (MgSO4) и концентрируют под вакуумом, что дает 3-пропилфенол (900 мг; 100%) в виде желтого масла.
Пример 495
A. 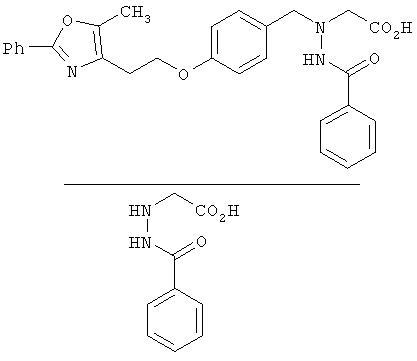
Смесь бензойной кислоты (1.22 г; 10 ммоль); метансульфонилхлорида (1.15 г; 10 ммоль), К2СО3 (5.52 г; 40 ммоль) и бензилтриэтиламмонийхлорида (0.23 г; 1 ммоль) в толуоле перемешивают при 80°С в течение 2 часов. Затем добавляют этилгидразинацетатгидрохлорид (1.55 г; 10 ммоль) и реакционную массу охлаждают в течение последующих 30 мин, затем охлаждают до комнатной температуры. Твердое вещество отфильтровывают и фильтрат концентрируют под вакуумом. Остаток хроматографируют (SiO2; постепенный градиент от 3:1 до 1:1, гексан:EtOAc), что дает соединение из части А (350 мг; 16%) в виде белого твердого вещества.
B. 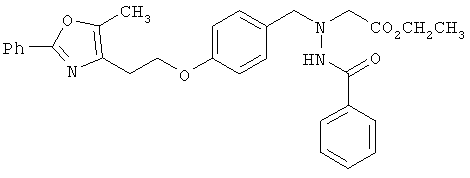
К охлажденному до 0°С раствору соединения из части А (49 мг; 0.22 ммоль) и альдегида (50 мг; 010 ммоль)
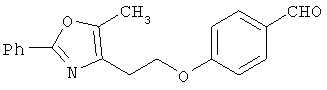
в DCE (3 мл) добавляют NaBH(ОАс)3 (30 мг; 0.42 ммоль), дают нагреться до комнатной температуры, а затем перемешивают при комнатной температуре в течение 2 часов, затем при 60°С в течение 18 часов. Смесь охлаждают до комнатной температуры и концентрируют под вакуумом. Остаток очищают препаративной ВЭЖХ (YMC S5 ODS 30×250 мм колонка; скорость потока 25 мл/мин; 20 мин непрерывный градиент от 70:30 В:А до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O:ТФК), что дает соединение из части В.
C. 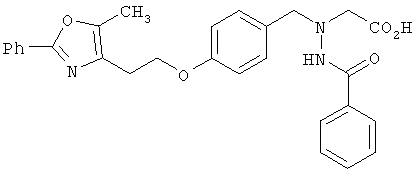
Раствор сырого соединения из части В в ТГФ (1 мл) и водный раствор LiOH (0.3 мл 1 М раствора; 0.3 ммоль) перемешивают при комнатной температуре в течение 3 часов, затем подкисляют до рН ˜3 водным раствором 1 М HCl. Водную фазу экстрагируют EtOAc (2x); объединенные органические экстракты концентрируют под вакуумом. Остаток очищают препаративной НРС (YMC S5 ODS 30×250 мм колонка; скорость потока 25 мл/мин; 22 мин непрерывный градиент от 70:30 В:А до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В = 90:10:0.1 метанол:H2O:ТФК), что дает названное соединение (26 мг; 33% выход после 2 стадий) в виде белого твердого вещества. [М+Н]+=486.3.
Пример 496
A. 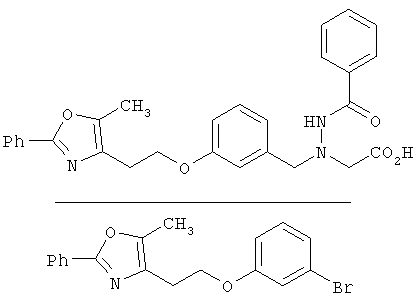
К охлажденному до 0°С раствору альдегида (200 мг; 0.65 ммоль)
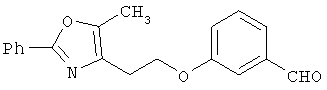
в метаноле (2 мл) добавляют порциями NaBH4 (24 мг; 0.65 ммоль), после чего реакции дают нагреться до комнатной температуры, а затем перемешивают при комнатной температуре в течение 1 часа. Летучие продукты удаляют под вакуумом и остаток разделяют между H2O и EtOAc. Органическую фазу сушат (Na2SO4) и концентрируют под вакуумом, что дает промежуточный спирт в виде масла. Раствор спирта в CH2Cl2 (2 мл) и PBr3 (1 мл 1 М раствора в CH2Cl2) перемешивают при комнатной температуре в течение 30 минут. Летучие продукты удаляют под вакуумом и остаток разделяют между насыщенным водным раствором NaHCO3 и EtOAc. Органическую фазу промывают H2O, сушат (Na2SO4) и концентрируют под вакуумом, что дает соединение из части А (150 мг; 62%) в виде масла.
B. 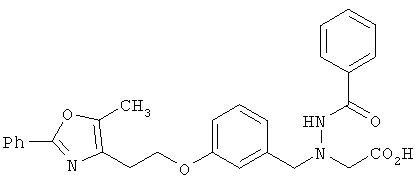
Раствор соединения из части А (42 мг; 0.11 ммоль) примера 500, соединение из части А (25 мг; 0.11 ммоль) и К2СО3 (100 мг; 0.71 ммоль) в ДМФА (1 мл) перемешивают при комнатной температуре в течение 3 дней. Реакционную смесь разделяют между EtOAc и H2O. Органическую фазу промывают H2O (2х) и концентрируют под вакуумом. Оставшееся масло растворяют в ТГФ (1 мл) и добавляют водный раствор LiOH (0.3 мл 1 М раствора). Реакционную массу перемешивают при комнатной температуре в течение 3 часов, затем подкисляют до рН ˜3 водным раствором 1 М HCl. Водную фазу экстрагируют EtOAc (2х); объединенные органические экстракты концентрируют под вакуумом. Остаток очищают препаративной НРС (YMC S5 CDS 30×250 мм колонка; скорость потока 25 мл/мин; 20 мин непрерывный градиент от 70:30 В:А до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O:ТФК), что дает названное соединение (15 мг; 27% выход после 2 стадий) в виде белого твердого вещества. [М+Н]+=486.4.
Пример 497
A. 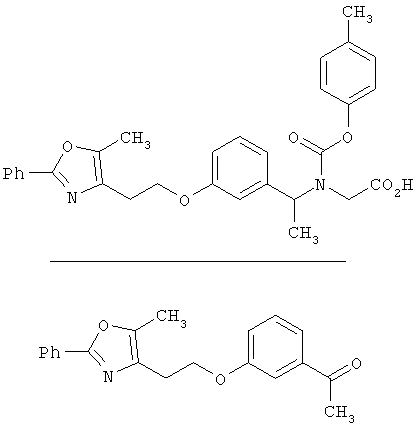
Смесь 3-гидроксиацетофенона (650 мг; 4.78 ммоль), К2СО3 (660 мг; 4.78 ммоль) и 2-фенил-5-метил-4-оксазолэтанолмезилата (1.12 г; 3.98 ммоль)
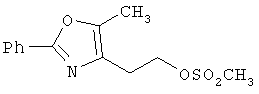
в MeCN (40 мл) нагревают с обратным холодильником в течение ночи. Летучие продукты удаляют под вакуумом и остаток разделяют между EtOAc (100 мл) и 1.0 М водным раствором NaOH (80 мл). Органическую фазу промывают рассолом (100 мл), сушат (MgSO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; гексан:EtOAc, 3:1), что дает соединение из части А (850 г; 67%) в виде желтого твердого вещества.
B.
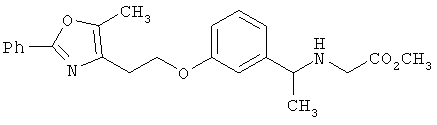
К раствору соединения из части А (850 мг, 2.65 ммоль) в DCE (15 мл) последовательно добавляют гидрохлорид глицинметилового эфира (333 мг, 2.65 ммоль), Et3N (554 мкл, 4.0 ммоль), NaBH(ОАс)3 (786 мг; 3.7 ммоль) и уксусной кислоты (152 мкл; 2.65 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 6 дней, затем разделяют между водным раствором 1 М NaOH и CH2Cl2. Водную фазу промывают H2O, затем концентрируют под вакуумом. Остаток разделяют между EtOAc и 1 М водным раствором HCl. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом, что дает возвращенный исходный продукт (соединение из части А). Значение рН водного раствора HCl фазы доводят до 10 избытком твердого вещества NaOH. Полученную водную фазу экстрагируют EtOAc (60 мл). Органический экстракт промывают рассолом (60 мл), сушат (MgSO4) и концентрируют под вакуумом, что дает сырое соединение, часть В (400 мг; 39%) в виде масла, которое используют на следующей стадии без дальнейшей очистки.
C. 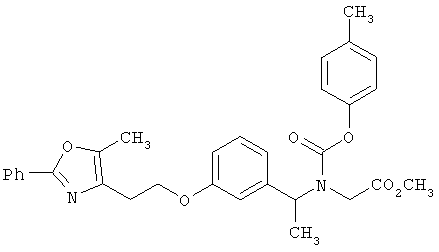
К раствору соединения из части В (29 мг; 0.074 ммоль) в пиридине (1.0 мл) добавляют 4-толилхлорформиат (14 мкл; 0.089 ммоль) и DMAP (10 мг). Раствор нагревают до 61°С в течение 2часов, затем охлаждают до комнатной температуры и концентрируют под вакуумом, что дает сырое соединение, часть С (36 мг) в виде сиропа.
D.
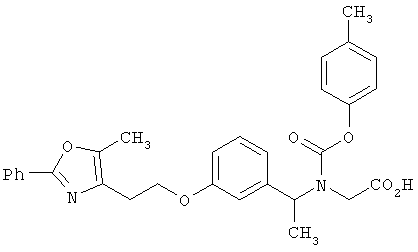
Раствор сырого соединения из части С (36 мг; 0.68 ммоль) и LiOH·H2O (12 мг; 0.28 ммоль) в ТГФ:метанол:H2O (1 мл 1:1:1 раствора) перемешивают при комнатной температуре в течение 2 часов. Летучие продукты удаляют под вакуумом и остаток подкисляют до рН 2 водным раствором 1 М HCl, затем экстрагируют EtOAc (3×40 мл). Объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом. Сырой продукт очищают препаративной ВЭЖХ (YMC S5 ODS 50×250 мм колонка; скорость потока 25 мл/мин; непрерывный 20 мин градиент от 70:30 В:А до 100% В, где А=90:10:0.1 H2O:метанол:ТФК и В=90:10:0.1 метанол:H2O:ТФК), что дает названное соединение (28 мг; 72% после 2 стадий) в виде белого твердого вещества. [М+H]+=515.3.
Пример 498

A.
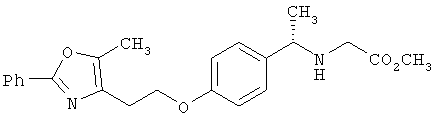
Раствор (S)-1-(4-метоксифенил)этиламина (11.9 г, 79 ммоль), метилбромацетата (11.5 г; 75 ммоль) и Et3N (12.6 мл; 90 ммоль) в ТГФ (156 мл) перемешивают при комнатной температуре в течение 15 часов. Реакционную массу разделяют между EtOAc и H2O. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом, что дает сырое соединение, часть А, которое используют на следующей стадии без дополнительной очистки.
B.
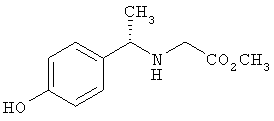
К охлажденному до 0°С раствору сырого соединения, часть А, полученного выше, в СН2Cl2 (198 мл) медленно добавляют по каплям BBr3 (12.0 мл; 127 ммоль), перемешивают при 0°С в течение 3 часов, затем выливают осторожно в охлажденную до 0°С смесь насыщенного водного раствора NH4Cl и EtOAc. Водную фазу нейтрализуют путем медленного добавления твердого NaHCO3, затем экстрагируют EtOAc (2х). Объединенные органические экстракты промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом, что приводит к соединению, часть В (7.29 г; 44% после 2 стадий).
C.
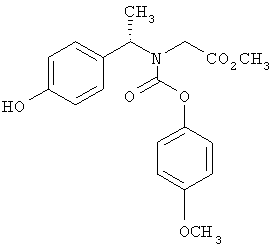
К раствору соединения из части В (6.13 г; 29.3 ммоль) в диоксане:H2O (98 мл 1:1 раствора) последовательно добавляют NaHCO3 (3.2 г; 38 ммоль) и 4-метоксифенилхлорформиат (3.92 мл; 26.4 ммоль). Реакционную массу перемешивают при комнатной температуре в течение 2 часов, затем разделяют между EtOAc и H2O. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом, что дает сырое соединение, часть С (10.0 г; 95%).
D.
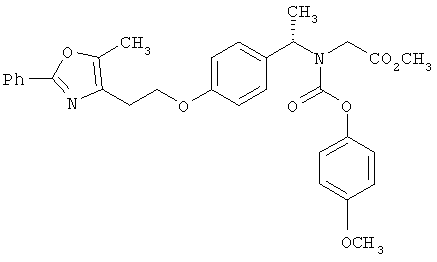
К раствору соединения из части С в MeCN (59 мл) последовательно добавляют К2СО3 (2.43 г; 17.6 ммоль) и мезилат (4.93 г; 17.6 ммоль).
Смесь нагревают до 90°С в течение 20 часов, затем охлаждают до комнатной температуры. Смесь разделяют между EtOAc и H2O. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; постепенный градиент от 8:1 - 3:1 до 1:1, гексан:EtOAc), что дает соединение, часть D (3.4 г; 36%).
E. 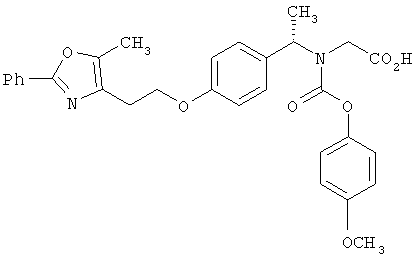
К раствору соединения из части D (3.4 г; 6.25 ммоль) в ТГФ:H2O (31 мл 2:1 раствора) добавляют LiOH·HiO (0.525 г; 125 ммоль). Реакционную массу перемешивают при комнатной температуре в течение ночи (14 часов). Добавляют EtOAc и раствор подкисляют 1 н. раствором HCl до рН ˜2. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом. Остаток очищают препаративной ВЭЖХ (YMC S5 ODS 30×250 мм колонка; скорость потока 25 мл/мин; 22 мин непрерывный градиент от 70:30 В:А до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O:ТФК; время задержки=17.8 мин), что дает названное соединение (2.1 г; 63% выход) в виде белого твердого вещества. [М+Н]+=531.3.
1H ЯМР (ДМСО-d6; 400 МГц): δ 1.50 (2д, J=6.6 Гц; 3H), 2.37 (с, 3H), 2.94 (т, J=7.0 Гц, 2Н), 3.74 (с, 3H), 3.81 (м; 2Н), 4.21 (т, J=6.2 Гц, 2Н), 5.36 (м, 1H), 6.93 (м, 6Н), 7.28 (м, 2Н), 7.50 (м, 3H), 7.91 (м, 2Н).
Пример 499
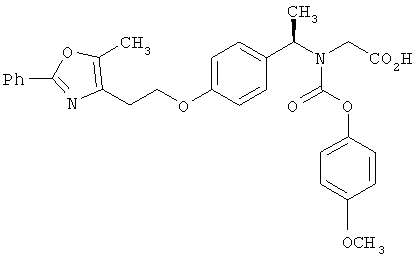
Синтез названного соединения осуществляют, используя ту же последовательность, которая раскрыта в примере 498, за исключением того, что (R)-4-метокси-α-метилбензиламин используют вместо (S) изомера. [М+Н]+=531.3.
1H ЯМР (ДМСО-d6; 400 МГц): δ 1.50 (2д, J=7.0 Гц; 3H), 2.37 (с, 3H), 2.94 (т, J=6.6 Гц, 2Н), 3.74 (с, 3H), 3.84 (м; 2Н). 4.21 (т, J = 6.6 Гц, 2Н), 5.35 (м, 1Н), 6.93 (м, 6Н), 7.29 (м, 2Н), 7.50 (м, 3H), 7.91 (м, 2Н).
Пример 500
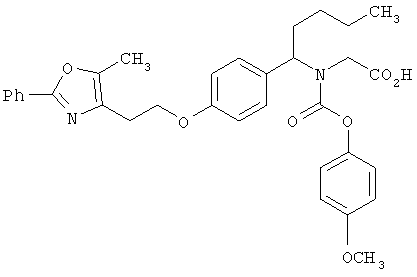
A.
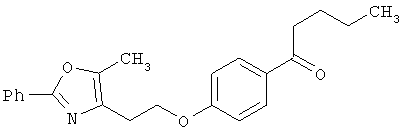
Смесь 4-гидроксифенилбутилкетона (2.50 г; 14.0 ммоль), 2-фенил-5-метилоксазол-4-этанолмезилата (3.30 г; 11.7 ммоль) и К2СО3 (1.94 г; 14.0 ммоль) в ацетонитриле (50 мл) нагревают с обратным холодильником в атмосфере Ar в течение 18 часов. Летучие продукты удаляют под вакуумом и остаток разделяют между H2O и EtOAc. Водную фазу экстрагируют EtOAc. Объединенные органические экстракты промывают водным раствором 1М NaOH и H2O, сушат (MgSO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; постепенный градиент от 3:1 до 9:1, гексан:EtOAc), что дает соединение из части А (3.42 г; 80%) в виде белого твердого вещества.
B. 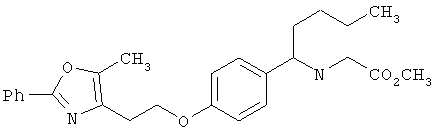
Смесь соединения из части А (3.42 г; 9.42 ммоль), глицинметиловый эфир, HCl соль (1.18 г; 9.42 ммоль), Et3N (1.97 мл; 14.1 ммоль), NaBH(OAc)3 (2.80 г; 13.2 ммоль) и НОАс (0.54 мл; 9.42 ммоль) в DCE (20 мл) перемешивают при комнатной температуре 6 дней. За это время реакция еще не завершена, но и не протекает дальше. Ее захолаживают насыщенным водным раствором NaHCO3 (6 мл), затем концентрируют под вакуумом. Остаток разделяют между насыщенным водным раствором NaHCO3 и EtOAc. Органическую фазу промывают насыщенным водным раствором NaHCO3 и H2O, затем экстрагируют 1 М водным раствором HCl (непрореагировавший исходный продукт остается в органической фазе). Водную фазу подщелачивают NaOH, затем экстрагируют EtOAc. Органическую фазу промывают H2O и рассолом, сушат (MgSO4) и концентрируют под вакуумом, что дает соединение, часть В (365 мг; 9%) в виде масла.
C. 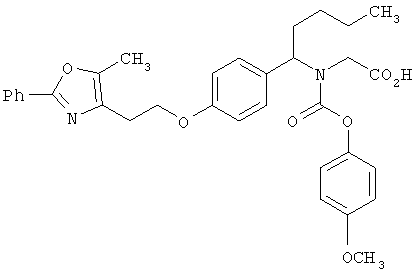
К раствору соединения из части С (50 мг; 0.11 ммоль) в пиридине (1 мл) добавляют 4 -метоксифенилхлорформиат (40 мкл) и DMAP (5 мг). Реакционную смесь нагревают до 60°С в течение 6 часов, затем охлаждают до комнатной температуры и летучие продукты удаляют под вакуумом. Остаток растворяют в ТГФ/метаноле/H2O (1 мл, 2:2:1 смесь) и добавляют LiOH (30 мг). Реакционную массу перемешивают при комнатной температуре в течение 18 часов, затем подкисляют водным раствором 1 М HCl до рН 2. Смесь экстрагируют EtOAc (30 мл), промывают H2O и рассолом (15 мл каждого), сушат (MgSO4) и концентрируют под вакуумом, что дает сырой продукт. Его очищают препаративной ВЭЖХ (YMC S5 ODS 30×250 мм колонка; непрерывный градиент от 60:40 А:В до 100% В через 30 мин), что дает, после лиофилизации из метанола/H2O, названное соединение (52 мг; 79%) в виде белого твердого вещества. [М+H]+=573.3.
Пример 501
A. 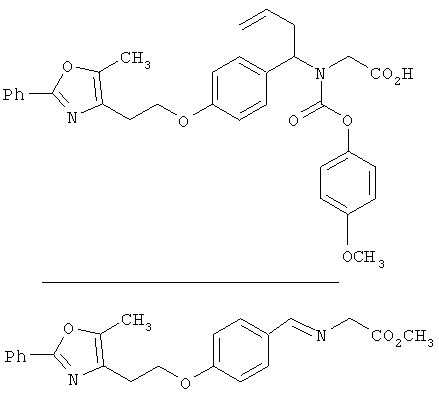
Смесь гидрохлорида глицинметилового эфира (245 мг; 1.95 ммоль), Et3N (271 мкл; 1.95 ммоль), альдегида
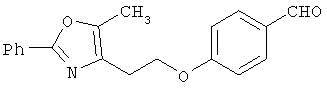
(400 мг; 1.3 ммоль) и безводного MgSO4 (200 мг) в ТГФ (4 мл) перемешивают при комнатной температуре в течение ночи, затем фильтруют. Фильтрат концентрируют под вакуумом, что дает сырое соединение из части А, которое используют на следующей стадии без дальнейшей очистки.
B. 
Смесь металлического индия (448 мг; 3.9 ммоль) и аллилбромида (334 мкл; 3.9 ммоль) в безводном ДМФА (2 мл) перемешивают при 0°С в течение 50 мин. Добавляют раствор сырого соединения из части А (см. выше) в безводном ДМФА (2 мл) к указанной смеси и реакцию энергично перемешивают при комнатной температуре в течение 3 часов. Анализ ВЭЖХ/MS показывает, что реакция завершена именно в этот момент. Реакционную массу разделяют между насыщенным водным раствором NH4Cl и EtOAc. Органическую фазу промывают H2O (образуется эмульсия) и рассолом, сушат (MgSO4) и концентрируют под вакуумом, что дает сырое соединение, часть В (300 мг; 55% для 2 стадий). Этот продукт используют на следующей стадии без дальнейшей очистки.
C. 
К охлажденному до 0°С раствору соединения из части В (150 мг; 0.36 ммоль) и Et3N (51 мкл; 0.36 ммоль) в CH2Cl2 (4 мл) добавляют по каплям 4-метоксифенилхлорформиат (53 (мкл; 0.36 ммоль), дают нагреться до комнатной температуры и затем ее перемешивают при комнатной температуре в течение 1 часа, а затем концентрируют под вакуумом. Остаток хроматографируют (SiO2; гексан:EtOAc, 2:1), что дает соединение, часть С (200 мг; 98%) в виде масла.
D. 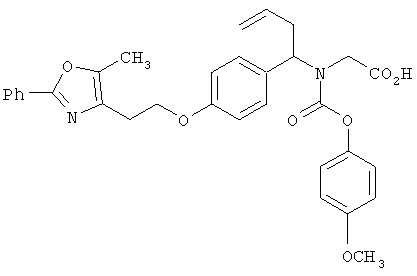
Раствор соединения из части С (100 мг, 0.18 ммоль) и LiOH·H2O (30 мг, 0.72 ммоль) в ТГФ:метаноле:H2O (1 мл, 1:1:1 раствора) перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь затем подкисляют до рН˜2 водным раствором 1 н. HCl. Водную фазу экстрагируют EtOAc (2х). Объединенные органические экстракты сушат (Na2SO4), концентрируют под вакуумом и лиофилизуют из диоксана, что обеспечивает названное соединение (80 мг; 82%) в виде белого твердого вещества. [М+Н]+=557.2.
Пример 502
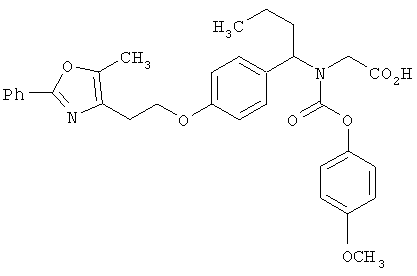
A.

