Изобретение относится к синтезу биологически активных веществ, в частности к синтезу экдистероидов, конкретно к синтезу 2,3-моноацетонида 20-гидроксиэкдизона (I).
2,3-Моноацетонид I в очень малых количествах присутствует в некоторых видах растений, например Rhaponticum carthamoides. [Ecdysteroids of the root bark of Vitex caescens. // Phytochemistry, 1997, 45, №6, 1149-1152].
Экдистероиды широко распространены в животном и растительном мире и выполняют функцию регуляторов линьки и метаморфоза насекомых и ракообразных. Являясь нетоксичными для млекопитающих, эти соединения и их аналоги представляют интерес для медицины.
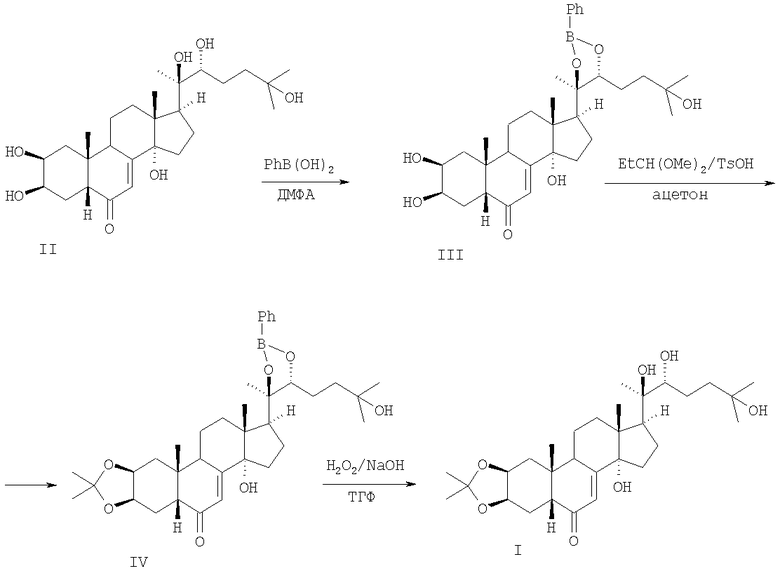
Как производное 20-гидроксиэкдизона (II), избирательно защищенное по 2,3-гидроксильным группам, 2,3-моноацетонид (I) удобен для последующих селективных трансформаций одного из наиболее представительных фитоэкдистероидов II в малораспространенные в природе экдистероиды. Например, в синтезе шидастерона 2,3-моноацетонид I получали путем трехстадийных трансформаций 20-гидроксиэкдизона II [1] [Roussel P.G., Turner N.J., Dinan L.N. Synthesis of Shidasdterone and the Unambiguous Determination of its Configuration at C-22. // J. Chem. Soc., Chem. Commun., 1995, 933-934]. При взаимодействии 20-гидроксиэкдизона II с фенилборной кислотой в диметилформамиде (ДМФА) был получен 20,22-фенилборонат 20-гидроксиэкдизона (III). При взаимодействии III с 2,2-диметоксипропаном и TsOH в ацетоне получен 2,3-ацетонид-20,22-фенилборонат 20-гидроксиэкдизона (IV). Обработка IV 30%-ным раствором перекиси водорода в присутствии NaOH в тетрагидрофуране привела к селективному снятию боронатной защиты и получению 2,3-моноацетонида I, суммарный выход которого составил 65% (схема 1).
Схема 1.
Недостатками данного способа являются многостадийность, использование специфических и дорогих реагентов, неполная конверсия исходных соединений на каждой стадии, что требует дополнительной очистки промежуточных и конечных продуктов.
Задачей предлагаемого изобретения является упрощение способа получения 2,3-моноацетонида 20-гидроксиэкдизона.
Существенным отличительным признаком предлагаемого способа, позволяющим решить поставленную задачу, является использование фосфорно-молибденовой кислоты в реакции 20-гидроксиэкдизона II с ацетоном с последующим разделением образовавшегося продукта колоночной хроматографией.
