Изобретение относится к области аналитической химии объектов окружающей среды и направлено на разработку средств аналитического контроля параметров экосистем и полиэлементного мониторинга природных вод и водных экосистем.
Известен способ вольтамперометрического определения цинка, кадмия, свинца и меди в почвах и биологических объектов [авторы Э.А.Захарова, Г.Б.Слепченко и другие], разработанный в лаборатории НИЛ микропримесей ТПУ и рекомендованный к реализации с помощью компьютерных вольтамперометрических анализаторов типа ТА 1, ТА 2, СТА и других (табл.1) внедренческой научно-производственной фирмой «ЮМХ».
Методика (аналог) основана на проведении инверсионно-вольтамперометрических измерений с накоплением цинка, кадмия, свинца и меди на индикаторном электроде (ртутно-пленочном, ртутно-графитовом) при заданном отрицательном потенциале электролиза - 1,4 В относительно хлоридсеребряного электрода сравнения. Процесс электрорастворения элементов с поверхности электрода и регистрация аналитических сигналов (анодных пиков) на вольтамперограмме проводится при ступенчатой развертке потенциала 100 мВ/с в анодном направлении от -1,2 В до +0,15 В относительно электрода. Потенциалы максимумов регистрируемых анодных пиков (аналитических сигналов) цинка, кадмия, свинца и меди на фоне муравьиной кислоты соответственно равны (-0,9±0,1) В; (-0,6±0,1) В; (-0,4±0,1) В; (-0,05±0,10) В.
Растворенный кислород удаляют инертным газом.
Подготовка индикаторного ртутно-пленочного или ртутно-графитового электрода перед аналитической процедурой включает стадию формирования ртутной пленки на поверхности соответствующего рабочего (серебряного, графитового) электрода. В случае природных вод и других жидких проб аналог требует длительной, около одного часа, трудоемкой подготовки аналитического образца, например ультрафиолетового облучения (УФО) с фотокатализатором - двуокисью титана. УФО может занимать до двух часов в зависимости от наличия в пробе органического вещества. Наличие в водах органического вещества в миллиграммовых количествах отрицательно влияет на аналитические сигналы элементов вследствие сорбции его на поверхности ртутной пленки или графите.
Сорбция органического вещества уменьшает величины предельных диффузионных токов элементов, с одной стороны, и смещает максимум токов пиков элементов из-за комплексующего действия растворенного органического вещества на определяемые элементы. Для разложения избыточного органического вещества в воде используют физическое воздействие: температуру, ультразвук и другие, либо химическое, например кислотную обработку сухого остатка воды после выпаривания. Подобного рода предварительная обработка водных образцов удлиняет аналитическую процедуру.
Недостатки аналога:
- решающим недостатком является сложность определения низких концентраций, менее 1 мкг/г свинца, кадмия и цинка в образцах,
- использование на стадии подготовки образцов природного материала кислотного озоления для химического разложения; такое разложение в смеси серной, азотной и хлористоводородной кислот требует соблюдения строгих правил техники безопасности при работе с агрессивными кислотами;
- в качестве активного коллектора аналита используется ртуть в виде металла или раствора; ртуть также по гигиеническим нормативам относится к веществу первого класса опасности;
- методика-аналог не позволяет определять железо, которое не образует амальгаму (ртутный сплав) и поэтому ионы железа не определяют на ртутных индикаторных электродах.
Недостаток аналога, заключающийся в сорбции органического вещества природных вод поверхностью индикаторного электрода (ртутно-пленочного на серебряной или графитовой подложке), заявляемый способ превращает в преимущество, так как реализует на этапе подготовки экстрагента введение тиоцианата в систему вода - антипирин - сульфосалициловая кислота для образования комплексных ионов цинком, кадмием, свинцом и медью при их экстрагировании из твердых природных объектов (взвесей, почв, частиц снежной массы, биологических или клинических объектов) органической фазой гидратотиоцианата сульфосалицилата антипириния.
Из известных технических решений наиболее близким по назначению и технической сущности к заявляемому объекту является экстракционный способ подготовки аналитических образцов (Патент РФ № 2232718) / С.В.Темерев, Л.С.Егорова // 20.07.2004, БИ № 20 (прототип), заключающийся в том, что для десорбции кадмия из твердых частиц природного материала используют расслаивающую систему, содержащую реагент тиопирин 0,002-0,003 моль, органическую трихлоруксусную кислоту 1,2-1,6 моль и неорганическую ортофосфорную кислоту 0,03-0,05 моль, и дистиллированную воду до литра при комнатной температуре 25°С в течение 30 мин. Данный способ обеспечивает максимальное извлечение кадмия за короткое время при комнатной температуре.
Недостатки прототипа:
- необходимость целевого синтеза тиопирина как комплексующего кадмий реагента и его недоступность для приобретения аналитиками для массовых мониторинговых исследований;
- агрессивность и большая токсичность трихлоруксусной кислоты;
- невозможность определения нескольких элементов в одном акте химического анализа в кварцевой печи, графитовой кювете или другом атомизаторе;
- невозможность повторного концентрирования аналита в атомно-абсорбционном методе в сравнении с электрохимическим групповым концентрированием элементов на электроде при потенциале накопления.
Вышеперечисленные недостатки устраняет заявляемый способ.
Экстракционно-вольтамперометрический способ определения цинка, кадмия, свинца, меди и железа в твердых образцах природных объектов заключается в извлечении металлов из твердых частиц природных материалов в органическую фазу (ОФ) расслаивающейся системы вода - антипирин - сульфосалициловая кислота - тиоцианат с последующим катодным концентрированием и получением анодной вольтамперограммы в виде пиков анодного окисления элементов, и полиэлементным количественным определением в режиме «ех situ» металлов в одном акте химического анализа.
В данном способе используют более доступные для аналитиков реагенты, смешивают 2 М растворы фармакопейного антипирина (HAnt - 2,3-диметил-1-фенил-3-пиразолин-5-он) и 2-водной сульфосалициловой кислоты (дигидрат HSSA×2H2O) марки ч.д.а. в объемном соотношении растворов реагентов 2:1 и вводят анионы тиоцианата в виде соли роданида калия марки ч.д.а. из расчета 0,1 моль KSCN на литр смеси реагентов.
В отличие от прототипа в качестве комплексующего определяемые элементы: Zn, Cd, Pb, Cu, Fe агента в заявляемом способе используется твердое вещество тиоцианат калия KSCN. Именно такая органическая фаза расслаивающейся системы вода - антипирин - сульфосалициловая кислота - тиоцианат калия используется в качестве экстрагента цинка, кадмия, свинца, меди и железа из твердых частиц природного материала, например твердой компоненты снежного покрова (particulate matter). Кроме того, сульфосалициловая кислота - фотометрический реагент на ионы железа.
Заявляемое изобретение реализует неразрушающий метод, дающий информацию о формах элементов в концентрате.
Осуществление изобретения.
Органическую фазу (ОФ) расслаивающейся системы H2O-HAnt-HSSA-KSCN для экстрагирования готовят следующим образом.
Сначала готовят двумолярные (2М) отдельно растворы реагентов:
- фармакопейного антипирина (брутто формула C11H12N2O, температура плавления 113°С, молекулярная масса 188,23 г/моль) и двухводной сульфосалициловой кислоты (ГОСТ 4478-78, брутто формула С7Н6О68×2H2O, молекулярная масса 254,21 г/моль).
Затем смешивают эти растворы HAnt и HSSA в объемном соотношении 2:1 и добавляют твердый тиоцианат калия из расчета 0,1 моль KSCN на литр смеси реагентов. Формирование нижней фазы системы ведут путем интенсивного встряхивания с последующим отстаиванием при комнатной температуре 25°С в высоком сосуде или центрифугированием. Такое отношение реагентов приводит к быстрому расслаиванию в течение 15-20 минут, при котором объем органической фазы (ОФ) занимает 1/4 от общего объема системы (4-кратное объемное концентрирование). Все приготовления ОФ занимают не более 30 минут. Навеску твердых частиц природного материала 0,1000-0,5000 г помещают в бюксы, добавляют по 2 мл органической фазы, тщательно перемешивают стеклянной палочкой 20-30 минут при 25°С. Затем отбирают аликвоту 5 мкл и равномерно наносят на торец чистого графитового электрода. Модифицированный графитовый электрод помещают в трехэлектродную ячейку с 0,1 моль/л KSCN, без задержки ведут электролиз при -1,4 В в течение 30 секунд. Затем без задержки регистрируют вольтамперограмму в пределах -1,00 до +1,00 В с пиками анодного окисления цинка, кадмия, свинца, меди и железа, наблюдаемым в виде максимумов предельных диффузионных токов примерно при тех же потенциалах, характерных для водных растворов (-0,900…-0,800 B)Zn, (-0,640…-0,610 B)Cd, (-0,450…-0,390 В)Pb, (-0,100…0,000 B)Cu, (+0,78…+0,79 B)Fe.