Раствор соединения из примера 501, часть С (100 мг; 0.18 ммоль) в метаноле (10 мл) в присутствии 10% Pd/C (50 мг) перемешивают в атмосфере Н2 в течение 2 часов при комнатной температуре. Затем катализатор отфильтровывают, используя подушку из Celite®. Фильтрат концентрируют под вакуумом, что дает соединение из части А (100 мг; 100%) в виде масла.
B.
Названное соединение (87 мг; 90%; белый твердый лиофилат) получают из соединения из части А так, как в примере 501, соединение синтезируют из соединения по примеру 501, часть С. [М+Н]+=559.2.
Пример 503
A. 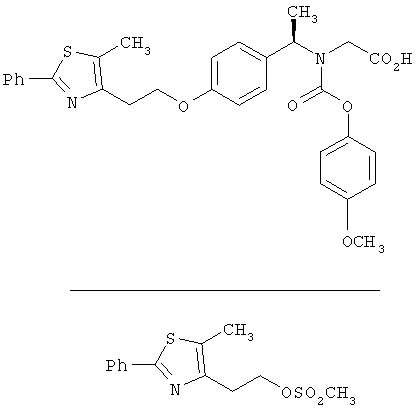
К раствору 5-метил-2-фенилтиазол-4-илэтанола (50 мг; 0.23 ммоль) в CH2Cl2 (3 мл) последовательно добавляют Et3Н (50 мкл; 0.36 ммоль) и метансульфонилхлорид (20 мл; 0.26 ммоль). Реакционную массу перемешивают при комнатной температуре в течение 2 часов, затем разделяют между СН2Cl2 и водным раствором 1 М HCl. Органическую фазу промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом, что дает соединение из части А (68 мг; 100%) в виде бесцветного масла. Этот продукт используют на следующей стадии без дальнейшей очистки.
B. 
Смесь фенола (получают, используя идентичный процесс, как раскрыто в примере 498 для синтеза соединения, часть С, за исключением того, что этилбромацетат используют вместо метилбромацетата)
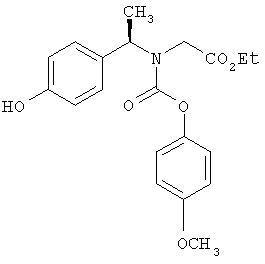
(18 мг; 0.048 ммоль) и К2СО3 (30 мг; 0.22 моль) в MeCN (2 мл) нагревают до 60°С в течение ночи, затем охлаждают до комнатной температуры и разделяют между EtOAc и избытком водного раствора 1 М HCl. Водную фазу экстрагируют с помощью EtOAc (2х); объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом. Остаток очищают препаративной ВЭЖХ (как раскрыто в примере 498), что обеспечивает соединение, часть В (12 мг; 43%).
C.
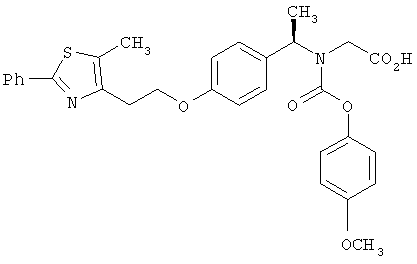
Раствор соединения из части В (12 мг; 0.02 ммоль) и LiOH·H2O (10 мг; 0.24 ммоль) в ТГФ (2 мл) и H2O (1 мл) перемешивают при комнатной температуре в течение 4 часов. Реакционную смесь подкисляют избытком водного раствора 1 М HCl и экстрагируют с помощью EtOAc (3x). Объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом; остаток очищают препаративной ВЭЖХ (как раскрыто в примере 498), что дает названное соединение (3 мг; 26%) в виде бесцветного масла. [М+Н]+=547.2.
Пример 504
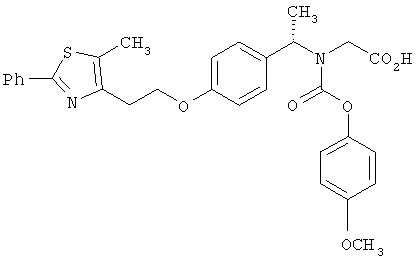
Названное соединение получают точно так же, как описано в примере 503, за исключением того, что [S]-энантиомер соединение А (100 мг; 47%) получают в виде масла. Этот полученный продукт используют на следующей стадии без дальнейшей очистки.
B. 
Раствор соединения из части А (100 мг; 0.61 ммоль), метилбромацетата (103 мг; 0.67 ммоль) и Et3N (102 мкл; 0.73 ммоль) в ТГФ перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь разделяют между EtOAc и H2O. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; СН2Cl2:метанол, 9:1), что дает соединение, часть В (90 мг; 62%) в виде масла.
C. 
К охлажденному до 0°С раствору соединения из части В (90 мг; 0.38 ммоль) в СН2Cl2 (12.7 мл) медленно добавляют чистый BBr3 (82 мкл; 0.87 ммоль). Реакционную массу перемешивают при 0°С в течение 3 часов, затем разделяют между охлажденным льдом насыщенным водным раствором NH4Cl и EtOAc. Органическую фазу отделяют и водный слой нейтрализуют добавлением NaHCO3, затем экстрагируют с помощью EtOAc (2х). Объединенные органические экстракты промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом, что дает соединение, часть С (50 мг; 59%).
D. 
Смесь соединения из части С (50 мг; 0.22 ммоль), 4-метоксифенилхлорформиат (33 мг; 0.22 моль) и NaHCO3 (25 мг; 0.29 ммоль) в (1:1) водном растворе диоксана (7.5 мл) перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь разделяют между EtOAc и H2O. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом, что дает соединение, часть D (45 мг; 52%).
E. 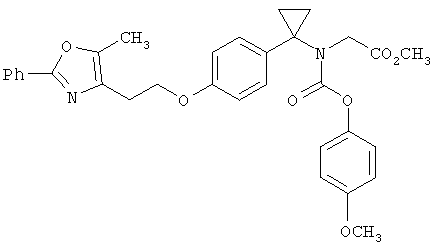
Смесь соединения из части D (45 мг; 0.12 ммоль), К2СО3 (30 мг; 0.22 моль) и мезилат (33 мг; 0.12 ммоль)
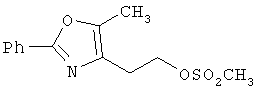
в MeCN (4 мл) нагревают до 90°С в течение 20 часов. Реакционную массу охлаждают до комнатной температуры и разделяют между EtOAc и H2O. Водную фазу экстрагируют с помощью EtOAc (2x); объединенные органические экстракты сушат (MgSO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; постепенный градиент от 9:1 до 1:1, гексан:EtOAc), что обеспечивает соединение, часть Е (42 мг; 65%).
F. 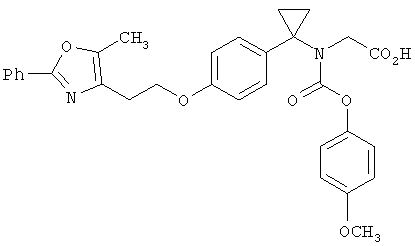
Раствор соединения из части Е (42 мг; 0.08 ммоль) и LiOH·H2O (6 мг; 0.15 ммоль) в ТГФ:H2O (3.8 мл, 2:1) перемешивают при комнатной температуре в течение ночи. Реакционную смесь подкисляют до рН 2 избытком водного раствора 1 М HCl и экстрагируют с помощью EtOAc (2x). Объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом; остаток очищают препаративной ВЭЖХ (как раскрыто в примере 498), что дает названное соединение (28 мг; 68%) в виде бесцветного масла. [M+H]+=543.2.
Следуя процессам, раскрытым выше, получают соединения по примерам с 506 по 518.
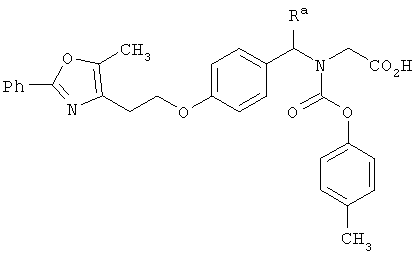
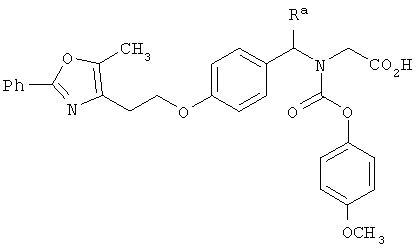
Пример 506
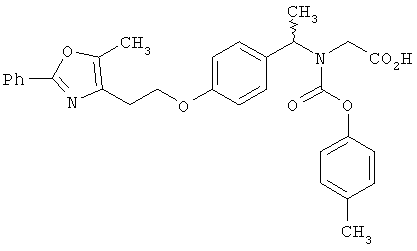
1H ЯМР (ДМСО-d6; 400 мГц): δ 1.47 и 1.54 (2д, J=7.5 Гц; 3H), 2.29 (с, 3H), 2.37 (с, 3H), 2.93 (т, J=6.6 Гц, 2Н). 3.81 (2д, J=18 Гц; 2Н), 4.21 (т, J=6.6 Гц, 2Н), 5.3 (м, 1Н), 6.94 (м, 4Н), 7.18 (д, J=8.4 Гц, 2Н), 7.31 (м, 2Н), 7.49 (м, 2Н).
Пример 508

1H ЯМР (ДМСО-d6; 400 МГц): δ 1.47 и 1.54 (2d, J=7.5Гц; 3H), 2.37 (с, 3H), 2.94 (т, J=6.6 Гц, 2Н), 3.74 (с, 3H), 3.81 (м, 2Н), 4.21 (т, J=6.6 Гц, 2Н), 5.36 (м, 1Н), 6.94 (м, 4Н), 7.29 (м, 2Н), 7.49 (м, 3H), 7.91 (м, 2Н).
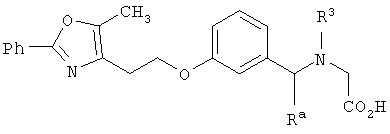

(±)
В синтезе по примерам 513-518 включено использоваание соединения по примеру 541, часть В, как алкилирующего агента.
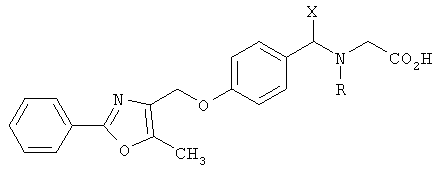
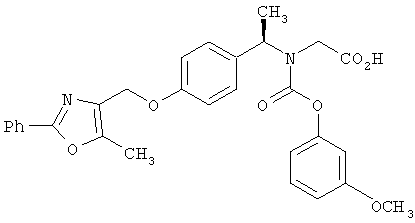
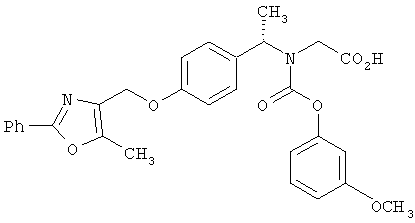
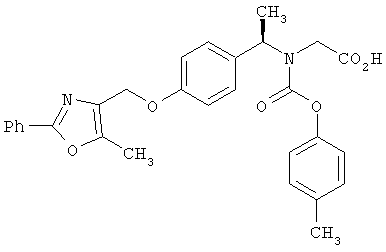

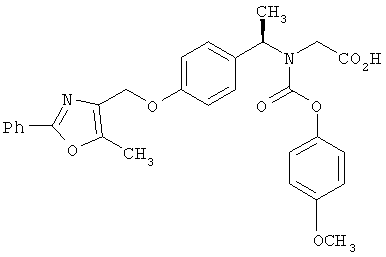

Пример 519
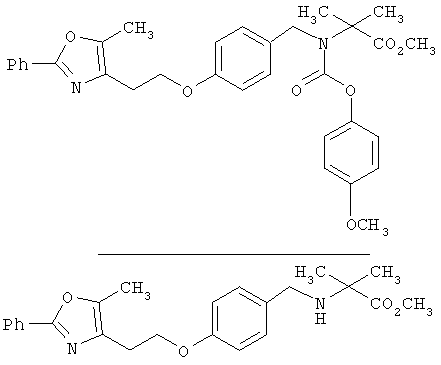
Смесь гидрохлорида метил-α-аминоизобутирата (108 мг; 0.7 ммоль), Et3N (146 мкл; 111 ммоль), NaBH(ОАс)3 (222 мг; 11 ммоль) и альдегида (215 мг; 07 ммоль)

в DCE (5 мл) перемешивают при комнатной температуре в течение 21 часа. Остается некоторое количество исходного продукта, поэтому его нагревают до 55°С в течение 4 часов (реакция не протекает). Добавляют насыщенный водный раствор NaHCO3 и летучие продукты удаляют под вакуумом. Остаток разделяют между H2O и EtOAc. Водную фазу экстрагируют с помощью EtOAc (2x). Объединенные органические экстракты промывают рассолом и экстрагируют водным раствором 1 М HCl. Водную фазу подщелачивают твердым NaOH и экстрагируют с помощью EtOAc (2x). Органические экстракты сушат (Na2SO4) и концентрируют под вакуумом, что дает сырое соединение из части А (174 мг; 61%).
B. 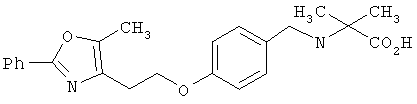
Раствор соединения из части А (120 мг; 0.29 ммоль), водный раствор LiOH (2.0 мл 0.3 М раствора 1:1:1 смесь ТГФ:метанол:H2O) перемешивают при комнатной температуре в течение ночи. Реакцию подкисляют до рН˜2 водным раствором 1 М HCl, затем концентрируют под вакуумом и очищают препаративной ВЭЖХ (YMC S5 ODS 30×250 мм колонка; скорость потока 25 мл/мин; непрерывный градиент от 40:60 В:А до 100% В в течение более 30 мин, где растворитель А=90:10:0.1 H2O:метанол:ТФК; растворитель В=90:10:0.1 метанол:H2O:ТФК), чтобы получить соединение, часть В (60 мг; 53%) в виде сиропа.
C. 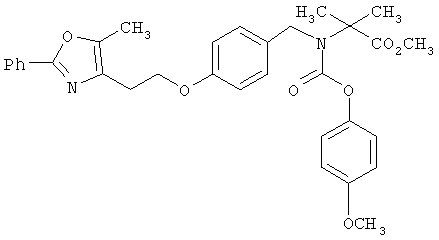
Раствор соединения из части В (25 мг; 0.06 ммоль), 4-метоксифенилхлорформиата (20 мкл) в пиридине (1 мл) нагревают до 60°С в течение 6 часов. Летучие продукты удаляют под вакуумом и остаток разделяют между EtOAc (2 мл) и водным раствором 1 М HCl (1 мл). Органическую фазу концентрируют под вакуумом и остаток очищают препаративной ВЭЖХ (YMC S5 ODS 30×250 мм колонка; скорость потока 25 мл/мин; непрерывный градиент от 40:60 В:А до 100% В в течение более 20 мин, где растворитель А=90:10:0.1 H2O:метанол: ТФК; растворитель В=90:10:0.1 метанол:H2О: ТФК), чтобы получить названное соединение (4 мг; 12%) в виде белой пены. [М+Н]+=545.3.
Следуя процессам, описанньм выше, получают соединения по следующим примерам с 520 по 535.
Примеры от 520 до 535

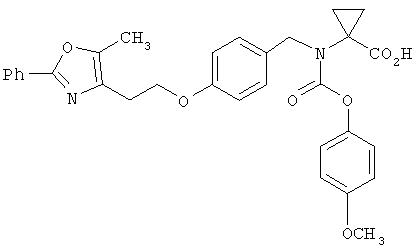
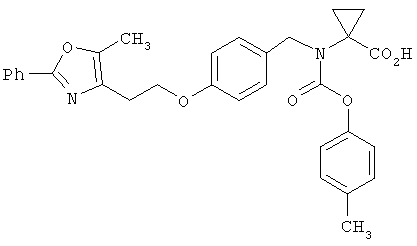


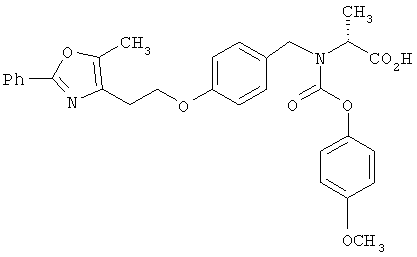
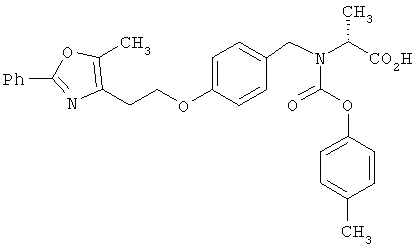

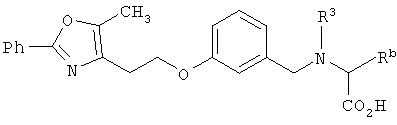
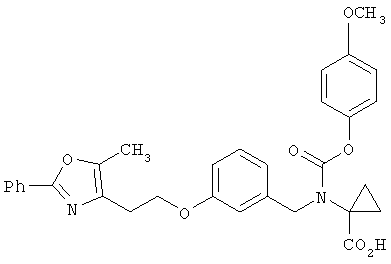
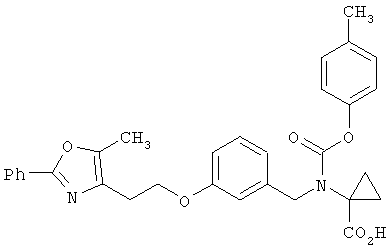

