Область техники, к которой относится изобретение
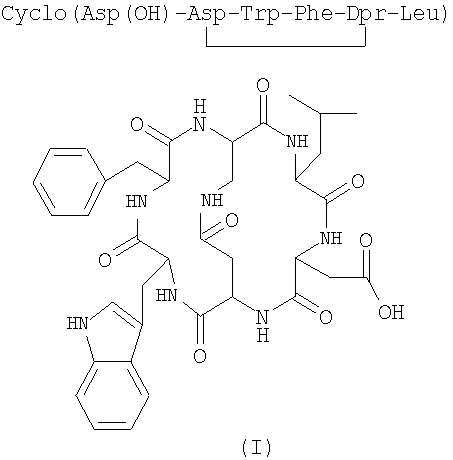
Настоящее изобретение относится к новому способу получения бициклических пептидных соединений формулы (I), указанной ниже, применимых в качестве промежуточных продуктов при получении фармакологически активных соединений, в частности при получении бициклических гликопептидов формулы (I-А), указанной ниже, которые обладает антагонистической активностью против рецептора NK2 тахикинина.
Уровень техники
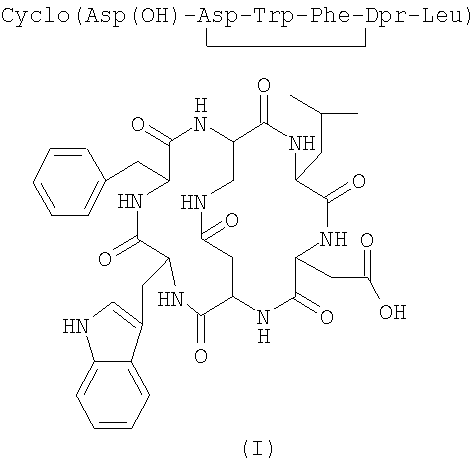
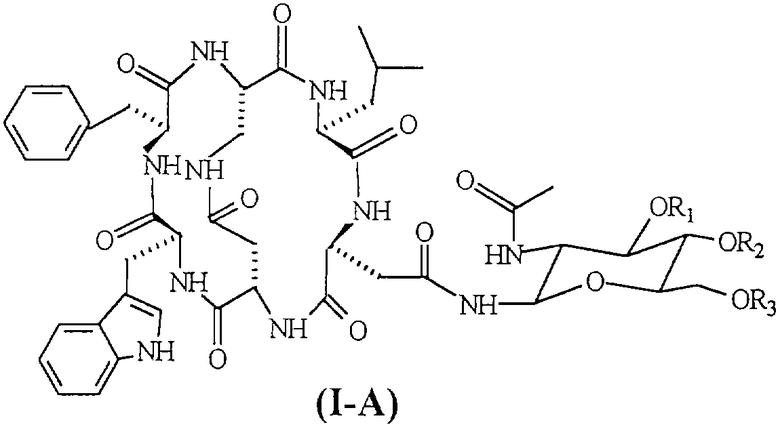
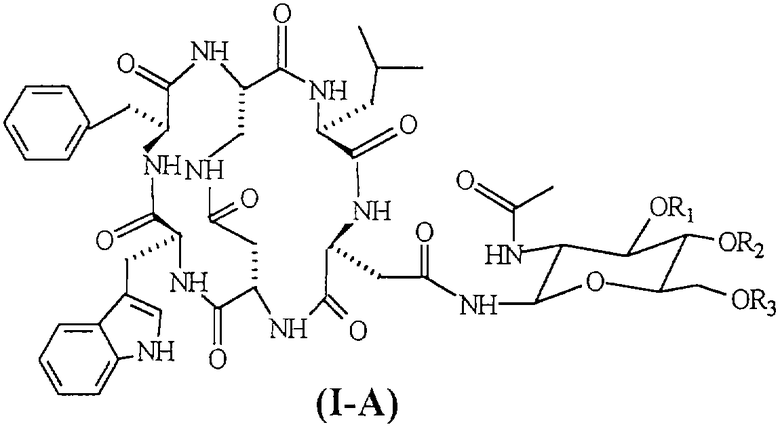
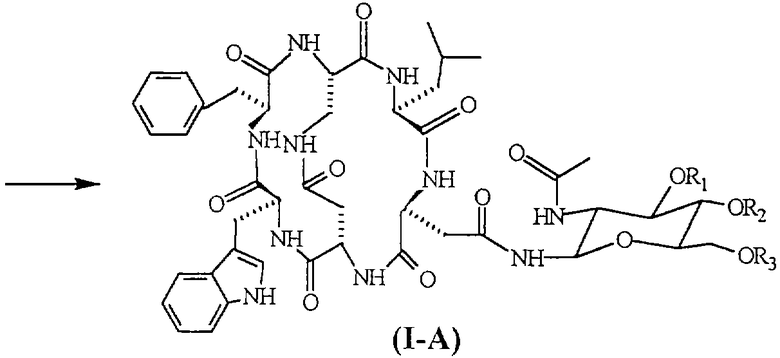
Соединения формулы (I-А), в частности [N-4-(2-ацетиламино-2-дезокси-β-D-глюкопиранозил)-L-аспарагинил-L-α-аспартил-L-триптофил-L-фенилаланил-L-2,3-диаминопропионил-L-лейцил]-С-4,2-N-3,5-лактам-С-1,6-N-2,1-лактам (соединение формулы (I-A), указанной ниже, в которой R1=R2=R3=H, известное под торговым названием «непадутант»), являются соединениями, которые имеют сильную антагонистическую активность против рецептора NK2 тахикинина и, следовательно, могут быть использованы для получения фармацевтических соединений для лечения заболеваний, и применимы при лечении и профилактике заболеваний, в которых тахикинины принимают участие в качестве нейромодуляторов.
Это соединение и некоторые из его промежуточных продуктов описаны в Европейском патенте №815126В1, особенно в примере 4. В указанном документе описаны на страницах 4 и 5 уже известные в литературе способы синтеза в растворе или твердой фазе линейных пептидов последовательным сочетанием подходящим образом защищенных аминокислот и их последующей конечной циклизацией для получения соединений общей формулы (I).
Эти способы описаны в очень общем виде, хотя более подробные детали предложены для получения соединений в примерах 1 и 2. В этих примерах используемым синтезом было сочетание Fmoc-аминокислот в твердой фазе до получения линейного пептида, который после отщепления от смолы циклизуют, очищают ВЭЖХ и снова циклизуют. Важно отметить, что по данному пути синтеза гликозидную боковую цепь вводят на стадии синтеза в твердой фазе линейного пептида на смоле, когда боковую цепь подходящим образом защищают аспарагином.
Сущность изобретения
Заявителем теперь неожиданно обнаружен новый и более эффективный способ получения бициклических пептидных соединений формулы (I), указанной ниже, применимых в качестве промежуточных продуктов для получения соединений с фармакологической активностью. Новый способ получения проводят полностью в растворе, а не в твердой фазе, что позволяет получить продукты с высокой чистотой и высокими выходами.
Следовательно, предметом изобретения является способ получения бициклических пептидных соединений формулы (I) (SEQ. ID. 1)
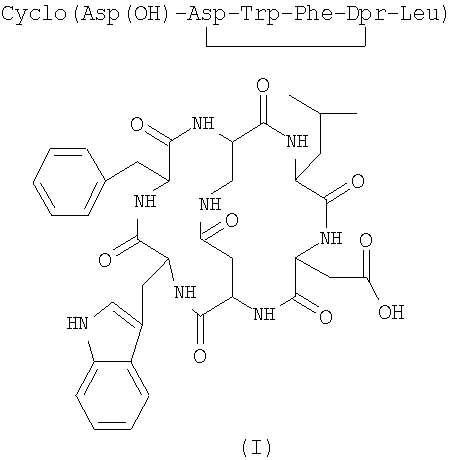
включающий в себя следующие стадии:
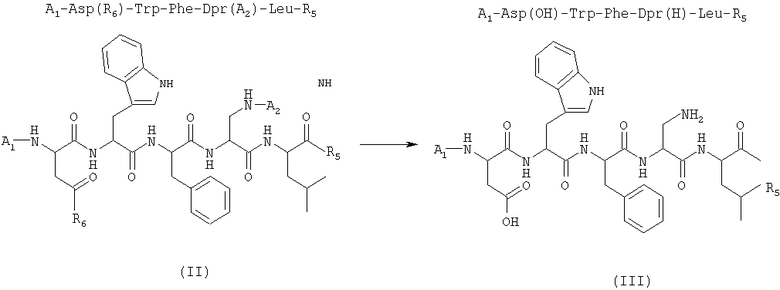
1) удаление защитной группы у линейного пентапептида формулы (II) (SEQ. ID. 2) в присутствии растворителя с получением соединения формулы (III)
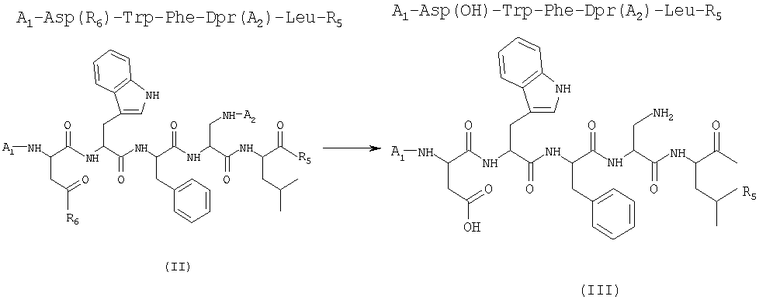
где A1 и A2 представляют собой две защитные группы атома азота, отличающиеся друг от друга, и R5 и R6, отличающиеся друг от друга, выбраны из бензилокси- и низших алкилоксигрупп, в которых алкильная часть представляет собой неразветвленную или разветвленную С1-С4-группу;
2) внутримолекулярную циклизацию соединения формулы (III), полученного на стадии 1), в присутствии растворителя и подходящего агента сочетания с получением соединения формулы (IV) (SEQ. ID. 3)
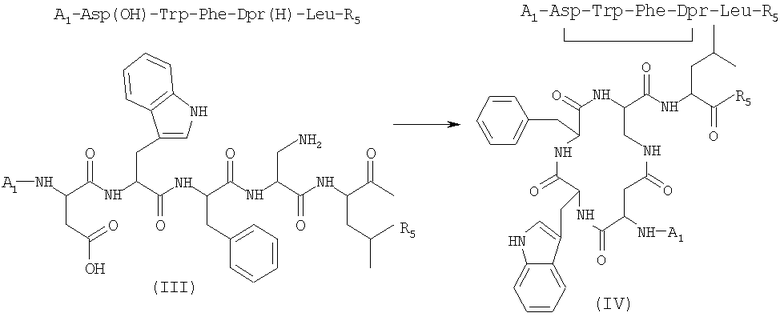
где R5 имеет указанные выше значения;
3) удаление защитной группы у соединения (IV), полученного на стадии 2), в присутствии растворителя с получением соединения формулы (V)
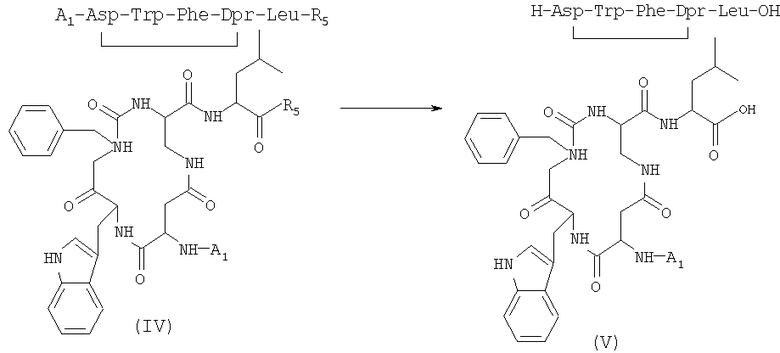
где R5 имеет указанные выше значения;
4) сочетание соединения формулы (V), полученного на стадии 3), и защищенной аминокислоты формулы (VIa) в присутствии растворителя с получением соединений формулы (VII) (SEQ. ID. 4)
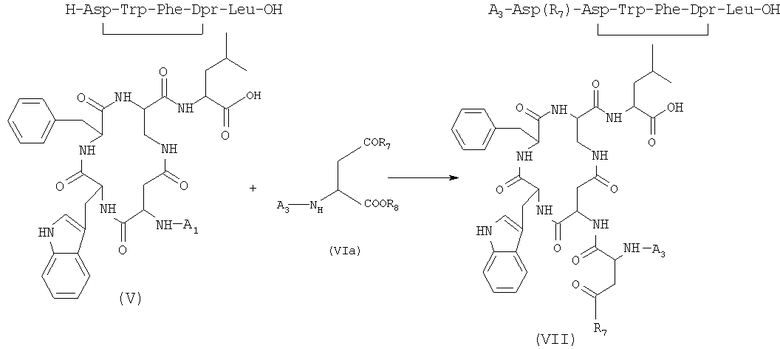
где A3 представляет собой защитную группу атома азота, R7 выбран из бензилокси- и низших алкилоксигрупп, в которых алкильная часть представляет собой неразветвленную или разветвленную С1-С4-группу; R8 представляет собой остаточную группу, образованную процедурой активации на карбоксильной группе;
5) удаление защитной группы у соединения формулы (VII), полученного на стадии 4), в присутствии растворителя с получением соединения формулы (VIII)
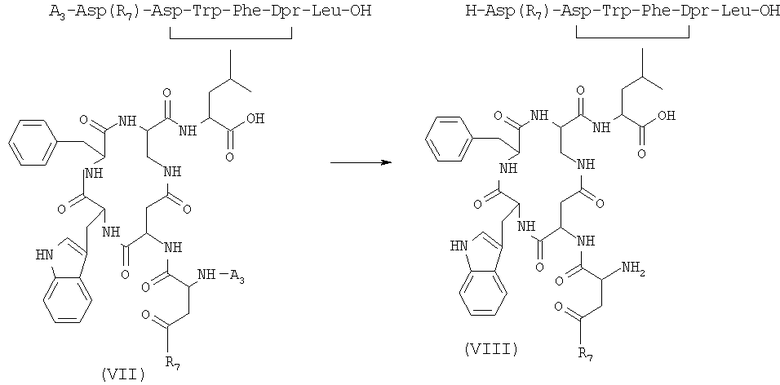
где R7 имеет указанные выше значения;
6) внутримолекулярную циклизацию, в присутствии растворителя и подходящего агента сочетания, соединения формулы (VIII), полученного на стадии 5), с получением бициклического соединения формулы (IX)
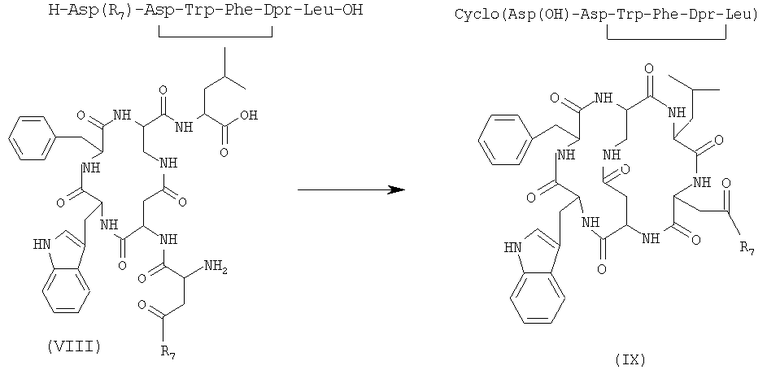
где R7 имеет указанные выше значения;
7) удаление защитной группы у бициклического соединения формулы (IX), полученного на стадии 6), в присутствии растворителя с получением соединения формулы (I)
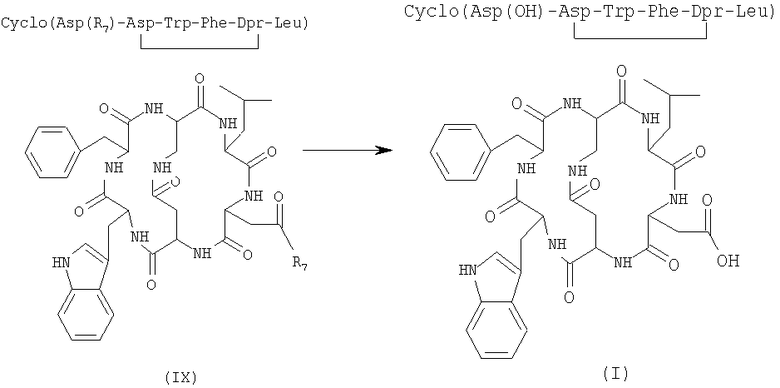
где R7 имеет указанные выше значения.
Соединение формулы (III) представлено SEQ. ID. 2, где соединения формулы (I) могут быть использованы, например, для получения бициклических гликопептидных соединений формулы (I-А), указанной ниже, которые обладают сильной антагонистической активностью для рецептора NK2 тахикинина; заявителем обнаружен новый способ получения, при помощи которого гликозидную боковую цепь вводят в соединения формулы (I) взаимодействием, проводимом в растворе, и очистка конечного продукта ВЭЖХ не является обязательной, так что получение этих соединений в больших масштабах может быть достигнуто при определенно более низких затратах, чем затраты современного способа получения.
Следующим предметом настоящего изобретения является способ получения бициклических гликопептидных соединений формулы (1-А) (SEQ. ID. 5)
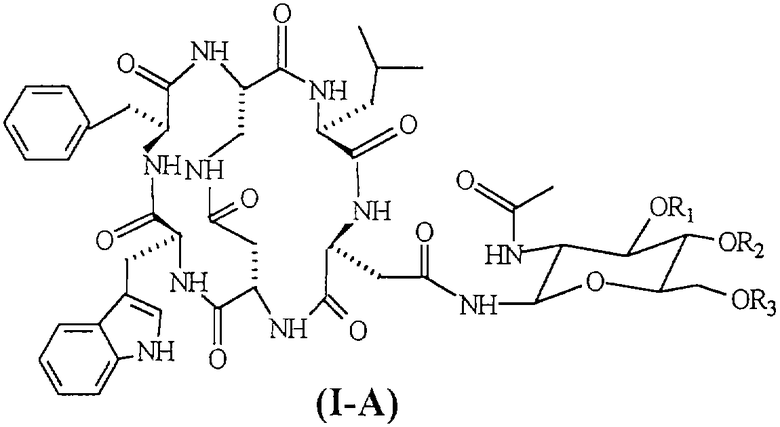
где R1, R2 и R3, одинаковые или отличные друг от друга, могут быть водородом или защитной группой атома кислорода,
включающий в себя следующие стадии:
1А) активацию бициклических пептидных соединений формулы (I) подходящим агентом сочетания с получением производного формулы (II-A)
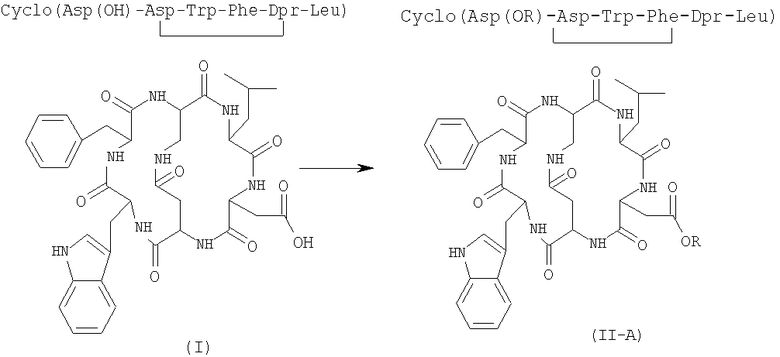
в которой R выбран из группы, состоящей из бензотриазола, возможно замещенного галогеном, азабензотриазола и сукцинимидила;
2А) взаимодействие соединения формулы (II-А), полученного на стадии 1А), в присутствии растворителя с гликозидным производным формулы (III-A)
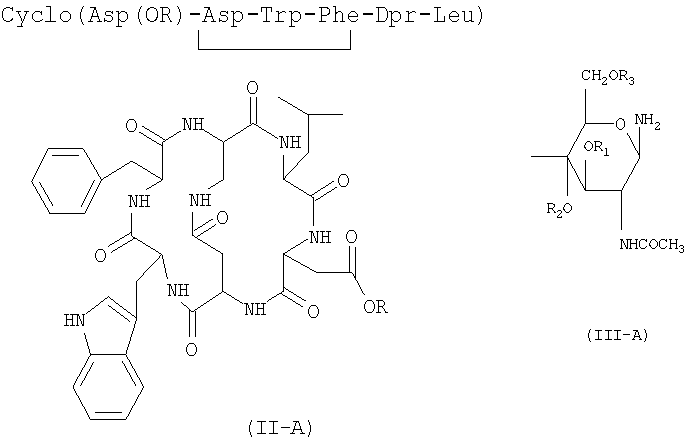
где R, R1, R2 и R3 имеют значения, указанные выше.
Следующим предметом изобретения является способ получения соединения формулы (I-A), начинающийся из соединений формулы (II) и формулы (III) и проходящий через образование соединения формулы (I), как описано в двух вышеуказанных способах.
Способы изобретения, проводимые полностью при помощи реакций в растворе, а не в твердой фазе, обнаруживают неожиданно высокие выходы и не требуют использования способов очистки ВЭЖХ, что, таким образом, позволяет значительно уменьшить затраты при получении и достичь получения продуктов в большом масштабе.
Подробное описание изобретения
Защитные группы азота, используемые в настоящих способах, могут быть выбраны из любых защитных групп, которые могут быть использованы для синтеза пептидов, таких как защитные группы, указанные в М.Bodansky, "Peptide Chemistry", Springer Verlag, 1988, или в J.Jones. "The Chemical Synthesis of Peptides", Clarendon Press. Oxford, 1994. В соответствии с данным изобретением защитные группы азота предпочтительно выбраны из группы, состоящей из бензилоксикарбонила и алкоксикарбонила, в котором алкильная часть представляет собой неразветвленную или разветвленную С1-С4-группу; более предпочтительно они выбраны из трет-бутоксикарбонила (Boc) и бензилоксикарбонила (Z).
R8 представляет собой остаточную группу, образованную процедурой активации, предпочтительно выбранную из группы, состоящей из бензилоксикарбонила, алкоксикарбонила, содержащего в алкильной части неразветвленную или разветвленную С1-С4-группу, сукцинимидила, бензотриазола, возможно замещенного галогеном, и азабензотриазола.
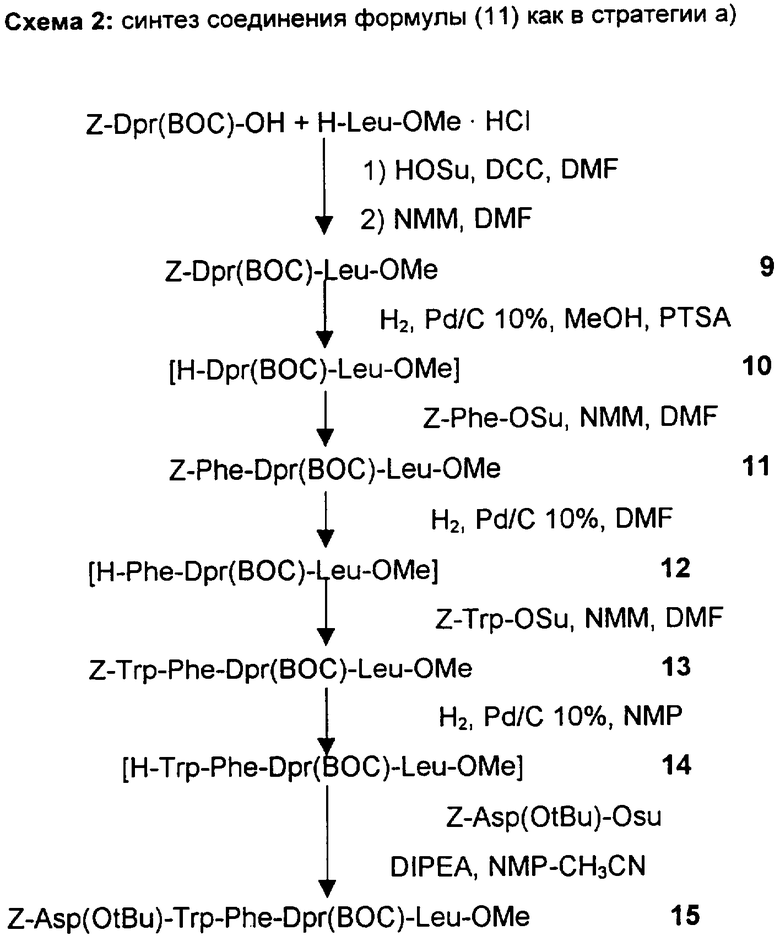
Линейные пептиды формулы (II) могут быть получены одной из следующих стратегий.
а) Постадийная стратегия: по этой стратегии аминокислоты, необходимые для получения пептида формулы (II), последовательно сочетают, исходя из производного аминокислоты Dpr формулы (X), защищенной у атома азота и полученной отдельно или генерированной in situ
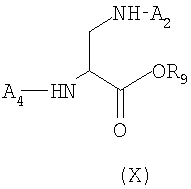
где A1 и A2, отличные друг от друга, представляют собой защитные группы атома азота, как определено выше;
R9 представляет собой остаточную группу, образованную процедурой активации, предпочтительно выбранную из группы, состоящей из бензилоксикарбонила, алкоксикарбонила, содержащего в алкильной части неразветвленную или разветвленную С1-С4-группу, и сукцинимидила;
производное формулы (X), указанной выше, подвергают взаимодействию с эфиром Leu (XI) в присутствии растворителя
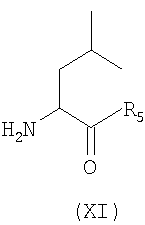
где R5 имеет значения, указанные выше,
таким образом получая дипептид A4-Drp(A2)-Leu-R5, который затем освобождают от защитной группы подходящим способом, зависящим от защитной группы на атоме азота, которая должна быть удалена, и совместимым с защитной группой, которая должна сохраняться.
Затем таким образом освобожденный от защитной группы дипептид сочетают с активированным эфиром аминокислоты Phe и так далее последовательно со звеньями Trp и Asp до получения соединения формулы (II).
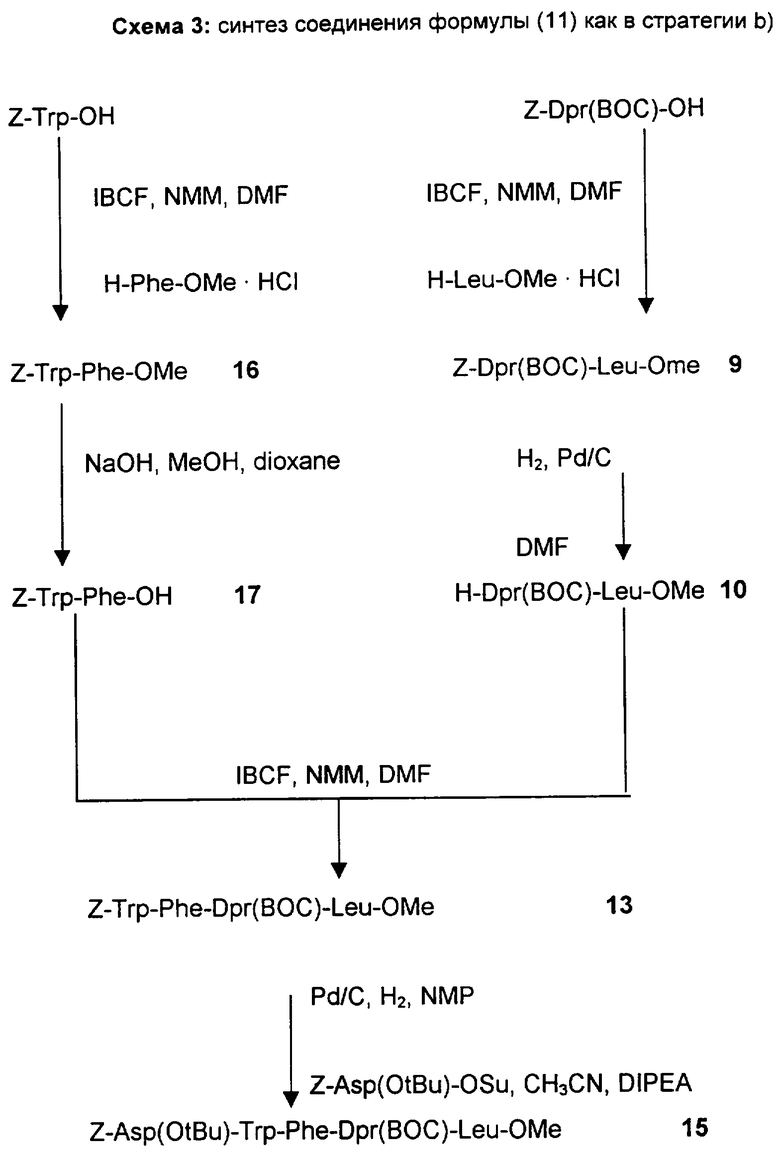
b) Стратегия 2+2+1: данная стратегия состоит в сочетании дипептида с одной удаленной защитной группой H-Dpr(A2)-Leu-R5, полученным, как описано выше по стратегии а), с активированным производным дипептида, имеющего нижеследующую формулу (XII)
A5-Trp-Phe-ОН
(XII)
где A2 и A5, отличающиеся друг от друга, представляют собой защитные группы атомов азота, как описано выше;
полученным отдельно или генерированным in situ сочетанием активированного эфира Trp, защищенного у атома азота, и полученного отдельно или генерированного in situ, с эфиром Phe, и последующим гидролизом эфирной группы.
Образовавшийся тетрапептид A5-Trp-Phe-Dpr(A2)-Leu-R5 подходящим образом освобождают от защитной группы, присоединенной к атому азота Trp, и сочетают с соединением формулы (VIb)
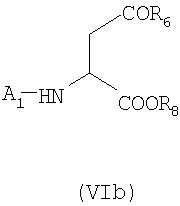
в которой A1, R6 и R8 имеет указанные выше значения.
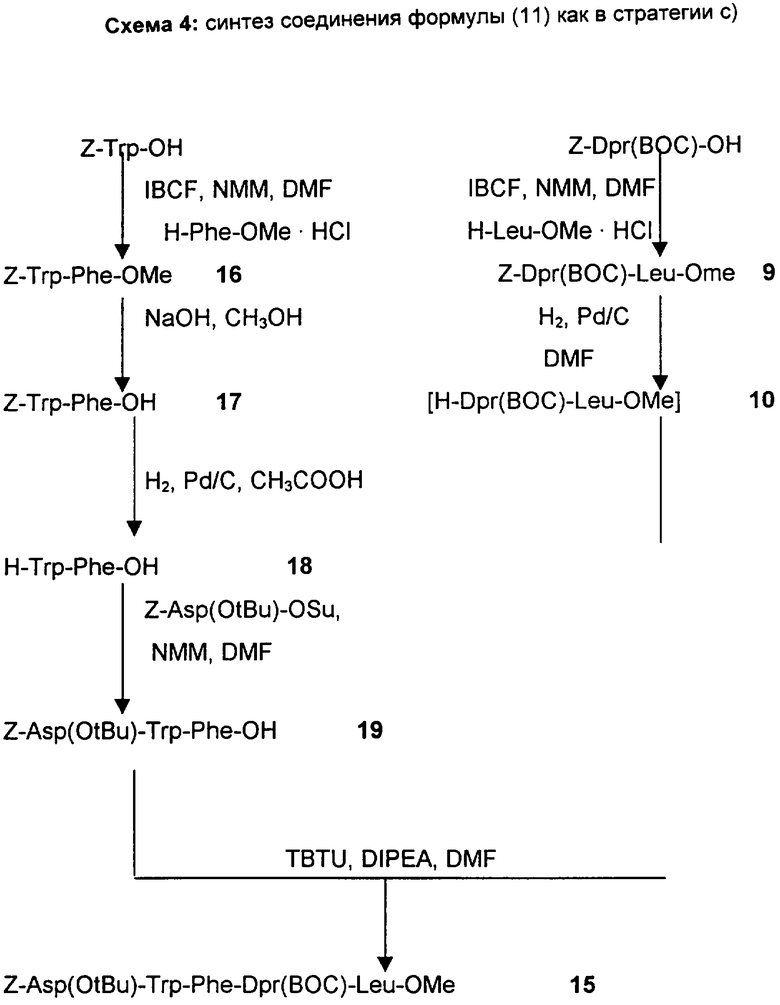
с) Стратегия 3+2: в соответствии с данной стратегией трипептид A1-Asp(R6)-Trp-Phe-OH, полученный удалением защитной группы атома азота у соединений формулы (XII), указанных выше, и последующим сочетанием с соединением формулы (VIb), указанным выше, затем сочетают с дипептидом с одной удаленной защитной группой H-Dpr-(A2)-Leu-R5, полученным, как описано в соответствии с процедурой стратегии а).
Используемый в настоящем изобретении термин «низшие алкоксильные группы» относится к тем алкоксильным группам, у которых алкильная часть содержит неразветвленную или разветвленную С1-С4-группу, предпочтительно выбранным из группы, состоящей из метила, этила, пропила, бутила, изопропила и трет-бутила. Это должно относится также к алкилоксикарбонильным группам изобретения, у которых алкильная часть содержит неразветвленную или разветвленную С1-С4-группу, предпочтительно выбранную из группы, состоящей из метила, этила, пропила, бутила, изопропила и трет-бутила.
Агент сочетания может быть выбран из любого из агентов сочетания, обычно используемых в синтезе пептидов, так чтобы получить активированное производное аминокислоты, такое как производные аминокислот, указанные, например, в М. Bodansky, "Peptide Chemistry", Springer Verlag, 1988, или в J. Jones, "The Chemical Synthesis of Peptides", Clarendon Press, Oxford, 1994.
Активированные производные, если они не являются коммерчески доступными, могут быть получены отдельно или in situ взаимодействием аминокислоты или пептида и одного или нескольких из многочисленных известных агентов сочетания, таких как изобутилхлорформиат (IBCF), карбодиимид, выбранный из дициклогексилкарбодиимида (DCC) и гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида (EDAC·HCI), возможно в комбинации с гидроксипроизводным, выбранным из 1-гидроксибензотриазола (HOBt), 1-гидрокси-7-азабензотриазола (HOAt), 6-хлор-1-гидроксибензотриазола (Cl-HOBt) и гидроксисукцинимида (HOSu); фосфониевой солью, солью N-оксида гуанидина или урониевой солью, такой как гексафторфосфат (бензотриазол-1-илокси)три(диметиламино)фосфония (ВОР), гексафторфосфат (бензотриазол-1-илокси)трипирролидинфосфония (РуВОР), гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1Н-бензотриазолия (HBTU), гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-5-хлор-1Н-бензотриазолия (HCTU), тетрафторборат 3-оксида 1-[бис(диметиламино)метилен]-1H-бензотриазолия (TBTU), гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазол[4,5-b]пиридиния (HATU), тетрафторборат 3-оксида 1-[бис(диметиламино)метилен]-5-хлор-1H-бензотриазолия (TCTU), тетрафторборат O-[(этоксикарбонил)цианометиленамино]-N,N,N',N'-тетраметилурония (TOTU), тетрафторборат O-(бицикло[2.2.1]гепт-5-ен-2,3-дикарбоксимидо)-N,N,N',N'-тетраметилурония (TNTU) или тетрафторборат O-(N-сукцинимидил)-N,N,N',N'-тетраметилурония (TSTU).
Когда указанное производное генерируют in situ, реакцию сочетания проводят сразу после добавления другого реагента, который в случае внутримолекулярных циклизаций явно соответствует свободной концевой аминогруппе, присутствующей в самой молекуле.
Реакцию сочетания обычно проводят в присутствии третичного амина, такого как N-метилморфолин (NMM), триэтиламин (TEA) или диизопропилэтиламин (DIPEA), в органическом растворителе, выбранном из растворителей, обычно используемых для синтеза пептидов. Предпочтительными растворителями для реакции сочетания являются этилацетат (AcOEt), диметилформамид (ДМФ) и N-метилпирролидон (NMP).
Реакции сочетания могут быть проведены при температуре, которая не должна вызывать деградации или сделать реакцию слишком медленной, причем температура предпочтительно находится между -20 и +50°C.
Удаления защитных групп в способах изобретения достигают подходящими способами для групп, которые нужно удалить, и совместимыми с группами, которые остаются; данные реакции удаления защитных групп обычно проводят способами каталитического гидрирования или обработками кислотами или основаниями.
Для гидрирований катализатор может быть выбран из тех рядов катализаторов, которые являются доступными и подходящими для этой цели, предпочтительным является 5% или 10% палладием. Растворитель для реакций удаления защитной группы каталитическим гидрированием может быть выбран из растворителей, которые растворяют соединения в реакции, в том числе кетоны, такие как ацетон, растворителей, которые отравляют катализатор, и растворителей, которые не взаимодействуют с самими компонентами реакции. Предпочтительными растворителями для реакции являются ДМФ, NMP, органические кислоты, такие как уксусная кислота и п-толуолсульфоновая кислота (PTSA), и спирты, такие как метанол, этанол и изопропанол, или их смеси. Температура реакции гидрирования находится между -20 и +50°C.
Для удаления защитных групп обработкой кислотой предпочтительно используют минеральные кислоты, такие как хлористоводородная кислота, или органические кислоты, такие как трифторуксусная кислота или муравьиная кислота, которые могут быть использованы отдельно или в смеси с другими растворителями. Температура составляет от -20 до +50°C.
Для удаления защитных групп обработкой основанием предпочтительно используют гидроксиды щелочных металлов и щелочноземельных металлов в присутствии растворителя, такого как вода, диоксан, ацетонитрил, метанол, этанол, изопропанол или их смеси; температура находится между -20 и +50°C.
Термин «защитная группа атома кислорода, используемый в настоящем изобретении, относится к защитной группе, выбранной из групп, обычно используемых для защиты групп -ОН и хорошо известных любому специалисту в данной области, выбранной, например, из группы, состоящей COR4, где R4 представляет собой неразветвленную или разветвленную алкильную группу, содержащую от 1 до 4 атомов углерода, фенил, возможно замещенный атомом галогена, бензил или бензоил; предпочтительной защитной группой атома кислорода является ацетил.
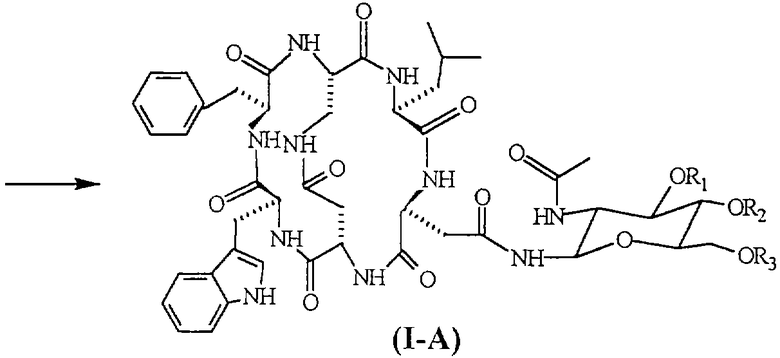
В соответствии с изобретением гликопептидные соединения формулы (I-А) могут быть получены взаимодействием гликозидного производного формулы (III-A) с активированным производным пептида формулы (II-A), полученным реакцией активации или генерированным in situ соединением формулы (I). Следовательно, в способе получения бициклических гликопептидных соединений формулы (I-A) гликозидную группу вводят не в линейный пептид, а в бициклическое пептидное соединение.
Если соединения формулы (III-A) подвергают реакции, когда R1, R2 и R3 не являются водородом, полученные соединения формулы (I-A) могут быть превращены в соответствующие соединения, в которой R1=R2=R3=H, посредством каталитического гидрирования или обработкой кислотой или основанием в соответствии с природой защитных групп R1, R2 и R3.
Гликозидные соединения формулы (III-A), предпочтительно используемые в способе изобретения, выбраны из группы, состоящей из 2-ацетамидо-2-дезокси-β-D-глюкопиранозиламина и 2-ацетамидо-3,4,6-три-O-ацетил-2-дезокси-β-D-глюкопиранозиламина, которые являются известными в литературе и могут быть получены, например, как описано соответственно в I. Shin. et al., Tetrahedron Letters, 42 (2001), 1325-1328, и D. Macmillan et al., Organic Letters, Vol.4, №9, 2002.
Нижеследующие примеры и схемы синтеза приведены для обеспечения неограничивающей иллюстрации изобретения.
В схеме 1 указан путь синтеза, который, начинаясь из соединений формулы (II), приводит к соединениям формулы (I-A), тогда как на схемах 2-4 показаны три разные стратегии получения соединений формулы (II).
Защитными группами, приведенными в качестве примеров, являются трет-бутоксикарбонил (ВОС) и бензилоксикарбонил (Z) для концевых аминогрупп и группа метилового эфира и трет-бутилового эфира для концевых карбоксильных групп.
Номера, указанные рядом с каждым соединением в нижеследующих схемах, соответствуют номерам, приписываемым соединениям в примерах.
Идентификацию и оценку чистоты для полученных соединений устанавливали элементным анализом, ВЭЖХ, 1H-ЯМР, ИК- и масс-спектральным анализом.
ПРИМЕР 1
Получение Z-Asp(OH)-Trp-Phe-DDr(H)-Leu-OMe (SEQ. ID. 1)
72 ммоль/л раствора Z-Asp(O-трет-Bu)-Trp-Phe-Dpr(BOC)-Leu-OMe, полученного, как описано в примере 15, в 95% муравьиной кислоте нагревают до 40°C в вакууме в течение 4 дней.
Реакционную смесь упаривают при пониженном давлении и остаток снова растворяют в смеси 8:2 CH3CN-H2O.
Суспензию охлаждают до 15-20°C и рН регулируют до 6 добавлением 20% водного раствора NMM.
Ацетонитрил выпаривают при пониженном давлении и образовавшуюся суспензию фильтруют.
Беловатое твердое вещество промывают H2O и сушат в вакууме при 30-40°C, получая продукт с выходом, равным 96,4%.
1H-ЯМР, диметилсульфоксид-d6 (ДМСО-d6) δ:
0,86 (2д; 6Н); 1,47-1,75 (м; 3H); 2,32-2,68 (м; 2Н); 2,79-3,55 (м; 6Н); 3,63 (с; 3H); 4,25-4,65 (м; 5Н); 4,99 (AB-Syst.; 2Н); 6,91-7,43 (м; 14Н); 7,48-7,60 (2д; 2Н); 7,82 (ушир.; 2Н), 8,03-8,43 (4д; 4Н); 10,83 (с; 1Н); 12,35 (ушир.; 1Н).
ПРИМЕР 2
Получение Z-Asp-Trp-Phe-Dpr-Leu-Ome (SEQ. ID. 2)
2,2 эквивалента NMM добавляют к 24 ммоль/л раствора Z-Asp(OH)-Trp-Phe-Dpr(NH2)-Leu-Ome в ДМФ и спустя 5-10 минут добавляют 1,2 эквивалента PyBOP.
После 2-3 часов перемешивания при комнатной температуре раствор упаривают при пониженном давлении до получения жидкого остатка, который добавляют по каплям в 0,5 М водный раствор NaHCO3.
Образовавшуюся суспензию фильтруют и полученное твердое вещество промывают смесью 4:6 ДМФ - Н2О, затем Н2O до достижения нейтрального значения рН и сушат в вакууме при 30-50°C, получая при этом продукт с выходом, равным 84,2%.
1H-ЯМР (ДМСО-d6) δ:
0,83 (2д; 6Н); 1,34-1,69 (м; 3H); 2,31-2,92 (м; 4Н); 3,03-3,91 (м; 4Н); 3,61 (с; 3H); 4,17-4,63 (м; 5Н); 5,01 (AB-Syst.; 2Н); 6,84-7,48 (м; 16Н); 7,60 (д; 1Н); 7,87 (д; 2Н); 8,01 (т; 1Н); 8,27 (д; 1Н); 10,81 (с; 1Н).
ПРИМЕР 3
Получение Z-Asp-Trp-Phe-Dpr-Leu-OH (SEQ. ID. 3)
Мутный раствор, содержащий 77 ммоль/л Z-Asp-Trp-Phe-Dpr-Leu-Ome в смеси 8:2 диоксан - H2O, нагревают до 35°C и выдерживают при рН 12,0-12,5 медленным и непрерывным добавлением 1,5 н. NaOH.
В конце реакции рН мутного раствора устанавливают до 9 добавлением 6 н. HCl, осветляют фильтрованием на слое для соадъювантного фильтрования и подкисляют до рН 3 снова добавлением 6 н. HCl.
Раствор концентрируют при пониженном давлении до получения фильтруемого раствора.
Полученное фильтрованием беловатое твердое вещество промывают смесью 1:1 диоксан - H2O, затем Н2О и сушат в вакууме при 30-40°C, получая при этом продукт с выходом, равным 97,7%.
1H-ЯМР (ДМСО-d6) δ:
0,84 (2д; 6Н); 1,42-1,76 (м; 3H); 2,29-3,48 (м; 7Н); 3,85 (м; 1Н); 4,10-4,65 (м; 5Н); 5,00 (AB-Syst.; 2Н); 6,86-7,47 (м; 16Н); 7,55-8,36 (4д+м; 5Н); 10,80 (д; 1Н); 12,65 (ушир.; 1Н).
ПРИМЕР 4
Получение Z-Asp(O-трет-Bu)-Asp-Trp-Phe-Dpr-Leu-OH (SEQ. ID. 4)
66 ммоль/л раствора Z-Asp-Trp-Phe-Dpr-Leu-OH в ДМФ гидрируют при комнатной температуре в присутствии 1 эквивалента NMM и каталитических количеств 10% Pd/C при 50% влаги.
После реакции в течение 6 часов суспензию фильтруют для удаления катализатора и фильтрат разбавляют ДМФ с получением 53 ммоль/л раствора Z-Asp-Trp-Phe-Dpr-Leu-OH, к которому добавляют 4 эквивалента NMM и 1,05 эквивалента Z-Asp(O-трет-Bu)Osu.
После перемешивания в течение 5 часов при комнатной температуре смесь упаривают при пониженном давлении до получения остатка, который прикапывают в 0,5 н. H2SO4. Образовавшуюся суспензию фильтруют и полученное твердое вещество промывают смесью 1:1 ДМФ - H2O, затем Н2О и сушат в вакууме при 30-40°C, получая при этом продукт с выходом, равным 93,7%.
1H-ЯМР (ДМСО-d6) δ:
0,84 (2д; 6Н); 1,35 (с; 9Н); 1,40-1,70 (м; 3H); 2,20-3,94 (м; 10Н); 4,10-4,81 (м; 6Н); 4,92-5,12 (AB-Syst; 2Н); 6,74-7,57 (м; 17Н); 7,71-8,35 (4д+1 т; 5Н); 10,70 (с; 1Н); 12,70 (ушир.; 1Н).
ПРИМЕР 5
Получение цикло[Asp(O-трет-Bu)-Asp-Trp-Phe-Dpr-Leu] (SEQ. ID. 5)
47 ммоль/л раствора Z-Asp(O-трет-Bu)-Asp-Trp-Phe-Dpr-Leu-OH в ДМФ гидрируют при комнатной температуре в присутствии 1 эквивалента DIPEA и каталитических количеств 10% Pd/C при 50% влаги.
После реакции в течение приблизительно 2 часов суспензию фильтруют для удаления катализатора и разбавляют ДМФ до получения 19 ммоль/л раствора H-Asp(O-трет-Bu)-Asp-Trp-Phe-Dpr-Leu-OH, к которому добавляют 1,4 эквивалента DIPEA и 1,2 эквивалента HATU.
После перемешивания в течение 30-60 минут при комнатной температуре раствор упаривают при пониженном давлении до получения остатка, который добавляют по каплям в 0,5 М водный раствор NaHCO3.
Образовавшуюся суспензию фильтруют и полученное твердое вещество промывают большим количеством H2O до нейтрального рН, сушат в вакууме при 30-50°C, получая при этом продукт с выходом, равным 94,1%.
1H-ЯМР (ДМСО-d6) δ:
0,88 (2д; 6Н); 1,38 (с; 9Н); 1,31-1,72 (м; 3H); 2,33-2,99 (м; 6Н); 3,20-3,63 (м; 3H); 3,87-4,62 (м; 7Н); 6,75-7,50 (м; 13Н); 8,04 (ушир.; 1Н); 8,56 (д; 1Н); 8,76 (д; 1Н); 9,18 (ушир.; 1Н); 10,84 (с; 1Н).
ПРИМЕР 6
Получение цикло[Asp(ОН)-Asp-Trp-Phe-Dpr-Leu] (SEQ. ID. 6)
83 ммоль/л раствора цикло[Asp(O-трет-Bu)-Asp-Trp-Phe-Dpr-Leu] в 90% муравьиной кислоте нагревают при 40°C в вакууме в течение 2 часов. Реакционную смесь упаривают при пониженном давлении до получения густого остатка, который снова растворяют в H2O.
Образовавшуюся суспензию фильтруют и полученное твердое вещество промывают H2O, сушат в вакууме при 30-40°C и, наконец, очищают при помощи колонки сефадекса® LH-20, элюируя метанолом.
Получают 314 г белого твердого вещества (титр 95,2%, выход 82,0%).
1H-ЯМР (ДМСО-d6) δ:
0,88 (2д; 6Н); 1,31-1,77 (м; 3H); 2,32-3,73 (м; 9Н); 3,80-4,65 (м; 7Н); 6,82-7,51 (м; 13Н); 7,94-9,19 (2д; 2 ушир.; 4Н); 10,85 (с; 1Н); 12,20 (с, 1Н).
ПРИМЕР 7
Получение цикло[Asp-Asp-Trp-Phe-Dpr-Leu] (SEQ. ID. 7)
3 эквивалента NMM, 1,2 эквивалента HATU и 2-ацетамидо-3,4,6-три-O-ацетил-2-дезокси-β-D-глюкопиранозиламина добавляют к 0,24 моль/л раствора цикло[Asp(OH)-Asp-Trp-Phe-Dpr-Leu] в ДМФ с интервалами 10 минут.
После перемешивания в течение 1 часа при 0-4°C реакционную смесь упаривают при пониженном давлении до получения жидкого остатка, который добавляют по каплям в 1% водный раствор NaHCO3.
Образовавшуюся суспензию фильтруют и полученное твердое вещество промывают H2O, сушат в вакууме при 30-40°C и очищают кристаллизацией из смеси EtOH - H2O.
Получают 117 г белого твердого вещества (титр 96,0%, выход 87,0%).
1H-ЯМР (ДМСО-d6) δ:
10,80 (д; 1Н); 8,90 (ушир.; 1Н), 8,72 (д; 1Н); 8,47 (д; 1Н); 8,46 (д; 1Н); 8,08 (ушир.; 1Н); 7,84 (д; 1Н); 7,43 (дд; 1н); 7,33 (дд; 1Н); 7,24 (ушир.; 1Н); 7,23 (м; 2Н); 7,16 (м; 3H); 7,14 (д; 1Н); 7,06 (дт; 1Н); 7,00 (д; 1Н); 6,98 (дт; 1Н); 6,90 (т; 1Н); 5,18 (дд; 1Н); 5,12 (дд; 1Н); 4,82 (дд; 1Н); 4,18 (дд; 1Н); 3,96 (дд; 1Н); 3,85 (ддд; 1Н); 3,80 (ддд; 1Н); 4,53 (м, 1Н); 4,47 (м; 1Н); 4,43 (м; 1Н); 4,39 (м; 1Н); 4,16 (м; 1Н); 4,08 (м; 1Н); 3,58 (м; 1Н); 3,30 (м; 1Н); 2,98 (м; 1Н); 2,88 (м; 1Н); 2,86 (м; 1Н); 2,70 (м; 1Н); 2,65 (м; 1Н); 2,60 (м; 1Н); 2,19 (м; 1Н); 2,00 (с; 3H); 1,96 (с; 3H); 1,90 (с; 3H); 1,73 (с; 3H); 1,65 (м; 1Н); 1,52 (м; 1Н); 1,37 (м; 1Н); 0,92 (д; 3H); 0,85 (д; 3H).
ПРИМЕР 8
Получение цикло[Asp-Asp-Trp-Pbe-Dpr-Leu] (непадутант) (SEQ. ID. 8)
Способ а)
2 эквивалента NMM и 1,3 эквивалента TBTU и 2-ацетамидо-2-дезокси-β-D-глюкопиранозиламина добавляют с интервалами 10 минут к 83 ммоль/л раствора в ДМФ цикло[Asp(ОН)-Asp-Trp-Phe-Dpr-Leu] (получен, как описано в примере 6).
После перемешивания в течение 1 часа при комнатной температуре реакционную смесь упаривают при пониженном давлении до получения густого маслянистого остатка, который снова растворяют в смеси 2:8 ацетонитрил-трет-бутоксиметан (ТВМЕ). Образовавшуюся суспензию энергично перемешивают в течение 30 минут при комнатной температуре и затем фильтруют.
Полученное твердое вещество промывают ТВМЕ, сушат в вакууме при 25-30°C и, наконец, очищают препаративной ВЭЖХ с использованием в качестве элюента смесей, состоящих из ацетонитрила и воды.
Получают 151 г белого твердого вещества (титр 93,0%, выход 89,3%).
1H-ЯМР (ДМСО-d6) δ:
0,85 (д; 3H); 0,92 (д; 3H); 1,36 (м; 1Н); 1,51 (м; 1Н); 1,65 (м; 1Н); 1,76 (с; 3H); 2,16 (дд; 1Н); 2,57 (дд; 1Н); 2,63 (дд, 1Н); 2,67 (дд; 1Н); 2,83 (дд; 1Н); 2,88 (дд; 1Н); 2,93 (м; 1Н); 3,04-3,09 (м; 2Н); 3,27-3,32 (м; 2Н); 3,42 (м; 1Н); 3,50 (ддд+ушир.; 2Н); 3,65 (дд; 1Н); 3,96 (ушир.; 1Н); 4,09 (м; 1Н); 4,12 (м; 1Н); 4,35 (м; 1Н); 4,43 (м; 1Н); 4,50 (м; 1Н); 4,53 (м+т; 2Н); 4,81 (дд; 1Н); 4,94 (д; 1Н); 4,98 (д; 1Н); 6,91 (ушир.; 1Н); 6,98 (т+ушир.; 2Н); 7,06 (т; 1Н); 7,14-7,17 (м; 4Н); 7,24 (т; 2Н); 7,27 (ушир.; 1Н); 7,33 (д; 1Н); 7,42 (д; 1Н); 7,77 (д; 1Н); 8,05 (ушир.; 1Н); 8,10 (д; 1Н); 8,51 (д; 1Н); 8,77 (д; 1Н); 9,00 (ушир.; 1Н); 10,84 (д; 1Н).
Способ b)
0,04 эквивалента 0,1 н. NaOMe в МеОН добавляют к 0,89 моль/л раствора в МеОН цикло[Asp-Asp-Trp-Phe-Dpr-Leu], полученного, как описано в примере 7.
После перемешивания в течение 3 часов при комнатной температуре рН регулируют до 6,5-7 и добавляют амберлист® 15. После удаления смолы раствор концентрируют при пониженном давлении до получения остатка, который разбавляет ТВМЕ.
Образовавшуюся суспензию фильтруют и полученное белое твердое вещество промывают ТВМЕ, сушат в вакууме при 35-40°C, получая при этом продукт с выходом, равным 94,8%.
ПРИМЕР 9
Получение Z-Dpr(BOC)-Leu-ОМе
Способ а)
1,2 эквивалента NMM добавляют к 0,66 моль/л раствора Z-Dpr(BOC)-OH в ДМФ. Раствор охлаждают до -25°C и 1 эквивалент IBCF добавляют по каплям при поддержании температуры ниже -20°C.
Спустя приблизительно 10 минут по каплям добавляют 0,78 моль/л предварительно охлажденного раствора, содержащего 1 эквивалент H-Leu-OMe·HCL и NMM в ДМФ, всегда поддерживая температуру ниже -15°C.
После перемешивания в течение одного часа реакционную смесь добавляют по каплям в 0,5 М водный раствор NaHCO3.
Образовавшуюся суспензию фильтруют и полученное твердое вещество промывают последовательно H2O, 0,05 М H2SO4 и Н2О до достижения нейтрального значения рН, сушат в вакууме при 30-50°C, получая при этом продукт с выходом, равным 89,0%. Точка плавления 122-125°C.
1H-ЯМР (ДМСО-d6) δ:
0,85 (2д; 6Н); 1,37 (с; 9Н); 1,40-1,71 (м; 3H); 3,01-3,36 (м; 2Н); 3,61 (с; 3H); 4,06-4,37 (м; 2Н); 5,03 (с; 2Н); 7,35 (с; 5Н); 6,66 (т; 1Н); 7,20 (д; 1Н); 8,29 (д; 1Н).
Способ b)
1 эквивалент DCC добавляют к 0,35 моль/л раствора Z-Dpr(BOC)-OH в ДМФ, содержащего 1 эквивалент HOSu и охлажденного до 0-5°C. Смесь нагревают до комнатной температуры и перемешивают в течение 1 часа. DCC удаляют фильтрованием и к прозрачному раствору добавляют 1,2 эквивалента H-Leu-ОМе·HCl и 2,6 эквивалента NMM. После перемешивания в течение 2-3 часов при комнатной температуре смесь разбавляют 0,5 н. NaHCO3, затем охлаждают до -5°C.
Образовавшуюся суспензию фильтруют и полученное твердое вещество последовательно промывают 0,5 н. NaHCO3, смесью 2:1 H2O - ДМФ и водой, затем сушат в вакууме при 30-40°C, получая при этом продукт с выходом, равным 93%.
ПРИМЕР 10
Получение H-Dpr(BOC)-Leu-OMe
0,14 моль/л раствора Z-Dpr(BOC)-Leu-OMe в МеОН, содержащего 1 эквивалент PTSA, гидрируют при комнатной температуре в присутствии каталитических количеств 10% Pd/C, 50% влаги.
После реакции в течение приблизительно 2 часов суспензию фильтруют для удаления катализатора и фильтрат разбавляют ДМФ.
МеОН и Н2O полностью выпаривают при пониженном давлении и остаточный раствор в ДМФ, содержащий дипептид, используют для последующего сочетания.
ПРИМЕР 11
Получение Z-Phe-Dpr(BOC)-Leu-OMe
Указанное соединение получают из дипептида H-Dpr(BOC)-Leu-OMe из примера 10 по способу, описанному в примере 9, с использованием Z-Phe-OH.
1H-ЯМР (ДМСО-d6) δ:
0,86 (2д; 6Н); 1,38 (с; 9Н); 1,40-1,74 (м; 3H); 2,73-3,02 (м; 2Н); 3,10-3,41 (м; 2Н); 3,62 (с; 3H); 4,17-4,46 (м; 3H); 4,94 (AB-Syst.; 2Н); 7,18-7,39 (м; 10Н); 6,52 (т; 1Н); 7,52(д; 1Н); 8,13(д; 1Н); 8,25(д; 1Н).
ПРИМЕР 12
Получение H-Phe-Dpr(BOC)-Leu-OMe
Данное соединение получают из защищенного производного из примера 11 по способу примера 10 с использованием ДМФ в качестве растворителя.
ПРИМЕР 13
Получение Z-Trp-Phe-Dpr(Boc)-Leu-OMe (SEQ. ID. 9)
Данное соединение получают с использованием способа в примере 9 из трипептида примера 12 и с использованием Z-Trp-OH или сочетанием двух пептидов Z-Trp-Phe-OH и H-Dpr(BOC)-Leu-OMe, полученных, как описано в примерах 17 и 10 соответственно.
1H-ЯМР (ДМСО-d6) δ:
0,86 (2д; 6Н); 1,37 (с; 9Н); 1,40-1,76 (м; 3H); 2,73-3,41 (м; 6Н); 3,62 (с; 3H); 4,16-4,67 (м; 4Н); 4,93 (AB-Syst.; 2Н); 6,89-7,65 (м; 16Н); 6,55 (т; 1Н); 8,07 (д; 1Н); 8,11 (д; 1Н); 8,29 (д; 1Н); 10,79 (с; 1Н).
ПРИМЕР 14
Получение H-Trp-Phe-Dpr(BOC)-Leu-OMe (SEQ. ID. 10)
Данное соединение получают из защищенного производного примера 13 по способу примера 10 с использованием NMP в качестве растворителя.
ПРИМЕР 15
Получение Z-Asp(O-трет-Bu)-Trp-Phe-Dpr(BOC)-Leu-OMe (SEQ. ID. 11)
Способ а)
1 объем CH3CN, 1,5 эквивалента DIPEA и 1,15 эквивалента 7-Asp(O-трет-Bu)-OSu добавляют к 0,16 моль/л раствора H-Trp-Phe-Dpr(BOC)-Leu-OMe в NMP, полученного реакцией гидрирования. После перемешивания в течение 3-4 часов при комнатной температуре реакционную смесь охлаждают до 5°C и разбавляют H2O. Образовавшуюся суспензию фильтруют и полученное твердое вещество промывают смесью 3:7 CH3CN - H2O и H2O и затем сушат в вакууме при 30-50°C, получая при этом продукт с выходом, равным 90%.
Способ b)
1 эквивалент DIPEA, 1,1 эквивалента TBTU и через 5 минут 1 эквивалент 0,25 моль/л раствора H-Dpr(BOC)-Leu-OMe в ДМФ, полученного реакцией гидрирования (пример 10), добавляют к 0,22 моль/л раствора Z-Asp-(O-трет-Bu)-Trp-Phe-OH в ДМФ, охлажденному до -5°C, при поддержании температуры ниже -5°C.
После перемешивания в течение приблизительно 2 часов реакционную смесь разбавляют 0,5 М водным раствором NaHCO3.
Образовавшуюся суспензию фильтруют и полученное твердое вещество промывают последовательно H2O, смесью 3:4 ДМФ - 0,5 М NaHCO3, Н2О и затем сушат в вакууме при 30-40°C, получая при этом продукт с выходом 84,4%. Точка плавления 215-218°C.
1H-ЯМР (ДМСО-d6) δ:
0,86 (2д; 6Н); 1,34 (с; 9Н); 1,37 (с; 9Н); 1,40-1,72 (м; 3H); 2,23-2,67 (м; 2Н); 2,71-3,39 (м; 6Н); 3,62 (с; 3H); 4,23-4,58 (м; 5Н); 5,01 (AB-Syst.; 2Н); 6,89-7,58 (м; 16Н); 6,50 (т; 1Н); 7,87-8,29 (4д; 4Н); 10,78 (с; 1Н).
ПРИМЕР 16
Получение Z-Trp-Phe-OMe
Соединение получают по способу примера 9 сочетанием двух аминокислот Z-Trp-OH и H-Phe-OMe
1H-ЯМР (CDCl3) δ:
2,88-2,98 (м; 2Н); 3,11 (дд; 1Н); 3,32 (дд; 1Н); 3,62 (с; 3H); 4,40-4,58 (м; 1Н); 4,16-4,30 (м; 1Н); 5,11 (с; 2Н); 5,45 (д; 1Н); 6,11 (д; 1Н); 6,72-6,85 (м; 2Н); 6,92-7,46 (м; 12Н); 7,67 (д; 1Н); 8,03 (с; 1Н).
ПРИМЕР 17
Получение Z-Trp-Phe-OH
Данное соединение получают из метилового эфира примера 16 по способу, описанному в примере 3.
1H-ЯМР (ДМСО-d6) δ:
2,70-3,15 (м; 4Н); 4,20-4,36 (м; 1Н); 4,38-4,55 (м; 1Н); 4,92 (с; 2Н); 6,85-7,42 (м; 15Н); 7,63 (д; 1Н); 8,26 (д; 1Н), 10,81 (с; 1Н); 12,30 (ушир.; 1Н).
ПРИМЕР 18
Получение H-Trp-Phe-OH
Данное соединение получают из защищенного производного примера 17 по способу примера 10 с использованием уксусной кислоты в качестве растворителя.
ПРИМЕР 19
Получение Z-Asp(O-трет-Bu)-Trp-Phe-OH
Соединение получают по способу примера 15 (способ а) из дипептида примера 18.
1H-ЯМР (ДМСО-d6) δ:
1,35 (с; 3H); 2,21-2,67 (м; 2Н); 2,71-3,18 (м; 4Н); 4,22-4,58 (м; 3H); 5,00 (AB-Syst; 2Н); 6,87-7,43 (м; 14Н); 7,55 (м; 2Н); 7,94 (д; 1Н), 8,17 (д; 1Н); 10,80 (с; 1Н); 12,25 (ушир.; 1Н).







| название | год | авторы | номер документа |
|---|---|---|---|
| СЕЛЕКТИВНЫЕ ЦИКЛОПЕПТИДЫ | 2001 |
|
RU2239642C1 |
| ЛИНКЕРЫ НА ОСНОВЕ СУЛЬФОМАЛЕИМИДА И СООТВЕТСТВУЮЩИЕ КОНЪЮГАТЫ | 2019 |
|
RU2815199C2 |
| СОЕДИНЕНИЯ АЗАЛАКТАМА В КАЧЕСТВЕ ИНГИБИТОРОВ HPK1 | 2021 |
|
RU2819642C1 |
| КОНЪЮГАТ АНТИТЕЛО-ПРОИЗВОДНОЕ ПИРРОЛОБЕНЗОДИАЗЕПИНА | 2018 |
|
RU2820928C2 |
| ПОЛУЧЕНИЕ ПЕПТИДОВ СОМАТОСТАТИНА | 2004 |
|
RU2360921C2 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПЕПТИДОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2092491C1 |
| НОВОЕ ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО ДИНУКЛЕОТИДА И ЕГО КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2019 |
|
RU2809547C2 |
| ДИПЕПТИДЫ В КАЧЕСТВЕ КОРМОВЫХ ДОБАВОК | 2010 |
|
RU2536467C2 |
| ПРОИЗВОДНЫЕ 5-АМИНО-4-ОКСИГЕКСАНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2067585C1 |
| ПРОИЗВОДНЫЕ МЕТАСТИНА И ИХ ПРИМЕНЕНИЕ | 2006 |
|
RU2430107C2 |
Настоящее изобретение относится к способу получения бициклических пептидных соединений формулы (I-A) через получение промежуточного продукта формулы (I) в жидкой фазе. Предложенный способ позволяет получать бициклические пептидные соединения с высоким выходом и высокой чистотой. 2 н. и 21 з.п. ф-лы.
посредством промежуточного продукта формулы (I)
включающий в себя следующие стадии:
1) удаление защитной группы у линейного пентапептида формулы (II) в присутствии растворителя с получением соединения формулы (III)
где A1 и А2 представляют собой две защитные группы атомов азота, отличные друг от друга, и R5 и R6, отличные друг от друга, выбраны из бензилокси- и низших алкилоксигрупп, в которых алкильная часть содержит неразветвленную или разветвленную С1-С4-группу;
2) внутримолекулярную циклизацию соединения формулы (III), полученного на стадии 1), в присутствии растворителя и подходящего агента сочетания с получением соединения формулы (IV)
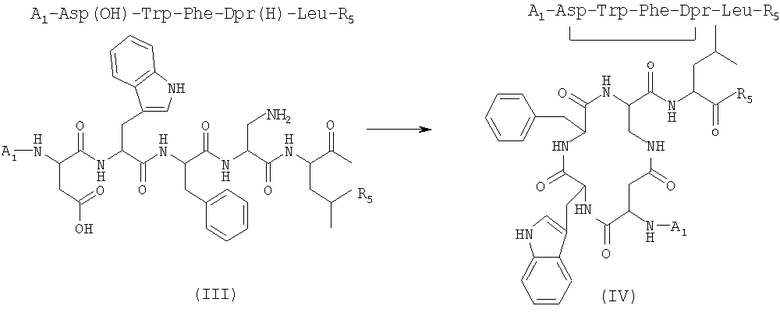
где R5 имеет указанные выше значения;
3) удаление защитной группы у соединения (IV), полученного на стадии 2), в присутствии растворителя с получением соединения формулы (V)
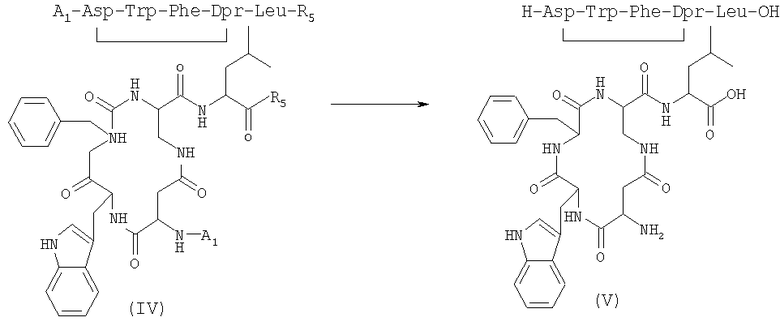
где R5 имеет указанные выше значения;
4) сочетание соединения формулы (V), полученного на стадии 3), и защищенной аминокислоты формулы (VIa) в присутствии растворителя с получением соединений формулы (VII)
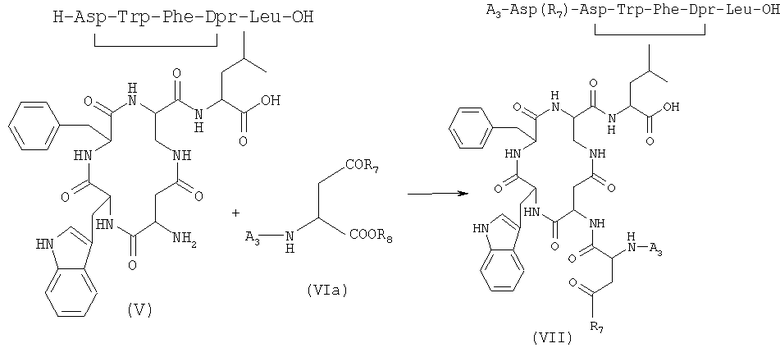
где А3 представляет собой защитную группу атома азота; R7 выбран из бензилокси- и низших алкилоксигрупп, в которых алкильная часть содержит неразветвленную или разветвленную С1-С4-группу; R8 представляет собой остаточную группу, образованную процедурой активации на карбоксильной группе;
5) удаление защитной группы у соединения формулы (VII), полученного на стадии 4), в присутствии растворителя с получением соединения формулы (VIII)
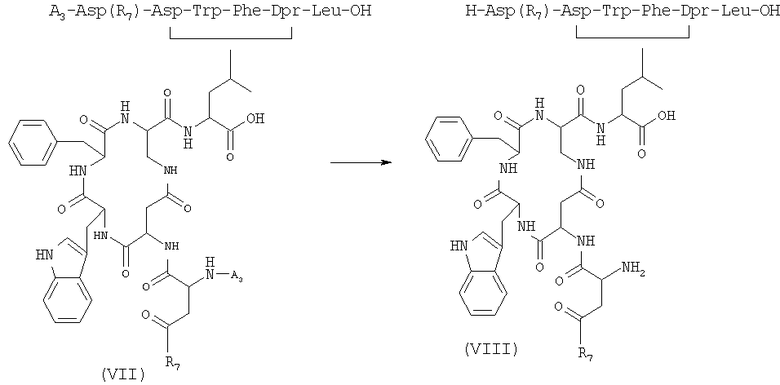
где R7 имеет указанные выше значения;
6) внутримолекулярную циклизацию, в присутствии растворителя и подходящего агента сочетания, соединения формулы (VIII), полученного на стадии 5), с получением бициклического соединения формулы (IX)
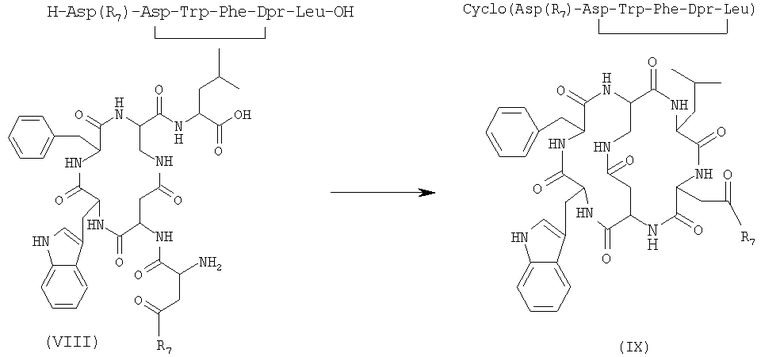
где R7 имеет указанные выше значения;
7) удаление защитной группы у бициклического соединения формулы (IX), полученного на стадии 6), в присутствии растворителя с получением соединения формулы (I)
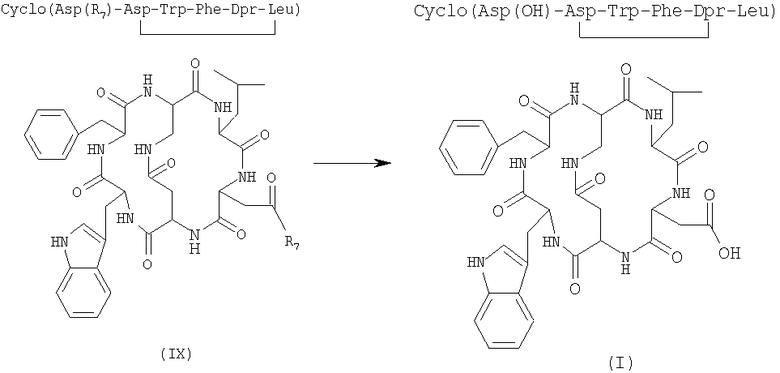
где R7 имеет указанные выше значения;
8) активация бициклических пептидных соединений формулы (I) с подходящим агентом сочетания с получением производного соединения формулы (II-A)
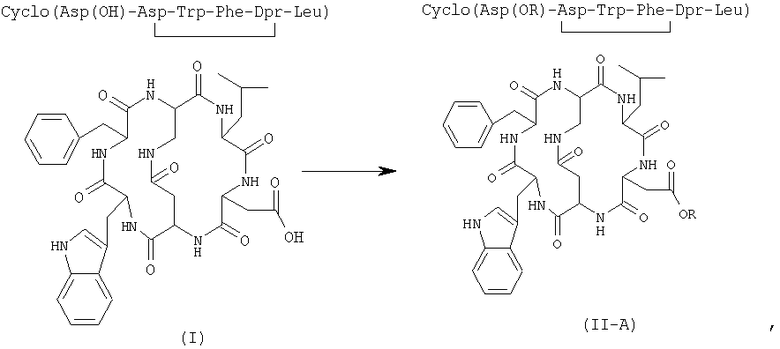
где R представляет собой группу, выбранную из бензотриазола, возможно замещенного галогеном, азабензотриазола и сукцинимидила;
9) взаимодействие соединения формулы (II-A), полученного на стадии 8), в присутствии растворителя с гликозидным производным формулы (III-A) с получением соединения формулы (I-A)
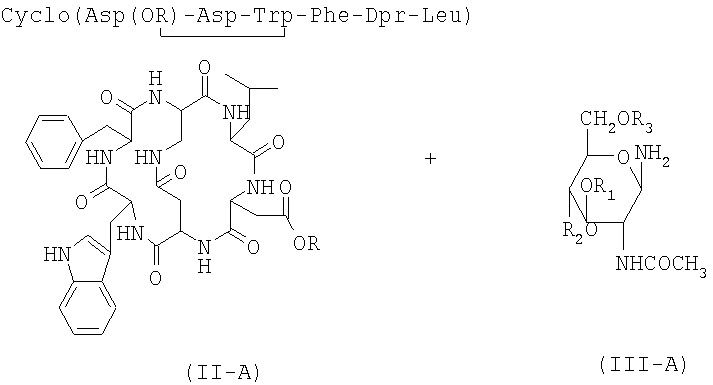
где R, R1, R2 и R3 имеют указанные выше значения.
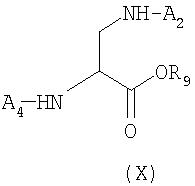
где А2 и А4, отличные друг от друга, представляют собой защитные группы атомов азота;
R9 представляет собой остаточную группу, образованную процедурой активации, предпочтительно выбранную из группы, состоящей из бензилоксикарбонила, алкоксикарбонила, содержащего в алкильной части неразветвленную или разветвленную С1-С4-группу, и сукцинимидила;
в соответствии со следующими стадиями:
взаимодействие производного формулы (X), приведенной выше, в присутствии растворителя с эфиром Leu формулы (XI)
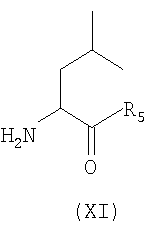
где R5 имеет значения, указанные в п.1, с получением дипептида A4-Dpr(A2)-Leu-R5,
удаление защитной группы у дипептида A4-Dpr(A2)-Leu-R5 с получением дипептида с одной удаленной защитной группой H-Dpr(A2)-Leu-R5;
сочетание дипептида с одной удаленной защитной группой Н-Dpr(А2)-Leu-R5 с активированным эфиром последующей аминокислоты Phe и затем последовательно с Trp и Asp до получения соединений формулы (II).
сочетание дипептида с одной удаленной защитной группой H-Dpr(A2)-Leu-R5, полученного, как описано в п.2, с активированным производным дипептида нижеследующей формулы (XII)
A5-Trp-Phe-OH,
(XII)
где А2 и А5, отличные друг от друга, представляют собой защитные группы атомов азота, полученных отдельно или генерированных in situ сочетанием активированного эфира Trp, защищенного у атома азота и полученного отдельно или генерированного in situ, с эфиром Phe, и последующим гидролизом эфирной группы с получением тетрапептида A5-Trp-Phe-Dpr(A2)-Leu-R5;
подходящее удаление защитной группы, присоединенной к атому азота у Trp, у тетрапептида A5-Trp-Phe-Dpr(A2)-Leu-R5;
сочетание тетрапептида с удаленной защитной группой с соединением формулы (VIb)
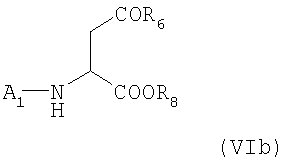
в которой A1, R6 и R8 имеют значения, указанные в п.1.
| Пароперегреватель для паровозов | 1925 |
|
SU697A1 |
| Hardy Weiβhoff et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Shimon T | |||
| Ansfield et al., "A Convergent Approach to the Chemical Synthesis of Asparagine-Linked Glycopeptides", J.Org | |||
| Chem | |||
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |