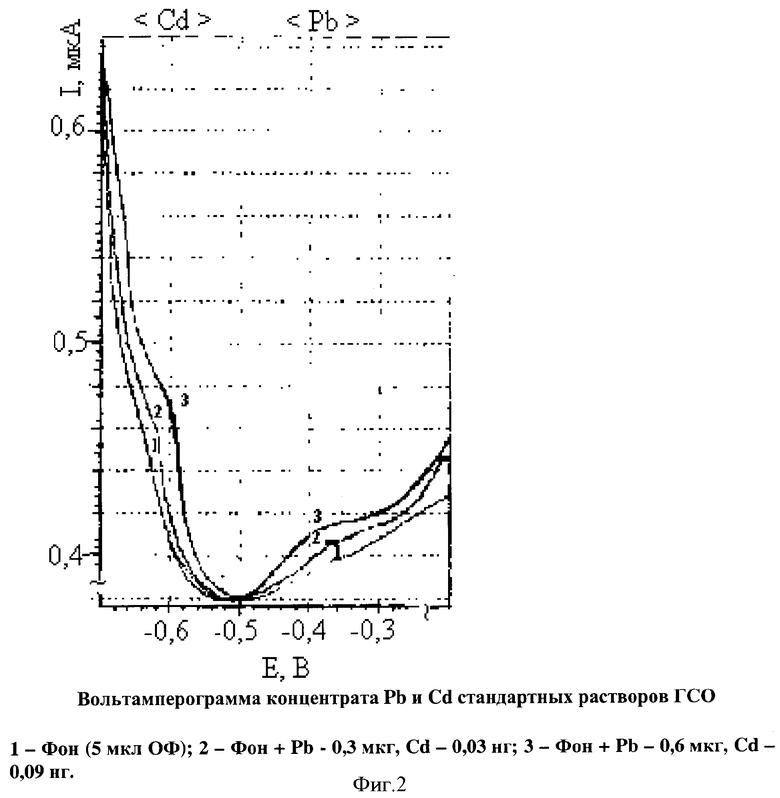
Таким образом, степень извлечения после определения независимым методом ААС в пламени представлена в табл.2.
Примечание: степень извлечения рассчитана по отношению к параллельным образцам частиц снега, озоленным в смеси 2 мл азотной +1 мл серной +1 мл хлористоводородной концентрированных кислот.
Образующаяся органическая фаза ОФ, состоящая из ионного ассоциата органической соли, сульфосалицилата антипириния и реагента антипирина, имеет кислую реакцию среды, обладает большой буферной емкостью и извлекает из частиц твердого природного материала железо и тяжелые металлы, вследствие образования сложного комплекса металлов с ионной по природе и органической по составу жидкости плотностью около 1,5 г/см3 с ограниченной растворимостью в воде. ОФ расслаивающейся системы, формирующаяся в нижней фазе при расслаивании в виде ионной жидкости желтого цвета, используется для модификации графитового рабочего электрода (фоновый концентрат ОФ). В присутствии железа в природном материале ОФ окрашивается в интенсивный малиновый цвет роданида железа.
Далее концентрат ГСО или экстракт природного материала (24 образца) «ех situ» отбирают хроматографическим шприцем 5 мкл нижней органической фазы ОФ и равномерно наносят на подготовленную поверхность графитового электрода. Перед каждой серией вольтамперограмм поверхность графитового электрода обновляют, полируют фильтровальной бумагой, обезжиривают в 10 мл 0,1 М HCl с добавкой 0,2 мл 3%-ного щелочного раствора борогидрида натрия и промывают в кипящей дистиллированной воде для удаления избытка кислорода с его поверхности. На сухую графитовую поверхность наносят 5 мкл органической фазы с помощью хроматографического микрошприца, равномерно распределяя по торцевой поверхности электрода (геометрическая площадь 0,13 см2). Такой пленочный электрод помещают в трехэлектродную ячейку с 10 мл 0,1 KSCN в качестве фонового раствора. Фоновый электролит не должен содержать растворенного кислорода. Растворенный в электролите кислород удаляют путем барботирования аргона в течение 3-5 минут. Далее без задержки подают потенциал накопления ртути -1,4 В в течение 30 с. Затем в режиме анодной развертки регистрируют вольтамперограмму в квадратно-волновом режиме регистрации со скоростью анодной развертки потенциала 150 мВ/с (ТА-2, Томск, ТПУ) с 0,1 М раствором KSCN, не содержащим растворенного кислорода, и накладывают перенапряжение -1,4 В на 30 секунд относительно хлоридсеребряного электрода сравнения, и регистрируют анодную вольтамперограмму в квадратно-волновом режиме в области потенциалов от -1,0 В до +1,0 В, типичные вольтамперограммы представлены на фиг.1.
Оптимальные условия регистрации вольтамперограмм: цинк, медь и железо определяются при квадратно-волновом режиме регистрации, скорости развертки, равной 150 мВ/с в диапазоне развертки от -1,0 В до 1,0 В, свинец и кадмий оптимально регистрировать в более узком диапазоне развертки потенциала от -0,7 В до -0,2 В и меньшей скорости 50 мВ/с (фиг.2).
Максимумы предельных диффузионных токов наблюдают примерно при тех же потенциалах, характерных для водных растворов (-0,900…-0,800 B)Zn, (-0,640…-0,610 B)Cd, (-0,450…-0,390 В)Pb, (-0,100…0,000 B)Cu, (+0,78…+0,79 B)Fe. Величины потенциалов соответствуют водным растворам электролитов потому, что вода служит единственным гомогенизирующим растворителем.
Эффективность и метрологические характеристики заявляемого способа иллюстрирует табл.1 на примере определения элементов в 12 пробах частиц снежного покрова, собранных на трековые мембраны диаметром пор 0,11 мкм, после фильтрования снеговых вод, собранных с территорий экосистемы реки Барнаулки с различным уровнем химической нагрузки.
Результаты статистического анализа, представленные в табл.2 показывают, что определение элементов в концентрате статистически незначимо и определяется эффективностью извлечения элементов в форме тиоцианатных комплексов определяемых элементов. Сравнение результатов заявляемым способом определения извлечения элементов в ОФ расслаивающейся системы (Э) с окончанием аналитической процедуры и методом подготовки образцов природного материала мокрым озолением в смеси концентрированных кислот с последующим анализом минерализатов пламенной атомно-абсорбционной спектрометрией представлено в табл.3.
Несмотря на неизбежные случайные погрешности результаты определений различаются в пределах доверительных интервалов и представительно характеризуют заявляемый способ экстракционной вольтамперометрии (табл.3). Величины предельных диффузионных токов линейно зависят от концентрации металлов в концентрате ОФ расслаивающейся системы.
Преимущества предлагаемого способа:
- удешевляет процедуру анализа из-за применения значительно более простого и недорогого оборудования, не имеющего источников повышенной опасности для аналитика,
- исключает стадии высокотемпературного разложения аналита с целью его атомизации.
- электрохимические анализаторы легче автоматизируются, значительно меньше по размерам и массе, а электрохимические методы позволяют дополнительно концентрировать аналит на поверхности рабочего электрода при потенциале накопления.
- вольтамперометрические анализаторы в полевом варианте позволяют контролировать элементы непосредственно на месте отбора проб воды.
- реализует определение железа по анодному пику тока при потенциале +0,78…+0,79 В.
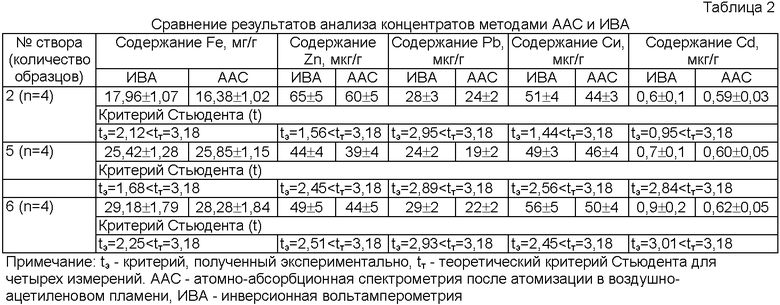
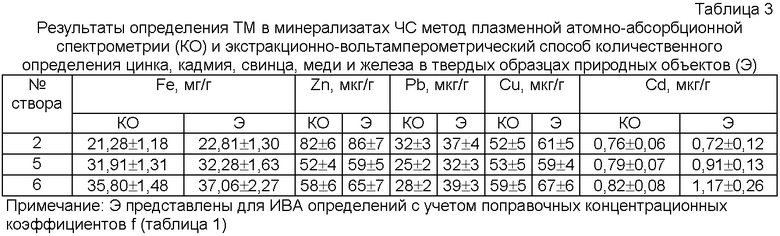
Способ заключается в том, что для экстрагирования металлов из твердых частиц природного материала используют водно-органическую расслаивающуюся систему, для приготовления экстракционного реагента смешивают 2 М растворы фармакопейного антипирина и 2-водной сульфосалициловой кислоты марки ч.д.а. в объемном соотношении растворов реагентов 2:1 и вводят анионы тиоцианата в виде соли роданида калия марки ч.д.а. из расчета 0,1 моль KSCN на литр смеси реагентов с последующим катодным концентрированием и поэлементным количественным определением металлов вольтамперометрическим методом в режиме «ex situ» в одном акте химического анализа. Изобретение обеспечивает возможность определения цинка, кадмия, свинца, меди и железа в одном акте химического анализа в атомизаторе, снижение токсичности, использования в массовых мониторинговых исследованиях. 3 табл., 2 ил.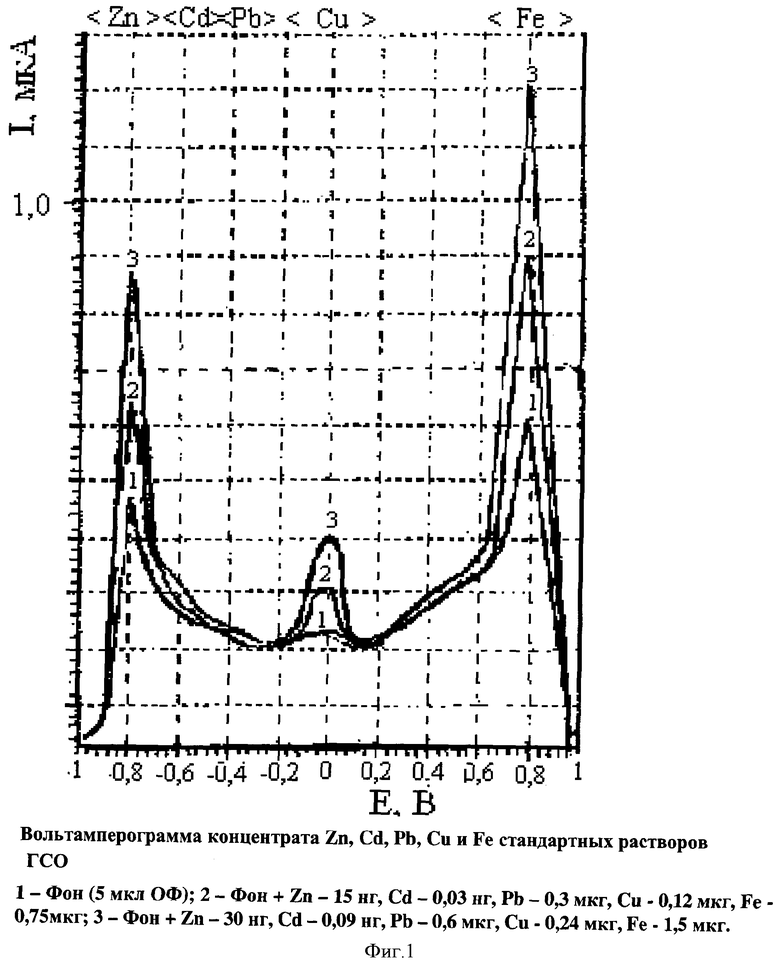
Экстракционно-вольтамперометрический способ определения цинка, кадмия, свинца, меди и железа в твердых образцах природных объектов, заключающийся в экстрагировании металлов из твердых частиц природного материала в органическую фазу расслаивающейся системы, отличающийся тем, что для приготовления экстракционного реагента смешивают 2 М растворы фармакопейного антипирина и 2-водной сульфосалициловой кислоты марки ч.д.а. в объемном соотношении растворов реагентов 2:1 и вводят анионы тиацианата в виде соли роданида калия марки ч.д.а. из расчета 0,1 моль KSCN на литр смеси реагентов с последующим катодным концентрированием и поэлементным количественным определением металлов вольтамперометрическим методом.
| ЭКСТРАКЦИОННЫЙ СПОСОБ ПОДГОТОВКИ АНАЛИТИЧЕСКИХ ОБРАЗЦОВ | 2003 |
|
RU2232718C1 |
| Способ экстракционно-вольтамперометрического определения этиленбисдитиокарбамата цинка (цинеба) | 1986 |
|
SU1377708A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СВИНЦА В КАДМИИ И ЕГО СОЛЯХ | 0 |
|
SU264759A1 |
| WO 9830738 A2, 16.07.1998. | |||