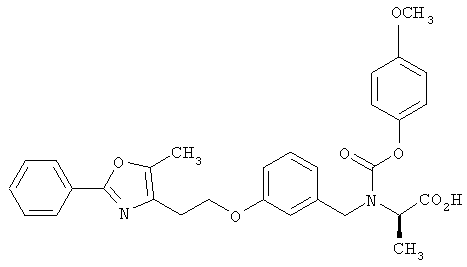

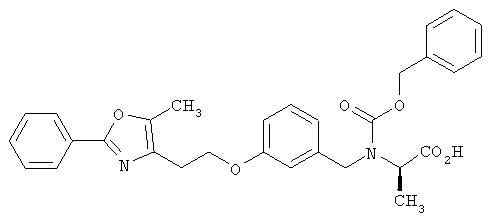
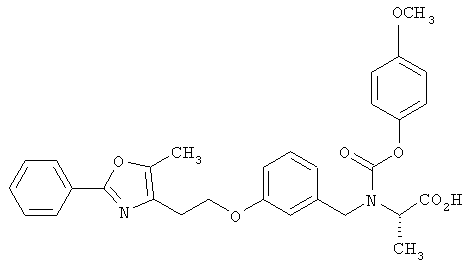
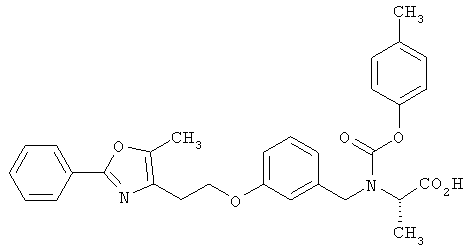
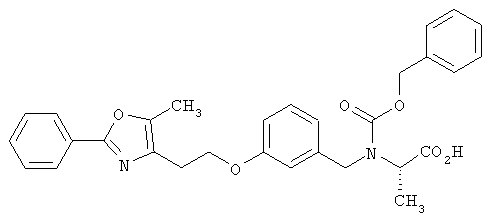
Пример 536
A. 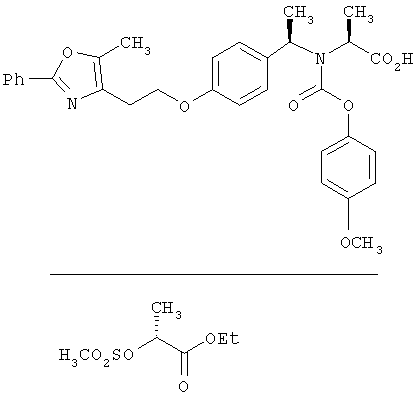
К охлажденному до 0°С раствору (R)-(-)-лактата (3.0 г; 29 ммоль) и Et3N (4.8 мл; 35 ммоль) в СН2Cl2 (60 мл) добавляют метансульфонилхлорид (2.67 мл; 35 ммоль). Смесь перемешивают при 0°С в течение 1 часа, затем разделяют между СН2Cl2 и 1 М водным раствором HCl (100 мл каждый). Органическую фазу промывают H2O и рассолом, сушат (MgSO4) и концентрируют под вакуумом без нагревания, что дает соединение, часть А, в виде масла (4.5 г; 86%), которое используют на следующей стадии без дальнейшей очистки.
B.
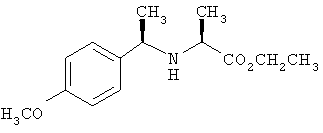
Смесь соединения из части А (1.42 г; 6.0 ммоль), (R)-4-метокси-α-метилбензиламина (300 мг, 2.0 ммоль) и К2СО3 (828 мг; 6.0 ммоль) в MeCN (20 мл) нагревают до 70°С в течение 17 часов (некоторое количество исходного амина еще остается). Реакцию охлаждают до комнатной температуры, фильтруют и летучие продукты удаляют под вакуумом. Остаток разделяют между EtOAc и H2O. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют в вакууме. Остаток хроматографируют (SiO2; постепенный градиент от 99:1 до 97:3, CHCl3:метанол), что дает соединение, часть В (330 мг; 70%) в виде масла.
C.
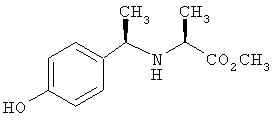
К охлажденному до 0°С раствору соединения из части В (330 мг; 1.39 ммоль) в CH2Cl2 (3 мл) медленно добавляют по каплям BBr3 (3.0 мл 1 М раствора в CH2Cl2; 30 ммоль). Реакцию перемешивают при 10°С в течение 3 часов, затем захолаживают, осторожно добавляя насыщенный водный раствор NH4Cl и CH2Cl2. Отделенную водную фазу нейтрализуют, добавляя твердый NaHCO3, затем экстрагируют с помощью EtOAc (2x). Объединенные органические экстракты промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом, чтобы получить сырое соединение, часть С (150 мг; 48%), которое используют в следующей реакции без дальнейшей очистки.
D.

К раствору соединения из части С (300 мг; 1.35 ммоль) в диоксане:H2О (6 мл, 1:1 раствор) последовательно добавляют NaHCO3 (500 мг; 5.95 ммоль) и медленно добавляют 4-метоксифенилхлорформиат (300 мкл; 2.0 ммоль). Реакционную массу перемешивают при комнатной температуре в течение 1 часа, затем разделяют между EtOAc и H2O. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом, что дает сырой остаток, который хроматографируют (SiO2; постепенный градиент от 3:1 до 1:1, гексан:EtOAc), чтобы получить соединение, часть D (330 мг; 66%).
E.
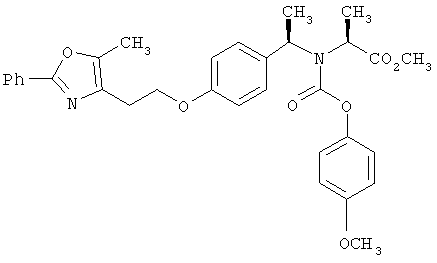
К раствору соединения из части D (330 мг; 0.88 ммоль) в MeCN (20 мл) последовательно добавляют К2СО3 (165 мг; 1.2 ммоль) и мезилат (337 мг; 1.2 ммоль)
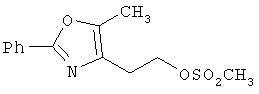
Реакционную смесь нагревают до 95°С в течение 16 часов, затем охлаждают до комнатной температуры и фильтруют. Фильтрат концентрируют под вакуумом и затем разделяют между EtOAc и H2O. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; 3:1, гексан : EtOAc) что дает соединение, часть Е (350 мг; 71%).
F.
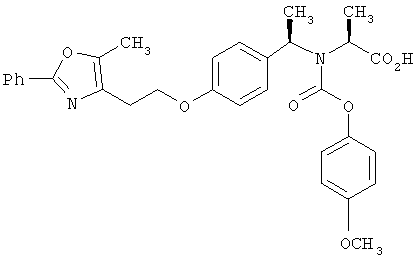
К раствору соединения из части Е (350 мг; 0.62 ммоль) в ТГФ:H2O (15 мл, 2:1 раствор) добавляют LiOH·H2O (52 мг; 1.2 ммоль). Реакционную массу перемешивают при комнатной температуре в течение ночи (14 часов), затем добавляют EtOAc и раствор подкисляют с помощью 1 н. раствора HCl до рН˜2. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом. Остаток очищают препаративной ВЭЖХ (YMC S5 ODS 30×250 мм колонка; скорость потока 25 мл/мин; 20 мин непрерывный градиент от 50:50 В:А до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O:ТФК; время задержки 26 мин) и лиофилизуют из диоксана, что дает названное соединение (208 мг; 61% выход) в виде белого твердого вещества. [М+Н]+=545.3.
Примеры от 537 до 539
Следуя процессам, приведенным выше, получают соединения по примерам с 537 по 539.
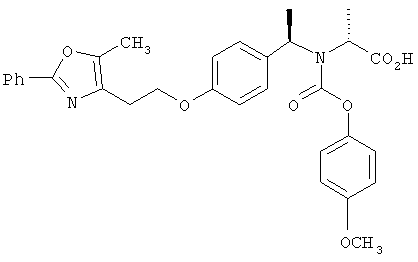
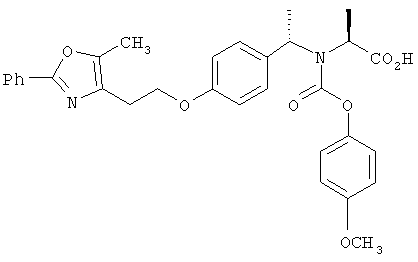
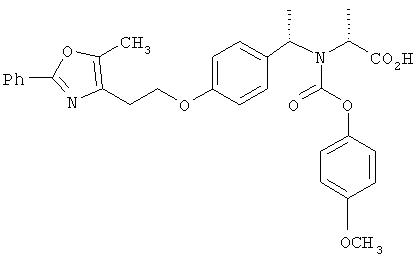
Пример 540
A. 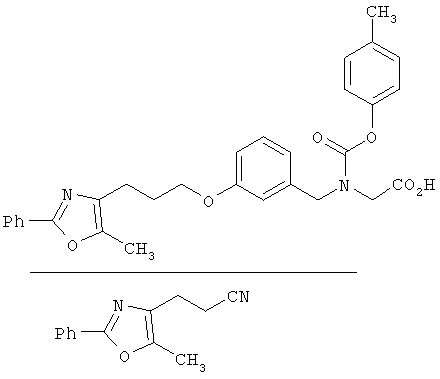
К охлажденному до 0°С раствору 5-метил-2-фенилоксазол-4-илэтанола (5.0 г; 24.6 ммоль), ацетонциангидрина (3.35 мл; 36.9 ммоль) и Ph3Р (7.5 г; 29.5 ммоль) в ТГФ (60 мл) добавляют по каплям DEAD (6.0 мл; 36.9 ммоль). После завершения прибавления реакционную смесь нагревают до комнатной температуры и перемешивают в течение ночи при комнатной температуре. Летучие продукты удаляют под вакуумом и остаток хроматографируют (SiO2; гексан:EtOAc, 2:1), что дает соединение из части А (4.5 г; 86%) в виде масла.
B. 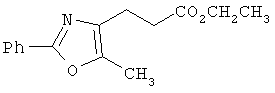
Раствор соединения из части А (4.5 г; 21.2 ммоль) и H2SO4 (концентрированной; 20 мл) в EtOH (100 мл) нагревают при кипении с обратным холодильником в течение ночи. Раствор концентрируют под вакуумом до 1/3 его исходного объема, затем осторожно добавляют EtOAc (150 мл) и H2O (100 мл). Органическую фазу промывают насыщенным водным раствором NaHCO3 (2×100 мл) и рассолом (150 мл), сушат (MgSO4) и концентрируют под вакуумом, что дает сырое масло. Полученный продукт хроматографируют (SiO2; гексан:EtOAc, 2:1) и получают соединение, часть В (2.1 г; 38%) в виде кристаллического твердого вещества.
С.
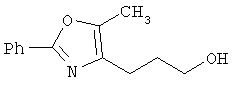
К охлажденному до -78°С раствору соединения из части В (2.1 г; 8.1 ммоль) в ТГФ (6 мл) добавляют по каплям LiAlH4 (16 мл 1.0 М раствора в ТГФ; 16 ммоль) в токе N2. Смеси дают нагреться до 0°С и перемешивают при 0°С в течение 30 мин, после чего реакция оказывается завершенной, как показывает ТЖХ (гексан:EtOAc, 1:1). Последовательно добавляют водный раствор HCl (1.0 мл 1 М раствора; 1 ммоль) и насыщенный водный раствор натрий-калийтартрата (10 мл) и смесь перемешивают при комнатной температуре в течение 30 мин. Смесь экстрагируют с помощью EtOAc (100 мл), промывают H2O и рассолом, сушат (Na2SO4) и концентрируют под вакуумом, что дает сырое соединение, часть С (1.78 г; 97%) в виде белого твердого вещества, которое используют на следующей стадии без дальнейшей очистки.
D. 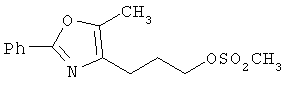
К раствору соединения из части С (670 мг; 3.09 ммоль) и Et3N (516 мкл; 3.71 ммоль) в CH2Cl2 (4 мл) добавляют метансульфонилхлорид (286 мкл; 3.71 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 30 мин, именно в этот момент реакция завершена, как показывает ТЖХ (гексан:EtOAc, 2:1). Смесь разделяют между CH2Cl2 (60 мл) и H2O (40 мл). Органическую фазу промывают рассолом (40 мл), сушат (MgSO4) и концентрируют под вакуумом, что дает соединение, часть D (910 мг; 100%), которое используют на следующей стадии без дальнейшей очистки.
E. 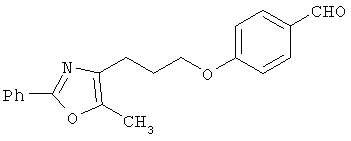
Смесь соединения из части D (380 мг; 1.29 ммоль), 4-гидроксибензальдегида (188 мг; 1.55 ммоль) и К2СО3 (214 мг; 1.55 мг) в MeCN (12 мл) нагревают с обратным холодильником на масляной бане в течение 17 часов. К этому моменту все исходное соединение из части D вступает в реакцию (но присутствует значительное количество гидролизованного побочного продукта, соединения из части С) по данным ВЭЖХ/МС. Реакцию охлаждают до комнатной температуры и твердый осадок отфильтровывают. Фильтрат концентрируют под вакуумом и разделяют между EtOAc (60 мл) и H2O (40 мл). Органическую фазу промывают рассолом (40 мл), сушат (MgSO4) и концентрируют под вакуумом, что дает сырой продукт. Его хроматографируют (SiO2; постепенный градиент от 4:1 до 1:2, гексан:EtOAc), что дает соединение, часть Е (150 мг; 36%) в виде масла, в дополнение к соединению из части С (100 мг; 36%).
F. 
Смесь соединения из части Е (150 мг; 0.50 ммоль), гидрохлорид глицинметилового эфира (75 мг; 0.60 ммоль) и Et3N (84 мкл; 0.60 ммоль) в метаноле (5 мл) перемешивают при комнатной температуре в течение 6 часов, после чего осторожно порциями добавляют NaBH4 (50 мг). Реакционную смесь перемешивают при комнатной температуре в течение ночи, затем летучие продукты удаляют под вакуумом. Остаток разделяют между EtOAc и H2O. Органическую фазу промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом, что дает соединение, часть F (180 мг; 97%) в виде масла.
G. 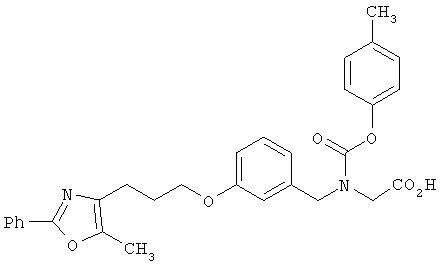
Смесь соединения из части F (23 мг; 0.060 ммоль), Et3N (10 мкл; 0.66 ммоль) и 4-толилхлорформиат (10 мкл; 0.066 ммоль) в СН2Cl2 (1 мл) перемешивают при комнатной температуре в течение 2 часов. Летучие продукты удаляют под вакуумом и остаток растворяют в растворе ТГФ:метаноле:H2O (1 мл, 2:2:1 смесь); добавляют LiOH·H2O (14 мг; 0.33 ммоль) и реакционную массу перемешивают при комнатной температуре в течение 2 часов. Летучие продукты удаляют под вакуумом и остаток разделяют между водным раствором 1 М HCl и EtOAc. Органический экстракт концентрируют под вакуумом и остаток очищают препаративной ВЭЖХ (YMC ODS S5 30 мм × 250 мм колонка, непрерывный 25-минутный градиент от 40% В:60% А до 100% В, выдержка при 100% В в течение 15 мин, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O:ТФК; скорость потока 25 мл/мин), что дает названное соединение в виде белого твердого вещества (13 мг; 45% после 2 стадий). [М+Н]+=515.3.
Пример 541
A. 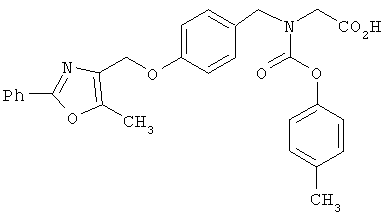
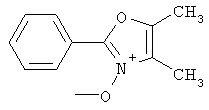
К раствору бензальдегида (23.8 г, 234 ммоль) в EtOAc (150 мл; предварительно насыщенный газом HCl) добавляют одной порцией 2,3-бутадионмонооксим (25.0 г, 234 ммоль) и полученный раствор перемешивают при комнатной температуре в течение 12 часов. Анализ ВЭЖХ показывает что весь исходный продукт вступил в реакцию. Реакционную смесь концентрируют под вакуумом, что дает соединение, часть А, в виде белого твердого вещества, которое используют на следующей стадии без дальнейшей очистки.
B. 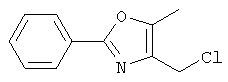
К раствору соединения из части А в CHCl3 (200 мл) добавляют по каплям POCl3 (30 мл, 320 ммоль). Реакционную смесь перемешивают в течение 12 часов при 50°С, затем концентрируют под вакуумом. Коричневый остаток разделяют между EtOAc (300 мл) и 1 н. водным раствором NaOH. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; Et2O), что дает соединение, часть В (41.5 г; 86%) в виде светло-коричневого твердого вещества (>95% чистотой, как показывает аналитическая ВЭЖХ и 1H-ЯМР анализ).
C. 
Раствор 4-гидроксибензальдегида (20 г, 160 ммоль), гидрохлорид глицинметилового эфира (22 г, 180 ммоль) и Et3N (25 мл, 180 ммоль) в метаноле (200 мл) перемешивают при комнатной температуре в течение 12 часов. Реакционную смесь охлаждают до 0°С и добавляют порциями NaBH4 (9.0 г, 240 ммоль), поддерживая температуру меньше комнатной. Реакционную смесь перемешивают в течение 5 часов, затем концентрируют под вакуумом, что дает сырое соединение, часть С, которое используют на следующей стадии без дальнейшей очистки.
D. 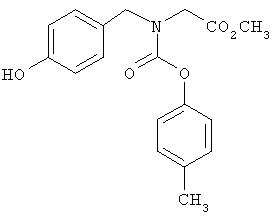
К раствору сырого соединения, часть С, в Et2O (300 мкл ) и H2O (200 мл) добавляют NaHCO3 (20 г, 240 ммоль, одной порцией) и 4-толилхлорформиат (15 мл, 150 ммоль; по каплям). Двухфазную реакционную смесь перемешивают в течение 12 часов при комнатной температуре. Водную фазу затем экстрагируют с помощью Et2O (2×200 мл). Объединенные органические экстракты промывают рассолом (2×50 мл), сушат (MgSO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; постепенный градиент от 3:1 до 1:1, гексан:EtOAc), что дает соединение, часть D (40.8 г; 76% после 2 стадий) в виде масла.
E. 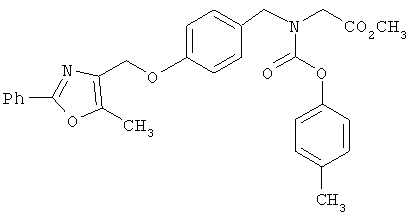
Раствор соединения из части В (14.5 г, 70 ммоль), соединение из части С (21.6 г, 67 ммоль) и К2СО3 (18.4 г, 134 ммоль) в СН3CN (150 мл) перемешивают при 80°С в течение 12 часов. Реакционную массу охлаждают до комнатной температуры и летучие продукты удаляют под вакуумом. Коричневый маслянистый остаток распределяют между EtOAc (250 мл) и рассолом (100 мл). Водный раствор (слой) экстрагируют с помощью EtOAc (3×100 мл). Объединенные органические экстракты сушат (MgSO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; постепенный градиент от 3:1 до 1:1, гексан:EtOAc), что дает соединение, часть D (23.6 г; 71%) в виде бесцветного масла.
F. 
Раствор соединения из части D (23.6 г, 47.4 ммоль) и LiOH·H2O (4.0 г, 95 ммоль) в ТГФ (200 мл) и H2O (120 мл) перемешивают при комнатной температуре в течение 4 часов. Реакционную смесь затем подкисляют до рН˜2 водным раствором 1 н. HCl. Водную фазу экстрагируют с помощью EtOAc (3×200 мл). Объединенные органические экстракты сушат (MgSO4) и концентрируют под вакуумом, что дает маслянистый остаток, который перекристаллизовывают из EtOAc, что обеспечивает названное соединение (19.4 г; 84%) в виде белого твердого вещества. [М+Н]+=487.23.
1H ЯМР (CD3OD; 400 МГц): δ 2.32 (с, 3H), 2.46 (с, 3H),3.99 и 4.04 (2с, 2Н), 4.47 и 4.54 (2с, 2Н), 5.01 и 5.00 (2с, 2Н), 6.99 (д, J=8.4 Гц, 2Н), 7.05 (м, 2Н), 7.17 (д, J=8.4 Гц, 2Н), 7.31 (м, 2Н); 7.49 (м, 3H), 8.01 (м, 2Н).
1H ЯМР (CDCl3; 400 МГц): δ 2.31 (с, 3H), 2.44 (с, 3H),4.00 (с, 2Н), 4.55 (2с, 2Н), 5.00 (2с, 2Н), 6.99 (м, 4Н), 7.13 (м, 2Н), 7.21 (д, J=8.8 Гц, 2Н), 7.31 (м, 2Н); 7.44 (с, 3Н), 8.01 (с, 2Н).
Пример 542
A. 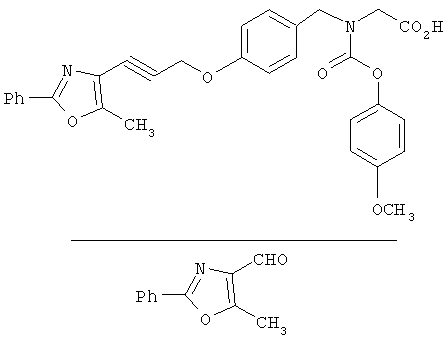
Смесь 2-фенил-5-метилоксазол-4-уксусной кислоты (470 мг; 2.17 ммоль; Maybridge), N-оксида пиридина (830 мг; 8.74 ммоль) и уксусного ангидрида (350 мг; 3.57 ммоль) в толуоле (10 мл) нагревают до 90°С в течение 12 часов, затем концентрируют под вакуумом. Остаток затем разделяют между EtOAc и 1 М водным раствором HCl. Органическую фазу промывают насыщенным водным раствором NaHCO3, рассолом, сушат (Na2SO4) и концентрируют под вакуумом, что дает темно-коричневое масло. Полученный продукт хроматографируют (SiO2; 4:1, гексан:EtOAc) с образованием соединения из части А (143 мг; 35%) в виде масла.
B. 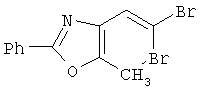
К охлажденному до 0°С раствору соединения из части А (600 мг; 3.21 ммоль) и Ph3Р (3.37 г; 12.9 ммоль) в CH2Cl2 (50 мл) добавляют по каплям раствор CBr4 (2.13 г; 6.4 ммоль) в CH2Cl2 (20 мл). Раствор перемешивают при 0°С в течение 2 часов, затем дают нагреться до комнатной температуры и перемешивают при комнатной температуре в течение ночи. Летучие продукты удаляют под вакуумом и остаток хроматографируют (85:15, гексан:EtOAc) с получением соединения, часть В (1.08 г; 98%) в виде бледно-желтого твердого вещества.
C. 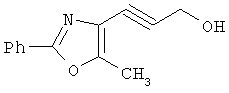
К охлажденному до -78°С раствору соединения из части В (1.12 г; 3.26 ммоль) в ТГФ (60 мл) добавляют по каплям н-бутиллитий (4.2 мл 1.6 М раствора в гексане; 6.72 ммоль) в течение более 25 мин, поддерживая внутреннюю температуру при ≤-71°С. Реакционную массу перемешивают при -78°С в течение 1 часа, затем дают нагреться медленно до 0°С. После этого вводят одной порцией параформальдегид (305 г), реакционную массу охлаждают при 0°С в течение 3 часов и затем захолаживают насыщенным водным раствором NH4Cl. Водную фазу экстрагируют с помощью EtOAc (2х); объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом, что дает темное масло. Полученный продукт хроматографируют (SiO2; 3:2, гексан:EtOAc) с образованием соединения из части С (466 мг; 67%) в виде желтого твердого вещества.
D. 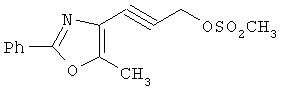
К охлажденному до 0°С раствору соединения из части С (466 мг; 2.19 ммоль) и Et3N в CH2Cl2 добавляют по каплям метансульфонилхлорид (2.45 ммоль) и реакционную массу охлаждают при 0°С в течение 1 часа. Затем смесь разделяют между CH2Cl2 и холодным 1 М водным раствором HCl. Органическую фазу промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом. Сырой продукт хроматографируют (SiO2; 3:2, гексан:EtOAc), что дает соединение, часть D (533 мг; 84%) в виде грязно-белого твердого вещества.
E. 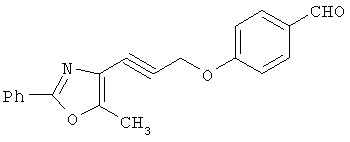
Смесь соединения из части D (198 мг; 0.68 ммоль), 4-гидроксибензальдегида (96 мг; 0.79 ммоль) и К2СО3 (141 мг; 1.02 ммоль) в CH3CN (13 мл) нагревают до 70°С в течение 3 часов, затем перемешивают при комнатной температуре в течение ночи. Летучие продукты удаляют под вакуумом и остаток разделяют между EtOAc и 1 М водным раствором NaOH. Органическую фазу промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом, что дает сырое соединение, часть Е (190 мг; 88%) в виде желтого масла, которое используют на следующей стадии без дальнейшей очистки.
F. 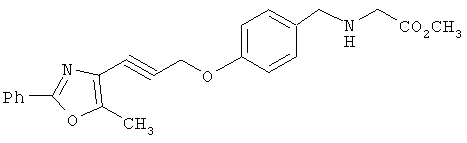
Смесь соединения из части Е (123 мг; 0.39 ммоль), гидрохлорида глицинметилового эфира (248 мг; 1.98 ммоль) и Et3N (600 мкл; 4.3 ммоль) в DCE (15 мл) перемешивают при комнатной температуре в течение 15 мин, после чего добавляют одной порцией NaBH(OAc)3H (262 мг; 1.2 ммоль). Реакционную массу охлаждают в течение 16 часов при комнатной температуре, затем вводят дополнительное количество NaBH(ОАс)3Н (200 мг; 0.94 ммоль). Перемешивание продолжают в течение 3 часов, после чего вводят еще NaBH(OAc)3H (200 мг; 0.94 ммоль). Реакционную массу перемешивают при комнатной температуре в течение 48 часов, после чего все соединение, часть Е, вступает в реакцию. Реакционную смесь разделяют между CH2Cl2 и водным раствором NaHCO3. Водную фазу экстрагируют с помощью CH2Cl2 (2х). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом. Сырой продукт хроматографируют (SiO2; постепенный градиент от 1:1 до 2:3, гексан:EtOAc), что дает соединение, часть F (120 мг; 79%) в виде бесцветного масла которое затвердевает при стоянии.
G. 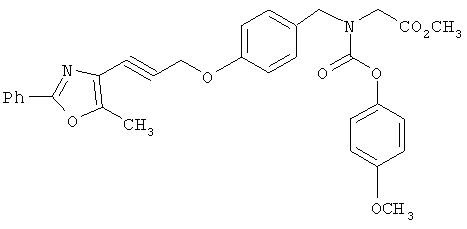
К раствору соединения из части F (180 мг; 0.46 ммоль) и пиридина (100 мкл; 1.24 ммоль) в CH2Cl2 (10 мл) добавляют 4-метоксифенилхлорформиат (105 мкл; 0.71 ммоль). Реакционную массу перемешивают при комнатной температуре в течение 3.5 часов, затем разделяют между водным раствором NaHCO3 и EtOAc. Водную фазу экстрагируют с помощью EtOAc (2x). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом. Сырой продукт хроматографируют (SiO2; гексан:EtOAc, 3:2), что дает соединение, часть G (232 мг; 93%) в виде бесцветного масла.
H. 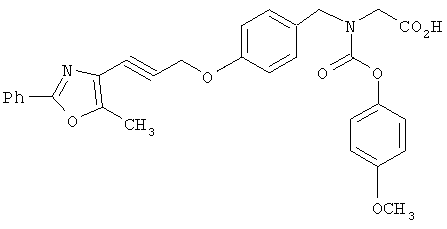
К раствору соединения из части G (232 мг; 0.43 ммоль) в ТГФ:H2O (12 мл, смесь 5:1) добавляют LiOH.H2O (1.3 ммоль). Раствор перемешивают при комнатной температуре в течение ночи, затем подкисляют водным раствором 1 М HCl и экстрагируют с помощью EtOAc (2x). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом. Сырой продукт очищают препаративной ВЭЖХ (YMC S5 ODS 30×75 мм колонка, скорость потока 20 мл/мин; непрерывный градиент от 70:30 В:А до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В 90:10:0.1 метанол: H2O: ТФК), что дает названное соединение (160 мг; 71%) в виде белого твердого вещества. [М+Н]+=527.2.
Пример 543
A. 
Раствор 5-метил-2-фенилоксазол-4-илуксусной кислоты (4.0 г; 18 ммоль) и концентрированной HCl (2 мл) в метаноле (60 мл) нагревают при кипении с обратным холодильником в течение ночи. Летучие продукты удаляют под вакуумом; остаток разделяют между H2O и EtOAc. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом, что дает сырое соединение, часть А, в виде бесцветного масла (4.00 г; 94%), которое используют на следующей стадии без дальнейшей очистки.
B. 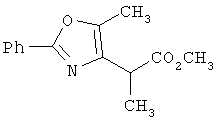
К охлажденному до -78°С раствору LDA (15.0 мл 2.0 М раствора гептана/ТГФ; 30 ммоль; Aldrich) последовательно добавляют по каплям раствор соединения из части А (2.3 г; 10 ммоль) в ТГФ (6 мл) и НМРА (500 мкл; 2.9 ммоль). Раствор перемешивают при -78°С 30 минут, после чего по каплям вводят йодистый метил (1.87 мл; 30 ммоль). Его перемешивают при -78°С в течение 1 часа, затем дают нагреться до 0°С и перемешивают при 0°С в течение 1 часа. Реакционный раствор разделяют между насыщенным водным раствором NH4Cl и EtOAc. Водную фазу экстрагируют с помощью EtOAc (2×50 мл). Объединенные органические экстракты сушат (MgSO4) и концентрируют под вакуумом, что дает сырое соединение, часть В (1.90 г; 78%) в виде бесцветного масла, которое используют на следующей стадии без дальнейшей очистки.
C. 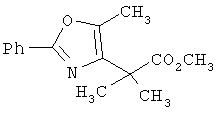
К охлажденному до -78°С раствору LDA (7.0 мл 2.0 М гептана/ТГФ; 14 ммоль; Aldrich) последовательно по каплям добавляют раствор соединения из части В (1.8 г; 7.3 ммоль) в ТГФ (5 мл) и НМРА (500 мкл; 2.9 ммоль). Раствор перемешивают при -78°С в течение 1 часа, затем по каплям добавляют раствор йодистого метила (1 мл; 11 ммоль). Раствор перемешивают при -78°С в течение 1 часа, затем дают нагреться до 0°С и перемешивают при 0°С в течение 1 часа. Затем его разделяют между насыщенным водным раствором NH4Cl и EtOAc. Водную фазу экстрагируют с помощью EtOAc (2×50 мл). Объединенные органические экстракты сушат (MgSO4) и концентрируют под вакуумом. Сырой продукт объединяют с продуктом из другой реакции (с 670 мг соединения из части В) и хроматографируют (SiO2; 9:1, гексан:EtOAc), что дает соединение, часть С (2.60 г; 95%) в виде бесцветного масла.
D. 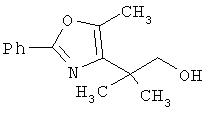
К охлажденному до -78°С раствору соединения из части С (1.2 г; 4.63 ммоль) в ТГФ (3 мл) в атмосфере N2 добавляют по каплям раствор LiAlH4 (1.0 мл 1.0 М раствора в ТГФ). Реакционную массу охлаждают при -78°С в течение 1 часа, затем дают нагреться до 0°С и перемешивают при 0°С в течение 30 минут. Реакцию захолаживают осторожным добавлением 1 М водного раствора натрий-калийтартрата, а затем H2O. Водную фазу экстрагируют с помощью EtOAc. Объединенные органические экстракты промывают H2O, сушат (MgSO4) и концентрируют под вакуумом, что дает сырое соединение, часть D (1.01 г; 94%) в виде масла, которое используют на следующей стадии без дальнейшей очистки.
E. 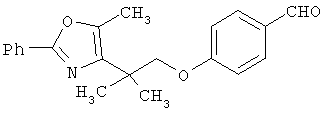
К нагретому до 80°С раствору соединения из части D (700 мг; 3.0 ммоль), Ph3Р (1.2 г; 4.6 ммоль) и 4-гидроксибензальдегида (406 мг; 3.3 ммоль) в ТГФ (10 мл) добавляют DEAD (720 мкл; ммоль) двумя порциями в течение более 5 минут. Раствор перемешивают при 80°С в течение 1 часа (исходный продукт еще остается), затем концентрируют под вакуумом. Остаток хроматографируют (SiO2; постепенный градиент от 9:1 до 5:1 гексан:EtOAc), что дает соединение, часть Е (160 мг; 16%).
F. 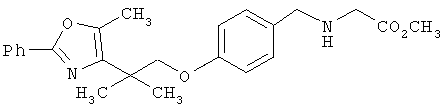
Раствор соединения из части Е (250 мг; 0.75 ммоль), гидрохлорид глицинметилового эфира (141 мг; 1.13 ммоль) и Et3N (157 мкл; 1.13 ммоль) в метаноле (30 мл) перемешивают при комнатной температуре в течение ночи. Осторожно добавляют избыток твердого NaBH4; реакционную массу охлаждают при комнатной температуре в течение 1 часа, затем концентрируют под вакуумом. Остаток разделяют между H2O и EtOAc. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом, что дает сырое соединение, часть F (300 мг; 98%), которое используют на следующей стадии реакции без дальнейшей очистки.
G. 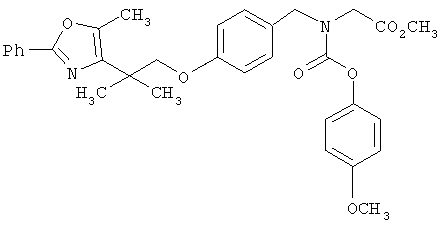
К охлажденному до 0°С раствору соединения из части F (150 мг; 0.37 ммоль) и Et3N (51 мкл; 0.37 ммоль) в CH2Cl2 (5 мл) добавляют 4-метоксифенилхлорформиат (55 мкл; 0.37 ммоль). Реакции дают нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 2 часов. Летучие продукты удаляют под вакуумом и остаток хроматографируют (SiO2; 5:1, гексан:EtOAc), что приводит к соединению, часть G (130 мг; 63%).
H. 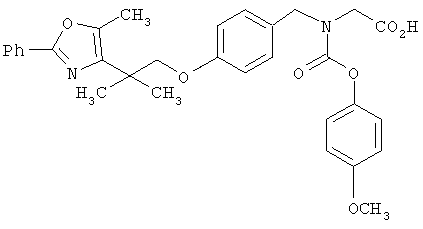
Раствор соединения из части G (130 мг) и LiOH·H2O (39 мг) в H2O/ТГФ/метаноле (2 мл, 1:2:2 смесь) перемешивают при комнатной температуре в течение 2 часов. Летучие продукты удаляют под вакуумом и остаток подкисляют 1.0 М водным раствором HCl, затем экстрагируют с помощью EtOAc. Органическую фазу сушат (Na2SO4) и концентрируют под вакуумом, что дает остаток, который очищают препаративной ВЭЖХ (YMC S5 ODS с обращенной фазой CIS, 30×250 мм колонка; скорость потока 25 мл/мин; непрерывный градиент от 50% А:В до 100% В в течение более 20 мин, где растворитель А=90:10:0.1 H2O:метанол:ТФК, В=90:10:0.1 метанол:H2O:ТФК), затем лиофилизуют из диоксана, что дает названное соединение (58 мг; 46%) в виде белого лиофилизата. [М+Н]+=545.4.
Пример 544
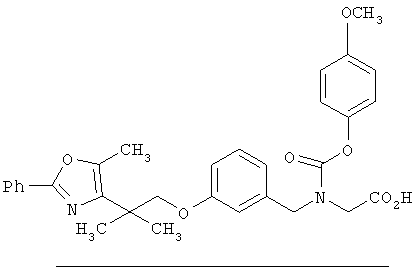
Названное соединение получают аналогично методике примера 543 за исключением того, что 3-гидроксибензальдегид используют вместо 4-гидроксибензальдегида (в приготовлении по примеру 543 соединения, часть Е). [М+Н]+=545.4.
Пример 545
A. 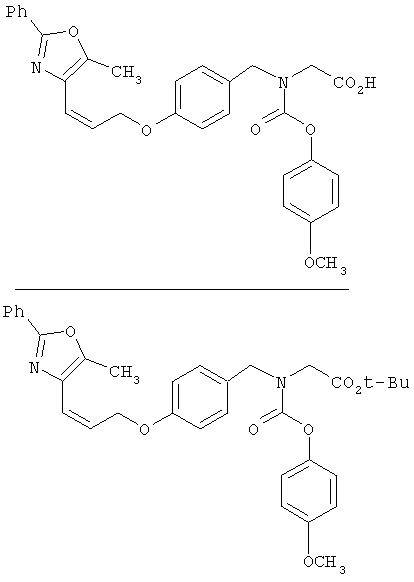
Смесь ацетилена (38 мг; 0.065 ммоль)
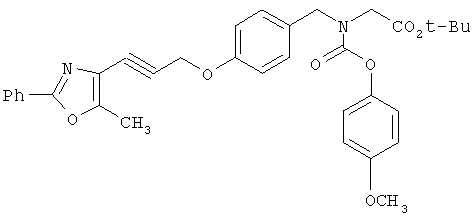
(синтезируют полностью аналогично методике примера 542, часть G, с гидрохлоридом глицин-трет-бутилового эфира вместо гидрохлорида глицинметилового эфира), хинолина (80 мг; 0.62 ммоль) и катализатора Линдлара (8 мг; Pd/СаСО3; Aldrich) в метаноле (8 мл) перемешивают в атмосфере Н2 при 0°С в течение 20 минут. Вводят дополнительное количество катализатора Линдлара (8 мг; Pd/СаСО3; Aldrich), затем продолжают перемешивание в атомсфере Н2 при 0°С в течение 25 минут, после чего реакция завершается. Смесь фильтруют и фильтрат концентрируют под вакуумом. Остаток очищают препаративной ВЭЖХ (YMC S5 CDS 20×100 мм колонка; скорость потока 20 мл/мин; непрерывный 20 мин градиент от 80:20 В:А до 100% В, где А=90:10:0.1 H2O:метанол:ТФК и В=90:10:0.1 метанол: H2O:ТФК), что дает соединение, часть А (22 мг; 56%) в виде бесцветного масла.
B. 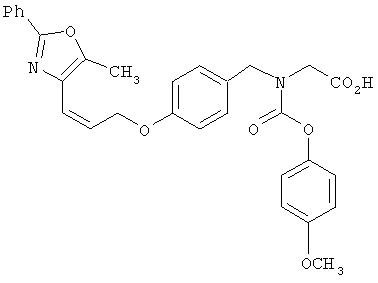
К раствору соединения из части А (3 мг; 0.005 ммоль) в CH2Cl2 (0.5 мл) добавляют по каплям ТФК (0.25 мл) и реакционную массу охлаждают в течение 2 часов при комнатной температуре. Летучие продукты удаляют под вакуумом; остаток растворяют в CDCl3, фильтруют через подушку из хлопка и концентрируют под вакуумом, что дает названное соединение (1.5 мг; 55%) в виде бесцветного масла. [М+Na]+=551.0.
Следуя процессам, приведенньм выше, получают соединения по примерам с 546 по 556.
Примеры с 546 по 556
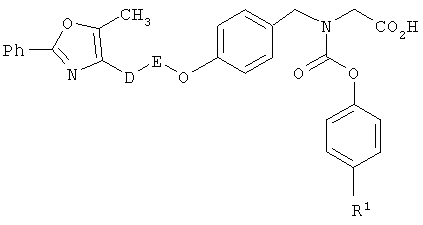
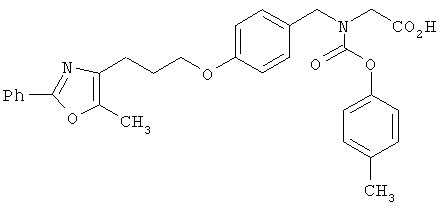
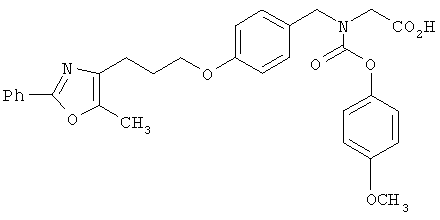
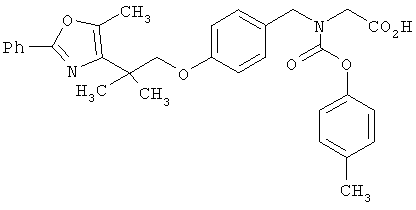
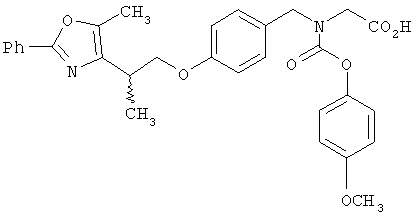
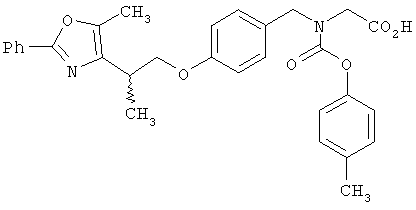
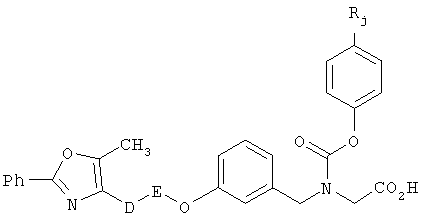
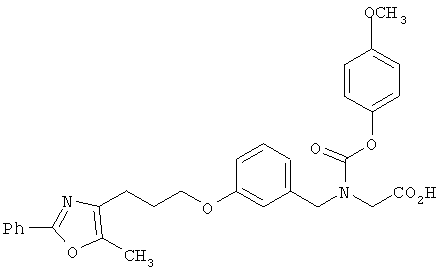
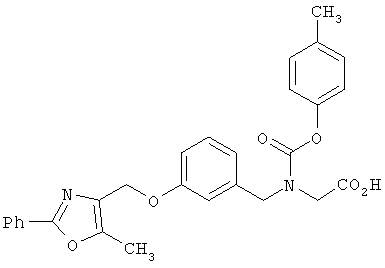
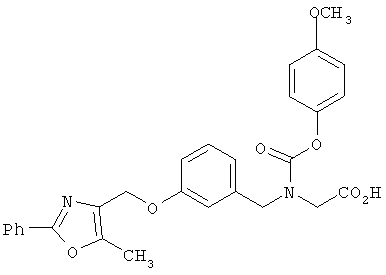
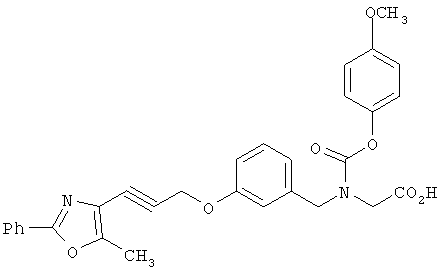
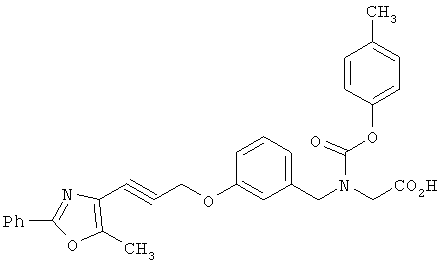
Пример 555
A. 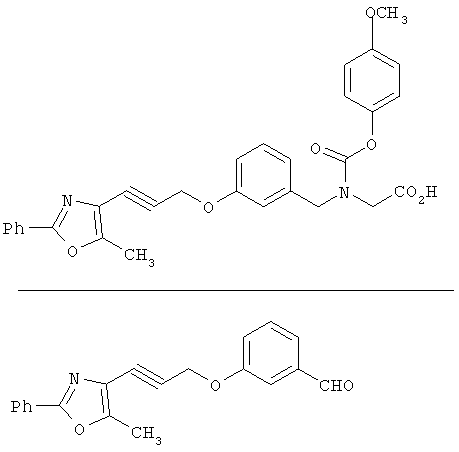
Смесь мезилата (124 мг; 0.43 ммоль)

3-гидроксибензальдегида (62 мг; 0.51 ммоль) и К2СО3 (94 мг; 0.68 ммоль) в СН3CN (10 мл) нагревают при 70°С в течение 48 часов. Реакционную массу охлаждают до комнатной температуры, EtOAc добавляют и смесь промывают водным раствором 1 М NaOH и рассолом. Органическую фазу сушат (Na2SO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; гексан:EtOAc, 4:1), что дает соединение из части А (71 мг; 52%) в виде бесцветного масла. [М+Н]+=318.2.
B.
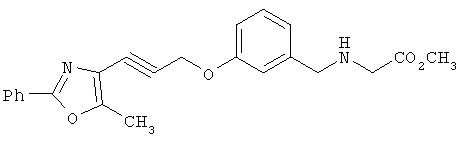
К смеси соединения из части А (71 мг; 0.22 ммоль), глицина·HCl (140 мг; 1.11 ммоль) и Et3N (0.3 мл; 2.16 ммоль) в 1,2-дихлорэтане (10 мл) добавляют NaBH(OAc)3 (150 мг). После перемешивания при комнатной температуре в течение 16 часов (реакция не завершена), добавляют еще NaBH(ОАс)3 (150 мг). Последнее добавление NaBH(ОАс)3 (150 мг; общее количество 2.12 ммоль) осуществляют после еще 3 часов и реакционную массу охлаждают в течение 48 часов при комнатной температуре. В этот момент реакция завершена; добавляют насыщенный водный раствор NaHCO3 и водную фазу экстрагируют с помощью CH2Cl2 (2x). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; гексан/EtOAc=4:6), что дает соединение, часть В (81 мг; 93%) в виде бесцветного масла.
C. 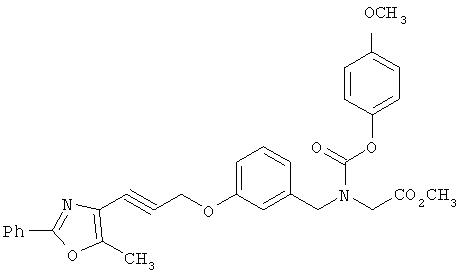
К раствору соединения из части В (10 мг; 0.026 ммоль) в CH2Cl2 (2 мл) последовательно добавляют пиридин (10 мкл; 0.12 ммоль) и 4-метоксифенилхлорформиат (10 мкл, 0.067 ммоль; каждый в 0.1 мл CH2Cl2). Реакционную массу перемешивают при комнатной температуре в течение 16 часов, затем разделяют между водным раствором 1 н. HCl и EtOAc. Органическую фазу промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом. Остаток очищают препаративной ВЭЖХ (YMC S5 ODS 30×75 мм колонка, скорость потока 20 мл/мин; непрерывный градиент от 70:30 А:В до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O:ТФК), что дает соединение, часть С (9 мг; 65%).
D. 
Раствор соединения из части С (9 мг; 0.017 ммоль) в ТГФ:H2О (3 мл, 2:1) добавляют LiOH (6 мг; 0.14 ммоль). Раствор перемешивают при комнатной температуре в течение 4 часов, затем подкисляют избытком 1 М HCl (водным раствором). Раствор экстрагируют с помощью EtOAc (2×5 мл). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом. Сырой продукт очищают препаративной ВЭЖХ, используя те же условия, которые описаны выше, что дает названное соединение (6 мг; 68%) в виде бесцветной пленки. [М+H]+=527.2.
Пример 556

Названное соединение синтезируют, используя ту же последовательность, что в примере 555, из соединения по примеру 555, часть В. Ацилирование 4-метилхлорформиата (67% после ВЭЖХ очистки) с последующим LiOH гидролизом приводит к названному соединению (5 мг; 57% после ВЭЖХ очистки). [М+Н]+=511.4.
Пример 557
A. 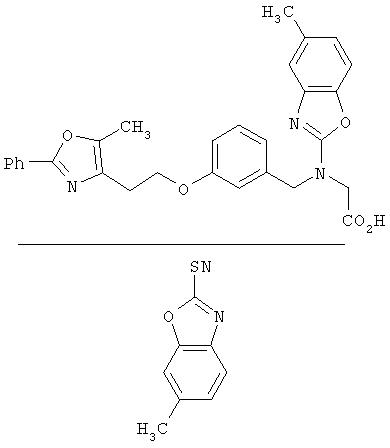
Раствор 2-амино-5-крезола (5.0 г; 40 ммоль), КОН (3.2 г; 57 ммоль) нагревают с обратным холодильником в EtOH (50 мл) и CS2 (40 мл) в течение 8 часов, после чего реакционную смесь концентрируют под вакуумом. Остаток разделяют между водным раствором 1 М HCl (100 мл) и EtOAc (200 мл). Органическую фазу промывают водой (2×100 мл), сушат (MgSO4) и концентрируют под вакуумом, что дает соединение из части А (4.0 г; 60%) в виде белого порошка.
В.
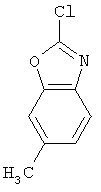
Раствор соединения из части А (3.2 г; 19 ммоль) и PCl5 (3.75 г; 19 ммоль) в толуоле (150 мл) нагревают до кипения с обратным холодильником в течение 2 часов. Реакционную смесь промывают последовательно водой и водным раствором NaHCO3,затем сушат (Na2SO4) и концентрируют под вакуумом, что дает соединение, часть В (4.0 г) в виде сырого масла. Полученный продукт используют на следующей стадии без дальнейшей очистки.
C.
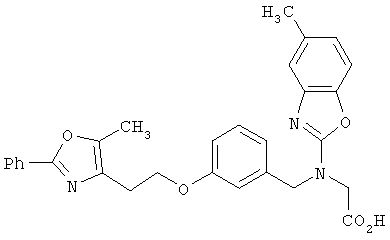
Раствор 1,3-бензилглицинового аминоэфира (150 мг; 0.39 ммоль), соединения части В (100 мг; 0.59 ммоль) и триэтиламина (0.2 мл; 1.98 ммоль) в ТГФ (5 мл) нагревают до 100°С в герметично закрываемой трубке в течение 4 дней. В этот момент ЖХ/МС показывает, что весь исходный продукт вступил в реакцию. Добавляют водный раствор LiOH (0.5 мл 1 М раствора) и перемешивают при комнатной температуре в течение 5 часов. Смесь концентрируют под вакуумом, что дает масло, которое очищают препаративной ВЭЖХ (как для примера 495) и получают названное соединение (72 мг; 37%) в виде твердого вещества.
Пример 558
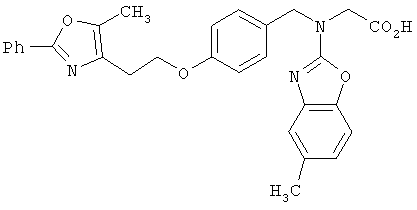
Раствор 1,4-бензилглицинового аминоэфира (50 мг; 0.13 ммоль), соединения по примеру 557, часть В (100 мг; 0.59 ммоль) и триэтиламина (0.2 мл; 1.98 ммоль) в ТГФ (5 мл) нагревают до 100°С в герметично закрываемой трубке в течение 4 дней. В этот момент ЖХ/МС показывает, что весь исходный продукт вступил в реакцию. Добавляют раствор LiOH (0.5 мл 1 М раствора) и перемешивают при комнатной температуре в течение 5 часов. Смесь концентрируют под вакуумом, что дает масло, которое очищают препаративной ВЭЖХ (как в примере 495), что дает названное соединение (26 мг; 40%) в виде твердого вещества.
Пример 559
A. 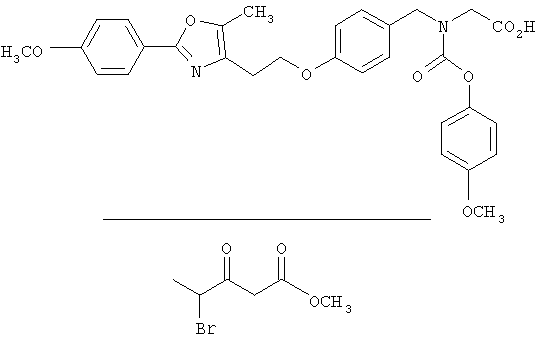
К раствору метилпропионилацетата (4.6 г, 35 ммоль) в CHCl3 (40 мл) добавляют по каплям раствор Br2 (5.6 г; 35 ммоль) в CHCl3 (10 мл) и полученную смесь перемешивают при 0°С в течение 0.5 часов. Реакции дают нагреться до комнатной температуры и затем в смесь пропускают воздух в течение 1 часа. Летучие продукты удаляют под вакуумом, что позволяет получить маслянистый остаток, который разделяют между EtOAc (100 мл) и насыщенным водным раствором NaHCO3. Органическую фазу промывают рассолом, сушат (MgSO4) и концентрируют под вакуумом, что обеспечивает сырое соединение из части А (7.4 г, >95% выход; >90% чистота) в виде масла, которое используют в следующей реакции без дальнейшей очистки.
B. 
Смесь соединения из части А (1.5 г, 7.2 ммоль) и 4-метоксибензамида (1.0 г, 6.6 ммоль) нагревают до 100°С в течение 2.5 часов. Реакционную смесь хроматографируют (SiO2; 5% ацетон/CH2Cl2) с получением соединения, часть В (0.57 г, 33%).
C. 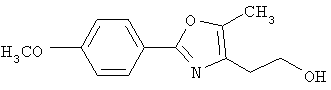
К раствору эфира (0.57 г, 2.3 ммоль) в ТГФ (10 мл) добавляют LiAH4 (2.5 мл 1 М раствора в ТГФ, 2.5 ммоль) по каплям в течение более 10 мин и реакционную массу охлаждают при комнатной температуре в течение 0.5 часов, охлаждают, добавляя несколько капель воды, и затем разделяют между EtOAc (50 мл) и рассолом (10 мл). Органическую фазу сушат (MgSO4) и концентрируют под вакуумом, что дает соединение, часть С (0.52 г, >95%) в виде масла, которое используют в следующий реакция без дальнейшей очистки.
D. 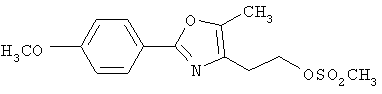
Смесь соединения из части С (0.52 г, 2.3 ммоль), СН3SO2Cl (0.25 мл, 3.3 ммоль) и Et3Н (0.5 мл, 3.6 ммоль) в CH2Cl2 (10 мл) перемешивают при комнатной температуре в течение 12 часов. Летучие продукты удаляют под вакуумом и остаток хроматографируют (SiO2; 4% ацетон/CH2Cl2), обеспечивая соединение, часть D (0.61 г, 85% для 2 стадий) в виде бесцветного масла.
E. 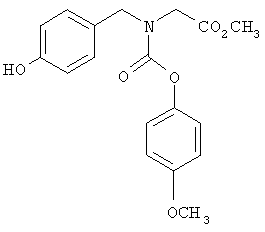
К смеси сырого соединения по примеру 541, часть С (синтезируют, используя 4-гидроксибензальдегид [2.0 г; 16 ммоль] и глицинметилового эфира гидрохлорид [2.3 г; 18 ммоль]) в диоксане:H2О (100 мл 1:1 смесь) последовательно добавляют NaHCO3 (2.5 г; 30 ммоль; одной порцией) и 4-метоксифенилхлорформиат (2.0 мл; 14 ммоль) по каплям. Реакционную массу перемешивают при комнатной температуре в течение 12 часов и затем экстрагируют с помощью EtOAc (4×150 мл). Объединенные органические экстракты сушат (MgSO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; 3% ацетон/CH2Cl2), обеспечивая соединение, часть Е (2.4 г; 44%) в виде бесцветного масла.
F. 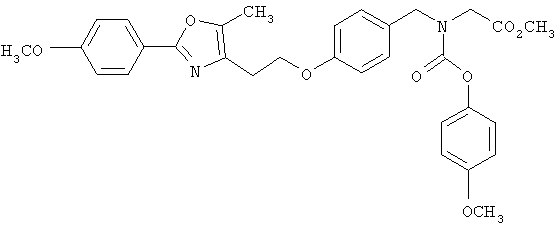
Смесь соединения из части Е (86 мг, 0.25 ммоль), соединение, часть D (60 мг, 0.20 ммоль) и К2СО3 (50 мг, 3.7 ммоль) в ДМФА (3 мл) нагревают до 80°С в течение 12 часов. Реакционную массу охлаждают до комнатной температуры и фильтруют. Летучие продукты удаляют под вакуумом и остаток хроматографируют (SiO2; 7:3, гексан:EtOAc), что обеспечивает названное соединение (41 мг; 36%) в виде бесцветного масла.
G. 
Раствор соединения из части F (41 мг, 0.071 ммоль) и LiOH·H2O (34 мг; 0.8 ммоль) в ТГФ:H2O (2 мл, 2:1 смесь) перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь подкисляют до рН˜2 1 М водным раствором HCl, затем экстрагируют с помощью EtOAc. Объединенные органические экстракты концентрируют под вакуумом и остаток очищают препаративной ВЭЖХ (YMC S5 ODS 30×250 мм колонка; скорость потока 25 мл/мин; 30 мин непрерывный градиент от 50% А:50% В до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O:ТФК) обеспечивает названное соединение (17 мг, 40%) в виде бесцветного масла. [М+Н]+=547.23.
Пример 560
A. 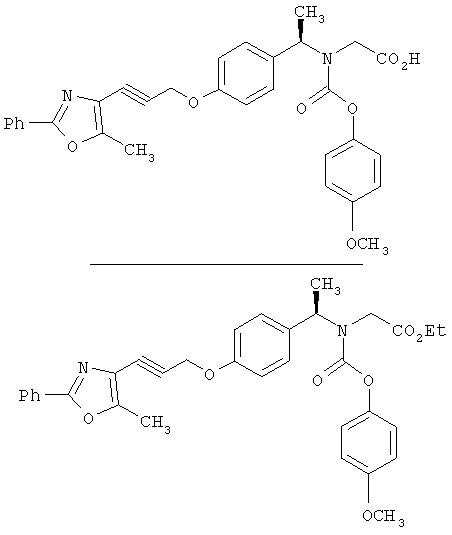
Смесь мезилата(18 мг; 0061 ммоль)
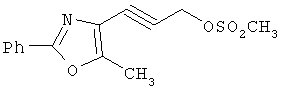
эфира
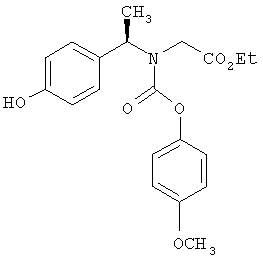
[описан в синтезе соединения по примеру 503, часть В (50 мг; 0.13 ммоль)], К2СО3 (17 мг; 0.34 ммоль) в CH3CN (1 мл) нагревают при 70°С в течение 24 часов. Вводят дополнительное количество К2СО3 (30 мг) и CH3CN (1 мл) и смесь нагревают до 75°С в течение еще 48 часов. Реакционную массу охлаждают до комнатной температуры, добавляют EtOAc и смесь промывают водным раствором 1 М NaOH и рассолом. Органическую фазу сушат (Na2SO4) и концентрируют под вакуумом, что дает сырой продукт. Его очищают препаративной ВЭЖХ (YMC S5 ODS 50×75 мм колонка; непрерывный градиент от 70:30 В:А до 100% В, где А=90:10:0.1 H2O:метанол:ТФК и В=90:10:0.1 метанол:H2O:ТФК), что дает соединение из части А (13 мг; 35%) в виде бесцветного масла.
B. 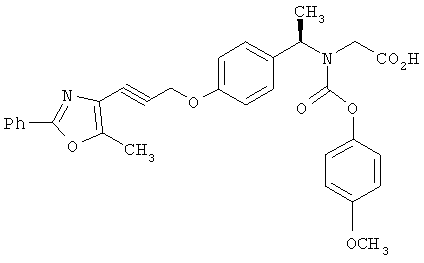
К раствору соединения из части А (12 мг; 0.021 ммоль) в ТГФ:H2О (1.5 мл, 2:1) добавляют LiOH (8 мг; 0.19 ммоль). Раствор перемешивают при комнатной температуре в течение 24 часов, затем подкисляют избытком 1 М HCl (водным раствором). Раствор экстрагируют с помощью EtOAc (2×5 мл). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом. Сырой продукт очищают препаративной ВЭЖХ, используя те же условия, что описаны выше, что дает названное соединение (6.4 мг) в виде бесцветной пленки. [М+Н]+=541.3.
Пример 561
A. 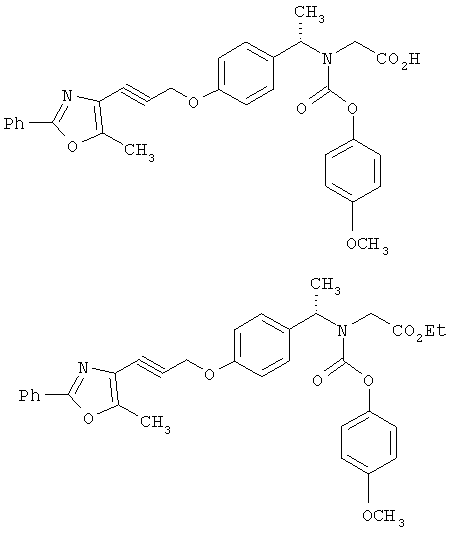
Смесь мезилата (18 мг; 0,061 ммоль)
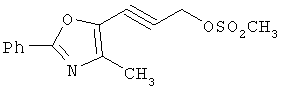
фенола (50 мг; 0.13 ммоль)
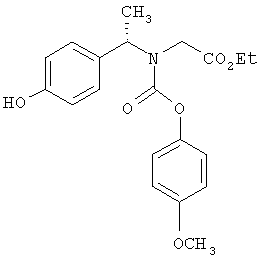
К2СО3 (17 мг; 0.34 ммоль) в СН3CN (1 мл) нагревают при 70°С в течение 24 часов. Вводят дополнительное количество К2СО3 (30 мг) и СН3CN (1 мл) и смесь нагревают до 75°С в течение еще 48 часов. Реакционную массу охлаждают до комнатной температуры, добавляют EtOAc и смесь промывают водным раствором 1 М NaOH и рассолом. Органическую фазу сушат (Na2SO4) и концентрируют под вакуумом, что дает сырой продукт. Его очищают препаративной ВЭЖХ (YMC 55 ODS 50×75 мм колонка; непрерывный градиент от 70:30 В:А до 100% В, где А=90:10:0.1 H2O:метанол:ТФК и В=90:10:0.1 метанол:H2O:ТФК), что дает соединение из части А (13 мг; 35%) в виде бесцветного масла.
B. 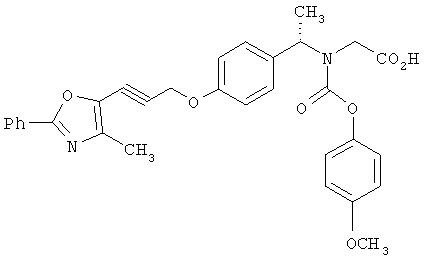
К раствору соединения из части А (12 мг; 0.021 ммоль) в ТГФ:H2O (1.5 мл, 2:1) добавляют LiOH (8 мг; 0.19 ммоль). Раствор перемешивают при комнатной температуре в течение 24 часов, затем подкисляют избытком 1 М HCl (водным раствором). Раствор экстрагируют с помощью EtOAc (2×5 мл). Объединенные органические экстракты промывают рассолом, сушат (Na2SO4) и концентрируют под вакуумом. Сырой продукт очищают препаративной ВЭЖХ, используя те же условия, что описаны выше, что дает названное соединение. [М+H]+=541.3.
Пример 562
A. 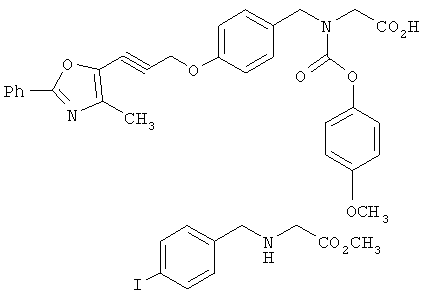
Раствор 4-йодбензальдегида (1.0 г; 4.31 ммоль) и гидрохлорида глицинметилового эфира (0.65 г; 5.17 ммоль) и Et3N (0.50 г; 4.95 ммоль) в метаноле (15 мл) перемешивают при комнатной температуре в течение 4 часов. Смесь охлаждают до 0°С и добавляют порциями раствор NaBH4 (230 мг; 6.0 ммоль) в метаноле. Смеси дают нагреться до комнатной температуры и перемешивают в течение ночи при комнатной температуре. Летучие продукты удаляют под вакуумом (без нагревания) и остаток разделяют между водным раствором NaHCO3 и EtOAc. Водную фазу экстрагируют с помощью EtOAc (3x). Объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом, что дает соединение, часть А, в виде масла. Полученный продукт используют на следующей стадии без дальнейшей очистки.
B. 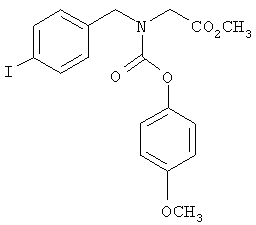
К раствору сырого соединения из части А и Et3N (0.80 г; 8.00 ммоль) в CH2Cl2 добавляют раствор 4-метоксифенилхлорформиата (0.93 г; 5.00 ммоль) в CH2Cl2. Реакционную смесь перемешивают при комнатной температуре в течение ночи, затем разделяют между насыщенный водным раствором NaHCO3 и EtOAc. Водную фазу экстрагируют с помощью EtOAc (2х); объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом, что дает остаток, который хроматографируют (SiO2; гексан:EtOAc, 3:1), что дает соединение, часть В (1.2 г; 61%) в виде масла.
C. 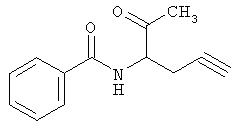
К охлажденному до 0°С раствору DL-пропаргилглицина (3.0 г; 26.5 ммоль) в пиридине (20 мл; 247 ммоль) добавляют по каплям бензоилхлорид (3.73 г; 26.5 ммоль). Раствору дают нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 1 часа. Добавляют уксусный ангидрид (10 мл) и смесь перемешивают при 90°С в течение 2 часов. Реакционную смесь разбавляют H2O (35 мл) и экстрагируют с помощью EtOAc (3x); объединенные органические экстракты промывают водным раствором 1 н. HCl, H2O, водным раствором NaHCO3 и наконец водой. Органическую фазу сушат (Na2SO4) и концентрируют под вакуумом. Сырой продукт хроматографируют (SiO2; постепенный градиент от 5:1 до 3:1, гексан:EtOAc), что дает соединение, часть С (1.0 г; 17%) в виде оранжевого твердого вещества.
D. 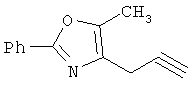
Раствор соединения из части С (1.0 г; 4.65 ммоль), трифторуксусный ангидрид (3 мл) и ТФК (3 мл) в герметично закрываемой трубке нагревают до 40°С в течение 8 часов. Летучие продукты удаляют под вакуумом и остаток растворяют в EtOAc (50 мл). Раствор промывают несколько раз насыщенным водным раствором NaHCO3 (пока вся кислота не будет удалена из органической фазы), затем рассолом, сушат (Na2SO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; 6:1, гексан:EtOAc), что дает соединение, часть D (800 мг, 87%; >98% чистота по ВЭЖХ) в виде масла.
E. 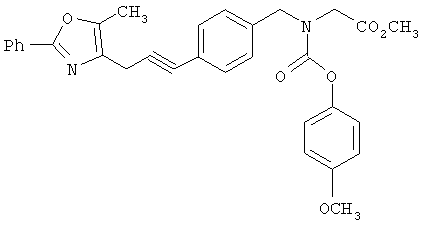
Смесь соединения из части D (100 мг; 0.507 ммоль), соединение, часть В (254 мг; 0.558 ммоль), Cul (2 мг; 0.01 ммоль) и (Ph3P)2PdCl2 (4 мг; 0.005 ммоль) в диэтиламине (2 мл) перемешивают при комнатной температуре в течение 3 часов в атмосфере N2. В этот момент ВЭЖХ/MS показывает, что весь исходный продукт вступил в реакцию, при этом наблюдается появление пика, который соответствует желаемому продукту. Реакционную смесь фильтруют и фильтрат концентрируют под вакуумом. Остаток хроматографируют (SlO2; постепенный градиент от 5:1 до 2:1, гексан:EtOAc), что обеспечивает соединение, часть Е (200 мг; 75%) в виде масла.
F. 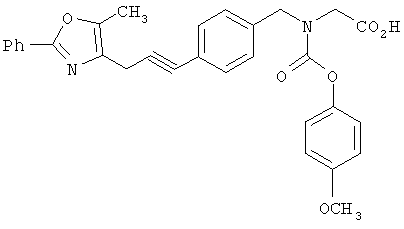
Раствор соединения из части Е (20 мг; 0.038 моль) в НОАс/конц. HCl (1 мл, 10:1 раствор) перемешивают при 45°С в течение ночи. Летучие продукты удаляют под вакуумом и остаток очищают препаративной ВЭЖХ (YMC S5 ODS, обращенная фаза, колонка 30×250 мм, скорость потока 25 мл/мин; 30 мин непрерывный градиент от 50:50 А:В до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O:ТФК), что дает названное соединение (6.8 мг; 35%) в виде лиофилата. [М+Н]+=511.2.
Пример 563
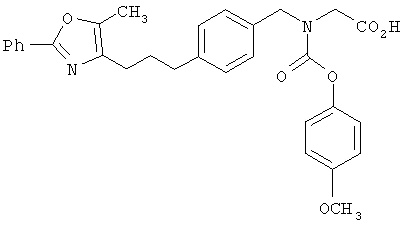
A.
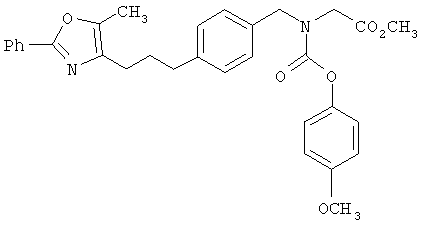
Раствор соединения по примеру 562, часть Е,
(38 мг; 0.072 ммоль) в метаноле (5 мл) перемешивают в атмосфере Н2 в присутствии 10% Pd/C катализатора (10 мг) при комнатной температуре в течение 2 часов. Катализатор отфильтровывают и фильтрат концентрируют под вакуумом, что дает соединение из части А (35 мг; 92%) в виде масла.
B.
Раствор соединения из части А (35 мг; 0.066 ммоль) в водном растворе LiOH (1 мл 1 M раствора) и ТГФ (5 мл) перемешивают при комнатной температуре в течение 2 часов. Реакцию подкисляют до рН 3 избытком водного раствора 1 M HCl и экстрагируют с помощью EtOAc (2×5 мл). Объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом. Остаток очищают препаративной ВЭЖХ (YMC S5 ODS колонка с обращенной фазой, 30×250 мм, скорость потока 25 мл/мин; 30 мин непрерывный градиент от 50:50 А:В до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O:ТФК), что дает, после лиофилизации из диоксана, названное соединение (31 мг; 87%) в виде белого твердого вещества. [М+Н]+=515.9.
Пример 564

Раствор соединения по примеру 562, часть Е,
(20 мг; 0.038 ммоль) и водный раствор LiOH (1 мл 1 М раствора; 1 ммоль) в ТГФ (2 мл) перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь подкисляют избытком водного раствора 1 М HCl и экстрагируют с помощью EtOAc. Объединенные органические экстракты концентрируют под вакуумом. Остаток очищают препаративной ВЭЖХ (YMC S5 ODS колонка с обращенной фазой, 30×250 мм, скорость потока 25 мл/мин; 30 мин непрерывный градиент от 50:50 А:В до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O:ТФК), что дает (9 мг; 46%) в виде белого твердого вещества. [М+Н]+=511,2.
Пример 565
A. 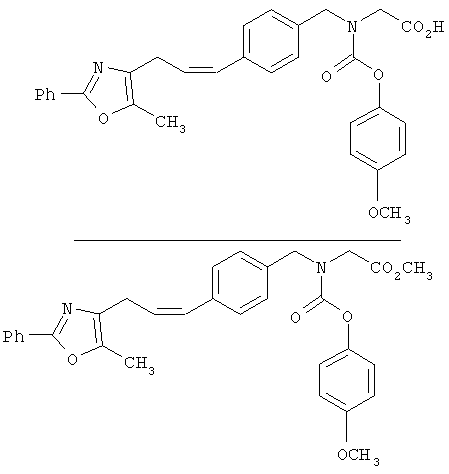
Смесь соединения по примеру 562, часть Е,
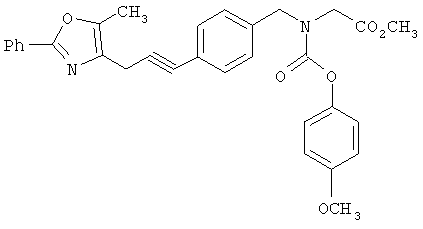
(80 мг; 0.15 ммоль), хинолина (2 мкл; 0.01 ммоль) и катализатора Линдлара (7 мг; 5% Pd/СаСО3) в толуоле (2 мл) перемешивают в атмосфере H2 в течение 2 часов. Вводят дополнительное количество катализатора Линдлара (20 мг) и продолжают перемешивание в атмосфере Н2 в течение еще 2 часов, после которого реакция завершена, как показывает аналитическая ВЭЖХ. Реакционную смесь фильтруют (Celite®) и фильтрат концентрируют под вакуумом. Остаток хроматографируют (SiO2; постепенный градиент от 3:1 до 2:1, гексан:EtOAc), что дает соединение из части А.
B. 
Раствор соединения из части А и водный раствор LiOH (1 мл 1 М раствора; 1 ммоль) в ТГФ перемешивают при комнатной температуре в течение ночи. Реакционную смесь подкисляют избытком водного раствора 1 М HCl и экстрагируют с помощью EtOAc (2х). Объединенные органические экстракты концентрируют под вакуумом. Остаток очищают препаративной ВЭЖХ (как в примере 495), что дает названное соединение (14 мг; 18%) в виде белого твердого вещества. [М+Н]+=513.3.
Пример 566 (рацемат)
A. 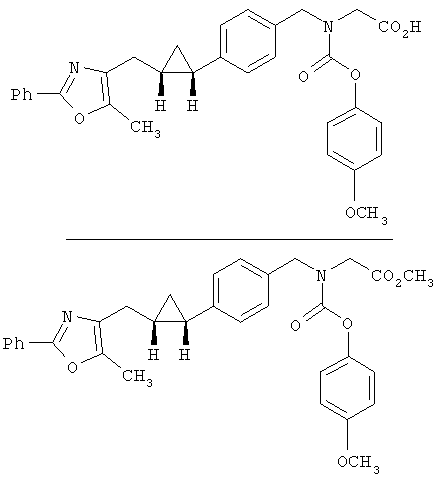
К охлажденному до 0°С раствору соединения по примеру 565, часть А,

(60 мг; 0.11 ммоль) в DCE (3 мл) добавляют по каплям диэтиловый цинк (43 мкл; 0.29 ммоль). Раствор перемешивают при 0°С в течение 10 мин и добавляют йодхлорметан (244 мкл; 0.57 ммоль), дают нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 3 часов, затем осторожно захолаживают, добавляя водный раствор HCl (1 мл 1 М раствора). Водный слой экстрагируют с помощью EtOAc (2x); объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом. Остаток хроматографируют (SiO2; постепенный градиент от 3:1 до 2:1, гексан: EtOAc ), что дает сырое соединение, часть А, которое используют на следующей стадии без дальнейшей очистки.
B. 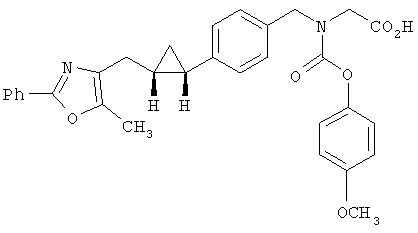
Раствор сырого соединения, часть А, и водного раствора LiOH (1 мл 1 М раствора; 1 ммоль) в ТГФ перемешивают при комнатной температуре в течение ночи. Реакционную смесь подкисляют избытком водного раствора 1 М HCl и экстрагируют с помощью EtOAc. Объединенные органические экстракты концентрируют под вакуумом. Остаток очищают препаративной ВЭЖХ (условия), что дает названное соединение (7 мг; 12% после 2 стадий) в виде белого твердого вещества. [М+Н]+=527.2.
Пример 567
A. 
Смесь соединения по примеру 562, часть D, и соединения
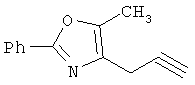
(300 мг; 1.52 ммоль), N-бромсукцинимида (297 мг; 1.67 ммоль) и AgNO3 (28 мг; 0.19 ммоль) в ацетоне (2 мл) перемешивают при комнатной температуре в течение 30 мин. Смесь фильтруют и фильтрат концентрируют под вакуумом. Остаток хроматографируют (SiO2; гексан:EtOAc, 5:1), что дает соединение из части А (320 мг; 76%) в виде желтых кристаллов.
B. 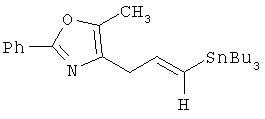
К раствору соединения из части А (320 мг; 1.2 ммоль), Ph3Р (13 мг; 0.05 ммоль) и трис(дибензилиденацетон)дипалладия(0) (5 мг; 0.006 ммоль) в ТГФ (1 мл) добавляют Bu3SnH (700 мкл; 2.5 ммоль) по каплям в атмосфере N2. Смесь перемешивают при комнатной температуре в течение 2 часов, затем захолаживают водным раствором KF (7 мл 1 М раствора). После чего ее энергично перемешивают в течение ночи, затем экстрагируют с помощью EtOAc (2x). Объединенные органические экстракты промывают H2O, сушат (Na2SO4) и концентрируют под вакуумом. Оставшееся масло хроматографируют (SiO2; гексан:EtOAc, 3:1), что дает соединение, часть В (200 мг; 35%) в виде масла. Кроме того, также получают побочный продукт, виниловое соединение
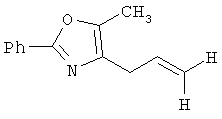
(100 мг; 43%).
C. 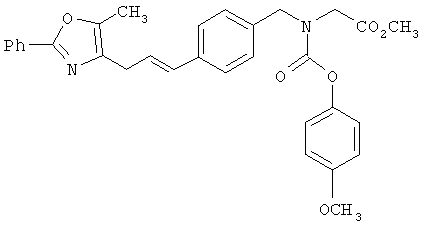
К раствору соединения из части В (100 мг; 0.020 ммоль) и соединения по примеру 562 из части В
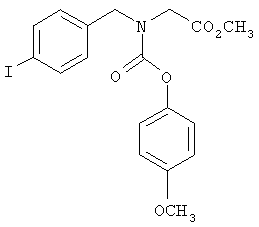
(100 мг; 0.22 ммоль) и (Ph3Р)4Pd° (3 мг; 0.002 ммоль) в толуоле нагревают до 100°С в течение ночи в атмосфере N2. Летучие продукты удаляют под вакуумом и остаток хроматографируют (SiO2; постепенный градиент от 3:1 до 2:1, гексан:EtOAc ), что дает соединение, часть С.
D. 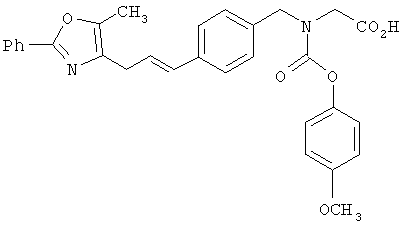
Раствор сырого соединения из части С в водном растворе LiOH (1 мл 1 М раствора) и ТГФ (5 мл) перемешивают при комнатной температуре в течение ночи. Реакционную массу подкисляют до рН 3 избытком водного раствора 1 М HCl и экстрагируют с помощью EtOAc (2×5 мл). Объединенные органические экстракты сушат (Na2SO4) и концентрируют под вакуумом. Остаток очищают препаративной ВЭЖХ (YMC S5 ODS колонка с обращенной фазой, 30×250 мм, скорость потока 25 мл/мин; 30 мин непрерывный градиент от 50:50 А:В до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O:ТФК) что дает, после лиофилизации из диоксана, названное соединение (23 мг; 20%) в виде белого твердого вещества. [M+H]+=513.3.
Примеры с 568 по 572
Следуя процессам, определенным выше и в рабочих примерах, получают следующие соединения.
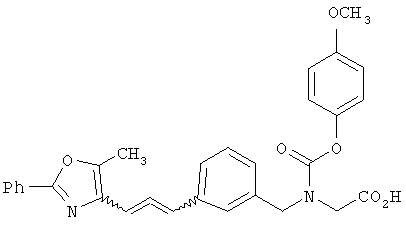
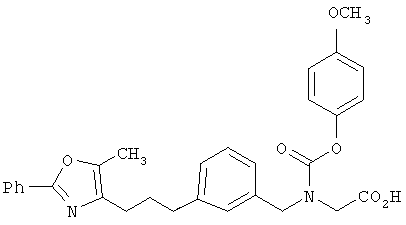
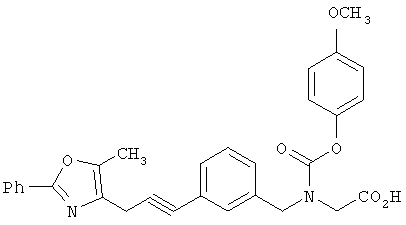
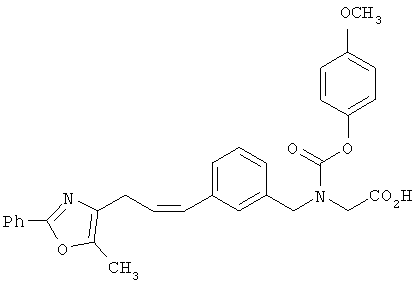
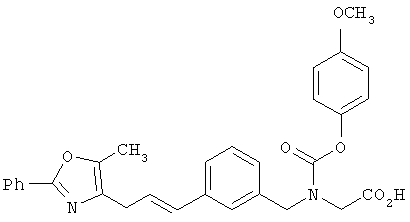
Пример 573
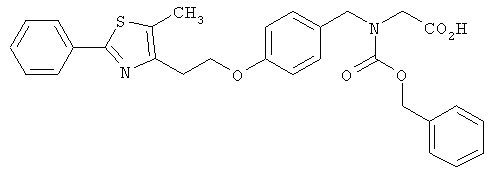
A.
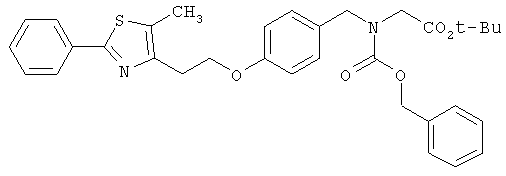
К смеси аминоэфира (27 мг; 0.073 ммоль)
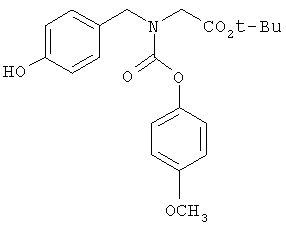
5-метил-2-фенилтиазол-4-илэтанола (25 мг; 0.11 ммоль; Maybridge), связанного со смолой Ph3Р (27 мг; 0.081 ммоль), в CH2Cl2 (0.5 мл) добавляют DEAD (20 мкл; 0.13 ммоль). Реакционную массу перемешивают при комнатной температуре в течение 6 часов, затем фильтруют. Фильтрат концентрируют под вакуумом и остаток очищают препаративной ВЭЖХ (YMC S5 ODS 30×100 мм колонка, скорость потока 50 мл/мин; непрерывный градиент от 30:70 В:А до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O:ТФК), что приводит к соединению из части А.
B.
Раствор соединения из части А в ТФК (1 мл) перемешивают при комнатной температуре в течение ночи, затем концентрируют под вакуумом, чтобы получить названное соединение (11 мг; 26%) в виде коричневого масла (94% чистота аналитическая по ВЭЖХ). [М+Н]+=517.2.
Пример 574
A. 
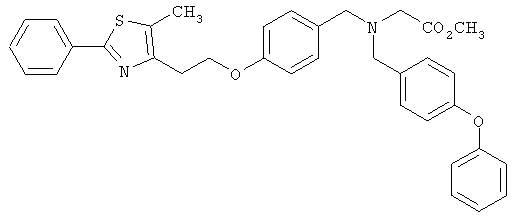
К смеси аминоэфира (31 мг; 0.082 ммоль)
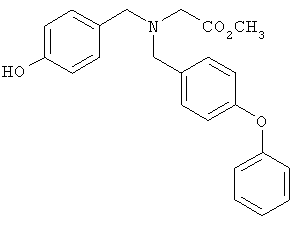
5-метил-2-фенилтиазол-4-илэтанола (25 мг; 0.11 ммоль; Maybridge), связанного со смолой Ph3Р (32 мг; 0.096 ммоль), в CH2Cl2 (0.5 мл) добавляют DEAD (20 мкл; 0.13 ммоль). Реакционную массу перемешивают при комнатной температуре в течение 6 часов, затем фильтруют. Фильтрат концентрируют под вакуумом, что дает сырое соединение, часть А.
B. 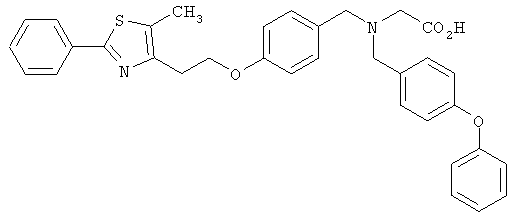
Раствор сырого соединения из части А и LiOH·H2O (20 мг; 0.48 ммоль) в ТГФ:метаноле:H2O (1 мл, 3:1:1 смесь) перемешивают при комнатной температуре в течение ночи. Реакцию подкисляют до рН˜4 водным раствором 1 н. HCl, затем экстрагируют с помощью EtOAc (2х). Объединенные органические экстракты концентрируют под вакуумом и остаток очищают препаративной ВЭЖХ (YMC S5 ODS 30×100 мм колонка, скорость потока 50 мл/мин; 10 мин непрерывный градиент от 30:70 В:А до 100% В, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O: ТФК), что позволяет получить названное соединение (16 мг; 34%) в виде коричневого масла (95% чистота аналитическая ВЭЖХ). [М+Н]+=565.2.
Пример 575
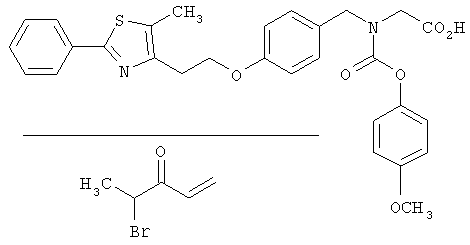
A.
К раствору 2,4-дибром-3-пентанона (Avocado Chemicals, 19.6 г, 80 ммоль) в CH2Cl2 (50 мл) добавляют по каплям Et3N (30 мл, 210 ммоль) в течение более 30 мин; полученный раствор нагревают при кипении с обратньм холодильником в течение 12 часов. Реакционную смесь охлаждают до комнатной температуры, затем выливают на лед и подкисляют концентрированной HCl. Органическую фазу концентрируют под вакуумом, что дает масло, которое перегоняют по фракциям (т.кип. 42-45°С при 13 мм рт.ст.), что дает соединение из части А (6.0 г, 46%; с ˜20% исходного продукта) в виде масла.
B. 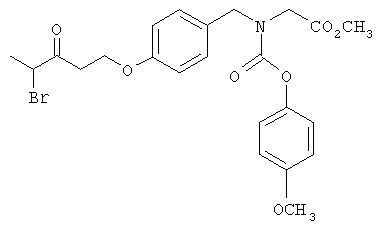
Смесь соединения по примеру 559, часть Е (0.60 г, 1.7 ммоль)
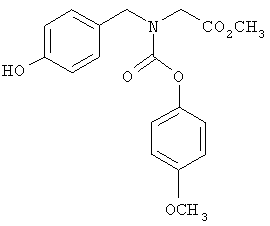
соединения из части А (0.60 г, 3.7 ммоль) и К2СО3 (1.0 г, 7.3 ммоль) в бензоле (20 мл) перемешивают при комнатной температуре в течение 12 часов. Как показывает ТСХ, в этот момент 50% исходного продукта вступает в реакцию и реакция прекращается. Реакционную смесь фильтруют и фильтрат концентрируют под вакуумом. Остаток хроматографируют (SiO2; 3% ацетон/CH2Cl2), что обеспечивает соединение, часть В (0.41 г; 47%) в виде масла.
C. 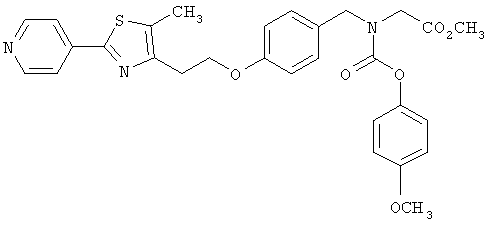
Раствор соединения из части В (40 мг, 0.080 ммоль) и тиоизоникотинамида (50 мг, 0.36 ммоль) в толуоле:EtOH (3 мл, 1:1 смесь) нагревают до 55°С в течение 12 часов. Реакционную массу охлаждают до комнатной температуры и летучие продукты удаляют под вакуумом. Сырой продукт очищают препаративной ВЭЖХ (YMC S5 ODS 30×250 мм, непрерывный 30 мин градиент от 30% В:70% A до 100% В 30 мин, где растворитель А=90:10:0.1 H2O:метанол:ТФК и растворитель В=90:10:0.1 метанол:H2O:ТФК), что дает соединение, часть С (17; 39%) в виде масла
D. 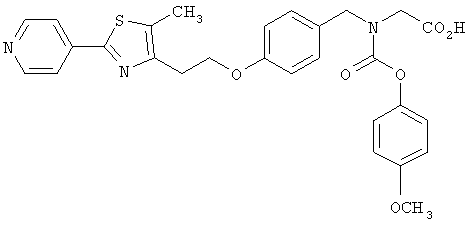
Раствор соединения из части С (17 мг, 0.031 ммоль) и LiOH·H2O (40 мг, 1 ммоль) в ТГФ:H2O (3 мл, 2:1 смесь) перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь подкисляют, добавляя уксусную кислоту, и затем разделяют между H2O (2 мл) и EtOAc (5 мл). Органическую фазу сушат (MgSO4) и концентрируют под вакуумом, что обеспечивает названное соединение (13.7 мг, 81%) в виде белого твердого вещества. [М+H]+=534.2.
Примеры с 576 по 580
Следуя процессам, приведенным выше и рабочим примерам, получают следующие соединения.

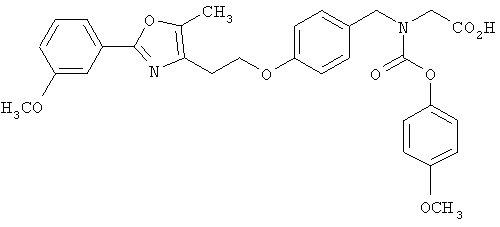
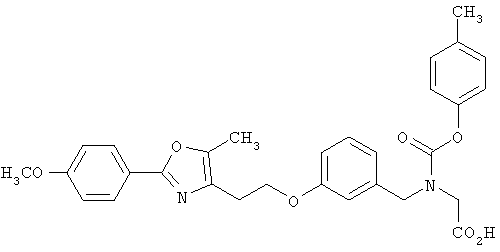
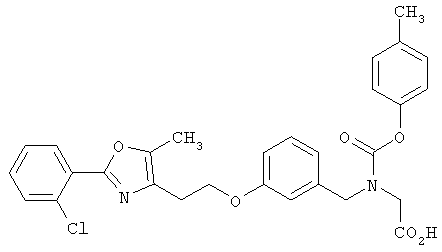
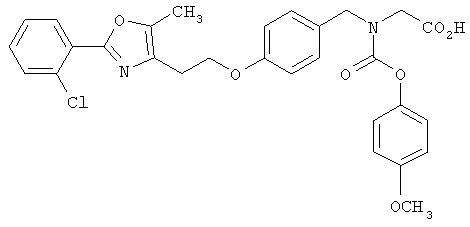
Соединения по примерам 581 и 582 синтезируют согласно общим процессам, раскрытым в примерах 313 и 314.

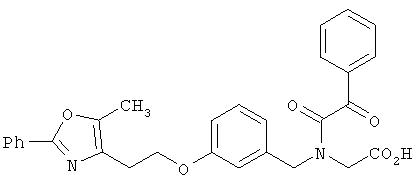
Соединения согласно примерам 583 и 584 синтезируют в соответствии с общими методиками, раскрытыми выше (например, для примера 139), используя 4-метокситиофенол.
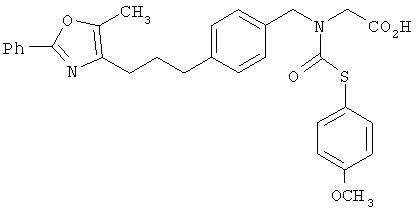
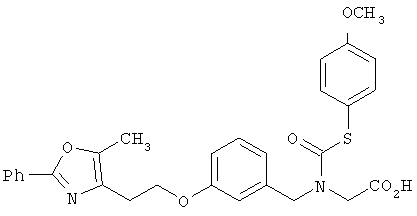
Пример 584
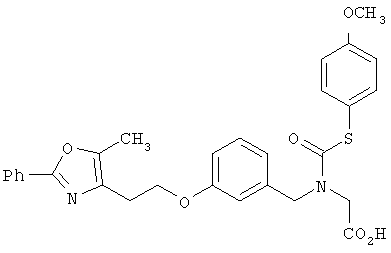
1H ЯМР (CDCl3; 400 МГц): δ 2.42 (с, 3H), 3.04 (расширенный с; 2Н), 3.79 (с, 3H), 4.03 (расширенный с, 2Н), 4.25 (расширенный с, 2Н), 4.70 (расширенный с, 2Н), 6.8-7.0 (м, 5Н), 7.15-7.30 (м, 1Н), 7.35-7.50 (м, 5Н), 7.95-8.05 (м, 2Н); 8.95 (расширенный с, 1Н).
Прилагаются данные, демонстрирующие PPARα и PPARγ активность соединений по изобретению, заявленные BMS 711939 и BMS 687453, имеющие структуры
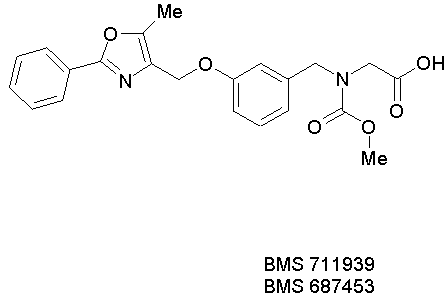
Данные получены с помощью GAL4 анализа и FP анализа, в которых BMS 711939 и BMS 687453 были проверены. Копия каждого из анализов и полученные данные прилагаются.
Термин "стандарт", относящийся к GAL4 анализу, представляет собой BMS 582989, имеющий структуру
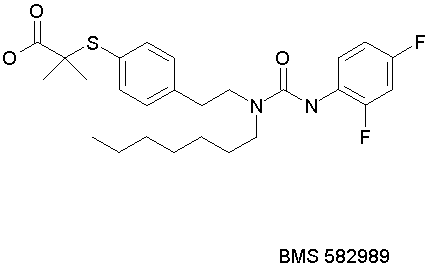
Данные FP и GAL4 анализов показывают, что вышеприведенные соединения обладают как PPARα, так и PPARγ активностью и, следовательно, пригодны для лечения диабета (PPARγ агонистическая активность), и аномалий липопротеинов (PPARα агонистическая активность).
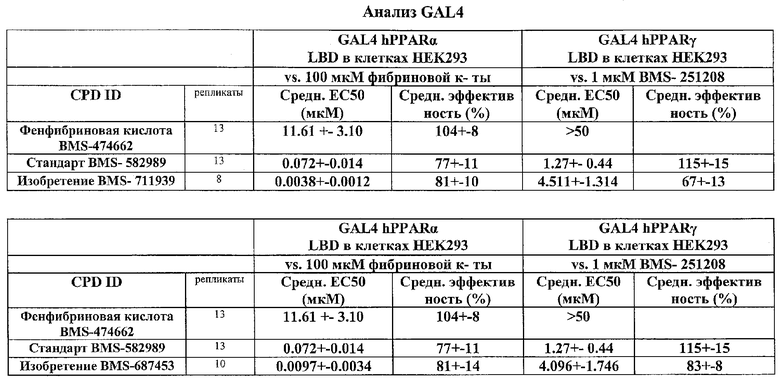

Краткое описание анализа
Соединения BMS-687453 и 711939 замещают меченый PPARα агонист, вытесняя его из очищенного PPARα белка, что измеряется флуоресцентной поляризацией, и, следовательно, являются лигандами для PPARα.
При анализе трансактивации в НЕК с применением лигандсвязывающего домена PPARα, гибридизованного с доменом связывания ДНК GAL4 дрожжей, оба соединения проявляют опосредованную PPARα индукцию гена в зависимости от дозы. Следовательно, BMS-687453 и 711939 являются агонистами PPARα.
Анализы трансфекции GAL4- PPARα
Материалы и методы
Среда OptiMEM I (Gibco/BRL #51985).
Клетки НЕК293 5×G4RE-Luc клона R2.8 (клетки).
Активированный уголь/декстран, обработанный фетальной телячьей сывороткой (csFCS) (Hyclone #SH-3006S.O3).
Белые 96-луночные планшеты Corning Costar с прозрачным дном (Corning #3903).
Белый 96-луночный планшет с U-образным дном (Becton Dickinson 353263).
Липофектамин 2000 (Invitrogen #11668).
Зеоцин (Invitrogen #R-250).
Пенициллин/стрептомицин (P/S) (Gibco/BRL #15140).
Трипсин EDTA (Gibco/BRL #25300).
DMEM (Gibco/BRL #12430).
DMEM без фенола красного (Gibco/BRL #21063).
Плазмидная ДНК GAL4 - внеклеточный рецептор LBD (1 мкг/мкл).
Аналитическая система Promega Steady-Glo Luciferase (E 2550).
Скотч Packard (#6005185).
Колбы Т-225 (Corning #431082).
Тестирование BMS-711939 и BMS-687453 методом анализа
НЕК293 GAL4-человеческий PPARα-LBD
BMS-711939 и BMS-687453 анализируют, используя устойчиво трансфицированные эмбриональные почечные клетки человека 293. Клетки устойчиво экспрессируют химерные конструкции, состоящие из синтетического промотора с пятью тандемными повторами сайта связывания GAL4 дрожжей, контролирующие экспрессию гена люциферазы Photinus pyralis (Американского светляка). Затем клетки транзиторно трансфицируют с применением плазмиды (pcDNA3.1), состоящей из химерной конструкции ДНК-связывающего домена GAL4 дрожжей (аминокислоты 1-441) в направлении 5' (upstream) от лигандсвязывающего домена рецептора, активированного пролифератором пероксисомы (PPAR)-α (аминокислоты 167-468) с последовательностью, зарегистрированной в GenBank под номером NM_005036.2. При стимуляции агонистами PPAR-α клетки экспрессируют белок люциферазы в зависимости от дозы (доза-эффект). Затем определяют люциферазную активность с помощью лизиса клеток и детектирования люминесценции побочного продукта катализа люциферазой субстрата люциферина. Транзиторно трансфицированные клетки стимулируют в присутствии и в отсутствие BMS-711939 и BMS-687453 в течение 20 часов, после чего клетки лизируют и анализируют на присутствие люциферазной ферментной активности.
Тестирование BMS-711939 и BMS-687453 методом анализа НЕК293 GAL4-человеческий PPAR-γ-LBD
BMS-711939 и BMS-687453 анализируют, используя устойчиво трансфицированные эмбриональные почечные клетки человека 293, как описано для анализа GAL4-человеческий PPARα-LBD. Клетки последовательно транзиторно трансфицируют с применением плазмиды (pcDNA3.1), состоящей из химерной конструкции ДНК-связывающего домена GAL4 дрожжей (аминокислоты 1-441) в направлении 5' (upstream) от лигандсвязывающего домена рецептора, активированного пролифератором пероксисомы (PPAR)-γ (аминокислоты 176-477) с последовательностью, зарегистрированной в GenBank под номером L40904. При стимуляции агонистами PPAR-γ клетки экспрессируют белок люциферазы в зависимости от дозы (доза-эффект). Затем определяют люциферазную активность с помощью лизиса клеток и детектирования люминесценции побочного продукта катализа люциферазой субстрата люциферина. Транзиторно трансфицированные клетки стимулируют в присутствии и в отсутствие BMS-711939 и BMS-687453 в течение 20 часов, после чего клетки лизируют и анализируют на присутствие люциферазной ферментной активности.
Культивирование и сохранение клеток
Клетки сохраняют в DMEM при 37°С и 5% CO2 в колбах Т-225 с 1% P/S, 500 мкг/мл зеоцина и 10% csFBS. Клетки при 90% монослоя обрабатывают трипсином. Клетки осторожно диспергируют, разводят в DMEM и центрифугируют при 1000 об/мин в течение 5 минут. Клеточный осадок (пеллеты) ресуспендируют в 3-5 мл DMEM. Клетки считают и исходный клеточный раствор разводят до 3.07×105 клеток/мл. Клетки засевают с помощью многоканальной пипетки (в DMEM без фенола красного с 10% csFBS, 1% P/S) в непрозрачные планшеты с прозрачным дном, 130 мкл/лунка, конечное число клеток 4×104 клеток/лунка.
Трансфекция
Все плазмиды берут в виде исходного раствора 1 мг/мл или делают поправку на вариант
Стадия 1. Разводят липофектамин 2000 в OptiMEM I до концентрации 20 мкл/мл. Готовят 24 мл на 96-луночный планшет.
Стадия 2. Разводят плазмидную ДНК в OptiMEM I до концентрации 8 мкг/мл. Готовят 2.4 мл на 96-луночный планшет.
Стадия 3. Инкубируют в растворе липофектамина 2000 при комнатной температуре в течение 5 мин, но не более 30 мин, перед тем как перейти к стадии 4.
Стадия 4. Смешивают ДНК и липофектамин вместе в соотношении 1:1.
Стадия 5. Инкубируют при комнатной температуре в течение 20 мин (производитель (Invitrogen) сообщает, что этот комплекс устойчив, по меньшей мере, в течение 6 часов после смешения).
Стадия 6. Добавляют 50 мкл смеси ДНК/липофектамин 2000 в каждую лунку клеточного планшета с помощью прибора для многоканального (multidrop) засевания (многоканальной пипеткой).
Стадия 7. Помещают клетки в инкубатор при 37°С и 5% CO2 на 4.5-5 часов.
Стадия 8. Добавляют 20 мкл соединения, разведенного в DMEM без фенола красного, но с 1% P/S и 10% csFBS (5% ДМСО).
Стадия 9. Помещают клетки в инкубатор при 37°С и 5% CO2 на 20-22 часа.
Стадия 10. См. следующий абзац.
Готовят реагенты Steady-Glo, как написано в инструкциях к набору, за исключением того, что раствор следует разводить 1:1 в DMEM без фенола красного. Кондиционированные среды во всех лунках отсасывают и добавляют 100 мкл смеси 1:1. (Примечание: при автоматическом анализе для отсасывания кондиционированных сред применяют автоматическое устройство для промывки планшетов (вошер)/аспиратор, оставляющее 60 мкл, к которым затем прибавляют 60 мкл 100% реагента Steady-Glo). Плотно закрывают планшеты с помощью прозрачной самоклеящейся ленты (скотча) Packard (или ее эквивалента) и оставляют стоять при комнатной температуре 20 минут, а затем считывают на Topcount со скоростью 5 сек/лунка.
Испытуемые соединения, 32, включая BMS-711939 и BMS-687453, серийно разводят 3 раза в чистом ДМСО, всего 10 разведений (10 точек). Соединения тестируют в двойном повторе в том же планшете и нормализуют, вычитая фоновое значение (носителя), а затем делят на активность общепризнанных (известных) агонистов для анализа (100 мкМ BMS-474662 (фенофибриновая кислота) для PPAR альфа и 1 мкМ BMS-251208 (BRL-49653) для PPAR гамма). Затем данные выражают в виде ЕС50, рассчитывая с помощью программы XLfit 4 подгонки параметра 205 и с плавающими всеми параметрами.
Флуоресцентная поляризация
Материалы и методы
Экспрессия и очистка белка
Белки PPAR-LBD экспрессируют и очищают с помощью GE&PB (Bryson group, LVL). Короче говоря, экспрессию проводят в клетках BL21(DE3) в минимальных средах, дополненных казаминокислотами и другими питательными веществами. Неиндуцированные клетки охлаждают на льду в течение 30 минут и при OD600 от 0.6 до 0.8 индуцируют, добавляя 0.4 мМ IPTG, и инкубируют в течение 16 часов при 20°С. Клетки собирают центрифугированием. Клеточный осадок (пеллеты) лизируют, затем очищают, комбинируя колонку с хелатирующим агентом (Ni2+ионы) и с Q-сефарозой HP после удаления His метки (хвоста) с помощью тромбина.
hPPAR-α-LBD
Соединения применяют в серийном разведении 1:3 в чистом ДМСО. 500 нл чистого исходного ДМСО раствора переносят непосредственно в планшет для анализа с помощью приспособления (роботизированной станции) для многоканального переноса жидкостей CyBi-Well (Cybio). Затем соединения инкубируют в течение 1 час в присутствии 10 нМ BMS-312590 и 119 нг/лунка hPPARα-LBD в буферном растворе, содержащем 10 мМ Tris-HCl (рН 8.0), 50 мМ KCl, 1 мМ CHAPS и 2 мМ DTT. Конечная концентрация ДМСО в анализе составляет 5.5%. Ингибирование в процентах рассчитывают относительно 250 мкМ немеченого GW2331, который используют в качестве неспецифического контроля. Данные флуоресцентной поляризации регистрируют на анализаторе LJL Analyst (Molecular Devices) при длине волны возбуждения 485 нм и длине волны испускания (эмиссии) 530 нм. Значения IC50, коэффициента Хилла, максимального и минимального ингибирования рассчитывают с помощью программы XLfit (IDBS).
hPPAR-γ-LBD
Соединения применяют в серийном разведении 1:3 в чистом ДМСО. 500 нл чистого исходного ДМСО раствора переносят непосредственно в планшет для анализа с помощью приспособления (роботизированной станции) для многоканального переноса жидкостей CyBi-Well (Cybio). Затем соединения инкубируют в течение 1 час в присутствии 25 нМ BMS-312590 и 11 нг/лунка hPPARγ-LBD в буферном растворе, содержащем 10 мМ Tris-HCl, (рН 8.0), 50 мМ KCl, 1 мМ CHAPS и 2 мМ DTT. Конечная концентрация ДМСО в анализе составляет 5.5%. Ингибирование в процентах рассчитывают относительно 50 мкМ немеченого розиглитазона, который используют в качестве неспецифического контроля. Данные флуоресцентной поляризации регистрируют на анализаторе LJL Analyst (Molecular Devices) при длине волны возбуждения 485 нм и длине волны испускания (эмиссии) 530 нм. Значения IC50, коэффициента Хилла, максимального и минимального ингибирования рассчитывают с помощью программы XLfit (IDBS).
| название | год | авторы | номер документа |
|---|---|---|---|
| ОКСА- И ТИАЗОЛПРОИЗВОДНЫЕ В КАЧЕСТВЕ АНТИДИАБЕТИЧЕСКИХ АГЕНТОВ И АГЕНТОВ ПРОТИВ ОЖИРЕНИЯ | 2000 |
|
RU2279427C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ПОВЫШЕННОЙ ЭФФЕКТИВНОСТЬЮ, СВЯЗЫВАЮЩИЕСЯ С РЕЦЕПТОРОМ ХЕМОКИНА | 2002 |
|
RU2325387C2 |
| С-АРИЛГЛЮКОЗИДНЫЕ ИНГИБИТОРЫ SGLT2 | 2000 |
|
RU2262507C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА КАК ИНГИБИТОРЫ ПРЕНИЛТРАНСФЕРАЗЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 1999 |
|
RU2241712C9 |
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV НА ОСНОВЕ КОНДЕНСИРОВАННЫХ ЦИКЛОПРОПИЛПИРРОЛИДИНОВ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2001 |
|
RU2286986C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИЙ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2290179C2 |
| НОВЫЕ ЦИКЛИЧЕСКИЕ ПЕПТИДНЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2436795C2 |
| 4-ФЕНИЛЗАМЕЩЕННЫЕ ТЕТРАГИДРОИЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2301808C2 |
| НОВЫЕ ГИДРОФИЛЬНЫЕ ПРОИЗВОДНЫЕ 2-АРИЛ-4-ХИНОЛОНОВ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2007 |
|
RU2424245C2 |
| ТРИЦИКЛИЧЕСКИЕ ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2293734C9 |
Изобретение относится к окса- и тиазолпроизводным общей формулы

и их стереоизомерам и фармацевтически приемлемым солям, обладающим PPARα и PPARγ активностью. Соединения могут найти применение для лечения заболеваний, опосредованных PPARα и PPARγ активностью, например диабета и аномалий липопротеинов. В общей формуле x имеет значение 1, 2, 3 или 4; m имеет значение 1 или 2; n имеет значение 1 или 2; Q представляет собой С или N; А представляет собой О или S; Z представляет собой О или связь; R1 представляет собой Н или С1-8алкил; X представляет собой СН; R2 представляет собой Н; R2a, R2b и R2c могут быть одинаковыми или различными и их выбирают из Н, алкокси, галогена; R3 представляет собой арилоксикарбонил, алкилоксикарбонил, алкил(галоген)арилоксикарбонил, алкилокси(галоген)арилоксикарбонил, циклоалкиларилоксикарбонил, циклоалкилоксиарилоксикарбонил, арилкарбониламино, алкилсульфонил, циклогетероалкилоксикарбонил, гетероарилалкенил, алкоксиарилоксикарбонил, арилалкилоксикарбонил, алкиларилоксикарбонил, галогеналкоксиарилоксикарбонил, алкоксикарбониларилоксикарбонил, арилалкенилоксикарбонил, арилоксиарилалкилоксикарбонил, арилалкенилсульфонил, гетероарилсульфонил, арилсульфонил, арилалкениларилалкил, арилалкоксикарбонилгетероарилалкил, гетероарилоксиарилалкил, где алкил представляет собой С1-8алкил; Y представляет собой CO2R4, где R4 представляет собой Н или С1-8алкил; включая все их стереоизомеры и фармацевтически приемлемые соли, при условии, что если А представляет собой О, тогда R3 не является арилоксикарбонилом или алкоксиарилоксикарбонилом. 1 н. и 9 з.п. ф-лы, 12 табл.
(57) 1. Соединение формулы
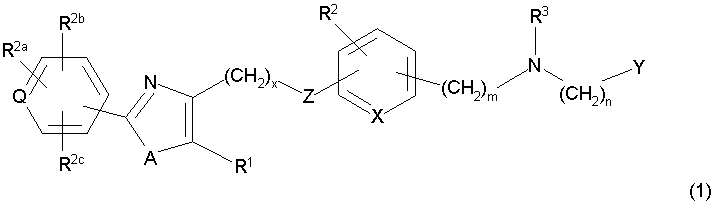
где x имеет значение 1, 2, 3 или 4; m имеет значение 1 или 2; п имеет значение 1 или 2;
Q представляет собой С или N;
А представляет собой О или S;
Z представляет собой О или связь;
R1 представляет собой Н или С1-8алкил;
X представляет собой СН;
R2 представляет собой Н;
R2a, R2b и R2c могут быть одинаковыми или различными и их выбирают из Н, алкокси, галогена;
R3 представляет собой арилоксикарбонил, алкилоксикарбонил, алкил(галоген)арилоксикарбонил, алкилокси(галоген)арилоксикарбонил, циклоалкиларилоксикарбонил, циклоалкилоксиарилоксикарбонил, арилкарбониламино, алкилсульфонил, циклогетероалкилоксикарбонил, гетероарилалкенил, алкоксиарилоксикарбонил, арилалкилоксикарбонил, алкиларилоксикарбонил, галогеналкоксиарилоксикарбонил, алкоксикарбониларилоксикарбонил, арилалкенилоксикарбонил, арилоксиарилалкилоксикарбонил, арилалкенилсульфонил, гетероарилсульфонил, арилсульфонил, арилалкениларилалкил, арилалкоксикарбонил-гетероарилалкил, гетероарилоксиарилалкил, где алкил представляет собой С1-8алкил;
циклоалкил обозначает насыщенные или частично ненасыщенные (содержащие 1 или 2 двойные связи) циклические углеводородные группы, содержащие от 1 до 3 колец, включая моноциклоалкил, бициклоалкил и трициклоалкил, содержащие в общем от 3 до 20 атомов углерода, образующих кольца, предпочтительно от 3 до 10 атомов углерода, образующих кольца;
арил обозначает моноциклические или бициклические ароматические группы, содержащие от 6 до 10 атомов углерода в кольце, выбранном из фенила или нафтила, включая 1-нафтил и 2-нафтил и могут необязательно включать от одного до трех дополнительных колец, сконденсированных с карбоциклическим или гетероциклическим кольцом, выбранным из арильного, циклоалкильного, гетероарильного или циклогетероалкильного кольца;
гетероарил обозначает 5- или 6- членное ароматическое кольцо, которое содержит 1, 2, 3 или 4 гетероатома, выбранных из азота, кислорода или серы, и такие кольца сконденсированы с арильным кольцом;
циклогетероалкилалкил обозначает циклогетероалкильную группу, 5- или 6-членное насыщенное или частично ненасыщенное гетероциклическое кольцо, которое содержит 1, 2 или 3 гетероатома, выбранных из N, О или S, связанное через С атом или гетероатом с (СН2)Р цепью; р имеет значение 0 или 1;
гетероарилалкенил обозначает гетероарильную группу, связанную через С атом или гетероатом с алкиленом или алкениленом;
Y представляет собой CO2R4, где R4 представляет собой Н или С1-8алкил;
где во всех указанных группах алкил представляет собой С1-8алкил, алкенил представляет собой С2-8алкенил, алкилен представляет собой С1-8алкилен, алкенилен представляет собой С2-8алкенилен;
включая все их стереоизомеры и фармацевтически приемлемые соли, при условии, что если А представляет собой О, тогда R3 не является арилоксикарбонилом или алкоксиарилоксикарбонилом.

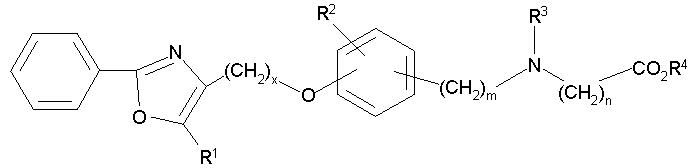
или

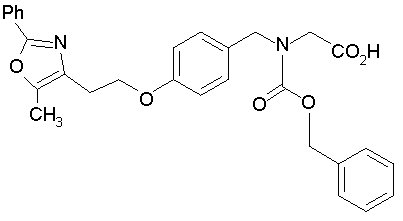 ,
, 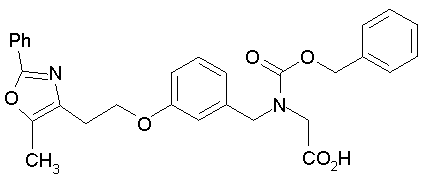
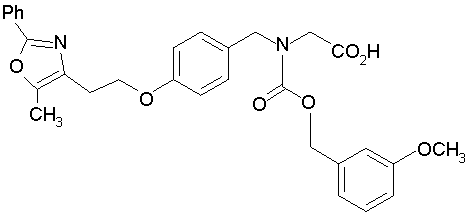
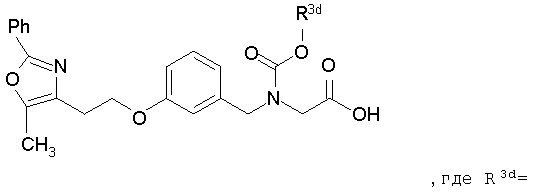
 ,
,  ,
,  ,
,  ,
,  ,
, 
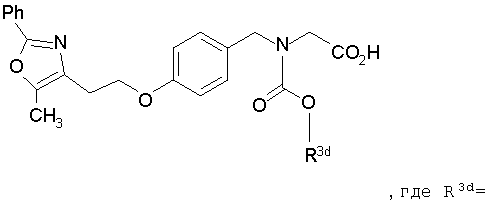
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,
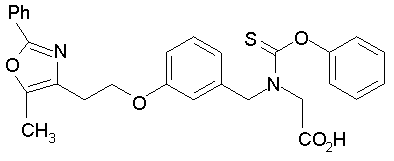 ,
, 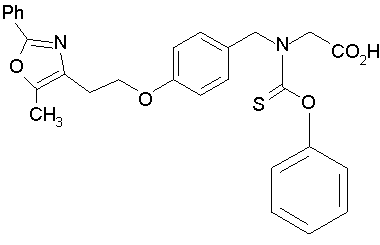 ,
,


 ,
,  ,
,  ,
,
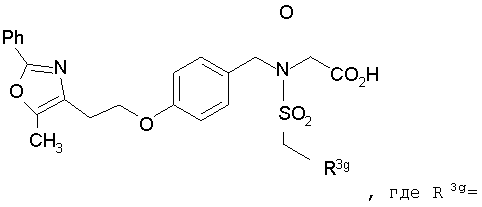
 ,
,  ,
,  , ,
, ,  ,
,  ,
,  ,
,
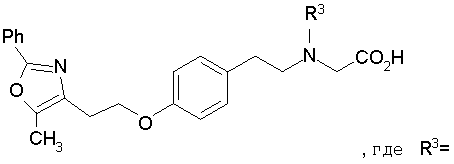
 ;
;

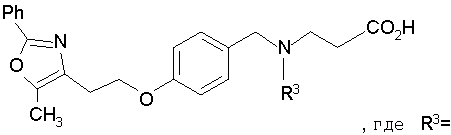 ;
;
 ,
,  ,
,  ,
, 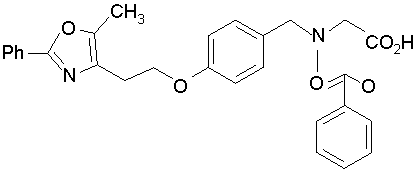 ,
, 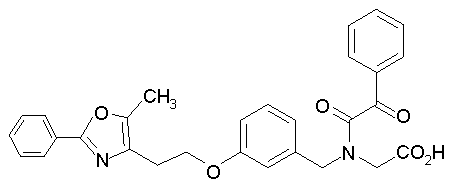
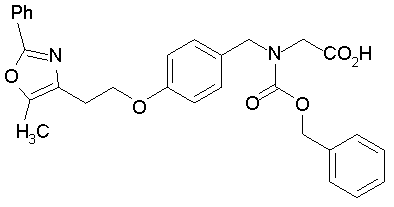

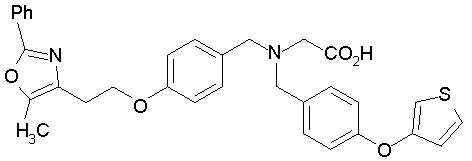
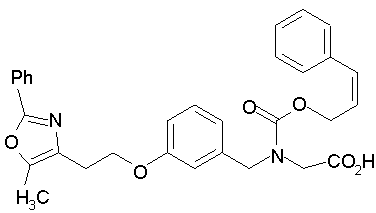
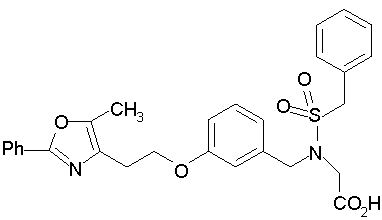
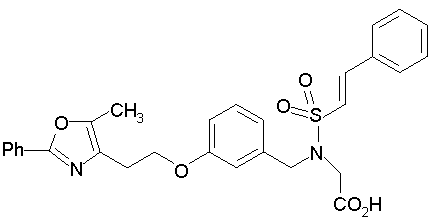
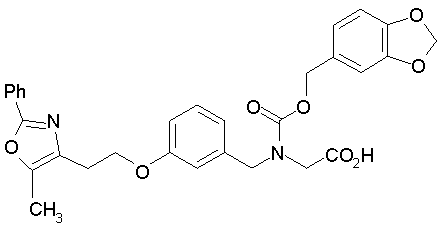
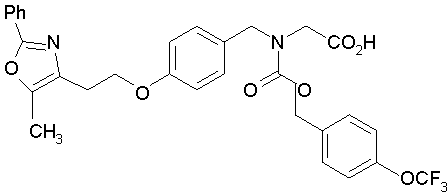
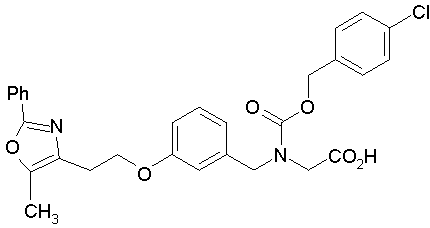

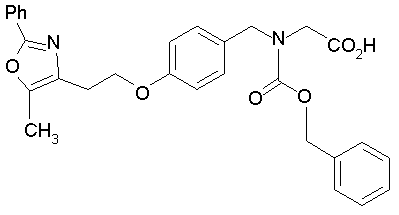 ,
, 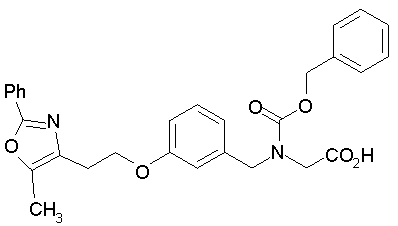
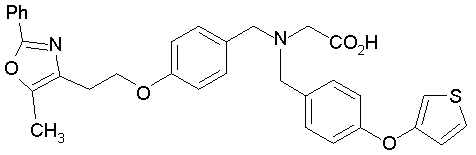 ,
, 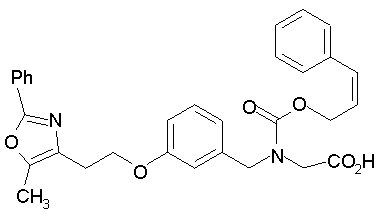 ,
,
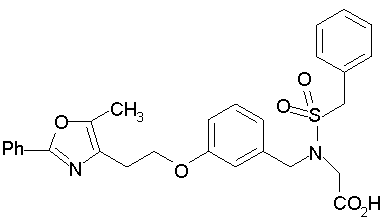
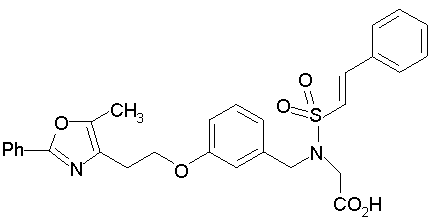

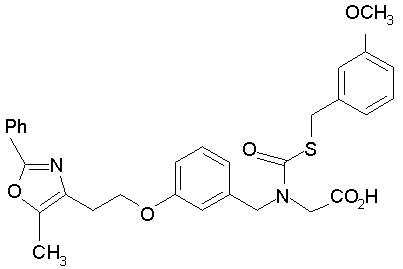
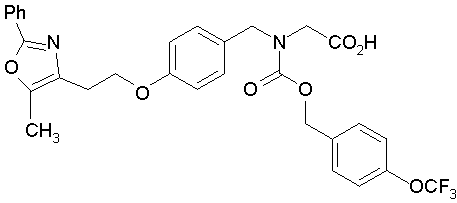
| ПРОИЗВОДНЫЕ ОКСАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КАТИОННЫЕ СОЛИ ИЛИ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ | 1992 |
|
RU2079497C1 |
| COBB J.E | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Structure-activity relationship and optimization of the N-aryl substituent" | |||
| Journal of medicinal chemistry, vol.41, no.25, 03.12.1998, p.5055-5069 | |||
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |