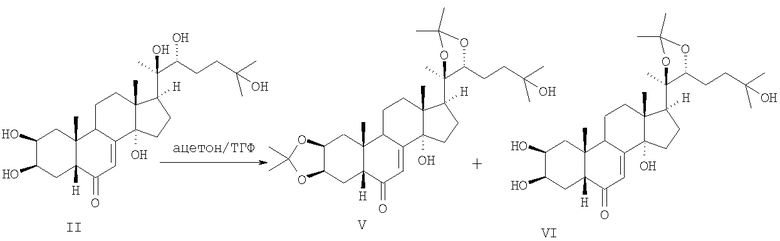
Весьма привлекательной представляется возможность получения соединения I путем взаимодействия 20-гидроксиэкдизона II с ацетоном в присутствии кислоты. Однако согласно известному способу [2] [Одиноков В.Н., Галяутдинов И.В., Недопекин Д.В., Халилов Л.М. Трифторацетилирование и дегидратация ацетонидов 20-гидроксиэкдизона. Синтез стахистерона В. // Изв. АН. Сер. Хим. - 2003. - №1. - С.220-224] реакция II с ацетоном в присутствии 0.1 мол.% фосфорно-молибденовой кислоты (ФМК) приводит к смеси (85:15) 2,3:20,22-диацетонида (V) и 20,22-моноацетонида 20-гидроксиэкдизона (VI):
Схема 2.
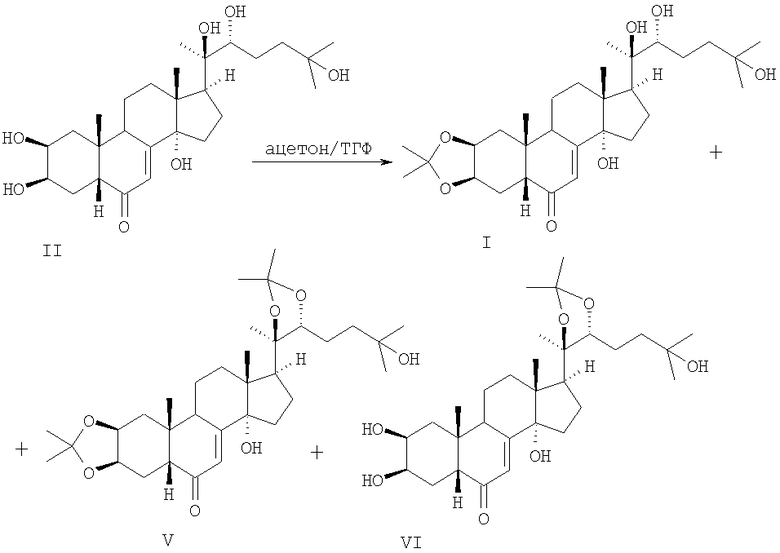
При исследовании взаимодействия 20-гидроксиэкдизона II с ацетоном в присутствии ФМК обнаружено, что при взаимодействии суспензии соединения II и 7-8 мол. % ФМК в ацетоне по истечении 5 мин наблюдалась гомогенизация и появлялась зеленая окраска реакционной смеси. Ее упариванием, нейтрализацией 0.1% раствором гидрокарбоната натрия с последующей экстракцией целевого продукта этилацетатом и хроматографированием был получен 2,3-моноацетонид, идентичный (ИК, УФ, ЯМР 1Н и 13С) 2,3-моноацетониду, описанному ранее в упомянутом 3-стадийном синтезе [1].
Схема 3.
Несомненным преимуществом предлагаемого синтеза является одностадийность процесса и доступность реагентов, что существенно упрощает способ получения целевого продукта.
Пример осуществления способа. К суспензии 1.0 г (2.08 ммоль) II в 200 мл ацетона добавили 0.3 г (0.16 ммоль, 7.7 мол.%) фосфорно-молибденовой кислоты. Реакционную массу перемешивали при комнатной температуре, пока реакционная смесь не станет гомогенной и не приобретет зеленоватый цвет (~5 мин). Реакционную смесь упарили до объема ~25 мл, добавили 15 мл 0.1% раствора NaHCO3 и экстрагировали этилацетатом (100 мл × 3). Экстракт упарили и хроматографировали на колонке с 40 г SiO2 (элюент - CHCl3-МеОН, 9:1), получив 0.35 г (32%) соединения I, т.пл. 135-137°С, [α]D 20+29.6° (с=2.53, МеОН), ИК, УФ, ЯМР 1Н и 13С идентичны приведенным в литературе [1].
Наряду с целевым соединением I из суммарного продукта реакции были выделены диацетонид V (выход 30%, т.пл. 134-135°С, [α]D 20+39.0° (с=1.1, CHCl3)) и 20,22-моноацетонид VI (выход 38%, т.пл. 223-224°С, [α]D 20+58.0° (с=0.9, CHCl3)).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ α-ЭКДИЗОНА | 2003 |
|
RU2246966C1 |
| СПОСОБ ПОЛУЧЕНИЯ ШИДАСТЕРОНА | 2001 |
|
RU2183641C1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛПЕРЕГРУППИРОВАННОГО ЭКДИСТЕРОИДА - 2,3:20,22-ДИАЦЕТОНИДА 9α,13α-ЭПОКСИ-14β-МЕТИЛ-13-ДЕМЕТИЛ-14-ДЕЗОКСИ-7,8-ДИГИДРО-20-ГИДРОКСИЭКДИЗОНА | 2010 |
|
RU2443709C1 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ УЛУЧШЕННОГО МЫШЕЧНОГО МЕТАБОЛИЗМА | 2016 |
|
RU2730853C2 |
| СПОСОБ ПОЛУЧЕНИЯ 7,8-ДИГИДРОАНАЛОГОВ ЭКДИСТЕРОИДОВ | 2010 |
|
RU2434877C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОНЪЮГАТА (6-ГИДРОКСИ-2,5,7,8-ТЕТРАМЕТИЛХРОМАН-2-ИЛ)АЦЕТАЛЬДЕГИДА С 20-ГИДРОКСИЭКДИЗОНОМ И ЕГО ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТИОКСИДАНТНОГО СРЕДСТВА, ИНГИБИРУЮЩЕГО ПРОЦЕСС ПЕРЕКИСНОГО ОКИСЛЕНИЯ ЛИПИДОВ | 2010 |
|
RU2490267C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2,3-ДЕЗОКСИ-ΔЭКДИСТЕРОИДОВ | 2020 |
|
RU2760001C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ДЕГИДРО-3-ЭПИ-20-ГИДРОКСИЭКДИЗОНА, МИНОРНОГО ЭКДИСТЕРОИДА СЕМЯН РАСТЕНИЙ Froelichia floridana | 2008 |
|
RU2397177C2 |
| АГЕНТ РЕГУЛИРОВАНИЯ ДИФФЕРЕНЦИАЦИИ КЛЕТОК КОЖИ, КУЛЬТУРАЛЬНАЯ СРЕДА ДЛЯ КЛЕТОК ИЛИ ТКАНЕЙ И СПОСОБ РЕГУЛИРОВАНИЯ ДИФФЕРЕНЦИАЦИИ КЛЕТОК КОЖИ | 1993 |
|
RU2143884C1 |
| ТОНИЗИРУЮЩЕЕ И АКТОПРОТЕКТОРНОЕ СРЕДСТВО "СЕРПИСТЕН" | 2005 |
|
RU2276991C1 |
Изобретение относится к синтезу биологически активных веществ, в частности, конкретно, к улучшенному способу получения 2,3-моноацетонида 20-гидроксиэкдизона формулы I, встречающегося в очень малых количествах в некоторых видах растений, например Rhaponticum carthamoides.
Способ осуществляют взаимодействием 20-гидроксиэкдизона (1.0 г, 2.08 ммоль) с ацетоном в присутствии фосфорно-молибденовой кислоты (ФМК), причем при действии на суспензию исходного соединения в ацетоне ФМК (0.3 г, 0.16 ммоль) по истечении 5 мин наблюдается гомогенизация реакционной смеси, после упаривания которой нейтрализацией 0.1% раствором гидрокарбоната натрия с последующей экстракцией целевого продукта этилацетатом и хроматографированием был выделен целевой 2,3-моноацетонид 20-гидроксиэкдизона с выходом 32%. Способ высокоселективен и одностадиен.
Способ получения 2,3-моноацетонида 20-гидроксиэкдизона формулы I
из 20-гидроксиэкдизона, отличающийся тем, что 20-гидроксиэкдизон подвергают взаимодействию с ацетоном в присутствии 7,7 мол.% фосфорно-молибденовой кислоты до гомогенизации реакционной смеси и появления зеленой окраски раствора при комнатной температуре в течение 5 мин, с последующим выделением целевого продукта известными приемами.
| СПОСОБ ПОЛУЧЕНИЯ α-ЭКДИЗОНА | 2003 |
|
RU2246966C1 |
| ROUSSEL P.G., et al | |||
| // J | |||
| Chem | |||
| Soc | |||
| Comm., 1995, 933-934 | |||
| ОДИНОКОВ B.H., ГАЛЯУТДИНОВ И.В., НЕДОПЕКИН Д.В., ХАЛИЛОВ Л.М | |||
| // Изв.АН | |||
| Сер | |||
| Хим., 2003, №1, с.220-224. | |||

