Область техники
Настоящее изобретение относится к области неорганической химии углерода, точнее к процессу получения стабильной суспензии кубической (алмазной) модификации углерода, образованной путем детонационного синтеза - детонационных наноалмазов (ДНА), которая может быть использована в гальванике; химическом осаждении металлов; в приготовлении полировальных систем; в полимерной химии; при получении магнитных носителей информации; при изготовлении режущего и абразивного инструмента; в плазменной химии; в качестве катализатора при синтезе любых алмазов, в гетерогенном и электрохимическом катализе; в качестве аккумулятора водорода; для изготовления полупроводниковых алмазов; для изготовления высокоактивных адсорбентов; для изготовления биологически активных композиций и в биохимическом синтезе; для приготовления коллоидных суспензий ДНА в неводных средах; для приготовления присадок для масел, смазок и смазочно-охлаждающих жидкостей.
Предшествующий уровень техники
Свойства алмазсодержащих материалов, полученных с использованием энергии взрыва, а при подрыве также в условиях их синтеза и отделения от примесей, известны в промышленности.
Свойства алмаза, полученного из химически связанного «лишнего» углерода взрывчатых веществ, описано К.В.Волковым и его соавторами [2]. Синтез производится во взрывной камере в атмосфере диоксида углерода и в водной оболочке. Размер частиц полученного алмаза - от 4 до 6 нм, форма частиц - круглая. Пикнометрическая плотность - 3,2 г/см3. Продукт на воздухе содержит 90% алмаза и до баланса адсорбированные газы. Продукт начинает окисляться при 623 К.
Для выделения конечного алмазного продукта применяется комплекс химических операций, направленных либо на растворение, либо на газификацию примесей, присутствующих в этом материале. Примеси, как правило, двух видов: не углеродные (металл, оксиды, соли и т.д.) и не алмазные формы углерода (графит, сажа, аморфный углерод).
Описан алмазсодержащий материал [4], имеющий следующий элементный состав в процентном соотношении по массе: углерод 75-90, водород 0,6-1,5, азот 1,0-4,5, кислород - остальное, следующий фазовый состав в массовых процентах: рентгеновская аморфная алмазоподобная фаза 10-30, алмаз кубической модификации - остальное, и имеющий пористую структуру. 10-20% поверхности материала состоит из метальных, нитрильных и гидроксильных групп и 1-2% поверхности состоит из углеродных атомов с некомпенсированными связями. Метод получения указанного материала состоит в детонации углеродсодержащего взрывчатого вещества с отрицательным кислородным балансом или смеси взрывчатых веществ в замкнутом объеме в атмосфере газов инертных к углероду с содержанием кислорода 0,1-6,0% по объему, при температуре 303-363 К и в присутствии ультрадисперсной углеродной фазы с концентрацией 0,01-0,15 кг/м. Однако все признаки, заявленные в этом патенте, широко известны из литературных источников. Так, п.1.:
1.1. Элементный состав, мас.%:
(известно из работ [5-7]).
1.2. Фазовый состав, мас.%:
(описан в работах [5, 8]).
1.3. Пористая структура заявляемого материала, имеющая поры с объемом пор в пределах от 0,6 до 1,0 см3/т (описана в работах [5, 9, 10]).
1.4. На 10-20% поверхности материала находятся метил, нитрил, первичная и вторичная нитрогруппы (известно из работ [5-7, 10, 11]); 1-2% поверхности материала занята атомами углерода с некомпенсированными связями (описано в работе [12]).
1.5. Удельная поверхность находится в пределах от 200 до 450 м2/г (описано в работах [5, 10]).
п.2.: синтетический алмазсодержащий материал по п.1., в котором диаметр пор в пределах от 7,5 до 12,5 нм (описано в работе [9]).
п.3.: синтетический алмазсодержащий материал по п.1., в котором постоянная (параметр) кристаллической решетки 0,03562±0,0004 нм (описано в работах [5, 14, 15]).
п.4. и п.5.: алмазсодержащий материал, полученный в процессе, состоящем по существу из подрыва в замкнутом объеме заряда (описано в работах [1-19]), состоящего по существу из углеродсодержащего ВВ или смеси ВВ (описано в работах [1-19]), имеющих отрицательный кислородный баланс (описано в работах [1-19]), и детонации заряда в присутствии углеродных частиц в концентрации примерно от 0,01 до 0,15 кг/м3 (обычное следствие периодически повторяющегося подрыва ВВ в замкнутом объеме - наиболее мелкие частицы углеродной фазы постоянно находятся во взвешенном состоянии) в среде, содержащей по существу от 0,1 до 6 об.% кислорода (обычный технический прием "выжигания" кислорода воздуха, которым до подрыва заполнена взрывная камера, указанное количество кислорода действительно мало сказывается на выход ДНА - очень подробно изучено и описано в [20]) и до баланса газов инертных к углероду при температуре от 303 до 363 К (описано в [14, 21, 22]).
п.6.: процесс приготовления синтетического алмазсодержащего материала, состоящего по существу из:
а) заполнения сосуда высокого давления зарядом, состоящим по существу, по крайней мере, из одного углеродсодержащего твердого ВВ или смеси углеродсодержащих твердых ВВ, указанный заряд, имеющий отрицательный кислородный баланс, и среды, состоящей по существу из газов и углеродных ультрадисперсных частиц в виде взвеси в газах, и концентрации от 0,01 до 0,15 кг/м3, указанные газы, состоящие по существу из кислорода в количестве от 0,1 до 6 об.% и до баланса азота или газов инертных к углероду (описано в [1-22]).
б) закрытые сосуды высокого давления и подрыв заряда, возникающий при температуре от 303 до 363 К в отсутствии углеродного материала (без загрузки (добавки) углеродного материала), исключая углеродсодержащее взрывчатое вещество или смеси взрывчатых веществ для того, чтобы образовать синтетический алмазсодержащий материал из продуктов разложения ВВ или смеси ВВ и не получить частицы углерода в среде (обычный технический прием при первом подрыве в газовой среде [1-22] или подрыве в воде [23, 24], или во льду).
с) извлечение синтетического алмазсодержащего материала (различные способы описаны во множестве источников [6, 9, 10].
п.7.: процесс, заявленный в п.6., в котором взрывчатое вещество или взрывчатые вещества выбираются из группы, состоящей из октогена, тринитротриаминобензола, гексогена и смеси гексогена и тротила (все эти ВВ и их смеси широко и тщательно исследовались и описаны в [1-24], особенно в работах [14, 15, 17].
Таким образом, все признаки изобретения, изложенные в патенте [4], широко известны и описаны как порознь, так и вместе задолго до подачи материалов, описанных в патенте [4].
В то же время, алмаз - содержащий материал, описанный в патенте [4], представляет собой обычные ультрадисперсные алмазы детонационного синтеза, полученные по стандартному способу.
Авторы приводят данные, что их очищенные ДНА могут иметь до 5% золы (несгораемого остатка) и, по их мнению, это не мешает использовать алмазы в различных областях применения. Однако это не так, для таких процессов как электрохимическое золочение, серебрение и платинирование наличие большого количества неорганических примесей нежелательно. Крайне нежелательно использование таких ДНА в операциях финишного и суперфинишного полирования, для модификации магнитозаписывающего слоя, в плазменном нанесении металл алмазного покрытия, в качестве катализаторов, для получения материалов для полупроводниковой техники, в медицине и биологии и т.д.
Для таких сложных иерархических структур чистота продукта в обычном понимании (количество и виды примесей) является, конечно, важным фактором для дальнейшего использования такого продукта, но только в очень простых технологиях. Для многих процессов имеет значение не только формальная чистота ДНА, но и агрегатное состояние, количество и соотношение поверхностных функциональных групп ДНА определяются отнюдь не только условиями синтеза, но, в определяющей степени, выбранным вариантом химической очистки и последующим модифицированием поверхности частиц ДНА.
Более того, сам процесс получения, достаточно подробно описанный в [4], достаточно прост, наиболее сложны как раз следующие операции. Низкое качество ДНА по этому патенту определяется, в первую очередь, неэффективной химической очисткой - кипячением алмазной шихты в соляной кислоте и последующим двухчасовым нагреванием при 250°С в смеси концентрированных азотной и серной кислот.
Несмотря на обилие работ об алмазсодержащих материалах, синтезированных с использованием энергии разложения взрывчатых веществ, а также особенных процессов получения этих материалов, они не могут раскрыть технические решения проблем, которые могли бы создать базу для эффективной, экономичной и экологически безопасной технологии промышленного производства устойчивых суспензий ДНА.
В качестве прототипа выбран способ стабилизации водных суспензий детонационных наноалмазов, образованной путем детонации углеродсодержащего взрывчатого вещества или смеси взрывчатых веществ с отрицательным кислородным балансом в замкнутом объеме в неокислительной среде, с последующей химической очисткой наноалмазов в окислительной среде, содержащей нитрат- и/или нитрит-ионы, и водной промывкой, с помощью диспергаторов [25]. В качестве диспергаторов были использованы поверхностно-активные вещества (ПАВ) - неонол (алкилфенол) и пирофосфат калия.
Использование диспергаторов не приводит к улучшению стабильности частиц детонационных наноалмазов. Добавка небольшого количества неола проводит к моментальной коагуляции частиц суспензии ДНА. Добавка пирофосфата калия при концентрациях от 0,1 до 0,0001% (по массе) способствует не стабилизации, а агрегации частиц ДНА. В настоящее время не существует стабильных водных суспензий наноалмазов. С течением времени, 1-6 месяцев, происходит укрупнение агрегатов и их выпадение в осадок.
Задача, на решение которой направлено предлагаемое изобретение, состоит в разработке способа стабилизации суспензий детонационных наноалмазов, позволяющего создать алмазсодержащий материал с повышенной стабибильностью частиц ДНА без их агрегации в суспензии длительное время.
Раскрытие изобретения.
Предложен способ получения стабильной суспензии детонационных наноалмазов, образованной путем детонации углеродсодержащего взрывчатого вещества или смеси взрывчатых веществ с отрицательным кислородным балансом в замкнутом объеме в неокислительной среде, с последующей химической очисткой наноалмазов в окислительной среде, содержащей нитрат- и/или нитрит-ионы, и водной промывкой, в котором, согласно изобретению, водную суспензию детонационных наноалмазов с рН 2,6-6,9 обрабатывают аммиаком до рН 7,1-12, нагревают до температуры 150-200°С в течение 1,0-120 минут с образованием в воде коллоидных частиц детонационных наноалмазов, 50-80% которых имеет размер 10-100 нм, а удельная площадь поверхности наноалмаза составляет 450-500 м2.
Анализ многочисленных работ [1-24], приведенных выше, убедительно доказывает, что размеры первичных кристаллов ДНА 4-6 нм (узкомодальное распределение в районе 4,3 нм) получаются всегда, когда есть условия для получения алмазной фазы вообще. Кроме того, на самом деле объем пор (0,3-1,0 мм3/г), их диаметр, форма кристаллов, параметр кристаллической решетки, уровень микронапряжений в кристалле алмаза (~10 ГПа), комбинация кубического кристалла алмаза с аморфной фазой, диэлектрические свойства поверхности ДНА, поверхностные и внутренние примеси практически не зависят от способа получения УДА и изменяются в довольно узком диапазоне (в отличие от утверждений в патенте [4]. А вот вид, количество функциональных групп, их соотношение, размер первичных агрегатов и последующая их организация и в целом - состояние поверхности ДНА, очень сильно зависят от метода химической очистки и последующих операций. По существу именно эти операции и определяют то или иное качество очищенных ДНА, степень их пригодности или напротив - непригодности для различных областей использования.
Так, при очистке в среде хлорной и соляной кислот появляются различные хлорпроизводные, включая хлорид-ион на поверхности ДНА; при обработке хромовыми смесями в серной кислоте - хромсодержащие группы, сульфаты, сульфиты и сульфогруппы; при обработке олеумно-азотными смесями - нитрат-, нитрит- и нитрогруппы, сульфогруппы различной природы; при обработке перекисью водорода и озоном - большое количество кислородсодержащих групп; а при обработке азотной кислотой - нитрат-, нитрит- и нитрогруппы. Кроме того, за счет сильного адсорбционного воздействия в сложной структуре агрегатов ДНА удерживаются кислотные остатки жидкофазного окислителя. Однако привитые на поверхности функциональные группы и адсорбционно удерживаемые кислотные остатки могут мешать использованию ДНА в ряде процессов: в некоторых процессах золочения, платинирования, серебрения и т.д., в некоторых процессах финишного полирования, в медицинской и биологической практике и т.д.
Наиболее совершенным, технологичным и экологичным является способ очистки ДНА от примесей с использованием разбавленной азотной кислоты при высокой температуре (до 280°С) и давлении (до 100 атм) [26]. Такая реакционная суспензия АШ в разбавленной азотной кислоте однородна, коллоидностабильна, высокодисперсна и маловязка. В ходе реакции обеспечивается свободный доступ окислителя к частицам твердой фазы и практически снимаются диффузионные ограничения на скорость процесса. Чистота ДНА после такого процесса может достичь 99,5%.
Чрезвычайно актуальным является избавиться как от избыточной кислотности ДНА, так и от присутствия нежелательных функциональных групп.
Кроме того, препятствием эффективного использования ДНА является относительно высокая агрегативность этих алмазов: в сухом состоянии (общепринятом для потребителей ДНА) агрегаты доходят до 2000-3000 мкм, в лучших из известных суспензий (без промежуточного выделения ДНА в виде сухого порошка) ~350-500 нм.
Т.о. две крупные проблемы были решены следующим образом: при обработке АШ любой окислительной средой, содержащей азотную кислоту или окислы азота, как уже отмечалось, происходит насыщение поверхности ДНА нитратными, нитритными и нитрогруппами, остаются адсорбционно связанными с поверхностью пор агрегатов ДНА кислотные азотсодержащие остатки и окислы азота.
После высокотемпературной обработки ДНА неоднократно промывают водой, однако адсорбционно связанные кислотные группировки всегда остаются в ДНА. Даже при очистке наиболее легко удаляемой азотной кислоты после 5-6 промывок устойчиво связанной азотной (и азотистой) кислоты в ДНА остается 0,3-0,4 мас.% от веса ДНА.
Если к водной суспензии ДНА с рН от 2,6 до 6,9 добавить такое количество газообразного или жидкого (в воде или в сжиженном виде) аммиака, чтобы после установления равновесия рН получившейся суспензии находился в рамках от 7,1 до 12, то все. кислотные остатки будут связаны в легко растворимые легко удаляемые соли аммиака.
А в случае очистки АШ только азотной кислотой или окислами азота - будут только нитрат аммония и нитрит аммония.
При нагревании такой суспензии при температуре от 150 до 200°С соли азотной и азотистой кислот начинают бурно разлагаться с выделением воды и газообразных продуктов по реакциям (1) и (2), закиси азота по реакции (3), а окислы азота по реакции (4), находясь в контакте с избыточным аммиаком, за счет химических реакций превращаются в азот и воду:
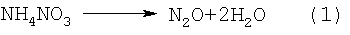
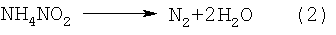
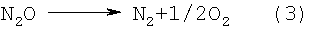
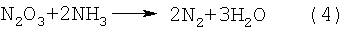
ДНА в этих процессах выступает еще в качестве катализатора процессов разложения аммония и его солей, сдвигая эти реакции в сторону более низких температур. Так, реакция интенсивного разложения NН4NО3 начинается не при 170°С (в обычных условиях), а уже при 150°С; реакция разложения NH4NO2 также начинается при 50°С, а не при 70°С. Разложение солей, поверхностных нитрогрупп в присутствии аммиака - по сути цепной процесс. В результате быстрого и бурного газовыделения происходит разрушение (разрыв) агрегатов УДА размером 350-500 нм на агрегаты от 10 до 100 нм - таких агрегатов в водной суспензии находится от 50 до 80% по количеству. При этом все продукты - безвредные газы (азот и лишний аммиак) или вода.
Удельная площадь таких ДНА, естественно, на 1-2 порядка больше удельной площади предыдущей суспензии. Однако общепринятая методика измерения, определяемая по изотермам по Brunauer-Emmet-Teller (BET) тепловой десорбции азота или аргона предполагает наличие ДНА в виде порошка. А УДА в виде порошка всегда агломерируется, но даже в этом случае удельная площадь полученных алмазов находится в диапазоне от 450 до 550 м2/г.
После охлаждения суспензии ДНА и ее 1-2 водных промывок получается очень устойчивая коллоидно-стабильная водная суспензия ДНА (осадок ДНА не выпадает в течение 24 месяцев), что чрезвычайно расширяет ряд ее использования во всех возможных процессах.
Использовать суспензию ДНА с рН менее 2,6 для нейтрализации аммиаком после химической очистки нецелесообразно из-за большого расхода аммиака на нейтрализацию кислот и большего времени выдержки при высоких температурах для разложения этих солей. В то же время добиваться максимально полной отмывки УДА от кислоты (непременно добиваясь рН ~7) тоже является нецелесообразным из-за больших затрат времени на этот процесс.
Нейтрализация аммиаком должна обеспечить создание щелочной среды в суспензии, гарантирующий небольшой избыток аммиака, т.е. рН 7,1-12, для того чтобы лишний аммиак, с одной стороны, поддерживал необратимость реакций (1), (2) и (4), а с другой стороны, его хватило бы на реакцию с кислотными остатками и кислыми газами, находящимися в глубоких порах ДНА, доступ к которым открывается за счет увеличения диффузии при высоких температурах (150-200°С) и, особенно, при разрушении газами агрегатов ДНА.
Создание рН более 12 при введении аммиака (в газообразном виде, в виде раствора в воде или в сжиженном виде) не целесообразно вследствие лишнего расхода аммиака и развития высоких давлений при ведении процесса.
Обычно давление при ведении процесса на 10-15% превышает давление водяного пара при соответствующей температуре.
Интенсивно процессы распада аммиачных солей и реакции окислов азота с аммиаком начинаются в наших условиях при 150°С. Поддерживать температуру свыше 200°С не целесообразно из-за нарастания давления в аппаратуре и, следовательно, повышенных требований к аппаратуре.
Если при 200°С процесс распада солей и реакции аммиака с остаточными окислами азота уже завершается в течение 1 мин, то при 150°С для гарантии завершения всех процессов необходимо время в 120 мин. При уменьшении времени ниже 1 мин при 200°С не успеют завершиться все необходимые нам реакции (1)-(4), а увеличение времени сверх 120 мин при 150°С не целесообразно по экономическим соображениям.
Для лучшего понимания настоящего изобретения по п.1 приводятся конкретные примеры.
Пример 1. Иллюстрирует типовое протекание процесса аммиачного термолиза ДНА в периодических условиях.
В титановый автоклав емкостью 400 мл, снабженный датчиком давления (ДТ), термопарой и системой стравливания газов, помещают 300 мл 8%-ной водной суспензии ДНА с рН 8,0, полученной предварительной нейтрализацией 300 мл 8%-ной водной кислой суспензии ДНА (фаза химической очистки - азотная кислота) с рН 2,6 газообразным аммиаком. Автоклав после герметизации помещают в баню со сплавом Розе, и поднимают температуру содержимого автоклава до 200°С, и выдерживают 5 минут. Давление в автоклаве в конце выдержки составляет 38 атм. После выдержки автоклав охлаждают до 20-30°С, избыточное давление газов стравливают. Получение ДНА в виде водной суспензии 2 раза промывают водой, понижая рН среды до 7,1. Полученная суспензия устойчива во времени (не появляется осадок агрегатированных ДНА) не менее 24 месяца. До обработки аммиаком при высокой температуре и давлении содержание несгораемых примесей в ДНА составляло 0,95 мас.%, а после аммиачной термообработки - 0,37 мас.%, 72% алмазов имеют размер от 10 до 100 нм, удельная поверхность сухого порошка ДНА (метод БЕТ) 515±10 нм.
Пример 2. Иллюстрирует типовое протекание процесса аммиачного термолиза ДНА и непрерывного осуществления способа.
Водную 9%-ную суспензию с рН 3, полученную после химической очистки алмазсодержащей шихты и частичной отмывки водой, нейтрализуют водным 25%-ным раствором аммиака до рН 7,2. Затем эту суспензию с помощью насоса-дозатора НД-2,5/400 прокачивают через однолитровый трубчатый реактор вытеснения со скоростью 1 л/час, обеспечивая одночасовое пребывание реакционной массы в зоне рабочих температур. Давление поддерживают на уровне 35 атм. После четырехлитрового титанового сборника реакционная масса дросселируется в открытую емкость, откуда суспензия НА с рН 7,05 отбирается на стадию протирки. Двухкратная протирка водой от солей приводит к покрытию коллоидно-устойчивой суспензии в течение 30 месяцев. До аммиачного термолиза содержание несгораемых примесей в ДНА составило 0,7 мас.%, а после обработки аммиаком согласно изображению содержание несгораемых примесей упало до 0,23 мас.% 61% алмазов имеют размер от 10 до 100 нм, удельная поверхность сухого порошка 490±10 нм.
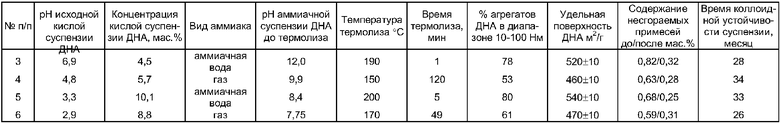
Остальные примеры приведены в таблице.
Алмазосодержащий материал, полученный по предлагаемому способу, обладает комплексом свойств, которые позволяют ему быть включенным в различные композиционные материалы и покрытия. Способ отличается безопасностью, надежностью, улучшенными техническими, экономическими и экологическими параметрами и позволяет организовать широкомасштабное производство стабильного алмазсодержащего материала.
Источники информации
1) Adadurov, G.A. et al. "Diamonds Obtained…, "The Physics of Pulse Pressures Proceedings, Research Institute of Physical and Radio Engineering Measurements, Moscow, 1979, p.157-161.
2) Волков К.В., Даниленко В.В., Един В.П. Синтез алмаза из углерода продуктов детонации ВВ, ФГВ, 1990, т.26, №3, с.123-125.
3) Британский патент 1154633, опубл. 1969.
4) Патент США 5861349, опубл. 1999.
5) А.Л.Верещагин, В.Ф.Комаров, В.М.Мастихин, В.В.Новоселов, Л.А.Петрова, И.И.Золотухина, Н.В.Бычин, К.С.Барабошкин, Е.А.Петров. "Исследование свойств алмазной фазы детонационного синтеза". В кн.: Докл.5 Всесоюзного совещания по детонации, т.1., Красноярск, 1991, с.99-103.
6) Т.М.Губаревич, Л.С.Кулагина, И.С.Ларионова. "Особенности элементного состава углеродных продуктов детонационного синтеза", там же, с.130-134.
7) Петрова Л.А., Верещагин А.Л., Новоселов В.В., Брыляков П.М., Шейн Н.В. "Исследование поверхностных групп алмазоподобной фазы углерода". Сверхтвердые материалы, 1989, №4, с.3-5.
8) Саввакин Г.И., Котко В.А., Островская Н.Ф., Курдюшов А.В. Структура ультрадисперсных углеродных фаз, образующихся из углеродсодержащих соединений в сильно неравновесных условиях. Порошковая металлургия, 1988, №10, с.78-82.
9) К.С.Барабошкин, Т.М.Губаревич, В.Ф.Комаров. "Особенности текстуры порошков конденсированного алмаз - содержащего углерода". Коллоидный журнал, 1992, т.54, №6, с.9-12.
10) Патент РФ. №2046094, МПК С01В 31/04, опубл. 1995.10.20.
11) Т.М.Губаревич, Н.М.Костюкова, P.P.Сатаев, Л.В.Фомина. "Исследование микропримесного состава ультрадисперсных алмазов". Сверхтвердые материалы, 1991, №5, с.30-34.
12) Л.А.Петрова, В.В.Новоселов, П.М.Брыляков и др. "Исследование химического состава поверхности ультрадисперсного алмаза детонационного синтеза". В сб. докл. 9 Всесоюзного симпозиума по горению и взрыву, г.Суздаль (СССР), 1989, с.14-18.
13) А.В.Ананьин, О.Н.Бреусов, В.Н.Дробышев, Г.Е.Ивангихин, А.И.Шунина. "Термографические и рентгенографические исследования свойств алмазов, синтеза". Сверхтвердые материалы, 1986, №5, C.11-14,
14) Лямкин А.И., Петров Е.А., Ершов А.П. и др. Получение алмазов из взрывчатых веществ, ДАН СССР, 1988, т.302, №3, с.611-613.
15) Титов В.М., Анисичкин В.М., Мальков И.Ю. Исследование процесса синтеза ультрадисперсного алмаза в детонационных ваннах. Физика горения и взрыва, 1989 г., №3, с.117-126.
16) В.Ф.Анисичкин. "Об особенностях ударно-волнового разложения и синтеза алмаза из ароматических соединений". В кн. Докл.5 Всесоюзного совещания по детонации, т.1, Красноярск, 1991, с.20-26.
17) В.Ф.Анисичкин, И.Ю.Мальков, Ф.А.Сагдеев. "Синтез алмаза при детонации ароматических нитросоединений", там же, с.27-30.
18) Н.В.Козырев, Г.В.Сакович, Сен Чел Су, М.С.Штейн. "Исследование процесса синтеза ультрадисперсных алмазов методом меченых атомов", там же, с.176-179.
19) А.Ю.Бабушкин, А.И.Лямкин, A.M.Ставер. "Влияние газов на выход конденсированных продуктов детонации углеродсодержащих взрывчатых веществ", там же, с.84-87.
20) И.Г.Кузьмин, А.И.Лямкин, A.M.Ставер. "Экспериментальное изучение состава газообразных продуктов детонации конденсированных ВВ в различных атмосферах". В кн.: "Ультрадисперсные материалы, получение и свойства", Межвузовский сборник, Красноярск, 1990, с.23-28
21) Л.И.Акимова, С.А.Губин, В.Д.Одинцов, В.И.Пилекин. "Детонация взрывчатых веществ с образованием алмаза". В кн.: Докл. 5 Всесоюзного совещания по детонации, Красноярск, 1991, т.1, с.14-19.
22) Бабушкин А.Ю., Лямкин А.И., Ставер A.M. "Особенности получения ультрадисперсного материала на основе углерода из взрывчатых веществ", там же, с.81-83.
23) Авторское свидетельство №1169304, от 28.04.82.
24) Патент РФ №2009999. МПК С01В 31/06, опубл. 1994.03.30.
25) Л.В.Агибалова, А.П.Возняковский, В.Ю.Долматов, В.В.Клюбин. «Структура суспензий ультрадисперсных алмазов взрывного синтеза (наноалмазов). - Сверхтвердые материалы, 1998, №4. С.87-95 - прототип.
26) Патент РФ №2109683, МПК С01В 31/06, опубл. 1998.04.27.
| название | год | авторы | номер документа |
|---|---|---|---|
| НАНОАЛМАЗ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2005 |
|
RU2348580C1 |
| УГЛЕРОДСОДЕРЖАЩАЯ НАНОЧАСТИЦА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2009 |
|
RU2424185C2 |
| АЛМАЗ-УГЛЕРОДНЫЙ МАТЕРИАЛ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2005 |
|
RU2359902C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭЛЕКТРОХИМИЧЕСКОГО СЕРЕБРЯНОГО ПОКРЫТИЯ | 2015 |
|
RU2599471C1 |
| Способ получения детонационных наноалмазов | 2019 |
|
RU2711599C1 |
| Способ получения детонационных наноалмазов | 2019 |
|
RU2703212C1 |
| СИНТЕТИЧЕСКИЙ УГЛЕРОДНЫЙ АЛМАЗСОДЕРЖАЩИЙ МАТЕРИАЛ | 1993 |
|
RU2046094C1 |
| Способ получения детонационных наноалмазов | 2019 |
|
RU2712551C1 |
| АЛМАЗСОДЕРЖАЩЕЕ ВЕЩЕСТВО И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1991 |
|
RU2051092C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОАЛМАЗОВ | 2003 |
|
RU2230702C1 |
Изобретение относится к области неорганической химии углерода и может быть использовано в гальванике, в приготовлении полировальных систем, в полимерной и плазменной химии, в электрохимическом катализе, при получении магнитных носителей информации, для изготовления высокоактивных адсорбентов, в биохимическом синтезе, для приготовления присадок для масел, смазок и смазочно-охлаждающих жидкостей. Суспензию детонационных наноалмазов (ДНА) получают путем детонации углеродсодержащего взрывчатого вещества или смеси взрывчатых веществ с отрицательным кислородным балансом в замкнутом объеме в неокислительной среде с последующей химической очисткой наноалмазов в окислительной среде, содержащей нитрат- и/или нитрит-ионы, и водной промывкой. После химической очистки водную суспензию детонационных наноалмазов с рН 2,6-6,9 обрабатывают аммиаком до рН 7,1-12, нагревают до температуры 150-200°С в течение 1,0-120 минут с образованием в воде коллоидных частиц детонационных наноалмазов, 50-80% которых имеет размер 10-100 нм при удельной площади поверхности наноалмазов 450-500 м2/г. Изобретение позволяет создать алмазосодержащий материал с повышенной стабильностью частиц ДНА без их агрегации в суспензии длительное время. 2 з.п. ф-лы, 1 табл.
1. Способ получения стабильной суспензии детонационных наноалмазов, образованной путем детонации углеродсодержащего взрывчатого вещества или смеси взрывчатых веществ с отрицательным кислородным балансом в замкнутом объеме в неокислительной среде с последующей химической очисткой наноалмазов в окислительной среде, содержащей нитрат- и/или нитрит-ионы, и водной промывкой, отличающийся тем, что после химической очистки водную суспензию детонационных наноалмазов с рН 2,6-6,9 обрабатывают аммиаком до рН 7,1-12, нагревают до температуры 150-200°С в течение 1,0-120 мин с образованием в воде коллоидных частиц детонационных наноалмазов, 50-80% которых имеет размер 10-100 нм при удельной площади поверхности наноалмазов 450-500 м2/г.
2. Способ по п.1, отличающийся тем, что используют газообразный аммиак.
3. Способ по п.1, отличающийся тем, что используют раствор водного аммиака.
| SPITSYN B.V | |||
| et al | |||
| Inroad to modification of detonation nanodiamond, "Diamond and Related Materials", 2006, vol.15, no.2-3, p.p.296-299 | |||
| АГИБАЛОВА Л.В | |||
| и др | |||
| Структура суспензий ультрадисперсных алмазов взрывного синтеза (наноалмазов) | |||
| Сверхтвердые материалы, 1998, №4, с.87-95 | |||
| СПОСОБ ПОДГОТОВКИ ПОРОШКОВ НАНОАЛМАЗА ДЛЯ ПОЛУЧЕНИЯ УСТОЙЧИВЫХ СУСПЕНЗИЙ | 2004 |
|
RU2302994C2 |

